
Sensitizing phosphorescent and radical emitters via triplet energy translation from CsPbBr3 nanocrystals†
Zongwei
Chen
ab,
Guijie
Liang
*c and
Kaifeng
Wu
 *b
*b
aHenan Institute of Advanced Technology, Zhengzhou University, Zhengzhou 450052, P. R. China
bState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, P. R. China. E-mail: kwu@dicp.ac.cn
cHubei Key Laboratory of Low Dimensional Optoelectronic Materials and Devices, Hubei University of Arts and Science, Xiangyang 441053, P. R. China. E-mail: guijie-liang@hbuas.edu.cn
First published on 18th October 2021
Abstract
Colloidal semiconductor nanocrystals can effectively sensitize surface-attached molecular species via triplet energy transfer. These sensitized molecular triplets are capable of triggering a variety of subsequent processes such as triplet-fusion upconversion, singlet oxygen generation and organic synthesis. Here we demonstrate that molecular triplets can also sensitize non-conventional light-emitting materials such as phosphorescent and radical molecules. Specifically, photoexcitation of CsPbBr3 perovskite nanocrystals resulted in triplet sensitization of naphthalene ligands, which further translated the energy into 2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphine platinum(II) or (4-(4′-triphenylamine-yl)-tetrachlorophenyl)bis-(pentachlorophenyl)-methyl in the bulk solution that can efficiently emit photons. As a result, the light absorption of phosphorescent and radical molecules is strongly enhanced and photon emission is effectively down-shifted from absorption. This new energy transduction scheme may prove useful in devices such as luminescent solar concentrators.
Introduction
Owing to their unique spin properties and long lifetimes, the spin-triplet excited states of molecules engage in many important applications.1,2 Because most of the molecular triplets are optically “dark” states, it is essential to develop triplet sensitizers that can translate their excitation energy to triplet acceptors.3,4 Recent studies have identified colloidal semiconductor nanocrystals (NCs) as efficient sensitizers.5–20 Compared to their organic counterparts, NCs have much smaller bright-dark state energy splitting because of a weak electron–hole exchange energy of only a few to 10s meV,21,22 which can in principle minimize the intersystem crossing (ISC) energy loss in sensitizers.10,15 Moreover, the facile spectral tunability of NCs across the ultraviolet (UV) to infrared (IR) regions through their compositions and sizes greatly expedites the choice and design of NC sensitizers for molecules with varying triplet state energies.8 Additionally, the large surface to volume ratio of NCs allows for their direct surface functionalization with molecular acceptors,7 which induces very efficient triplet energy transfer.Molecular triplet acceptors anchored on NC surfaces act as triplet transmitters that can trigger a variety of subsequent physical and chemical processes, such as triplet-fusion photon upconversion,5–20 thermally activated delayed photoluminescence,23,24 singlet oxygen generation6,25 and organic transformations.26,27 Still, there is ample room for exploring new interaction schemes and/or functionalities based on these NC–transmitter complexes. For example, they can be utilized to sensitize various generations of materials developed for organic light-emitting diodes (OLEDs), including phosphorescent molecules,28,29 thermally active delayed fluorescence (TADF) molecules30,31 and radical molecules,32–34 through Dexter-like electron-exchange interactions (Fig. 1). Phosphorescent molecules for OLEDs are often organometallic complexes made from porphine derivatives and their light absorption spectrum typically displays a so-called Q-band in the visible region and a Soret-band in the UV region;35 the large gap between these two bands can be filled by the absorption of NC–transmitter complexes. Similarly, efficient radical emitters reported to date usually contains a triphenylmethyl unit whose absorption spectrum displays a weak doublet absorption band in the visible region and the other strong one in the UV region.34 Therefore, sensitization using NC–transmitter complexes can in general strongly enhance the light-harvesting capability of these OLED molecules. Meanwhile, photons emitted by these molecules are significantly red-shifted compared to photons absorbed by NC–transmitter complexes, a property highly desired for applications such as luminescent solar concentrators35–38 and radiation detectors (scintillators).39
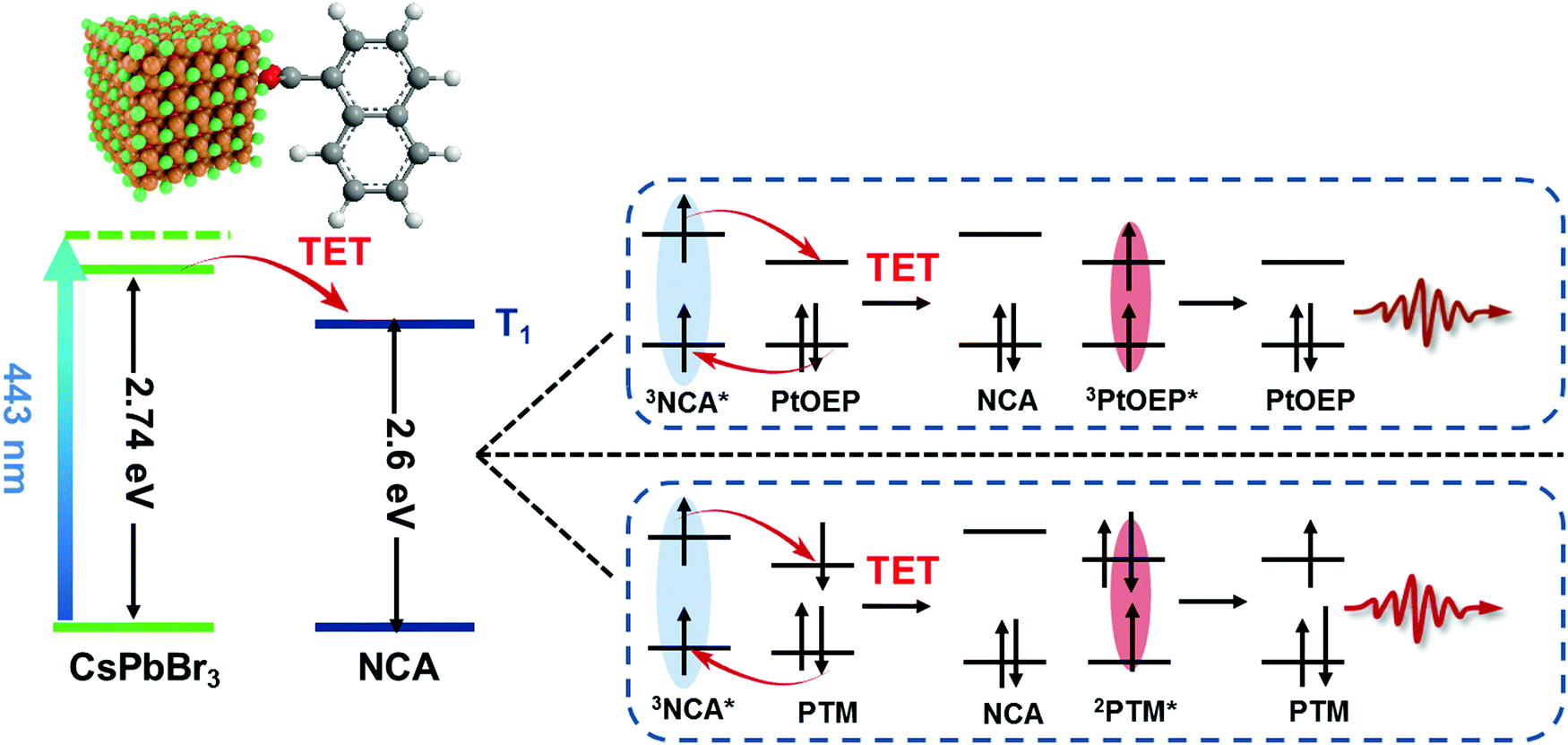 | ||
| Fig. 1 Sensitization of phosphorescent and radical emitters using NC–transmitter complexes. Photoexcitation of CsPbBr3 NCs resulted in efficient triplet energy transfer (TET) to the NCA ligands which further translated the energy into PtOEP or PTM-TPA molecules in the bulk solution. Energy transfer from 3NCA* to PtOEP or PTM-TPA occurs through electron exchange. | ||
Here we demonstrate using static and time-resolved spectroscopy that photoexcitation of CsPbBr3 perovskite nanocrystals resulted in triplet energy transfer to surface-anchored naphthalene transmitters that further translated the energy to a prototypical phosphorescent molecule, 2,3,7,8,12,13,17,18-octaethyl-21H,23H-porphine platinum(II) (PtOEP), or a radical emitter, (4-(4′-triphenylamine-yl)-tetrachlorophenyl)-bis-(pentachlorophenyl)-methyl (PTM-TPA), in the bulk solution. For both systems, the efficiencies of the first and second triplet energy transfer steps were 74% and 98%, respectively, amounting to an overall sensitization efficiency of 72%.
Results and discussion
Triplet energy transfer from CsPbBr3 to NCA
We synthesized CsPbBr3 NCs with an average diameter of 3.2 nm using literature methods;40 see the ESI† for details. The size is much smaller than the Bohr exciton diameter of ∼7 nm for CsPbBr3,41 resulting in a strong quantum confinement effect. Our recent studies have established that quantum confinement can facilitate triplet energy transfer (TET) from perovskite NCs mainly by enhancing the donor–acceptor electronic coupling.13–15 The first exciton absorption peak of these NCs is at ∼453 nm (Fig. 2a), which is strongly blue-shifted compared to the absorption onset of ∼520 nm for bulk CsPbBr3. The triplet transmitter used in this study is 1-naphthalene carboxylic acid (NCA) with a triplet energy of 2.6 eV, which has been employed as efficient triplet acceptors for CsPbBr3 NCs in previous studies.14,15,42 NCA transmitters were functionalized onto NC surfaces using a simple ligand exchange method; see the ESI.† On the basis of the absorption spectrum in Fig. 2a, the molar ratio of NCA:NC was 561 using the extinction coefficients of NCA and CsPbBr3 NCs. The average number of NCA bound to each NC is lower than this number, due to the slight solubility of NCA in NC-toluene solution.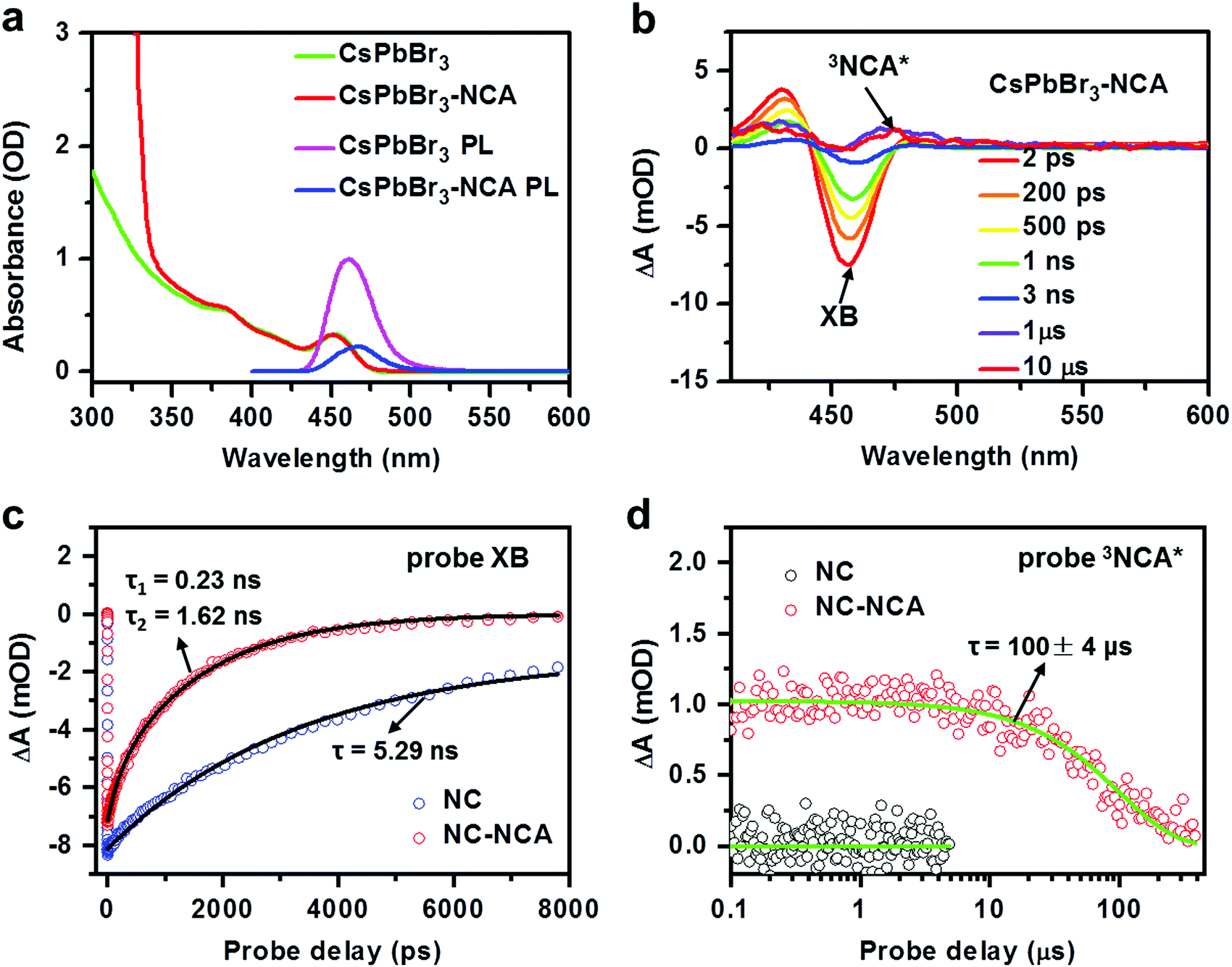 | ||
| Fig. 2 (a) Absorption spectra of CsPbBr3NCs with (red) and without (green) surface-functionalized NCA molecules dispersed in toluene. PL spectra of CsPbBr3NCs with (blue) and without (pink) surface-functionalized NCA molecules dispersed in toluene acquired using 442 nm excitation. (b) TA spectra of CsPbBr3–NCA complexes at selected delays from 2 ps to 10 μs following 400 nm excitation. The NC exciton bleach (XB) and NCA triplet (3NCA*) features are indicated. (c) TA kinetics of the NC (blue open circles) and NC–NCA (red open circles) probed at the XB center (∼450 nm). The black solid lines are their exponential fits. (d) TA kinetics of NC (black open circles) and NC–NCA (red open circles) probed at the 3NCA* feature (470 nm). The green solid lines are their fits. | ||
The photoluminescence (PL) quantum yield of the CsPbBr3 NCs at 442 nm excitation was estimated as 61% (see the ESI† for details). Upon NCA functionalization, the PL was quenched by 78% (Fig. 2a). In our prior studies, we have systematically investigated the mechanisms of TET from perovskite NCs to surface-anchored acceptors, and revealed the influences of the NC size and composition and NC-acceptor interfacial energetics on TET.13,43 According to these studies, the quenching observed here is caused by TET from CsPbBr3 NCs to NCA, while charge transfer and singlet energy transfer pathways are energetically disallowed,43 although a possibility of endothermic charge-transfer-mediated TET cannot be excluded.44 In order to directly observe TET, we performed transient absorption measurements from sub-ps to sub-ms timescales; see the ESI† for details. Fig. 2b presents the TA spectra of NC–NCA complexes at selected delays from 2 ps to 10 μs following 400 nm excitation which selectively excites the NCs. Within a few ns, the TA spectra are dominated by a NC exciton bleach (XB) feature at ∼453 nm, arising from state-filling effects, as well as some photoinduced absorption features at higher energy induced by exciton-activated forbidden transitions and/or the biexcitonic Stark effect.13,43 These features gradually evolve into broad-band absorption features assignable to the NCA triplets (3NCA*).15,43 The kinetics plotted in Fig. 2c further confirm the accelerated XB recovery in NC–NCA complexes as compared to free NCs. By fitting the XB kinetics, we find a single-exponential lifetime of 5.3 ± 0.2 ns for free NCs and double-exponential lifetimes of 0.23 ± 0.02 ns (20%) and 1.62 ± 0.03 ns (80%) for NC–NCA complexes. The amplitude-averaged lifetime of NC–NCA is 1.35 ± 0.03 ns. From the above constants, we can calculate an average TET time of 1.8 ± 0.1 ns and a TET yield of 74%; see the ESI† for details. The TET yield is consistent with the PL quenching efficiency obtained in Fig. 2a (∼78%).
The lifetime of 3NCA* is 100 ± 4 μs (Fig. 2d). This lifetime is shorter than the millisecond-lifetime of free NCA triplets reported in the literature.1 A possible reason is that the triplet energy of NCA is only ∼0.14 eV lower than the NC exciton energy, and therefore there exists a possibility of thermally activated reverse TET from NCA triplets to NCs, shortening the triplet lifetime.23,45 Indeed, we can observe a persistent weak dip at ∼450 nm in the μs-TA spectra in Fig. 2b, which can be attributed to a quasi-equilibrium between the NC and NCA excited states established through the bi-directional TET. Relatedly, our recent studies reported TET and reverse TET between CsPbBr3 NCs and phenanthrene transmitters (triplet energy of 2.64 eV, similar to NCA), and all the details of using TA and transient PL to establish rTET can be found therein.46,47 Nevertheless, a lifetime of ∼100 μs is still four orders-of-magnitude longer than the exciton lifetime of free NCs. The short exciton lifetime of perovskite NCs is insufficient for diffusion-controlled charge/energy transfer involved in many applications. This issue is now overcome by storing their exciton energy in the form of long-lived NCA triplets,6 which is the essential motivation of using triplet transmitters for perovskite NCs. In the following, we will utilize the long-lived NC–transmitter triplet excited states to sensitize PtOEP and PTM-TPA emitters in the bulk solution.
Triplet energy transfer from NC–NCA to PtOEP
Fig. 3a plots the absorption spectra of CsPbBr3 NC–NCA complexes, PtOEP molecules and their mixture (NC–NCA//PtOEP) in toluene. The concentrations of NC–NCA complexes and PtOEP molecules are ∼0.66 μM and ∼0.21 mM, respectively. The two absorption peaks of PtOEP at ∼533 and 500 nm are the vibronic progressions of the Q-band, while the Soret-band is situated in the UV (<400 nm). Clearly, the absorption of CsPbBr3NCs complements the transparency window of PtOEP within the range of 420–480 nm. The PL spectrum of PtOEP shows vibronic progressions in the range of ∼620–750 nm (Fig. 3a). The emission peak at 642 nm is strongly red-shifted from the absorption peak of the Q-band (533 nm), because the emission results from the triplet rather than the singlet state of the Q-band. The triplet state is barely discernable on the absorption emission because of its very weak oscillator strength. Strong spin–orbit coupling enabled by the Pt atom results in near unity-yield ISC from the singlet to the triplet excited state. For the same reason, phosphorescence emission from the triplet excited state to the singlet ground state is also made efficient. The emission quantum yield was estimated to be 41% (see the ESI† for details). It is the efficient phosphorescence emission that has made PtOEP a prototype for modern OLED materials;28 in OLEDs, current injection leads to a statistical ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for triplets and singlets, and hence utilizing triplet emission is a unique route to obtain efficient OLEDs.
:1 for triplets and singlets, and hence utilizing triplet emission is a unique route to obtain efficient OLEDs.
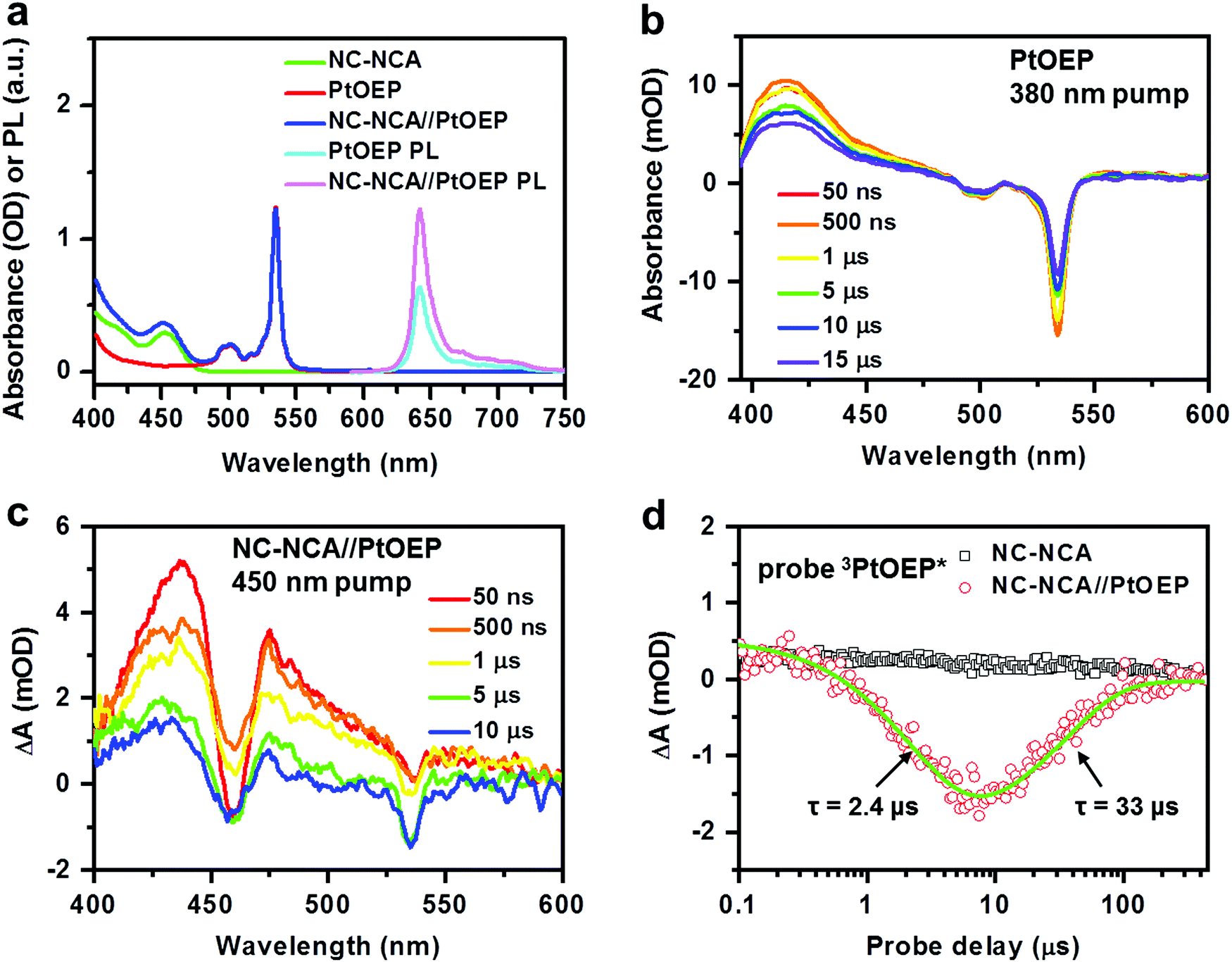 | ||
| Fig. 3 (a) Absorption spectra of CsPbBr3 NC–NCA complexes (green), PtOEP molecules (red) and their mixture (blue) in toluene. The PL spectra of PtOEP (cyan) and NC–NCA//PtOEP (pink) excited at 442 nm are also shown. (b) TA spectra of PtOEP at indicated delays following 380 nm excitation. (c) NC–NCA//PtOEPat indicated delays following 450 nm excitation. (d) TA kinetics probed at 535 nm (the GSB of PtOEP) for NC–NCA (black open square) and NC–NCA//PtOEP (red open circle). The green solid line is a multi-exponential fit to the formation and decay of 3PtOEP* in NC–NCA//PtOEP. | ||
Importantly, when the PtOEP and NC–NCA//PtOEP samples with the same concentration of PtOEP are excited at 440 nm, the mixture displays an enhanced PtOEP emission as compared to pure PtOEP (Fig. 3a). This observation directly evidences energy transfer from CsPbBr3 NC–NCA complexes to PtOEP in the solution. Direct singlet or triplet energy transfer from CsPbBr3 NCs to PtOEP can be excluded, as simple mixing of these two species does not quench the PL of NCs at all, which is consistent with our expectation that the 5 ns exciton lifetime of CsPbBr3 NCs is insufficient for diffusion-controlled energy transfer under typical concentrations.
We performed TA measurements to directly observe energy transfer from CsPbBr3 NC–NCA complexes to PtOEP. The TA spectra of pure PtOEP in acetonitrile under 380 nm excitation are presented in Fig. 3b, which show sharp ground state bleach (GSB) features at 535 and 500 nm and a broad absorptive feature from UV to ∼450 nm that can be assigned to the absorption of PtOEP triplets (3PtOEP*) generated by efficient ISC. The lifetime of 3PtOEP* can be fitted to a single-exponential time constant of 36.8 μs (Fig. S2, ESI†). For the NC–NCA//PtOEP sample excited under the same conditions as those used for NCs, we observe TA spectra that are initially dominated by the absorptive features of 3NCA* and gradually evolve into 3PtOEP*-like spectra (Fig. 3c). The TA kinetics probed at 535 nm for the NC–NCA and NC–NCA//PtOEP samples are compared in Fig. 3d, which clearly reveals the growth and decay of the PtOEP GSB on the μs timescale. A multi-exponential fit indicates the growth and decay time constants of 2.4 and 33 μs, respectively, with the latter being consistent with the lifetime of 3PtOEP*. The 2.4 μs time can be attributed to TET from 3NCA* to ground-state PtOEP. The kinetics probed at the 3NCA* feature (∼470 nm) reveals a consistent result (Fig. S3, ESI†). This energy transfer rate is much smaller than the estimated collision rate of PtOEP molecules with NC–NCA complexes in toluene under our experimental concentrations (see the ESI† for details). Therefore, energy transfer from NC–NCA complexes to PtOEP molecules is limited by the TET step rather than the diffusion process. Nevertheless, because of a long lifetime of 100 μs for 3NCA*, the TET efficiency is near unity (97.6%).
Triplet energy transfer from NC–NCA to PTM-TPA
A similar experiment using PTM-TPA radical emitters as the acceptors has been performed. Fig. 4a shows the absorption spectra of CsPbBr3 NC–NCA complexes, PTM-TPA molecules and their mixture (NC–NCA//PTM-TPA) in toluene. The concentrations of NC–NCA complexes and PTM-TPA molecules are ∼0.66 μM and 0.11 mM, respectively. PTM-TPA displays a weak, broad absorption band centered at ∼650 nm and the another strong absorption band in the UV. According to recent studies, the two bands can be assigned transitions from the ground-state doublet (D0) to the first and second excited-state doublets (D1 and D2, respectively).33,34 The fact that D2 has a much stronger transition intensity than D1 is a direct consequence of structural alternate symmetry.34 Radical units like triphenyl-methyl are alternant hydrocarbon systems in which the singly occupied molecular orbital (SOMO) to lowest unoccupied molecular orbital (LUMO) transition and the highest occupied molecular orbital (HOMO) to SUMO transition are nearly degenerate. For the D1 band, these two transitions are out-of-phase and hence interfere destructively, resulting in a vanishing oscillator strength, whereas the D2 band is a constructive interference with a high oscillator strength.34 The addition of a TPA unit enhances D1 by borrowing the intensity from D2.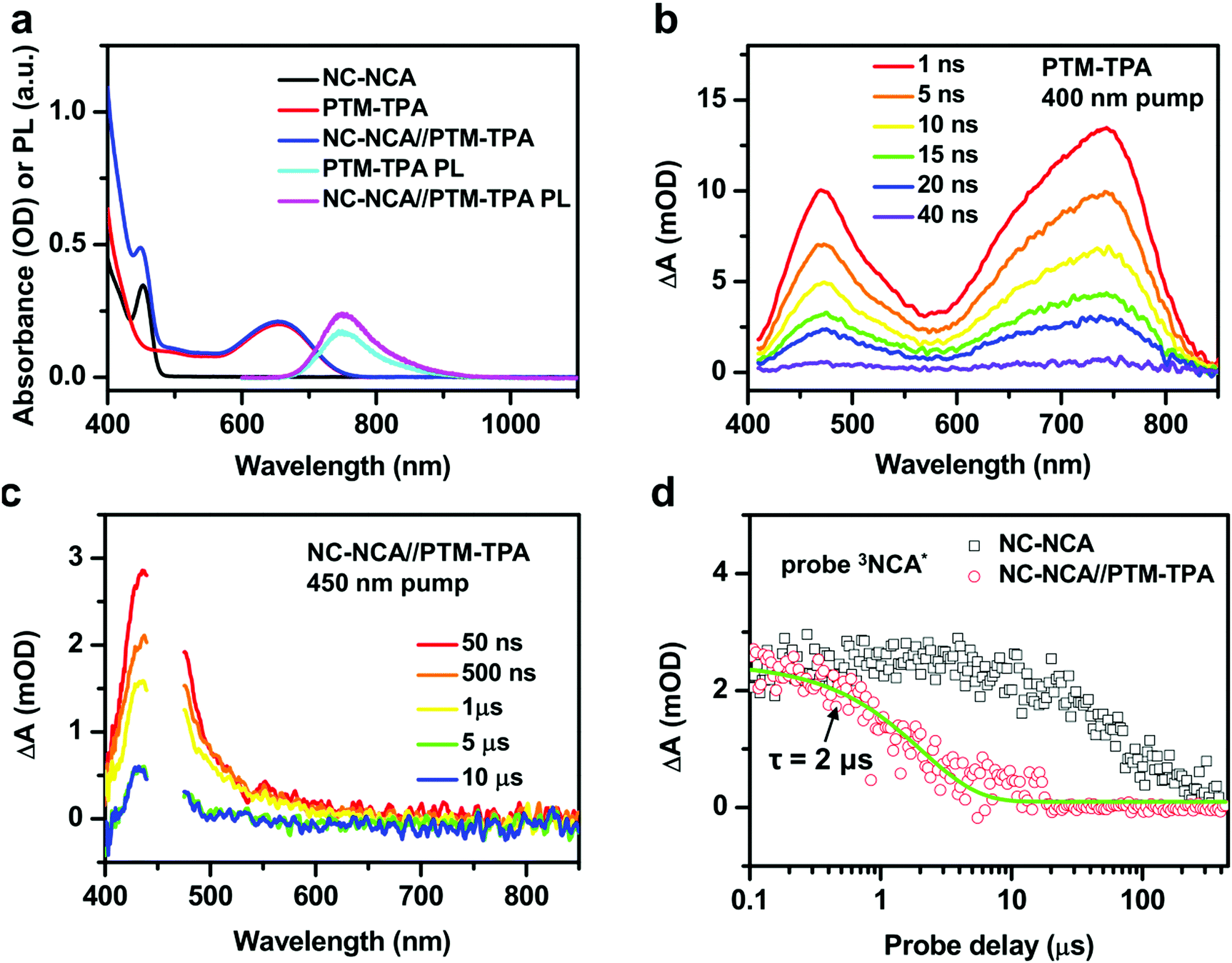 | ||
| Fig. 4 (a) Absorption spectra of CsPbBr3 NC–NCA complexes (black), PTM-TPA molecules (red) and their mixture (blue) in toluene. The PL spectra of PTM-TPA (cyan) and NC–NCA//PTM-TPA (pink) excited at 442 nm are also shown. (b) TA spectra of PTM-TPA at indicated delays following 400 nm excitation. (c) NC–NCA//PTM-TPA at indicated delays following 450 nm excitation. (d) TA kinetics probed at 470 nm (the absorption of 3NCA*) for NC–NCA (black open square) and NC–NCA//PTM-TPA (red open circle). The green solid line is a single-exponential fit to the decay of 3NCA* in NC–NCA//PTM-TPA. | ||
In the NC–NCA//PTM-TPA sample, the gap between D1 and D2 is partially filled by the absorption of CsPbBr3 NCs. The PL spectrum of PTM-TPA is a broad band within ∼700–850 nm (Fig. 4a), arising from the D1 to D0 transition. The emission quantum yield of PTM-TPA was estimated to be 33% (see the ESI† for details). Because of their radical nature, the issue of spin statistics (triplets to singlets 3:1) is circumvented for the radical emitters and the D1 to D0 transition is spin-conserving. For this reason, these radical emitters have attracted strong attention as next-generation materials for efficient OLEDs.32–34,48 The emission of the NC–NCA//PTM-TPA sample is enhanced than that of pure PTM-TPA excited under the same conditions (Fig. 4a), evidencing energy transfer from NC–NCA complexes to PTM-TPA molecules in the solution.
TA was also applied to directly observe energy transfer from CsPbBr3 NC–NCA complexes to PTM-TPA. Fig. 4b shows the TA spectra of pure PTM-TPA in acetonitrile under 400 nm excitation. The spectra are dominated by two absorptive features centered at around 475 and 740 nm which can be assigned to the D1 excited state absorption (ESA) of PTM-TPA (2PTM-TPA*). The excited state lifetime can be fitted to a single-exponential constant of 12.8 ns (Fig. S4, ESI†). This emission lifetime is much shorter than PtOEP because it is fully spin-allowed. In the NC–NCA//PTM-TPA sample, we only observe the absorptive features of 3NCA* that decay with time, without the formation of PTM-TPA features (Fig. 4c). This observation is consistent with the short lifetime of 2PTM-TPA*. The formation of 2PTM-TPA* via energy transfer is slower than its decay, and hence there is no population accumulation for the 2PTM-TPA* species; see the ESI† for details. The energy transfer time is also ∼2 μs (Fig. 4d). Similarly, this TET rate is also limited by the energy transfer step rather than the diffusion process (see the ESI† for details).
While the energy transfer from 3NCA* to PtOEP is a typical Dexter-type TET process, the one associated with PTM-TPA is worth-mentioning. In this system (Fig. 1), energy transfer presumably also occurs by electron exchange,49i.e., electron transfer from the LUMO of 3NCA* to the SOMO of PTM-TPA is accompanied by electron transfer from the HOMO of PTM-TPA to the HOMO of 3NCA*. A notable change is that the spin of the electron in PTM-TPA is flipped upon energy transfer and radiative recombination, a property that can potentially be harnessed for spin injection and manipulation using radical emitters.
Conclusions
In summary, we demonstrated that the triplet energy extracted from CsPbBr3 perovskite NCs and stored in naphthalene surface ligands further translated into phosphorescent and radical light-emitting molecules in the bulk solution. Through this energy propagation scheme, light harvesting of the system is strongly enhanced by CsPbBr3 NCs and strongly red-shifted photons are emitted from the phosphorescent and radical molecules. These properties are highly desired for applications like luminescent solar concentrators (LSCs). However, the performances of these systems in LSCs will be limited by the emission quantum yields of the phosphorescent (PtOEP ∼41%) and radical (PTM-TPA ∼33%) emitters. Therefore, molecular tailoring will be needed to improve the quantum yields. Moreover, current systems are demonstrated in the solution phase for the sake of clean spectroscopic studies. For future real-life applications, these systems need to be integrated into solid-state devices. A possible solution is to embed the NC–ligand complexes and light-emitting molecules in a rubbery host polymer which permits triplet energy migration at temperatures higher than the glass transition temperature of the polymer.50 Alternatively, the phosphorescent and radical molecules can be functionalized with anchoring groups and directly attached onto NC surfaces for an efficient interfacial triplet energy transfer, thus enabling direct integration into various device configurations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (21975253, 21803070). The PTM-TPA molecule in this study was a general gift from Prof. Feng Li at Jilin University.Notes and references
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of photochemistry, CRC Press, CRC, Taylor & Francis, Boca Raton, 3rd edn, 2006 Search PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Modern molecular photochemistry of organic molecules, University Science Books, 2010 Search PubMed.
- T. N. Singh-Rachford and F. N. Castellano, Photon upconversion based on sensitized triplet–triplet annihilation, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS.
- J. Zhao, W. Wu, J. Sun and S. Guo, Triplet photosensitizers: from molecular design to applications, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- M. Wu, D. N. Congreve, M. W. B. Wilson, J. Jean, N. Geva, M. Welborn, T. Van Voorhis, V. Bulović, M. G. Bawendi and M. A. Baldo, Solid-state infrared-to-visible upconversion sensitized by colloidal nanocrystals, Nat. Photonics, 2015, 10, 31–34 CrossRef.
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Direct observation of triplet energy transfer from semiconductor nanocrystals, Science, 2016, 351, 369–372 CrossRef CAS.
- Z. Huang and M. L. Tang, Designing Transmitter Ligands That Mediate Energy Transfer between Semiconductor Nanocrystals and Molecules, J. Am. Chem. Soc., 2017, 139, 9412–9418 CrossRef CAS.
- Z. Huang, X. Li, M. Mahboub, K. M. Hanson, V. M. Nichols, H. Le, M. L. Tang and C. J. Bardeen, Hybrid Molecule–Nanocrystal Photon Upconversion Across the Visible and Near-Infrared, Nano Lett., 2015, 15, 5552–5557 CrossRef CAS.
- K. Okumura, K. Mase, N. Yanai and N. Kimizuka, Employing Core-Shell Quantum Dots as Triplet Sensitizers for Photon Upconversion, Chem. – Eur. J., 2016, 22, 7721–7726 CrossRef CAS PubMed.
- N. Yanai and N. Kimizuka, New Triplet Sensitization Routes for Photon Upconversion: Thermally Activated Delayed Fluorescence Molecules, Inorganic Nanocrystals, and Singlet-to-Triplet Absorption, Acc. Chem. Res., 2017, 50, 2487–2495 CrossRef CAS.
- Z. Huang and M. L. Tang, Semiconductor Nanocrystal Light Absorbers for Photon Upconversion, J. Phys. Chem. Lett., 2018, 9, 6198–6206 CrossRef CAS PubMed.
- L. Nienhaus, M. Wu, V. Bulović, M. A. Baldo and M. G. Bawendi, Using lead chalcogenide nanocrystals as spin mixers: a perspective on near-infrared-to-visible upconversion, Dalton Trans., 2018, 47, 8509–8516 RSC.
- X. Luo, R. Lai, Y. Li, Y. Han, G. Liang, X. Liu, T. Ding, J. Wang and K. Wu, Triplet Energy Transfer from CsPbBr3 Nanocrystals Enabled by Quantum Confinement, J. Am. Chem. Soc., 2019, 141, 4186–4190 CrossRef CAS PubMed.
- S. He, X. Luo, X. Liu, Y. Li and K. Wu, Visible-to-Ultraviolet Upconversion Efficiency above 10% Sensitized by Quantum-Confined Perovskite Nanocrystals, J. Phys. Chem. Lett., 2019, 10, 5036–5040 CrossRef CAS.
- Y. Han, X. Luo, R. Lai, Y. Li, G. Liang and K. Wu, Visible-Light-Driven Sensitization of Naphthalene Triplets Using Quantum-Confined CsPbBr3 Nanocrystals, J. Phys. Chem. Lett., 2019, 10, 1457–1463 CrossRef CAS.
- Y. Han, S. He, X. Luo, Y. Li, Z. Chen, W. Kang, X. Wang and K. Wu, Triplet Sensitization by “Self-Trapped” Excitons of Nontoxic CuInS2 Nanocrystals for Efficient Photon Upconversion, J. Am. Chem. Soc., 2019, 141, 13033–13037 CrossRef CAS.
- P. Xia, E. K. Raulerson, D. Coleman, C. S. Gerke, L. Mangolini, M. L. Tang and S. T. Roberts, Achieving spin-triplet exciton transfer between silicon and molecular acceptors for photon upconversion, Nat. Chem., 2020, 12, 137–144 CrossRef CAS.
- K. Okumura, N. Yanai and N. Kimizuka, Visible-to-UV Photon Upconversion Sensitized by Lead Halide Perovskite Nanocrystals, Chem. Lett., 2019, 48, 1347–1350 CrossRef CAS.
- K. Mase, K. Okumura, N. Yanai and N. Kimizuka, Triplet sensitization by perovskite nanocrystals for photon upconversion, Chem. Commun., 2017, 53, 8261–8264 RSC.
- Z. Xu, T. Jin, Y. Huang, K. Mulla, F. A. Evangelista, E. Egap and T. Lian, Direct triplet sensitization of oligothiophene by quantum dots, Chem. Sci., 2019, 10, 6120–6124 RSC.
- A. L. Efros and M. Rosen, The Electronic Structure of Semiconductor Nanocrystals, Annu. Rev. Mater. Sci., 2000, 30, 475–521 CrossRef CAS.
- J. M. Pietryga, Y.-S. Park, J. Lim, A. F. Fidler, W. K. Bae, S. Brovelli and V. I. Klimov, Spectroscopic and Device Aspects of Nanocrystal Quantum Dots, Chem. Rev., 2016, 116, 10513–10622 CrossRef CAS.
- C. Mongin, P. Moroz, M. Zamkov and F. N. Castellano, Thermally activated delayed photoluminescence from pyrenyl-functionalized CdSe quantum dots, Nat. Chem., 2018, 10, 225–230 CrossRef CAS.
- D. M. Kroupa, D. H. Arias, J. L. Blackburn, G. M. Carroll, D. B. Granger, J. E. Anthony, M. C. Beard and J. C. Johnson, Control of Energy Flow Dynamics between Tetracene Ligands and PbS Quantum Dots by Size Tuning and Ligand Coverage, Nano Lett., 2018, 18, 865–873 CrossRef CAS PubMed.
- R. Lai and K. Wu, Red-to-blue photon upconversion based on a triplet energy transfer process not retarded but enabled by shell-coated quantum dots, J. Chem. Phys., 2020, 153, 114701 CrossRef CAS.
- Y. Jiang, C. Wang, C. R. Rogers, M. S. Kodaimati and E. A. Weiss, Regio- and diastereoselective intermolecular [2 + 2] cycloadditions photocatalysed by quantum dots, Nat. Chem., 2019, 11, 1034–1040 CrossRef CAS PubMed.
- K. Wu and L.-Z. Wu, Semiconductor nanoparticles photocatalyze precise organic cycloaddition, Chem, 2021, 7, 842–844 CAS.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Highly efficient phosphorescent emission from organic electroluminescent devices, Nature, 1998, 395, 151 CrossRef CAS.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Thompson and S. R. Forrest, Very high-efficiency green organic light-emitting devices based on electrophosphorescence, Appl. Phys. Lett., 1999, 75, 4–6 CrossRef CAS.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Organic light-emitting diodes employing efficient reverse intersystem crossing for triplet-to-singlet state conversion, Nat. Photonics, 2012, 6, 253 CrossRef CAS.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Highly efficient organic light-emitting diodes from delayed fluorescence, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Organic Light-Emitting Diodes Using a Neutral π Radical as Emitter: The Emission from a Doublet, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS PubMed.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Efficient radical-based light-emitting diodes with doublet emission, Nature, 2018, 563, 536–540 CrossRef CAS.
- A. Abdurahman, T. J. H. Hele, Q. Gu, J. Zhang, Q. Peng, M. Zhang, R. H. Friend, F. Li and E. W. Evans, Understanding the luminescent nature of organic radicals for efficient doublet emitters and pure-red light-emitting diodes, Nat. Mater., 2020, 19, 1224–1229 CrossRef CAS.
- M. J. Currie, J. K. Mapel, T. D. Heidel, S. Goffri and M. A. Baldo, High-efficiency organic solar concentrators for photovoltaics, Science, 2008, 321, 226–228 CrossRef CAS.
- W. H. Weber and J. Lambe, Luminescent greenhouse collector for solar radiation, Appl. Opt., 1976, 15, 2299–2300 CrossRef CAS PubMed.
- C. Tummeltshammer, M. Portnoi, S. A. Mitchell, A.-T. Lee, A. J. Kenyon, A. B. Tabor and I. Papakonstantinou, On the ability of Förster resonance energy transfer to enhance luminescent solar concentrator efficiency, Nano Energy, 2017, 32, 263–270 CrossRef CAS.
- K. Wu, H. Li and V. I. Klimov, Tandem luminescent solar concentrators based on engineered quantum dots, Nat. Photonics, 2018, 12, 105–110 CrossRef CAS.
- M. Gandini, I. Villa, M. Beretta, C. Gotti, M. Imran, F. Carulli, E. Fantuzzi, M. Sassi, M. Zaffalon, C. Brofferio, L. Manna, L. Beverina, A. Vedda, M. Fasoli, L. Gironi and S. Brovelli, Efficient, fast and reabsorption-free perovskite nanocrystal-based sensitized plastic scintillators, Nat. Nanotechnol., 2020, 15, 462–468 CrossRef CAS.
- Y. Dong, T. Qiao, D. Kim, D. Parobek, D. Rossi and D. H. Son, Precise Control of Quantum Confinement in Cesium Lead Halide Perovskite Quantum Dots via Thermodynamic Equilibrium, Nano Lett., 2018, 18, 3716–3722 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nanocrystals of Cesium Lead Halide Perovskites (CsPbX3, X = Cl, Br, and I): Novel Optoelectronic Materials Showing Bright Emission with Wide Color Gamut, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS.
- K. Okumura, N. Yanai and N. Kimizuka, Visible-to-UV Photon Upconversion Sensitized by Lead Halide Perovskite Nanocrystals, Chem. Lett., 2019, 48, 1347–1350 CrossRef CAS.
- X. Luo, Y. Han, Z. Chen, Y. Li, G. Liang, X. Liu, T. Ding, C. Nie, M. Wang, F. N. Castellano and K. Wu, Mechanisms of triplet energy transfer across the inorganic nanocrystal/organic molecule interface, Nat. Commun., 2020, 11, 28 CrossRef CAS.
- R. Lai, Y. Liu, X. Luo, L. Chen, Y. Han, M. Lv, G. Liang, J. Chen, C. Zhang, D. Di, G. D. Scholes, F. N. Castellano and K. Wu, Shallow distance-dependent triplet energy migration mediated by endothermic charge-transfer, Nat. Commun., 2021, 12, 1532 CrossRef CAS PubMed.
- D. T. Yonemoto, C. M. Papa, C. Mongin and F. N. Castellano, Thermally Activated Delayed Photoluminescence: Deterministic Control of Excited State Decay, J. Am. Chem. Soc., 2020, 142, 10883–10893 CrossRef CAS PubMed.
- S. He, Y. Han, J. Guo and K. Wu, Long-Lived Delayed Emission from CsPbBr3 Perovskite Nanocrystals for Enhanced Photochemical Reactivity, ACS Energy Lett., 2021, 6, 2786–2791 CrossRef CAS.
- S. He, Y. Han, J. Guo and K. Wu, Entropy-Gated Thermally Activated Delayed Emission Lifetime in Phenanthrene-Functionalized CsPbBr3 Perovskite Nanocrystals, J. Phys. Chem. Lett., 2021, 12, 8598–8604 CrossRef CAS.
- H. Guo, Q. Peng, X.-K. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Credgington, V. Coropceanu, R. H. Friend, J.-L. Brédas and F. Li, High stability and luminescence efficiency in donor–acceptor neutral radicals not following the Aufbau principle, Nat. Mater., 2019, 18, 977–984 CrossRef CAS PubMed.
- J. Han, Y. Jiang, A. Obolda, P. Duan, F. Li and M. Liu, Doublet–Triplet Energy Transfer-Dominated Photon Upconversion, J. Phys. Chem. Lett., 2017, 8, 5865–5870 CrossRef CAS.
- R. R. Islangulov, J. Lott, C. Weder and F. N. Castellano, Noncoherent Low-Power Upconversion in Solid Polymer Films, J. Am. Chem. Soc., 2007, 129, 12652–12653 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc04086c |
| This journal is © The Royal Society of Chemistry 2022 |