
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA strategy to enhance recyclability of degradable block copolymers by introducing low-temperature formability†
Ikuo
Taniguchi‡
 *a,
Thao Thi Thu Nguyen
b,
Kae
Kinugasa
a and
Kazunari
Masutani
c
*a,
Thao Thi Thu Nguyen
b,
Kae
Kinugasa
a and
Kazunari
Masutani
c
aInternational Institute for Carbon-Neutral Energy Research (WPI-I2CNER), Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
bDepartment of Automotive Science, Graduate School of Integrated Frontier Sciences, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
cCenter for Fiber and Textile Science, Kyoto Institute of Technology, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan
First published on 19th October 2022
Abstract
Most thermoplastics are processed by melt-molding, which consumes energy and often results in thermal degradation of polymers and reduction of recycling opportunities. A potential solution is to introduce low-temperature formability in polymeric materials. Degradable block copolymers composed of poly(trimethylene carbonate) and polylactide are derived from renewables and processable at temperatures as low as ambient temperature through the application of pressure, thereby suppressing thermal degradation during processing and keeping the mechanical properties unchanged. An upper order-to-disorder transition phase behavior for the block copolymers can be predicted by the compressible regular solution (CRS) model. The block copolymers undergo a phase transition from an ordered (solid) state to a disordered (melt/solid) state by pressurizing to show fluidity. The melt/solid state is then reversibly solidified by removing the applied pressure. The low-temperature formability is dependent on the composition and molecular weight of the block copolymers. The “pressure plastics” substantially reduce energy consumption during processing with mitigating CO2 emissions and enhancing recyclability. The elastomeric properties obtained allow the bock copolymers to be alternative to petroleum-based thermoplastic elastomers, such as polyethylene. In addition, the degradable nature reduces environmental impact even though escaping the collection system of the end-of-life plastics.
Introduction
Plastics that are currently indispensable in our daily life are manufactured for use in packaging, construction, automotive, and a variety of other sectors. The surging amount of plastic production exceeded 380 million tons in 2015.1 In addition, the vast majority of plastic materials are derived from fossil resources and thus accumulate in landfills or the natural environment.2 Although collections of the end-of-life plastics have been launched in many countries, only 14% of plastic packaging (11 million tons) were collected for recycling, and 25 million tons escaped the collection system in 2013.3 At least 8 million tons are deposited in the ocean annually and threaten marine life, resulting in microplastic pollution.4 Recent investigations also warn airborne microplastics, widespread contaminants in the atmosphere.5 Elimination of the end-of-life plastics is thus an urgent task to be tackled. To address this issue, the campaign to Reduce, Reuse, and Recycle plastic materials plus use Renewables, 4R (3R+1R), is recommended. Herein, the focus is to investigate polymers developed from Renewables with enhanced Recyclability.Most thermoplastic materials are processed by melt-molding with energy-intensive resin-heating and mold-cooling processes.6 One reason to suppress plastic recycling is thermal degradation of polymers during the heating process, which is especially severe for polyesters, such as poly(ethylene terephthalate) and polylactide (PLA),7,8 although various parameters were adjusted to minimize thermal degradation in the molding.9 Therefore, the processed materials are scarcely converted back to the original ones by material recycling. On the other hand, chemical recycling has gained attention as a potential option for effective recycling of polymeric materials, in which polymers are depolymerized to the corresponding monomers followed by repolymerization, such as for polymers obtained by ring-opening polymerizations (ROPs).10,11 While the chemical recycling manner gives polymers with the same material properties as the virgin ones, the recycling system integration would be required with cost reduction.
Semicrystalline poly(L-lactide) (PLLA) has a melting temperature of 170–180 °C and decomposes above 200 °C.12 However, PLA is commonly processed at 200–210 °C to obtain sufficient fluidity in the molding,8 causing thermal degradation of the polyester backbone. Mayes et al. introduced low-temperature processable polymeric materials by pressure-induced flow, termed baroplastics, which substantially suppressed polymer degradation with a reduction of energy consumption during processing.13 Baroplastics are nanophase polymeric materials composed of a multi-component polymeric system. For a binary case, baroplastic block copolymers of a low-Tg (glass transition temperature) soft segment and a high-Tg hard segment, such as poly(n-alkyl acrylates)-b-polystyrene, can be processed at ambient temperature through the application of pressure.13–16 The microphase-separated structure of the block copolymers at ambient conditions is miscibilized by pressurizing to induce flow. Taniguchi and Lovell developed low-temperature processable block copolyesters of amorphous low-Tg (<−30 °C) poly(ε-caprolactone) (PCL) derivatives and semicrystalline high-Tg (55 °C) PLLA, revealing the mechanism of low-temperature formability upon a reversible pressure-induced phase transition by small-angle X-ray scattering (SAXS).17 The soft segment PCL derivatives are synthesized from fossil feedstock, while PLLA is from renewables. Herein, we introduce a low-temperature formable block copolymer derived from renewables, being composed of poly(trimethylene carbonate) (PTMC) and PLLA or poly(D-lactide) (PDLA). 1,3-Propanediol (PDO) has been manufactured from corn starch as Zemea®, and a facile one-pot synthetic route of the cyclic TMC monomer from PDO and CO2 was recently explored.18 The flow properties of the block copolymers under pressure are also investigated.
Prediction of a polymer pair
The CRS model is derived by Mayes et al. through an extension of the Flory–Huggins regular solution model for polymer mixtures to compressible systems, taking volume and density changes into account.13–16 In a binary compressible polymer system, a change in the free energy per unit volume Δgmix can be expressed as shown in eqn (1),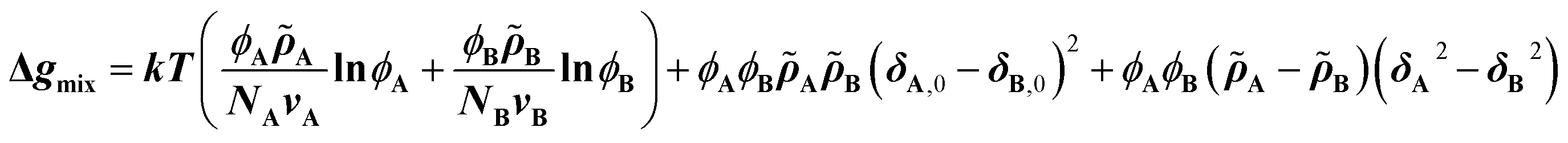 | (1) |
![[small rho, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e4.gif) is defined as ρ ρ*−1, where ρ is the mass density at T K and ρ* (= MuN0−1v−1; N0: Avogadro's number) is the hard-core density (0 K) for a monomer of molecular weight Mu. While the first term on the right-hand side expresses conventional entropy mixing, the second term corresponds to the χ term of the Flory–Huggins regular solution model. The third term can switch the sign depending on the solubility parameters or reduced densities, the so-called compressible term. Several requirements for the pressure-induced miscibility were discussed, such as combinations of a low-Tg and a high-Tg polymeric block with similar reduced densities (0.94 < AB−1 < 1.06).17,19,20
is defined as ρ ρ*−1, where ρ is the mass density at T K and ρ* (= MuN0−1v−1; N0: Avogadro's number) is the hard-core density (0 K) for a monomer of molecular weight Mu. While the first term on the right-hand side expresses conventional entropy mixing, the second term corresponds to the χ term of the Flory–Huggins regular solution model. The third term can switch the sign depending on the solubility parameters or reduced densities, the so-called compressible term. Several requirements for the pressure-induced miscibility were discussed, such as combinations of a low-Tg and a high-Tg polymeric block with similar reduced densities (0.94 < AB−1 < 1.06).17,19,20
Eqn (2) is the second derivative of eqn (1) with respect to composition to yield the stability criterion using pure component properties as input, and this expression equals zero at the spinodal temperature. A polymer pair of PTMC (Tg: −20 °C) and PLA is considered, because amorphous PTMC flows at ambient conditions and can be a suitable soft segment in the block copolymers. Fig. 1a represents a predicted phase diagram for a block copolymer of PTMC and PLA (PTMC-b-PLA) from pure group contribution parameters, and an upper disorder-to-order transition phase behaviour is plotted. The detailed calculations are described in the ESI.† The ratio of reduced density (PTMCPLA−1) was between 1.02 and 1.05 in the temperature range calculated, and thus, block copolymers of PTMC and PLA are expected to show pressure-induced flow upon phase mixing.
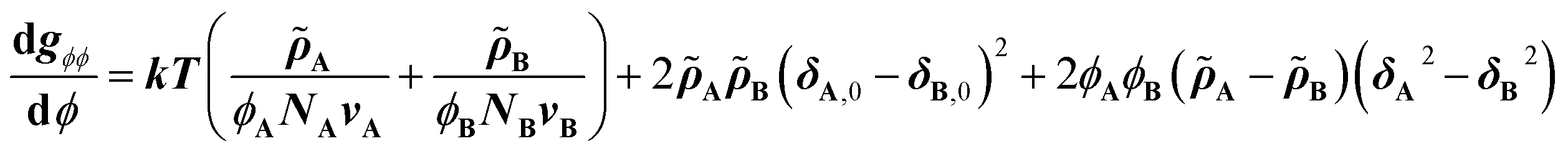 | (2) |
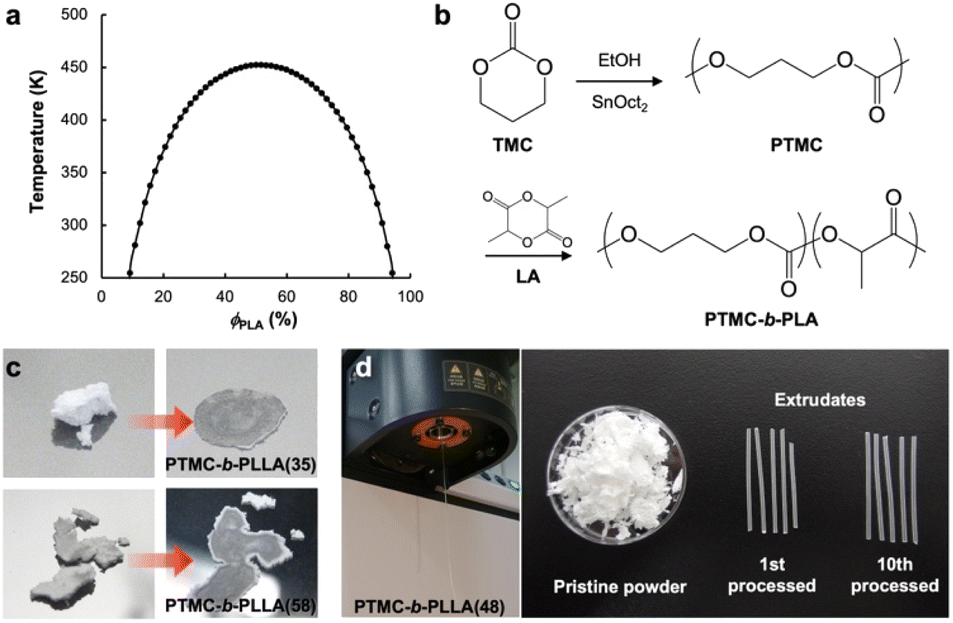 | ||
| Fig. 1 (a) Spinodal behavior of PTMC and PLLA derived from a second derivative of the CRS model. (b) Synthetic scheme of PTMC-b-PLA by two-step ring-opening polymerization of TMC and then LA. (c) Compression molding of PTMC-b-PLLA(35) (Mw 44 kDa) under 5 MPa for 10 s and PTMC-b-PLLA(58) (Mw 85 kDa) under 50 MPa for 5 min at ambient temperature by a hand press. (d) Extrusion of PTMC-b-PLLA(48) (Mw 45 kDa) under 49 MPa at 50 °C through a die (1.0 mm in orifice diameter and 1.0 mm in length), and the extrudates after the first and 10-times processing. | ||
Synthesis of block copolymers
Based on the above prediction, PTMC-b-PLAs can be synthesized by two-step ROP of TMC and then L-LA or D-LA with various stoichiometries (Fig. 1b and Table S1†). Block copolymer formation was confirmed by 13C-NMR and differential scanning calorimetry (DSC). In 13C-NMR, only two single resonances at 154.9 ppm and 169.8 ppm in the carbonyl region indicate the formation of PTMC and PLA homoblocks, respectively (Fig. S1A and S1B†).21,22 In addition, the block copolymers display two distinct Tgs at ca. −18 °C and 50 °C in DSC, which shows the existence of PTMC and PLA domains upon microphase separation (Fig. S2A–S2F,† Table S2†).23 In contrast, copolymerization of TMC and L-LA exhibits multiple peaks in the carbonyl region in 13C-NMR and a single Tg at 11 °C in DSC, indicating the random sequence of the monomers along the polymer chain to form PTMC-r-PLLA (Fig. S1C and S2G, Table S2†). Due to the lower Tg than ambient temperature, PTMC-r-PLLA flows under ambient conditions.Low-temperature formability
The low-temperature formability of PTMC-b-PLAs was at first examined under pressure of up to 50 MPa at ambient temperature using a hydraulic hand press. White precipitates of the block copolymers can be processed to a transparent film, if the polymers flow under the applied pressure. Block copolymers with a LA fraction of 33–57 wt% and weight average molecular weight (Mw) of 34–100 kDa were successfully processable under 30 MPa at ambient temperature, while PTMC-b-PLLA(58) (LA fraction: 58 wt%) with Mw of 84.8 kDa showed incomplete formability under the pressurized conditions in Fig. 1c.In plastic melt-molding, flow or rheological properties at elevated temperatures are critical in determining the processability of the polymeric materials6,24 and also in understanding the pressure-induced flow. The flow properties of PTMC-b-PLAs were then examined on a capillary rheometer under various pressures. Fig. 1d represents a typical pressure-induced flow of the block copolymer, PTMC-b-PLLA(48) with Mw of 45 kDa, at 50 °C under 49 MPa. Immediately after the application of pressure, the polymer was extruded through a die hole to form a transparent fibrous extrudate (Movie S1†). The flow properties, mass flow rate, viscosity and shear rate under various pressures are presented in Fig. 2a. A pressurizing piston on the block copolymer specimen did not move below 40 °C under 49 MPa, meaning that the polymer did not flow. However, the mass flow rate and shear rate started to increase gradually at 40 °C and then suddenly increased above 45 °C, reaching 6.03 × 10−3 cm3 s−1 and 61.4 s−1 at 56 °C, respectively. On the other hand, the viscosity dropped down rapidly by two orders of magnitude from 3.73 × 107 Pa s at 40 °C to 2.00 × 105 Pa s at 56 °C, which suggested that the block copolymer was able to be processed above 40 °C under 49 MPa. The flow temperature increased with reduction in applied pressure and reaches 80 °C under 9 MPa. However, the temperature is still far below the melting temperature of the PLA crystal at 170 °C (Fig. S2 and Table S2†), and thus, thermal degradation can be negligible.
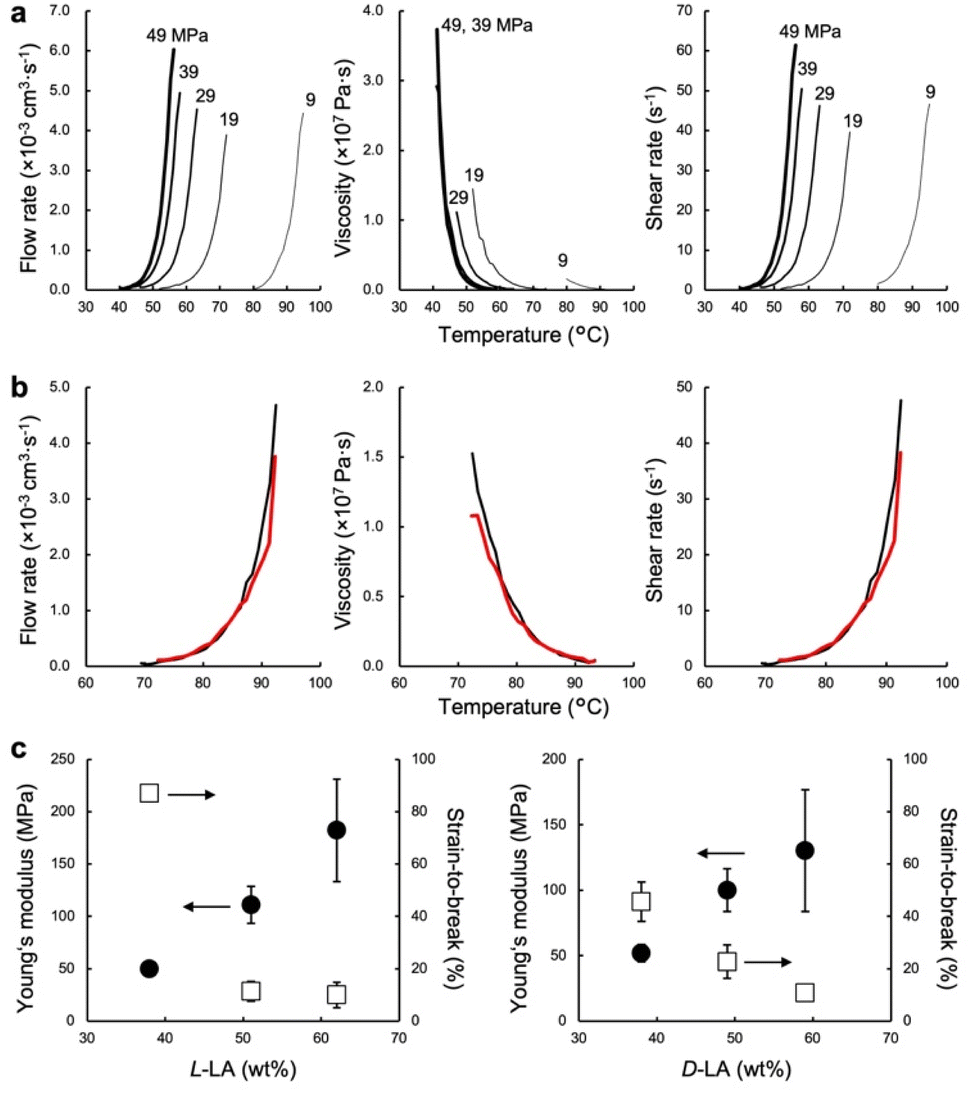 | ||
| Fig. 2 (a) Effect of the applied pressure on flow properties of PTMC-b-PLLA(48) (Mw 45 kDa) on pressure-processing. (b) Effect of the optical isomerism of the lactyl unit of PTMC-b-PLAs on flow properties under 49 MPa. Black and red lines are PTMC-b-PLLA(46) (Mw 121 kDa) and PTMC-b-PDLA(46) (Mw 105 kDa), respectively. (c) Tensile properties of PTMC-b-PLLA (left) and PTMC-b-PDLA (right) extrudates through a capillary rheometer as a function of LA content under 49 MPa. | ||
The pressure-processing of other PTMC-b-PLAs was conducted to study the effect of composition and molecular weight of the block copolymers and optical isomerism of the lactyl unit in PLA blocks on processability. In Table 1, PTMC-b-PDLA(37) (Mw: 119 kDa) flows at or above 57 °C under 49 MPa, while PTMC-b-PDLA(46) (105 kDa) and PTMC-b-PDLA(56) (122 kDa) start to flow under the pressure at 75 °C and 92 °C, respectively. The flow temperature increases with an increase of hard segment weight fraction when the block copolymers have similar molecular weights. With the same hard segment PLLA fraction, PTMC-b-PLLA(46) (39.9 kDa) flows at 40 °C, which is much lower than the temperature for the copolymer with Mw of 121 kDa (at 74 °C). Thus, the higher the hard segment fraction and molecular weight of the block copolymers, the higher the flow temperature.
| Block copolymers | PLAa (wt%) | M w b (kDa) | Flow temperaturec (°C) | X c,PLA (%) |
|---|---|---|---|---|
| a Determined by 1H NMR. b Determined by GPC. c Determined by capillary rheometer. d Determined by DSC. | ||||
| PTMC-b-PLLA | 35 | 44.3 | 36 | 21.4 |
| 38 | 66.6 | 54 | 18.2 | |
| 46 | 39.9 | 40 | 30.9 | |
| 121 | 74 | 25.2 | ||
| 51 | 103 | 71 | 31.0 | |
| 58 | 84.8 | 79 | 23.5 | |
| 62 | 95.1 | 93 | 34.1 | |
| PTMC-b-PDLA | 37 | 119 | 57 | 23.8 |
| 38 | 88.6 | 54 | n.t. | |
| 46 | 105 | 75 | n.t. | |
| 49 | 100 | 79 | 29.3 | |
| 56 | 122 | 92 | 18.6 | |
| 59 | 101 | 95 | 32.7 | |
PTMC-b-PDLAs show a similar flow tendency in the pressure-processing, and the effect of the optical isomerism was validated. Fig. 2b represents the flow profiles under 49 MPa of PTMC-b-PLLA(46) (121 kDa) and PTMC-b-PDLA(46) (105 kDa), having the same PLA fraction and similar molecular weight. Both the block copolymers flew above 74 °C and showed indistinguishable flow properties. Optical isomerism of the lactyl unit in the PLA block is thus not related to the pressure-processability.
Recyclability
Mechanical properties of the block copolymers studied by tensile testing demonstrate elastomeric properties depending on the composition of soft and hard segments rather than optical isomerism of the lactyl unit in Fig. 2c. For PTMC-b-PLLA, Young's modulus increases from 49.9 ± 1.3 to 182 ± 49 MPa with an increment of the hard segment fraction from 38 to 62 wt%, while the strain-to-break decreases from 87 ± 2.8 to 10 ± 4.9%. The tensile moduli found are similar to that of low-density PE.25 Pressure-processing of PTMC-b-PLLA(48) was then repeated 10 times under 49 MPa at 70 °C, and changes in flow and tensile properties are summarized in Table 2. In the first processing, the powder-like polymer gave a lower melt flow rate (MFR) of 5.1 g 10 min−1 in comparison to that in the repeated processing due to a larger void of the powder-like specimen. However, the MFR became almost identical and 7.0 g 10 min−1 in the multiple processing of the extrudate, which was similar to that of polyolefins at much higher temperatures, such as at 200 °C.26 The tensile properties of the fibrous extrudates were substantially unchanged by repeating the pressure-processing. For example, the Young's modulus was 59.5 ± 2.0 MPa over 10 times processing. In addition, changes in molecular weight and dispersity were not found at all by gel permeation chromatography (GPC) (Fig. S3†), which confirmed no degradation in repeating the pressure-processing, presenting infinite recyclability. On the other hand, the extrudate processed at 200 °C exhibited higher tensile properties than that at 70 °C. The block copolymer could melt entirely at higher temperature, which resulted in providing higher entanglement of the polymer chains. However, the high-temperature processing caused thermal degradation of the polymer chain.| Process time | MFR (g 10 min−1) | Young's modulus (MPa) | Strain-to-break (%) | Tensile strength (MPa) | M w (kDa) | PDIa (−) | |
|---|---|---|---|---|---|---|---|
| a Determined by GPC, PDI: Mw/Mn; ± denotes standard deviation (n = at least 3). | |||||||
| 0 | At 70 °C | — | — | — | — | 44.9 | 1.63 |
| 1 | 5.14 ± 0.16 | 59.3 ± 2.9 | 79.3 ± 11.1 | 7.51 ± 0.31 | 44.9 | 1.64 | |
| 3 | 7.01 ± 0.27 | 57.0 ± 1.8 | 67.1 ± 5.9 | 6.45 ± 0.47 | 44.6 | 1.65 | |
| 5 | 7.11 ± 0.22 | 59.6 ± 4.4 | 70.2 ± 1.9 | 6.12 ± 0.44 | 44.5 | 1.61 | |
| 10 | 7.00 ± 0.03 | 61.9 ± 1.4 | 82.1 ± 7.0 | 6.12 ± 0.22 | 45.1 | 1.62 | |
| At 200 °C | n.d. | 77.5 ± 9.5 | 87.4 ± 6.4 | 8.41 ± 0.16 | 41.8 | 1.81 | |
| LDPE | — | 203 ± 64.9 | 164 ± 27.7 | 7.68 ± 0.91 | n.t. | n.t. | |
Mechanism of pressure-induced flow
The significant changes in flow properties under pressure of the block copolymers would be due to a pressure-induced phase transition from an ordered (solid) state to a disordered (melt/solid) state.27 Two distinct Tgs of PTMC and PLA in DSC indicate microphase separation at ambient pressure (Fig. S2 and Table S2†).To verify the pressure-induced flow, PTMC-b-PLLA(46) (121 kDa) and PTMC-b-PDLA(46) (105 kDa) were co-processed at 80 °C under 49 MPa on a capillary rheometer, and the extrudate was further processed into a flat sheet by a hand press at 80 °C under 50 MPa for 10 min for the following DSC and X-ray diffraction (XRD) studies. When the soft and hard blocks are miscibilized by pressuring, rearrangement of polymer chains takes place at the interphase between the PTMC and PLA domains. That triggers PLA stereocomplex formation between PLLA and PDLA blocks through a backward disordered-to-ordered phase transition in removing the applied pressure as illustrated in Fig. 3.
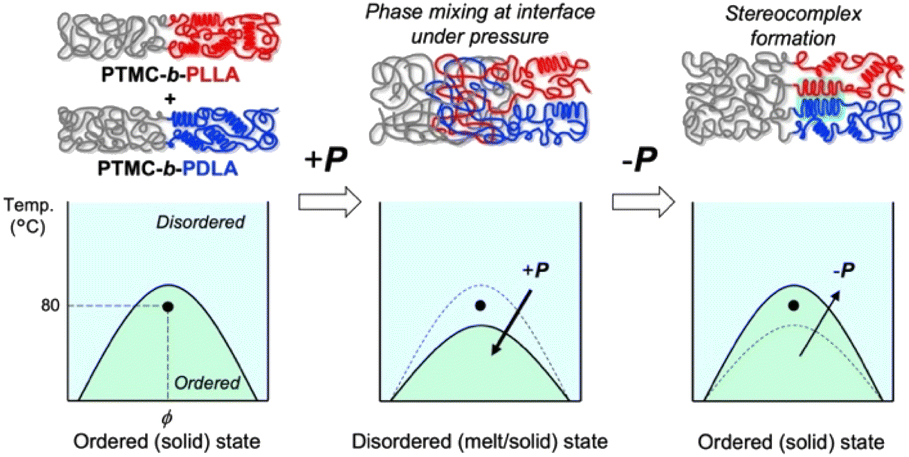 | ||
| Fig. 3 Schematic drawings of PLA stereocomplex formation upon pressure-induced phase transition (top) and the phase diagrams at ambient and high pressures (bottom). | ||
A DSC profile of the co-processed specimen presents an exothermic peak at 195 °C after melting of PLA homocrystals at 170 °C and a broad endothermic peak at 220–230 °C in Fig. 4a, which indicates crystallization and melting of the PLA stereocomplex, respectively. However, the PLA stereocomplex formation may be induced during heating at temperature above the melting point of PLA homocrystals in the DSC measurement,28 although the heat of fusion ΔHm (−20.0 J g−1) of the PLA stereocomplex is greater than the heat of crystallization ΔHc (6.6 J g−1). The DSC result is thus insufficient to prove the stereocomplex formation by pressure-processing.
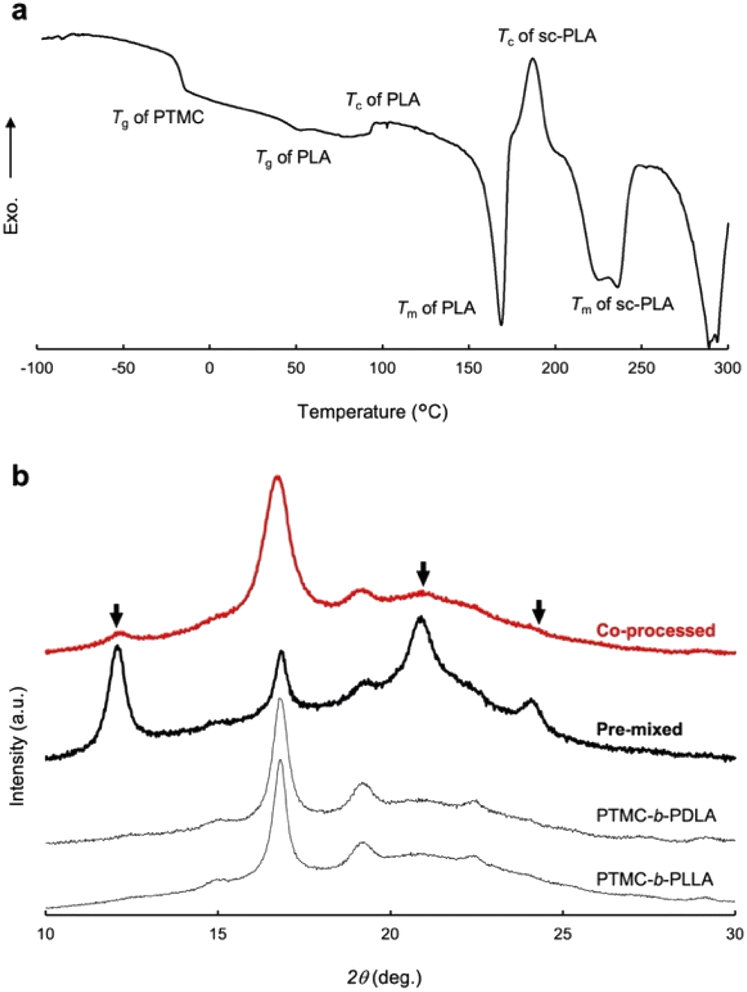 | ||
| Fig. 4 (a) DSC thermograph of a co-processed extrudate of PTMC-b-PLLA(46) (Mw 121 kDa) and PTMC-b-PDLA(46) (Mw 105 kDa). (b) XRD spectra of various degradable block copolymers. Top to bottom: co-processed PTMC-b-PLLA(46) and PTMC-b-PDLA(46), pre-mixed PTMC-b-PLLA(46) and PTMC-b-PDLA(46), and PTMC-b-PLLA(46). | ||
Stereocomplex formation was then investigated by XRD. As a reference, the PLA stereocomplex was intentionally assembled. The PLLA and PDLA block copolymers were dissolved together in CHCl3, and the block copolymer mixture was recovered by reprecipitation as a pre-mixed specimen. The resulting precipitate was processed by the same procedures for the XRD measurements. Different from the XRD spectra of PTMC-b-PLLA(46) and PTMC-b-PDLA(46) homopolymers, the copolymer pre-mixed displays diffraction peaks at 2θ of 12, 21, and 24°, which are derived from the PLA stereocomplex in Fig. 4b.28 For the copolymer co-processed, the peaks at 2θ of 12° and 21° can be confirmed, although the peak intensity is smaller than those of PLA homocrystals at 2θ of 17° and 19°.26 In Fig. 3, a dot in the phase diagram explains that the PLLA and PDLA block copolymers are in an ordered state at 80 °C and ambient pressure. Then, a binodal line of the block copolymers would be lowered by pressurizing, driving the polymers to move to a disordered state or to be miscible at the temperature. Here, both the block copolymers would show the same phase transition because of the same pressure-–processability in Fig. 2b. The increased fluidity of the polymers facilitates rearrangement of the polymer chains in the miscible state, resulting in the formation of the PLA stereocomplex on removing the applied pressure. The obtained XRD results in Fig. 4b are evidence to prove the pressure-induced phase mixing.
Similar to the effect of repetition times of the pressure-processing in Table 2, applied pressures in the processing are not a significant factor to characterize the tensile properties of the processed polymers. DSC measurements reveal that the extrudates processed at different pressures of 9 to 49 MPa have very similar crystallinity to PLLA blocks Xc,PLLA of 32.4 ± 0.9% as shown in Table 3. In pressure-processing, disordering even at the interface between PTMC and PLA domains allows the entire polymers to flow, while bulk domains of PTMC and PLA remain intact. The hard PLA domains move in the resulting fluidized medium under pressure. Such partial mixing or melt/solid flow can be the reason why the XRD gives small peaks of the PLA stereocomplex in the co-processed enantiomeric block copolymers in Fig. 4b.
| Processing pressure (MPa) | Young's modulus (MPa) | Strain-to-break (%) | Tensile strength (MPa) | X c,PLLA (%) |
|---|---|---|---|---|
| a ± denotes standard deviation (n = at least 3). | ||||
| 9 | 55.8 ± 7.2 | 179 ± 23 | 5.02 ± 0.27 | 33.4 |
| 19 | 51.2 ± 10.4 | 247 ± 26 | 4.97 ± 0.05 | 32.8 |
| 29 | 63.7 ± 15.6 | 219 ± 45 | 5.48 ± 0.38 | 32.5 |
| 39 | 41.8 ± 6.8 | 199 ± 56 | 6.05 ± 0.41 | 32.6 |
| 49 | 52.2 ± 9.8 | 301 ± 38 | 6.36 ± 0.12 | 30.9 |
Experimental
Synthesis of copolymers
PTMC-b-PLAs were prepared by two-step ROP of TMC and then L-LA or D-LA with tin(II) 2-ethylhexanoate initiated by ethanol. The ROP was conducted at 100 °C to restrain the ester/carbonate-exchanging reactions, causing disarrangement of the monomer sequence along the polymer backbone.29 The detailed procedures are described in the ESI.†Analyses
1H NMR and 13C NMR measurements were conducted in deuterated chloroform at 25 °C on a Bruker AVANCE III HD operating at 600 and 150 MHz, respectively. Molecular weight and the dispersity were measured by gel permeation chromatography (GPC). The GPC system consisted of a PU-2080 pump, a CO-2060 column oven, and an RI-2031 detector (JASCO). A TSKgel G3000HxL column (Tosoh) was used, and the samples were eluted out with chloroform at a flow rate of 1.0 mL min−1 at 40 °C. The molecular weight was calibrated with polystyrene standards (Sigma-Aldrich). Modulated temperature DSC of PTMC-b-PLAs was conducted on a PerkinElmer Pyris diamond DSC, while other DSC measurements were carried out on a Netzsch DSC 204 F1. The 2nd heating profiles were recorded at a heating rate of 10 °C min−1 after rapid cooling from a melt state at 200 °C. The crystallinity of PLLA Xc,PLLA was determined from eqn (3).| Xc,PLLA = (ΔHc + ΔHm)/ΔHm,100 × 100 (%) | (3) |
Conclusions
Block copolymers of PTMC and PLAs expressed low-temperature formability under pressure, and the mechanism of pressure-induced flow was investigated. The block copolymers can be processed at much lower temperatures and pressures than the commonly-used polymer resins, which are generally processed at >200 °C and under >100 MPa in the conventional melt-molding manner.6,31 Although the window of a soft/hard segment ratio of the block copolymers is not wide and from 5.4 × 10−1 to 1.8 in weight/weight to obtain pressure-processability, the mechanical properties are readily tuned to provide elastomeric nature like PE, which accounts for the largest portion of current plastic production.1,4 The flow properties under pressure in the extrusion experiments allow use of the existing molding machines without modifications for the processing. While a few research groups have studied theoretical aspects of block copolymers with baroplasticity,27,32,33 the rheological properties under pressure have scarcely been discussed, despite being one of the important parameters in material use.The low-energy processing with enhanced recyclability of the block copolymers can accelerate substitution for commodity plastic elastomers and thus suppresses fossil resource depletion. In addition, similar to a previous report,34 the developed polymers are composed of renewable compounds and CO2. Recent advances in incorporation of CO2 into polymers,18,35 such as to develop polycarbonates, are of great importance from viewpoints of plastic circular economy and CO2 capture and utilization toward the climate change control.
Author contributions
I. T. conceptualized and performed the research and wrote the manuscript. N. T. T. T., K. K., and K. M. validated the experimental results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Ogasawara foundation for the Promotion of Science & Engineering, JSPS KAKENHI Grant Number JP20K05633, and JST CREST Grant Number JPMJCR21L4. The WPI-I2CNER is supported by the World Premier International Research Center Initiative (WPI), MEXT, Japan. We thank Mr Masataka Hino for contributing to preliminary block copolymer syntheses, Prof. (emerit.) Dr Yoshiharu Kimura for providing D-LA, Dr Fuji Kodera and Ms. Mai Yoshizawa for helping in capillary rheometer experiments, and Prof. Dr Kazukiyo Nagai for modulated DSC measurement.Notes and references
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- D. K. A. Barnes, F. Galgani, R. C. Thompson and M. Barlaz, Philos. Trans. R. Soc. London, Ser. B, 2009, 364, 1985–1998 CrossRef CAS PubMed.
- Ellen MacArthur Foundation, The New Plastics Economy: Rethinking the Future of Plastics & Catalysing Action, https://www.ellenmacarthurfoundation.org/publications/the-new-plastics-economy-rethinking-the-future-of-plastics-catalysing-action, accessed: Jul. 2022 Search PubMed.
- J. R. Jambeck, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 CrossRef CAS PubMed.
- L. E. Revell, P. Kuma, E. C. Le Ru, W. R. C. Somerville and S. Gaw, Nature, 2021, 598, 462–467 CrossRef CAS PubMed.
- Z. Tadmor and C. G. Gogos, Principles of Polymer Processing, Wiley, Hoboken, NJ, USA, 2006 Search PubMed.
- G. Montaudo, C. Puglisi and F. Samperi, Polym. Degrad. Stab., 1993, 42, 13–28 CrossRef CAS.
- L.-T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS.
- Z. Shuidong, T. Lingcao, L. Jizhao, H. Hanxiong and J. Guo, Polym. Degrad. Stab., 2014, 105, 140–149 CrossRef.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- C. Li, L. Wang, Q. Yan, F. Liu, Y. Shen and Z. Li, Angew. Chem., Int. Ed., 2020, 61, e20221407 Search PubMed.
- F. D. Kopinke, M. Remmler, K. Mackenzie, M. Moder and O. Wachsen, Polym. Degrad. Stab., 1996, 53, 329–342 CrossRef CAS.
- J. A. Gonzalez-Leon, M. H. Acar, S.-W. Ryu, A.-V. G. Ruzette and A. M. Mayes, Nature, 2003, 426, 424–428 CrossRef CAS PubMed.
- A.-V. G. Ruzette and A. M. Mayes, Macromolecules, 2001, 34, 1894–1907 CrossRef CAS.
- J. A. Gonzalez-Leon and A. M. Mayes, Macromolecules, 2003, 36, 2508–2515 CrossRef CAS.
- A.-V. G. Ruzette, A. M. Mayes, M. Pollard, T. P. Russell and B. Hammouda, Macromolecules, 2003, 36, 3351–3356 CrossRef CAS.
- I. Taniguchi and N. G. Lovell, Macromolecules, 2012, 45, 7420–7428 CrossRef CAS PubMed.
- M. Honda, M. Tamura, K. Nakao, K. Suzuki, Y. Nakagawa and K. Tomishige, ACS Catal., 2014, 4, 1893–1896 CrossRef CAS.
- A.-V. G. Ruzette, P. Banerjee, A. M. Mayes and T. P. Russell, J. Chem. Phys., 2001, 114, 8205–8209 CrossRef CAS.
- A.-V. G. Ruzette, P. Banerjee, A. M. Mayes, M. Pollard, T. P. Russell, R. Jerome, T. Slawecki, R. Hjelm and P. Thiyagarajan, Macromolecules, 1998, 31, 8509–8516 CrossRef CAS.
- S. Matsumura, K. Tsukada and K. Toshima, Int. J. Biol. Macromol., 1999, 25, 161–162 CrossRef CAS PubMed.
- N. Andronova and A.-C. Albertsson, Biomacromolecules, 2006, 7, 1489–1495 CrossRef CAS PubMed.
- A.-C. Albertsson and M. Sjoling, J. Macromol. Sci., Part A: Pure Appl.Chem., 1992, 29, 43–54 CrossRef.
- S. G. Hatzikiriakos and J. M. Dealy, J. Rheol., 1992, 36, 703–741 CrossRef CAS.
- P. T. DeLassis and N. F. Whiteman, in Polymer Handbook, eds J. Brandrup, E. H. Immergut and E. A. Grulke, Wiley-VCH, New Jersey, USA, 1999, Ch. 2, vol. 1 Search PubMed.
- H. Mavridis and R. N. Shroff, Polym. Eng. Sci., 1992, 32, 1778–1791 CrossRef CAS.
- J. A. Gonzalez-Leon, S.-W. Ryu, S. A. Hewlett, S. H. Ibrahim and A. M. Mayes, Macromolecules, 2005, 38, 8036–8044 CrossRef CAS.
- H. Tsuji, Adv. Drug. Deliver. Rev., 2016, 107, 97–135 CrossRef CAS PubMed.
- H. R. Kricheldorf, C. Boettcher and K.-U. Tönnes, Polymer, 1992, 33, 2817–2824 CrossRef CAS.
- T. Miyata and T. Masuko, Polymer, 1998, 39, 5515–5521 CrossRef CAS.
- P. C. Wu, C. F. Huang and C. G. Gogos, Polym. Eng. Sci., 1974, 14, 223–230 CrossRef.
- D. Y. Ryu, U. Jeong, J. K. Kim and T. P. Russell, Nat. Mater., 2002, 1, 114–117 CrossRef CAS PubMed.
- D. Y. Ryu, C. Shin, J. Cho, D. H. Lee, J. K. Kim, K. A. Lavery and T. P. Russell, Macromolecules, 2007, 40, 7644–7655 CrossRef CAS.
- Y. Iwasaki, K. Takemoto, S. Tanaka and I. Taniguchi, Biomacromolecules, 2016, 17, 2466–2471 CrossRef CAS PubMed.
- M. Tamura, K. Ito, M. Honda, Y. Nakagawa, H. Sugimoto and K. Tomishige, Sci. Rep., 2019, 6, 24038 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta06036a |
| ‡ Current address: Faculty of Science and Engineering, Kyoto Institute of Technology, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan. E-mail: ikuot@kit.ac.jp |
| This journal is © The Royal Society of Chemistry 2022 |