
Simple upcycling of virgin and waste polyethylene into covalent adaptable networks: catalyst-free, radical-based reactive processing with dialkylamino disulfide bonds†
Logan M.
Fenimore
 a,
Boran
Chen
a and
John M.
Torkelson
*ab
a,
Boran
Chen
a and
John M.
Torkelson
*ab
aDepartment of Chemical and Biological Engineering, Northwestern University, Evanston, IL 60208, USA. E-mail: j-torkelson@northwestern.edu
bDepartment of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA
First published on 8th November 2022
Abstract
Polyethylene (PE) is a ubiquitous commodity polymer that faces significant barriers to efficient recycling despite its thermoplastic nature. PE can be permanently cross-linked to enhance its properties and expand its applicability, but conventionally cross-linked PE (PEX) is not reprocessable in the melt-state and thus cannot be recycled for high-value use. Here, we have transformed thermoplastic PE into PE covalent adaptable networks (CANs) via reactive radical-based, melt-state processing with 1 wt% dicumyl peroxide and 5 wt% bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide (BiTEMPS methacrylate), a dynamic covalent cross-linker. The simple, catalyst-free, one-pot reactive process employing dialkylamino disulfide dynamic chemistry was used to upcycle both commodity and waste thermoplastic PE into thermally stable and reprocessable PE CANs, and the thermomechanical properties of resulting CANs are tunable without sacrificing their recyclability. Low-density PE CANs and high-density PE CANs fully recover cross-link densities and associated properties after multiple reprocessing steps, resist creep deformation at elevated temperature relative to their thermoplastic precursors, and, like PEX but unlike some PE vitrimers, exhibit no phase separation. This novel procedure opens the door to the development of CANs based on reactive processing of ethylene-based copolymers as well as cross-linked PE nanocomposites and foams.
1. Introduction
Polyethylene (PE) is an essential material based on its utility and ubiquity. From mulches and agricultural films to pipes and cable wiring and even single-use disposables, PE has a diverse range of applications and comprises 36% of global plastics production, enabling the conveniences of modern life.1 The pervasiveness of PE ushers in considerable sustainability concerns, as 353 million metric tons of plastic waste were generated globally in 2019 alone.1,2 In theory, PE and other thermoplastic materials can be recycled as they soften and become moldable at elevated processing temperatures.3 Mechanical recycling methods for thermoplastics such as injection molding and re-extrusion with additives are widespread and economically viable, yet they often result in inferior polymers with reduced properties due to thermomechanical degradation with enough reprocessing.3–8 Consequently, only 9% of plastic waste was recycled in 2019 while the rest was incinerated, landfilled, or littered.2 In light of these issues, some researchers have turned toward developing controlled chemical recycling methods in which polymers may be degraded into their constituent monomers and repolymerized into new plastics. Despite the promise of these methods to develop into precise techniques that result in high-quality materials, progress is still needed to decrease energy demands and generalize methods across polymer classes.7–10Thermoplastics are sometimes cross-linked into thermoset materials to enhance properties and expand their applicability.11,12 For example, PE can be processed into cross-linked PE (PEX, also called XLPE) by radical-based reactive extrusion of high-density PE (HDPE) or low-density PE (LDPE).11–13 Compared to their thermoplastic counterparts, thermosets exhibit more robust mechanical properties, outstanding resistance to chemical attack, and excellent heat stability owing to their irreversible covalent cross-links and resulting three-dimensional network architecture. These advantages suit thermosets to an assortment of applications; PEX is commonly utilized as infrastructure piping for plumbing and transport of natural gas as well as insulation for electrical wires and cables.12,13 However, permanent cross-links restrict the flow of polymer chains upon heating, preventing thermosets from being remolded. Thus, once spent, cross-linked materials like PEX cannot be recycled for high-value applications, further worsening polymer pollution.14,15
A promising avenue by which these recyclability challenges can be addressed involves the incorporation of dynamic covalent bonds during polymer network syntheses.16–23 When integrated into polymer structures as cross-links, dynamic covalent bonds transform polymer networks previously incapable of flowing at high temperature into covalent adaptable networks (CANs), also called dynamic covalent polymer networks (DCPNs), that may be recycled via melt-state processing.20–22 Such bonds in CANs undergo reversible reactions that reconfigure their chemical arrangements and enable chain mobility upon the input of a stimulus such as heat or light. Associative dynamic chemistries involve the simultaneous formation and breaking of bonds such that the total number of bonds at any point remains unchanged, maintaining the theoretical cross-link density of the network. Dissociative dynamic chemistries involve the reversion of bonds with the input of a stimulus and their reassociation after removal of the stimulus; above a particular temperature, there is a reduction in bonds contributing to network behavior with increasing temperature.23 Polymer networks containing dynamic covalent cross-links function as conventional thermosets with enhanced properties at service temperatures and have the potential to be recycled, like conventional thermoplastics, via melt processing at higher temperatures. As such, CANs bridge the gap between thermoset and thermoplastic polymers. Recently, the library of dynamic chemistries available for CANs has expanded to include associative chemistries such as transesterification,24–26 transamination,27–29 boronic ester exchange,30–32 and disulfide exchange33–35 as well as dissociative chemistries such as the Diels–Alder reaction,16,36–40 alkoxyamine chemistry,41–43 hindered urea exchange,44–47 and dialkylamino disulfide chemistry.48–52 Some CANs may exhibit one or several concurrent dynamic chemistries;53–57 CANs based exclusively on associative dynamic chemistries are referred to as vitrimers.58,59
While many polymer species have been developed as CANs, a significant majority of CAN studies have focused on step-growth or condensation-type polymers or have employed step-growth reactions of functional groups to synthesize CANs. These include step-growth reactions between polymers made by free-radical polymerization (FRP) and small-molecule additives serving as cross-linkers.60,61 Preparing dynamic networks with robust properties by exclusively addition-type polymerization methods, e.g., FRP and its variants32,42,43,48–52,62–64 or radical-based reactive processing,65–79 is a relatively untapped approach in CAN technology. It is important that methods to develop CANs via FRP and radical-based reactive processing are investigated because many thermosets with limited recyclability, e.g., PEX, are synthesized by such methods.
In 2016, Jin et al.42 produced CANs from polybutadiene, styrene, 4-methacryloyloxy-2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO methacrylate), and a radical initiator using alkoxyamine dynamic chemistry associated with nitroxide-mediated polymerization (NMP), a variant of FRP. CANs synthesized at 120 °C led to robust networks at use conditions and reprocessability with full recovery of cross-link density within experimental uncertainty after several reprocessing steps.42 Later, Li et al.43 showed that network composites produced by this NMP approach, including CANs made from cis-polyisoprene and carbon black, exhibited reprocessability with full cross-link density recovery and excellent elevated-temperature creep resistance. However, this NMP-based approach does not lead to full cross-link density recovery after reprocessing for CANs made by reacting only monomer with TEMPO methacrylate and a radical initiator because of the loss of reactivity of carbon-based radicals during reprocessing.43
In 2017, Otsuka and co-workers demonstrated that the bis(2,2,6,6-tetramethylpiperidin-1-yl)disulfide (BiTEMPS) functionality imparted polyurethane and polymethacrylate networks with dynamic covalent character leading to some reprocessability.48,49 The dialkylamino disulfide functionality present in BiTEMPS undergoes reversible homolytic dissociation upon heating above 80 °C, leading to stable sulfur-based (thiyl or, more specifically, TEMPS) radicals which do not suffer from disproportionation reactions or oxygen sensitivity that occur with carbon-based radicals.48–50,80–83 TEMPS radicals also do not abstract hydrogens appreciably from good donors such as hindered phenols and are unreactive toward molecular substrates such as olefins and phosphites that react readily with free radicals.82 Otsuka and co-workers later demonstrated the utility of BiTEMPS-containing molecules with a trisulfide bridge (dialkylamino trisulfide) as dynamic cross-linkers in the synthesis of polymethacrylate CANs.62,64 Dialkylamino trisulfides dissociate asymmetrically upon heating into stable thiyl and dithiyl radicals.62 Compared to the disulfide bond in BiTEMPS, the S–S bonds in dialkylamino trisulfides were found to have nearly identical bond dissociation energies (BDEs, 108.4 kJ mol−1 for trisulfide vs. 109.6 kJ mol−1 for disulfide) and a slightly larger propensity for chain transfer due to a reduction in steric hindrance.62,84 Any generation of permanent cross-links in their CANs from chain transfer had little effect on the processability of their CANs, as the networks exhibited nearly quantitative damage healability after compression molding at 110 °C for 24 h. Their disulfide-containing CANs exhibited similar healability after compression molding at 100 °C for 24 h.62
Recently, Bin Rusayyis and Torkelson50,52 showed that the dynamic covalent cross-linker BiTEMPS methacrylate (Scheme 1), which is designed for use in the FRP of monomer resulting in CANs, leads to full cross-link density recovery in reprocessed networks and excellent elevated-temperature creep suppression some tens of degrees below the reprocessing temperature. Outstanding creep suppression at elevated temperatures exhibited by the BiTEMPS-based CANs is owed to the relatively high BDE of the disulfide bond in BiTEMPS reported by Otsuka and co-workers (109.6 kJ mol−1).48,52 Because the viscous flow and stress relaxation activation energies of 106.7 kJ mol−1 and 106.2 kJ mol−1, respectively, of the BiTEMPS-based poly(hexyl methacrylate) (PHMA) CANs agree well with the BiTEMPS disulfide BDE, it is evident that the PHMA CAN elevated-temperature creep and stress relaxation responses are dominated by the BiTEMPS unit dynamic chemistry rather than the network viscoelasticity.52 Given the utility of BiTEMPS methacrylate in synthesizing creep-resistant, addition-type CANs that recover cross-link density after reprocessing, it stands to reason that BiTEMPS methacrylate could be used in formulating CAN versions of some other thermosets that have poor recyclability, such as PEX.
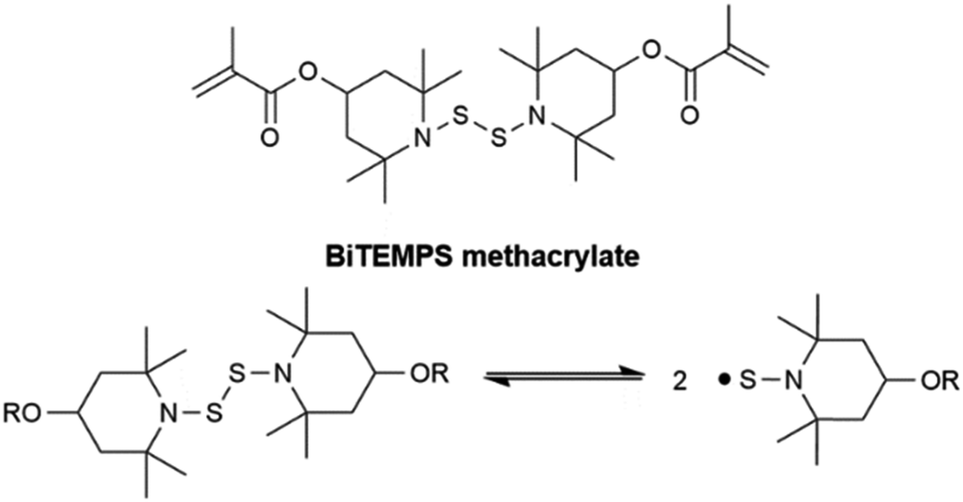 | ||
| Scheme 1 Dynamic activity of dialkylamino disulfides: BiTEMPS methacrylate undergoing homolytic dissociation of its disulfide bond to form sulfur-centered (TEMPS) radicals. | ||
Radical-based reactive processing to modify polymers with dynamic grafting units has recently gained traction as a method by which commodity plastics may be upcycled into CANs with enhanced properties and reprocessability.65–79,85,86 Following the 2017 seminal study by Röttger et al.66 on the production of HDPE vitrimers by transesterification of boronic esters grafted onto HDPE during reactive processing, a number of studies have been reported on polyethylene (PE) CANs.67–76,78,79,85,86 However, few studies address the myriad of challenges that remain before the potential of PE CANs may be realized. For instance, most studies rely on external catalysis for bond exchange, use functionalized polyolefins as precursors rather than virgin PE, or involve complex multistep syntheses. Among studies on CANs that avoid these issues, other problems have been reported such as poor thermal stability of cross-linking units67 or the emergence of nanophase separation70,71,87 that is not present in PEX.12,71
A few studies have described simple methods for producing PE CANs by reactive processing of PE without external catalysis.67,70,76 As a follow-up to the study by Röttger et al.,66 Caffy and Nicolaÿ67 demonstrated single-step functionalization of a dinitroxide bis(dioxaborolane) cross-linker onto HDPE via reactive extrusion to produce HDPE CANs. Maaz et al.70 later noted the poor thermal stability of this dinitroxide bis(dioxaborolane) cross-linking unit at conventional processing temperatures and introduced their own single-step reactive extrusion of HDPE with a dimaleimide bis(dioxaborolane) cross-linker to produce HDPE CANs. However, these CANs were not reprocessed, and no property recovery information was reported.70 Nanophase separation has also been reported in these and other PE CANs with analogous bis(dioxaborolane) cross-linking units because of the thermodynamic incompatibility of the grafted bis(dioxaborolane) moiety with PE.71,87 Reprocessing nanophase-separated PE CANs could exacerbate the heterogeneity to a macrolevel, thereby compromising properties intended to parallel those of PEX. Wang et al.76 designed a one-step procedure for LDPE CAN production from both virgin and waste LDPE via the simultaneous grafting of maleic anhydride onto LDPE backbones and cross-linking with butanediol in an extruder. To the best of our knowledge, this is the only study that has demonstrated the upcycling of post-consumer PE waste into reprocessable PE CANs that recover their thermomechanical properties after reprocessing. Saed et al.77 described their failures to produce polypropylene (PP) CANs using the Wang et al.76 procedure due to the premature reaction of maleic anhydride and butanediol prior to grafting. They instead refer to Maaz et al.70 for their superior one-step protocol using a premade exchangeable cross-linker.
Studies of PE CANs, and CAN studies in general, sometimes fail to recycle or reprocess and property test the dynamic covalent networks. Insufficient assessment of cross-link density and associated mechanical properties after reprocessing calls into question the utility of the recycled materials. A simple, addition-type CAN technology is needed that can turn both virgin and waste polyolefins like PE into dynamic networks that demonstrate property recovery after recycling.
Here, we have developed a simple, catalyst-free protocol to prepare PE CANs from virgin and waste LDPE and HDPE via radical-based reactive melt-processing. BiTEMPS methacrylate was used as a pre-synthesized exchangeable cross-linker and grafted onto PE backbones to endow PE CANs with dissociative dynamic character. We establish ranges in which loading conditions of this cross-linker and a radical initiator may be varied to tune the resulting thermomechanical properties of reprocessable PE CANs. The BiTEMPS-based PE CANs are thermally stable up to 200 °C, are reprocessable by compression molding, and recover cross-link densities and associated properties within experimental error after three successive (re)processing cycles. We demonstrate that LDPE and HDPE CANs are resistant to creep similarly to LDPE and HDPE when subjected to continuous stress below their melt transitions. Above their melting range, we show that the dissociative dynamic chemistry of BiTEMPS dominates the temperature dependence of the LDPE CAN creep response. Owing to their robust, dynamic cross-links, LDPE and HDPE CANs exhibit significant reductions in viscous creep above their melt transitions compared to their thermoplastic precursors. We also provide evidence of the absence of nanophase separation in PE CANs with our low loadings of BiTEMPS cross-linker (<0.13 mol% based on mol of ethylene repeat units). Lastly, we extend our upcycling method to waste plastics by synthesizing PE CANs from post-consumer LDPE and HDPE waste (PE-W CANs). This is the first report of reprocessable PE CANs upcycled from post-consumer PE waste by exclusively free-radical reactive processing. Similarly to upcycled commodity PE CANs, these CANs from PE waste are reprocessable and recover their thermomechanical properties after multiple compression molding cycles.
2. Experimental
2.1. Materials
All chemicals were used as received unless otherwise stated. 2,2,6,6-tetramethyl-4-piperidyl methacrylate (TMPM) was purchased from TCI America. Petroleum ether (anhydrous), sulfur monochloride (S2Cl2, 98%), o-xylene (98%), and dicumyl peroxide (DCP, 98%) were purchased from Sigma-Aldrich. Two LDPE samples (LD 103 Series, MFI of 1.1 g/10 min and LD 654, MFI of 70 g/10 min) were from ExxonMobil. Another LDPE sample (5004I, MFI of 4.2 g/10 min) was from The Dow Chemical Company. HDPE (Petrothene LM600700, MFI of 0.8 g/10 min) was from Equistar Chemicals. Waste LDPE and HDPE samples (plastic bags and a milk jug, respectively) were obtained from Jewel-Osco. Petroleum ether was dried over activated 4 Å molecular sieves for at least 48 h before use. After its initial use as received, S2Cl2 was distilled and dried over activated 4 Å molecular sieves for at least 48 h before further use.2.2. Synthesis of cross-linker
Bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide (BiTEMPS methacrylate) was synthesized, purified, and characterized according to literature procedures.50,52 Commonly, 2,2,6,6-tetramethyl-4-piperidyl methacrylate (TMPM) (∼8.78 g, 39.0 mmol) was dissolved in ∼100 mL of dry petroleum ether. After chilling this solution in a dry ice/acetone bath to −70 °C, a second solution of new or freshly distilled and dry S2Cl2 (∼1.3 g, 9.7 mmol) in ∼1.25 mL of dry petroleum ether was added dropwise with stirring. The reaction vessel was then stirred for 15 min at −70 °C and for 30 min at room temperature before its contents were washed in distilled water. Subsequent washes in water, brine, and 1 M HCl helped to remove excess and protonated TMPM (generated after the production of HCl in situ) as well as the creamy, off-white color in the resulting product. After recrystallization in methanol, vacuum filtering, and oven drying at 50 °C for 48 h, BiTEMPS methacrylate was obtained (∼2.4 g) without need for further purification.50,522.3. Synthesis of PE CANs, PE-W CANs, and PEX
PE pellets (1.0 to 2.0 g basis) were added with crushed powders of BiTEMPS methacrylate and DCP by spatula into an Atlas Laboratory Mixing Molder (flushed twice with PE prior to synthesis) with three steel balls to emulate the chaotic mixing of reactive extrusion.88 Various loadings of BiTEMPS methacrylate and DCP were employed during this study; the wt% of each loading was calculated based on the initial mass of PE added to the mixer. The LDPE batches were mixed at 130 °C and 120 rpm for 3–5 min to homogenize blends in the melt state without substantially initiating DCP. The HDPE batches were mixed at 140 °C and 120 rpm for 3–5 min to homogenize blends. The mixer temperature was then ramped to 160 °C to commence grafting of BiTEMPS methacrylate and cross-linking of PE, and this mixing continued for ∼20 min. The mixer rotor was cycled upward and downward manually to aid in mixing during cross-linking. Mixtures had 28–30 min residence times in the mixer to ensure cross-linking of PE CANs. CANs were removed from the mixer post-synthesis via spatula. Both LDPEX and HDPEX were synthesized with 1 wt% DCP by the same procedure without BiTEMPS methacrylate.2.4. Compression molding and reprocessing of PE CANs
As-synthesized network blends were cooled to room temperature, cut into millimeter-sized pieces, and processed using a PHI press (Model 0230C-X1) to complete cross-linking and produce healed films. To prepare samples used in dynamic mechanical analysis experiments, network pieces were hot pressed into ∼0.6 mm-thick films at 160 °C with a 10-ton ram force (∼8 MPa) for 30 min. Such films are considered to be 1st-molded samples. To reprocess films, 1st-molded samples were cut into small pieces and hot pressed at the same conditions to generate 2nd-molded samples. Likewise, 2nd-molded samples were cut into pieces and reprocessed at the same conditions to generate 3rd-molded samples. To mold the samples used in creep experiments, network pieces were hot pressed into discs ∼2 mm in thickness and ∼23 mm in diameter at the same conditions.2.5. Dynamic mechanical analysis (DMA)
DMA was conducted on rectangular PE and PE CAN samples cut from processed films using a TA Instruments RSA-G2 Solids Analyzer or a TA Instruments Discovery DMA 850 to measure storage modulus (E′), loss modulus (E′′), and damping ratio (tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ = E′′/E′) as functions of temperature under nitrogen flow. Typically, the tension-mode experiments were done at a 1 Hz frequency and 0.03% oscillatory strain (or 0.2% oscillatory strain for softer samples) with a heating ramp from 25 °C to 160 °C at a heating rate of 3 °C min−1. DMA experiments for thermal stability had an extended temperature window of 25 °C to 500 °C with the same heating rate.
δ = E′′/E′) as functions of temperature under nitrogen flow. Typically, the tension-mode experiments were done at a 1 Hz frequency and 0.03% oscillatory strain (or 0.2% oscillatory strain for softer samples) with a heating ramp from 25 °C to 160 °C at a heating rate of 3 °C min−1. DMA experiments for thermal stability had an extended temperature window of 25 °C to 500 °C with the same heating rate.
2.6. Gel content determination
Network pieces were massed (md, typically 1–1.5 g total) and placed into Growing Labs cellulose Soxhlet extraction thimbles. Pieces were immersed in boiling xylene under reflux at 165 °C for 72 h. Subsequently, the insoluble network fraction was dried in a vacuum oven for 48 h and massed (mo). Gel content was determined as follows: gel content (%) = 100(mo/md).2.7. Fourier transform infrared (FTIR) spectroscopy
Attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy was done via a Bruker Tensor 37 MiD FTIR spectrophotometer equipped with a diamond/ZnSe ATR attachment. Sixteen scans were collected at room temperature over the 4000 to 600 cm−1 range at 4 cm−1 resolution. FTIR spectroscopy was used to confirm the presence of BiTEMPS methacrylate (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1725 cm−1) as the cross-linker in PE CANs before and after washing in boiling xylene.
O stretch at 1725 cm−1) as the cross-linker in PE CANs before and after washing in boiling xylene.
2.8. Differential scanning calorimetry (DSC)
Relevant melting and crystallization temperatures for PE and PE CANs were determined by DSC using a Mettler Toledo DSC822e under nitrogen flow. 5–10 mg samples were tested in hermetically sealed aluminum pans. In the first cycle, samples were heated from room temperature to 180 °C at a rate of 10 °C min−1, held at 180 °C for 5 min, and cooled back to room temperature at a rate of 10 °C min−1. Melting and crystallization peak temperatures were determined from the respective endothermic and exothermic peaks of the heating and cooling scans, respectively, as were the melting endpoint and crystallization onset temperatures. Percent crystallinities were determined by integrating crystallization peaks to obtain latent heats of fusion of samples and calculating the ratio of these latent heats against the latent heat of fusion for fully crystalline PE of 293 J g−1.89,902.9. Thermogravimetric analysis (TGA)
TGA experiments were conducted on BiTEMPS methacrylate, PE samples, and PE CANs using a Mettler Toledo TGA/DSC3+. Samples were heated under nitrogen flow from 25 °C to 600 °C at a rate of 5 °C min−1. The weight of the sample was monitored as a function of temperature.2.10. Creep
Shear creep experiments were conducted with a 3.0 kPa stress (unless otherwise noted) on molded disc samples using an Anton-Paar MCR 302 rheometer and a 25 mm parallel plate fixture. Samples were equilibrated at the temperature of interest until normal forces stabilized (∼10 min) before commencing experiments. ∼10 N of normal force were used for samples at temperatures above their melt transitions, and ∼35 N of normal force were used for samples at temperatures below their melt transitions. Creep tests on PE CANs were carried out for 18000 s at 90 °C and for 10000 s at 120–150 °C. Shear strain rates (![[small gamma, Greek, dot above]](https://www.rsc.org/images/entities/i_char_e0a2.gif) ) were calculated at each temperature from the fitted slope of the linear part of the creep curves employing data between t = 16 200 s to t = 18000 s for 90 °C and between t = 8000 s to t = 10 000 s for 120–150 °C. Creep strain values were obtained by extrapolating these fitted lines to t = 0 s and subtracting these y-intercepts from the corresponding final strain values to capture and estimate the creep deformation contributed solely by the viscous component of the CANs. Network viscosities (η) in Pa s were calculated at each temperature as: η = σ/, where σ is the shear stress applied to the disc. Creep tests on thermoplastic PE samples were carried out for 18000 s at 90 °C and for 1000 s at 120–150 °C. values were calculated at each temperature from the fitted slope of the linear part of the creep curves employing data between t = 16200 s to t = 18000 s for 90 °C and between various t ranges for 120–150 °C, as linear segments of thermoplastic PE creep curves occurred at different times depending on temperature and sample. Creep strain values and η values were calculated for PE samples similarly to PE CAN samples.
) were calculated at each temperature from the fitted slope of the linear part of the creep curves employing data between t = 16 200 s to t = 18000 s for 90 °C and between t = 8000 s to t = 10 000 s for 120–150 °C. Creep strain values were obtained by extrapolating these fitted lines to t = 0 s and subtracting these y-intercepts from the corresponding final strain values to capture and estimate the creep deformation contributed solely by the viscous component of the CANs. Network viscosities (η) in Pa s were calculated at each temperature as: η = σ/, where σ is the shear stress applied to the disc. Creep tests on thermoplastic PE samples were carried out for 18000 s at 90 °C and for 1000 s at 120–150 °C. values were calculated at each temperature from the fitted slope of the linear part of the creep curves employing data between t = 16200 s to t = 18000 s for 90 °C and between various t ranges for 120–150 °C, as linear segments of thermoplastic PE creep curves occurred at different times depending on temperature and sample. Creep strain values and η values were calculated for PE samples similarly to PE CAN samples.
Tensile creep experiments were conducted with a 3.5 MPa stress on molded dog-bone shaped samples (∼0.65 mm × 4.6 mm) by hanging a 1.055 kg weight onto clamped samples in an oven. Temperatures were 60 °C for LDPE-based samples and 90 °C for HDPE-based samples. Initial sample lengths and final sample lengths after 30 min were measured using a caliper, and strains were calculated from these lengths as follows: (final length − initial length)/initial length.
2.11. Small-angle X-ray scattering (SAXS)
SAXS experiments employed a Rigaku S-MAX 3000 SAXS system in transmission mode. An X-ray generator emitting Cu Kα radiation (λ = 0.154 nm) was operated at 45 kV and 0.88 mA. The system was calibrated with silver behenate to a sample-to-detector distance of 1619.8 mm. The scattering angle (2θ) ranged from 0.4248° to 2.3839° with a 0.0035° step. Neat PE and PE CAN samples were cut from 1st-molded films. PEX SAXS samples were obtained by compression molding single pieces of as-synthesized PEX. Datasqueeze software was used to average 2D scattering patterns azimuthally to produce 1D plots of intensity versus scattering vector q, where q = 4πsin(θ)/λ. Experimental methods for Guinier analyses are given in ESI.†3. Results and discussion
3.1. Upcycling virgin LDPE and HDPE into CANs by reactive processing
LDPE and HDPE CANs were synthesized from LDPE and HDPE in the presence of BiTEMPS methacrylate as dynamic cross-linker and dicumyl peroxide (DCP) as radical initiator. LDPE (MFI of 1.1 g/10 min) and HDPE (MFI of 0.8 g/10 min) were used in this study. BiTEMPS methacrylate was synthesized using literature procedures from 2,2,6,6-tetramethyl-4-piperidyl methacrylate (TMPM) and sulfur monochloride (S2Cl2).50,52 TMPM reacts with S2Cl2 at −70 °C in dry petroleum ether to give bis(2,2,6,6-tetramethyl-4-piperidyl methacrylate) disulfide and small amounts of polysulfide derivatives such as trisulfide and tetrasulfide after quenching the reaction and washing the crude product in distilled water. We note that the formation of polysulfide analogues of BiTEMPS methacrylate is possible given other syntheses involving the reaction of tetramethylpiperidine derivatives and S2Cl2 (and in other syntheses involving the use of S2Cl2).62,64,80,91–96 S2Cl2 reacts with water to produce elemental sulfur which dissolves in and reacts with S2Cl2 to give organic polysulfanes with the formula SnCl2 (n > 2).97 Polysulfanes present in S2Cl2 will predispose the synthesis of BiTEMPS methacrylate (and other desired compounds) to forming higher order polysulfides as major and minor products.95 Thus, S2Cl2 should be new or freshly distilled and dried before its use in the synthesis of BiTEMPS methacrylate. Performing the synthesis in an inert environment will also limit the exposure of S2Cl2 to water vapor in air. In addition to quenching and washing the reaction mixture in distilled water, the product may be further purified by multiple liquid–liquid extractions with brine and 1 M HCl to remove unreacted and protonated TMPM and by column chromatography and recrystallization from methanol to give BiTEMPS methacrylate with the desired sulfur bridge length.48,62,64 We do not discount the small presence of polysulfides resulting from the cross-linker synthesis even after rigorous organic workup. Polysulfide derivatives of BiTEMPS-containing molecules and the dynamic polymer networks synthesized with them have been shown previously to have similar dynamic character to BiTEMPS-containing molecules and their dynamic covalent polymer networks with disulfide bridges.62,64 As we demonstrate further, any polysulfide presence in BiTEMPS methacrylate has negligible effect on the thermomechanical properties and reprocessability of PE CANs.Our PE CAN upcycling procedure involved reactive processing in a benchtop melt mixer of PE with BiTEMPS methacrylate and DCP. PE CAN syntheses were conducted on a 1.0–2.0 g basis of neat PE pellets with the wt% of BiTEMPS methacrylate and DCP calculated with respect to neat PE. PE pellets and solid powder mixtures of BiTEMPS methacrylate and DCP were added uniformly to the melt mixer via spatula. LDPE batches were mixed at 130 °C and 120 RPM to homogenize the blends above the melt transition of LDPE without appreciably initiating free-radical grafting. This step was similarly done for HDPE batches at 140 °C. After this initial homogenization, cross-linking was commenced by ramping the mixer temperature to 160 °C. Reaction times of 40–60 min at 160 °C are adequate to achieve high PE cross-link densities with DCP.13,98 After a total mixer residence time of 28–30 min with consistent blending, our as-synthesized PE CANs were removed from the mixer via spatula and compression molded at 160 °C and 8 MPa for 30 min to complete cross-linking and obtain 1st-molded samples.
During our procedure, cross-linking in PE CANs begins when the peroxide bonds in DCP are cleaved at high temperature to form free radicals which subsequently abstract hydrogen atoms from PE backbones. PE macroradicals are likely to attack the amenable carbon–carbon double bonds on BiTEMPS methacrylate to occupy a stable tertiary carbon position in the methacrylate. This process effectively grafts BiTEMPS methacrylate to a PE backbone chain. Such is what occurs during the grafting of similar molecules, e.g., glycidyl methacrylate, onto PE.99 The resulting radicals can continue to propagate through either carbon–carbon double bond on additional BiTEMPS methacrylate units. Dynamic cross-links are formed upon the grafting of both ends of BiTEMPS methacrylate units or chains (a run of two of more BiTEMPS methacrylate units). Radicals on BiTEMPS methacrylate may also transfer to PE to form another macroradical or terminate with one another or a PE macroradical by disproportionation. Permanent cross-links can form during this reactive process when PE radicals or other carbon-centered radicals present on a chain lacking a dynamic bond terminate by combination. See Scheme 2 for the preparation of PE CANs including some of these chemistries. Considering that the dynamic moiety in BiTEMPS methacrylate is exchangeable at processing temperatures to form sulfur-centered radicals, it is possible to form permanent cross-links through their irreversible termination onto the PE backbone.62,84 Although we do not discount that this happens in small amounts given the possibility of chain transfer from any polysulfide derivatives of BiTEMPS methacrylate leftover after purification, sufficiently hindered disulfides have limited chain transfer capabilities in general.100,101 The bulky nature of the dialkylaminos surrounding the sulfur bridge in BiTEMPS methacrylate and the low wt% of BiTEMPS methacrylate relative to PE during PE CAN synthesis limit the possibility of side reactions occurring over disulfide recombination. As described further below, no deleterious effects within error on PE CAN reprocessability occur due to side reactions.
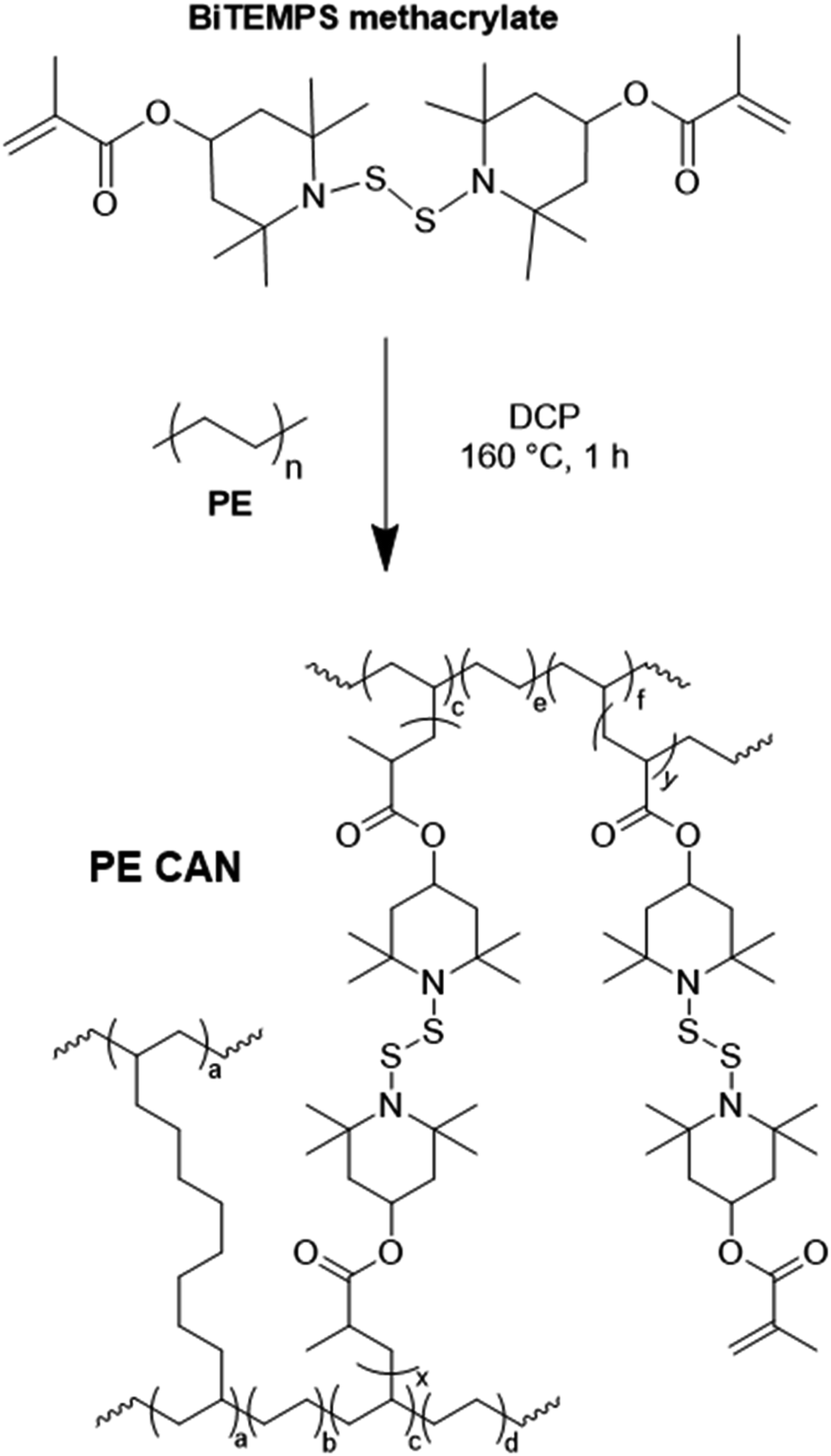 | ||
| Scheme 2 Preparation of PE CANs by reactive melt-state mixing of PE with BiTEMPS methacrylate and DCP with several possible chemistries shown. | ||
3.2. Tunability of BiTEMPS methacrylate and DCP contents to achieve reprocessability
Given the complexity of the PE dynamic cross-linking process, it is important to establish conditions in which PE CANs are robustly cross-linked compared to their thermoplastic precursors yet still processable from dynamic cross-links and uncross-linked PE chains. With insufficient amounts of cross-linking, PE materials will be processable but possess thermomechanical properties that are not a major improvement over thermoplastic PE or characteristic of cross-linked network behavior. On the other hand, too many permanent cross-links formed from an insufficient level of dynamic cross-linker or an excess of DCP will not allow for PE CANs to be (re)processed. Hence, permanent cross-links should be limited to a percolation threshold such that PE CANs may be (re)processed and recover their robust thermomechanical properties.A range of BiTEMPS methacrylate and DCP loadings were tested during the synthesis of LDPE and HDPE CANs. First, the healability of samples after compression molding at 160 °C for 30 min was used to assess (re)processability for each set of BiTEMPS methacrylate and DCP loadings in PE CANs. As control samples, it was observed that neither LDPE nor HDPE could be processed into healed films when cross-linked with 1 wt% DCP and no BiTEMPS methacrylate to give LDPEX and HDPEX. This was expected, as PEX cannot be processed at high temperature. BiTEMPS methacrylate loadings were varied at a constant loading of 1 wt% DCP to find the minimum amount of dynamic cross-linker required to allow for PE CAN processability. LDPE CANs required as little as 1 wt% BiTEMPS methacrylate in the presence of 1 wt% DCP to produce healed films after compression molding. HDPE CANs required at least 2.25 wt% BiTEMPS methacrylate to heal fully in the presence of 1 wt% DCP at the same processing conditions. Loadings of BiTEMPS methacrylate less than these amounts resulted in unhealed films, meaning that the levels of permanent cross-links formed were too large to allow for dynamic processing. See Fig. 1a, b, S1, and S2† for images of the healed and unhealed LDPE and HDPE films.
BiTEMPS methacrylate was increased incrementally to 10 wt% in LDPE and HDPE CANs in the presence of 1 wt% DCP, and the thermomechanical properties of these healed 1st-molded samples were characterized by DMA. Per Flory's ideal rubber elasticity theory,102 the rubbery plateau modulus E is linearly proportional to cross-link density and absolute temperature. In the rubbery plateau regime, the DMA loss modulus (E′′) is much lower in magnitude than E′, allowing for E in the rubbery plateau regime to be approximated by E′. Thus, the rubbery plateau E′ values of the PE CANs above their melt transitions were used to assess their extents of cross-linking. Fig. 1c, d, Tables S1 and S2† display the DMA properties of LDPE and HDPE CANs as a function of BiTEMPS methacrylate loading and temperature. (In the legends and for simplicity in text, PE CANs are subsequently referred to by their BiTEMPS methacrylate and DCP loadings using the convention “BiTEMPS methacrylate wt%”–“DCP wt%”, oftentimes before “PE CAN”.) PE CANs exhibit E′ rubbery plateaus above their melt transitions characteristic of cross-linked polymers. As BiTEMPS methacrylate loading is increased from 1 wt% to 5 wt% in LDPE CANs, E′ rubbery plateaus increase in magnitude. Beyond 5 wt%, E′ rubbery plateaus decrease in magnitude. It is expected that increasing the loading of cross-linker beyond 10 wt% will further decrease E′ rubbery plateau values in LDPE CANs. From this assessment, 5 wt% BiTEMPS methacrylate with 1 wt% DCP is sufficient to cross-link LDPE into robust CANs. 2.25-1 and 3-1 HDPE CANs give the highest E′ rubbery plateaus, and further increases in BiTEMPS methacrylate loading generally decrease the E′ rubbery plateau value. Despite not giving the highest E′ rubbery plateau value, a 5 wt% BiTEMPS methacrylate loading was used here for consistency when conducting further tests on LDPE and HDPE CANs. It is also worth noting that most LDPE and HDPE CANs exhibit a slight decrease in the rubbery plateau E′ with increasing temperature. An ideal, cross-linked rubber exhibits a linear increase in E′ with increasing temperature (T) in its rubbery plateau.102 Contrary to this, it has been shown that CANs with exclusively dissociative dynamic chemistry exhibit decreasing T-dependent rubbery plateau behavior.50,52 The BiTEMPS units endow PE CANs with dissociative dynamic chemistry, which is the origin of the decreasing T-dependent rubbery plateau.
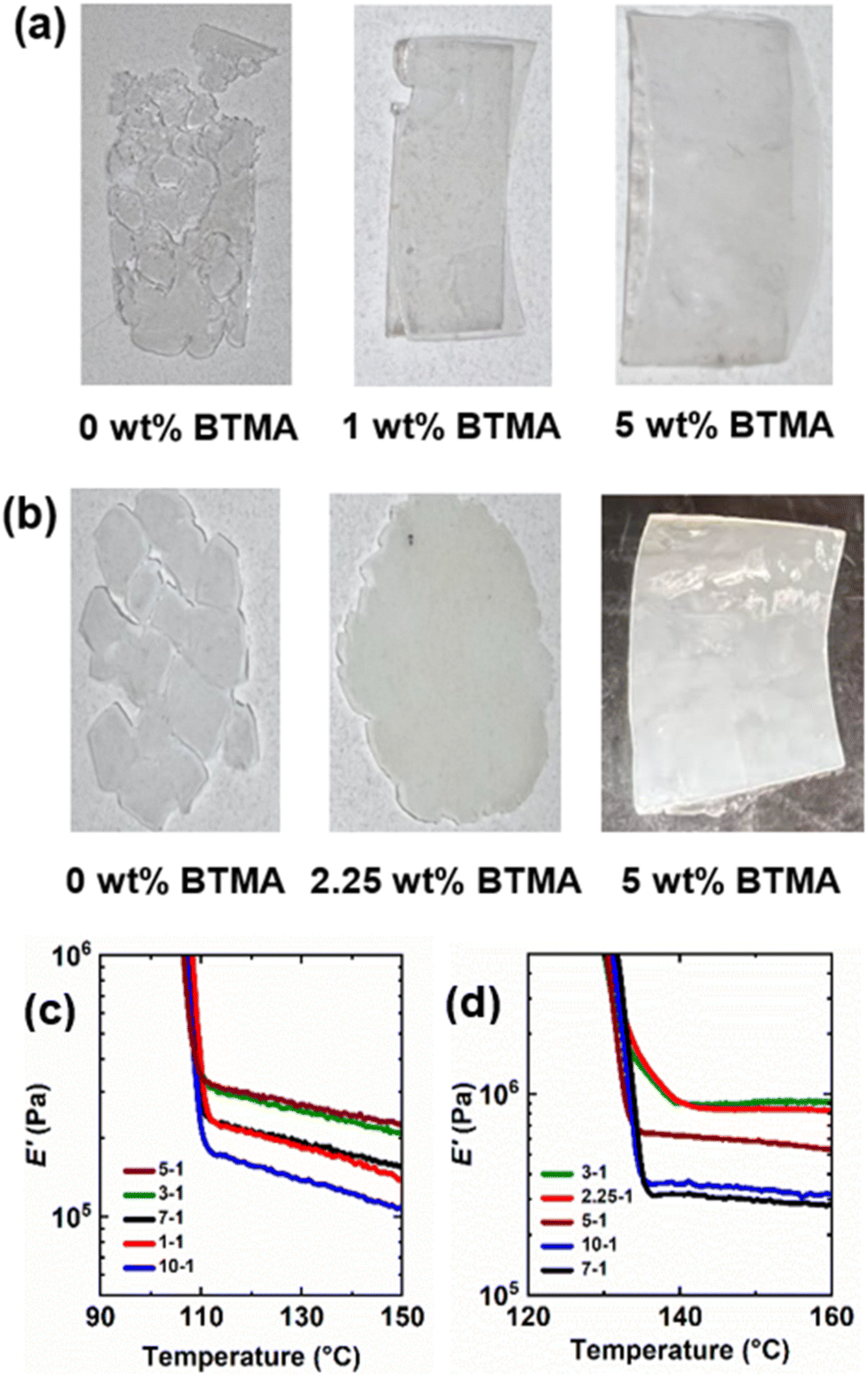 | ||
| Fig. 1 (a) Films of LDPE CANs synthesized with 1 wt% DCP and (left to right) 0 wt%, 1 wt%, and 5 wt% BiTEMPS methacrylate cross-linker. (b) Films of HDPE CANs synthesized with 1 wt% DCP and (left to right) 0 wt%, 2.25 wt%, and 5 wt% BiTEMPS methacrylate cross-linker. (c and d) Tensile storage modulus (E′) as a function of temperature and BiTEMPS methacrylate loading (wt%) for (c) LDPE CANs and (d) HDPE CANs. Legends are read as “BiTEMPS methacrylate wt%”–“DCP wt%”. | ||
Increasing the DCP loading in the presence of a constant amount of BiTEMPS methacrylate is expected to increase the E′ rubbery plateau until the networks can no longer heal after compression molding due to the increase of permanent cross-links beyond a percolation threshold. To understand these limits in PE CANs, loadings of DCP were varied in the presence of 5 wt% BiTEMPS methacrylate. The healability of 1st-molded samples after compression molding at 160 °C for 30 min was used to assess the upper DCP loading limit. For LDPE, 2.5 wt% DCP in the presence of 5 wt% BiTEMPS methacrylate led to unhealed films, whereas loadings of 2.25 wt% DCP and below led to healed films. For HDPE, the upper limit for healed films in the presence of 5 wt% BiTEMPS methacrylate was 2 wt% DCP; 2.25 wt% DCP loadings and above led to unhealed films. See Fig. 2a, b, S3 and S4† for images of healed and unhealed PE films. E′ rubbery plateaus for the healed films were characterized by DMA and are given in Fig. 2c, d, Tables S1 and S2.† As DCP loading in LDPE CANs increases from 0.25 wt% to 2.25 wt%, their E′ rubbery plateaus generally increase in magnitude. At 140 °C, E′ values range from 0.011 MPa for 5-0.25 LDPE CAN to 0.55 MPa for 5-2.25 LDPE CAN. Interestingly, 5-2 and 5-2.25 LDPE CANs possess larger E′ rubbery plateaus than LDPEX (0.52 MPa for 5-2 LDPE CAN and 0.55 MPa for 5-2.25 LDPE CAN at 140 °C vs. 0.46 MPa for LDPEX at 140 °C). Thus, it is possible to synthesize processable PE CANs with greater thermomechanical properties than permanently cross-linked PE that cannot be (re)processed. In HDPE CANs, increasing DCP loading from 0.25 wt% to 2 wt% while holding BiTEMPS methacrylate loading constant at 5 wt% also generally increases E′ rubbery plateaus. At 160 °C, E′ in HDPE CANs vary by nearly an order of magnitude between 0.094 MPa and 0.90 MPa with different DCP loadings. These E′ ranges for processable LDPE and HDPE CANs establish the thermomechanical properties that could be achieved by tuning DCP loading with a constant BiTEMPS methacrylate loading of 5 wt%. HDPE CANs could not replicate the E′ of HDPEX synthesized with 1 wt% DCP and no BiTEMPS methacrylate in its rubbery plateau; HDPEX possesses an E′ at 160 °C of 1.33 MPa. Nonetheless, unlike HPDE CANs, HDPEX cannot be processed into a healed film.
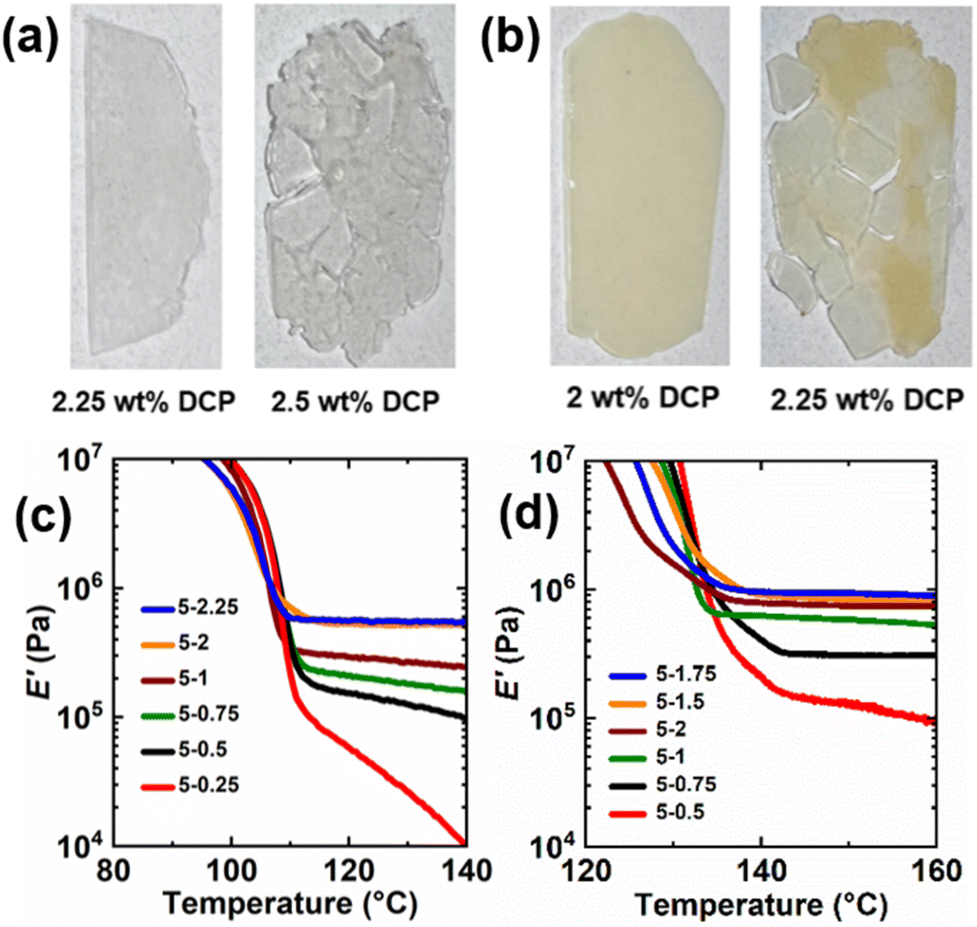 | ||
| Fig. 2 (a) Films of LDPE CANs synthesized with 5 wt% BiTEMPS methacrylate cross-linker and (left to right) 2.25 wt% and 2.5 wt% DCP. (b) Films of HDPE CANs synthesized with 5 wt% BiTEMPS methacrylate cross-linker and (left to right) 2 wt% and 2.25 wt% DCP. (c and d) Tensile storage modulus (E′) as a function of temperature and DCP loading (wt%) for (c) LDPE CANs and (d) HDPE CANs. Legends are read as “BiTEMPS methacrylate wt%”–“DCP wt%”. | ||
3.3. Characterization of 5-1 PE CANs as a function of reprocessing
Further characterizations were completed for PE CANs synthesized in the presence of 5 wt% BiTEMPS methacrylate and 1 wt% DCP. The cross-linked natures of the 5-1 PE CANs were further confirmed via Soxhlet extractions in boiling xylene at 165 °C for 72 h. After vacuum oven drying for 48 h, gel contents of 5-1 LDPE and HDPE CANs were determined to be 61% and 95%, respectively. During extraction, PE CANs are not only melted but also possess dissociating cross-links due to the dynamic activity of the BiTEMPS unit, and yet the calculated gel contents of 5-1 PE CANs are similar to the gel contents of their permanently cross-linked counterparts (68% for LDPEX and 97% for HDPEX). Thus, given the similar, high gel contents, PE CANs synthesized using our reactive processing method contain robust cross-links like those in PEX.Notably, the 5-1 HDPE CAN possesses a higher gel content than the 5-1 LDPE CAN. This observation aligns with the differences in the E′ rubbery plateaus of 5-1 HDPE and LDPE CANs; 1st-molded 5-1 HDPE CAN generally exhibits a rubbery plateau E′ value about three times that of the 1st-molded LDPE CAN (0.57 ± 0.06 MPa vs. 0.19 ± 0.02 MPa at 140 °C). See Table S3† for E′ data for a range of temperatures and for comparisons of 5-1 LDPE and HDPE CANs as well as LDPE and HDPE. According to Flory's ideal rubber elasticity theory,102 for E′ values at 140 °C, 1st-molded HDPE CAN possesses three times the cross-link density of 1st-molded LDPE CAN at 140 °C (5.5 × 10−5 ± 0.6 × 10−5 mol cm−3 in 5-1 HDPE CAN vs. 1.8 × 10−5 ± 0.2 × 10−5 mol cm−3 in 5-1 LDPE CAN). Clearly, HDPE can cross-link to a greater extent than LDPE. One reason for this could be the greater presence of tertiary carbons due to the side-chain branches in LDPE. Not only are these branching points more stable positions for radicals that could otherwise occupy the tertiary carbons of BiTEMPS methacrylate after attacking their carbon–carbon double bonds, but they are prone to β-scission if they abstract a proton from elsewhere. This action will cleave the PE chain and decrease the propensity for PE to cross-link dynamically or permanently.11,13,99 The abundance of secondary carbons that are less prone to β-scission in HDPE relative to LDPE likely improves the propagation of backbone radicals onto the tertiary carbons of BiTEMPS methacrylate, leading to higher cross-link density and gel content.13 Branching in LDPE could also sterically hinder radicals and prevent them from propagating efficiently during cross-linking.
Additionally, the HDPE and LDPE used here also possess different melt flow indices (0.8 g/10 min MFI for HDPE vs. 1.1 g/10 min MFI for LDPE). PE with higher weight-average molecular weight (Mw) is expected to cross-link to a greater extent due to the presence of fewer chain ends that are unable to cross-link.11 As MFI is influenced by Mw of PE, differing MFI could be a factor influencing the extent of cross-linking. To test this, two additional LDPE samples of much higher MFI (4.2 g/10 min and 70 g/10 min) were upcycled into 5-1 LDPE CANs, and their thermomechanical properties were characterized by DMA and DSC and are presented in Fig. S5 and Table S4.† As MFI of LDPE decreases (corresponding to an increase in Mw), the E′ rubbery plateau of the resulting 5-1 LDPE CAN increases in magnitude. This corroborates the hypothesis that higher Mw increases the extent of cross-linking, other things being roughly equal. It is uncertain whether a 5-1 LDPE CAN synthesized from LDPE of 0.8 MFI would yield an E′ rubbery plateau of equal magnitude to the 5-1 HDPE CAN synthesized from HDPE of 0.8 MFI given that side-chain branching is predicted to affect cross-linking negatively.11,13,99 However, it is likely that the E′ rubbery plateau of the 5-1 LDPE CAN would increase with a further decrease in MFI.
The presence of BiTEMPS methacrylate in 5-1 PE CANs as a grafted cross-linker before and after washing in boiling xylene was confirmed by FTIR spectroscopy. The 1720–1725 cm−1 peak in the spectra in Fig. 3a and S6† corresponds to the carbonyl stretch of the BiTEMPS methacrylate cross-linker. This peak is not present for neat PE but exists for PE CANs before and after washing in boiling xylene to remove sol fractions, demonstrating the successful grafting of BiTEMPS methacrylate to the PE backbone instead of its inert dispersion in the PE matrix.
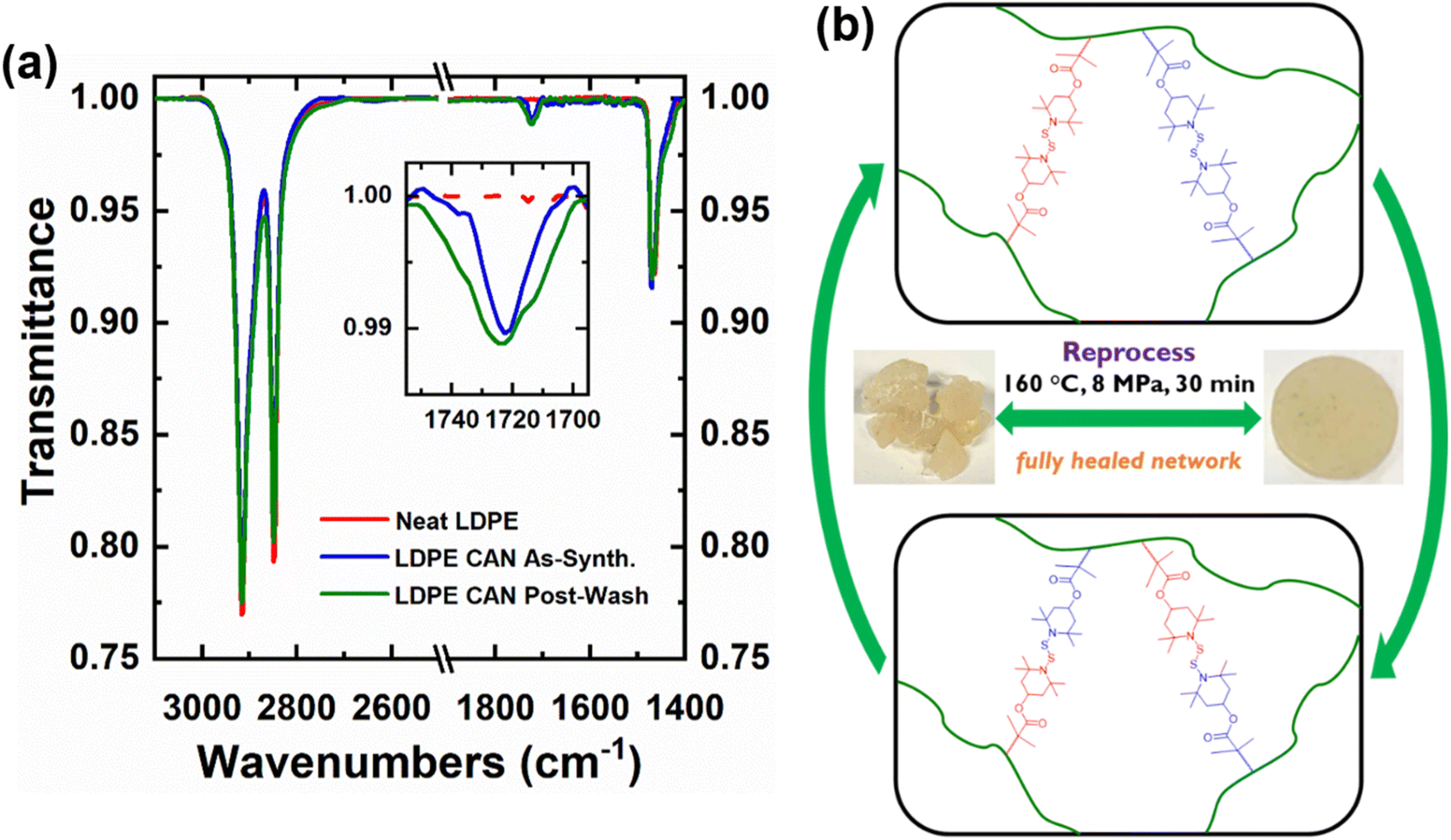 | ||
| Fig. 3 (a) FTIR spectra of neat LDPE and 5-1 LDPE CANs before and after washing in boiling xylene. Carbonyl stretch at ∼1725 cm−1 indicates successful grafting of BiTEMPS methacrylate during reactive processing. (b) Dissociative exchange of disulfide linkages during reprocessing of PE CANs. | ||
5-1 PE CANs were reprocessed successively by hot compression molding after being cut into pieces. Fig. 3b displays an idealized visual of the dissociative dynamic chemistry and the exchange of BiTEMPS units on PE chains during reprocessing. Neat PE and reprocessed 5-1 PE CANs were analyzed by DSC; see Table 1 and Fig. S7.† It is expected that cross-linking PE will diminish the lamellae thickness of crystals formed after cooling from processing temperatures, resulting in lower crystallinities in PE CANs.13 1st-molded 5-1 LDPE and HDPE CAN samples exhibited crystallinities of 29% and 59%, respectively. These values decreased from 32% for neat LDPE and 69% for neat HDPE, as expected. Crystallinity reduction for 5-1 HDPE CAN was greater than for 5-1 LDPE CAN. The melt transitions for the PE CANs occurred at slightly lower temperatures or were unchanged within error relative to neat LDPE and neat HDPE. 2nd-molded and 3rd-molded 5-1 LDPE CANs reproduced the crystallinities and thermal transitions of 1st-molded 5-1 LDPE CAN, indicating that successive reprocessing of the networks did not have significant effects on their thermal properties. 2nd-molded 5-1 HDPE CAN reproduced the crystallinity and melt transitions of 1st-molded 5-1 HDPE CAN. 3rd-molded 5-1 HDPE CAN exhibited a crystallinity of 57% and reproduced the melt transitions of 1st-molded 5-1 HDPE CAN. Given that the uncertainty in reported crystallinity is 1%, this value is within experimental error the same as that for the 1st-molded sample. We note that it has been shown previously that a narrow window of applied pressure can maximize crystallinity of PE,103 which suggests further refinement of our upcycling procedure may allow us to achieve somewhat greater crystallinity.
| Sample | Mold | T m,peak (°C) | T m,endpoint (°C) | T c,onset (°C) | T c,peak (°C) | Crystallinitya (%) |
|---|---|---|---|---|---|---|
| a Determined by DSC. Listed values are ±1 °C or %. | ||||||
| Neat LDPE | — | 108 | 113 | 97 | 90 | 32 |
| 5-1 LDPE CAN | 1st | 106 | 112 | 96 | 88 | 29 |
| 2nd | 106 | 112 | 96 | 88 | 29 | |
| 3rd | 106 | 112 | 96 | 88 | 30 | |
| Neat HDPE | — | 133 | 139 | 121 | 111 | 69 |
| 5-1 HDPE CAN | 1st | 131 | 138 | 122 | 110 | 59 |
| 2nd | 131 | 140 | 122 | 110 | 59 | |
| 3rd | 132 | 139 | 121 | 110 | 57 | |
Fig. 4 and S8† and Tables 2 and S3† show the DMA properties of 5-1 PE CANs as a function of temperature and molding step. All molds of 5-1 LDPE and HDPE CANs exhibit E′ rubbery plateaus above their melt transitions characteristic of cross-linked polymers; in contrast, their neat counterparts flow above their melt transitions. Average E′ values in the rubbery plateaus of the PE CANs also decrease with increasing temperature, consistent with the dissociative dynamic chemistry of the BiTEMPS cross-linker.50,52 As shown in Tables 2 and S3,† 2nd-molded and 3rd-molded 5-1 PE CANs exhibit average E′ values at several temperatures in and below their rubbery plateaus that are, within experimental error, equal to the average E′ values of the 1st-molded PE CANs at the same temperatures. Based on Flory's ideal rubber elasticity theory,102 5-1 LDPE and HDPE CANs fully recover their cross-link densities. Thus, 5-1 PE CANs are reprocessable with full recovery of properties within experimental error after successive compression molding cycles. 3rd-molded 5-1 LDPE and HDPE CANs also exhibit gel contents of 64% and 96%, respectively, after Soxhlet extraction in boiling xylene, further confirming that robust cross-links are present in near equal amounts before and after reprocessing. 1st-molded 5-1 LDPE CAN could also be reprocessed into a 2nd mold by cutting and melt mixing at 160 °C for 10 min before compression molding the blend at 160 °C for 5 min. After melt mixing, the resulting 2nd-molded sample recovered within experimental uncertainty the E′ rubbery plateau and thus the cross-link density of its 1st-molded sample. This E′ rubbery plateau reproduction is shown in Fig. S9a† with a corresponding image of the film in Fig. S9b.† 1st-molded 5-1 HDPE CAN could not be reprocessed by this method into a fully healed, 2nd-molded film. Evidence of the partial healing and damage to the film are shown in Fig. S9c.† It is possible that the strong shearing forces during mixing damaged the highly cross-linked HDPE network structure, inhibiting the sample from fully healing under mild compression molding conditions.
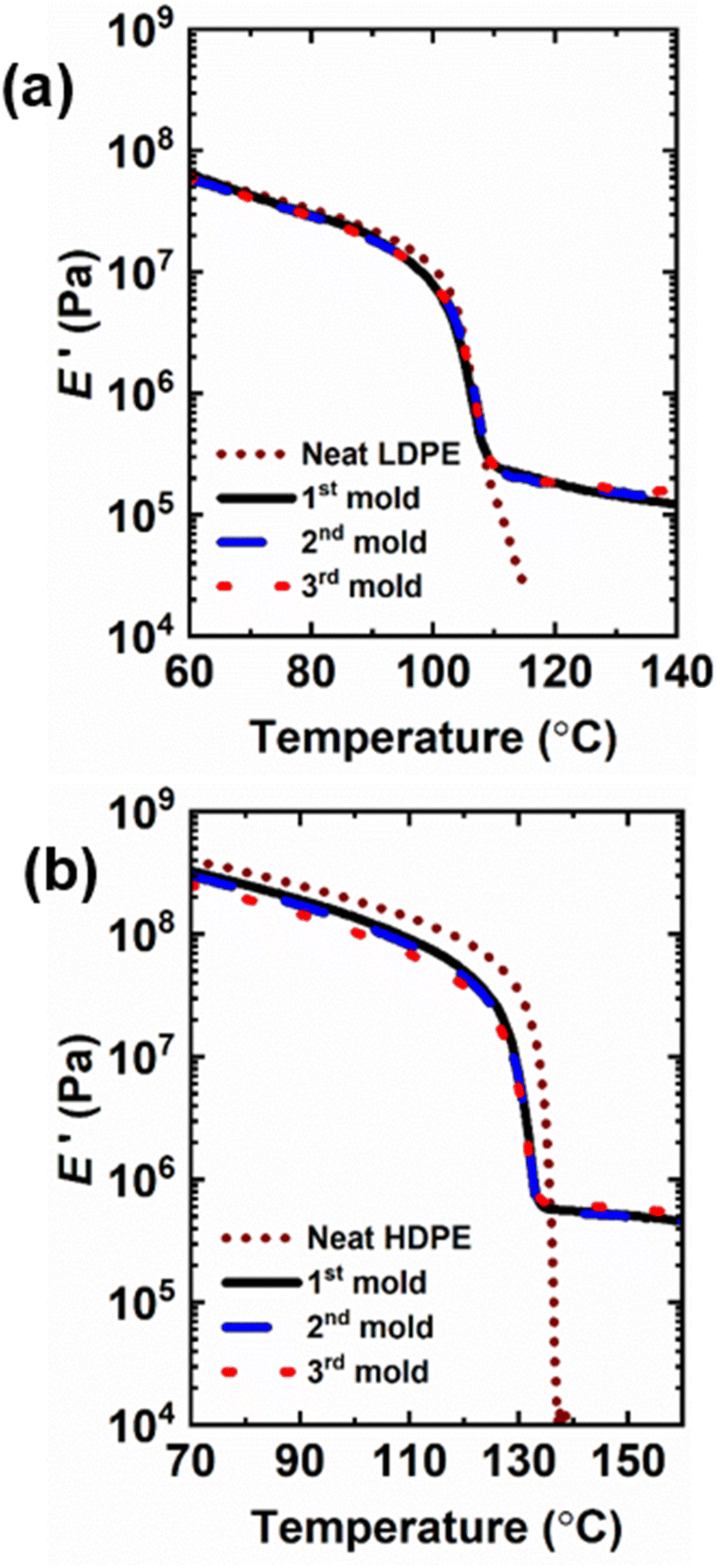 | ||
| Fig. 4 Tensile storage modulus (E′) as a function of temperature of 1st mold, 2nd mold, and 3rd mold (a) 5-1 LDPE CAN and (b) 5-1 HDPE CAN samples with their neat counterparts. | ||
| Sample | Mold | E′a (MPa) | |||
|---|---|---|---|---|---|
| 30 °C | 90 °C | 140 °C | 150 °C | ||
| a Determined by DMA. Error bars represent ± one standard deviation of three or four measurements. | |||||
| Neat LDPE | — | 253 | 22 | 0.005 | 0.003 |
| 5-1 LDPE CAN | 1st | 220 ± 40 | 20.0 ± 1.4 | 0.19 ± 0.02 | 0.17 ± 0.01 |
| 2nd | 220 ± 60 | 19.1 ± 1.4 | 0.21 ± 0.08 | 0.19 ± 0.08 | |
| 3rd | 200 ± 20 | 18.2 ± 0.5 | 0.30 ± 0.11 | 0.29 ± 0.11 | |
| Neat HDPE | — | 892 | 249 | 0.006 | 0.001 |
| 5-1 HDPE CAN | 1st | 810 ± 60 | 177 ± 14 | 0.57 ± 0.06 | 0.52 ± 0.08 |
| 2nd | 760 ± 100 | 174 ± 23 | 0.57 ± 0.04 | 0.54 ± 0.05 | |
| 3rd | 630 ± 30 | 147 ± 8 | 0.61 ± 0.04 | 0.58 ± 0.04 | |
Thermal stabilities of 5-1 LDPE and HDPE CANs, their thermoplastic precursors, and BiTEMPS methacrylate were assessed using thermogravimetric analysis (TGA). A heating ramp from 20 °C to 600 °C was applied at 5 °C min−1, and sample masses were monitored as shown in Fig. 5a. LDPE and HDPE begin to lose mass near 370 °C and 400 °C, respectively. The control samples exhibit significant weight loss near 420 °C and 440 °C, respectively; at these temperatures, ∼95 wt% of the initial sample remains (∼Td,95%). 5-1 LDPE and HDPE CANs experience abrupt weight loss at the same respective temperatures as LDPE and HDPE but exhibit different Td,95%s owing to the weight loss of BiTEMPS methacrylate cross-linker at lower temperatures. Td,95% values for 5-1 LDPE and HDPE CANs are 388 °C and 424 °C, respectively, but these CANs begin to lose mass near 185 °C. This is also the temperature at which BiTEMPS methacrylate begins to lose mass. This alignment of the initial mass losses for BiTEMPS methacrylate and the 5-1 LDPE and HDPE CANs in the TGA curves is expected. BiTEMPS methacrylate exhibits a Td,95% of 202 °C above which it degrades rapidly. Above 322 °C, its mass loss reduces to a slow decline. Near this temperature, 5-1 PE CANs begin to experience a corresponding plateau in weight loss until PE further degrades at higher temperature. These results indicate that BiTEMPS methacrylate and PE CANs made with BiTEMPS methacrylate are thermally stable up to 185–200 °C. Above 200 °C, the cross-linker is not likely to be stable, and PE CANs will begin to degrade.
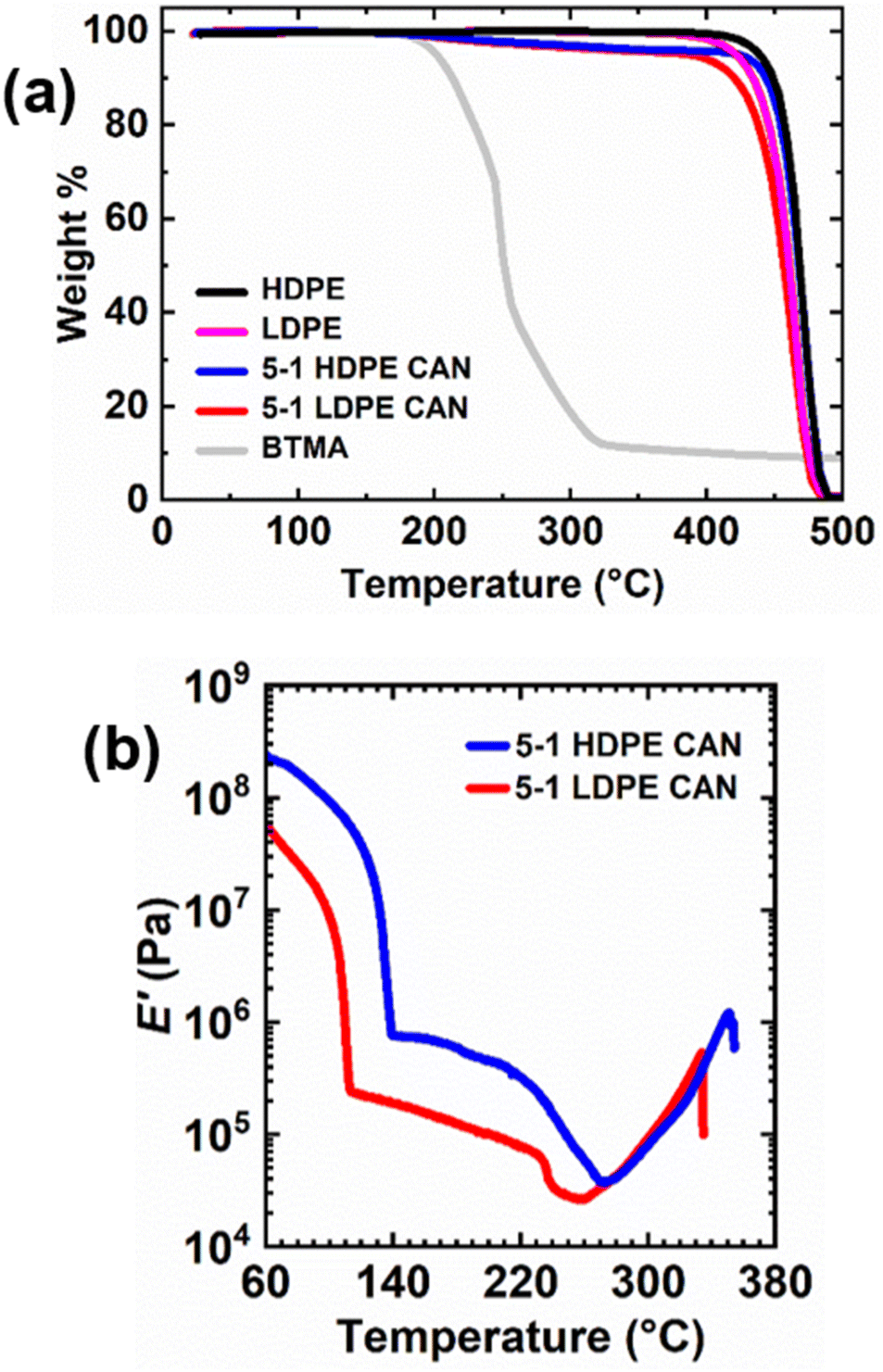 | ||
| Fig. 5 (a) Weight losses as a function of temperature given by TGA for BiTEMPS methacrylate (BTMA), PE, and 5-1 PE CANs. (b) E′ as a function of temperature to monitor thermal stability and property degradation in 5-1 PE CANs at high temperatures. | ||
To accompany the TGA results as metrics for thermal stability, DMA was conducted on 5-1 LDPE and HDPE CANs; see Fig. 5b. 5-1 LDPE CAN exhibits a steady decline in its E′ rubbery plateau up to 232 °C. Above 232 °C, 5-1 LDPE CAN experiences softening and property degradation evidenced by an abrupt loss in E′ until 260 °C. This abrupt loss in E′ likely corresponds to the onset of irreversible dissociation of BiTEMPS dynamic cross-links, a behavior shown previously in CANs.67 The sample stiffens as degradation continues, subsequently increasing E′ until the sample breaks near 334 °C. As previously noted, 5-1 HDPE CAN exhibits a declining E′ rubbery plateau above its melt transition; unlike that of 5-1 LDPE CAN, this plateau does not appear to diminish in a steady manner. Instead, the declining E′ rubbery plateau of 5-1 HDPE CAN shows greater downward curvature. 5-1 HDPE CAN exhibits a greater reduction in E′ near 220 °C that likely corresponds to irreversible dissociation of BiTEMPS units, and this reduction continues until 273 °C when the sample begins to stiffen during degradation until it breaks near 350 °C. The stiffening during degradation that is observed at high temperature in 5-1 PE CANs could result from the irreversible termination of sulfur-centered radicals forming permanent cross-links. This suggests that this behavior is minimal and does not affect thermomechanical properties at temperatures where degradation is absent. DMA results indicate that 5-1 PE CANs do not experience abrupt property degradation at the BiTEMPS degradation temperature shown in TGA. Instead, major property degradation in 5-1 PE CANs begins near 220 °C. Below this temperature, E′ reduction with increasing temperature occurs due to the increasing exchangeability of the BiTEMPS dynamic bonds. The generally very good thermal stability and robust properties of the PE CANs suggest that our approach could also be used to develop reprocessable CAN nanocomposites75,104,105 and foams106,107 with PE as the base polymer.
3.4. Elevated-temperature creep response of 5-1 PE CANs
Creep responses for 5-1 PE CANs were studied under low stress for long times and high stress for short times to assess creep resistances and behavior relative to their neat counterparts. As shown in Fig. 6, creep deformations in neat PE precursors and 5-1 PE CANs were monitored at 90 °C under a constant shear stress of 3.0 kPa for 5 h. At 90 °C, the crystallinities of PE materials are expected to dominate creep responses. Additionally, BiTEMPS dissociation has been shown to be nearly absent and useful for arresting creep near 90 °C in CANs.52 The time-dependent viscous creep responses were estimated by extrapolating the fit of the linear segment of each curve (modelled by a dashpot in the Burgers model108) to t = 0 h and subtracting this y-intercept strain from the strain at t = 5 h. Tables 3 and S5† display the quantitative results. 5-1 LDPE CAN exhibited a very low viscous creep strain of 0.000043 (i.e., 0.0043%), confirming its high resistance to creep and excellent dimensional stability at 90 °C. LDPE exhibited a viscous creep strain of 0.00029 (i.e., 0.029%). While this viscous creep strain is more than 6 times greater than that experienced by 5-1 LDPE CAN, both creep strains are very small and could converge further at longer experimental times. LDPE and 5-1 LDPE CAN experienced similar total strains after 5 h of 0.0043 and 0.0046 (i.e., 0.43% and 0.46%), respectively. 5-1 HDPE CAN also displayed excellent creep resistance and dimensional stability at 90 °C with a low viscous creep strain of 0.0013 (i.e., 0.13%) after 5 h at 3.0 kPa. This result confirms that the creep responses of 5-1 PE CANs are comparable to those of static networks at this temperature.109–111 Neat HDPE exhibited a low viscous creep strain of magnitude similar to that of the 5-1 HDPE CAN of 0.0012 (i.e., 0.12%). While the high crystallinities of HDPE and 5-1 HDPE CANs governed their 90 °C viscous creep responses, resulting in very similar values, the high cross-link density of 5-1 HDPE CAN likely contributed to the reduction of the delayed elastic response in 5-1 HDPE CAN relative to HDPE, as seen in Fig. 6b, resulting in a lower total strain of 0.0038 (i.e., 0.38%) relative to 0.0043 (i.e., 0.43%) in HDPE. It is noteworthy that the 5-1 HDPE CAN experienced a higher viscous creep strain than the 5-1 LDPE CAN despite having higher crystallinity and cross-link density. We attribute this behavior to experimentation time. The HDPE materials were transitioning between delayed elastic and viscous creep responses near the end of the 5 h window and require more time under stress to achieve pure viscous creep. Extending experimentation time for HDPE materials under the 3.0 kPa stress is likely to lead to smaller calculated viscous creep strains relative to LDPE materials.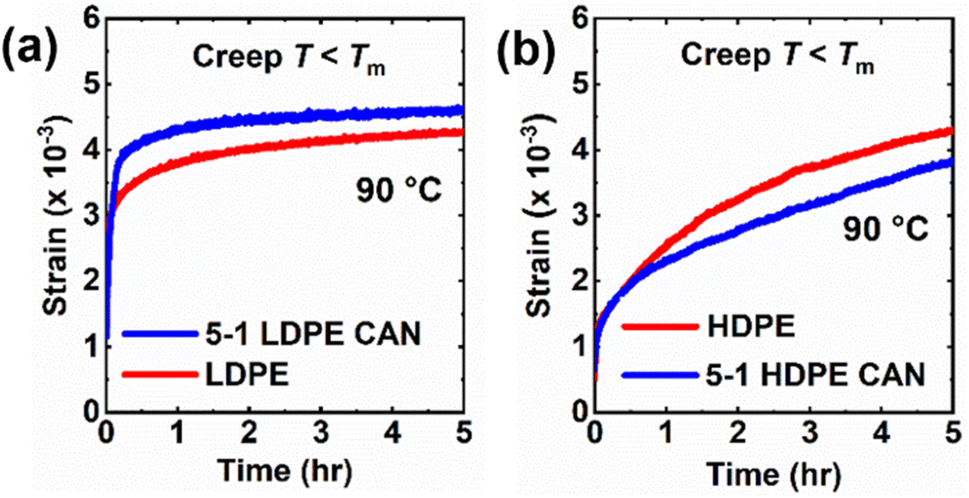 | ||
| Fig. 6 Creep curves of (a) LDPE and 5-1 LDPE CAN as well as (b) HDPE and 5-1 HDPE CAN at 90 °C under a constant shear stress of 3 kPa. | ||
| Sample | Creep at 3000 Pa, 5 h, 90 °C | Creep at 3.5 MPa, 30 min, 60 °Ca/90 °Cb | |
|---|---|---|---|
| Viscous creep strainc | Total straind | Total straine | |
| a 60 °C used for LDPE and 5-1 LDPE CANs samples. b 90 °C used for HDPE and 5-1 HDPE CANs samples. c Viscous creep strains were each calculated by extrapolating the linear fit of the creep curve to t = 0 s and subtracting the y-intercept from the total strain value. d Total strain at t = 5 h. e Total strain at t = 30 min. Error bars represent ± one standard deviation of four measurements. | |||
| Neat LDPE | 0.00029 | 0.0043 | 0.16 ± 0.01 |
| 5-1 LDPE CAN | 0.000043 | 0.0046 | 0.165 ± 0.006 |
| Neat HDPE | 0.0012 | 0.0043 | 0.08 ± 0.01 |
| 5-1 HDPE CAN | 0.0013 | 0.0038 | 0.046 ± 0.007 |
At low stresses in which linear viscoelasticity is maintained, neat PE and 5-1 PE CANs exhibit similarly low creep responses below their melt transitions. To investigate the differences in creep response between neat PE and 5-1 PE CANs outside their linear viscoelastic regimes, short-term creep experiments were conducted under a high tensile stress of 3.5 MPa for 30 min at 60 °C for LDPE materials and 90 °C for HDPE materials. See Table 3 for strain data from these experiments. The 5-1 LDPE CANs exhibited a strain of 0.165 ± 0.006 (i.e., 16.5 ± 0.6%) under the applied stress after 30 min. This strain is within the experimental uncertainty of the strain exhibited by neat LDPE samples of 0.16 ± 0.01 (i.e., 16 ± 1%). From both the long-term, low-stress and short-term, high-stress creep experiments, it is evident that crystallinity dictates the creep response of semi-crystalline, LDPE-based materials and that 5-1 LDPE CANs do not gain an advantage in creep resistance over LDPE at this level of cross-linking (61% gel content).
The 5-1 HDPE CANs exhibited a strain of 0.046 ± 0.007 (i.e., 4.6 ± 0.7%) under the 3.5 MPa stress after 30 min at 90 °C. Compared to neat HDPE, which exhibited a strain of 0.08 ± 0.01 (i.e., 8 ± 1%) after 30 min of constant stress, 5-1 HDPE CAN saw a nearly 42% reduction in total strain. These results indicate that the level of cross-linking in 5-1 HDPE CANs (95% gel content) lessens high-stress, nonlinear viscoelastic creep in HDPE in its semi-crystalline state. Dynamic cross-linking of HDPE and the low activity of BiTEMPS at 90 °C in short time intervals effectively compensate for any negative effect on 5-1 HDPE CAN creep resistance caused by the reduction in crystallinity from cross-linking relative to neat HDPE. It is plausible that LDPE would see a similar reduction in its semi-crystalline creep response to high stress with a similar level of cross-linking. The fact that the 90 °C HDPE and 5-1 HDPE CAN strains are lower than the 60 °C LDPE and 5-1 LDPE CAN strains also confirms that longer low-stress creep studies are needed to obtain pure viscous creep responses in HDPE and 5-1 HDPE CANs.
Creep measurements were also conducted at several temperatures above the melt transition of LDPE and 5-1 LDPE CANs to characterize their T-dependent viscosity and viscous flow activation energy (Ea,η). This is a worthwhile study, as BiTEMPS chemistry has been shown to dominate network creep and stress relaxation responses in CANs at high temperatures, with viscous flow and stress relaxation activation energies very close to the BiTEMPS BDE.49 Creep deformation was monitored in 5-1 LDPE CANs at 120 °C to 150 °C under a 3.0 kPa shear stress for 10000 s. To draw comparisons between the 5-1 LDPE CANs and their thermoplastic precursors, creep deformation was monitored in LDPE control samples at the same temperatures under a 3.0 kPa shear stress for 1000 s. Fig. 7a and b show the resulting creep strain curves for 5-1 LDPE CAN and LDPE, respectively. Tables 4, S6 and S7† show the calculated viscous creep strains and network viscosities at each temperature. Creep tests for LDPE were conducted for shorter times, as these samples flow more readily above their melt transition and take less time to reach the viscous creep regime. At every temperature, 5-1 LDPE CAN experiences reduced creep relative to LDPE, which is expected due to the presence of dynamic cross-links. 5-1 LDPE CAN exhibits a 97% reduction in viscous creep at 120 °C compared to LDPE (0.35 or 35% strain for 5-1 LDPE CAN vs. 12.3 or 1230% for LDPE). At 150 °C, this reduction in viscous creep is 63% (4.05 or 405% strain for 5-1 LDPE CAN vs. 11.0 or 1100% for LDPE).
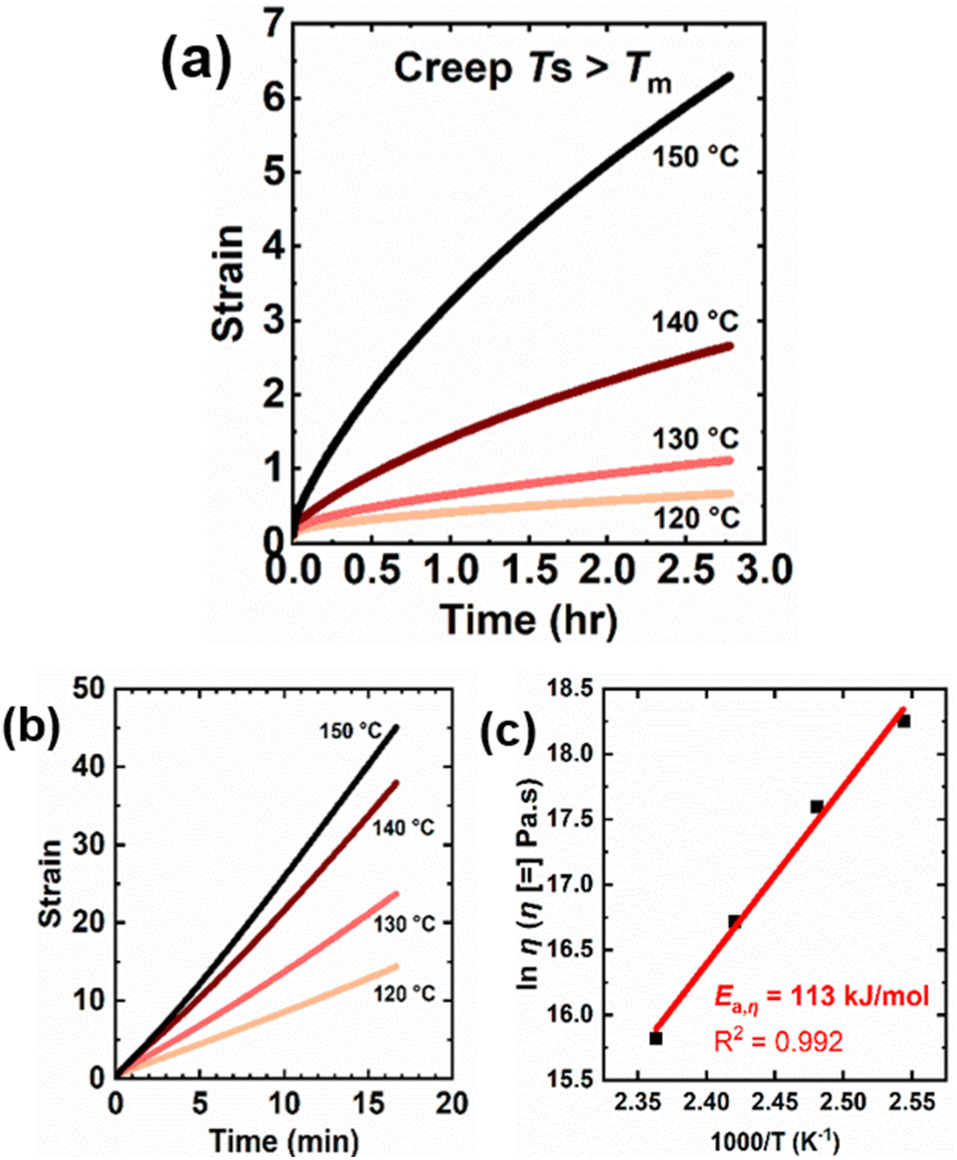 | ||
| Fig. 7 Creep curves at different temperatures above the melt transition of (a) 5-1 LDPE CANs and (b) LDPE under a constant shear stress of 3 kPa. (c) Arrhenius activation energy of viscosity for 5-1 LDPE CANs as calculated from creep data taken over a T-range of 120–150 °C. | ||
| CAN sample | Temperature (°C) | η (Pa s) | Viscous creep strainb | Total strainc |
|---|---|---|---|---|
|
a Viscosities (η) were each calculated from a constant shear rate of 3000 Pa and strain rates calculated from the slope of the linear fit of the creep curve between t = 8000 s and t = 10000 s.
b Viscous creep strains were each calculated by extrapolating the linear fit of the creep curve to t = 0 s and subtracting the y-intercept from the total strain value.
c Total strains measured at t = 10000 s.
|
||||
| 5-1 LDPE CAN | 120 | 8.49 × 107 | 0.35 | 0.67 |
| 130 | 4.38 × 107 | 0.68 | 1.12 | |
| 140 | 1.82 × 107 | 1.65 | 2.26 | |
| 150 | 7.42 × 106 | 4.05 | 6.30 | |
| 5-1 HDPE CAN | 145 | 1.71 × 109 | 0.018 | 0.052 |
| 150 | 6.32 × 108 | 0.047 | 0.083 | |
Fig. 7c displays the temperature dependence of 5-1 LDPE CAN viscosity in an Arrhenius plot. An Ea,η of 113 ± 7 kJ mol−1 was calculated from the slope of the linear fit in the Arrhenius plot. This value agrees within error with the previously reported S–S BDE of BiTEMPS with a disulfide bridge (109.6 kJ mol−1)48 as well as the BDE of the S–S bonds in BiTEMPS with a trisulfide bridge (108.4 kJ mol−1).62 It is also notable that the Ea,η of our 5-1 LDPE CANs synthesized with BiTEMPS methacrylate agrees within error with the viscous flow and stress relaxation activation energies of previously reported poly(hexyl methacrylate) (PHMA) CANs synthesized with BiTEMPS methacrylate as dynamic cross-linker (106.7 kJ mol−1 and 106.2 kJ mol−1, respectively).52 In contrast, thermoplastic LDPE has a lower Ea,η of 56.5 ± 0.4 kJ mol−1 estimated from the slope of the linear fit of its Arrhenius plot, as shown in Fig. S10.† This value aligns well with previously reported values for branched PE.112 In all, this evidence indicates that the T-dependence of the creep response of 5-1 LDPE CANs in their melt state is dominated by the dissociative BiTEMPS chemistry rather than their viscoelasticity, further confirming the role of BiTEMPS methacrylate as a dynamic cross-linker in the CANs. It also suggests the applicability and utility of this dissociative dynamic chemistry as the determinant for creep response in CANs of different viscoelasticities (e.g., PHMA vs. LDPE), a result that had yet to be fully demonstrated. Studies are warranted to consider how BiTEMPS chemistry governs the stress relaxation response of 5-1 LDPE CANs relative to previously reported BiTEMPS-based CANs.
Creep measurements were also conducted on 5-1 HDPE CANs at 145 °C and 150 °C for 10 000 s under a 3.0 kPa shear stress as well as control HDPE samples at the same temperatures and shear stress for 1000 s. See Fig. 8. Tables 4 and S5† display the corresponding creep data for 5-1 HDPE CANs and HDPE, respectively. For the same reason as LDPE samples, thermoplastic HDPE requires less time to reach its viscous creep regime. 5-1 HDPE CANs exhibit substantially reduced creep at 145 and 150 °C relative to HDPE due to their sufficiently robust cross-links. At 145 °C, 5-1 HDPE CAN had a viscous creep strain of 0.018 (i.e., 1.8%), a 99.98% decrease in viscous creep strain relative to HDPE at the same temperature (84.4 or 8440% strain). At 150 °C, 5-1 HDPE CAN exhibited a viscous creep strain of 0.047 (i.e., 4.7%), a 99.95% decrease relative to that exhibited by HDPE (91.4 or 9140%). A reliable Ea,η could not be extracted from just these two temperatures; 150 °C is the upper temperature bound for our shear rheometer. Future viscous creep studies above 150 °C are needed to assess how the BiTEMPS methacrylate dissociative dynamic chemistry and the HDPE viscoelasticity dictate creep response.
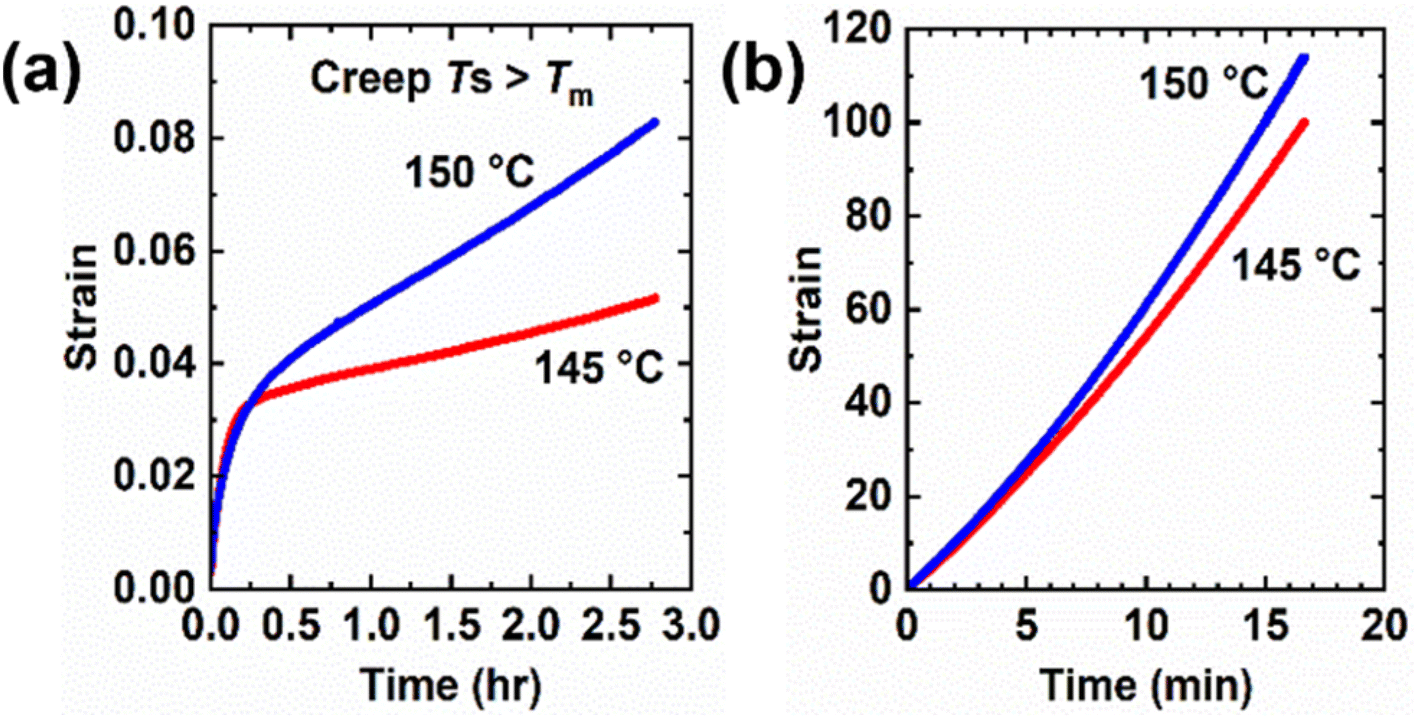 | ||
| Fig. 8 Creep curves at different temperatures above the melt transition of (a) 5-1 HDPE CANs and (b) HDPE under a constant shear stress of 3 kPa. | ||
3.5. Absence of phase separation in 5-1 PE CANs
As phase separation has been an issue for PE CANs synthesized by reactive processing with thermodynamically incompatible cross-linkers,70,71,87 it is worth addressing the presence or absence of phase separation in PE CANs synthesized using BiTEMPS methacrylate. Given the small level of cross-linker present in 5-1 PE CANs (<0.13 mol% based on mol of ethylene repeat units), phase separation in any form is not expected for 5-1 PE CANs. Macroscopic phase separation can arise in polymers and presents itself as opacity through simple visual inspection.71 This turbidity can be difficult to assess as phase separation in PE materials at room temperature, as crystals reduce the optical transmission and transparency of materials. Thus, it is worth visually inspecting semi-crystalline samples above their melt transitions to assess macrophase separation at high temperatures. Fig. 9a shows 0.65 mm-thick samples of molten PE, permanently cross-linked PE (PEX), and 5-1 PE CANs placed over logos to aid visualization at 165 °C. These samples are largely transparent and not turbid. This result was expected for molten PE and PEX, as these polymers do not conventionally exhibit phase separation.13,71 5-1 PE CAN samples demonstrating similar transparencies by visual inspection suggests that they also do not contain appreciable macrophase separation at this temperature. Of course, thinner samples will be more transparent by visual inspection than thicker samples. However, molten PE CAN samples as thin as 0.3 mm have been previously shown to be turbid and therefore macrophase separated via this method.71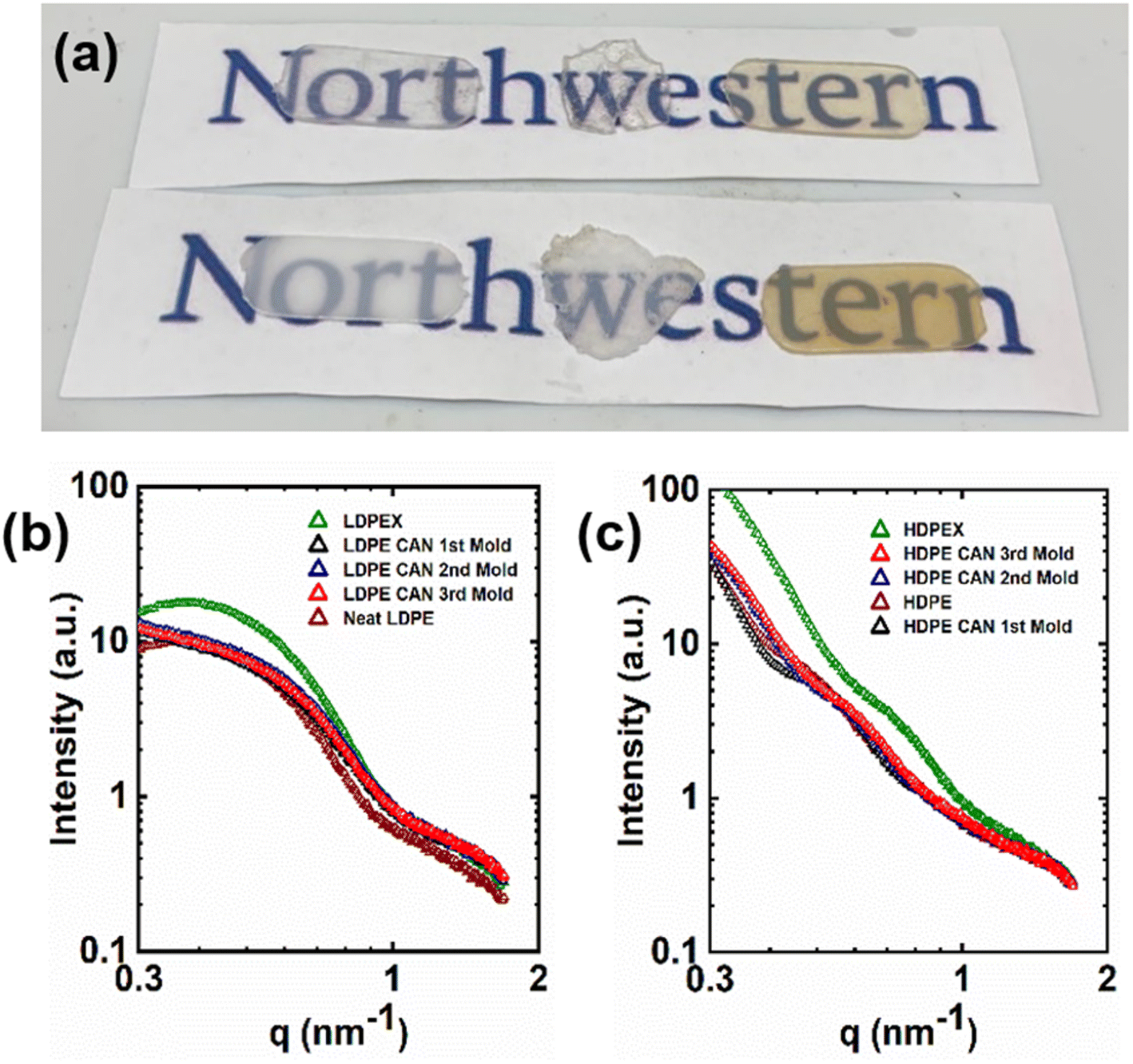 | ||
| Fig. 9 (a) Image of molten PE samples at 165 °C. Samples were placed on logos to aid visualization of transparency. Top row, left to right: neat LDPE, LDPEX, LDPE CAN; bottom row, left to right: neat HDPE, HDPEX, HDPE CAN. (b) SAXS patterns of semi-crystalline LDPE-based samples. (c) SAXS patterns of semi-crystalline HDPE-based samples. | ||
Macrophase separation has also been assessed in semi-crystalline PE CANs by comparing normalized crystallinities (relative to neat PE crystallinity) before and after washing away soluble (uncross-linked) fractions by Soxhlet extraction in boiling xylene. Ricarte et al.71 found that macrophase-separated PE CANs show a reduction in this normalized crystallinity upwards of 10% after removal of the soluble PE fraction. This phenomenon has been hypothesized to result from dynamic cross-links slowing crystallization kinetics in PE CANs, leading to lower crystallinities in regions of phase-separating and aggregated cross-links.71 Cross-linking LDPE into a 1st-molded 5-1 LDPE CAN reduces crystallinity from 32% to 29% (normalized crystallinity of 91%). After washing away the soluble PE fraction, 1st-molded 5-1 LDPE CAN displays a crystallinity of 30% (normalized crystallinity of 94%). In this case, normalized crystallinity did not decrease, providing further evidence that 5-1 LDPE CAN is not macrophase separated. Cross-linking HDPE into a 1st-molded 5-1 HDPE CAN reduces crystallinity from 69% to 59% (normalized crystallinity of 86%), and subsequently washing away its soluble PE fraction gives a slightly reduced crystallinity of 58% (normalized crystallinity of 84%). As non-normalized crystallinity decreased only 1% and normalized crystallinity 2% rather than 10%, there is no significant evidence that macrophase separation exists in 5-1 HDPE CANs. Phase separation in other HDPE CANs has also presented itself by low gel contents (<50%) given cross-linker incompatibility and its negative effect on cross-linking.71 As previously mentioned, 5-1 HDPE CAN exhibits a very high gel content of 95%, near the 97% gel content of HDPEX which is not phase separated. Furthermore, there is an absence of evidence indicating macrophase separation in 5-1 LDPE and HDPE CANs.
Nanophase separation can be studied via SAXS. The nanostructures of semi-crystalline PE, PEX, and 5-1 PE CAN samples were probed using SAXS at room temperature, and the resulting patterns are present in Fig. 9b and c. The SAXS patterns of molded 5-1 LDPE CANs exhibited the same general shape featuring two shoulders at q = ∼0.6 nm−1 and ∼1.5 nm−1. Neat LDPE and LDPEX SAXS patterns exhibited similar shapes with a peak at q = ∼0.4 nm−1 and a shoulder at q = ∼1.5 nm−1, shifted in intensity. The presence of these peaks and shoulders are attributed to the crystal lamellae structure factor, not nanophase separation.71,113 Oriented crystal formation in LDPEX after compression molding increased sample anisotropy and light scattering; this effect resulted in increased SAXS intensities relative to neat LDPE and 5-1 LDPE CAN patterns in the low-q region.113 5-1 LDPE CANs scatter with intensity similar to LDPE at low q, as the dynamic cross-linking did not noticeably orient crystals in 5-1 LDPE CANs relative to neat LDPE. At high q, 5-1 LDPE CANs scatter with similar intensity to LDPEX due to the similar number of crystal lamellae stacks that formed after cross-linking.113 Inferences about scatterer interactions can be made using Guinier analysis of low-q SAXS patterns; nonlinearity in Guinier plots is attributed to the presence of scatterer aggregation or repulsion or a lack of monodispersity in scattering bodies.113 Guinier parameters and plots as well as an analysis of the low-q behavior in the SAXS patterns of LDPE materials are presented in ESI (Table S8 and Fig. S11†). The linear behavior in the Guinier plots of 5-1 LDPE CANs indicates that no particle aggregation or repulsion are present in the samples, further corroborating that dynamic cross-links of 5-1 LDPE CANs do not aggregate or phase separate from PE chains.
SAXS patterns of neat HDPE, HDPEX, and HDPE CANs are similar in general shape and curvature, only shifted in intensity and q. Again, these patterns are comparable in shape because of the crystal lamellae structure factor.71,113 The small variability seen in neat HDPE and 5-1 HDPE CAN patterns is attributed to their slight differences in crystallinity and resulting lamellae scattering after cross-linking (or in the absence of cross-linking with neat HDPE), not by the presence of nanophase separation.71,113 As with LDPEX, HDPEX experienced oriented crystal formation after conventional cross-linking and compression molding that increased scattering intensities relative to neat HDPE and 5-1 HDPE CANs at low q. In the high-q region, neat HDPE, HDPEX, and 5-1 HDPE CANs scatter similarly. These results suggest that 5-1 HDPE CANs do not exhibit phase separation in the studied nanoscopic range compared to neat HDPE and HDPEX. SAXS studies conducted on melt-state PE and 5-1 PE CANs at lower q values are warranted to investigate aggregation and phase separation without the scattering influence of crystal structures.
3.6. Recycling waste PE by upcycling into PE CANs
We have extended our reactive processing method for synthesizing reprocessable PE CANs to upcycle PE waste. See Fig. 10. Post-consumer LDPE plastic bags were cut into pieces, homogenized at 130 °C in the presence of 5 wt% BiTEMPS methacrylate and 1 wt% DCP, and cross-linked at 160 °C to produce 1st-molded, 5-1 LDPE waste CANs. Nearly the same was done with a post-consumer HDPE milk jug in the presence of 5 wt% BiTEMPS methacrylate and 1 wt% DCP to produce 1st-molded, 5-1 HDPE waste CANs. The PE waste CANs are designated “LDPE-W CANs” and “HDPE-W CANs”. PE-W CANs were reprocessed by compression molding at 160 °C and 8 MPa for 30 min into 2nd- and 3rd-molded films. PE waste products were also permanently cross-linked with 1 wt% DCP to produce samples (LDPEX-W and HDPEX-W) that could not be processed into healed films after compression molding. The preparation route of PE-W CANs and images of these samples are given in Fig. 10a and b.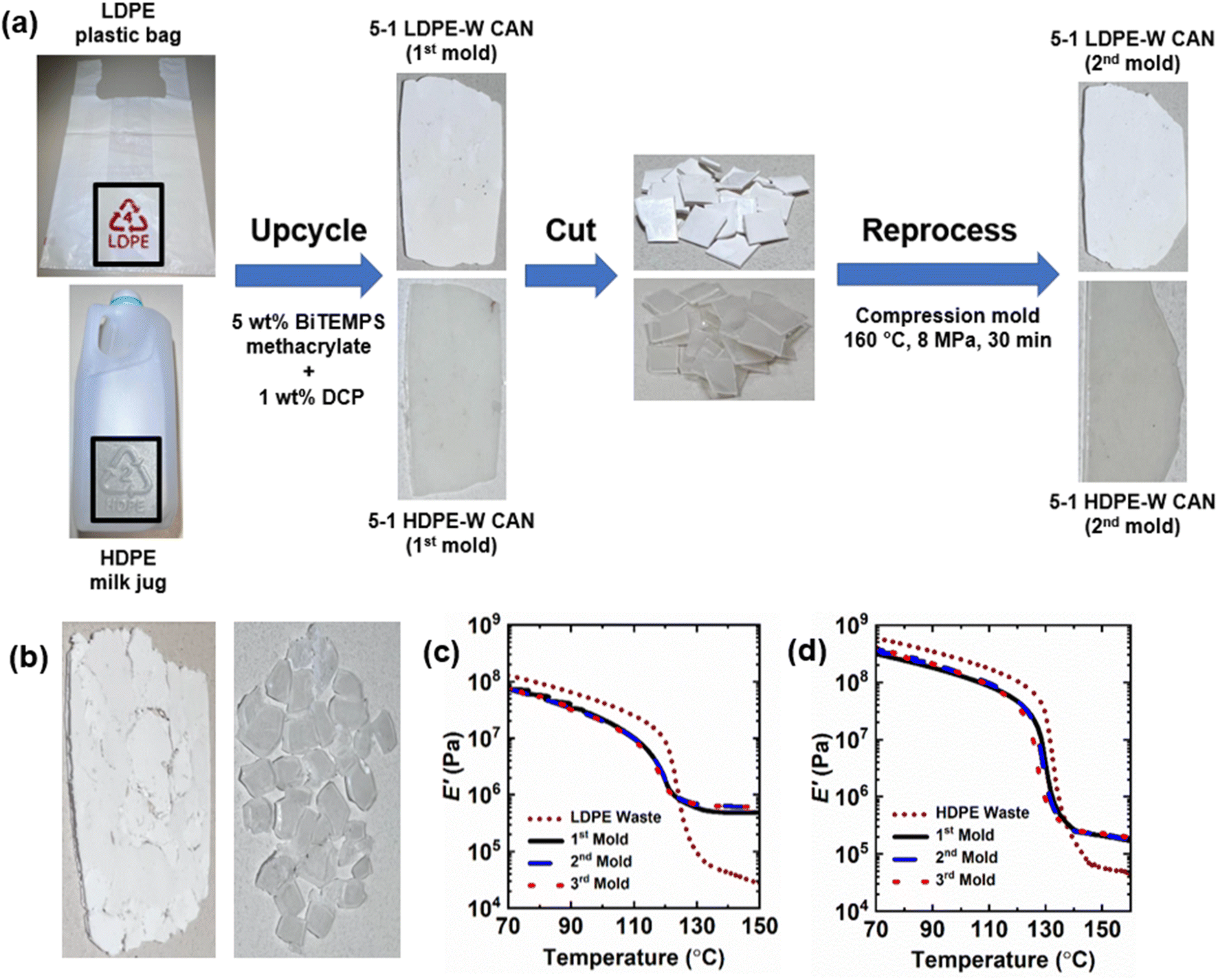 | ||
| Fig. 10 (a) Preparation route and reprocessing of PE-W CANs from plastic bags and a milk jug. (b) (Left) Unhealed LDPEX-W film. (Right) Unhealed HDPEX-W film. (c and d) Tensile storage modulus (E′) as a function of temperature of 1st mold, 2nd mold, and 3rd mold (c) 5-1 LDPE-W CAN and (d) 5-1 HDPE-W CAN samples with their PE waste precursor counterparts. | ||
The thermal properties of all PE-W CAN molds and their waste precursors were characterized by DSC; see Table 5. The LDPE plastic bags and HDPE milk jug exhibited crystallinities of 34% and 83%, respectively. After dynamic cross-linking into 1st-molded 5-1 PE-W CANs, these respective crystallinities expectedly decreased to 32% and 73%. The reduction in crystallinity from waste HDPE to 5-1 HDPE-W CAN is larger than the crystallinity reduction from waste LDPE to 5-1 LDPE-W CAN. In line with the reduction in crystallinities, melt transitions of PE-W CANs occurred at slightly lower temperatures. After reprocessing, 2nd-molded and 3rd-molded 5-1 LDPE-W and HDPE-W CANs reproduced within error the melt transitions and crystallinities of the 1st-molded PE-W CANs. This indicates that the successive processing cycles did not have significant effects on the thermal properties of the CANs.
| Sample | Mold | T m,peak (°C) | T m,endpoint (°C) | T c,onset (°C) | T c,peak (°C) | Crystallinitya (%) | E′ at 150 °Cb (MPa) |
|---|---|---|---|---|---|---|---|
| a Determined by DSC. Values are ±1 °C or %. b Determined by DMA based on a single sample run. | |||||||
| LDPE plastic bag | — | 125 | 131 | 111 | 106 | 34 | 0.028 |
| 5-1 LDPE-W CAN | 1st | 124 | 129 | 113 | 103 | 32 | 0.49 |
| 2nd | 124 | 130 | 114 | 100 | 32 | 0.59 | |
| 3rd | 124 | 130 | 114 | 101 | 31 | 0.61 | |
| HDPE milk jug | — | 136 | 143 | 117 | 107 | 83 | 0.059 |
| 5-1 HDPE-W CAN | 1st | 134 | 139 | 123 | 108 | 73 | 0.21 |
| 2nd | 133 | 139 | 122 | 108 | 71 | 0.22 | |
| 3rd | 133 | 139 | 122 | 108 | 73 | 0.23 | |
Fig. 10c, d and Table 5 show the DMA properties of 5-1 PE-W CANs and their precursors as a function of temperature and molding step. 5-1 PE-W CANs exhibit E′ rubbery plateaus of significantly greater magnitude than the post-consumer waste products, indicating the upcycled and cross-linked nature of the CANs. 5-1 LDPE-W CAN improves on the E′ rubbery plateau of waste LDPE plastic bags by over an order of magnitude (0.49 MPa for 5-1 LDPE-W CAN vs. 0.028 MPa for waste LDPE at 150 °C). 5-1 HDPE-W CAN exhibited an E′ of 0.21 MPa at 150 °C, over 3.5 times that of the waste HDPE milk jug (0.059 MPa at 150 °C). After reprocessing, 2nd and 3rd molds of 5-1 LDPE-W and HDPE-W CANs reproduce the E′ rubbery plateau values of their respective 1st molds at several temperatures above their melt transitions. Thus, waste LDPE and HDPE products were upcycled into reprocessable CANs that fully recover their cross-link densities and associated thermomechanical properties. These results demonstrate the applicability of our upcycling method for producing robust CANs not only from virgin commodity plastics but also from post-consumer waste polymers. These results also represent a critical advance in upcycling plastic waste.114
4. Conclusions
Using a simple, one-pot procedure, we upcycled LDPE and HDPE into reprocessable CANs by incorporating dynamic cross-links based on the dissociative reversible chemistry of BiTEMPS methacrylate through melt-state reactive grafting. The thermomechanical properties of PE CANs are easily tuned by varying the loadings of BiTEMPS methacrylate and DCP without sacrificing their (re)processability. Relative bounds for these processing limits have been established for PE CANs. PE CANs synthesized with 5 wt% BiTEMPS methacrylate and 1 wt% DCP had high gel contents and rubbery plateau storage moduli characteristic of conventionally cross-linked networks. 5-1 LDPE and HDPE CANs recovered their thermal properties as well as their cross-link densities and associated mechanical properties within experimental error after three processing cycles, indicating their recyclability. 5-1 PE CANs are thermally stable up to ∼200 °C.Semi-crystalline 5-1 PE CANs exhibited excellent linear viscoelastic creep resistance over 5 h under a 3.0 kPa shear stress at 90 °C like their neat counterparts. Compared to neat HDPE, semi-crystalline 5-1 HDPE CANs also exhibited enhanced nonlinear viscoelastic creep resistance over 30 min under a 3.5 MPa tensile stress at 90 °C. At these conditions, 5-1 HDPE CANs saw a 42% reduction in total strain relative to neat HDPE. The dialkylamino disulfide chemistry responsible for the dynamic nature of the PE CANs was confirmed using FTIR spectroscopy and an Arrhenius analysis of linear viscoelastic creep responses of 5-1 LDPE CAN above its melt transition.49 The Ea,η of 5-1 LDPE CAN (∼113 kJ mol−1) agrees well with the previously reported S–S bond dissociation energies of both the disulfide bond in BiTEMPS methacrylate and the trisulfide bond derivatives of BiTEMPS methacrylate. Additionally, the Ea,η of the 5-1 LDPE CAN parallels the viscous flow and stress relaxation activation energies of previously reported PHMA CANs with BiTEMPS cross-links. These results indicate the utility of BiTEMPS chemistry in making reprocessable and creep-resistant polymer networks with different viscoelasticities. 5-1 LDPE and HDPE CANs do not exhibit phase separation of the grafted BiTEMPS units from PE chains at their small loading. This was evidenced by the lack of molten turbidity, minimal or no reductions in normalized crystallinities after removing soluble PE fractions, and the absence of phase separation signatures in room-temperature SAXS experiments.
Post-consumer PE waste products were upcycled using our simple method into 5-1 PE CANs by exclusively free-radical methods. Plastic bags and a milk jug were modified with robust, dynamic cross-links allowing for recovery of cross-link densities and thermomechanical properties after reprocessing. Given their robust mechanical and thermal properties, PE CANs synthesized with this one-pot, melt-mixing procedure have potential for use in high-temperature applications typical of PEX such as piping and cable wiring with the advantage of being recyclable. We envision that this method for upcycling PE can be extended to upcycle ethylene-based copolymers and other addition-type polymers containing side chains with ample CH2 groups into CANs. Dynamically cross-linked nanocomposites and foams synthesized from virgin or waste PE are worth studying using similar upcycling procedures with BiTEMPS methacrylate, as well.
Author contributions
Logan M. Fenimore: conceptualization, data curation, formal analysis, investigation, methodology, visualization, writing – original draft, writing – review & editing. Boran Chen: conceptualization, data curation, formal analysis, investigation, methodology, visualization. John M. Torkelson: conceptualization, funding acquisition, project administration, resources, supervision, validation, writing – review & editing.Conflicts of interest
Two US patent applications have been filed that are related to the research described in this manuscript.Acknowledgements
This research was supported by the University Partnership Initiative between Northwestern University and The Dow Chemical Company. We also gratefully acknowledge the support from an NSF Graduate Research Fellowship (L. M. F.) and Northwestern University via discretionary funds associated with a Walter P. Murphy Professorship (J. M. T.).References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- Organisation for Economic Co-operation and Development Environment Statistics, accessed June 2022, DOI:10.1787/a92f5ea3-en.
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef CAS PubMed.
- F. Gu, J. Guo, W. Zhang, P. A. Summers and P. Hall, Sci. Total Environ., 2017, 601–602, 1192–1207 CrossRef CAS PubMed.
- B. J. Luijsterburg and J. G. P. Goossens, Resour., Conserv. Recycl., 2014, 85, 88–97 CrossRef.
- M. K. Eriksen, J. D. Christiansen, A. E. Daugaard and T. F. Astrup, Waste Manage., 2019, 96, 75–85 CrossRef CAS PubMed.
- K. Khait and J. M. Torkelson, Polym.-Plast. Technol. Eng., 1999, 38, 445–457 CrossRef CAS.
- K. Ragaert, L. Delva and K. Van Geem, Waste Manage., 2017, 69, 24–58 CrossRef CAS PubMed.
- M. G. Davidson, R. A. Furlong and M. C. McManus, J. Cleaner Prod., 2021, 293, 126–163 CrossRef.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- M. Lazár, R. Rado and J. Rychlý, Adv. Polym. Sci., 1990, 95, 149–197 CrossRef.
- R. Patterson, A. Kandelbauer, U. Müller and H. Lammer, Crosslinked Thermoplastics, in Handbook of Thermoset Plastics, ed. H. Dodiuk, Elsevier, Amsterdam, 2014, vol. 3, pp. 697–737 Search PubMed.
- S. M. Tamboli, S. T. Mhaske and D. D. Kale, Indian J. Chem. Technol., 2004, 11, 853–864 CAS.
- S. Nilsson, T. Hjertberg and A. Smedberg, Eur. Polym. J., 2010, 46, 1759–1769 CrossRef CAS.
- L. Imbernon and S. Norvez, Eur. Polym. J., 2016, 82, 347–376 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- M. Podgórski, B. D. Fairbanks, B. E. Kirkpatrick, M. McBride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Adv. Mater., 2020, 32, 1906876 CrossRef PubMed.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 1606100 CrossRef PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Chem. Sci., 2016, 7, 30–38 RSC.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, J. Am. Chem. Soc., 2012, 134, 7664–7667 CrossRef CAS PubMed.
- Z. Pei, Y. Yang, Q. Chen, Y. Wei and Y. Ji, Adv. Mater., 2016, 28, 156–160 CrossRef CAS PubMed.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Macromolecules, 2018, 51, 5537–5546 CrossRef CAS.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Adv. Funct. Mater., 2015, 25, 2451–2457 CrossRef CAS.
- M. M. Obadia, A. Jourdain, P. Cassagnau, D. Montarnal and E. Drockenmuller, Adv. Funct. Mater., 2017, 27, 1703258 CrossRef.
- J. J. Lessard, L. F. Garcia, C. P. Easterling, M. B. Sims, K. C. Bentz, S. Arencibia, D. A. Savin and B. S. Sumerlin, Macromolecules, 2019, 52, 2105–2111 CrossRef CAS.
- Y. Chen, Z. Tang, X. Zhang, Y. Liu, S. Wu and B. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 24224–24231 CrossRef CAS PubMed.
- B. Soman and C. M. Evans, Soft Matter, 2021, 17, 3569–3577 RSC.
- S. Tajbakhsh, F. Hajiali and M. Marić, ACS Appl. Polym. Mater., 2021, 3, 3402–3415 CrossRef CAS.
- J. Deng, X. Kuang, R. Liu, W. Ding, A. C. Wang, Y. C. Lai, K. Dong, Z. Wen, Y. Wang, L. Wang, H. J. Qi, T. Zhang and Z. L. Wang, Adv. Mater., 2018, 30, 1705918 CrossRef PubMed.
- H. Si, L. Zhou, Y. Wu, L. Song, M. Kang, X. Zhao and M. Chen, Composites, Part B, 2020, 199, 108278 CrossRef CAS.
- L. Li, X. Chen and J. M. Torkelson, ACS Appl. Polym. Mater., 2020, 2, 4658–4665 CrossRef CAS.
- K. K. Oehlenschlaeger, J. O. Mueller, J. Brandt, S. Hilf, A. Lederer, M. Wilhelm, R. Graf, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Adv. Mater., 2014, 26, 3561–3566 CrossRef CAS PubMed.
- J. Bai, H. Li, Z. Shi and J. Yin, Macromolecules, 2015, 48, 3539–3546 CrossRef CAS.
- L. M. Polgar, M. van Duin, A. A. Broekhuis and F. Picchioni, Macromolecules, 2015, 48, 7096–7105 CrossRef CAS.
- C. Shao, M. Wang, H. Chang, F. Xu and J. Yang, ACS Sustainable Chem. Eng., 2017, 5, 6167–6174 CrossRef CAS.
- X. Xu, S. Ma, S. Wang, B. Wang, H. Feng, P. Li, Y. Liu, Z. Yu and J. Zhu, Macromol. Rapid Commun., 2022, 43, e2100777 CrossRef PubMed.
- H. Otsuka, Polym. J., 2013, 45, 879–891 CrossRef CAS.
- K. Jin, L. Li and J. M. Torkelson, Adv. Mater., 2016, 28, 6746–6750 CrossRef CAS PubMed.
- L. Li, X. Chen, K. Jin, M. A. Bin Rusayyis and J. M. Torkelson, Macromolecules, 2021, 54, 1452–1464 CrossRef CAS.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- Y. Zhang, H. Ying, K. R. Hart, Y. Wu, A. J. Hsu, A. M. Coppola, T. A. Kim, K. Yang, N. R. Sottos, S. R. White and J. Cheng, Adv. Mater., 2016, 28, 7646–7651 CrossRef CAS PubMed.
- Q. Zhang, S. Wang, B. Rao, X. Chen, L. Ma, C. Cui, Q. Zhong, Z. Li, Y. Cheng and Y. Zhang, React. Funct. Polym., 2021, 159, 104807 CrossRef CAS.
- M. A. Bin Rusayyis and J. M. Torkelson, ACS Macro Lett., 2022, 11, 568–574 CrossRef CAS PubMed.
- A. Takahashi, R. Goseki and H. Otsuka, Angew. Chem., Int. Ed., 2017, 56, 2016–2021 CrossRef CAS PubMed.
- A. Takahashi, R. Goseki, K. Ito and H. Otsuka, ACS Macro Lett., 2017, 6, 1280–1284 CrossRef CAS PubMed.
- M. A. Bin Rusayyis and J. M. Torkelson, Macromolecules, 2020, 53, 8367–8373 CrossRef CAS.
- S. Kataoka, A. Tsuruoka, D. Aoki and H. Otsuka, ACS Appl. Polym. Mater., 2021, 3, 888–895 CrossRef CAS.
- M. A. Bin Rusayyis and J. M. Torkelson, Polym. Chem., 2021, 21, 2760–2771 RSC.
- X. Chen, L. Li, K. Jin and J. M. Torkelson, Polym. Chem., 2017, 8, 6349–6355 RSC.
- L. Li, X. Chen and J. M. Torkelson, Macromolecules, 2019, 52, 8207–8216 CrossRef CAS.
- X. Chen, S. Hu, L. Li and J. M. Torkelson, ACS Appl. Polym. Mater., 2020, 2, 2093–2101 CrossRef CAS.
- S. Hu, X. Chen and J. M. Torkelson, ACS Sustainable Chem. Eng., 2019, 7, 10025–10034 CrossRef CAS.
- C. Zhang, X. Wang, D. Liang, H. Deng, Z. Lin, P. Feng and Q. Wang, J. Mater. Chem. A, 2021, 9, 18431–18439 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS PubMed.
- J. S. A. Ishibashi, I. C. Pierce, A. B. Chang, A. Zografos, B. M. El-Zaatari, Y. Fang, S. J. Weigand, F. S. Bates and J. A. Kalow, Macromolecules, 2021, 54, 3972–3986 CrossRef CAS.
- J. J. Lessard, G. M. Scheutz, R. W. Hughes and B. S. Sumerlin, ACS Appl. Polym. Mater., 2020, 2, 3044–3048 CrossRef CAS.
- M. Aiba, T. Koizumi, M. Futamura, K. Okamoto, M. Yamanaka, Y. Ishigaki, M. Oda, C. Ooka, A. Tsuruoka, A. Takahashi and H. Otsuka, ACS Appl. Polym. Mater., 2020, 2, 4054–4061 CrossRef CAS.
- Z. Zhou, S. Chen, X. Xu, Y. Chen, L. Xu, Y. Zeng and F. Zhang, Prog. Org. Coat., 2021, 154, 106213 CrossRef CAS.
- M. Aiba, T. Koizumi, K. Okamoto, M. Yamanaka, M. Futamura, Y. Ishigaki, M. Oda, C. Ooka, A. Takahashi and H. Otsuka, Mater. Adv., 2021, 2, 7709–7714 RSC.
- T. Raidt, R. Hoeher, F. Katzenberg and J. C. Tiller, Macromol. Rapid Commun., 2015, 36, 744–749 CrossRef CAS PubMed.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 CrossRef PubMed.
- F. Caffy and R. Nicolaÿ, Polym. Chem., 2019, 10, 3107–3115 RSC.
- F. Ji, X. Liu, C. Lin, Y. Zhou, L. Dong, S. Xu, D. Sheng and Y. Yang, Macromol. Mater. Eng., 2019, 304, 1800528 CrossRef.
- G. P. Kar, M. O. Saed and E. M. Terentjev, J. Mater. Chem. A, 2020, 8, 24137–24147 RSC.
- M. Maaz, A. Riba-Bremerch, C. Guibert, N. J. Van Zee and R. Nicolaÿ, Macromolecules, 2021, 54, 2213–2225 CrossRef CAS.
- R. G. Ricarte, F. Tournilhac and L. Leibler, Macromolecules, 2019, 52, 432–443 CrossRef CAS.
- J. Tellers, R. Pinalli, M. Soliman, J. Vachon and E. Dalcanale, Polym. Chem., 2019, 10, 5534–5542 RSC.
- Z. Wang, Y. Gu, M. Ma, Y. Liu and M. Chen, Macromolecules, 2021, 54, 1760–1766 CrossRef CAS.
- A. Zych, R. Pinalli, M. Soliman, J. Vachon and E. Dalcanale, Polymer, 2020, 199, 122567 CrossRef CAS.
- B. Zhao, G. Hang, L. Li and S. Zheng, Mater. Today Chem., 2022, 24, 100759 CrossRef CAS.
- W.-Y. Wang, X.-J. Zha, R.-Y. Bao, K. Ke, Z.-Y. Liu, M.-B. Yang and W. Yang, J. Polym. Res., 2021, 28, 210 CrossRef CAS.
- M. O. Saed, X. Lin and E. M. Terentjev, ACS Appl. Mater. Interfaces, 2021, 13, 42044–42051 CrossRef CAS PubMed.
- M. C. Montoya-Ospina, H. Verhoogt and T. A. Osswald, SPE Polymers, 2021, 3, 25–40 CrossRef.
- S. Wang, S. Ma, J. Qui, A. Tian, Q. Li, X. Xu, B. Wang, N. Lu, Y. Liu and J. Zhu, Green Chem., 2021, 23, 2931–2937 RSC.
- J. E. Bennett, H. Sieper and P. Tavs, Tetrahedron, 1967, 23, 1697–1699 CrossRef CAS.
- W. C. Danen and D. D. Newkirk, J. Am. Chem. Soc., 1976, 98, 516–520 CrossRef CAS.
- B. Maillard and K. U. Ingold, J. Am. Chem. Soc., 1976, 98, 520–523 CrossRef CAS.
- G. A. O'Neil and J. M. Torkelson, Macromolecules, 1999, 32, 411–422 CrossRef.
- T. Sugimura, Y. Ogata and T. Minoura, J. Polym. Sci., Part A: Polym. Chem., 1966, 4, 2747–2756 CrossRef CAS.
- F. Cantamessa, G. Damonte, O. Monticelli, R. Arrigo and A. Fina, ACS Appl. Polym. Mater., 2022, 4, 4796–4807 CrossRef CAS PubMed.
- L. Odenwald, F. P. Wimmer, N. K. Mast, M. G. Schußmann, M. Wilhelm and S. Mecking, J. Am. Chem. Soc., 2022, 144, 13226–13233 CrossRef CAS PubMed.
- R. G. Ricarte, F. Tournilhac, M. Cloitre and L. Leibler, Macromolecules, 2020, 53, 1852–1866 CrossRef CAS.
- M. Marić and C. W. Macosko, Polym. Eng. Sci., 2004, 41, 118–130 CrossRef.
- S. Alapati, J. T. Meledath and A. Karmarkar, IET Sci., Meas. Technol., 2014, 8, 60–68 CrossRef CAS.
- D. Li, L. Zhou, X. Wang, L. He and X. Yang, Materials, 2019, 12, 1746 CrossRef CAS PubMed.
- S. Morimura, H. Horiuchi, C. Tamura and T. Yoshioka, Bull. Chem. Soc. Jpn., 1980, 53, 1666–1669 CrossRef CAS.
- P. Jaitner, K. Jäger, A. Dorfer and K.-E. Schwarzhans, J. Organomet. Chem., 2001, 621, 173–176 CrossRef CAS.
- Y. Hou, I. A. Abu-Yousef, Y. Doung and D. N. Harpp, Tetrahedron Lett., 2001, 42, 8607–8610 CrossRef CAS.
- E. Zysman-Colman and D. N. Harpp, J. Org. Chem., 2003, 68, 2487–2489 CrossRef CAS PubMed.
- L. S. Konstantinova and O. A. Rakitin, Russ. Chem. Rev., 2014, 83, 225–250 CrossRef.
- J. P. Grace and N. B. Bowden, ACS Omega, 2021, 6, 17741–17747 CrossRef CAS PubMed.
- H. L. Olin, J. Am. Chem. Soc., 1926, 48, 167–168 CrossRef CAS.
- D. H. Kim and S. C. Kim, Polym. Korea, 1984, 8, 44–52 CAS.
- N. Torres, J. J. Robin and B. Boutevin, J. Appl. Polym. Sci., 2001, 81, 581–590 CrossRef CAS.
- W. A. Pryor and T. L. Pickering, J. Am. Chem. Soc., 1962, 84, 2705–2711 CrossRef CAS.
- W. A. Pryor, Tetrahedron Lett., 1963, 4, 1201–1204 CrossRef.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, 1953 Search PubMed.
- N. C. Parasnis and K. Ramani, J. Mater. Sci.: Mater. Med., 1998, 9, 165–172 CrossRef CAS PubMed.
- X. Chen, L. Li, T. Wei, D. C. Venerus and J. M. Torkelson, ACS Appl. Mater. Interfaces, 2018, 11, 2398–2407 CrossRef PubMed.
- S. Hu, X. Chen, M. A. Bin Rusayyis, N. S. Purwanto and J. M. Torkelson, Polymer, 2022, 252, 124971 CrossRef CAS.
- L. Cheng, S. Liu and W. Yu, Polymer, 2021, 222, 123662 CrossRef CAS.
- L. Cheng, B. Li, S. Liu and W. Yu, Polymer, 2021, 232, 124159 CrossRef CAS.
- W. N. Findley, J. S. Lai and K. Onaran, Creep and Relaxation of Nonlinear Viscoelastic Materials, North-Holland Publishing Company, Oxford, 1976 Search PubMed.
- K. Watanabe, Rubber Chem. Technol., 1962, 35, 182–199 CrossRef.
- D. J. Plazek, J. Polym. Sci., Part A-2: Polym. Chem., 1966, 4, 745–763 CrossRef CAS.
- L. E. Nielsen, J. Macromol. Sci., Part C, 1969, 3, 69–103 CrossRef CAS.
- R. S. Porter, J. P. Knox and J. F. Johnson, Trans. Soc. Rheol., 1968, 12, 409–419 CAS.
- A. Guinier and G. Fournet, Small-angle Scattering of X-Rays, John Wiley & Sons, New York, 1955 Search PubMed.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta06364f |
| This journal is © The Royal Society of Chemistry 2022 |