
Construction of DNA ligase-mimicking nanozymes via molecular imprinting†
Xinpei
He
,
Qi
Luo
,
Zhanchen
Guo
,
Ying
Li
and
Zhen
Liu
 *
*
State Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, 163 Xianlin Avenue, Nanjing 210023, China. E-mail: zhenliu@nju.edu.cn
First published on 31st January 2022
Abstract
Enzyme mimics are of significant importance due to their facile preparation, low cost and stability to rigorous environments. Molecularly imprinted polymers (MIPs) have been important synthetic mimics of enzymes. However, effective strategies for the rational design of enzyme-mimicking MIPs have still remained limited. Herein, we report a new strategy, termed affinity gathering-enhanced coupling and thermal cycling amplification (AGEC-TCA), for the rational design and engineering of molecularly imprinted mesoporous silica nanoparticles (MSNs) that are capable of ligating short ssDNA fragments. This strategy relied on enhancing the effective collision probability via binding substrates into highly favorable orientation by product-imprinted MSNs as well as product release via thermal cycling which enabled successive product amplification. Using modified and natural hexadeoxyribonucleotide as templates, the prepared product-imprinted MSNs exhibited a remarkably enhanced reaction speed (by up to 63-fold) as well as excellent sequence specificity towards substrate trideoxyribonucleotides. Thus, this strategy opened up a new avenue to access enzyme mimics via molecular imprinting.
Introduction
Enzymes, as biological catalysts, exhibit significant advantages including efficiency, specificity and mild reaction conditions. However, enzymes suffer from some intrinsic drawbacks, such as ease of denaturation, laborious preparation, high cost, difficulty of recycling, and so on. In addition to engineering enzymes with desired properties via directed molecular evolution,1–3 numerous efforts have been devoted to the development of artificial enzymes or enzyme mimics.4–17 The design of artificial enzymes is usually based on some supramolecules and materials with intrinsic catalytic potential, such as cyclodextrins,4 carbon-based materials,5–7 metal materials,8–10 metal oxide-based materials,11,12 and so on. Particularly, enzyme-like nanomaterials or nanozymes have gained increasing attention in recent years.13–15 However, most of these materials encounter an intrinsic problem: lack of specificity.16,17 Meanwhile, general strategies for the rational design and controllable synthesis of enzyme mimics still remain to be developed.Molecularly imprinted polymers (MIPs), as synthetic mimics of enzymes and antibodies, have shown great potential for important applications due to the predetermined selectivity and affinity.18–26 The major strategy for the design of enzyme-mimicking MIPs relies on the imprinting of transition state analogues (TSAs),27–30 which facilitates the transformation of substrates into products through reducing the activation energy. However, this strategy is associated with two severe drawbacks. On one hand, the structures of transition states of many reactions have remained to be elucidated. On the other hand, even if the structure of a transition state has been known, the design and synthesis of TSAs are yet rather challenging. Another strategy is imprinting with the reaction product or an analogue of the product,31–34 but the reaction efficiency is suppressed by the product inhibition effect.35 Recently, the introduction of molecular imprinting for the surface modification of inorganic nanoenzymes has allowed significant enhancement of the reaction specificity.36 On the other hand, engineering catalytic MIPs into the nanoparticle format has exhibited unique advantages in biomolecular catalysis such as peptide cyclization37 and guided peptide folding.38 Due to the lack of sound theoretical guidance for rational design and controllable fabrication, however, the development of enzyme-mimicking MIPs has still largely lagged behind their counterpart, antibody-mimicking MIPs, which have already found applications in many important areas, such as affinity separation,39,40 disease diagnosis,41–43 drug delivery,44–46 targeted bioimaging,47 and cancer therapy.48–51
Enzymes that catalyse reactions of nucleic acid substrates may provide straightforward models for the fundamental investigation of enzyme mimicking, because of the relatively simple structural diversity of nucleic acids (only four building blocks) as compared with other biomolecules such as proteins and saccharides as well as the simplicity of interactions governing the molecular recognition (predominantly hydrogen bonding). Particularly, DNA ligases, which catalyse the ligation of two DNA fragments by joining 3′-OH and 5′-PO4 termini to form a phosphodiester and are of paramount importance in DNA replication/repair and genetic engineering, can be an ideal model for this purpose. Meanwhile, the mimicking of DNA ligases may shed light on the origin and evolution of earlier nucleic acids in the prebiotic environment in which some minerals might have functioned as natural and primordial catalysts. However, attempts on the imprinting of ssDNA have still remained limited, though a few studies on the imprinting of dsDNA have been reported.52–54 So far, to the best of our knowledge, there have been only one publication on the imprinting of ssDNA, which reported hybrid DNA–polymer nanoparticles via introducing modified thymine bases into oligonucleotide sequences so that the hybrid oligomer imprinted polymeric nanoparticles (oligoMIP NPs) could specifically recognize their complementary DNA strands.55 Such a recognition mechanism is similar to conventional base pairing-based DNA recognition. Therefore, the mimicking of DNA ligases via molecular imprinting may provide a new paradigm of molecular imprinting for ssDNA recognition.
Herein, we report a novel strategy, called affinity gathering-enhanced coupling and thermal cycling amplification (AGEC-TCA), for the rational design and engineering of imprinted nanoparticles towards the mimicking of DNA ligases. Different from the conventional strategy of TSA imprinting that aims to reduce the activation energy,27–30 this strategy turned to enhancing effective collision probability through affinity adsorbing substrate molecules together with highly favourable spatial orientation by product-imprinted MIPs. To make such product-imprinted MIPs function effectively, mesoporous silica nanoparticles (MSNs) were employed as the base platform in this study because of their high specific surface area56,57 as well as great potential in enhancing the effective collision probability.58 Meanwhile, to make the enzyme-mimicking material return to its original state at the completion of the reaction, the product was released by elevating the temperature to a certain level that can disrupt the hydrogen bonding that governs the binding and effectively release the product from mesopores. To do this, the AGEC reaction was carried out on a PCR thermocycler with a special TCA program; the coupling reaction was implemented at a low temperature (room temperature or lower), while the product release was performed at a high temperature, and such thermocycling was repeated until adequate product was generated. To the best of our knowledge, to date such a TCA program has never been employed in the design of enzyme-mimicking materials, but it can be used as a simple but effective way to eliminate the effect of product inhibition. The principle and procedure of the AGEC-TCA strategy are illustrated in Fig. 1. As a proof of the principle, a modified hexadeoxyribonucleotide was first employed as the imprinting template, and its imprinted MSNs with desired structure and properties were successfully prepared. With this imprinted nanomaterial, the effectiveness of the AGEC-TCA strategy was verified. The imprinted MSNs exhibited affinity towards the substrate ssDNA with the dissociation constant at the 10−4 M level. The imprinted MSNs could endure thermal cycling for at least several hours, permitting successive generation of the product. The molecularly imprinted MSN-based AGEC-TCA reaction exhibited high specificity and a significantly enhanced reaction rate as compared with the control reaction in the absence of the imprinted MSNs without temperature cycling. Using a naturally occurring hexadeoxyribonucleotide as the template, we finally achieved the ligation of naturally occurring trideoxyribonucleotides with excellent specificity and an enhanced reaction rate by up to 63 fold. Thus, the AGEC-TCA strategy opened a new avenue to the rational design and controllable synthesis of enzyme-mimicking MIPs.
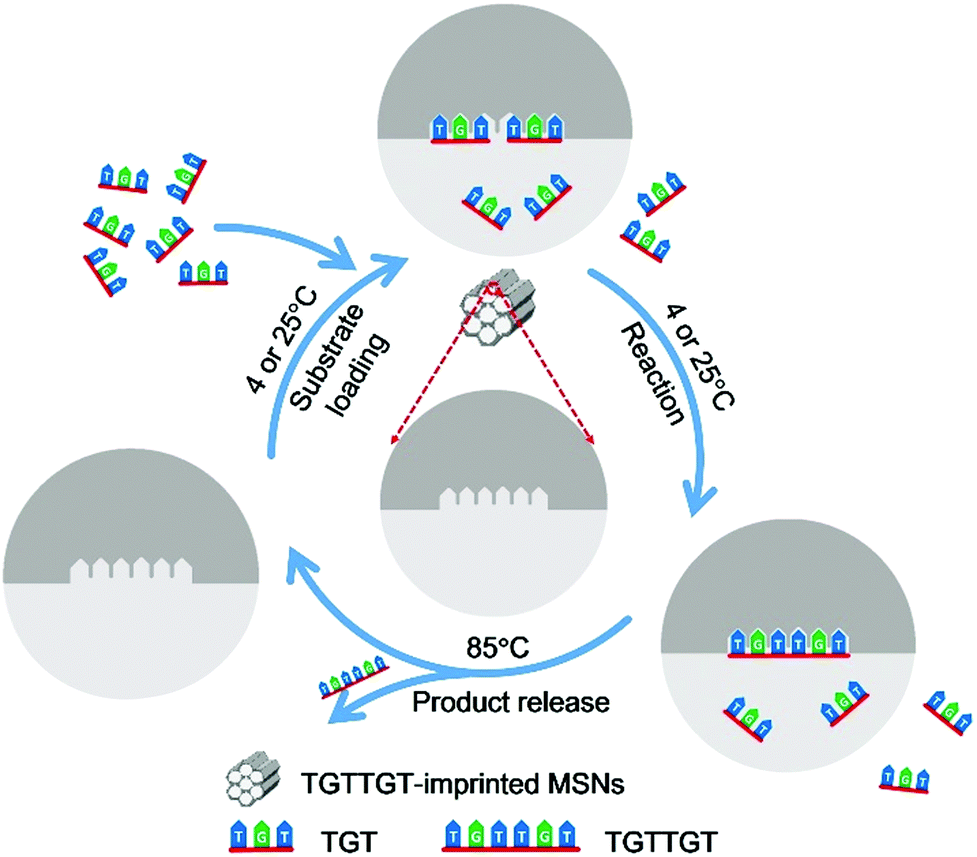 | ||
| Fig. 1 Schematic diagram of the AGEC-TCA strategy. The temperature for substrate loading and reaction was set at 4 and 25 °C for natural and modified ssDNA, respectively. | ||
Experimental
Synthesis of hexadeoxyribonucleotide-imprinted MSNs
Hexadeoxyribonucleotide-imprinted MSNs were synthesized according to the dual-template docking oriented molecular imprinting (DTD-OMI) approach59 with major modifications using modified or unmodified hexadeoxyribonucleotides as the template. Briefly, 6.0 mg cetyltrimethylammonium bromide (CTAB), 1.68 mg NaOH, and 2.88 mL H2O were added into a 10 mL reaction tube. After the mixture was mixed for 15 min, 20 OD (2.28 μmol) of the template (HOTGTTGT or TGTTGT) was added and stirred for 15 min. Then a mixture of tetraethoxysilane (TEOS) (98 μmol, 25 μL) and 3-aminopropyltriethoxysilane (APTES) (2 μmol, 0.5 μL) was added dropwise to the solution under vigorous stirring and the resulting mixture was allowed to react at room temperature for 2 h to produce a white precipitate. The solid crude product was filtered and dried under a high vacuum to yield the as-synthesized materials.To remove the surfactant template and imprinting template, the Soxhlet extraction method was employed. Briefly, a methanolic acid solution prepared by mixing 30 μL HCl (37.2%) with 3 mL methanol was used as the extraction solvent. After extracting for 24 h, the material was placed under a high vacuum with heating at 60 °C to remove residual solvent within the mesopores to get molecularly imprinted MSNs.
AGEC-TCA reaction of natural trideoxyribonucleotides
For the ligation of TGT, molecularly imprinted mesoporous silica (1 mg) was shaken in a 5 μL solution of TGT (0.2 μmol) in 0.1 M MES buffer (pH 6.15) containing 50 mM MgCl2 at 4 °C. Then the solution was added with 5 μL of 0.4 M 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC). The mixture was then subjected to a temperature-circled AGEC reaction. The temperature circle was 4 °C for 5 min and 85 °C for 3 min per cycle. Since the time required for lowering the temperature from 85 °C to 4 °C was also 1 min, the total reaction time needed per round was also 9 min. After reacting for 36, 72, 108, 180, 216, 243, 288, 315, 378, 405, and 450 min, the mixture was centrifuged and the supernatant was collected. The MSNs were washed with 10 μL of 100 mM acetic acid solution and centrifuged again. The supernatant was pooled with that obtained from the previous centrifugation. The amount of product obtained from the HEPES solution and that recovered by acetic acid wash were quantified by capillary electrophoresis (CE) analysis.For the control reaction, a 5 μL solution of TGT (0.2 μmol) in 0.1 M MES (pH 6.15) buffer containing 50 mM MgCl2 was added with 5 μL of 2 μmol EDC and then reacted at 4 °C using an ice bath. After reacting for 24, 48, 66, 96, 120, 144, 192, 240, 264, 288, and 336 h, the mixture was quantified by CE analysis.
Results and discussion
To prepare ssDNA-imprinted MSNs, the DTD-OMI approach59 we previously developed was adopted because it has been well developed and widely validated. It has proved to be efficient and facile, providing easy template removal, high template usage and excellent binding properties. This approach has enabled successful preparation of imprinted MSNs for the recognition of nucleotides,59 phosphorylated peptides60 and Amadori compounds.61 It has also been adopted and extended by others for the imprinting of various templates.62–64As a proof of concept, we first selected an unusual oligo-deoxyribonucleotide with an even number of base and a hydroxyl group at the 5’ end (such as HOTGTTGT) as the imprinting template and selected two of their half sequences with modified terminals (such as HOTGTp and HOTGT) as the reactants. This selection was due to the fact that such modified deoxyribonucleotides exhibited relatively high reaction activity.65 Taking modified trideoxyribonucleotides (HOTGTp and HOTGT) and their normal deoxyribonucleotide analogues (TGT) as examples, the reaction between these unusual trideoxyribonucleotides and the regular reaction between normal trideoxyribonucleotides are illustrated in Fig. S1 (ESI†). It should be noted that both the reaction activities of these natural and modified deoxyribonucleotides are low so that activation by an activator such as EDC as well as relatively high substrate concentrations is required. By using APTES and TEOS as the functional monomer and cross-linker, respectively, ssDNA-imprinted MSNs were prepared according to our previous DTD-OMI approach,59 which was specially designed for imprinting within mesoporous materials. In this method, the surfactant CTAB was employed to form rod-like positively charged micelles that further functioned as mesopore templates. Since the template ssDNA was negatively charged, once it was added to the micellar solution, the template molecules were docked onto the surface of the micelles due to electrostatic attraction. For the imprinting, a mixture containing TEOS and APTES was added to the solution. APTES provided a hydrogen binding moiety towards the template. It should be noted that, due to the limited size of the mesopores, MSNs are unsuitable for the imprinting of ssDNA with a long length, for which other nanomaterial formats need to be explored. According to the van’t Hoff equation, as a general trend, the equilibrium constant of binding will decrease with increasing temperature; therefore, TCA can be a general strategy for effectively eliminating the effect of product inhibition, regardless of the interaction types involved in the binding between the product and MIPs.
Because the imprinting protocol adopted herein generated a pore diameter around 2.5 nm,59,60 while mesoporous nanomaterials are associated with the nanoconfinement effect,66 we first investigated the effect of the sequence length of the ssDNA template to ensure that the template/product ssDNA can diffuse out of the pore freely to the largest extent. As shown in Fig. S2 (ESI†), among three modified oligo-deoxyribonucleotides with no secondary structure (HOTGTTGT, HOGTTGTTGT and HOGTGTTGTTGT), MSNs imprinted with the shortest one showed the best imprinting effect, providing the highest binding towards the template, while the corresponding non-imprinted MSNs (NIP) showed the lowest binding. Particularly, non-imprinted MSNs generated much higher binding towards longer oligonucleotide templates. The high non-specific adsorption of longer oligonucleotides with non-imprinted MSNs can be attributed to the confinement effect of mesopores. Although shorter ssDNA templates may experience much less confinement, they are too short to be ideal for the investigation on the reaction specificity of the substrate. Therefore, we selected HOTGTTGT as the imprinting template for further investigations. However, it is noteworthy that a longer ssDNA template is also applicable, provided that the diameter of mesopore can be appropriately enlarged.
The ratio of the functional monomer and crosslinker was optimized in terms of the imprinting factor (IF), which is an essential parameter that reflects the binding capability of the prepared MIP towards the template as compared with the non-imprinted polymer (NIP) prepared under otherwise identical conditions. As shown in Fig. S3 (ESI†), with the monomer/crosslinker ratio at 0.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9.8, the highest IF value was obtained (4.3), which is well acceptable. Fig. 2 shows the transmission electron microscopy (TEM) image, X-ray diffraction (XRD) pattern, N2 adsorption–desorption isotherms and pore size distribution of the imprinted MSNs. The mesoporous structure was clearly confirmed by the TEM image and the XRD pattern. It can be seen from the TEM images that the obtained MSNs exhibited typical MCM-41-like morphology. The average particle size was about 90 nm as estimated from the TEM image. The average pore diameter was measured to be 2.5 nm, while the BET surface area was measured to be 915 m2 g−1, indicating that the material had a typical mesoporous structure. Such a high specific surface area would highly favour the substrate binding capacity. The Fourier transform infrared (FTIR) spectrum confirmed the plentiful presence of the Si–O bond and amino group (Fig. S4, ESI†), which suggests successful preparation of MSNs with desired functionalities.
:9.8, the highest IF value was obtained (4.3), which is well acceptable. Fig. 2 shows the transmission electron microscopy (TEM) image, X-ray diffraction (XRD) pattern, N2 adsorption–desorption isotherms and pore size distribution of the imprinted MSNs. The mesoporous structure was clearly confirmed by the TEM image and the XRD pattern. It can be seen from the TEM images that the obtained MSNs exhibited typical MCM-41-like morphology. The average particle size was about 90 nm as estimated from the TEM image. The average pore diameter was measured to be 2.5 nm, while the BET surface area was measured to be 915 m2 g−1, indicating that the material had a typical mesoporous structure. Such a high specific surface area would highly favour the substrate binding capacity. The Fourier transform infrared (FTIR) spectrum confirmed the plentiful presence of the Si–O bond and amino group (Fig. S4, ESI†), which suggests successful preparation of MSNs with desired functionalities.
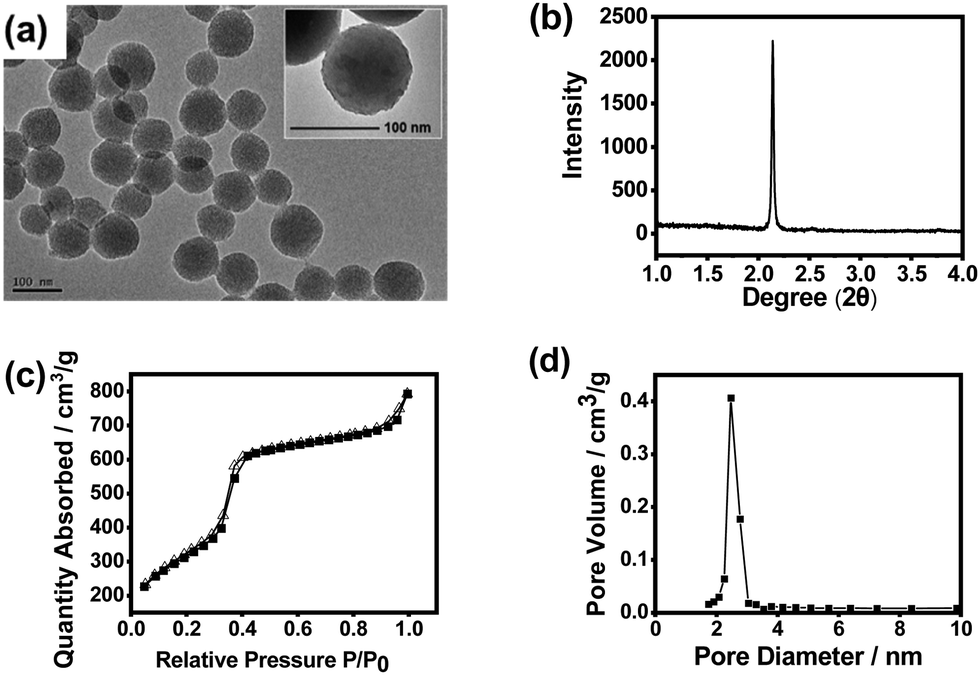 | ||
| Fig. 2 Characterization of the structure and properties of HOTGTTGT-imprinted MSNs. (a) TEM image (inset: a high-resolution image), (b) XRD pattern, (c) N2 adsorption–desorption isotherms, and (d) pore size distribution. | ||
The affinity of the HOTGTTGT-imprinted MSNs towards the template (also the product) as well as the reactants was investigated. The dissociation constant (Kd) between the imprinted MSNs and the template was about 120 μM, while the Kd values were about 410 and 760 μM for HOTGTp and HOTGT, respectively (Fig. S5–S7, ESI†). Given the hydrogen bonding capability of the template and reactants, this affinity level was reasonable.
We checked the possibility of spontaneous ligation of the substrate ssDNA in the absence of imprinted MSNs as well as the possibility of template leakage from the MIP. As shown in Fig. S8 (ESI†), even when imprinted MSNs were absent, the mixture of the substrate ssDNA reacted under the TCA mode or the constant temperature mode both generated a small peak of product, which means that the ligation of the ssDNA substrate was spontaneous but the speed was low. Meanwhile, the peak areas for the product under the two modes were almost the same, indicating that processing at a high temperature in the absence of MIP could not facilitate the ligation reaction. Moreover, when the reactants were not added, the processing of imprinted MSNs with the buffer solution under the TCA mode did not generate any product of HOTGTTGT. As shown in Fig. S9 (ESI†), no phosphorus element was observed on the prepared imprinted MSNs, indicating that there were no template molecules (which are high in phosphor) left behind in the material after template removal. These results suggest that the template used for the imprinting had been completely removed during the MIP synthesis process and there was no template leakage during the processing, indicating that the imprinted MSNs were well constructed.
We further investigated the effectiveness of the AGEC strategy and the TCA strategy using the prepared HOTGTTGT-imprinted MSNs as an ssDNA ligase mimic, with a series of control reactions performed under different conditions for comparison. As shown in Fig. S10 (ESI†), when imprinted or non-imprinted MSNs were absent, whatever the reaction was carried out under the TCA mode or the constant temperature mode, the peak area for HOTGTTGT was limited, indicating again that spontaneous ligation was possible but with a very low speed. When imprinted MSNs were present and the reaction was performed under the constant temperature mode, the peak area for HOTGTTGT apparently increased, confirming the effectiveness of the AGEC strategy. More obviously, when imprinted MSNs were present and the reaction was performed under the TCA mode, the peak area for HOTGTTGT dramatically increased, further verifying the effectiveness of the TCA program. As a comparison, when non-imprinted MSNs were present and the reaction was performed under the TCA mode, the peak area for HOTGTTGT was comparable to that observed for the reaction under the TCA mode without the presence of imprinted or non-imprinted MSNs. This indicates that the TCA strategy was effective only when imprinted MSNs were present.
The effect of product release temperature in the TCA program was optimized. As shown in Fig. S11 (ESI†), although high temperatures ranging from 75 to 95 °C worked effectively, product release at 85 °C generated the highest product amount. Therefore, 85 °C was used for further investigations. The temperature cycling program was further optimized through investigating the maximum reaction time and product release time needed for each round. The product amount produced by the imprinted MSNs reached its maximum when the substrates were reacted for 5 min at 25 °C in a single round (Fig. S12, ESI†), while the amount of product released from the imprinted MSNs reached the maximum when the reaction vial was heated for 3 min at 85 °C (Fig. S13, ESI†). With this optimized temperature-cycling program, the thermal stability and reusability of the imprinted MSNs were investigated through a continuous AGEC-TCA reaction and quantitative analysis of the product generated in each round. As shown in Fig. 3a, considering possible measurement errors, the product amount generated in each round was almost the same. This indicates that the imprinted MSNs were thermally stable and reusable. The nanomaterial could endure temperature circling between 25 and 85 °C for at least 28 rounds (totally 252 min), allowing amplification of the product by increasing the TCA round number. This finding implies that 3 min of product release at 85 °C could effectively release the product from the mesoporous nanoreactors. Meanwhile, this also implies that the product release temperature was high enough to induce convection within the mesopores to facilitate mass transfer between the mesopores and outside solution. Therefore, for further reactions, the reaction time and product release time were set at 5 and 3 min, respectively.
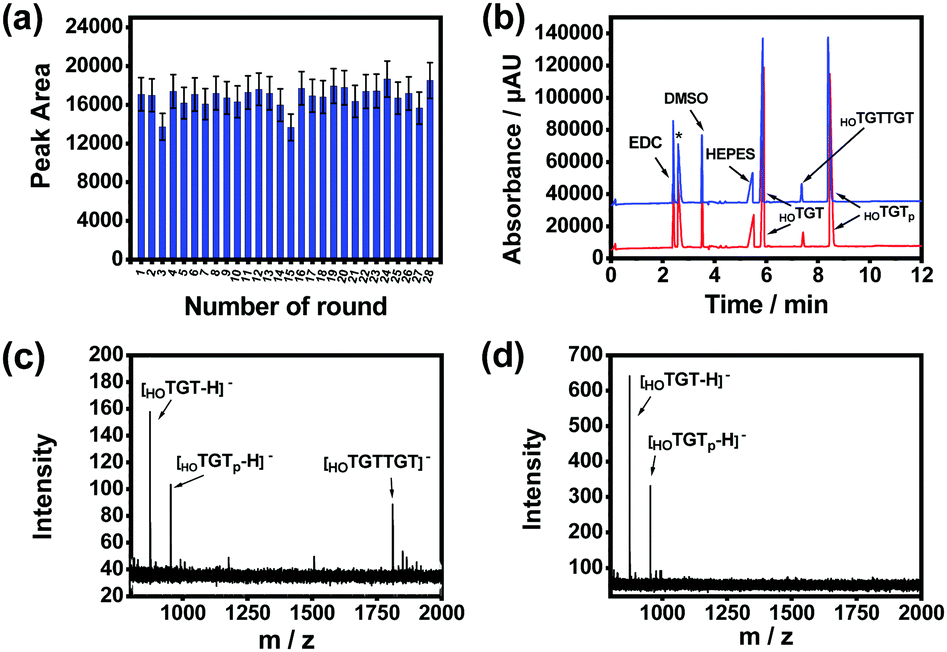 | ||
| Fig. 3 Reusability of HOTGTTGT-imprinted MSNs and characterization of the product of the AGEC-TCA reaction of modified trideoxyribonucleotides. (a) The peak area for the reaction product generated by HOTGTTGT-imprinted MSNs in different rounds within consecutive 28 rounds. (b) Capillary electropherograms of the products generated by the HOTGTTGT-imprinted MSNs for 108 min (temperature-circled) (blue) and reaction for 66 h at room temperature in the absence of the HOTGTTGT-imprinted MSNs (red). The peak with asterisk is the system peak. MALDI-TOF MS spectra of the substrates and reaction product in the presence (c) and absence (d) of HOTGTTGT-imprinted MSNs after reacting for 24 h. | ||
We investigated the catalytic activity of the imprinted MSNs through CE analysis. As shown in Fig. 3b, the components in the reaction mixture including the buffering species were well separated by CE and the imprinted MSN-based AGEC-TCA reaction for a much shorter time produced a much higher amount of product as compared with the control reaction for a much longer time. This finding further confirmed the effectiveness of the proposed AGEC-TCA strategy and the imprinted MSNs. We further investigated the catalytic activity of the imprinted MSNs using matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS). As shown in Fig. 3c and d, when the reactants interacted in the presence of the imprinted MSNs under the optimized TCA program for 24 h, an apparent peak for the expected reaction product was observed; as a comparison, no product was observed for the control reaction in the absence of the imprinted MSNs at a constant temperature (25 °C) for the same period. These results reveal that the imprinted MSNs could effectively facilitate the ligation of the reactant ssDNA fragments.
We comparatively investigated the reaction equilibriums for the imprinted MSN-based AGEC-TCA reaction and control reaction. As shown in Fig. S14 (ESI†), the control reaction at room temperature took 66 h to reach equilibrium, whereas the AGEC-TCA reaction took only 108 min (1.8 h). Clearly, the AGEC-TCA reaction was remarkably faster. We further investigated the performance of the imprinted MSN-based AGEC-TCA reaction and its comparison with the control reaction at three different substrate concentrations (10, 2 and 1 mM for each substrate). The concentration of the generated product was quantified in terms of the UV absorbance of the product according to a standard curve (Fig. S15, ESI†). At these substrate concentrations, the AGEC reaction exhibited significantly enhanced performance with reaction rate enhancement by 46.4–59.3 fold (Table S1, ESI†). Interestingly, a higher enhancement factor of reaction rate was obtained at a lower substrate concentration, indicating the AGEC-TCA reaction favoured the reaction at a low substrate concentration at which the collision probability of substrate molecules was low.
For the enzymatic reaction, substrate specificity is an important property. Prior to the investigation of substrate specificity, we investigated the specificity of the MIP towards the template as well as four template analogues. As shown in Fig. S16 (ESI†), the imprinted MSNs exhibited reasonably acceptable selectivity towards the template over their analogues, considering the number of mismatched nucleotides. We further observed the specificity of the imprinted MSN-based AGEC-TCA reaction towards the modified trideoxyribonucleotide substrates and their analogues (structures shown in Fig. S17, ESI†). As shown in Fig. S18 (ESI†), the imprinted MSNs exhibited excellent substrate specificity towards the predetermined substrates, while the control reaction exhibited no specificity at all.
Finally, we prepared MSNs imprinted with a naturally occurring hexadeoxyribonucleotide (TGTTGT) using the above established conditions. Since it is highly similar to the modified hexadeoxyribonucleotide in structure, the obtained TGTTGT-imprinted MSNs were expected to exhibit similar properties and catalytic capability. As shown in Fig. S19 (ESI†), the TGTTGT-imprinted MSNs showed similar binding affinity towards the template TGTTGT (Kd ≈ 1.2 × 10−4 M). The material also exhibited apparent affinity towards the trideoxyribonucleotide substrate (TGT), yielding a Kd value at the mM level (Fig. 4a and b). It gave a maximum binding capacity to the substrate that is about 2-fold higher than that to the template, which is reasonable since a TGTTGT-imprinted cavity can accommodate two TGT molecules. As expected, the imprinted MSNs facilitated the ligation of the substrate ssDNA molecules with a significantly enhanced reaction speed (Fig. 4c). Since the reaction activity of the unmodified trideoxyribonucleotide is lower than that of its modified analogues,65 its reaction speeds in the AGEC-TCA reaction and control reaction were apparently lower than those of modified analogues. The AGEC-TCA reaction reached equilibrium in 405 min (6.75 h), while the control reaction reached its maximum in 288 h (Fig. 4d and e). Through the calibration curve shown in Fig. S20 (ESI†), the enhancement factor of the average reaction rate was found to be within 53.3–63.0 (Table 1). Meanwhile, the TGTTGT-imprinted MSN-based AGEC-TCA reaction exhibited excellent specificity (Fig. 4f). Thus, the ligation of natural ssDNA fragments was achieved through the AGEC-TCA strategy-based molecular imprinting technology.
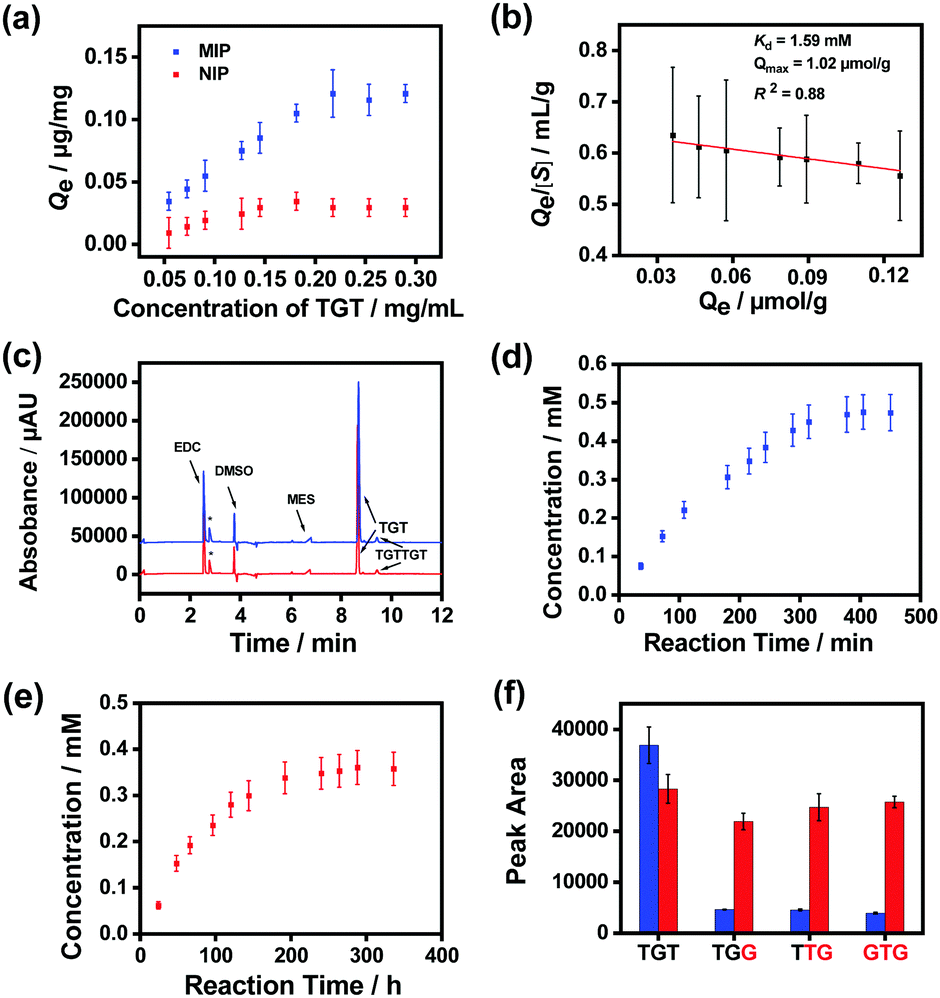 | ||
| Fig. 4 Performance of TGTTGT-imprinted MSNs for ssDNA ligation. (a) Binding isotherms of the TGTTGT-imprinted MSNs and non-imprinted MSNs towards the substrate trideoxynucleotide (TGT). (b) Scatchard plot for the measurement of the binding constant of the TGTTGT-imprinted MSNs. (c) Comparison of the product of the AGEC reaction for 108 min (blue) and the control reaction for 66 h (red). The peaks marked with asterisks were undefined. (d) Dependence of product concentration on reaction time for the TGTTGT-imprinted MSN-based AGEC-TCA reaction. (e) Dependence of product concentration on reaction time for the control reaction in the absence of TGTTGT-imprinted MSNs. (f) Comparison of the selectivity of the TGTTGT-imprinted MSN-based AGEC-TCA reaction (blue) and control reaction (red) towards different substrates. Red letters indicate the mismatched nucleotides. | ||
| Substrate conc. (mM) | Reaction type | Product conc. (μM) | Average reaction rate (μM h−1) | Enhancement factor |
|---|---|---|---|---|
| a Substrate: TGT; TCA program: reaction at 4 °C for 5 min, product release at 85 °C for 3 min; temperature cooling back in 1 min; control reaction: reaction at 4 °C in the absence of TGTTGT-imprinted MSNs. The average reaction rates for the AGEC-TCA reaction were averaged for a reaction duration of 6.75 h, while those for the control reaction were averaged for 288 h. | ||||
| 20 | AGEC-TCA | 49.0 | 7.26 | 55.8 |
| Control | 38.1 | 0.13 | ||
| 4 | AGEC-TCA | 10.8 | 1.60 | 53.3 |
| Control | 7.9 | 0.03 | ||
| 2 | AGEC-TCA | 5.6 | 0.82 | 63.0 |
| Control | 3.8 | 0.013 | ||
In our previous study,58 non-imprinted boronate affinity MSNs, which were employed as nanoreactors for efficient solid-phase labelling of saccharides, exhibited an over 200-fold enhancement of the reaction speed. The relatively poor enhancement (46–63 fold) in this study can be attributed to the lower reaction activities of the substrates as well as the activation step involved that took extra time. However, through the introduction of molecular imprinting, the imprinted MSNs exhibited excellent substrate specificity. As a comparison, the non-imprinted affinity MSNs did not exhibit such excellent reactant specificity.58 On the other hand, from the structures of the reactants used in this study as well as the reaction formulas shown in Fig. S1 (ESI†), we once worried that possible by-products such as HOTGTTGTTGT and TGTTGTTGT would be formed if the binding pockets were not well formed. This was because poorly-formed binding cavities might allow the termini groups of the product extruding out of the pocket, being accessible to the reactants to form by-products. However, no such by-products were observed (Fig. 3b and 4c), which in turn indicates that the binding cavities were well formed within the mesopores.
Conclusion
In conclusion, we have developed a novel strategy called AGEC-TCA for the rational design and controllable synthesis of enzyme-mimicking MIPs in this study. By imprinting the product ssDNA within mesopores, the imprinted MSNs could specifically recognize and bind the substrate ssDNA fragments, drawing them together into a highly favorable orientation and therefore greatly enhancing the effective collision probability. On the other hand, the reaction was implemented in a temperature-cycling manner, effectively avoiding the product inhibition effect and thereby enabling successive generation of the product. Based on the AGEC-TCA strategy, both types of the imprinted MSNs constructed in this study allowed selective ligation of short ssDNA fragments with an enhanced reaction speed and excellent sequence specificity. To the best of our knowledge, this is the first time of introducing the AGEC strategy and the temperature-cycling program into enzyme-mimicking studies. Although the catalytic ability of the current enzyme-mimicking MIPs is still far from that of natural enzymes and many inorganic nanozymes, the AGEC-TCA strategy opened up new avenues to the rational design and engineering of artificial enzymes towards highly desirable performance.Author contributions
Z. L. designed the research and wrote the manuscript. X. P. H. conducted most of the experimental work and data analysis, and prepared the draft of the manuscript. Q. L., Y. L. and Z. C. G. carried out partial experimental work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the National Science Fund for Distinguished Young Scholars (21425520) and the Key Grant (21834003) from the National Natural Science Foundation of China as well as the Excellent Research Program (ZYJH004) from Nanjing University to ZL.References
- K. Chen and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 5618–5622 CrossRef CAS PubMed.
- H. Joo, Z. Lin and F. H. Arnold, Nature, 1999, 399, 670–673 CrossRef CAS PubMed.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- R. Breslow, A. W. Czarnik, M. Lauer, R. Leppkes, J. Winkler and S. Zimmerman, J. Am. Chem. Soc., 1986, 108, 1969–1979 CrossRef CAS.
- H. Tokuyama, S. Yamago, E. Nakamura, T. Shiraki and Y. Sugiura, J. Am. Chem. Soc., 1993, 115, 7918–7919 CrossRef CAS.
- H. J. Sun, A. D. Zhao, N. Gao, K. Li, J. S. Ren and X. G. Qu, Angew. Chem., Int. Ed., 2015, 54, 7176–7180 CrossRef CAS PubMed.
- Y. Song, X. H. Wang, C. Zhao, K. G. Qu, J. S. Ren and X. G. Qu, Chem. – Eur. J., 2010, 16, 3617–3621 CrossRef CAS.
- R. Bonomi, F. Selvestrel, V. Lombardo, C. Sissi, S. Polizzi, F. Mancin, U. Tonellato and P. Scrimin, J. Am. Chem. Soc., 2008, 130, 15744–15745 CrossRef CAS PubMed.
- M. Comotti, C. Della Pina, R. Matarrese and M. Rossi, Angew. Chem., Int. Ed., 2004, 43, 5812–5815 CrossRef CAS PubMed.
- X. Shen, W. Liu, X. Gao, Z. Lu, X. Wu and X. Gao, J. Am. Chem. Soc., 2015, 137, 15882–15891 CrossRef CAS PubMed.
- H. L. Tan, C. J. Ma, L. Gao, Q. Li, Y. H. Song, F. G. Xu, T. Wang and L. Wang, Chem. – Eur. J., 2014, 20, 16377–16383 CrossRef CAS PubMed.
- C. Korsvik, S. Patil, S. Seal and W. T. Self, Chem. Commun., 2007, 1056–1058 RSC.
- F. Manea, F. B. Houillon, L. Pasquato and P. Scrimin, Angew. Chem., Int. Ed., 2004, 43, 6165–6169 CrossRef CAS PubMed.
- B. Liu and J. Liu, Nano Res., 2017, 10, 1125–1148 CrossRef CAS.
- L. Huang, J. Chen, L. Gan, J. Wang and S. Dong, Sci. Adv., 2019, 5, eaav5490 CrossRef CAS PubMed.
- H. Wei and E. K. Wang, Chem. Soc. Rev., 2013, 42, 6060–6093 RSC.
- D. W. Jiang, D. L. Ni, Z. T. Rosenkrans, P. Huang, X. Y. Yan and W. B. Cai, Chem. Soc. Rev., 2019, 48, 3683–3704 RSC.
- G. Wulff and A. Sarhan, Angew. Chem., Int. Ed. Engl., 1972, 11, 341 CAS.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812–1832 CrossRef CAS.
- G. Vlatakis, L. I. Andersson, R. Müller and K. Mosbach, Nature, 1993, 361, 645–647 CrossRef CAS.
- X. T. Shen, J. Svensson Bonde, T. Kamra, L. Bülow, J. C. Leo, D. Linke and L. Ye, Angew. Chem., Int. Ed., 2014, 53, 10687–10690 CrossRef CAS.
- K. J. Shea, D. A. Spivak and B. Sellergren, J. Am. Chem. Soc., 1993, 115, 3368–3369 CrossRef CAS.
- J. Svenson, N. Zheng and I. A. Nicholls, J. Am. Chem. Soc., 2004, 126, 8554–8560 CrossRef CAS.
- H. Shi, W.-B. Tsai, M. D. Garrison, S. Ferrari and B. D. Ratner, Nature, 1999, 398, 593–597 CrossRef CAS.
- L. Li, Y. Lu, Z. J. Bie, H. Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2013, 52, 7451–7454 CrossRef CAS PubMed.
- J. K. Awino and Y. Zhao, J. Am. Chem. Soc., 2013, 135, 12552–12555 CrossRef CAS PubMed.
- D. K. Robinson and K. Mosbach, J. Chem. Soc., Chem. Commun., 1989, 14, 969–970 RSC.
- K. Ohkubo, Y. Urata, S. Hirota, Y. Honda, Y.-I. Fujishita and T. Sagawa, J. Mol. Catal., 1994, 93, 189–193 CrossRef CAS.
- J. Q. Liu and G. Wulff, J. Am. Chem. Soc., 2004, 126, 7452–7453 CrossRef CAS PubMed.
- G. Wulff and J. Q. Liu, Acc. Chem. Res., 2012, 45, 239–247 CrossRef CAS PubMed.
- J. Damen and D. C. Neckers, J. Am. Chem. Soc., 1980, 102, 3265–3267 CrossRef CAS.
- K. J. Shea, E. A. Thompson, S. D. Pandey and P. S. Beauchamp, J. Am. Chem. Soc., 1980, 102, 3149–3155 CrossRef CAS.
- L. I. Andersson and K. Mosbach, Macromol. Rapid Commun., 1989, 10, 491–495 CrossRef CAS.
- S. E. Bystroem, A. Boerje and B. Akermark, J. Am. Chem. Soc., 1993, 115, 2081–2083 CrossRef CAS.
- G. Wulff, Chem. Rev., 2002, 102, 1–28 CrossRef CAS PubMed.
- Z. Zhang, X. Zhang, B. Liu and J. Liu, J. Am. Chem. Soc., 2017, 139, 5412–5419 CrossRef CAS PubMed.
- X. T. Shen, C. X. Huang, S. Shinde, K. K. Jagadeesan, S. Ekstrom, E. Fritz and B. Sellergren, ACS Appl. Mater. Interfaces, 2016, 8, 30484–30491 CrossRef CAS PubMed.
- L. Cenci, G. Guella, E. Andreetto, E. Ambrosi, A. Anesi and A. M. Bossi, Nanoscale, 2016, 8, 15665–15670 RSC.
- B. R. Hart, D. J. Rush and K. J. Shea, J. Am. Chem. Soc., 2000, 122, 460–465 CrossRef CAS.
- A. Nematollahzadeh, W. Sun, C. S. A. Aureliano, D. Lütkemeyer, J. Stute, M. J. Abdekhodaie, A. Shojaei and B. Sellergren, Angew. Chem., Int. Ed., 2011, 50, 495–498 CrossRef CAS PubMed.
- A. M. Bossi, P. S. Sharma, L. Montana, G. Zoccatelli, O. Laub and R. Levi, Anal. Chem., 2012, 84, 4036–4041 CrossRef CAS.
- J. Ye, Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2014, 53, 10386–10389 CrossRef CAS PubMed.
- R. R. Xing, Y. R. Wen, Y. R. Dong, Y. J. Wang, Q. Zhang and Z. Liu, Anal. Chem., 2019, 91, 9993–10000 CrossRef CAS PubMed.
- B. Sellergren and C. J. Allender, Adv. Drug Delivery Rev., 2005, 57, 1733–1741 CrossRef CAS PubMed.
- F. Canfarotta, L. Lezina, A. Guerreiro, J. Czulak, A. Petukhov, A. Daks, K. Smolinska-Kempisty, A. Poma, S. Piletsky and N. A. Barlev, Nano Lett., 2018, 18, 4641–4646 CrossRef CAS.
- Z. K. Gu, Y. R. Dong, S. X. Xu, L. S. Wang and Z. Liu, Angew. Chem., Int. Ed., 2021, 60, 2663–2667 CrossRef CAS.
- M. Panagiotopoulou, Y. Salinas, S. Beyazit, S. Kunath, L. Duma, E. Prost, A. G. Mayes, M. Resmini, B. Tse Sum Bui and K. Haupt, Angew. Chem., Int. Ed., 2016, 55, 8244–8248 CrossRef CAS.
- X. S. Tang, F. Li, J. Jia, C. Yang, W. Liu, B. Jin, X. Y. Wang, R. X. Gao, D. L. He and P. Guo, Int. J. Nanomed., 2017, 12, 2979–2993 CrossRef CAS PubMed.
- Y. R. Dong, W. Li, Z. K. Gu, R. R. Xing, Y. Y. Ma, Q. Zhang and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 10621–10625 CrossRef CAS PubMed.
- P. X. Medina Rangel, E. Moroni, F. Merlier, L. A. Gheber, R. Vago, B. Tse Sum Bui and K. Haupt, Angew. Chem., Int. Ed., 2020, 59, 2816–2822 CrossRef CAS PubMed.
- S. X. Xu, L. S. Wang and Z. Liu, Angew. Chem., Int. Ed., 2021, 60, 3858–3869 CrossRef CAS PubMed.
- O. Slinchenko, A. Rachkov, H. Miyachi, M. Ogiso and N. Minoura, Biosens. Bioelectron., 2004, 20, 1091–1097 CrossRef CAS PubMed.
- M. Ogiso, N. Minoura, T. Shinbo and T. Shimizu, Biomaterials, 2006, 27, 4177–4182 CrossRef CAS PubMed.
- V. Ratautaite, S. N. Topkaya, L. Mikoliunaite, M. Ozsoz, Y. Oztekin, A. Ramanaviciene and A. Ramanavicius, Electroanalysis, 2013, 25, 1169–1177 CrossRef CAS.
- H. Brahmbhatt, A. Poma, H. M. Pendergraff, J. K. Watts and N. W. Turner, Biomater. Sci., 2016, 4, 281–287 RSC.
- P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 368, 321–323 CrossRef CAS PubMed.
- Y. Wan and D. Y. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed.
- X. H. Pan, Y. Chen, P. X. Zhao, D. J. Li and Z. Liu, Angew. Chem., Int. Ed., 2015, 54, 6173–6176 CrossRef CAS PubMed.
- Y. Chen, X. L. Li, D. Y. Yin, D. J. Li, Z. J. Bie and Z. Liu, Chem. Commun., 2015, 51, 10929–10932 RSC.
- Y. Chen, D. J. Li, Z. J. Bie, X. P. He and Z. Liu, Anal. Chem., 2016, 88, 1447–1454 CrossRef CAS PubMed.
- X. H. Pan, X. P. He and Z. Liu, Anal. Chim. Acta, 2018, 1019, 65–73 CrossRef CAS PubMed.
- X. D. Zheng, F. S. Zhang, E. L. Liu, X. C. Xu and Y. S. Yan, ACS Appl. Mater. Interfaces, 2017, 9, 730–739 CrossRef CAS PubMed.
- N. Lv, J. L. Zhang, G. M. Li, X. Wang and J. Z. Ni, ChemistrySelect, 2017, 2, 5089–5094 CrossRef CAS.
- S. C. Ding, Z. L. Li, Y. Cheng, C. B. Du, J. F. Gao, Y. W. Zhang, N. Zhang, Z. T. Li, N. H. Chang and X. L. Hu, Nanotechnology, 2018, 29, 375604 CrossRef PubMed.
- G. Vonkiedrowski, Angew. Chem., Int. Ed. Engl., 1986, 25, 932–935 CrossRef.
- Y. Chen, S. S. Wang, J. Ye, Z. Liu and X. C. Wu, Nanoscale, 2014, 6, 9563–9567 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb02325j |
| This journal is © The Royal Society of Chemistry 2022 |