
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA 2D copper-imidazolate framework without thermal treatment as an efficient ORR electrocatalyst for Zn–air batteries†
Ana
Franco‡
a,
José Ángel
Salatti-Dorado‡
 b,
Valentín
García-Caballero
b,
Sebastián
Lorca
c,
Luis
Camacho
b,
Manuel
Cano
*b,
Antonio J.
Fernández-Romero
c,
Juan J.
Delgado
d,
Juan J.
Giner-Casares
b and
Carolina
Carrillo-Carrión
*ef
b,
Valentín
García-Caballero
b,
Sebastián
Lorca
c,
Luis
Camacho
b,
Manuel
Cano
*b,
Antonio J.
Fernández-Romero
c,
Juan J.
Delgado
d,
Juan J.
Giner-Casares
b and
Carolina
Carrillo-Carrión
*ef
aDepartment of Organic Chemistry, University of Córdoba, E-14014 Córdoba, Spain
bDepartment of Physical Chemistry and Applied Thermodynamics, Institute of Fine Chemistry and Nanochemistry, University of Córdoba, E-14014 Córdoba, Spain. E-mail: q82calum@uco.es
cGrupo de Materiales Avanzados para la Producción y Almacenamiento de Energía, Universidad Politécnica de Cartagena, Aulario II, Campus de Alfonso XIII, 30203 Cartagena, Spain
dDepartamento de Ciencia de los Materiales e Ingeniería Metalúrgica y Química Inorgánica, Universidad de Cádiz, 11510 Puerto Real, Cádiz, Spain
eInstitute for Chemical Research (IIQ), CSIC-University of Seville, 41092 Sevilla, Spain. E-mail: carolina.carrillo@csic.es
fDepartment of Organic Chemistry, University of Seville, 41012 Sevilla, Spain
First published on 2nd November 2022
Abstract
To face unmet energy demands, the search for more stable, low-cost, and scalable electrocatalyst materials is imperative. Within this context, single-atom catalysts (SACs) have drawn considerable attention due to their maximum atom utilization. With this idea in mind, we have synthesized a new ultrathin and water-stable 2D Cu-based metal–organic framework (2DCIF), which presents a notable electrocatalytic activity for the oxygen reduction reaction (ORR) in alkaline media without the need for calcination, which makes the difference when compared to most MOF-based electrocatalysts. The designed MOF-based SAC consists of single-atom sites (isolated and accessible Cu) coordinated to imidazole carboxylic ligands, giving rise to Cu–N4O actives sites confined in a 2D-nanostructured network. This unique structure, along with the ultrathin nanosheets that favor mass transport and electrical conductivity, and the high chemical stability of these 2DCIFs are the key features that endow them with excellent performance in the ORR, which occurs via a direct four-electron transfer pathway, with an onset potential of 0.86 V vs. RHE and a maximum current density of 6.4 mA cm−2. These good catalytic properties of 2DCIFs have allowed their use as efficient air electrodes in alkaline flooded and all-solid-state Zn–air batteries. In the former case, 2DCIF-based air electrodes presented a specific power density of 91.2 kW cm−2 kg−1 and a specific capacity of 296.2 A h g−1, significantly exceeding the specific capacity values reported previously for other Cu-based catalysts. Besides, the specific capacity increased to 389.1 A h g−1 when 2DCIFs were tested in an all-solid-state Zn–air battery.
Introduction
Nowadays the depletion of natural resources and the overuse of fossil fuels have led not only to climate change issues, but also to a current energy crisis. Therefore, the global shift in energy business towards renewable energy and energy efficiency is an urgent need.1,2 Within this context, the search for low-cost, more stable, and scalable electrocatalyst materials for the development of clean and renewable energy devices is believed to be the key to meeting the challenges in our future energy landscape. The oxygen reduction reaction (ORR) is a pivotal process for diverse electrochemical energy conversion and storage technologies, such as regenerative fuel cells, rechargeable Zn–air batteries, and hydrogen peroxide production.3–7 Owing to the intrinsically sluggish kinetics of the ORR, which results in high overpotentials, the overall energy generation efficiency is often low, and thus the use of high-performance catalysts to accelerate the reaction is critical.8 So far, platinum- or palladium-derived catalysts9–11 have been demonstrated to be able to accelerate the ORR kinetics; however, their high cost and limited availability make these materials not economically feasible for large-scale deployment. Therefore, designing new non-noble-metal catalysts as alternatives for the ORR is of utmost importance. Apart from the efficiency of the catalyst, which is generally quite high for Pt- and Pd-based materials, durability and long-term performance are often critical bottlenecks, not solved yet even for such noble-metal catalysts. On the other hand, the use of green, cost-effective, and scalable synthetic methods for the preparation of those electrocatalytic materials is an important challenge, often overlooked, that must be considered in line with the objectives set in the 2030 Agenda for Sustainable Development.12Single-atom catalysts (SACs), with maximized utilization of metal atoms, offer a great opportunity for energy-related catalysis, serving as a potential bridge to connect heterogeneous and homogeneous catalysts.13–16 However, single atoms tend to aggregate during the synthetic and catalytic processes due to their high surface energy. To face this issue, metal–organic frameworks (MOFs), featuring well-defined porous structures, excellent tailorability, and high surface areas, have been recognized as very promising candidates for producing SACs.17–19 MOFs can be rationally designed to incorporate electrochemically active or redox-active metal sites (Co, Cu, Fe, Pt) and/or electroactive organic linkers (π-conjugated ligands such as the derivatives of benzene, pyridine, imidazole, and thiophene). This exceptional tunability of MOFs is one of the major advantages in comparison with other conventional porous materials employed commonly for anchoring SACs. In particular, SACs with metal–nitrogen coordinated moieties (M–Nx) in the carbon matrix have shown superior electrocatalytic activities toward the ORR.20–22 A recent study demonstrated that well-defined Cu–N4 coordination sites, which were stabilized by covalent attachment to graphene sheets, acted as highly efficient catalytic sites for the ORR.23 However, the synthesis of materials containing such active Cu–N4 moieties from single molecular building blocks, and with precise synthetic control, remains one of the most difficult challenges. In this sense, the high designability of MOFs could allow us to easily introduce these Cu–N4 into the framework, without the need for additional substrates to anchor and stabilize such SACs. On the negative side, most pristine MOFs cannot be used directly as electrocatalysts owing to their inherent low electrical conductivity and poor stability under electrocatalytic conditions.24 One of the most common strategies to increase both the conductivity and chemical stability is the use of MOFs as sacrificial templates for the synthesis of carbon-based porous materials after thermal treatments (calcination or pyrolysis).25,26 However, the high temperature required in these processes leads to the collapse of the MOF crystalline structure, with the consequent loss of the well-defined ordered structure, agglomeration of the metal centers, and limited accessibility to the active sites.
Apart from the composition of the MOF-based SAC, the morphology of the material is known to play a key role in boosting the catalytic activity. 2D MOF nanosheets have aroused great interest in electrochemistry due to some particular features not found in conventional 3D or 2D bulk MOF crystals,27–29 such as: (i) a higher density of exposed coordinatively unsaturated metal sites on the surface, which can act as active sites, and (ii) the nanosheet structure, which may allow faster mass transport and electron transfer. The first example of a 2D MOF for electrochemical ORR with remarkable activity was reported by Dincă and co-workers30 in 2016; they prepared an intrinsically conductive Ni3(hexaiminotriphenylene)2 2D MOF with Ni–N4 active sites and demonstrated its notable ORR activity in alkaline media. Although a few more 2D MOFs for the ORR have been reported to date,30–33 it still remains a great challenge to directly synthesize uniform ultrathin 2D MOF nanosheets with a thickness of sub-10 nm and without the need for calcination to achieve a good electrocatalytic response.
By keeping these mentioned issues and challenges in mind, we have herein developed a simple and green method for preparing an ultrathin water-stable 2D Cu-based MOF (in particular, 2D Copper–Imidazolate Framework nanosheets, denoted as 2DCIFs) as an efficient and sustainable electrocatalyst for the ORR. The resulting 2DCIFs were applied as air electrodes in two types of Zn–air battery prototypes. As far as we know this is the first 2D Cu-based MOF that presents a notable electrocatalytic activity in its pristine form, that is, without the need for further thermal treatments to achieve catalytically active sites.
Results and discussion
2DCIFs were prepared under mild conditions (in water and at room temperature) by coordinating copper ions (Cu2+) with imidazole-2-carboxaldehyde (ICA) ligands in the presence of a cationic surfactant, specifically cetyl trimethyl ammonium bromide (CTAB). The synthetic strategy of 2DCIFs is schematically illustrated in Fig. 1A. The CTAB acts as a stabilizing agent by decreasing the growth rate of the nanocrystals, as previously reported for other types of MOFs.34,35 This surfactant-assisted method for the synthesis of 2DCIFs led to highly homogeneous and ultrathin nanosheets. It must be noted that the CTAB plays a role in the formation of the nanosheets, but once removed after purification with methanol (as demonstrated by Fourier-transform infrared spectroscopy), the 2DCIFs retained their stability without signs of aggregation. Synthetic parameters were selected through a systematic experimental study (ESI† for details) and we found the molar ICA/Cu ratio of 2, CTAB concentration of 0.1 mM, and growth time of 15 h to be the optimal. It is worth noting that when using CuCl2 and Cu(NO3)2 as Cu(II) salts, sheet particles were produced, while Cu(OAc)2 did not produce sheet particles; the non-appearance of turbidity with time in the latter case indicated the absence of MOF formation. This finding is not surprising since the large influence of the metal source in the MOF formation process (mainly in the nucleation stage) has already been reported for other MOFs.36 Under optimized conditions, the bluish initial precursor mixture turned purplish after a few minutes, which indicated the successful nucleation of the crystals, and the turbidity of the solution increased with time as a consequence of the crystal growth; no further change was observed after ca. 12 h (Fig. 1B).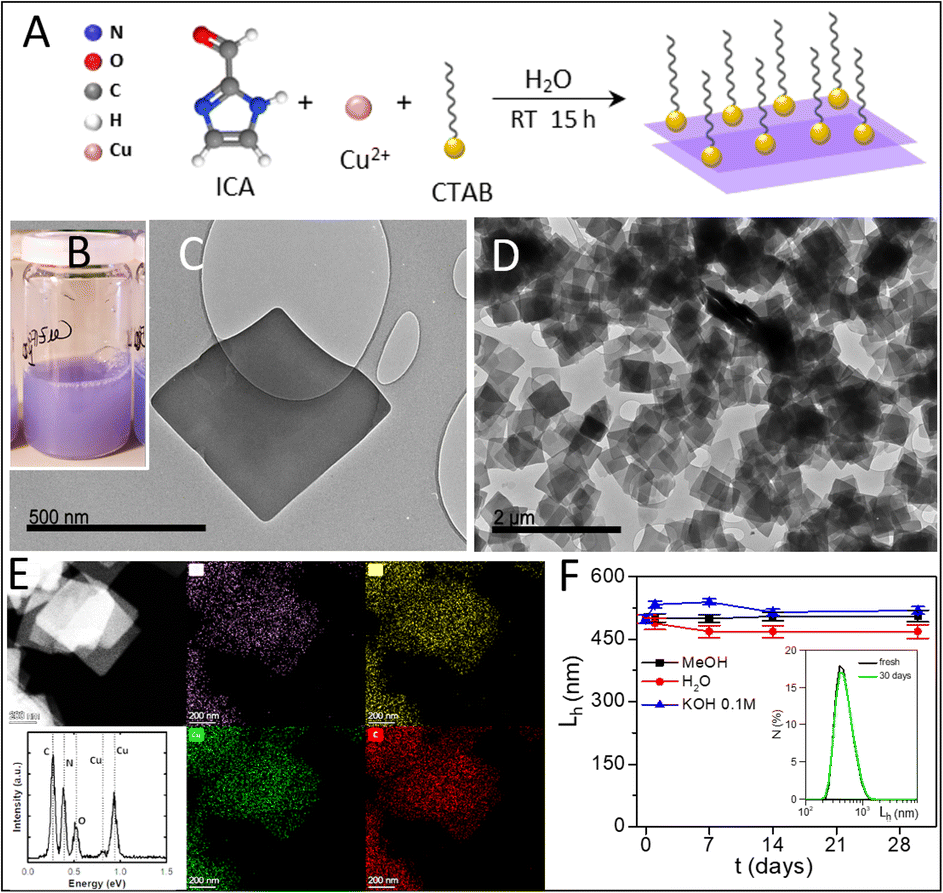 | ||
| Fig. 1 (A) Synthetic scheme of 2DCIF nanosheets. (B) Photograph of 2DCIFs dispersed in methanol. (C and D) TEM micrographs of the as-prepared nanosheets. (E) HAADF-STEM image and EDX elemental map displaying the uniform distribution of O (purple), Cu (green), N (yellow), and C (red) in the nanosheets (scale bar 200 nm). (F) Mean hydrodynamic lateral length Lh (mean value ± SD) of 2DCIFs over time in different media (MeOH, Milli-Q water, and KOH 0.1 M). The inset shows the DLS spectra of 2DCIFs freshly prepared and after 1 month storage in MeOH at 4 °C. | ||
The resulting 2DCIF particles were quite homogeneous, having a layered sheet morphology with an average lateral size (average sheet length) of 470 ± 30 nm as determined by transmission and scanning electron microscopy (TEM and SEM; Fig. 1C and D and S4†), and a thickness of just a few nanometers as suggested by the transparency of the overlayed sheets in the TEM image, along with the vertically deposited sheets observed in the SEM image (Fig. S4-A1†). In order to have more information at the atomic scale, we performed energy high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) in combination with energy-dispersive X-ray spectroscopy (EDX), observing a homogeneous distribution of all the elements (Cu, N, C, O) in the nanosheets (Fig. 1E). Deep analysis of the accumulative energy dispersive spectrum of a single sheet allowed the chemical composition of the sample to be determined (Table S1†), which was really consistent with results from X-ray photoelectron spectroscopy (XPS, Table S4†). Moreover, HAADF-STEM images recorded at high magnification (Fig. S9†) showed the presence of small brilliant dots, demonstrating the atomic dispersion of the Cu atoms in the nanosheets. On the other hand, an advantageous feature of the as-obtained 2DCIFs compared to other reported 2D MOFs is the extremely good colloidal stability, not only in organic solvents such as methanol but also in aqueous media including water (Milli-Q, pH = 6.5) or alkaline medium (KOH 0.1 M), as determined by dynamic light scattering (DLS; hydrodynamic lateral length, Lh ∼ 500 nm freshly prepared in methanol, and no significant changes were observed after 1 month in the different media tested; Fig. 1F, S19 and Table S5†). 2DCIFs dispersed in water were positively charged, having a ζ-potential of 7.9 ± 0.4 mV as determined by laser Doppler anemometry (LDA). No less important is the demonstrated potential to scale-up the preparation of these 2DCIF nanosheets, overcoming one of the major challenges in the field of MOF-based catalysis. This scalable strategy yielded 105 mg of nanosheets (Fig. S3†), which means a very large amount of material considering that for each ORR measurement only 0.05 mg is used.
The crystalline structure of the as-synthesized 2DCIFs (Fig. 2A) was deduced from powder X-ray diffraction (PXRD) data and confirmed by Rietveld refinement (ESI† for details). The assignment of the diffraction peak is summarized in Table S8.† This was possible thanks to the high crystallinity of the sample, presenting well resolved and intense peaks that matched well with the simulated XRD pattern (Fig. 2B). Data revealed that 2DCIFs crystallizes in the monoclinic No. 7 P1c1 space group, where the coordination number of Cu atoms is five with square pyramidal geometry. Each Cu center of the SBU unit is coordinated with four ICA ligands; specifically, three ICA ligands are only interacting with the metal ion through a N atom while the fourth ICA is interacting with Cu through one N atom (from the imidazole ring) and the O atom (from its aldehyde group), Fig. 2A-a1. Perspectives along each axis of the structure (Fig. 2A-a2 and S22†) revealed the existence of sheets, confirming the 2D layer structure of 2DCIFs. As can be seen in Fig. 2A-a3, Cu atoms remain relatively exposed on one side of the layer where all the O atoms are coordinated. However, on the opposite side of the layer, the O atoms do not participate in the coordination. Cu–N and Cu–O distances are in the range of other reported copper MOFs.37,38 The interaction of the metal center not only with N atoms but also with the O atom may be the key for stabilizing the structure, as it was proposed and confirmed by single crystal X-ray diffraction analysis for a few Co-based MOFs using pyridine-N-oxides as ligands.39
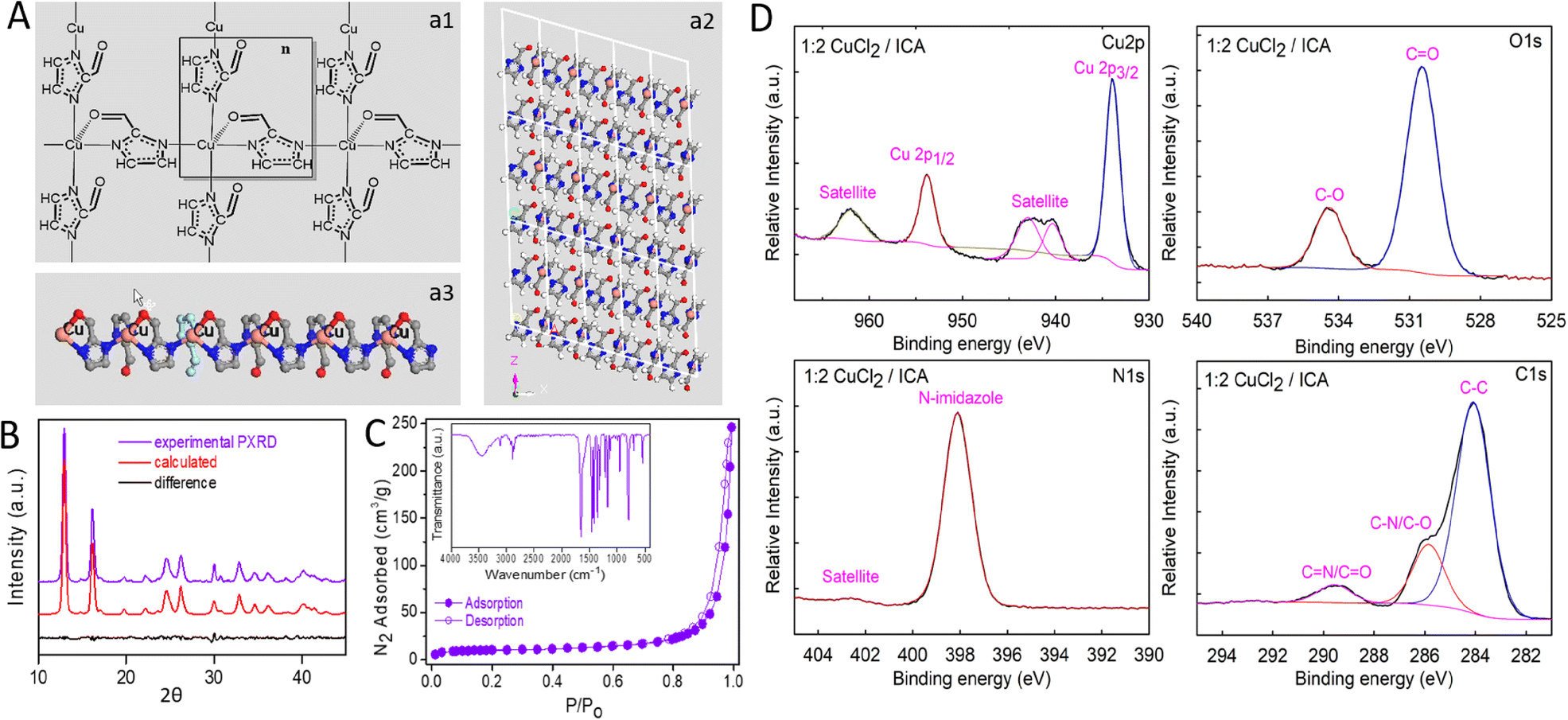 | ||
Fig. 2 (A) Crystal structure of 2DCIFs: (a1) coordination mode of the Cu ion in the structure; (a2) representation of the 2DCIFs along the y axis confirms the 2D layer structure of the nanocrystals; (a3) representation of one 2D layer, showing that Cu atoms are relatively exposed on one side of the nanosheet, O atoms are coordinated to Cu atoms on one side but not coordinated on the opposite site. Color code: pink Cu, gray C, blue N, red O, white H. (B) Experimental PXRD (violet) from powder 2DCIFs, the calculated XRD pattern (red) from the structure model, and their difference (black). R factors in the Rietveld refinement: Rwp = 5.33%, Rp = 10.17%. (C) Nitrogen adsorption (filled circles)–desorption (open circles) isotherms and the FTIR spectrum (inset) of the 2DCIFs. (D) High-resolution XPS spectra of Cu 2p, N 1s, O 1s and C 1s for as-synthesized 2DCIFs under optimized conditions (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 CuCl2/ICA). :2 CuCl2/ICA). | ||
Other characterization techniques were used to provide additional information of the as-obtained 2DCIF nanosheets. The FTIR spectrum (Fig. 2C-inset) showed evidence that the imidazole ring is retained upon the nanosheet formation, which is believed to be an important structural unit for facilitating the electron transfer. The absorption peak at 3110 cm−1 is assigned to the N–H stretching vibration of the imidazole ring while peaks at 1455 and 1419 cm−1 are due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N vibration modes of the imidazole moiety. Several peaks at around 1360 and 1130 cm−1 are ascribed to C–N stretching vibrations of the ring. Furthermore, the sharp peak at 1645 cm−1 corresponds to the CO stretching of the aldehyde group of the ICA linker. Most importantly, the Cu–N band is found at around 695 cm−1, confirming the successful coordination of Cu and N atoms.40 Also the disappearance of the ligand NH signals at 3150 cm−1 further demonstrates the coordination of N to Cu. Notably, the same FTIR peaks (Fig. S13A†) were found for 2DCIF samples prepared from different copper salts (CuCl2 and Cu(NO3)2) as well as at different reaction volumes (small- and large-scale), proving that all these samples had the same bond structure, consistent with the same morphology shown by SEM. It should be noted that the absence of peaks related to CTAB in the FTIR spectrum of 2DCIFs (Fig. S13B†) revealed the successful removal of this structuring agent during the purification step.
N vibration modes of the imidazole moiety. Several peaks at around 1360 and 1130 cm−1 are ascribed to C–N stretching vibrations of the ring. Furthermore, the sharp peak at 1645 cm−1 corresponds to the CO stretching of the aldehyde group of the ICA linker. Most importantly, the Cu–N band is found at around 695 cm−1, confirming the successful coordination of Cu and N atoms.40 Also the disappearance of the ligand NH signals at 3150 cm−1 further demonstrates the coordination of N to Cu. Notably, the same FTIR peaks (Fig. S13A†) were found for 2DCIF samples prepared from different copper salts (CuCl2 and Cu(NO3)2) as well as at different reaction volumes (small- and large-scale), proving that all these samples had the same bond structure, consistent with the same morphology shown by SEM. It should be noted that the absence of peaks related to CTAB in the FTIR spectrum of 2DCIFs (Fig. S13B†) revealed the successful removal of this structuring agent during the purification step.
Next, N2 adsorption–desorption measurements were performed to analyze the specific surface area and porous nature of as-synthesized 2DCIF particles. As shown in Fig. 2C, the 2DCIF nanosheets displayed a typical type IV isotherm with a small hysteresis loop at high relative pressures, suggesting a mesoporous structure. The Brunauer−Emmett−Teller specific surface area (SBET) was 34.5 m2 g−1 (total surface area), with a total pore volume of 0.18 cm3 g−1. Pore size analysis by the Barrett–Joyner–Halenda (BJH) method gave an average pore size of 29 nm (mesoporous). The external surface area was 18.4 m2 g−1 as determined by t-plot analysis (Fig. S14 and Table S3†). This high external surface area (up to 53% of total surface area), typical of thin films, ensures that most of the active catalytic sites are on the surface and therefore easily accessible, which provides a rapid transport pathway for O2 and benefits ORR kinetics. On the other hand, the finding of a mesoporous structure is most likely due to the formation of stacking laminar plate-like aggregates after drying the sample, which is also observed in the SEM and TEM images of concentrated samples (Fig. S3 and S4†). The deposition of the nanosheets on the electrode by forming such stacking structures could explain the extremely good stability of 2DCIFs under electrochemical operating conditions as indicated by the durability tests performed (ESI† for details). However, it must be remarked that when dispersed in solution the nanosheets are present as “individual colloidal particles” as confirmed by DLS measurements (Fig. 1F), as well as by TEM images of samples prepared from very diluted suspensions (Fig. 1C). Regarding the thermal properties, thermogravimetric analysis (TGA) under air conditions revealed that the 2DCIF nanosheets are thermally stable up to 280 °C (Fig. S15†).
To study the chemical composition and the oxidation state of Cu in the nanosheets, X-ray photoelectron spectroscopy (XPS) was employed (Fig. 2D). The survey XPS spectrum (Fig. S16B†) confirmed the existence of Cu, O, N, and C elements in the 2DCIF nanosheets. Accordingly, the high-resolution Cu 2p core-level photoelectron spectrum (HR-XPS) indicated the presence of the typical Cu 2p3/2 and Cu 2p1/2 peaks at binding energies (BEs) of 934.3 and 954.2 eV, respectively, together with two satellite peaks that further support the majority presence of Cu(II) in 2DCIFs.41 The HR-XPS O 1s spectra could be fitted to two peaks located at 530.9 and 534.7 eV, belonging to O–Cu and C–O, respectively.42 The latter peak with a higher binding energy was related to the presence of oxygen vacancies,43 which may display a key role for ionic conduction and dominated the ORR electrochemical performance of electrocatalyst materials.44 Moreover, the HR-XPS N 1s spectrum showed only one peak at 398.5 eV and its aromatic satellite assigned to N-imidazole, confirming the presence of a single type of nitrogen species, whilst the HR-XPS C 1s spectrum could be deconvoluted into three peaks at BEs of 284.4, 286.2, and 289.9 eV, which are assigned to C–C, C–N/C–O, and CN/CO, respectively.45 Furthermore, XPS analyses were performed to confirm the stoichiometry of the Cu(ICA)2 structure (Table S4†), and to demonstrate the absence of chloride anions in the network through inferring the chemical composition and states of the 2DCIFs (Fig. S17 and S18†).
With these already characterized 2DCIF nanosheets at hand, we wanted to explore if their unique features, mainly the 2D well-defined structure containing isolated Cu–N4O catalytic sites and a high number of stabilized open metal sites on the surface, could boost the electrochemical ORR performance of pristine (non-calcinated) Cu-based MOFs. Cyclic voltametric (CV) measurements of a glassy carbon electrode (GCE) modified with 2DCIFs were performed under static conditions in O2- or N2-saturated 0.1 M aqueous KOH solution (Fig. 3A). The response of the 2DCIFs in N2-saturated electrolyte revealed the electrochemical reduction of Cu(II) to Cu(I) (∼0.70 V vs. RHE), and subsequently to Cu(0) (∼0.33 V), indicating the presence of available Cu(II) active sites.46 Similar results have been reported with Cu–N4–C electrocatalyst materials containing a Cu–N4 coordinative structure.23 The experiments carried out under an O2 atmosphere in 0.1 M KOH showed a significant increase of the current densities of both reduction peaks, suggesting that 2DCIFs possess a notable electrocatalytic activity toward the ORR. Linear sweep voltammetry (LSV) curves at 2500 rpm of 2DCIFs and commercial 10 wt% Pt/C catalysts for comparison are presented in Fig. 3B. It is noteworthy that the limiting current density of the 2DCIF catalyst, about 6.4 mA cm−2 at 0.0 V, is notably superior to the values deduced for the Pt/C catalyst (4.1 mA cm−2 at 0.0 V), reflecting the superior mass transfer property of 2DCIFs. More importantly, the limiting current density obtained with 2DCIFs is the highest reported to date for pristine (non-calcinated) Cu-based MOFs (Table S11†). On the other hand, whilst the ORR onset potential (0.86 V) determined for 2DCIFs is similar to the best values reported to date for other MOF-derived Cu/C materials, it is still slightly lower than that of 10 wt% Pt/C (0.98 V vs. RHE).
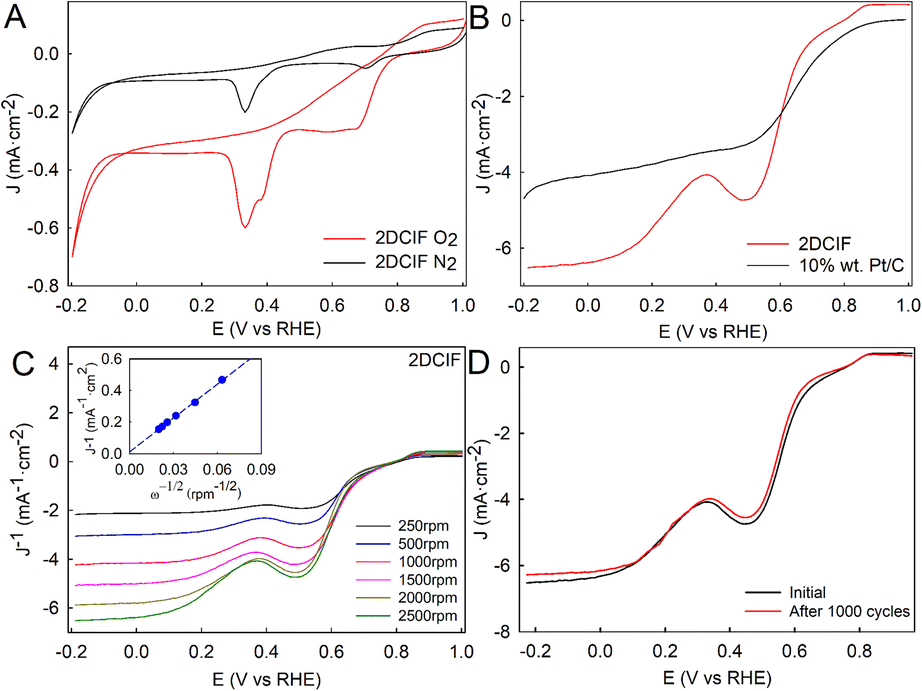 | ||
| Fig. 3 (A) CV curves obtained for the GCE modified with 2DCIFs in O2-saturated and N2-saturated 0.1 M KOH. (B) RDV curves of GCE/2DCIFs and GCE/10 wt% Pt/C at the rotation rate of 2500 rpm in O2-saturated 0.1 M KOH. (C) RDV curves of GCE/2DCIFs at different rates (scan rate: 10 mV s−1). Inset: K–L plot (J−1vs. ω−1/2) for the 2DCIFs at 0.00 V. (D) CV curves of GCE modified of GCE/2DCIFs before and after 1000 cycles of the AAT, in O2-saturated 0.1 M KOH at a scan rate of 10 mV s−1 and using a rotating rate of 2500 rpm. | ||
The ORR kinetics and reaction pathways in 2DCIFs were evaluated by LSV measurements at different rotation rates (from 250 to 2500 rpm) and using the Koutecky–Levich (K–L) equations. The limited diffusion current densities of 2DCIFs linearly increased with increasing rotation rates (Fig. 3C), revealing a typical diffusion-controlled electrochemical process. The K–L plot at 0.00 V (Fig. 3C-inset) showed a good linear relationship between the reduction peak current and the square root of scan rates, indicating a first-order reaction kinetics toward dissolved O2. The average number of electrons transferred (n) per oxygen molecule, calculated from the slope of the K−L plot, gave a value of 3.93, suggesting a dominant four-electron pathway. Other calculated ORR kinetic parameters, in particular, Jk = 88.1 mA cm−2 and k = 0.761 cm s−1, further confirmed the excellent catalytic ORR performance of these novel 2DCIF nanosheets. The presence of both pyrrolic and pyridinic N (coming from the ICA linker) may reduce the energy barrier of O2 adsorption, thus facilitating the first rate-limiting step in the ORR by increasing electron transfer. Based on the theoretical 2DCIF structure obtained by the powder refinement module (Fig. S25A†) together with the derived electrochemical results, and inspired by a mechanism previously reported for a similar 2D MOF,47 we proposed a plausible ORR mechanism for our 2DCIF catalyst (Fig. S25B†).
It is well known that the durability of electrocatalysts is one of the most important issues that has to be addressed before the commercialization of electrochemical devices for energy storage and conversion. Therefore, to evaluate the actual feasibility of the as-synthesized 2DCIFs as an effective electrocatalyst for the ORR, their stability was investigated using an accelerated aging test (AAT), which consisted of 1000 continuous potential cycles performed by CV measurements in O2-saturated 0.1 M KOH solution. The results (Fig. 3D and S26†) demonstrated the excellent stability of the 2DCIF electrocatalyst material under both static and rotating conditions, where the slight decrease in current density at 2500 rpm would be associated with the loss of material deposited on the GCE surface after the AAT. The high stability of these nanosheets can be attributed to several factors: (i) the presence of N-anchor-metal, Cu–N in this case, which is known to promote the corrosion resistance;48 (ii) the high chemical stability of 2DCIFs in water and alkaline media, as demonstrated by DLS analysis; and (iii) the deposition of the nanosheets on the electrode by forming stabilized stacking structures, which may mitigate the dissolution of the active species in the electrolyte.
Post-ORR characterization on the 2DCIFs was performed using SEM, XRD and XPS analysis to check whether the structure of the nanosheets was affected during the ORR process. SEM images of the 2DCIFs supported on the ITO electrode before and after the ORR measurements (Fig. S27†) indicated that neither the size nor the shape was significatively affected after the ORR process, confirming the good stability under working conditions in alkaline electrolytes. Similarly, XRD analysis was performed using the 2DCIF-modified ITO electrode, showing that both the position (∼13°) and the FWHM of the most intense peak remain constant (Fig. S28†). This indicates that the crystal structure (actually the inter-planar spacing c = 1.425 nm) is preserved after the ORR cycles. Comparing the survey XPS spectra of 2DCIFs before and after the ORR (Fig. S29†), the presence of potassium (K 3s peak) from the electrolyte solution can be highlighted in the latter case, and the appearance of some new peaks from the ITO electrode is due to the material loss during ORR measurements. In addition, the HR-XPS Cu 2p demonstrated that the copper state did not change after the ORR (Fig. S30A†) as well as the N-imidazole remained intact as shown in HR-XPS N 1s (Fig. S31A†). However, the HR-XPS O 1s spectrum (Fig. S30B†) showed the appearance of a new peak at 528 eV, attributed to the presence of metallic oxygen (assigned to adsorbed O2 during the ORR process), whilst in the case of HR-XPS C 1s two new peaks at 291 and 294 eV associated with the CO2 chemisorption were observed (Fig. S31B†), which is in agreement with previous results reported for other electrocatalysts.49 All this together pointed out the notable ORR activity and exceptional stability of our 2DCIFs in its pristine form during the ORR process, that is, without the need for post-synthetic thermal treatments, whereas MOFs designed for electrocatalysis require usually calcination to increase their stability and electrocatalytic activity.
Once the catalytic activity of 2DCIFs was confirmed by RDV measurements, we wanted to investigate their potential application in real devices, such as Zn–air batteries. Thus, air electrodes were prepared by adding the 2DCIFs ink onto carbon paper and they were first tested in a flooded Zn–air battery (Fig. S32A†) using a Zn plate anode and 6 M KOH electrolyte. With the aim of working under the most economical conditions while achieving comparable efficiency to that achieved with conventional Pt/C electrodes, the amount of 2DCIF catalyst used for these experiments (0.375 mg) was reduced to the minimum required for covering the carbon paper area (0.63 cm2) in contact with the electrolyte (Fig. 4A); the high surface area of the nanosheets is beneficial for this aspect. In contrast, a higher amount of Pt (0.750 mg) was needed to cover the carbon paper. Fig. 4B and C show the polarization curves using the 2DCIF-modified electrode compared to the conventional Pt/C-based electrode. It can be clearly seen that the potential and power density values of 2DCIFs exceeded those obtained for Pt/C even when working with half the amount of catalyst, which is in agreement with the results obtained for RDV measurements (Fig. 3B). A maximum power density of 34.2 mW cm−2 at 53.7 mA cm−2 was obtained for the battery with an air electrode containing 0.375 mg of 2DCIFs, which revealed an excellent specific power density value of 91.2 kW cm−2 kg−1 (considering only the catalyst mass) or 0.98 kW cm−2 kg−1 (taking into account the mass of the entire air electrode, carbon paper + catalyst). It should be noted that the battery using Pt/C as the catalyst provided a similar maximum power density of 32.8 at 51.8 mA cm−2, but considering that 0.750 mg of Pt was added to the air electrode, the specific power density for the Pt/C air electrode was less than half of that obtained when 2DCIFs were used. Fig. S33† compares the power density obtained for the battery using the 2DCIF-based cathode with those adding the same mass amount of Pt/C catalyst and the double mass of this, confirming the best behaviour of the 2DCIF-modified electrodes. Importantly, the stability of the 2DCIF-based air electrode was confirmed by monitoring the evolution of voltages at different density currents from 1 to 50 mA cm−2 (Fig. 4D); although the voltage was not maintained stable at 60 mA cm−2, probably due to the passivation of the Zn electrode, the battery recovered the initial potential values after applying 5, 2 and 1 mA cm−2.
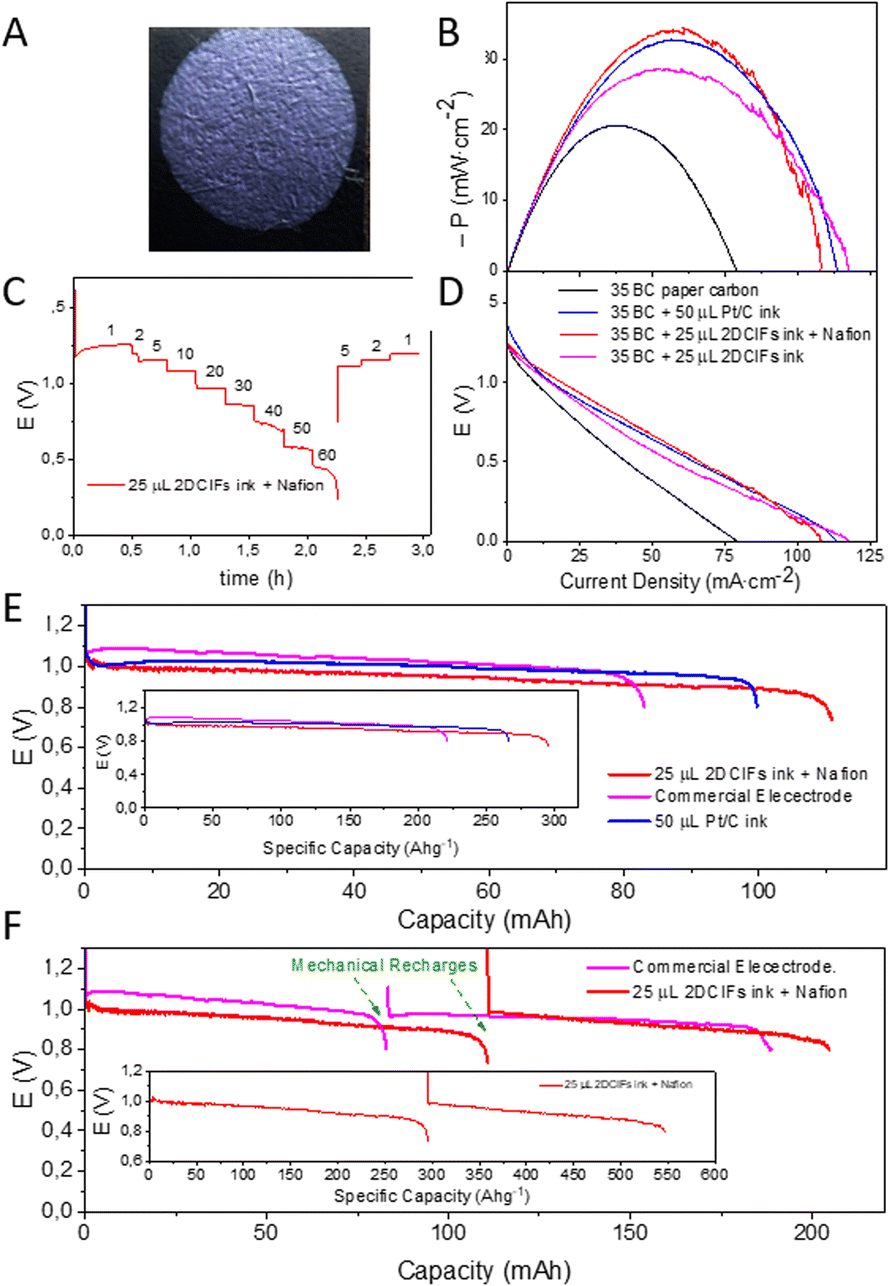 | ||
| Fig. 4 (A) Photograph of the air electrode prepared by depositing 2DCIFs onto carbon paper. (B) Power density plot of the pristine carbon paper and modified with either 2DCIFs or Pt/C. (C) Voltage versus current density plot. (D) Voltage versus time at different current densities. (E) Galvanostatic discharge curve of the Zn/6 M KOH/air battery using the 2DCIF-based air electrode and compared against the Pt/C-based electrode and a commercial air electrode. (F) Galvanostatic discharge curves obtained for the Zn/6 M KOH/air battery with the 2DCIF-based electrode and a commercial air electrode carrying out a “mechanical recharge” of the Zn electrode. The insets in E and F show the corresponding galvanostatic discharge curves as a function of the specific capacities. | ||
Regarding the galvanostatic discharge test, the 2DCIF-based air electrode presented a minimal lower voltage value compared with the Pt/C-electrode, as well as with the Gaskatel commercial air electrode (Fig. 4E). However, the maximum discharge capacity was reached for the 2DCIF-based air electrode, achieving a specific capacity of 296.2 A h g−1 (with respect to the catalyst mass) or 3.17 A h g−1 (with respect to the entire electrode mass). This result exceeds greatly the specific capacity values reported previously for other Cu-based catalysts, such as 100 A h g−1 (with respect to the Cu–N/C catalyst mass).50 In addition, a “mechanical recharge” of the Zn electrode allowed us to continue with the discharge process as shown in Fig. 4F. After a “mechanical recharge” of the negative electrode, the battery reached a specific capacity of 548.0 A h g−1 (with respect to the catalyst mass) or 5.85 A h g−1 (with respect to the entire electrode mass). All these results demonstrated the possibility of using this novel 2DCIF material as a highly efficient catalyst to prepare air electrodes for metal–air batteries.
Furthermore, the 2DCIF catalyst was also tested in an all-solid battery, using a PVA-KOH gel polymer electrolyte (Fig. S32B–S35†).51 Galvanostatic discharge and polarization curves obtained for these batteries are shown in Fig. S34†. Once again, the highest power density was found for the battery using the 2DCIF catalyst in the air electrode, with a value of 91.7 mW cm−2, when compared with the Pt/C-based electrode. Besides, the obtained specific capacities, 389.1 A h g−1 (with respect to the catalyst mass) or 3.61 A h g−1 (with respect to the entire electrode mass), were notably higher than values obtained for the flooded battery, supporting the good properties of the 2DCIFs to be used as air electrodes for all-solid metal–air batteries.
Conclusions
In summary, we have developed a simple, green and scalable synthetic method for the preparation of a novel 2D Cu-based MOF (2DCIF), featuring well-defined Cu–N4O coordination centers, ultrathin nanosheets, and high chemical and colloidal stability in alkaline media. The proposed surfactant-assisted synthetic approach was able to achieve synergistically a good control of the morphology, composition, and electronic structure of the 2DCIFs. These nanosheets exhibited a surprisingly high ORR activity in their pristine form, without the need for thermal treatment as required for most of the MOF-based electrocatalysts. The ORR performance of these nanosheets resulted in an onset potential of 0.86 V vs. RHE, n = 3.93, and Jk = 88.1 mA cm−2 in 0.1 M KOH, together with an exceptional electrochemical stability, outperforming most of the pristine Cu-based MOFs reported to date for electrocatalysis.The successful application of 2DCIFs as air electrodes in two types of Zn–air batteries (i.e. flooded Zn/6 M KOH/air and all-solid-state Zn/PVA-KOH/air), achieving excellent specific capacity and power density values, confirmed the great features of the 2D Cu-based MOFs as electrocatalysts for energy storage devices. The existence of uniformly distributed Cu–N4O units confined in a 2D stable framework, as determined from the crystal structure and computational studies, is strongly believed to be the key feature for the exceptional ORR performance of these pristine 2DCIFs.
Overall, the simplicity and low-cost of the synthetic method endow 2DCIFs with great promise for large-scale MOF-based catalyst preparation. Further studies are ongoing to investigate the potential of the here developed synthetic approach for preparing other transition metal–nitrogen frameworks for various energy-related applications.
Data availability
All the datasets supporting the findings of this work are available within the manuscript and in the ESI.†Author contributions
C. C.-C. and M. C. conceived the idea and designed the research; C. C.-C. and A. F. synthesized and characterized the materials; J. A. S.-D., V. G.-C., M. C., and J. J. G.-C. carried out the electrocatalytic experiments; L. C. performed the theoretical studies; J. J. D. carried out HAADF-STEM analysis; S. L. and A. J. F. R. performed the Zn–air battery tests; C. C.-C. drafted the manuscript. All authors have discussed the results and given approval to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. C.-C. acknowledges the financial support by Spanish Ministry of Science, Innovation and Universities (MICINN) and the Spanish Research Agency (AEI) through the “Ramón y Cajal” programme (RYC-2019-027527-I) and PID2019-107665RJ-I00 project. C. C.-C. also acknowledges funding of the University of Seville through the “VI Plan Propio de Investigación y Transferencia de la US” (VIPPIT-2020-IV.2). A. F. acknowledges MICINN for an FPI contract associated with CTQ2016-78289-P. J. J. G.-C. also received funding from MICINN and AEI through the PID2020-112744GB-I00/AEI/10.13039/501100011033 project. J. J. D thanks MICINN and the Andalusian Government for funding through projects PID2020-113809RB-C33 and the ELECMIC facilities. A. J. F. R. and S. L. thank MICINN and AEI for funding through the PID2019-104272RB-C55/AEI/10.13039/501100011033 project, and Fundación Séneca-Agencia de Ciencia y Tecnología de la Región de Murcia for financial support through the 20985/PI/18 project.References
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner and R. Gorini, Energy Strategy Rev., 2019, 24, 38–50 CrossRef.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- Y. Li, Q. Li, H. Wang, L. Zhang, D. P. Wilkinson and J. Zhang, Electrochem. Energy Rev., 2019, 2, 518–538 CrossRef CAS.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 6321 CrossRef PubMed.
- X. Feng, Y. Bai, M. Liu, Y. Li, H. Yang, X. Wanga and C. Wu, Energy Environ. Sci., 2021, 14, 2036–2089 RSC.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2021, 143, 9423–9428 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- J. Ying, Front. Chem., 2021, 719 Search PubMed.
- J. Liu, M. Jiao, L. Lu, H. M. Barkholtz, Y. Li, Y. Wang, L. Jiang, Z. Wu, D.-J. Liu, L. Zhuang, C. Ma, J. Zeng, B. Zhang, D. Su, P. Song, W. Xing, W. Xu, Y. Wang, Z. Jiang and G. Sun, Nat. Commun., 2017, 8, 15938 CrossRef CAS PubMed.
- T. Wang, A. Chutia, D. J. Brett, P. R. Shearing, G. He, G. Chai and I. P. Parkin, Energy Environ. Sci., 2021, 14, 2639–2669 RSC.
- E. Bontempi, G. P. Sorrentino, A. Zanoletti, I. Alessandri, L. E. Depero and A. Caneschi, Molecules, 2021, 26, 1407 CrossRef CAS PubMed.
- X. F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- J. Liu, D. Zhu, C. Guo, A. Vasileff and S. -Z. Qiao, Adv. Energy Mater., 2017, 7, 1700518 CrossRef.
- Y. C. Lin, P. Y. Liu, E. Velasco, G. Yao, Z. Q. Tian, L. J. Zhang and J. Chen, Adv. Mater., 2019, 31, 1808193 CrossRef PubMed.
- H. Huang, K. Shen, F. Chen and Y. Li, ACS Catal., 2020, 10, 6579–6586 CrossRef CAS.
- L. Jiao and H. L. Jiang, Chem, 2019, 5, 786–804 CAS.
- A. Kumar, S. Ibraheem, T. A. Nguyen, R. K. Gupta, T. Maiyalagan and G. Yasin, Coord. Chem. Rev., 2021, 446, 214122 CrossRef CAS.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005 CrossRef CAS PubMed.
- R. Zhou, M. Jaroniec and S. -Z. Qiao, ChemCatChem, 2015, 7, 3808–3817 CrossRef CAS.
- W. Li, C. Min, F. Tan, Z. Li, B. Zhang, R. Si, M. Xu, W. Liu, L. Zhou, Q. Wei, Y. Zhang and X. Yang, ACS Nano, 2019, 13, 3177–3187 CrossRef CAS PubMed.
- L. Sun, M. G. Campbell and M. Dincă, Angew. Chem., Int. Ed., 2016, 55, 3566–3579 CrossRef CAS PubMed.
- Z. Liang, T. Qiu, S. Gao, R. Zhong and R. Zou, Adv. Energy Mater., 2021, 2003410 Search PubMed.
- Y. Ying, B. Khezri, J. Kosina and M. Pumera, ChemSusChem, 2021, 14, 3402–3412 CrossRef CAS PubMed.
- W. Zheng, C.-S. Tsang, L. Y. S. Lee and K.-Y. Wong, Mater. Today Chem., 2019, 12, 34–60 CrossRef CAS.
- J. Ran, J. Qu, H. Zhang, T. Wen, H. Wang, S. Chen, L. Song, X. Zhang, L. Jing, R. Zheng and S. -Z. Qiao, Adv. Energy Mater., 2019, 9, 1803402 CrossRef.
- J. Duan, S. Chen and C. Zhao, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- E. Miner, T. Fukushima, D. Sheberla, L. Sun, Y. Surendranath and M. Dincă, Nat. Commun., 2016, 7, 10942 CrossRef CAS PubMed.
- H. Zhong, K. H. Ly, M. Wang, Y. Krupskaya, X. Han, J. Zhang, J. Zhang, V. Kataev, B. Büchner, I. M. Weidinger, S. Kaskel, P. Liu, M. Chen, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 10677 CrossRef CAS PubMed.
- L. Huang, X. Zhang, Y. Han, Q. Wang, Y. Fang and S. Dong, J. Mater. Chem. A, 2017, 5, 18610–18617 RSC.
- J. Li, W. Xia, J. Tang, H. Tan, J. Wang, Y. V. Kaneti, Y. Bando, T. Wang, J. He and Y. Yamauchi, Nanoscale Horiz., 2019, 4, 1006–1013 RSC.
- Y. Pan, D. Heryadi, F. Zhou, L. Zhao, G. Lestari, H. Su and Z. Lai, CrystEngComm, 2011, 13, 6937–6940 RSC.
- M. Zhao, Y. Wang, Q. Ma, Y. Huang, X. Zhang, J. Ping, Z. Zhang, Q. Lu, Y. Yu, H. Xu, Y. Zhao and H. Zhang, Adv. Mater., 2015, 27, 7372–7378 CrossRef CAS PubMed.
- A. Schejn, L. Balan, V. Falk, L. Aranda, G. Medjahdi and R. Schneider, CrystEngComm, 2014, 16, 4493–4500 RSC.
- J. Luo, L. F. Ying, F. Zhang, Z. Zhou and Y. G. Zhang, ACS Omega, 2021, 6, 5856–5864 CrossRef CAS PubMed.
- Z. L. Ma, P. X. Liu, Z. Y. Liu, J. J. Wang, L. B. Li and L. Tian, Inorg. Chem., 2021, 60, 6550–6558 CrossRef CAS PubMed.
- A. S. Munn, S. Amabilino, T. W. Stevens, L. M. Daniels, G. J. Clarkson, F. Millange, M. J. Lennox, T. Düren, S. Bourelly, P. L. Llewellyn and R. I. Walton, Cryst. Growth Des., 2015, 15, 891–899 CrossRef CAS.
- H. Zhong, K. Liu, Q. Zhang, F. Meng, D. Bao and X. Zhang, Nano Sel., 2020, 1, 311–319 CrossRef.
- C. Chen, T. Wu, D. Yang, P. Zhang, H. Liu, Y. Yang, Y. Guanying and B. Han, Chem. Commun., 2018, 54, 5984–5987 RSC.
- L. Guo, J. Sun, W. Zhang, L. Hou, L. Liang, Y. Liu and C. Yuan, ChemSusChem, 2019, 12, 5051–5058 CrossRef CAS PubMed.
- P. Lackner, Z. Zou, S. Mayr, U. Diebold and M. Schmid, Phys. Chem. Chem. Phys., 2019, 21, 17613–17620 RSC.
- Q. Ji, L. Bi, J. Zhang, H. Cao and X. S. Zhao, Energy Environ. Sci., 2020, 13, 1408–1428 RSC.
- S. M. You, W. El Rouby, L. Assaud, R. A. Doong and P. Millet, Hydrogen, 2021, 2, 58–75 CrossRef CAS.
- C. O. Ania, M. Seredych, E. Rodriguez-Castellon and T. J. Bandosz, Appl. Catal., B, 2015, 163, 424–435 CrossRef CAS.
- B. You, N. Jiang, M. Sheng, W. S. Drisdell, J. Yano and Y. Sun, ACS Catal., 2015, 5, 7068–7076 CrossRef CAS.
- H. Zhong, K. H. Ly, M. Wang, Y. Krupskaya, X. Han, J. Zhang, J. Zhang, V. Kataev, B. Büchner, I. M. Weidinger, S. Kaskel, P. Liu, M. Chen, R. Dong and X. Feng, Angew. Chem., Int. Ed., 2019, 58, 10677–10682 CrossRef CAS.
- D. Alba-Molina, A. R. P. Santiago, J. J. Giner-Casares, E. Rodríguez-Castellón, M. T. Martín-Romero, L. Camacho, R. Luque and M. Cano, J. Mater. Chem. A, 2019, 7, 20425–20434 RSC.
- Q. Lai, J. Zhu, Y. Zhao, Y. Liang, J. He and J. Chen, Small, 2017, 13, 1700740 CrossRef PubMed.
- F. Santos, J. P. Tafur, J. Abad and A. J. Fernández Romero, J. Electroanal. Chem., 2019, 850, 113380 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Procedures for the synthesis of 2DCIFs, optimization and characterization data, crystal structure determination, stability studies, and additional electrocatalytic data and battery studies. See DOI: https://doi.org/10.1039/d2ta05988f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |