
Recent advances in protein-imprinted polymers: synthesis, applications and challenges
Yanting
He
ab and
Zian
Lin
 *b
*b
aSchool of Pharmacy, Bengbu Medical University, 2600 Donghai Avenue, Bengbu, Anhui 233000, China
bMinistry of Education Key Laboratory of Analytical Science of Food Safety and Biology, Fujian Provincial Key Laboratory of Analysis and Detection Technology for Food Safety, College of Chemistry, Fuzhou University, Fuzhou, Fujian 350108, China. E-mail: zianlin@fzu.edu.cn; Fax: +86-591-22866165
First published on 19th April 2022
Abstract
The molecular imprinting technique (MIT), also described as the “lock to key” method, has been demonstrated as an effective tool for the creation of synthetic polymers with antibody-like sites to specifically recognize target molecules. To date, most successful molecular imprinting researches were limited to small molecules (<1500 Da); biomacromolecule (especially protein) imprinting remains a serious challenge due to their large size, chemical and structural complexity, and environmental instability. Nevertheless, protein imprinting has achieved some significant breakthroughs in imprinting methods and applications over the past decade. Some special protein-imprinted materials with outstanding properties have been developed and exhibited excellent potential in several advanced fields such as separation and purification, proteomics, biomarker detection, bioimaging and therapy. In this review, we critically and comprehensively surveyed the recent advances in protein imprinting, particularly emphasizing the significant progress in imprinting methods and highlighted applications. Finally, we summarize the major challenges remaining in protein imprinting and propose its development direction in the near future.
1. Introduction
Molecular recognition, which is the most extensive and subtle recognition mechanism in nature, plays a vital role in various biological processes such as DNA replication, cell division and differentiation, and immune responses in the immune system.1 Because of the precise recognition properties, considerable attempts have been made to create synthetic recognition systems with high specificity and selectivity for certain molecules by utilizing biological macromolecules such as antibodies, receptors, enzymes and aptamers.1–4 Despite their high selectivity and broad range of applications, these biomolecules always suffer from shortcomings such as low long-term stability and high manufacturing cost.5,6 Therefore, developing stable, scalable, reusable and low-cost synthetic recognition systems remains an urgent need. To this end, molecular imprinting technique (MIT), which could obtain synthetic receptors with high selectivity toward target molecules, has attracted tremendous attention and has been proposed as an alternative for synthetic recognition systems.7–9Molecular imprinting (MI) is a well-known technique that can fabricate synthetic polymers with tailor-made recognition sites toward certain template molecules or similar structured molecules. The idea of molecular imprinting originated from the natural recognition mechanism between antibodies and antigens, which was presented by Breinl and Haurowitz in 193010 and evolved with Mudd in 1932,11 and was first proposed by Polyakov in 1931.12 Based on Pauling's antibody theory13 that antibody and antigen showed a specific three-dimensional space complementarity when contacting with each other, Dickey14 reported in 1949 silica gels with specific affinity for dye molecules by preparing silica gels in the presence of template dyes. In 1972, Wulff and Sarhan15 successfully prepared organic polymeric materials through a “covalent molecular imprinting” method, and the concept of molecular imprinting was really established. However, after that, molecular imprinting did not attract much attention. Until 1994, Mosach16 introduced a “non-covalent molecular imprinting” method to synthesize crosslinked polymers in the presence of template molecules. Since then, the molecular imprinting technique began to develop rapidly and has achieved significant progress.
Generally, the synthetic polymers, defined as molecularly imprinted polymers (MIPs), are synthesized through three steps: (1) forming a complex between a target molecule (template) and functional monomers by self-assembly; (2) copolymerization of the complex and cross-linkers through various polymerization methods; (3) removal of the template using a rational washing protocol, as shown in Fig. 1.17 The resulting MIPs show specific “memory” ability to recognize and bind template molecules, which is similar to the molecular recognition such as in antibodies and enzymes in biological systems. Thus, MIPs have often been termed “synthetic/artificial/plastic antibodies” or “antibody mimics”. However, compared to natural antibodies, MIPs possess several unique advantages.18,19 In addition to the good affinity and selectivity comparable to antibodies, MIPs are more stable over time in harsh conditions such as under refrigeration or in acidity, resulting from long-term storage and have a wider range of applications. Besides, MIPs can be obtained through a simpler, faster, low-cost and even scalable method. Actually, the most attractive advantage of MIPs is to create synthetic receptors for target molecules whose natural binding partners and even the structures are unknown. Because of these features, MIPs have attracted strong interest among researchers and have been applied in various applications such as separation and purification,20,21 target drug delivery,22,23 chemical sensors,24,25 proteomics,26,27 artificial antibodies,28,29 and medical diagnosis.30,31
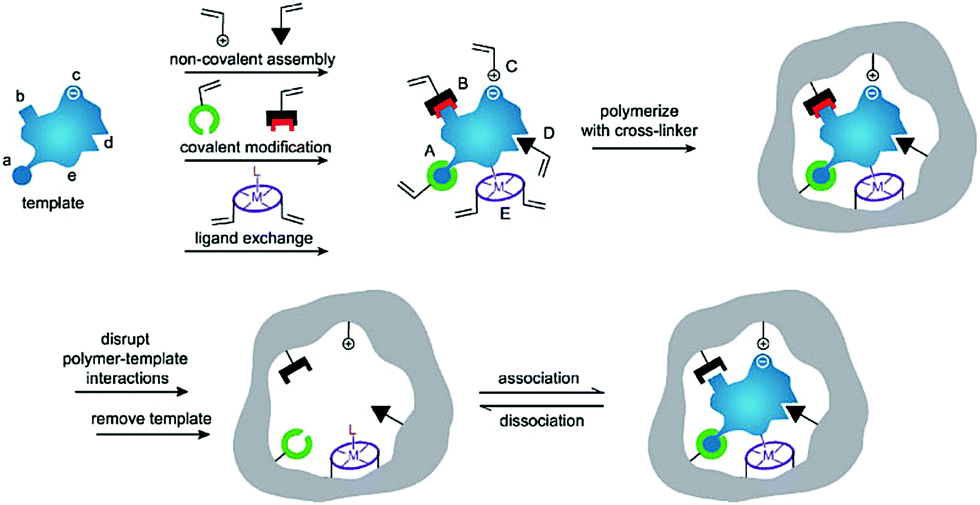 | ||
| Fig. 1 Schematic illustration of the molecular imprinting process. Reprinted from ref. 17. | ||
To date, a wide range size of template molecules, including inorganic ions, organics, nucleic acids, peptides, proteins, viruses and whole cells have been extensively applied in the synthesis of various specific MIPs.31,32 Among them, the majority of the publications and successful applications in molecular imprinting are aimed at the recognition and separation of small molecules. The development of MIPs for imprinting biological macromolecules, especially proteins, has been relatively slow.33,34 Nevertheless, due to the tantalizing prospect of creating protein-imprinted polymers (PIPs) for biomedical and biodiagnostic applications,28,29 much attention has been focused on the preparation of PIPs for specific and selective protein separation from complex biological samples. However, several obstacles pose great challenges to creating PIPs by traditional imprinting methods.35,36 First of all, the large size made proteins difficult to diffuse into or out of the imprinted cavities, resulting in poor mass transfer and low protein desorption efficiency. To date, this issue has not been resolved but overcome to some extent by surface imprinting or epitope imprinting. Second, the structural and chemical complexity of proteins may cause non-specific binding and heterogeneous binding sites, subsequently affecting the binding behavior of PIPs. Besides, the protein's flexible structure and conformation, which are tightly related to the environmental changes, such as temperature, pH, ion strength and surfactant, may lead to the direct failure of the synthesis of PIPs.
Despite the great challenges in protein imprinting, the past decade has witnessed substantial progress in the design and application of PIPs. In addition to the improvement and upgrading of traditional imprinting methods, several novel protein imprinting methods, including boronate affinity-based molecular imprinting, solid-phase synthesis and post-imprinting modification, have emerged and further spawned into several advanced applications, including separation and purification, proteomics, biomarker detection, bioimaging and therapy (Fig. 2). Although some reviews have focused on a certain protein imprinting method or application have been published,37–40 a critical and comprehensive review is relatively scarce for summarizing the recent development of protein imprinting. In this review, we mainly focused on the recent developments in the imprinting strategies and highlighted applications for PIPs. Moreover, the challenges still existed in protein imprinting are discussed in detail and future prospects are also proposed.
 | ||
| Fig. 2 Overview of the imprinting methods for PIPs and their related applications. | ||
2. Protein imprinting methods
Although molecular imprinting with proteins as the templates was first reported by Glad et al.41 as early as 1985, the development of protein imprinting was still far behind small molecules. Due to the inherent properties of proteins, the traditional imprinting methods such as bulk imprinting, which were effective for small molecules, were not applicable to large proteins. However, after great efforts made over the past several decades, protein imprinting also has achieved significant progress. In addition to the improvement and upgrading of traditional imprinting methods, some advanced protein imprinting methods have emerged. Meanwhile, the successful examples of PIPs always integrated several imprinting methods together, rather than using only a single imprinting method. Up to now, protein imprinting methods can be mainly divided into traditional imprinting methods (bulk imprinting, surface imprinting and epitope imprinting) and emerging special imprinting methods (boronate affinity-based molecular imprinting, solid-phase synthesis and post-imprinting modification) (Fig. 2). In this section, we emphasized on reviewing the recent development of the protein imprinting methods and also discussed their merits and drawbacks (Table 1).| Synthesis methods for PIPs | Merits | Drawbacks |
|---|---|---|
| Bulk imprinting | 1. The most straight-forward method | 1. Long binding equilibrium time caused by limited diffusibility |
| 2. High-density imprinting sites | 2. Non-specific adsorption for homologous peptides | |
| 3. Limited imprinting efficiency derived from the conformational changes and solubility of proteins | ||
| 4. Binding sites will be destroyed by the mechanical crushing and grinding processes | ||
| Surface imprinting | 1. Quick binding kinetics derived from the favourable accessibility of proteins in and out of the imprinted sites | 1. Limited binding capacity due to the relatively low amount of template proteins for imprinting |
| 2. Multiple advanced functionalities by using various nanomaterials as substrates | ||
| Epitope imprinting | 1. The epitope peptides with simple structures could facilitate the immobilization and removal of templates; | 1. Ineffective when the amino acid sequence of protein is unknown |
| 2. Also, decrease the non-specific binding sites | 2. Suffer from limited accessibility to imprinted sites in the rebinding process | |
| 3. Epitope peptides were more stable, more available and cost-effective | ||
| Boronate affinity-based molecular imprinting | 1. The reversible boronate affinity could facilitate the immobilization and removal of glycosylation templates | 1. Mostly used for glycoprotein imprinting |
| 2. Enhanced specificity for glycoproteins | ||
| Solid-phase synthesis | 1. Automated operation and short production time | 1. Heterogeneous recognition sites from the variation of protein orientation |
| 2. High purity of nanoMIPs with high-affinity | 2. Low amount of templates immobilized on the solid support | |
| 3. Templates were reusable | ||
| 4. More homogeneous binding sites and high specific affinity | ||
| 5. High stability and good solubility of nanoMIPs | ||
| 6. Advanced functionalities by introducing functional nanomaterials | ||
| Post-imprinting modification | 1. Post-modification of the imprinted cavity | 1. Rational design of complex functional monomers |
| 2. Introduce more functionalities by various chemical derivatization | 2. A tedious chemical synthesis procedure | |
| 3. The effect of chemical modifications on the rebinding process is unknown | ||
2.1. Traditional imprinting methods
Although the bulk imprinting of proteins is far behind that of small molecules, it is still the most simplest method for protein imprinting. In recent years, the traditional bulk imprinting of proteins has also shown some progress. Several special bulk imprinting forms have been reported to imprint proteins.49–52 For instance, Boitard et al.50 exploited a fast and simple polymerization method using a grafting process onto magnetic nanoparticles to construct bovine serum albumin(BSA)-imprinted polymers by bulk imprinting. The resulting PIPs showed specific recognition and high adsorption capacity toward BSA. Notably, this imprinting process occurred in water media at room temperature, which might have the potential to imprint other proteins. This is the first example of a combination of bulk imprinting and grafting polymerization to synthesize PIPs. In addition, Tan et al.52 prepared a novel type of polymer-assisted hierarchically porous PDA/CaCO3 microparticles with a specific recognition ability toward lysozyme (Lyz). The authors called this method “hierarchically bulk imprinting”, which could increase the binding capacity of PIPs by improving the accessibility of recognition sites to proteins. Although alternative imprinting methods have been highly recommended to solve, these drawbacks remained in the bulk imprinting of proteins in the past few years, the above recent progress indicates the possibility of improving this method by integrating the advantages of other imprinting methods.
Silica nanoparticles (NPs) are the most common substrates for surface imprinting of proteins mainly due to their chemical/mechanical stability, biocompatibility, controllable sizes, ease of preparation and functionalization. Fu et al.63 synthesized core–shell surface-imprinted polymers of Lyz with modified silica NPs as the substrate. The silica NPs were pre-functionalized with double bonds and carboxylic acid groups using 3-aminopropyltrimethoxysilane and maleic anhydride. The double bonds could polymerize with crosslinkers and the carboxylic acid groups provided hydrogen bonds to interact with proteins. The resulted PIPs showed a high imprinting factor and large adsorption capacity toward Lyz. Then, our group64 prepared highly monodisperse and uniform core–shell surface imprinted polymers of Lyz with vinyl modified silica NPs as the substrate. This method was simple and the resultant Lyz-MIP silica NPs could rebind Lyz from human serum, indicating good selectivity toward Lyz. Dopamine (DA), which could provide non-covalent interactions with proteins, is an ideal monomer for protein imprinting. Interestingly, it can self-polymerize under the basic conditions at room temperature. Based on this, our group65 developed a facile method to synthesize polydopamine (PDA)-coated surface imprinted polymers of bovine hemoglobin (BHb) with non-modified silica NPs as the substrate. A thickness of only ∼5 nm of the surface imprinted PDA layer was obtained, which endowed the PIPs with rapid adsorption kinetics and high binding capacity. The successful application of the PIPs in deleting highly abundant BHb from cattle whole blood revealed their good potential in practical applications. Immobilized metal ion affinity chromatography (IMAC) is an effective method for the affinity separation and purification of proteins. Combining the IMAC and surface imprinting, Liu et al.66 fabricated a new type of core–shell structured surface-imprinted polymers of porcine serum albumin (PSA) with Cu2+-immobilized silica NPs as the substrate. The pre-modified Cu2+ could not only help to immobilize and remove the templates, but also provide recognition sites for rebinding PSA. The obtained PIPs displayed excellent selectivity and quick binding kinetics toward PSA. Hierarchical imprinting is a special surface imprinting strategy that creates imprinted polymers on the surface of the substrate and then sacrifices the substrate to expose the binding sites. Nematollahzadeh et al.67 synthesized hierarchically imprinted polymers of human serum albumin (HSA) using wide pore silica particles as the substrate. Then, the silica matrix was further etched, which not only helped to remove the templates but also exposed the binding sites on the surface of imprinted polymers. The obtained PIPs possessed a high binding capacity toward HSA and could purify the functional HSA from blood serum, indicating their promising potential in biotechnology. Based on the inherent recognition mechanism of MIPs, Bhakta et al.68 developed surface-imprinted silica NPs (AAHSA) to rebind HSA and glucose oxidase (GOx) by hydrophobic, hydrophilic and hydrogen bonding interactions, which were similar to those between the antibody and antigen (Fig. 3). In this study, four organosilane monomers with amino acid-like residues were used to form antibody-like interactions with target proteins. Unfortunately, the AAHSA showed a low adsorption capacity (5.9 mg g−1) toward HSA. Recently, Zhang et al.69 applied large pore silica as the substrate to successfully synthesize surface-imprinted polymers of BSA (MI-LPSPs). Similarly, two organosilane monomers were used to mimic antibody-like interactions to rebind BSA. Due to the introduction of large pore silica, the MI-LPSPs displayed a high adsorption capacity of 162.82 mg g−1 and fast adsorption kinetics toward BSA.
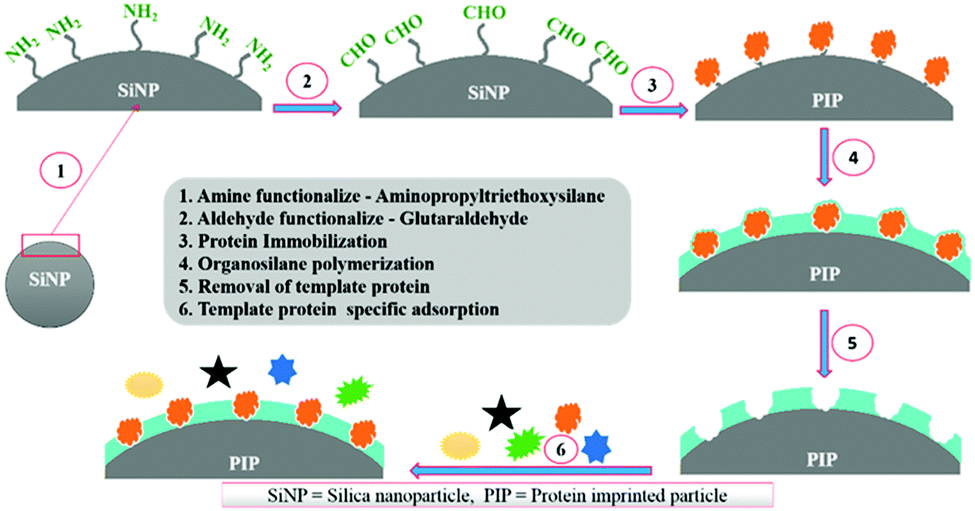 | ||
| Fig. 3 The synthesis procedure and recognition process of artificial antibody sites. Reprinted from ref. 68. | ||
Magnetic NPs such as Fe3O4 NPs are the most often used solid supports for sample preparation due to their magnetic responsiveness, which could make the synthesis and separation procedure very convenient using an external magnet. Meanwhile, magnetic NPs are easy to prepare and post-functionalize, and also possess good biocompatibility and reusability. Thus, magnetic NPs were another attractive substrate for the surface imprinting of proteins.37,40 Zhou et al.70 simply prepared MIPs on the Fe3O4 NPs by self-polymerization of DA, and resultant magnetic PIPs showed good recognition abilities and high adsorption capacities toward target proteins. Gao et al.71 successfully synthesized core–shell surface imprinted polymers with Fe3O4 NPs as the substrate. This imprinting method was demonstrated to be general for four proteins (BSA, BHb, Lyz, bovine pancreas ribonuclease A (RNase A)) with different isoelectric points. Among these proteins, the BHb–surface imprinted Fe3O4 NPs exhibited the best imprinting effect and highest adsorption capacity. The rebinding of BHb from the bovine blood demonstrated its good potential in practical applications. Based on their previous work,68 Bhakta et al.72 introduced the silica-coated Fe2O3 NPs as the substrate to construct HSA–surface imprinted polymers with antibody-like recognition sites toward target proteins. The magnetic cores endowed the PIPs with convenient separation and good reusability. Besides, ∼88% of albumin could be extracted from human serum using these new HSA-MIPs, which revealed the great potential for practical use. Recently, combining the IMAC and self-polymerization of DA, zhou et al.73 synthesized magnetic PDA-coated BSA-imprinted materials by a Ni2+-BSA directional coordination strategy. Hollow Fe3O4@mSiO2 microspheres served as the substrate. These well-designed PIPs possess high binding capacity, enhancing the imprinting factor and fast adsorption kinetics, and were successfully applied to rebind BSA from bovine serum samples. Generally, the major advantage of the introduction of magnetic NPs in the surface imprinting of proteins is the convenient separation procedure, which could extend the practical application of PIPs. In fact, their imprinting strategies did not have great differences from those of PIPs with silica NPs as the substrate.
Li et al.74 synthesized surface imprinted nanowires with a general imprinting effect on several target proteins (albumin, hemoglobin, and cytochrome c (Cyt c)) for the first time. In a typical synthesis procedure, the N,N′-methylenebisacrylamide and acrylamide were polymerized on the nanoporous alumina membrane with the template proteins modified on their pore walls. After the alumina membrane was etched, the templates were removed and imprinting sites were exposed on the surface of the imprinted nanowires. Because of the large surface area of nanowires, these imprinted nanowires exhibited high binding capacity toward target proteins. Based on this study, Ouyang et al.75 used the same method to synthesize surface-imprinted nanowires by self-polymerization of DA. Although the above two works exhibited a satisfactory imprinting effect, their synthesis procedures were tedious. To solve this problem, Chen et al.76 directly fabricated BHb-imprinted PDA polymers on the surface of silica nanowires. Without the etching step, the surface-imprinted silica nanowires still displayed large binding capacity, quick binding kinetics, and outstanding reusability. Inspired by the excellent performance of nanowires, another one-dimensional substrate of carbon nanotubes was then widely applied to construct surface-imprinted polymers of proteins.77–83 Zhang et al.77 described the synthesis of BSA-imprinted polymers on multiwalled carbon nanotubes (MWCNTs) by the polymerization of acrylamide and N,N′-methylenebisacrylamide for the first time. Unfortunately, these BSA-surface imprinted MWCNTs showed low adsorption capacity and slow binding kinetics toward template proteins. Xu et al.81 prepared Lyz-surface imprinted MWCNTs by self-polymerization of DA. Compared with other MIPs of Lyz, the highest binding capacity, excellent selectivity and good reproducibility were obtained in this study. To achieve high adsorption capacity and rapid adsorption kinetics, BSA–surface imprinted tubular carbon nanofibers (SIPTCFs) with self-driven properties were constructed by Yang et al.82 Self-polymerization of DA occurred on the substrate of TCFs–COOH (Fig. 4). Because of the high specific surface area, cavity and porous tube wall, SIPTCFs could reach an excellent adsorption capacity of 541.99 mg g−1 within 1 h and showed high selectivity toward BSA. Based on this work, the same group83 recently integrated the magnetic responsiveness with SIPTCFs to simplify the synthesis and separation procedure. The resultant surface imprinted magnetic tubular carbon nanofibers (SIPMTFs) still showed high capacity and good selectivity toward target proteins.
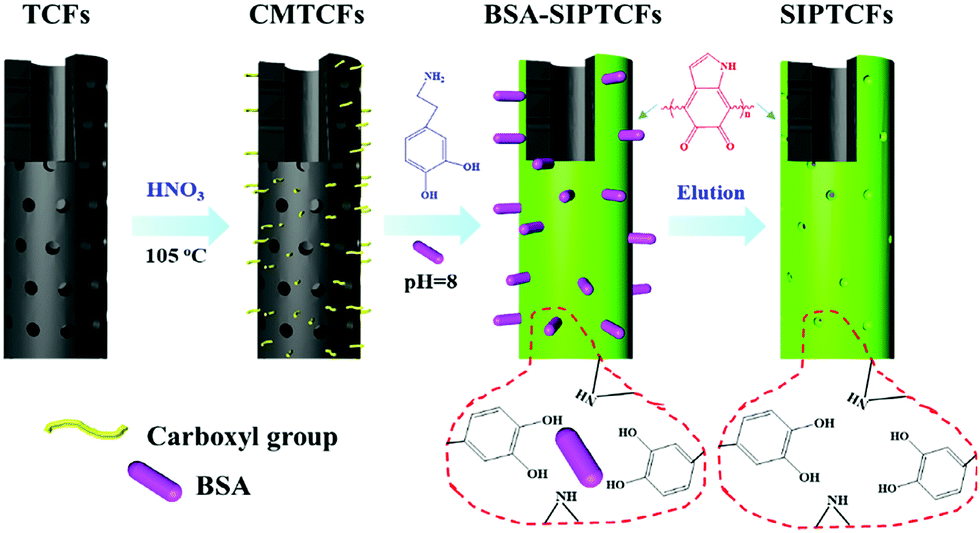 | ||
| Fig. 4 Preparation of self-driven surface BSA imprinted tubular nanofibers. Reprinted from ref. 82. | ||
Compared with the above nanomaterials, graphene with a two-dimensional plane structure has a larger surface area, which is considered an ideal substrate to create surface-imprinted polymers with higher protein rebinding capacity.84–87 For instance, graphene oxide (GO) nanosheets served as a novel substrate to yield BHb imprinted PDA@RGO.84 The as-prepared BHb imprinted PDA@RGO presented an excellent binding capacity of 198 mg g−1 toward BHb and fast adsorption kinetics to achieve the 89% of the maximum capacity within 5 min. The imprinting factor of 4.95 was obtained, indicating a good selectivity of this imprinted material. This study demonstrated that the two-dimensional structure and large surface area of GO could provide MIPs with enhanced binding capacity and more accessible recognition sites for target proteins. However, this type of graphene-based MIPs is tedious in the synthesis and separation processes. To this end, Fe3O4–graphene composites were then utilized as the substrate for the surface imprinting of proteins.85 Fe3O4–GO composites were first obtained by co-precipitation method and then used as the supporting substrate to fabricate Fe3O4–graphene PDA-based 2D MIPs with DA as the monomers and BSA as the templates. Benefiting from the large surface area of GO, the resulting PIPs displayed a high binding capacity of 117.1 mg g−1 and good selectivity for BSA and could be successfully applied in bovine blood samples.
Semiconductor fluorescent nanomaterials often termed “quantum dots (QDs)” are featured by their outstanding optical and electrical properties. Optical biosensors with increased stability, sensitivity and selective fluorescence response could be constructed by integrated surface imprinted polymers with QDs.60,61,88,89 For the first time, Tang et al.90 demonstrated the validity of the combination of CdS QDs and MIT to synthesize BSA–surface imprinted polymers for target protein recognition. The CdS QDs not only provided large specific surface areas, which were beneficial to high binding capacity but also were responsive to the binding with a template, which led to the quenching of the photoluminescence emission of CdS QDs. Zhang et al.91 then successfully prepared fluorescent Cyt c-imprinted CdTe QDs composites with tetraethoxysilane as the crosslinker and 3-aminopropyltriethoxysilane as the functional monomer. CdTe QDs were stabilized by 3-mercaptopropionic acid (MPA) and functionalized with carboxylic acid groups at the same time. The luminescence of CdTe QDs was quenched when Cyt c was rebound by the MIP-coated CdTe QDs. A linear range was from 0.97 μM to 24 μM, and the detection limit was 0.41 μM when using MIP-coated CdTe QDs to recognize Cyt c. The same imprinting method was further utilized to develop MIP-coated CdTe QDs for three template proteins (Lyz, Cyt c, and methylated BSA).92 However, the difference was that the denatured BSA-stabilized CdTe QDs were used, and also could provide recognition sites for target proteins. To enhance the binding capacity and accessibility of imprinting sites, MWCNT-QDs were later introduced to synthesize BSA–surface imprinted fluorescent polymers.93 Benefiting from the large surface area of MWCNTs, the resultant BMIP-coated MWCNT-QDs exhibited a fast binding response time of 25 min and high binding selectivity toward BSA. The linear range was 5.0 × 10−7–35.0 × 10−7 M, and the detection limit was 80 nM.
Although QDs have been successfully applied in PIPs, the toxicity and chemical instability greatly limit their wide applications. In contrast, upconversion nanoparticles (UCNPs) are more advantageous fluorescent materials due to their low toxicity, low photo-bleaching, long lifetimes, and lack of auto-fluorescence. Guo et al.94 demonstrated the feasibility of using UCNPs as the fluorescent substrate to construct the protein surface imprinted fluorescent materials. A sol–gel reaction occurred around Cyt c (template protein) on the surface of UCNPs. The obtained UCNPs@MIP exhibited strong fluorescence changes for the rebinding with Cyt c and the imprinting factor was 3.19, indicating a good selectivity of UCNPs@MIP. Encouraged by this study, the same group combined MIT with UCNPs and metal-organic frameworks (MOFs) to fabricate UCNPs/MOFs/MIP composites.95 In this study, BHb was chosen as the template, which has a metal coordination interaction with the Cu2+ in MOFs (HKUST-1). MOFs are attractive porous framework materials in various applications because of their fascinating properties including high specific surface area, tunable size and post-functionalization. Thus, the introduction of MOFs could increase the number of recognition sites and facilitate the mass transfer of UCNPs/MOFs/MIP toward target proteins. Meanwhile, a functional monomer of N-isopropyl acrylamide (NIPAAM) was used, resulting in the swelling and shrinking when the temperature changed. Finally, UCNPs/MOFs/MIP exhibited strong fluorescence changes toward the rebinding with BHb and achieved a high adsorption capacity of 167.6 mg g−1.
Furthermore, MOFs have also been introduced as a single substrate to synthesize surface-imprinted polymers of proteins. Taking advantage of MOFs including high specific surface area and porous structure, this type of PIPs always showed outstanding binding capacity and kinetics. Li et al.96 applied MOF-74(Ni) as the substrate to form surface-imprinted polymers of Lyz via the self-polymerization of DA. The MOF-74(Ni) not only provided a high specific surface area of 150.0 m2 g−1, but also facilitated the immobilization and rebinding of Lyz through the metal chelation of Ni2+. A thickness of 10 nm for the imprinted layers was obtained, which enabled a fast binding kinetic of 10 min. Encouragingly, the resulting MOF@PDA-MIP achieved a significant binding capacity of 313.5 mg g−1 and imprinting factor of 7.8, demonstrating that MOFs are promising materials for the surface imprinting of proteins. Inspired by this study, Qian et al.97 utilized MOFs/carbon nanoparticle (CN) composites (CN@UIO-66) as the substrate to construct the protein surface imprinted polymers. Interestingly, the resulting CN@UIO-66@MIPs possessed a high specific surface area of 551.4 m2 g−1, which led to an extremely high binding capacity (815 mg g−1) toward Cyt c. Meanwhile, a high imprinting factor of 6.1 and rapid adsorption kinetics of 40 min were obtained.
As discussed above, compared with bulk imprinting, surface imprinting has become an advantageous method for creating PIPs due to their favourable accessibility to target proteins. Just as a coin has two sides, surface imprinting for proteins easily suffers from low binding capacity because of the decrease in recognition sites. Nanomaterials, featured by their high specific surface area, could be introduced as the imprinting substrate to solve this problem. Thus far, various nanomaterials, including silica NPs, magnetic NPs, MWCNTs, graphene, QDs, UCNPs and MOFs, have been applied as the substrate to fabricate protein–surface imprinted polymers. It should be noted that, in addition to the improvement of binding performance, the introduction of nanomaterials could endow protein–surface imprinted polymers with some special functionalities such as magnetic responsiveness and fluorescence properties, which have the potential to extend their wide applications. With the rapid upgrade of nanomaterials, more protein surface imprinted polymers with excellent adsorption performance and advanced functionalities will be developed in the future.
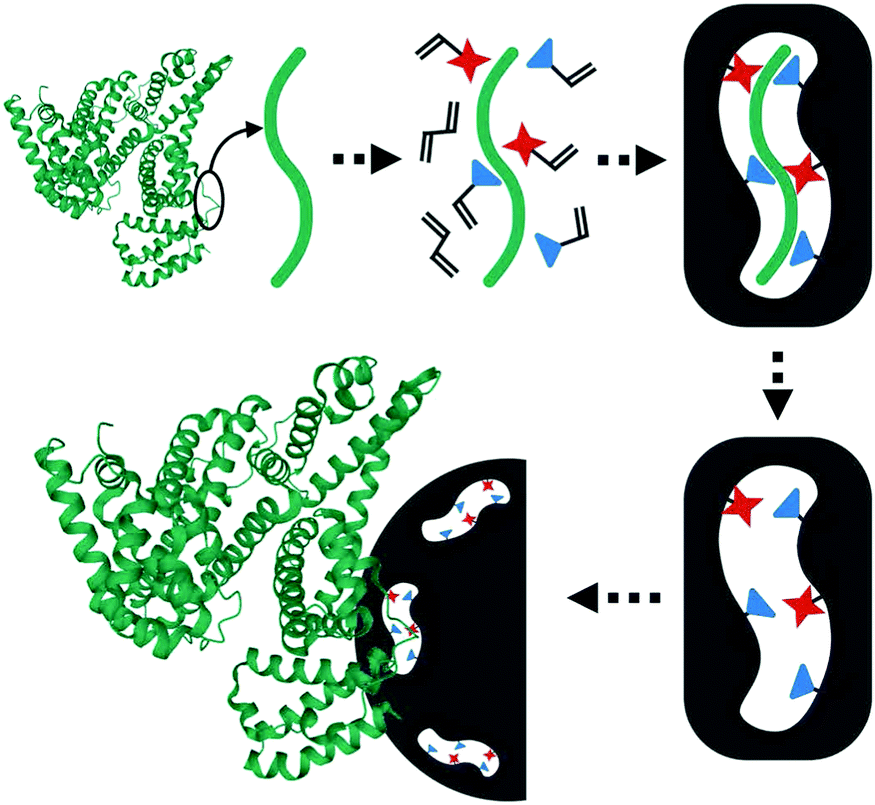 | ||
| Fig. 5 Rationale of the epitope imprinting concept. Reprinted from ref. 99. | ||
Nishino et al.101 developed a general method to create surface-imprinted polymers toward three proteins (Cyt c, Lyz and alcohol dehydrogenase (ADH)) by epitope imprinting. Unique epitope peptides (nine amino acid sequences) exposed on the surface of three proteins were used as the templates. After the epitope peptides were immobilized on the pretreated glass or silicon surface, the polymers were formed by free radical polymerization. Then, the imprinted sites were exposed by the etching of the glass or silicon to yield the protein-imprinted polymer film. Yang et al.102 synthesized transferrin (TRF) epitope imprinted polyethersulfone (PES) beads by self-assembly polymerization around the epitope template of the N-terminal sequence (MRLAVGALL). The transferrin epitope imprinted particles showed excellent binding capacity and selectivity toward transferrin epitope and transferrin, and were successfully applied in a real sample of human plasma. Then, the same group103 tried to develop a multiepitope imprinting strategy to synthesize multiepitope imprinted particles using the same PES self-assembly method. Three epitope peptides of proteins (HSA, TRF, and immunoglobulin G (IgG)) were selected as the templates. The resulting multiepitope imprinted particles could simultaneously capture three proteins from the complex human plasma, indicating the promising potential of this multiepitope imprinting strategy in practical applications. Zhao et al.104 synthesized magnetic surface epitope-imprinted polymers with a core–shell structure (Fe3O4@EMIPs) for the separation of BSA by combining epitope imprinting, surface imprinting, and Fe3O4 NPs. The introduction of Fe3O4 NPs made the synthesis and separation procedure more convenient. The as-prepared Fe3O4@EMIPs showed high selectivity toward BSA from bovine blood, indicating their good potential in real use. Li et al.105 applied His-tag-anchored epitope from HSA as a template to provide a new oriented surface imprinting method. The surface imprinting occurred on the Fe3O4@SiO2@IDA@Ni2+ substrate. The introduction of strong metal coordination between Ni2+ and His-tag-anchored epitope could make the immobilization and removal of templates more convenient. After optimizing the utilization efficiency of templates and thickness of the imprinted layer, the obtained epitope-oriented surface-imprinted nanoparticles exhibited specific recognition ability toward HSA. Based on this study, the same group106 then prepared epitope-oriented surface-imprinted nanoparticles using His-tag (HHHHHH) as the template and were further applied to purify His-tagged proteins. In this example, the IMAC could not only help to immobilize and remove templates but also provide affinity sites for rebinding His-tagged proteins. Due to the high selectivity of MIT, the authors called this method “Epitope imprinting enhanced IMAC (EI-IMAC)”. Qin et al.107 created epitope-imprinted polymers with magnetic carbon nanotubes (MCNTs) as the substrate, epitope peptide (AYLKKATNE) derived from Cyt c as the template (Fig. 6). MCNTs@EMIP was fabricated by free radical polymerization using ethylene glycol dimethacrylate (EGDMA) as a cross-linker and zinc acrylate as a functional monomer. The hydroxyl and amino groups of the epitope could form a five-membered ring with the Zn2+. Together with the large surface area of CNTs, these PIPs achieved an extremely high binding capacity of 780.0 mg g−1 and an imprinting factor of 11.7. The outstanding binding performance showed the successful separation of Cyt c from a real bovine blood sample, indicating the excellent selectivity of MCNTs@EMIP and promising potential in real use. Li et al.108 synthesized thermo-sensitive epitope surface imprinted polymers by combining carbon dots (CDs) and SiO2. Double templates of epitope peptides derived from C- and N-terminals of Cyt c were used. The resulted CDs/SiO2/MIP exhibited fluorescence quenching when rebinding with the target protein of Cyt c. Then, a linear range of 0.1–40 μM and a detection limit of 89 nM were obtained. Meanwhile, the temperature-sensitive monomer endowed the CDs/SiO2/MIP with swelling and shrinking capabilities with temperature changes.
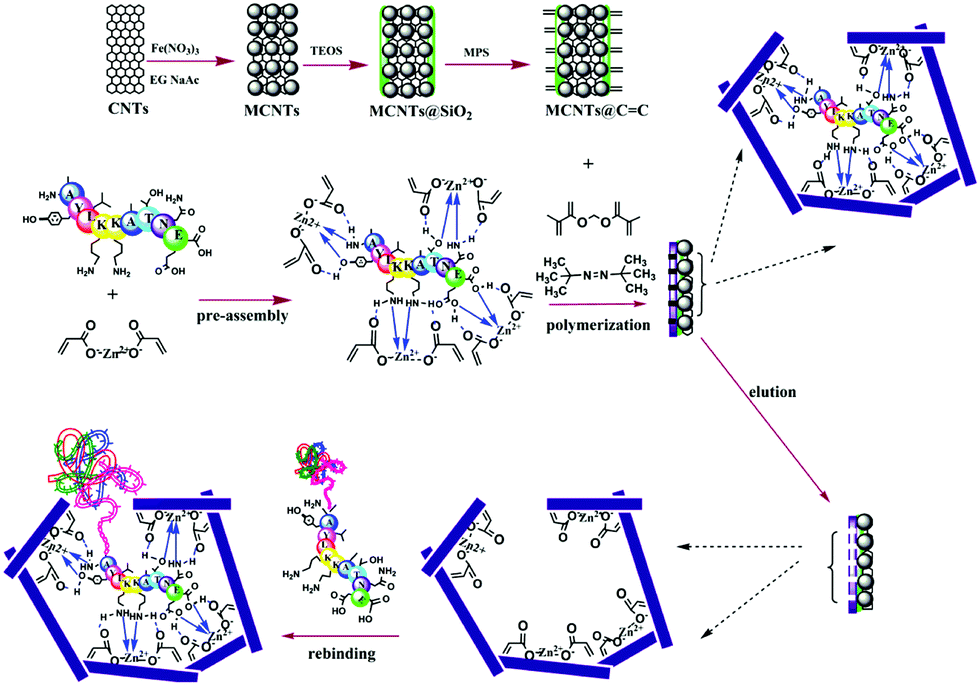 | ||
| Fig. 6 Synthesis protocol of MCNTs@EMIP via surface imprinting and epitope imprinting. Reprinted from ref. 107. | ||
Generally, epitope imprinting is the most potent protein imprinting method because they are based on the recognition mechanism between antibody and antigen is consistent with MIT, which is also the most attractive advantage of MIT. In the past few years, epitope imprinting of proteins has achieved significant progress. Many new types of epitope-imprinted polymers with excellent properties and multifunctionalities were continuously developed by integrating the advantages of surface imprinting and advanced nanomaterials. If the amino acid sequence exposed on the surface of the protein is known, the corresponding imprinted polymers could be created by using the peptide as a template. In contrast, epitope imprinting will fail when the amino acid sequence is unknown.
2.2. Emerging special imprinting methods
Our group111 synthesized a molecularly-imprinted monolithic column for HRP by combining boronate affinity and surface imprinting. HRP was first modified on the boronate-functionalized monolithic column and then self-polymerization of DA occurred. After the templates were removed, the resulting imprinted monolith exhibited high recognition ability toward HRP and was successfully applied in human serum, indicating a good potential in practical use. Then, our group112 developed surface-imprinted polymers of HRP with 3-acrylamidophenylboronic acid-immobilized silica nanoparticles as the substrate. A sol–gel reaction occurred on the HRP-immobilized SiO2@AAPBA NPs. The obtained PIPs showed good adsorption ability and high selectivity for HRP with an imprinted factor of 2.71, and further successfully separated HRP from human serum. Later, the “thiol–ene” click reaction was applied to construct PDA-coated boronate affinity-imprinted silica NPs for HRP recognition.113 Liu et al.114 applied boronic acid-modified graphene oxide (GO-APBA) as the substrate to create surface-imprinted polymers of ovalbumin (OVA). The template OVA was first modified on the GO-APBA substrate and then a sol–gel reaction occurred. To the combination of boronic acid and surface imprinting, together with a large surface area of GO, the resulting PIPs exhibited a high binding capacity of 278 mg g−1, rapid adsorption kinetics of 40 min, and imprinted factor of 9.5. Sun et al.115 introduced Fe3O4 NPs as the substrate to synthesize HRP-surface imprinted polymers with a convenient synthesis and separation procedure.
Li et al.116 developed a “photolithographic boronate affinity molecular imprinting” strategy to successfully create surface-imprinted polymers toward five different glycoproteins. UV-initiated free radical polymerization occurred between the functional monomer of APBA and a crosslinker around the template glycoproteins. This method is fast, tolerant of interference, and applicable to a wide range of pH. The successful detection of a-fetoprotein (AFP) with low concentrations from human serum by MIP array-based enzyme-linked immunosorbent assay (ELISA) demonstrated the great potential for practical use in complex real samples. Then, the sample group117 developed another facile and general “boronate affinity-based controllable oriented surface imprinting” strategy for the recognition of glycoproteins. The boronic acid pre-functionalized substrate was first used to immobilize templates and then a thickness-controllable imprinting layer was formed on the substrate by self-polymerization of DA and APBA. After the templates were removed, boronate affinity-based controllable oriented surface imprinted polymers were obtained. The affinity of such MIPs could be tuned by controlling the strength of the boronate affinity interaction by adjusting the pH. Xing et al.118 introduced a new and universal strategy called “controllable oriented surface imprinting of boronate affinity-anchored epitopes” (Fig. 7). This work is the successful integration of surface imprinting, epitope imprinting, and boronate affinity. The template of glycated epitope peptide was used, which could facilitate the immobilization and removal of templates on the surface of the boronic acid-functionalized substrate. The resulting MIPs could recognize both the peptides and proteins. This method is efficient and general for creating MIPs with an affinity to proteins. Based on this work, the same group119 proposed a new strategy called molecular imprinting and cladding (MIC) to solve the dilemma between the best affinity and best specificity for MIPs. The key to this strategy is the chemically inert cladding thin layer generated after molecular imprinting to cover the non-imprinted area. Using this strategy, a special approach termed boronate affinity-anchored epitope-oriented surface imprinting and cladding (BOSIC) was developed. The resulting cMIPs were general for the recognition of TRF, TRF receptor (TfR), AFP and carcinoembryonic antigen (CEA), and were successfully applied for fluorescence imaging of cancer cells against normal cells by targeting TfR by encapsulating fluorophore and the diagnosis of diabetes by detecting C-peptide in human urine using a dual cMIPs-based surface-enhanced Raman scattering (SERS) assay.
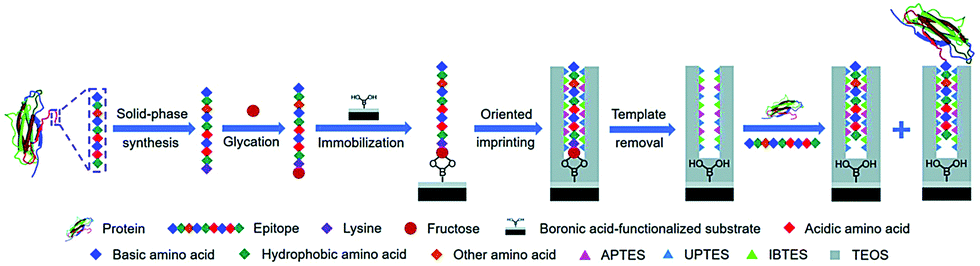 | ||
| Fig. 7 Schematic of the principle and procedure of controllable oriented surface imprinting of boronate affinity-anchored epitopes. Reprinted from ref. 118. | ||
Overall, the boronate affinity-based molecular imprinting method is successful and tailor-made for glycosylated proteins. By integrating the advantages of other imprinting methods, a series of novel, special and general imprinting strategies based on boronate affinity interaction for glycoproteins has been developed and applied in various advanced fields such as clinical disease diagnosis and biological imaging. Although this method is only effective for glycoproteins, as a successful example of glycoprotein imprinting, its success could undoubtedly provide a lot of inspiration and thoughts for the development of protein imprinting methods, which is a general method for different proteins.
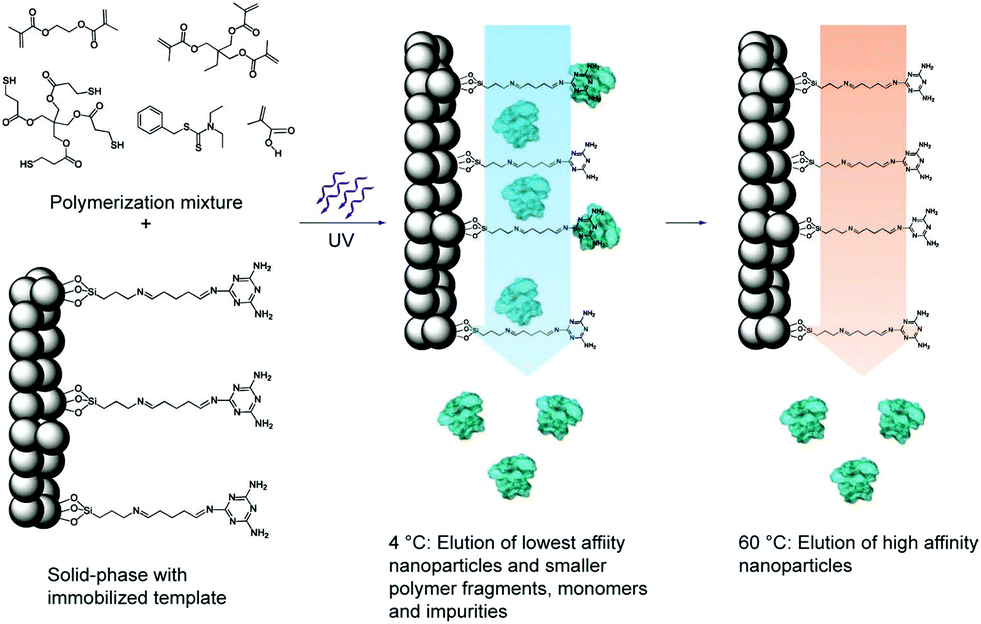 | ||
| Fig. 8 Schematic representation of the solid-phase synthesis of MIP nanoparticles. The monomer mixture is injected onto the column reactor with an immobilized template and polymerization is initiated by UV-irradiation. The low-affinity particles, as well as unreacted monomers, are eluted at low temperatures. The temperature is then increased and high-affinity particles are eluted from the column for collection. Reprinted from ref. 120. | ||
By using this method, Poma et al.122 constructed an automated solid-phase synthesis method for synthesizing nanoMIPs under computer control in aqueous media. Using this method, they successfully prepared nanoMIPs with specific recognition ability toward three proteins (pepsin A, trypsin and α-amylase), indicating a good potential in industrial manufacturing. However, the template proteins were modified on the solid support by covalent coupling of glutaraldehyde, which resulted in the heterogeneous recognition sites from the variation in protein orientation. Thus, Ambrosini et al.123 presented a novel method to prepare nanoMIPs by using an affinity ligand (p-aminobenzamidine (PAB)) of protein to immobilize the template trypsin. Different from the covalent coupling, the affinity ligand endowed all the binding sites with the same orientation, thus leading to more homogeneous binding sites. The obtained nanoMIPs exhibited high specificity and selectivity toward trypsin. Based on the same strategy, Xu et al.124 first formed polymer NPs on the trypsin–PAB–solid support and then modified fluorescein isothiocyanate (FITC) on the polymers NPs. After being removed from the solid support, fluorescent MIP NPs were obtained with high affinity toward trypsin and showed almost no cross-reactivity with other proteins. Finally, the fluorescent MIP NPs were employed in a sandwich fluoroimmunoassay for the detection of trypsin with a low concentration of 50 pM spiked in human serum. The same group125 introduced metal chelation (Cu2+), inspired by PAB, to immobilize proteins with surface histidines for the fabrication of nanoMIPs by solid-phase synthesis. The IMAC interaction helped form oriented homogeneous imprinting sites on the surface of nanoMIPs. This study provides a general method for solid-phase synthesis of nanoMIPs for proteins. Then, Xu et al.126 applied a cyclic 3S epitope (CGSWSNKSC) immobilized GBs as the solid support to fabricate water-soluble nanoMIPs with oriented homogeneous imprinting sites by solid-phase synthesis. The 3S cyclic epitope is the motif of the envelope glycoprotein 41 (gp41) of human immunodeficiency virus type 1 (HIV-1) and thus, the resulting anti-3S antibodies could be applied to target and block the 3S peptide of HIV-1 to prevent CD4+ T cells from declining.
In the above works, microsized glass beads served as the solid support. Their low specific surface area leads to a small amount of immobilization of template proteins and the low yield of nanoMIPs. Thus, Ashley et al.127 employed epoxide magnetic microspheres (FeOx@SiO2-epoxide, 600–700 nm) as the solid support to immobilize trypsin. Because of the high specific surface area, highly abundant templates could be immobilized. Compared with conventional solid-phase synthesis, a high yield of nanoMIPs with increased 83–167 folds could be achieved by using magnetic microspheres as the solid support. Meanwhile, magnetic responsiveness facilitates the synthesis and separation procedure. This method was called “dispersive solid-phase imprinting” because the imprinting process was conducted on the surface of FeOx@SiO2-epoxide, which was dispersive in the mixture. Mahajan et al.128 used amino-functionalized sol–gel coated magnetic nanoparticles (magNPs) as solid support to immobilize the templates of enzymes trypsin and pepsin. A high-dilution polymerization of monomers occurred around the template on the solid support. A monomer of N-fluoresceinylacrylamide was added to yield fluorescent nanoMIPs with high specificity and selectivity toward the respective template proteins.
For the first time, Cecchini et al.129 developed fluorescent nanoMIPs by combining solid-phase synthesis, surface imprinting, epitope imprinting and QDs. The template of the epitope peptide of human vascular endothelial growth factor (hVEGF) was immobilized on the glass beads. The resulting fluorescent nanoMIPs exhibited excellent specificity toward hVEGF, and were successfully applied in the target imaging of overexpressing hVEGF. Based on the same strategy, Gómez-Caballero et al.130 synthesized nanoMIPs by using epitope peptide (C-terminus 15 amino acids sequence) of the CB1 receptor. The sizes of nanoMIPs were found to be related to temperature changes. The nanoMIPs were successfully demonstrated as artificial anti-CB1 antibodies to high selectively recognize the target proteins with 15 amino acids epitope.
Solid-phase synthesis is the most promising method to create MIPs as artificial antibodies with comparable size, specificity and solubility to antibodies. Due to its facile and automated operation, it has significant potential in industrial production. In the past few years, solid-phase synthesis has achieved some great progress, mainly emphasizing the increase of nanoMIPs yields, more homogeneous binding sites, advanced functionalities and further extending their biological applications. Many excellent solid-phase synthesis approaches have been developed to create advanced nanoMIPs by combining the advantages of other imprinting methods.
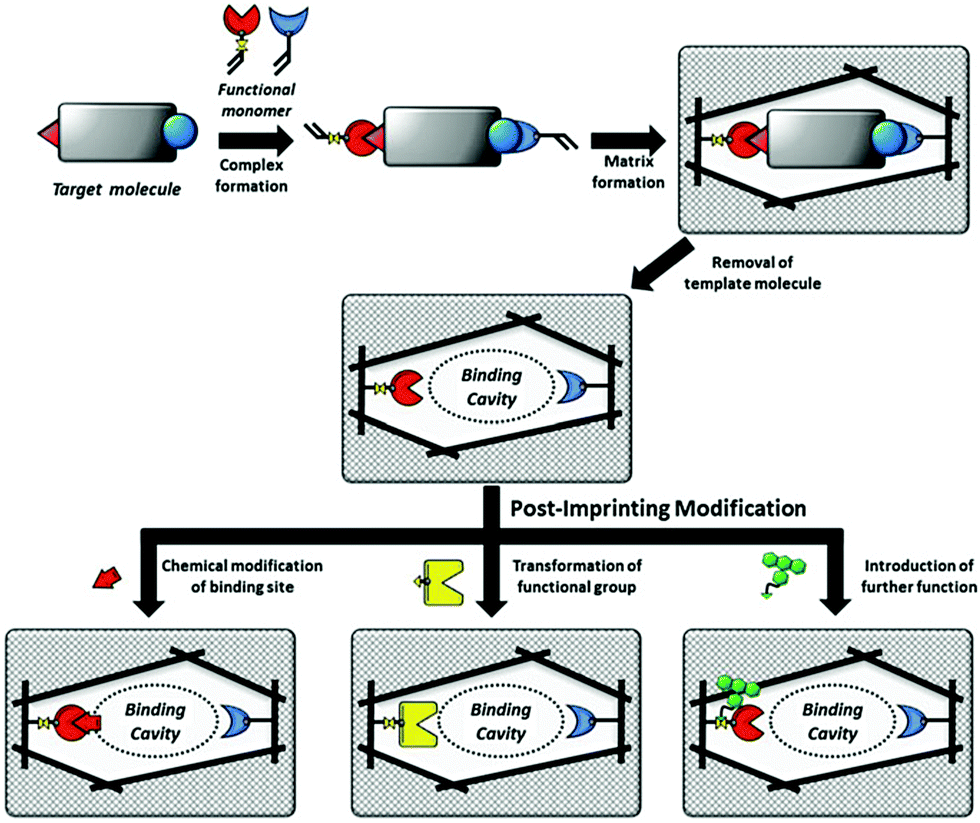 | ||
| Fig. 9 Schematic illustration of MIPs and PIMs of the binding cavities of MIPs. Reprinted from ref. 132. | ||
Tao et al.137 developed a one-step PIM strategy to fabricate a biosensor to characterize the specific recognition of glycoprotein (HRP). After the removal of the template HRP, the exposed thiol residues in the imprinted cavities were modified with boronic acid ligand by the “thiol–ene” click reaction. Due to the fluorescence response of the boronic acid ligand, the resulting PIPs could act as a biosensor for HRP detection by the transduction of binding events into fluorescence change. Based on their previous work,136 Morishige et al.138 created a new recognition system for AFP by PIMs. In this work, AFP was first immobilized on the surface of 4-carboxy-3-fluorophenylboronic acid (CFPBA)-functionalized support. After polymerized, AFP was removed by cleavage of disulfide bonds and cyclic diesters. Then, fluorescent dyes were functionalized with the exposed thiol groups to yield a fluorescent AFP-imprinted biosensor. The introduction of CFBA could not only facilitate the oriented immobilization of template AFP but also provide recognition sites for AFP. The successful detection of AFP in human serum demonstrated good potential in biomedical application. Wang et al.139 presented a ratiometric nanosensor by PIMs for the fluorescence determination of OVA. Meanwhile, the binding result could be transformed into the visual identification on fluorescent test papers under UV (365 nm). OVA-imprinted polymers were first constructed on the SiO2 NPs. FITC was post-modified in the imprinted cavities after the templates were removed. The resulting ratiometric nanosensor showed a detection limit of as low as 15.4 nM and could be successfully applied in real samples of human urine and chicken egg white, indicating its good potential in real applications.
The major advantage of the PIM method is to impart new functionalities to PIPs. Diverse functionalities could be introduced by various chemical derivatization based on the excellent stability of the imprinted polymers. These new functionalities will transform the recognition behaviour toward template proteins into optical or other responses, which greatly exceed more wide applications of PIPs. As discussed above, solid-phase synthesis could create nanoMIPs as artificial antibodies with comparable properties to those of natural antibodies. By using the PIM strategy, sophisticated MIP-based artificial antibodies with functionalities have the potential to go beyond natural antibodies. However, PIM also suffered from some drawbacks such as the design of complex functional monomers and tedious chemical synthesis procedures. Meanwhile, due to the unclear recognition mechanism of PIPs, the rational design of chemical modifications in the imprinted cavities should be further explored.
3. Applications of PIPs
Over the past decade, protein imprinting has achieved significant progress. Several novel imprinting methods have emerged and many protein-imprinted polymers with advanced properties have been developed, which extend their wide applications, including separation and purification, proteomics, biomarker detection, bioimaging and therapy. In this section, we focus on the recent development of PIPs in their highlighted applications.3.1. Separation and purification
The high interferences derived from the complex components of biological samples pose great challenges for the separation of target proteins. Based on the rapid development of surface imprinting and nanomaterials, various PIPs with excellent binding capacity and selectivity were developed and applied for the separation and purification of proteins from complex biological samples such as human serum. Our group64 synthesized Lyz–surface imprinted Silica NPs with high selectivity toward Lyz in human serum. Then, we created a thin BHb-imprinted polymer on silica NPs, which possessed a large binding capacity, high adsorption selectivity and fast binding kinetics for BHb.65 Inspired by the excellent binding performance, the as-prepared surface-imprinted silica NPs could successfully separate BHb from cattle whole blood samples with high selectivity. Tan et al.52 synthesized poly(styrenesulfonate sodium) (PSS)-assisted hierarchical bulky imprinted microparticles (denoted as PSS–PDA–MIP) of Lyz by a novel polymer-assisted hierarchically bulk imprinting strategy. Then, the as-prepared PSS–PDA–MIP possessing ultrahigh adsorption capacity of 1203.4 mg g−1 and high selectivity for Lyz was successfully applied for the highly selective separation of Lyz from diluted egg whites and spiked human serum. Pan et al.140 fabricated multi-responsive rattle-type magnetic hollow molecular imprinted poly (ionic liquids) nanospheres with BSA as the template. The resulting Fe3O4@void@PILMIP were confirmed to show specific recognition ability toward BSA and were successfully applied in the separation of BSA from bovine calf serum. Yang et al.141 synthesized thermo-sensitive surface-imprinted hollow nanocages with ZIF-67@Co–Fe as the substrate (Co–Fe@CBMA–MIPs) for the recognition of BSA. ZIF-67@Co–Fe provided a high specific surface area and Co–Fe double ions facilitated the immobilization and removal of the templates. Under optimum adsorption conditions, the Co–Fe@CBMA–MIPs displayed a high binding capacity of 520.35 mg g−1, rapid binding kinetics of 50 min, and imprinting factor of 8.55. Encouraged by their excellent binding performance, these PIPs could successfully separate BSA from fetal bovine serum. Bhakta et al.72 applied antibody-like magnetic surface imprinted polymers to separate HSA. Benefiting from the magnetic responsiveness and high binding capacity, the resulting PIPs achieved the removal of ∼88% of albumin from human serum. Yang et al.102 successfully applied transferrin (TRF) epitope-imprinted PES beads for a human plasma proteome analysis and quantification for the first time. Then, the same group103 fabricated multiepitope imprinted PES particles to simultaneously capture three proteins (HSA, TRF, and IgG) from the complex human plasma, indicating the promising potential in practical applications (Fig. 10). Li et al.106 further proposed an EI-IMAC strategy to design surface epitope imprinting polymers with His-tag epitope as a template. The resulting PIPs possessed fast adsorption kinetics (15 min) and a high imprinting factor (7.1). Compared with the IMAC strategy, this strategy exhibited increased purity (∼5%) for His-tagged recombinant proteins separated from crude cell lysis. Weerasuriya et al.142 presented a new strategy to create surface-imprinted silica-coated magnetic NPs with protein (PrA)-like nanopockets for the purification of IgG antibodies. Mouse IgG2a, which is the binding partner of PrA, was used as the template. A sol–gel reaction of organosilane monomers occurred around the templates. After capping the nonbinding sites and removing the templates, the resulting PIPs showed PrA-like specific recognition of IgG. Rough core–shell NPs were found to possess a high binding capacity and better selectivity than the commercial PrA magnetic beads. These PIPs are cost-effective, reusable, stable, and the first report to mimic PrA recognition to purify antibodies.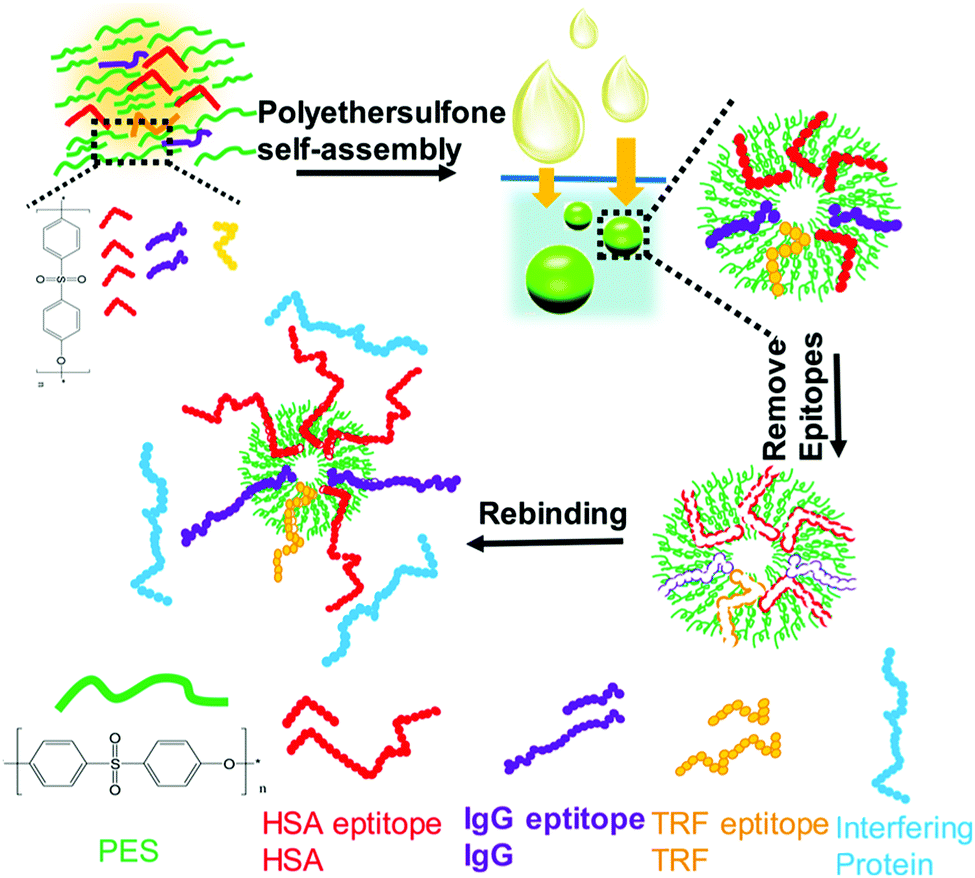 | ||
| Fig. 10 Fabrication of multiepitope templates imprinted particles via PES self-assembly and application in the simultaneous capture of various target proteins. Reprinted from ref. 103. | ||
3.2. Proteomics
To date, mass spectrometry (MS) has become a powerful and popular tool for proteomic analysis because of its high resolution, high throughput, high sensitivity, and high accuracy. However, the analysis of complex biological samples by MS still remains a great challenge. This is because the complex components of biological samples often lead to signal interference, and greatly suppress the MS ionization of target proteins. Thus, prior to MS analysis, highly selective separation of target proteins from complex biological samples is urgently needed. Protein imprinted polymers featured by excellent selectivity have great potential for MS analysis of complex biological samples.Wan et al.143 for the first time-developed PIPs to highly separate target proteins for matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) analysis. The self-polymerization of DA occurred around the template Lyz. After the templates were removed, thin Lyz-imprinted polymers were introduced on the Fe3O4@SiO2 NPs to yield Lyz-surface imprinted magnetic NPs (Lyz-MIPs). Lyz-MIPs showed excellent binding capacity and high selectivity for Lyz, then applied to highly selectively separate Lyz from diluted egg white samples for MALDI-TOF MS analysis. The enhanced effect of MALDI-TOF MS signals for Lyz was demonstrated after the selective separation procedure via Lyz-MIPs. Bertolla et al.144 proposed a fast and easy online method for target protein analysis by integrating PIPs with MALDI-TOF MS. Poly-(acrylamido)-derivative (PAD) nanoMIPs were synthesized using human serum transferrin (HTR) as the template. The resulting solvent-responsive PAD-nanoMIPs not only showed specific recognition ability toward HTR but also could release in situ the bound HTR from the PAD-nanoMIPs-target-plate by adding acetonitrile for MALDI-TOF MS analysis. This PAD-nanoMIPs/MALDI-TOF MS analysis platform could successfully detect HTR from a real sample of serum, indicating its great potential in clinical diagnosis and targeted proteomics. Cyt c, which is important in cell apoptosis and can interact with anti-apoptosis proteins, which leads to the effect of functions. Two pivotal anti-apoptosis proteins of HSP27 and Bcl-xL could interact with Cyt c. Based on these findings, to study Cyt c-HSP27 and Cyt c-Bcl-xL interactions, Zhang et al.145 developed a simultaneous quantification method by combining protein imprinting and an LC-MS/MS method (Fig. 11). The template of the epitope peptide of Cyt c was used to synthesize Cyt c-surface imprinted silica NPs. The binding performance, including adsorption capacity, binding kinetics and selectivity was confirmed in detail. Compared with the Co-IP/Western Blotting, this combinational approach could successfully quantitatively analyse the protein–protein interactions within Cyt c and two anti-apoptosis proteins in a different complex biological sample of breast cancer cells.
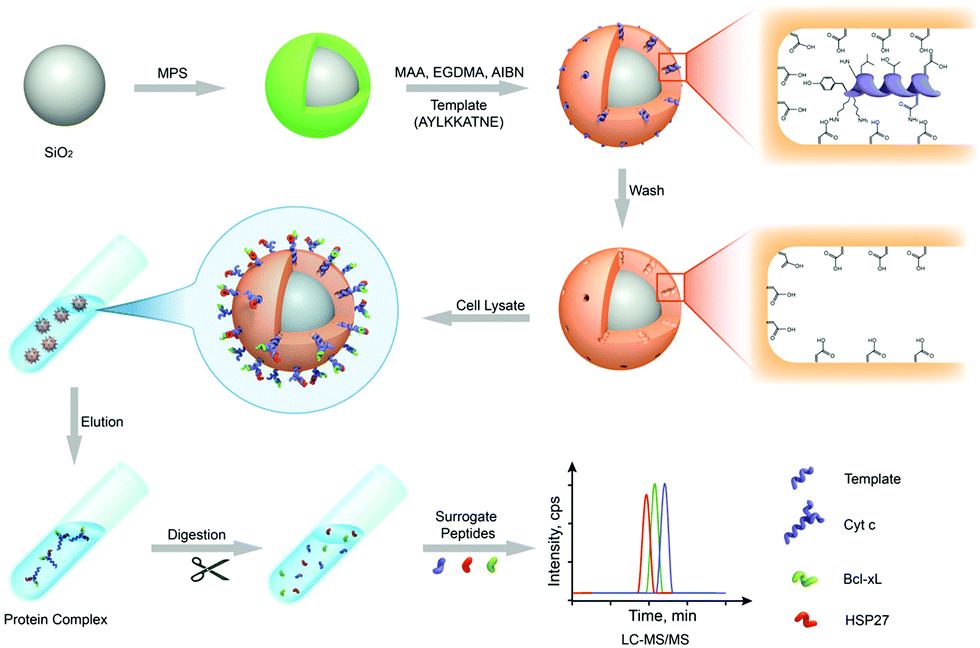 | ||
| Fig. 11 Schematic representation of MIPs coupled with LC-MS/MS-based targeted proteomics for the simultaneous quantification of Cyt c interactions with HSP27 and Bcl-xL. Reprinted from ref. 145. | ||
3.3. Biomarker detection
PIPs, which are considered synthetic antibodies, have also been used as recognition materials for protein biomarker detection from complex pathological samples. Based on their previous work,117 Bi et al.146 developed boronate affinity imprinted microplates as ELISA to detect AFP. 4-Formylphenylboronic acid (FPBA) was first functionalized on the bottom of the well and walls of a 96-well microplate. Then, the template glycoproteins were immobilized on the basis of the boronate affinity interactions, and self-copolymerization of aniline in water occurred around the templates. After the removal of the templates in acid media, glycoprotein-imprinted microplates were obtained with the appropriate thickness of imprinted layers. This strategy was first demonstrated by the excellent binding performance of PIPs with standard HRP as an imprinting template. Finally, a PIPs-based sandwich ELISA was constructed for the detection of AFP by integrating with AFP-imprinted microplates. Under the optimal analysis condition, a concentration of 12 ± 2.0 ng mL−1 for AFP in human serum was detected by this ELISA, which corresponded to the result (10 ng mL−1) obtained by radioimmunoassay. However, because of the use of specific antibodies and biological reagents, this ELISA was not stable and expensive. Thus, based on their previous work,116 the same group147 further developed a novel boronate-affinity sandwich assay (BASA) for AFP analysis by integrating photolithographic boronate-affinity molecular imprinting with boronate-affinity based SERS probe (AgNPs) (Fig. 12). Standard HRP was first selected as the template to evaluate the binding performance of BASA. The practical potential of this BASA was then demonstrated by the AFP-imprinted BASA. To the human serum containing AFP with a known concentration of 12.0 ± 2.0 ng mL−1, the concentration of 13.8 ± 3.3 ng mL−1 was determined by the AFP-imprinted BASA. This approach is stable, fast, cost-effective, and has great potential in clinical diagnostics with high throughput. Patra et al.148 developed a trace prostate-specific antigen (PSA) electrochemical sensor by combining protein–surface imprinted polymers and nanomaterials. MnO2-Nanoparticles were modified on the surface of MWCNTs to yield MnO2-nanoparticle decorated MWCNTs-iniferter. This iniferter was attached to a pencil graphite electrode (PGE) and then a controlled radical polymerization occurred with the existence of the template PSA. After the templates were removed, a PSA-imprinted electrochemical sensor was obtained. The detecting limit of this PSA-sensor by the square wave stripping voltammetric (SWSV) technique was 0.25 pg L−1, and 3.04 pg L−1 by the differential pulse stripping voltammetric (DPSV) technique. The successful detection of PSA in men and women samples of serum, urine, and forensic by the as-prepared sensor was demonstrated using the commercially available ELISA, indicating the great potential in the clinical diagnosis of PSA. Shumyantseva et al.149 synthesized myoglobin (Mb)-imprinted polymers on the MWCNTs-modified electrodes for the sensitive detection of Mb in undiluted human plasma samples. Based on this Mb-electrochemical sensor, plasma samples could be accurately classified and referred to related groups of healthy donors (HDs) and patients with acute myocardial infarction (AMI) by combining the multi-parameter electrochemical analysis and computational cluster assay. Compared with other standard immunochemical methods for Mb detection, this approach is facile, fast, cost-effective, and showed excellent potential in personalized medicine such as “point-of-care” biosensors. Karami150 constructed a novel and low-cost dual-modality immunosensor for the simultaneous detection of PSA and Mb by combining protein–surface imprinting and nanocomposite (NCP)-based biosensing layer. Protein–surface imprinted polymers were fabricated with a gold screen-printed electrode (SPE) as the substrate, on which radical polymerization occurred with ethylene glycol dimethacrylate as a crosslinker, methyl acrylate as the monomer, PSA and Mb as dual-templates. After the templates were removed, PIP-SPE was formed with specific sensing for both PSA and Mb. Fe3O4 NPs were successively modified with MWCNTs, GO and specific antibodies for PSA (Ab) to yield the NCP-based biosensing material. To simultaneously detect PSA and Mb, PIP-SPE was first used to recognize PSA and Mb, and their binding signal was sent as an output to electrochemical impedance spectroscopy (EIS). This was followed by adding the NCP-based sensing material to PIP-SPE, another EIS signal was obtained from the immune reaction between PSA-bound SPE and NCP. The EIS signal of Mb was calculated by the differences in the EIS signal of the two steps. This dual-modality immunosensor exhibited high specificity and selectivity toward PSA and Mb and possessed a low detecting limit of 5.4 pg mL−1 for PSA and 0.83 ng mL−1 for Mb. Finally, this novel immunosensor was successfully applied to simultaneously analyse PSA and Mb in human serum and urine samples, indicating a great potential in disease diagnosis of multi-biomarkers.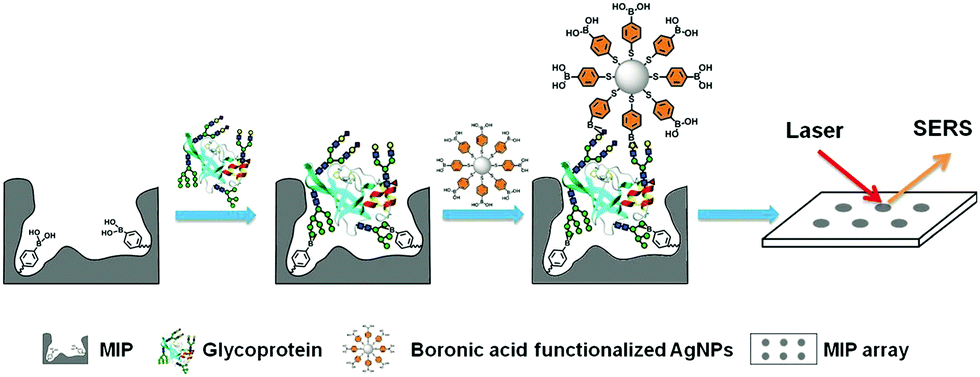 | ||
| Fig. 12 Schematic representation of the boronate-affinity sandwich assay of glycoproteins. Reprinted from ref. 147. | ||
3.4. Bioimaging and therapy
PIPs are alternatives to natural antibodies for biological recognition, but also possess a lot of advantages including high stability, low cost, and ease of functionalization, which can introduce fluorescence or better biocompatibility. Thus, PIPs hold good potential in target bioimaging and therapy, in which PIPs could specifically recognize corresponding biomarkers in cancer cells and release the loaded drugs.Liu et al.151 made the first attempt to employ PIPs as antibody-like NPs to specifically sequestrate target proteins in living cells. With an initiator of 4,4′-azobis-(4-cyanopentanoic acid) (ACPA) functionalized Fe3O4@SiO2 NPs as the substrate, a facile polymerization occurred around the template protein (DNase I) to yield protein–surface imprinted polymers with high hydrophilicity and biocompatibility. The use of NIPAm endowed the PIPs with thermo-responsiveness toward temperature changes, and the fluorescent monomer provided fluoresce quenching when PIPs rebound with target proteins. The resulting Fe3O4@SiO2@MIP ANPs presented excellent aqueous dispersion stability with an average size (85 nm). The introduction of magnetic cores not only facilitated the synthesis and separation but also offered the magnetic manipulation of PIPs within cells. After confirming the good adsorption specificity and selectivity, the as-prepared Fe3O4@SiO2@MIP ANPs were successfully applied to the specific sequestration of target proteins in living cells without disruption. This study has good potential in the study of protein functions in cells and medical diagnosis. Cecchini et al.129 developed fluorescent nanoMIPs for the specific recognition of hVEGF and further targeting imaging of its overexpression (Fig. 13). The integration of QDs in the PIP-hybrid nanoprobes provided fluorescence quenching against the recognition of nanoMIPs toward hVEGF. The resulting QD-PIPs hybrid nanoprobes exhibited specific recognition and good binding selectivity toward hVEGF in vitro and were finally applied to target imaging of overexpressing hVEGF in zebrafish embryos for the xenotransplantation of human melanoma cells. Zhang et al.152 prepared core–shell protein–surface imprinted polymers to specifically recognize epidermal growth factor receptor (EGFR) by integrating surface imprinting with epitope imprinting. The template of the epitope peptide of EGFR was bonded with palmitic acid, then a reverse microemulsion polymerization occurred on the surface of CD-embedded silica NPs with acrylamide as the functional monomer and N,N-methylenebisacrylamide as the crosslinker. The resulting CD-embedded PIPs exhibited fluorescence quenching against the rebinding with EGFR and showed high adsorption selectivity and sensitivity for EGFR. In the in vitro experiments, HeLa cells with high expression of EGFR could be accurately targeted and distinguished by the CDs-embedded PIPs, which showed stronger fluorescence than that in MCF-7 cells with low high expression of EGFR. Inspired by the good performance in vitro experiment, the CD-embedded PIPs were further successfully used for the target imaging of tumour cells with overexpressing EGFR in mice. Guo et al.153 developed a general approach called reverse microemulsion-confined epitope-oriented surface imprinting and cladding (ROSIC) to fabricate coreless and core/shell NPs with specific target capability toward proteins and peptides. A series of NPs, including QDs, SPMNPs (superparamagnetic NPs), AgNPs and UCNPs were used as substrates to obtain a variety of size controllable dual-functional single-core@cMIP NPs. Finally, QD@cMIP NPs were applied to demonstrate the potential in cancer cell target imaging. With specificity toward two typical cancer biomarkers, including human epidermal growth factor receptor-2 (HER2) and transmembrane glycoprotein non-metastatic gene B (GPNMB), QD@cMIP NPs could successfully differentiate triple-negative breast cancer (TNBC) cells from other cell lines via fluorescence imaging. The practical potential was demonstrated by in vivo target imaging of TNBC-bearing mice.
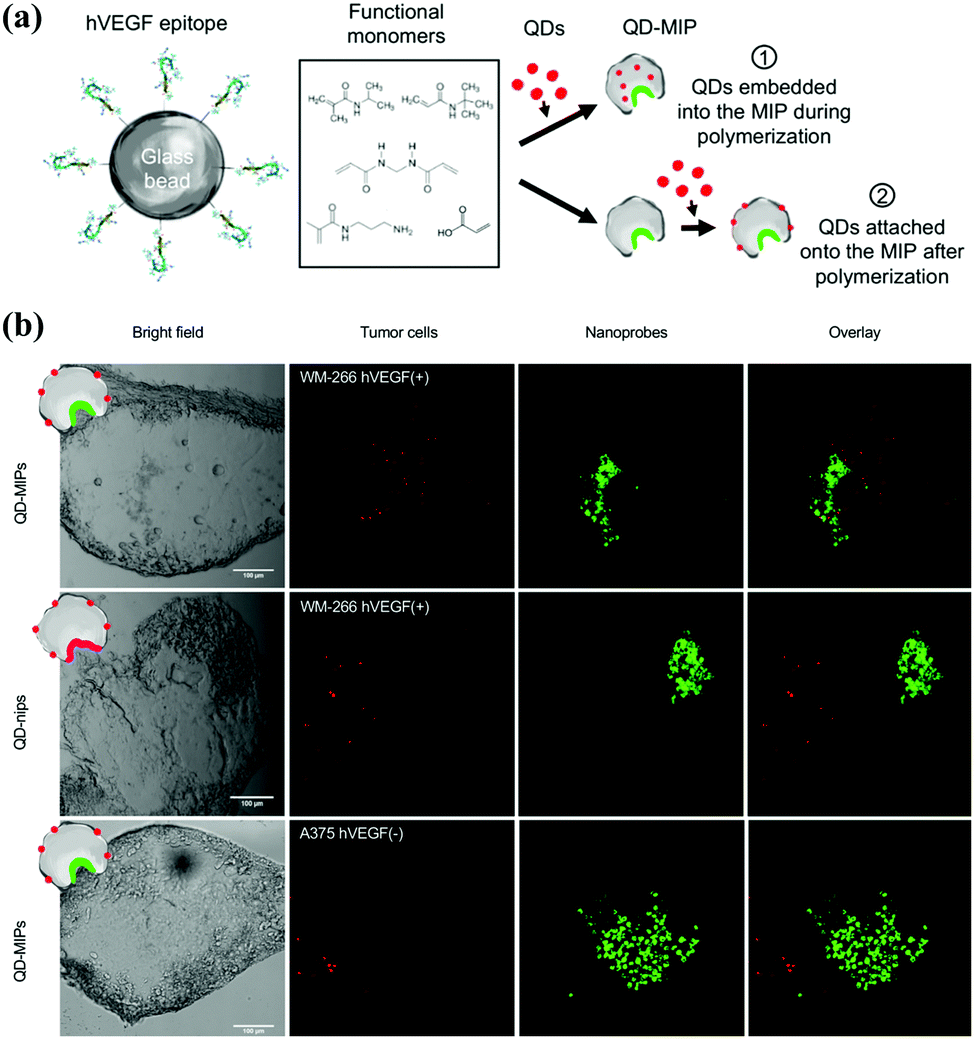 | ||
| Fig. 13 (a) Schematic of the two strategies exploited to produce QD-MIPs, based on solid-phase synthesis: (i) embedding the QDs in the MIP matrix during its polymerization; (ii) attaching the QDs onto the nanoMIPs after the polymerization process. (b) A panel of bright field and fluorescence images of human melanoma cells (WM-266 hVEGF(+) model and A-375 hVEGF(−) model) (green) and the fluorescent nanoprobes (red), acquired with a confocal microscope Leica SP2 (scale bar 100 μm), and the overlay of the two signal. Reprinted from ref. 129. | ||
In addition to target cell imaging, PIPs also could load drugs and control their release for cancer therapies.154–163 Canfarotta et al.154 developed dual-template imprinted polymers to achieve the specific recognition of cancer cells and further result in specific drug release. Solid-phase synthesis was applied to prepare doxorubicin (DOX)-loaded nanoMIPs with an epitope peptide of EGFR and doxorubicin as the dual-templates. The specific recognition ability and adsorption selectivity of the nanoMIPs was first confirmed. Based on the specific recognition of EGFR on cancer cells, the DOX-loaded nanoMIPs could specifically release DOX in cancer cells with overexpressing EGFR to elicit their cytotoxicity and apoptosis. Qin et al.155 then synthesized imprinted polymers of dual-templates for the target recognition toward cell membrane proteins to achieve simultaneous target imaging and therapy by specific drug release for cells. The DOX-loaded fluorescent PIPs (FMIPs@DOX) were synthesized by using fluorescent Si@SiO2@MPS NPs as the substrate, epitope peptide of membrane protein (P32) and DOX as the double templates. In an in vitro experiment, the FMIPs@DOX was successfully applied for the target imaging of 4T1 cancer cells based on the specific recognition between FMIPs@DOX and high expression P32 proteins. Moreover, only the 4T1 cancer cells with overexpressing P32 proteins were elicited to apoptosis after the release of DOX. Then, FMIPs@DOX was further intravenously injected into tumour-bearing mice for therapy, which was demonstrated to exhibit almost the same anti-tumour effects compared with the intratumoral injection. Peng et al.156 prepared double-template imprinted polymers with multifunctionalities, including specific recognition of cancer cells, target fluorescence imaging, magnetic resonance (MR) imaging, specific drug release, chemotherapy and photodynamic (PD) therapy (Fig. 14). The DOX-loaded PIPs (MIPs@DOX) were formed with FSiO2@MPS encapsulating gadolinium-doped silicon quantum dots (SiGdQDs) and chlorin e6 (Ce6) as the substrate and using the epitope peptide of CD59 protein and DOX as the dual templates. The introduction of SiGdQDs led to fluorescence and MR imaging, and the Ce6 could produce reactive oxygen species (ROS) for PD therapy. The MIPs@DOX were confirmed to specifically recognize cancer cells with high expression of CD59 for target imaging, and the DOX released from the MIPs@DOX and high abundance of ROS generated by light irradiation were integrated as a synergistic therapy effect for cancer cells. The target imaging and synergistic therapy were both demonstrated by in vitro and in vivo tests. The same group157 then developed novel DOX-loaded PIPs (FZIF-8/DOX-MIPs) with biodegradable effects in the tumour microenvironment. The FZIF-8/DOX-MIPs were fabricated on the surface of ZIF-8, which encapsulated CDs and DOX, and applied epitope peptide of protein (CD59) as the template. The disulfide bond in the crosslinker of N,N′-diacrylylcystamine (BAC) will be broken at a high concentration of glutathione, and the monomer of dimethylaminoethyl methacrylate (DMAEMA) will swell at low pH. Together with the instability of ZIF-8 in weak acid, GSH/pH dual-stimulation was formed to achieve controllable drug release. Finally, the FZIF-8/DOX-MIPs exhibited target fluorescence imaging, and further specific and controllable drug release toward cancer cells with overexpressing CD59, which were validated by in vitro and in vivo tests. Lu et al.163 synthesized sialic acid (SA)-imprinted biodegradable NPs (BS-NPs) by the boronate-affinity-controllable oriented surface imprinting approach. Cytotoxic ribonuclease A (RNase A) was pre-caged in the matrix of disulfide-hybridized silica NPs as nanovectors, which could be degraded under the GSH triggering. The prepared SA-imprinted RNase A@BS-NPs thus could selectively target SA-overexpressed tumor cells and was subsequently biodegraded by GSH to release RNase A to enhance cell cytotoxicity. The in vitro and in vivo experiments demonstrated the practical potential of SA-imprinted RNase A@BS-NPs in specific targeting and therapeutic efficiency toward cancer cells.
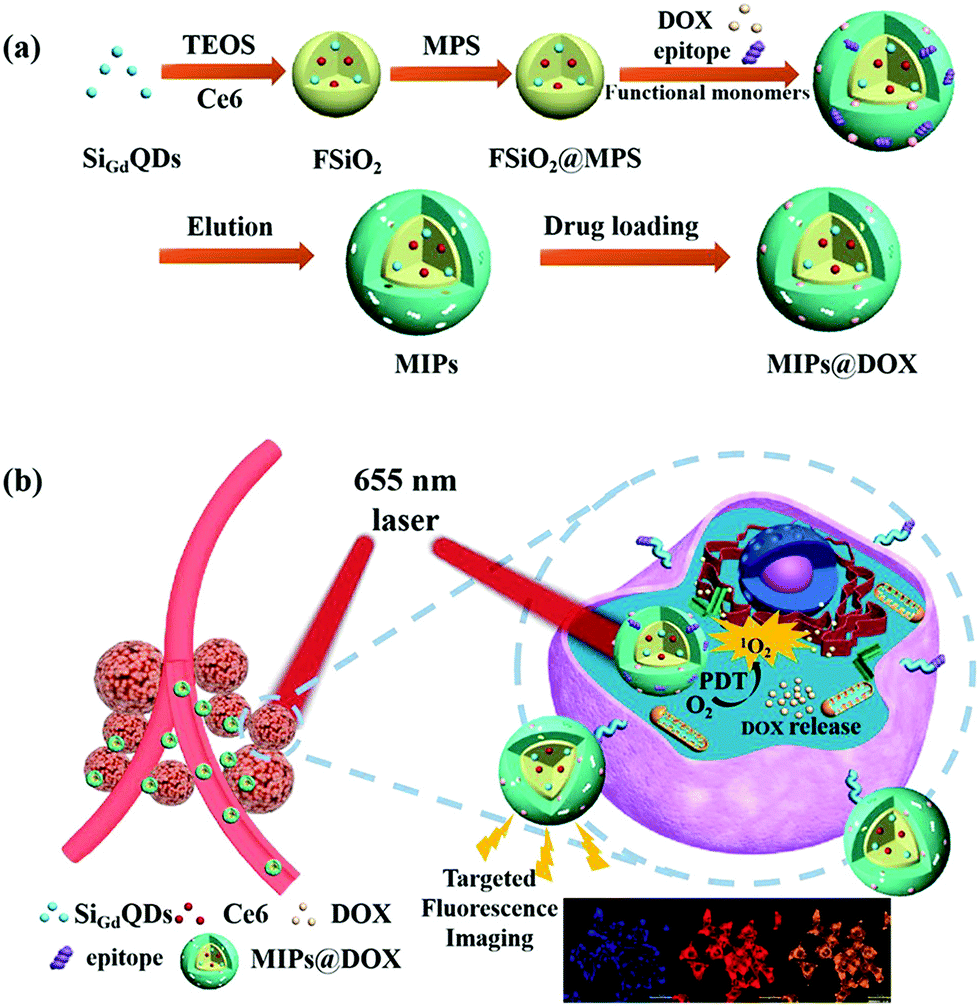 | ||
| Fig. 14 (a) Schematic illustration for the preparation of MIPs@DOX; (b) Schematic illustration of MIPs@DOX for targeted chemo-photodynamic synergistic treatment of tumour in vivo. Reprinted from ref. 156. | ||
4. Conclusion and future perspectives
In summary, a comprehensive review of the recent advances in protein imprinting methods and their highlighted applications is presented. Various novel protein imprinting methods have been developed to solve the remaining bottlenecks that traditional imprinting methods could not address. Nanomaterial-based surface imprinting of proteins could improve the binding performance of PIPs, including adsorption capacity, binding kinetics, sensitivity and selectivity. The use of epitope templates instead of the whole proteins in epitope imprinting could almost address all the problems derived from the inherent properties of proteins, which greatly increased the success and applicability of molecular imprinting for proteins. Solid-phase synthesis provides a great potential in the scalable synthesis of nanoMIPs with properties comparable to natural antibodies. Meanwhile, advanced multifunctionalities could be imparted into PIPs through the introduction of functional nanomaterials and post-imprinting modification, which make PIPs possible to go beyond natural antibodies. Combining the advantages of these protein imprinting methods, special boronate affinity-based molecular imprinting has been successful for glycoproteins. Based on the excellent properties and advanced multifunctionalities, the traditional application of PIPs in separation and purification has evolved into several advanced fields, including proteomics, biomarker detection, target bioimaging and therapy.Despite making significant progress, there are still several challenges that are remaining to be resolved in protein imprinting. First, the recognition mechanism between the templates and functional monomers is still obscure and rarely explored. More attention and designed experiments are needed to study the recognition process, which will be beneficial to the synthesis of PIPs and greatly stimulate their development. Second, some problems in the synthesis process also limit the development of protein imprinting. For example, synthesis conditions must be optimised to maintain the conformation and solubility of proteins. As for the use of epitope peptides as a template, the acquisition of epitope peptides of proteins with an unknown amino acid sequence is an unavoidable issue. The immobilization of templates with different orientations will also cause non-specific adsorption derived from the heterogeneous binding sites. Third, the reported PIPs were mostly limited to a standard protein, a general protein imprinting method, which is applicable to different proteins should be explored. Finally, exploring the potential of PIPs as synthetic receptor/antibody in biomedical applications is still in an early stage.
According to the above discussion, it can be anticipated that the development of protein imprinting in the near future will be carried out in two aspects: on the one hand, more PIPs with excellent binding performances will be designed for the separation and recognition of proteins by the introduction of advanced nanomaterials and novel protein imprinting methods. On the other hand, various multifunctional nanoMIPs will be continuously created for biomedical applications by combining the advantages of several imprinting methods such as solid-phase imprinting, epitope imprinting, and surface imprinting, and thus demonstrating the potential of PIPs as synthetic antibodies to replace or even go beyond natural antibodies. As an interdisciplinary field, the rapid development of material science, omics, and life science will provide great opportunities for the development of protein imprinting and will further extend their advanced application fields.
Author contributions
Yanting He: conceptualization and writing the original draft preparation; Zian Lin: funding acquisition, conceptualization, editing and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21974021 and 22036001), and the Major Project of Science and Technology of Fujian Province (2020HZ06006).Notes and references
- N. M. Bergmann and N. A. Peppas, Prog. Polym. Sci., 2008, 33, 271–288 CrossRef CAS.
- Y. J. Cao, M. Zheng, W. H. Cai and Z. C. Wang, Chin. Chem. Lett., 2020, 31, 463–467 CrossRef CAS.
- H. K. Li, H. L. Ye, X. X. Zhao, X. L. Sun, Q. Q. Zhu, Z. Y. Han, R. R. Yuan and H. M. He, Chin. Chem. Lett., 2021, 32, 2851–2855 CrossRef CAS.
- J. R. Clegg and N. A. Peppas, Soft Matter, 2020, 16, 856–869 RSC.
- A. A. Shukla and J. Thoemmes, Trends Biotechnol., 2010, 28, 253–261 CrossRef CAS PubMed.
- A. L. Bole and P. Manesiotis, Adv. Mater., 2016, 28, 5349–5366 CrossRef CAS PubMed.
- M. Cieplak and W. Kutner, Trends Biotechnol., 2016, 34, 922–941 CrossRef CAS PubMed.
- H. Asanuma, T. Hishiya and M. Komiyama, Adv. Mater., 2000, 12, 1019–1030 CrossRef CAS.
- B. Sellergren, TrAC, Trends Anal. Chem., 1997, 16, 310–320 CrossRef CAS.
- F. Breinl and F. Haurowitz, Physiol. Chem., 1930, 192, 45–57 CrossRef CAS.
- S. Mudd, J. Immunol., 1932, 23, 423–427 CAS.
- M. V. Polyakov, Zhur. Fiz. Khim., 1931, 2, 799–805 Search PubMed.
- L. Pauling, J. Am. Chem. Soc., 2002, 62, 2643–2657 CrossRef.
- F. H. Dickey, Proc. Natl. Acad. Sci. U. S. A., 1949, 35, 227–229 CrossRef CAS PubMed.
- G. Wulff and A. Sarhan, Angew. Chem., Int. Ed. Engl., 1972, 11, 341–346 CAS.
- K. Mosbach, Trends Biochem. Sci., 1994, 19, 9–14 CrossRef CAS PubMed.
- C. Alexander, H. S. Andersson, L. I. Andersson, R. J. Ansell, N. Kirsch, I. A. Nicholls, J. O’Mahony and M. J. Whitcombe, J. Mol. Recognit., 2006, 19, 106–180 CrossRef CAS PubMed.
- G. Vasapollo, R. Del Sole, L. Mergola, M. R. Lazzoi, A. Scardino, S. Scorrano and G. Mele, Int. J. Mol. Sci., 2011, 12, 5908–5945 CrossRef CAS PubMed.
- L. X. Chen, S. F. Xu and J. H. Li, Chem. Soc. Rev., 2011, 40, 2922–2942 RSC.
- R. Kecili and C. M. Hussain, Int. J. Anal. Chem., 2018, 2018, 8503853 Search PubMed.
- E. Turiel and A. Martin-Esteban, Anal. Chim. Acta, 2010, 668, 87–99 CrossRef CAS PubMed.
- J. P. Schillemans and C. F. van Nostrum, Nanomedicine, 2006, 1, 437–447 CrossRef CAS PubMed.
- S. N. He, L. P. Zhang, S. K. Bai, H. Yang, Z. Cui, X. F. Zhang and Y. P. Li, Eur. Polym. J., 2021, 143, 110179 CrossRef CAS.
- N. Leibl, K. Haupt, C. Gonzato and L. Duma, Chemosensors, 2021, 9, 123 CrossRef CAS.
- J. Wackerlig and P. A. Lieberzeit, Sens. Actuators, B, 2015, 46, 144–157 CrossRef.
- B. Amma, B. Sjb, B. Spw and B. Jb, TrAC, Trends Anal. Chem., 2021, 144, 116431 CrossRef.
- R. R. Xing, Y. R. Wen, H. He, Z. C. Guo and Z. Liu, TrAC, Trends Anal. Chem., 2019, 110, 417–428 CrossRef CAS.
- J. J. Xu, H. H. Miao, J. X. Wang and G. Q. Pan, Small, 2020, 16, 1906644 CrossRef CAS PubMed.
- K. Haupt, P. Ra Ngel and B. Bui, Chem. Rev., 2020, 120, 9554–9582 CrossRef CAS PubMed.
- H. Q. Zhang, Adv. Mater., 2020, 32, 1806328 CrossRef CAS PubMed.
- Z. El-Schich, Y. Zhang, M. Feith, S. Beyer and A. G. Wingren, Biotechniques, 2020, 69, 406–419 CrossRef CAS PubMed.
- N. Zhang, Y. R. Xu, Z. L. Li, C. R. Yan, K. Mei, M. L. Ding, S. C. Ding, P. Guan, L. W. Qian, C. B. Du and X. L. Hu, Macromol. Rapid Commun., 2019, 40, 1900096 CrossRef PubMed.
- A. N. Peppas and R. D. Kryscio, Acta Biomater., 2012, 8, 461–473 CrossRef PubMed.
- X. H. Ma, J. P. Li, C. Wang and G. B. Xu, Chin. J. Anal. Chem., 2016, 44, 152–159 CAS.
- S. Li, S. Cao, M. J. Whitcombe and S. A. Piletsky, Prog. Polym. Sci., 2014, 39, 145–163 CrossRef CAS.
- H. R. Culver and N. A. Peppas, Chem. Mater., 2017, 29, 5753–5761 CrossRef CAS PubMed.
- M. Dinc, C. Esen and B. Mizaikoff, TrAC, Trends Anal. Chem., 2019, 114, 202–217 CrossRef CAS.
- X. D. Wang, G. Chen, P. Zhang and Q. Jia, Anal. Methods, 2021, 13, 1660–1671 RSC.
- T. Khumsap, A. Corpuz and L. T. Nguyen, RSC Adv., 2021, 11, 11403–11414 RSC.
- C. Boitard, A. Bée, C. Menager and N. Griffete, J. Mater. Chem. B, 2018, 6, 1563–1580 RSC.
- M. Glad, O. Norrlöw, B. Sellergren, N. Siegbahn and K. Mosbach, J. Chromatogr. A, 1985, 347, 11–23 CrossRef CAS.
- O. Brüggemann, K. Haupt, Y. Lei, E. Yilmaz and K. Mosbach, J. Chromatogr. A, 2000, 889, 15–24 CrossRef.
- P. A. Lieberzeit, S. Gazda-Miarecka, K. Halikias, C. Schirk, J. Kauling and F. L. Dickert, Sens. Actuators, B, 2005, 111, 259–263 CrossRef.
- E. Gizem and M. Bo, Sensors, 2017, 17, 288 CrossRef PubMed.
- A. Mujahid, N. Iqbal and A. Afzal, Biotechnol. Adv., 2013, 31, 1435–1447 CrossRef CAS PubMed.
- A. L. Hillberg and M. Tabrizian, IRBM, 2008, 29, 89–104 CrossRef.
- M. J. Whitcombe, I. Chianella, L. Larcombe, S. A. Piletsky, J. Noble, R. Porter and A. Horgan, Chem. Soc. Rev., 2011, 40, 1547–1571 RSC.
- K. G. Yang, S. W. Li, L. K. Liu, Y. W. Chen and W. Zhou, Adv. Mater., 2019, 31, 1902048 CrossRef CAS PubMed.
- N. Zhang, X. L. Hu, P. Guan, C. B. Du, J. Li, L. W. Qian, X. Y. Zhang, S. C. Ding and B. P. Li, Chem. Eng. J., 2017, 317, 356–367 CrossRef CAS.
- C. Boitard, A. L. Rollet, C. Ménager and N. Griffete, Chem. Commun., 2017, 53, 8846–8849 RSC.
- Y. N. Yuan, C. L. Yang, T. W. Lv, F. X. Qiao, Y. Zhou and H. Y. Yan, Anal. Chim. Acta, 2018, 1033, 213–220 CrossRef CAS PubMed.
- S. Y. Tan, Y. Long, Q. Han, J. D. Wang, Q. L. Liang and M. Y. Ding, ACS Appl. Bio Mater., 2018, 2, 388–396 CrossRef PubMed.
- H. Shi, W. B. Tsai, M. D. Garrison, S. Ferrari and B. D. Ratner, Nature, 1999, 398, 593–597 CrossRef CAS PubMed.
- J. Kalecki, Z. Iskierko, M. Cieplak and P. S. Sharma, ACS Sens., 2020, 5, 3710–3720 CrossRef CAS PubMed.
- P. Zahedi, M. Ziaee, M. Abdouss, A. Farazin and B. Mizaikoff, Poly. Adv. Technol., 2016, 27, 1124–1142 CrossRef CAS.
- M. Pan, L. Hong, X. Xie, K. Liu, J. Yang and S. Wang, Macromol. Chem. Phys., 2021, 222, 2000222 CrossRef CAS.
- B. D. Gupta, A. M. Shrivastav and S. P. Usha, Sensors, 2016, 16, 1381 CrossRef PubMed.
- X. Guo, J. Li, M. Arabi, X. Wang, Y. Wang and L. Chen, ACS Sens., 2020, 5, 601–619 CrossRef CAS PubMed.
- H. R. Culver, S. D. Steichen and N. A. Peppas, Biomacromolecules, 2016, 17, 4045–4053 CrossRef CAS PubMed.
- Y. Lv, T. Tan and F. Svec, Biotechnol. Adv., 2013, 31, 1172–1186 CrossRef CAS PubMed.
- X. Ding and P. A. Heiden, Macromol. Mater. Eng., 2014, 299, 268–282 CrossRef CAS.
- M. Niu, C. Pham-Huy and H. He, Microchim. Acta, 2016, 183, 2677–2695 CrossRef CAS.
- G. Fu, H. He, Z. Chai, H. Chen, J. Kong, Y. Wang and Y. Jiang, Anal. Chem., 2011, 83, 1431–1436 CrossRef CAS PubMed.
- Z. Lin, Z. Xia, J. Zheng, D. Zheng, L. Zhang, H. Yang and G. Chen, J. Mater. Chem., 2012, 22, 17914–17922 RSC.
- Z. Xia, Z. Lin, Y. Xiao, L. Wang, J. Zheng, H. Yang and G. Chen, Biosens. Bioelectron., 2013, 47, 120–126 CrossRef CAS PubMed.
- J. Liu, K. Yang, Q. Deng, Q. Li, L. Zhang, Z. Liang and Y. Zhang, Chem. Commun., 2011, 47, 3969–3971 RSC.
- A. Nematollahzadeh, W. Sun, C. S. Aureliano, D. Lütkemeyer, J. Stute, M. J. Abdekhodaie, A. Shojaei and B. Sellergren, Angew. Chem., Int. Ed., 2011, 50, 495–498 CrossRef CAS PubMed.
- S. Bhakta, M. S. I. Seraji, S. L. Suib and J. F. Rusling, ACS Appl. Mater. Interfaces, 2015, 7, 28197–28206 CrossRef CAS PubMed.
- Z. Zhang, X. Zhang, D. Niu, Y. Li and J. Shi, J. Mater. Chem. B, 2017, 5, 4214–4220 RSC.
- W. H. Zhou, C. H. Lu, X. C. Guo, F. R. Chen, H. H. Yang and X. R. Wang, J. Mater. Chem., 2010, 20, 880–883 RSC.
- R. Gao, X. Kong, X. Wang, X. He, L. Chen and Y. Zhang, J. Mater. Chem., 2011, 21, 17863–17871 RSC.
- S. Bhakta, C. K. Dixit, I. Bist, J. Macharia, M. Shen, K. Kadimisetty, J. He, B. Dutta, S. L. Suib and J. F. Rusling, Chem. Commun., 2017, 53, 9254–9257 RSC.
- J. Zhou, Y. Wang, J. Bu, B. Zhang and Q. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 25682–25690 CrossRef CAS PubMed.
- Y. Li, H. H. Yang, Q. H. You, Z. X. Zhuang and X. R. Wang, Anal. Chem., 2006, 78, 317–320 CrossRef CAS PubMed.
- R. Ouyang, J. Lei and H. Ju, Chem. Commun., 2008, 5761–5763 RSC.
- T. Chen, M. Shao, H. Xu, S. Zhuo, S. Liu and S. T. Lee, J. Mater. Chem., 2012, 22, 3990–3996 RSC.
- M. Zhang, J. Huang, P. Yu and X. Chen, Talanta, 2010, 81, 162–166 CrossRef CAS PubMed.
- C. Zheng, X. L. Zhang, W. Liu, B. Liu, H. H. Yang, Z. A. Lin and G. N. Chen, Adv. Mater., 2013, 25, 5922–5927 CrossRef CAS PubMed.
- M. Liu, J. Pi, X. Wang, R. Huang, Y. Du, X. Yu, W. Tan, F. Liu and K. J. Shea, Anal. Chim. Acta, 2016, 932, 29–40 CrossRef CAS PubMed.
- Y. P. Qin, D. Y. Li, X. W. He, W. Y. Li and Y. K. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 10155–10163 CrossRef CAS PubMed.
- X. Xu, P. Guo, Z. Luo, Y. Ge, Y. Zhou, R. Chang, W. Du, C. Chang and Q. Fu, RSC Adv., 2017, 7, 18765–18774 RSC.
- Z. Yang, J. Xu, J. Wang, Q. Zhang and B. Zhang, Chem. Eng. J., 2019, 373, 923–934 CrossRef CAS.
- Z. Yang, J. Chen, J. Wang, Q. Zhang and B. Zhang, ACS Sustainble. Chem. Eng., 2020, 8, 3241–3252 CrossRef CAS.
- J. Luo, S. Jiang and X. Liu, J. Phys. Chem. C, 2013, 117, 18448–18456 CrossRef CAS.
- F. Chen, W. Zhao, J. Zhang and J. Kong, Phys. Chem. Chem. Phys., 2016, 18, 718–725 RSC.
- J. Guo, Y. Wang, Y. Liu, C. Zhang and Y. Zhou, Talanta, 2015, 144, 411–419 CrossRef CAS PubMed.
- J. Luo, J. Huang, J. Cong, W. Wei and X. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 7735–7744 CrossRef CAS PubMed.
- M. Sobiech, P. Bujak, P. Luliński and A. Pron, Nanoscale, 2019, 11, 12030–12074 RSC.
- M. Díaz-Álvarez and A. Martín-Esteban, Biosensors, 2021, 11, 79 CrossRef PubMed.
- P. P. Tang and J. B. Cai, Chin. J. Chem. Phys., 2010, 23, 195 CrossRef CAS.
- W. Zhang, X. W. He, Y. Chen, W. Y. Li and Y. K. Zhang, Biosens. Bioelectron., 2011, 26, 2553–2558 CrossRef CAS PubMed.
- W. Zhang, X. W. He, Y. Chen, W. Y. Li and Y. K. Zhang, Biosens. Bioelectron., 2012, 31, 84–89 CrossRef CAS PubMed.
- Z. Ding, S. A. Bligh, L. Tao, J. Quan, H. Nie, L. Zhu and X. Gong, Mater. Sci. Eng., C, 2015, 48, 469–479 CrossRef CAS PubMed.
- T. Guo, Q. Deng, G. Fang, C. Liu, X. Huang and S. Wang, Biosens. Bioelectron., 2015, 74, 498–503 CrossRef CAS PubMed.
- T. Guo, Q. Deng, G. Fang, D. Gu, Y. Yang and S. Wang, Biosens. Bioelectron., 2016, 79, 341–346 CrossRef CAS PubMed.
- J. Li, M. Ma, C. Zhang, R. Lu, L. Zhang and W. Zhang, Anal. Bioanal. Chem., 2020, 412, 7227–7236 CrossRef CAS PubMed.
- L. Qian, W. Liu, H. Liu, V. Nica, S. Zhang, Q. Zhou, W. Song and Q. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 31010–31020 CrossRef CAS PubMed.
- A. Rachkov and N. Minoura, Biochem. Biophys. Acta, 2001, 1544, 255–266 CrossRef CAS.
- S. P. Teixeira, R. L. Reis, N. A. Peppas, M. E. Gomes and R. M. Domingues, Sci. Adv., 2021, 7, eabi9884 CrossRef CAS PubMed.
- S. Dietl, H. Sobek and B. Mizaikoff, TrAC, Trends Anal. Chem., 2021, 143, 116414 CrossRef CAS.
- H. Nishino, C. S. Huang and K. J. Shea, Angew. Chem., Int. Ed., 2006, 118, 2452–2456 CrossRef.
- K. Yang, J. Liu, S. Li, Q. Li, Q. Wu, Y. Zhou, Q. Zhao, N. Deng, Z. Liang, L. Zhang and Y. Zhang, Chem. Commun., 2014, 50, 9521–9524 RSC.
- K. Yang, S. Li, J. Liu, L. Liu, L. Zhang and Y. Zhang, Anal. Chem., 2016, 88, 5621–5625 CrossRef CAS PubMed.
- X. L. Zhao, D. Y. Li, X. W. He, W. Y. Li and Y. K. Zhang, J. Mater. Chem. B, 2014, 2, 7575–7582 RSC.
- S. Li, K. Yang, J. Liu, B. Jiang, L. Zhang and Y. Zhang, Anal. Chem., 2015, 87, 4617–4620 CrossRef CAS PubMed.
- S. Li, K. Yang, B. Zhao, X. Li, L. Liu, Y. Chen, L. Zhang and Y. Zhang, J. Mater. Chem. B, 2016, 4, 1960–1967 RSC.
- Y. P. Qin, D. Y. Li, X. W. He, W. Y. Li and Y. K. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 10155–10163 CrossRef CAS PubMed.
- D. Y. Li, X. M. Zhang, Y. J. Yan, X. W. He, W. Y. Li and Y. K. Zhang, Biosens. Bioelectron., 2016, 79, 187–192 CrossRef CAS PubMed.
- R. Xing, S. Wang, Z. Bie, H. He and Z. Liu, Nat. Protoc., 2017, 12, 964–987 CrossRef CAS PubMed.
- D. Li, Y. Chen and Z. Liu, Chem. Soc. Rev., 2015, 44, 8097–8123 RSC.
- Z. Lin, J. Wang, X. Tan, L. Sun, R. Yu, H. Yang and G. Chen, J. Chromatogr. A, 2013, 1319, 141–147 CrossRef CAS PubMed.
- Z. Lin, L. Sun, W. Liu, Z. Xia, H. Yang and G. Chen, J. Mater. Chem. B, 2014, 2, 637–643 RSC.
- L. Sun, D. Lin, G. Lin, L. Wang and Z. Lin, Anal. Methods, 2015, 7, 10026–10031 RSC.
- J. Luo, J. Huang, J. Cong, W. Wei and X. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 7735–7744 CrossRef CAS PubMed.
- X. Y. Sun, R. T. Ma, J. Chen and Y. P. Shi, Microchim. Acta, 2017, 184, 3729–3737 CrossRef CAS.
- L. Li, Y. Lu, Z. Bie, H. Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2013, 52, 7451–7454 CrossRef CAS PubMed.
- S. Wang, J. Ye, Z. Bie and Z. Liu, Chem. Sci., 2014, 5, 1135–1140 RSC.
- R. R. Xing, Y. Ma, Y. Wang, Y. Wen and Z. Liu, Chem. Sci., 2019, 10, 1831–1835 RSC.
- R. R. Xing, Z. C. Guo, H. F. Lu, Q. Zhang and Z. Liu, Sci. Bull., 2022, 67, 278–287 CrossRef CAS.
- A. Poma, A. Guerreiro, M. J. Whitcombe, E. V. Piletska, A. P. F. Turner and S. A. Piletsky, Adv. Funct. Mater., 2013, 23, 2821–2827 CrossRef CAS PubMed.
- F. Canfarotta, A. Poma, A. Guerreiro and S. Piletsky, Nat. Protoc., 2016, 11, 443–455 CrossRef CAS PubMed.
- A. Poma, A. Guerreiro, S. Caygill, E. Moczko and S. Piletsky, RSC Adv., 2014, 4, 4203–4206 RSC.
- S. Ambrosini, S. Beyazit, K. Haupt and B. Tse Sum Bui, Chem. Commun., 2013, 49, 6746–6748 RSC.
- J. Xu, K. Haupt and B. Tse Sum Bui, ACS Appl. Mater. Interfaces, 2017, 9, 24476–24483 CrossRef CAS PubMed.
- J. J. Xu, S. Ambrosini, E. Tamahkar, C. Rossi, K. Haupt and B. Tse Sum Bui, Biomacromolecules, 2016, 17, 345–353 CrossRef CAS PubMed.
- J. J. Xu, F. Merlier, B. Avalle, V. Vieilard, P. Debre, K. Haupt and B. T. S. Bui, ACS Appl. Mater. Interfaces, 2019, 11, 9824–9831 CrossRef CAS PubMed.
- J. Ashley, X. Feng, A. Halder, T. Zhou and Y. Sun, Chem. Commun., 2018, 54, 3355–3358 RSC.
- R. Mahajan, M. Rouhi, S. Shinde, T. Bedwell, A. Incel, L. Mavliutova, S. Piletsky, I. A. Nicholls and B. Sellergren, Angew. Chem., Int. Ed., 2019, 58, 727–730 CrossRef CAS PubMed.
- A. Cecchini, V. Raffa, F. Canfarotta, G. Signore, S. Piletsky, M. P. MacDonald and A. Cuschieri, Nano Lett., 2017, 17, 2307–2312 CrossRef CAS PubMed.
- A. Gómez-Caballero, A. Elejaga-Jimeno, G. García del Caño, N. Unceta, A. Guerreiro, M. Saumell-Esnaola, J. Sallés, M. A. Goicolea and R. J. Barrio, Microchim. Acta, 2021, 188, 368 CrossRef PubMed.
- H. Sunayama, T. Ooya and T. Takeuchi, Biosens. Bioelectron., 2010, 26, 458–462 CrossRef CAS PubMed.
- T. Takeuchi, H. Sunayama, E. Takano and Y. Kitayama, Adv. Biochem. Eng. Biot., 2015, 150, 95–106 CAS.
- T. Takeuchi and H. Sunayama, Chem. Commun., 2018, 54, 6243–6251 RSC.
- Y. Suga, H. Sunayama, T. Ooya and T. Takeuchi, Chem. Commun., 2013, 49, 8450–8452 RSC.
- H. Sunayama, T. Ooya and T. Takeuchi, Chem. Commun., 2014, 50, 1347–1349 RSC.
- R. Horikawa, H. Sunayama, Y. Kitayama, E. Takano and T. Takeuchi, Angew. Chem., Int. Ed., 2016, 55, 13023–13027 CrossRef CAS PubMed.
- T. Zhao, J. Wang, J. He, Q. Deng and S. Wang, Biosens. Bioelectron., 2017, 91, 756–761 CrossRef CAS PubMed.
- T. Morishige, E. Takano, H. Sunayama, Y. Kitayama and T. Takeuchi, ChemNanoMat, 2019, 5, 224–229 CrossRef CAS.
- X. Wang, S. Yu, J. Wang, J. Yu, M. Arabi, L. Fu, B. Li, J. Li and L. Chen, Talanta, 2020, 211, 120727 CrossRef CAS PubMed.
- J. P. Fan, J. X. Yu, X. M. Yang, X. H. Zhang, T. T. Yuan and H. L. Peng, Chem. Eng. J., 2018, 337, 722–732 CrossRef CAS.
- Z. Yang, Y. Zhang, J. Ren, Q. Zhang and B. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 34829–34842 CrossRef CAS PubMed.
- D. R. K. Weerasuriya, S. Bhakta, K. Hiniduma, C. K. Dixit, M. Shen, Z. Tobin, J. He, S. L. Suib and J. F. Rusling, ACS Appl. Bio Mater., 2021, 4, 6157–6166 CrossRef CAS PubMed.
- W. Wan, Q. Han, X. Zhang, Y. Xie, J. Sun and M. Ding, Chem. Commun., 2015, 51, 3541–3544 RSC.
- M. Bertolla, L. Cenci, A. Anesi, E. Ambrosi, F. Tagliaro, L. Vanzetti, G. Guella and A. M. Bossi, ACS Appl. Mater. Interfaces, 2017, 9, 6908–6915 CrossRef CAS PubMed.
- W. Zhang, T. Zhang and Y. Chen, J. Proteomics, 2019, 192, 188–195 CrossRef CAS PubMed.
- X. Bi and Z. Liu, Anal. Chem., 2014, 86, 959–966 CrossRef CAS PubMed.
- J. Ye, Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2014, 53, 10386–10389 CrossRef CAS PubMed.
- S. Patra, E. Roy, R. Madhuri and P. K. Sharma, Biosens. Bioelectron., 2015, 66, 1–10 CrossRef CAS PubMed.
- V. V. Shumyantseva, T. V. Bulko, L. V. Sigolaeva, A. V. Kuzikov, P. V. Pogodin and A. I. Archakov, Biosens. Bioelectron., 2018, 99, 216–222 CrossRef CAS PubMed.
- P. Karami, H. Bagheri, M. Johari-Ahar, H. Khoshsafar, F. Arduini and A. Afkhami, Talanta, 2019, 202, 111–122 CrossRef CAS PubMed.
- Y. Liu, S. Fang, J. Zhai and M. Zhao, Nanoscale, 2015, 7, 7162–7167 RSC.
- Y. Zhang, S. Li, X. T. Ma, X. W. He, W. Y. Li and Y. K. Zhang, Microchim. Acta, 2020, 187, 228 CrossRef CAS PubMed.
- Z. C. Guo, R. R. Xing, M. H. Zhao, Y. Li, H. F. Lu and Z. Liu, Adv. Sci., 2021, 8, 2101713 CrossRef CAS PubMed.
- F. Canfarotta, L. Lezina, A. Guerreiro, J. Czulak, A. Petukhov, A. Daks, K. Smolinska-Kempisty, A. Poma, S. Piletsky and N. A. Barlev, Nano Lett., 2018, 18, 4641–4646 CrossRef CAS PubMed.
- Y. T. Qin, H. Peng, X. W. He, W. Y. Li and Y. K. Zhang, Anal. Chem., 2019, 91, 12696–12703 CrossRef CAS PubMed.
- H. Peng, Y. T. Qin, X. W. He, W. Y. Li and Y. K. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 13360–13370 CrossRef CAS PubMed.
- Y. T. Qin, Y. S. Feng, Y. J. Ma, X. W. He, W. Y. Li and Y. K. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 24585–24598 CrossRef CAS PubMed.
- H. Y. Wang, P. P. Cao, Z. Y. He, X. W. He, W. Y. Li, Y. H. Li and Y. K. Zhang, Nanoscale, 2019, 11, 17018–17030 RSC.
- C. Jia, M. Zhang, Y. Zhang, Z. B. Ma, N. N. Xiao, X. W. He, W. Y. Li and Y. K. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 32431–32440 CrossRef CAS PubMed.
- Y. R. Dong, W. Li, Z. K. Gu, R. R. Xing, Y. Y. Ma, Q. Zhang and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 10621–10625 CrossRef CAS PubMed.
- Z. K. Gu, Y. R. Dong, S. X. Xu, L. S. Wang and Z. Liu, Angew. Chem., Int. Ed., 2021, 133, 2695–2699 CrossRef.
- P. X. M. Rangel, E. Moroni, F. Merlier, L. A. Gheber, R. Vago, B. T. S. Bui and K. Haupt, Angew. Chem., Int. Ed., 2020, 59, 2816–2822 CrossRef PubMed.
- H. F. Lu, S. X. Xu, Z. C. Guo, M. H. Zhao and Z. Liu, ACS Nano, 2021, 15, 18214–18225 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |