
Applying intermolecular hydrogen bonding to exploit TADF emitters for high-performance orange-red non-doped OLEDs†
Yi-Zhong
Shi
 ,
Kai
Wang
*,
Xiao-Chun
Fan
,
Hao
Wu
,
Xue-Mei
Ou
,
Jia
Yu
,
Jian-Sheng
Jie
and
Xiao-Hong
Zhang
*
,
Kai
Wang
*,
Xiao-Chun
Fan
,
Hao
Wu
,
Xue-Mei
Ou
,
Jia
Yu
,
Jian-Sheng
Jie
and
Xiao-Hong
Zhang
*
Institute of Functional Nano & Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, 199 Ren’ai Road, Suzhou, 215123, Jiangsu, P. R. China. E-mail: xiaohong_zhang@suda.edu.cn; wkai@suda.edu.cn
First published on 4th November 2021
Abstract
Exploiting novel TADF emitters for highly efficient orange-red non-doped organic light-emitting devices (OLEDs) is a research hotspot. Nevertheless, serious non-radiative transitions of the electrogenerated excitons significantly hinder the pace of the development. Herein, two novel donor–π–acceptor (D–π–A) type TADF emitters 10-(4-(2,6-di(pyridin-3-yl)pyrimidin-4-yl)phenyl)-10H-phenoxazine (PyPmP) and 10-(4-(2-(pyridin-3-yl)-[4,5′-bipyrimidin]-6-yl)phenyl)-10H-phenoxazine (PmPmP) were designed and synthesized. In their crystals, suitable intermolecular hydrogen bonding interactions are observed that enable head-against-tail 3D supramolecular frameworks. Such frameworks not only prefer a high horizontal molecular orientation but also strictly restrict the non-radiative process in non-doped conditions. Their non-doped OLEDs realize decent orange-red electroluminescence with maximum current efficiencies (CE), power efficiencies (PE), and external quantum efficiencies (EQE) of 50.5 cd A−1, 53.5 lm W−1, and 18.8% for PyPmP, 23.2 cd A−1, 22.1 lm W−1, and 11.3% for PmPmP, respectively. Moreover, both devices can still retain high efficiencies as luminance increases, and exhibit a neglectable efficiency roll-off at 1000 cd m−2. These results are among the best of ever reported orange-red non-doped OLEDs. These results demonstrate that intermolecular hydrogen bonding interactions can play important roles in exploiting TADF emitters for high-performance orange-red non-doped OLEDs.
Introduction
Organic light-emitting devices (OLEDs) are regarded as the next-generation display and lighting resources owing to their advantages such as low power consumption, ultra-thin, self-lighting, and flexible characteristics. According to the spin-statistics, only a quarter of electron-generated excitons are singlets, and conventional fluorescent emitters can only utilize them for a theoretical 25% internal quantum efficiency (IQE). To effectively utilize both singlet and the rest three quarters of triplet excitons, phosphorescence and thermally activated delayed fluorescence (TADF) based OLEDs are two most successful mechanisms.1,2 In particular, TADF emitters, which are usually pure organic molecules, have attracted increasing attention within recent years. They utilize triplet excitons via a reverse intersystem crossing (RISC) process and a small energy splitting (ΔEST) between the lowest singlet (S1) and triplet (T1) excited states is one of the key factors.3–8 This is the main consideration for TADF emitters that generally employ highly twisted electron-donor (D)–electron-acceptor (A) structures. Another key parameter is the singlet fluorescence radiation rate (kr). Its competitive relationship with the corresponding non-radiative transition rate (kSnr) determines the eventual fluorescence quantum yield (ϕF). For orange-red emitters, kSnr is generally very large according to the energy-gap law, comparable or even surpasses kr.9 Such a kinetic reverse would lead to a dramatic efficiency decline. To avoid this, rigid aromatic skeletons are widely employed in high-efficiency orange-red TADF emitters since they can suppress non-radiative decays.10–12 Nevertheless, such emitters generally have strong π–π stacking tendencies and have to be doped in suitable host matrixes with precisely controlling doping concentrations.13,14Compared with doped OLEDs, non-doped counterparts are more attractive toward commercial productions due to the enhanced device fabrication repeatability and promising stabilities.15,16 However, those above-mentioned emitters that contain rigid aromatic skeletons cannot maintain decent performances in non-doped devices due to their close intermolecular π–π interactions that would induce serious quenching of the long-lived triplet excitons.17 On the other hand, current TADF molecular design strategies toward non-doped OLEDs are basically introducing steric hindrance,18–21 aggregation-induced emission (AIE),22–25 and intermolecular charge-transfer transition26,27 to inhibit electron–exchange interactions. Utilizing TADF emitters based on these strategies, highly efficient non-doped OLEDs have been achieved in the blue to yellow region. However, in the orange-red region, the corresponding efficiencies OLEDs are still poor because the above-mentioned strategies are unable to address the serious non-radiative issues.15
Recently, we have proposed a new molecular design concept by using suitable intermolecular hydrogen bonding to modulate molecular packing to develop high-performance non-doped OLEDs.28,29 Suitable intermolecular hydrogen bonding interactions favor forming supramolecular frameworks, which can not only strictly suppress the exciton quenching via suppressing intermolecular close π–π stacking, but also help to increase the rigidity of the supramolecular framework and thus relieve non-radiative decays. In this study, with an aim to develop high-performance orange-red non-doped OLEDs with intermolecular hydrogen bonds, we used phenoxazine (PXZ) as the D segment, and joint pyridine (Py)/pyrimidine (Pm) series as the A segments and constructed two novel TADF emitters 10-(4-(2,6-di(pyridin-3-yl)pyrimidin-4-yl)phenyl)-10H-phenoxazine (PyPmP) and 10-(4-(2-(pyridin-3-yl)-[4,5′-bipyrimidin]-6-yl)phenyl)-10H-phenoxazine (PmPmP) (Fig. 1). With suitable intermolecular hydrogen bonding, head-against-tail 3D supramolecular frameworks can be observed, which not only prefer a high horizontal molecular orientation but also strictly restrict the non-radiative process in non-doped conditions. In non-doped OLEDs, PyPmP and PmPmP succeed in decent orange-red electroluminescence (EL) performances with maximum current efficiencies (CE), power efficiencies (PE), and external quantum efficiencies (EQE) are 50.5 cd A−1, 53.5 lm W−1, and 18.8% for PyPmP, 23.2 cd A−1, 22.1 lm W−1, and 11.3% for PmPmP respectively, with negligible efficiency roll-offs at 1000 cd m−2. To the best of our knowledge, these results are among the best of ever reported orange-red non-doped OLEDs. These results demonstrate that intermolecular hydrogen bonding interactions can play important roles in exploiting TADF emitters for high-performance orange-red non-doped OLEDs.
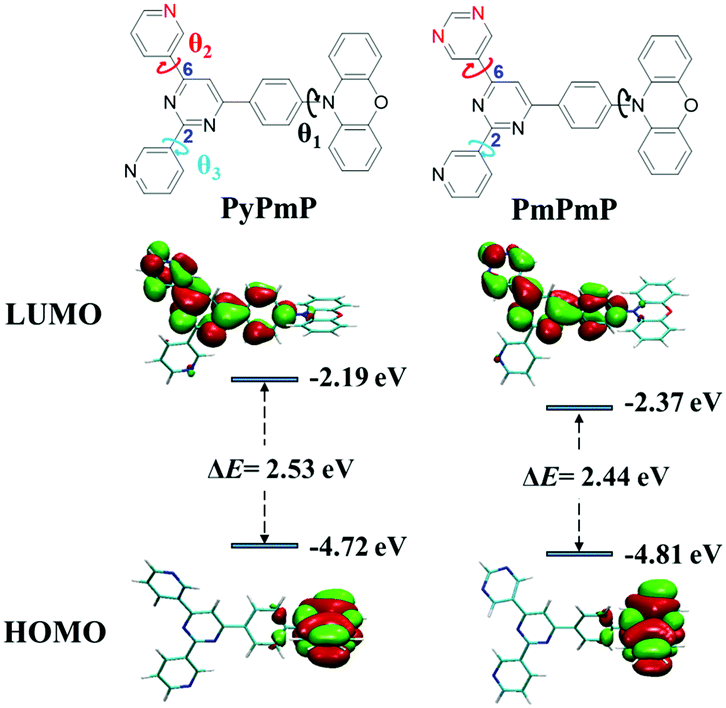 | ||
| Fig. 1 Molecular structures of PyPmP and PmPmP and their simulated HOMO/LUMO energy levels and spatial distributions. The isovalue is 0.02. | ||
Results and discussion
Synthesis and characterization
Our previous studies demonstrated that inside films that contain PXZ and Py/Pm series intermolecular hydrogen bonding is facilely formed, which is beneficial to improve the performances of non-doped OLEDs.28,29 Herein, we used two novel A segments, namely 2,6-di(pyridin-3-yl)pyrimidine (PyPm) and 2-(pyridin-3-yl)-4,5′-bipyrimidine (PmPm) that have stronger electron-withdrawing abilities than previously employed A segments, namely TPy and DPmP; while the D segments are still PXZ to realize non-doped orange-red systems. The only difference of PyPm and PmPm is the peri-azaaromatic ring (the substituent labeled number 6 in Fig. 1) to study in detail the influence of active sites to the molecular stacking. Further applying a typical D–π–A molecular structure, two novel TADF emitters (PyPmP and PmPmP) were thus designed and synthesized. Detailed experimental procedures of PyPmP and PmPmP are described in Scheme S1 (ESI†). Chemical structures of the intermediates and final products were confirmed via nuclear magnetic resonance (NMR) and mass spectroscopy. As shown in Fig. S1 (ESI†), PyPmP and PmPmP both show good thermostability with decomposition temperatures (Td, 5%) of 453 °C and 394 °C and glass transition temperature (Tg) of 98 °C and 110 °C, respectively, which fulfill the requirement of the vacuum-deposition process.Theoretical calculations
In order to study the structure–property relationships of PyPmP and PmPmP at the single-molecule level, their optimized molecular geometries at ground states (S0) under vacuum conditions were estimated via the density functional theory (DFT) with the B3LPY/6-31G* basis set. As displayed in Fig. 1 and summarized in Table S1 (ESI†), geometries of both emitters are very similar. The twists between PXZ and phenyl bridges (marked as θ1 in Fig. 1) are both nearly orthogonal due to their evident steric hindrances. Inside the A sides, the torsions between the core Pm and 6-substituted rings (marked as θ2 in Fig. 1) and between the core Pm and 2-substituted Py rings (marked as θ3 in Fig. 1) are respectively ∼6° and ∼22°. The different torsions between cores and peripheral rings (θ2 and θ3) should be ascribed to the different intramolecular hydrogen bonds. Owing to their similar optimized geometries, frontier molecular orbital (FMO) distributions of both emitters are very similar. The highest occupied molecular orbitals (HOMOs) are well confined in the electron-rich PXZ segment; while the lowest unoccupied molecular orbitals (LUMOs) are spread over the whole A moieties except the 2-substituted Py rings, which can be ascribed to the interrupted molecular conjugations.30,31 Based on the HOMO and LUMO energy levels, PmPmP is calculated to own a narrower bandgap (ΔE) of 2.44 eV than PyPmP (2.53 eV), which suggests replacing the Py moiety by Pm can help in redshifting emissions. We also calculated their corresponding lowest adiabatic excited states to estimate their TADF properties. As shown in Fig. S2 (ESI†), S1 and T1 states of both emitters exhibit strong intramolecular charge-transfer (ICT) characteristics, and both ΔESTs are estimated to be only 0.0044 eV, benefiting their RISC process.Electrochemical properties
Cyclic voltammetry (CV) measurements were first applied on both compounds to compare the electron-donating and electron-withdrawing abilities of their consisting segments. As depicted in Fig. 2a, due to their same D segments, both emitters exhibit similar oxidation curves. The HOMO energy levels are estimated to be −5.30 eV for PyPmP and −5.36 eV for PmPmP from the onset. While from the onset of their reduction curves, the LUMO energy levels of PyPmP and PmPmP are respectively estimated to be −3.06 eV and −3.19 eV. The shallower LUMO of PmPmP than that of PyPmP reveals that the electron-withdrawing ability of PmPm is stronger than that of PyPm, which is well-consistent with the previous theoretical simulation results.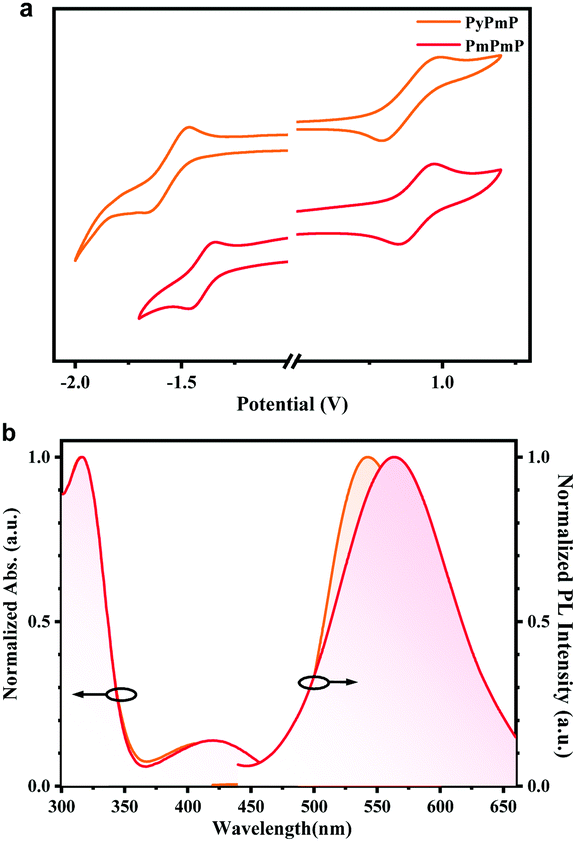 | ||
| Fig. 2 (a) Cyclic voltammetry curves of PyPmP and PmPmP measured in DMF, and (b) their normalized absorption and emission spectra in dilute toluene at ambient temperature Molecular structures of PyPmP and PmPmP and their simulated HOMO/LUMO energy levels and spatial distributions. The isovalue is 0.02. | ||
Photophysical properties in the dilute solution state
Photophysical properties of PyPmP and PmPmP in dilute toluene were then studied at room temperature. As depicted in Fig. 2b, both emitters display similar locally excited absorption bands at around 320 nm due to their similar consisting components. However, the weak bands in the range from ∼360 to ∼500 nm should be assigned to their ICT transitions. Particularly, with stronger electron-withdrawing ability of the PmPm segment, the absorption cutoff of PmPmP shows a significant bathochromic shift compared to that of PyPmP. Likewise, in their emission spectra, broad and structureless ICT bands are observed both in a similar yellowish-green region; while the emission peak of PmPmP (563 nm) is redshifted compared to that of PyPmP (542 nm) because of the stronger electron-withdrawing ability of PmPm.Single-crystal structure analysis
Single crystals of both compounds were then cultivated via the classical vapor phase diffusion method between tetrahydrofuran (THF) and petroleum ether (PE), and further analyzed by X-ray diffraction to investigate their molecular packing and intermolecular interactions. As depicted in Fig. 3, the dihedral angles between the PXZ moieties and phenyl bridges are both around 75°, sufficient to ensure small ΔESTs. Moreover, in terms of intermolecular levels, it can be noticed that for both molecules the active sites that cause intermolecular hydrogen bonding are identical. In both crystals, there are two pairs of hydrogen bonds observed, which are respectively between C–H and O (2.62 Å for PyPmP and 2.65 Å for PmPmP, respectively, marked as green-dashed line) and between C–H and N (both 2.57 Å, marked as red-dashed line). With similar geometries and intermolecular interactions, their ultimate packing modes are determined to be very similar with head-against-tail 3D supramolecular frameworks. Such supramolecular frameworks are expected to increase the overall rigidities and thus suppress the molecular vibrations and rotations that bring about non-radiative decays. It is notable that in the supramolecular frameworks dominated by intermolecular hydrogen bonding, π–π stackings are also accompanied. For A moieties, stackings are observed with distances of ∼3.4 Å (seen in the blue box in Fig. 3), which may favor the electron mobility and further carrier balances.28 While on the other hand, the electron-rich PXZ moieties are also observed to show close packing (seen in the red box in Fig. 3) with a distance of ∼3.4 Å. Such close stackings would increase the possibilities of the concentration quenching particularly related to triplet excitons.28,32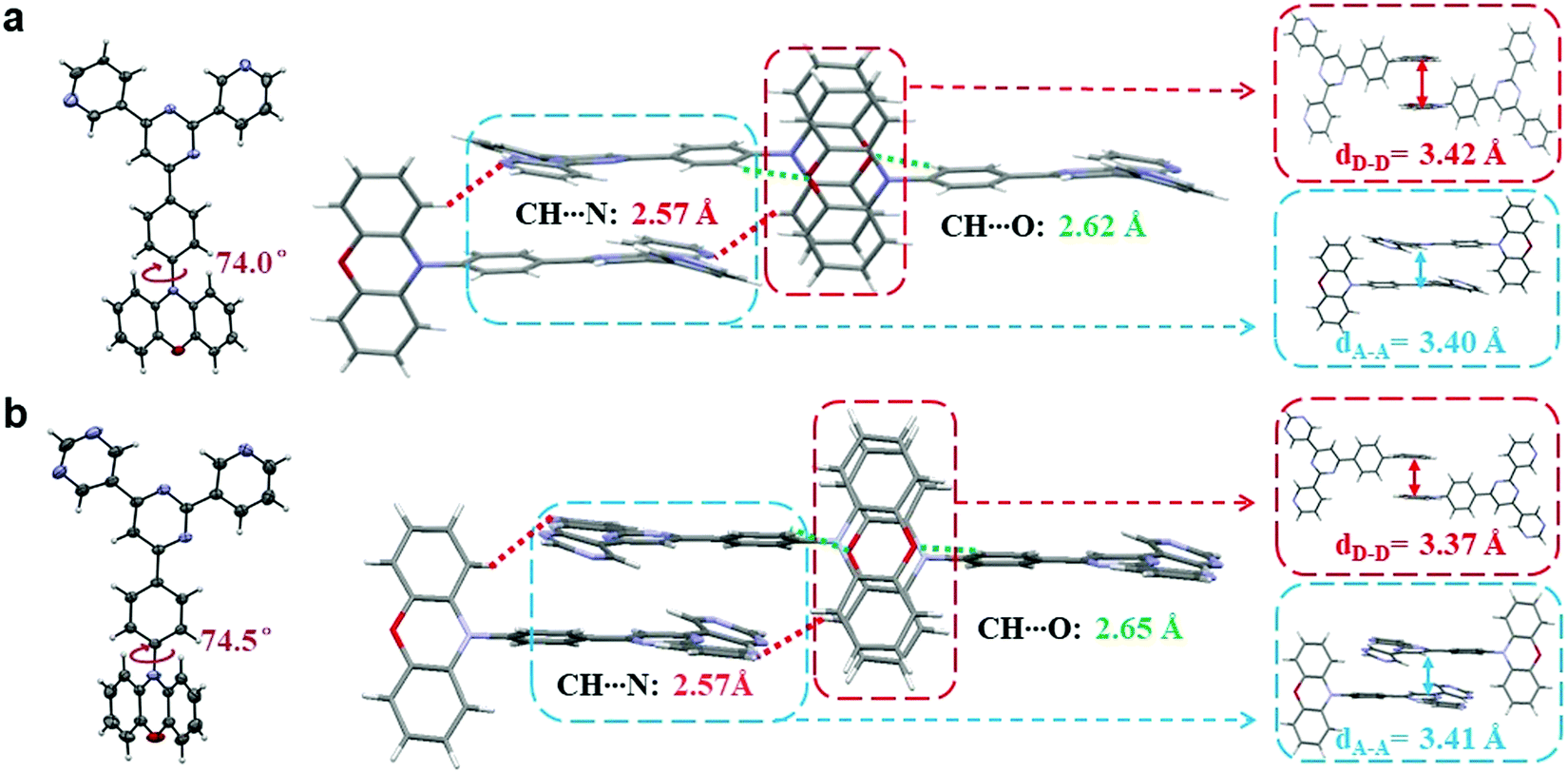 | ||
| Fig. 3 Thermal ellipsoid drawing structures and intermolecular geometries of (a) PyPmP and (b) PmPmP in the single crystals determined by X-ray analysis. Abbreviations: dD–D and dA–A are the identity distances between the neighbor D and A moieties, respectively. | ||
Photophysical properties in amorphous neat film state
With the information obtained from the single crystals, we further studied the photophysics of PyPmP and PmPmP pristine films to exploit the influence of molecular packing in amorphous states. At room temperature, both films exhibit orange-red emission with peaks at 570 nm for PyPmP and 591 nm for PmPmP, respectively. (Seen in Fig. S3, ESI†) The corresponding ϕF are respectively measured to be 65.9% and 42.9%. These results obtained are decent values among the orange-red non-doped films. Fluorescence and phosphorescence spectra of both films are further measured at 77 K to characterize the S1 and T1 states. As shown in Fig. S3 (ESI†), both films exhibit typical broad and structureless bands indicating they are from CT transitions. Derived from the corresponding onsets of their fluorescence and phosphorescence spectra, their ΔESTs are estimated to be only 0.04 and 0.05 eV for PyPmP and PmPmP, respectively (Summarized in Table S2, ESI†). Such small ΔESTs can significantly enhance the RISC process and benefit triplet exciton utilization. Transient PL decays were then carried out to confirm their TADF characteristics. As shown in Fig. S4 (ESI†), both PyPmP and PmPmP pristine films exhibit not only prompt emissions in the nanosecond range, but also delayed signals in microseconds. With an increase in temperature from 100 to 300 K, the delayed lifetimes are slightly decreased. Moreover, in terms of time-resolved spectroscopy, the delayed emission spectra of both films show only slight redshifts compared to the corresponding prompt ones.33,34 These results demonstrate that both emitters are with TADF characteristics. As shown in Fig. S5 (ESI†) and summarized in Table S2 (ESI†), it is worth noting that their delayed lifetimes are just 0.5 and 0.7 μs, respectively, at 300 K, which are evidently shorter than the control films by 10 wt% doping them in 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) films, benefiting to suppress efficiency roll-offs at high current densities. Moreover, the prompt lifetimes of neat films are longer than those of the control ones, indicating that the intermolecular hydrogen bonding modulated 3D supramolecular frameworks can help to retrain singlet non-radiative decays.Carrier transport and molecular orientation
Besides photophysical properties, molecular packings are expected to significantly affect the carrier transporting performances as well. Thus, we further fabricated the hole-only devices (HODs) and electron-only devices (EODs) based on PyPmP and PmPmP non-doped films to investigate their carrier balances. Fig. S6 (ESI†) shows the J–V characteristic of HOD and EODs. Both emitters in pristine amorphous films behave as electron-preferred materials, which is well-consistent with their single crystal results that contain close stacking of electron-withdrawing A segments.The 3D frameworks dominated by intermolecular hydrogen bonding may also affect molecular orderliness of the films. To convince their influences, molecular orientations of both emitters in the amorphous state were quantified via the variable angle spectroscopic ellipsometry (VASE). As expected, PyPmP and PmPmP neat films both exhibit relatively high horizontal molecular orientation with order parameter S of −0.25 and −0.23, respectively.35,36 (Fig. S7, ESI†) These preferred horizontal orientation would benefit the out-coupling efficiencies and ultimate performances of OLEDs.
Electroluminescence properties
To evaluate their electroluminescent (EL) performances, non-doped OLEDs are fabricated with device structures of: ITO/TAPC (40 nm)/TCTA (10 nm)/PyPmP and PmPmP (20 nm)/TmPyPb (45 nm)/LiF (1 nm)/Al (depicted in Fig. S9, ESI†). In these devices, ITO (indium tin oxide) was used as the anode, and LiF/Al was used as the cathode; TAPC (1,1-bis[4-[N,N-bis(p-tolyl)amino]phenyl]cyclohexane) acted as the hole transporting layer; TCTA (4,4′,4′′-tris(carbazol-9-yl)triphenylamine) was utilized as the exciton-blocking layer; and TmPyPb (1,3,5-tris[(3-pyridyl)benzene-3-yl]benzene) was utilized as the electron-transporting, hole-blocking, and exciton-blocking layer. The key performances of OLEDs are listed in Table 1.| Emitter | Device | λ Max. (nm) | CE/PE/EQE (cd A−1/lm W−1/%) | CIE (x, y) | ||
|---|---|---|---|---|---|---|
| Max | @103 cd m−2 | @104 cd m−2 | ||||
| PyPmP | 10 wt% doped | 536 | 77.3/71.6/23.4 | 62.7/37.6/19.1 | 39.5/17.1/12.0 | (0.35, 0.58) |
| Non-doped | 576 | 50.5/53.5/18.8 | 45.1/33.3/16.7 | 4.3/28.2/15.0 | (0.50, 0.49) | |
| PmPmP | 10 wt% doped | 548 | 69.1/55.6/21.0 | 63.8/40.3/19.2 | 44.6/20.3/13.5 | (0.39, 0.56) |
| Non-doped | 596 | 23.3/22.1/11.3 | 20.4/14.6/10.0 | 18.2/11.0/8.6 | (0.56, 0.44) | |
As displayed in Fig. 4a and c, the non-doped devices based on PyPmP and PmPmP exhibited orange-red emission with peaks at 576 nm and 596 nm, respectively. More importantly, as shown in Fig. 4b and d, their maximum CEs, PEs and EQEs are achieved to be 50.5 cd A−1, 53.5 lm W−1, and 18.8% for PyPmP and 23.2 cd A−1, 22.1 lm W−1, and 11.3% for PmPmP, respectively. To the best of our knowledge, these are among the best ever reported orange-red non-doped devices (summarized in Table S3, ESI†). Moreover, both devices retain high efficiencies with smooth efficiency roll-offs at 1000 cd m−2. Even at high brightness of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cd m−2 EQE of PyPmP and PmPmP based devices can still remain as high as 15.0% and 8.6%, respectively, corresponding to their relative roll-offs of only 20.0% and 24.5%, respectively.
000 cd m−2 EQE of PyPmP and PmPmP based devices can still remain as high as 15.0% and 8.6%, respectively, corresponding to their relative roll-offs of only 20.0% and 24.5%, respectively.
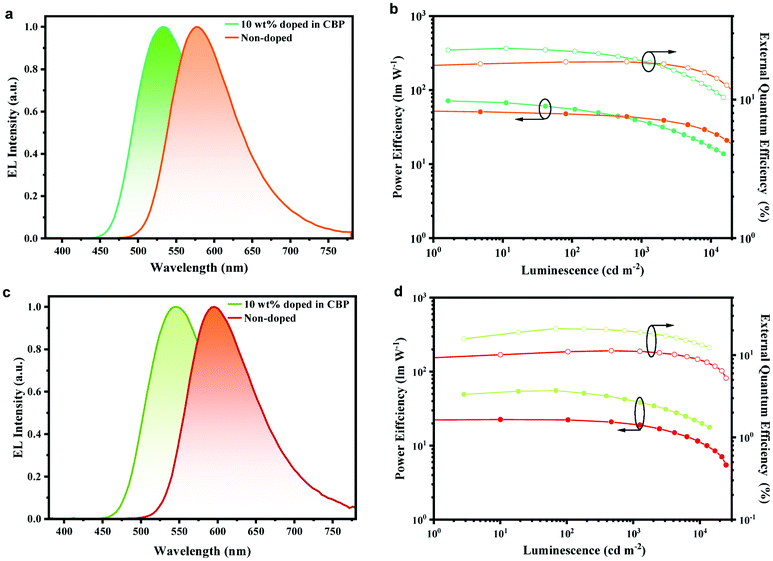 | ||
| Fig. 4 EL spectra and the PE–luminance-EQE characteristics of the doped and non-doped devices based on PyPmP (a and b) and PmPmP (c and d). | ||
Finally, to better understand the roles of the 3D framework based on PyPmP and PmPmP in devices, we fabricated series of OLEDs with structures like the above ones except introducing CBP as the host matrix to sensitize PyPmP and PmPmP with various doping ratios. The corresponding device performances are displayed in Fig. S10 (ESI†) and key parameters are summarized in Table S4 (ESI†). With an increase in the doping weights of TADF emitters from 5 wt% to 100% (i.e. non-doped), both EL spectra series exhibit an evident bathochromic shift gradually from the yellowish-green to the orange-red region. Therefore, it is expected that the energy-gap law induced singlet non-radiatve decays would be significantly magnified. Fig. 4 shows the optimized device performances in which the doping ratios were optimized to be 10 wt%. The OLEDs based on PyPmP and PmPmP exhibit yellowish-green emission with peaks at 536 nm and 548 nm, and maximum EQEs of 23.4% and 21.0%, respectively. While with further increase in doping ratios, only mild efficiency declines can be observed. Apparently, triplet quenching mainly due to the close π–π stacking of the electron-rich PXZ moiety contributes a lot to the efficiency declines. While on the other hand, owing to the improved rigidity due to the intermolecular hydrogen bonds, the radiation is not totally surpassed by the non-radiative channel and the efficiency is not dramatically dropped in the orange-red region. These results further confirm that intermolecular hydrogen bonding can play important roles to help with an improved efficiency for non-doped OLEDs in the orange-red region.37–39
Conclusions
In summary, we present that suitable intermolecular hydrogen bonding can effectively enhance system rigidities and suppress exciton losses, which can help to exploit novel TADF emitters for highly efficient orange-red non-doped OLEDs. Two newly designed D–π–A TADF emitters PyPmP and PmPmP both exhibit head-against-tail 3D supramolecular frameworks in crystals due to their suitable intermolecular hydrogen bonding. These not only determine a high horizontal molecular orientation but also strictly restrict the non-radiative process in their non-doped conditions. As in return, the corresponding non-doped OLEDs by using PyPmP and PmPmP as the emitting layers realize decent orange-red EL performances with maximum EQEs of 18.8% and 11.3% respectively. Moreover, a neglectable efficiency roll-off at 1000 cd m−2 can also be observed which are among the best of every reported orange-red non-doped OLEDs. All these results demonstrate that intermolecular hydrogen bonding interactions can play important roles in exploiting TADF emitters for high-performance orange-red non-doped OLEDs.Author contributions
Yi-Zhong Shi: conceptualization, writing – original draft; Kai Wang: writing – review & editing, supervision; Xiao-Chun Fan: investigation; Hao Wu: investigation; Xue-Mei Ou: resources; Jia Yu: visualization; Jian-Sheng Jie: resources; Xiao-Hong Zhang: project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research & Development Program of China (Grant No. 2020YFA0714601, 2020YFA0714604), the National Natural Science Foundation of China (Grant No. Grant No. 52130304, 51821002, 52003185, 52003186), the China Postdoctoral Science Foundation (Grant No. 2019M661924), Suzhou Key Laboratory of Functional Nano & Soft Materials, Collaborative Innovation Center of Suzhou Nano Science & Technology, the 111 Project, Joint International Research Laboratory of Carbon-Based Functional Materials and Devices.References
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef.
- S. L. Zhang, Y. Z. Shi, K. Wang, X. C. Fan, J. Yu, X. M. Ou and X. H. Zhang, Mater. Today Energy, 2020, 20, 100581 CrossRef.
- K. Wang, C. J. Zheng, W. Liu, K. Liang, Y. Z. Shi, S. L. Tao, C. S. Lee, X. M. Ou and X. H. Zhang, Adv. Mater., 2017, 29, 1701476 CrossRef.
- M. Zhang, W. Liu, C. J. Zheng, K. Wang, Y. Z. Shi, X. Li, H. Lin, S. L. Tao and X. H. Zhang, Adv. Sci., 2019, 6, 1801938 CrossRef PubMed.
- F.-Y. Hao, Y.-Z. Shi, K. Wang, X.-C. Fan, L. Wu, J. Ye, C.-J. Zheng, Y.-Q. Li, X.-M. Ou and X.-H. Zhang, J. Mater. Chem. C, 2020, 8, 10416–10421 RSC.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bassler, D. Beljonne, M. Buck, Y. Olivier, A. Kohler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588–6599 CrossRef CAS PubMed.
- J. A. Knöller, G. Meng, X. Wang, D. Hall, A. Pershin, D. Beljonne, Y. Olivier, S. Laschat, E. Zysman-Colman and S. Wang, Angew. Chem., Int. Ed., 2019, 59, 3156–3160 CrossRef.
- Q. Zhang, H. Kuwabara, W. J. Potscavage, S. Huang, Y. Hatae, T. Shibata and C. Adachi, J. Am. Chem. Soc., 2014, 136, 18070–18081 CrossRef CAS.
- J. X. Chen, K. Wang, C. J. Zheng, M. Zhang, Y. Z. Shi, S. L. Tao, H. Lin, W. Liu, W. W. Tao, X. M. Ou and X. H. Zhang, Adv. Sci., 2018, 5, 1800436 CrossRef.
- Y. L. Zhang, Q. Ran, Q. Wang, Y. Liu, C. Hanisch, S. Reineke, J. Fan and L. S. Liao, Adv. Mater., 2019, 31, e1902368 CrossRef.
- Y.-J. Yu, Y. Hu, S.-Y. Yang, W. Luo, Y. Yuan, C.-C. Peng, J.-F. Liu, A. Khan, Z.-Q. Jiang and L.-S. Liao, Angew. Chem., Int. Ed., 2020, 59, 21578–21584 CrossRef CAS.
- H. Xu, B. Zhao, H. Wang, C. Han, P. Ma, Z. Li and P. Chang, Angew. Chem., Int. Ed., 2020, 59, 19042–19047 CrossRef PubMed.
- J. X. Chen, W. W. Tao, W. C. Chen, Y. F. Xiao, K. Wang, C. Cao, J. Yu, S. Li, F. X. Geng, C. Adachi, C. S. Lee and X. H. Zhang, Angew. Chem., Int. Ed., 2019, 58, 14660–14665 CrossRef CAS PubMed.
- M. Godumala, S. Choi, M. J. Cho and D. H. Choi, J. Mater. Chem. C, 2019, 7, 2172–2198 RSC.
- S. Jhulki, M. W. Cooper, S. Barlow and S. R. Marder, Mater. Chem. Front., 2019, 3, 1699–1721 RSC.
- J. Lee, N. Aizawa, M. Numata, C. Adachi and T. Yasuda, Adv. Mater., 2017, 29, 1604856 CrossRef.
- K. Matsuo and T. Yasuda, Chem. Sci., 2019, 10, 10687–10697 RSC.
- W. Li, B. Li, X. Cai, L. Gan, Z. Xu, W. Li, K. Liu, D. Chen and S. J. Su, Angew. Chem., Int. Ed., 2019, 58, 11301–11305 CrossRef CAS.
- I. S. Park, K. Matsuo, N. Aizawa and T. Yasuda, Adv. Funct. Mater., 2018, 28, 1802031 CrossRef.
- L. Wu, K. Wang, C. Wang, X.-C. Fan, Y.-Z. Shi, X. Zhang, S.-L. Zhang, J. Ye, C.-J. Zheng, Y.-Q. Li, J. Yu, X.-M. Ou and X.-H. Zhang, Chem. Sci., 2021, 12, 1495–1502 RSC.
- H. Liu, J. Zeng, J. Guo, H. Nie, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2018, 57, 9290–9294 CrossRef CAS.
- J. Guo, J. Fan, L. Lin, J. Zeng, H. Liu, C. K. Wang, Z. Zhao and B. Z. Tang, Adv. Sci., 2019, 6, 1801629 CrossRef.
- L. Yu, Z. Wu, G. Xie, C. Zhong, Z. Zhu, D. Ma and C. Yang, Chem. Commun., 2018, 54, 1379–1382 RSC.
- J. Luo, S. Gong, Y. Gu, T. Chen, Y. Li, C. Zhong, G. Xie and C. Yang, J. Mater. Chem. C, 2016, 4, 2442–2446 RSC.
- Y. Z. Shi, K. Wang, X. Li, G. L. Dai, W. Liu, K. Ke, M. Zhang, S. L. Tao, C. J. Zheng, X. M. Ou and X. H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9480–9484 CrossRef CAS PubMed.
- X. Zhang, J.-X. Chen, K. Wang, Y.-Z. Shi, X.-C. Fan, S.-L. Zhang, L. Wu, Y.-Q. Li, X.-M. Ou and X.-H. Zhang, J. Mater. Chem. C, 2020, 8, 17457–17463 RSC.
- Y. Shi, K. Wang, Y. Tsuchiya, W. Liu, T. Komino, X. Fan, D. Sun, G. Dai, J. Chen, M. Zhang, C. Zheng, S. Xiong, X. Ou, J. Yu, J. Jie, C.-S. Lee, C. Adachi and X. Zhang, Mater. Horiz., 2020, 7, 2734–2740 RSC.
- Y. Z. Shi, K. Wang, X. C. Fan, J. X. Chen, X. M. Ou, J. Yu, J. S. Jie, C. S. Lee and X. H. Zhang, Adv. Opt. Mater., 2021, 9(16), 2100461 CrossRef CAS.
- S. Kang, S. H. Jeon, Y. M. Cho, Y. J. Kim, T. Kim and J. Y. Lee, Org. Electron., 2020, 78, 105595 CrossRef CAS.
- P. Ganesan, R. Ranganathan, Y. Chi, X.-K. Liu, C.-S. Lee, S.-H. Liu, G.-H. Lee, T.-C. Lin, Y.-T. Chen and P.-T. Chou, Chem. – Eur. J., 2016, 23, 2858–2866 CrossRef PubMed.
- W.-L. Tsai, M.-H. Huang, W.-K. Lee, Y.-J. Hsu, K.-C. Pan, Y.-H. Huang, H.-C. Ting, M. Sarma, Y.-Y. Ho, H.-C. Hu, C.-C. Chen, M.-T. Lee, K.-T. Wong and C.-C. Wu, Chem. Commun., 2015, 51, 13662–13665 RSC.
- K. Masui, H. Nakanotani and C. Adachi, Org. Electron., 2013, 14, 2721–2726 CrossRef CAS.
- K. Nasu, T. Nakagawa, H. Nomura, C. J. Lin, C. H. Cheng, M. R. Tseng, T. Yasuda and C. Adachi, Chem. Commun., 2013, 49, 10385–10387 RSC.
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476–8483 CrossRef CAS.
- D. Yokoyama, J. Mater. Chem. C, 2011, 21, 19187–19202 RSC.
- C. M. Han, Z. Zhang, D. X. Ding and H. Xu, Chemistry, 2018, 4, 2154–2167 CrossRef CAS.
- X. Zheng, R. Huang, C. Zhong, G. Xie, W. Ning, M. Huang, F. Ni, F. B. Dias and C. Yang, Adv. Sci., 2020, 7, 1902087 CrossRef CAS PubMed.
- M. Zhang, C. J. Zheng, K. Wang, Y. Z. Shi, D. Q. Wang, X. Li, H. Lin, S. L. Tao and X. H. Zhang, Adv. Funct. Mater., 2021, 31, 2010100 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis; thermal properties; theoretical simulations; photophysical properties; carrier mobility and molecular orientation; crystal results and electroluminescence performances. CCDC 2093258 and 2093260. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1tc03803f |
| This journal is © The Royal Society of Chemistry 2022 |