
Approaching a stable oxygen redox reaction in lithium-rich cathode materials: structural perspectives from mechanism to optimization
Zijia
Yin†
a,
He
Zhu†
a,
Yalan
Huang
a,
Dong
Luo
a,
Yang
Ren
a,
Si
Lan
bc and
Qi
Liu
 *acde
*acde
aDepartment of Physics, City University of Hong Kong, Hong Kong 999077, China. E-mail: qiliu63@cityu.edu.hk
bSchool of Materials Science and Engineering, Nanjing University of Science and Technology, Nanjing 210094, China
cShenzhen Research Institute, City University of Hong Kong, Shenzhen 518057, China
dCenter for Neutron Scattering, City University of Hong Kong, Hong Kong 999077, China
eHong Kong Institute for Clean Energy, City University of Hong Kong, Hong Kong 999077, China
First published on 13th July 2022
Abstract
Oxygen redox (OR) chemistry has been an attractive topic in the field of high-energy lithium-ion batteries, as it enables extra storage of charge and boosts the capacity of highly potential layered Li-rich oxide (LLO) cathode materials. However, the OR reaction is usually irreversible during the electrochemical process, inducing severe performance degradation that sets an impenetrable barrier to the LLO applications. Over the last two decades, great efforts have been made to fundamentally understand the irreversibility of OR, finally reaching a consensus that it is deeply rooted in the structural features of LLOs. Although the structural mechanism is complex and still remains to be further clarified, the current findings of the structure–OR coupling have already inspired blooming optimistic expectations from structural perspectives. Herein, we systematically review the recent progress of the OR investigations in LLOs, with a special emphasis on deciphering the structure–OR coupling. Moreover, efficient structural control strategies for promoting the reversibility of OR are also introduced, followed by an outlook on future rational design and development of LLO materials. This comprehensive summary and perspective are expected to be helpful to promote further OR and LLO research.
 Zijia Yin | Zijia YIN is currently a doctoral candidate in the Department of Physics, City University of Hong Kong, supervised by Dr Qi Liu. She received her B.S. degree and M.S. degree from the School of Chemical Engineering, Tianjin University. Her current research interests are structural design and performance optimization of Li-rich cathode materials. |
 He Zhu | Dr He Zhu is currently a postdoctoral fellow in the Department of Physics, City University of Hong Kong. He received his PhD from the Department of Physical Chemistry, University of Science and Technology Beijing, focusing on local structures and related properties of nanomaterials. His current research interests are the relationships between multiple-scale structures and electrochemical response in energy storage materials investigated with synchrotron- and neutron-based techniques. His broader research interests include solid phase transition behaviors and related triggering mechanisms with regard to electrons, phonons and magnetic moments. |
 Qi Liu | Qi Liu is currently an assistant professor in the Department of Physics, City University of Hong Kong. He obtained his PhD from Purdue University in 2014. Before joining CityU, he worked as a postdoctoral fellow at Argonne National Laboratory. His current research interests focus on the structure–property studies of functional materials via multiple neutron- and synchrotron-based techniques. His broader research activities include the design and synthesis of novel energy storage materials, phase transition mechanisms and neutron-/synchrotron physics. |
1. Introduction
Driven by the carbon-neutral pressure on the electric-vehicle (EV) industry, the worldwide demand for rechargeable lithium-ion batteries (LIBs) is becoming greater than ever. However, today's automakers are facing two critical challenges—the insufficient energy density and high price of LIBs, mostly limited by conventional cobalt-dependent cathode materials (e.g., LiCoO2).1 As a better cathode of choice, layered Li-rich oxides (LLOs), xLi2TMO3 (1 − x)LiTMO2 (TM stands for 3d, 4d or 5d transition metals), recording exceptionally high capacity (>250 mA h g−1) have drawn wide attention.2 Along with the superiority with respect to capacity, excluding expansive and less-abundant cobalt can also lower the overall cost and reduce the cobalt-dependency risks in EV manufacturing.3Despite the great prospects, the practical uses of LLOs are hindered by inherent drawbacks such as severe voltage decay, low coulombic efficiency, irreversible capacity loss, and so on, while most of them could be attributed to the irreversible oxygen redox (OR) reaction upon charge–discharge processes.4,5 Typically, the OR chemistry refers to oxidation or reduction of oxygen under high chemical potential (e.g., vs. O2, H2/H+ or AM/AM+, AM = alkali metal).6 For the LLO cathodes in particular, the OR reaction mainly occurs at voltages higher than 4.0 V (vs. Li/Li+), offering substantial capacity but meanwhile bringing about a host of irreversible charge–discharge behaviors. Although many aspects are involved, basically, the key factor that causes the OR irreversibility in the LLOs could trace back to the inherent Li2TMO3 “honeycomb” structure, which is unstable at high cut-off voltages and cannot be fully maintained upon cycling.7 The “honeycomb” structure mentioned here means a special Li/TM ordered distribution in the TM slabs, where Li ions are weakly bonded to the adjacent O to enable either O2−/O2n− or O2−/O2 redox couple during high-voltage cycling (>4.0 V).8 Once O2 gas is released, along with structural degradation and safety threats, the generated oxygen vacancies will weaken the Mn–O bonding, promoting Mn migration to Li layers that terminates with irreversible phase transitions.9 Even oxygen redox occurs between O2− and O−, high-voltage charge loss will promote the reduction of Mn4+ towards Mn3+, and the generated Mn3+ ions not only distort the local octahedral structure, but further weaken the Mn–O bonding and in turn promote Mn migration and oxygen release.10 All these structural changes are closely connected and persistently occur upon prolonged cycling, finally leading to a shallowed Fermi level (EF), lowered operating voltage, and fast capacity drop in the LLO cathodes.
Review articles in recent years have summarized LLOs from the perspectives of reaction mechanism,11 research progress,12,13 advanced characterization techniques,11,14–16 and commercialization challenges.17 The summaries of these predecessors are meaningful and provide important directions for the further improvement of LLO cathode properties. However, there are still limited summaries to systematically interpret the origin and evolution of OR processes in LLOs from a structural perspective. Based on the strong structure–OR coupling, many efforts have been made to reveal fundamental insights into the irreversibility of OR (Fig. 1), followed by blooming-research activities aiming to structurally approach reversible OR chemistry in the LLO-based cathodes.10,18–29 All these great efforts have motivated us to make a summary from structural perspectives. In this review, we will first provide a systematic overview of the recent progress of the structure–OR coupling mechanisms. After addressing the structural principles to the OR reversibility, the efficient structural control strategies for optimizing the LLO cathode materials will also be introduced. Lastly, an outlook on future rational design and development of LLO materials will be presented.
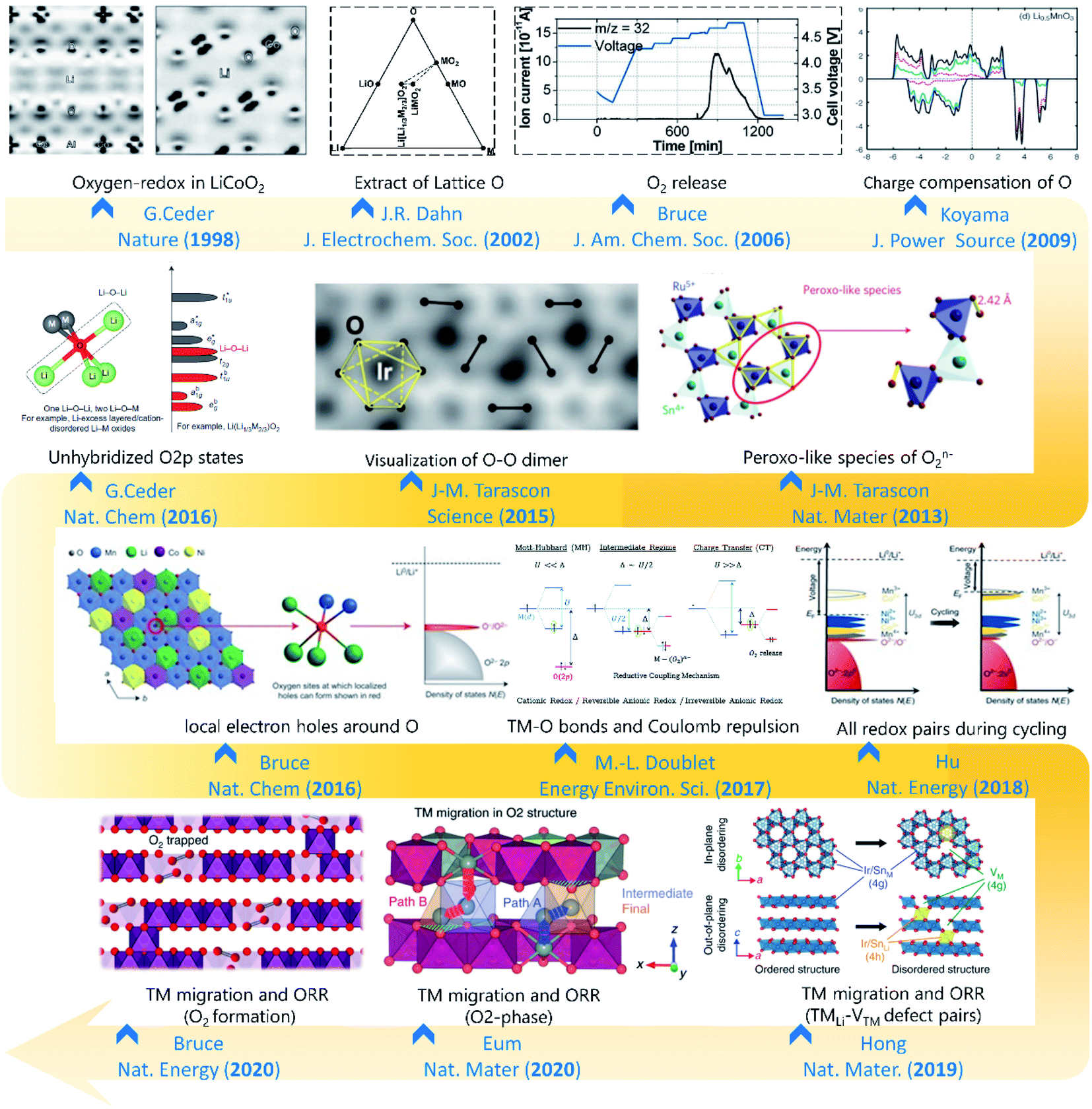 | ||
| Fig. 1 The historical graph of structure investigations on the oxygen redox reaction. Reproduced with permission from ref. 18. Copyright 1998, Springer Nature. Reproduced with permission from ref. 19. Copyright 2002, The electrochemical society. Reproduced with permission from ref. 20. Copyright 2006, the American Chemical Society. Reproduced with permission from ref. 21. Copyright 2009, Elsevier. Reproduced with permission from ref. 22. Copyright 2013, Springer Nature. Reproduced with permission from ref. 23. Copyright 2015, AAAS. Reproduced with permission from ref. 24. Copyright 2016, Springer Nature. Reproduced with permission from ref. 25. Copyright 2016, Springer Nature. Reproduced with permission from ref. 26. Copyright 2017, Royal Society of Chemistry. Reproduced with permission from ref. 10. Copyright 2018, Springer Nature. Reproduced with permission from ref. 27. Copyright 2018, Springer Nature. Reproduced with permission from ref. 28. Copyright 2016, Springer Nature. Reproduced with permission from ref. 29. Copyright 2016, Springer Nature. | ||
2. Structure and mechanisms
2.1 General principles
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) structure, where the TM-O2 octahedra are sandwiched by Li-ion slabs (Fig. 2a). As for the Li2MnO3 component, an ordered substitution of one Li for every three Mn is arranged in the Mn layers, and the space group also is transformed from hexagonal Rm into monoclinic C2/m (Fig. 2b). Viewed along the [001] direction, this Li/Mn ordered distribution seems to be “honeycomb-like”, which is the most distinctive structural feature of the LLO materials (Fig. 2c).30
m) structure, where the TM-O2 octahedra are sandwiched by Li-ion slabs (Fig. 2a). As for the Li2MnO3 component, an ordered substitution of one Li for every three Mn is arranged in the Mn layers, and the space group also is transformed from hexagonal Rm into monoclinic C2/m (Fig. 2b). Viewed along the [001] direction, this Li/Mn ordered distribution seems to be “honeycomb-like”, which is the most distinctive structural feature of the LLO materials (Fig. 2c).30
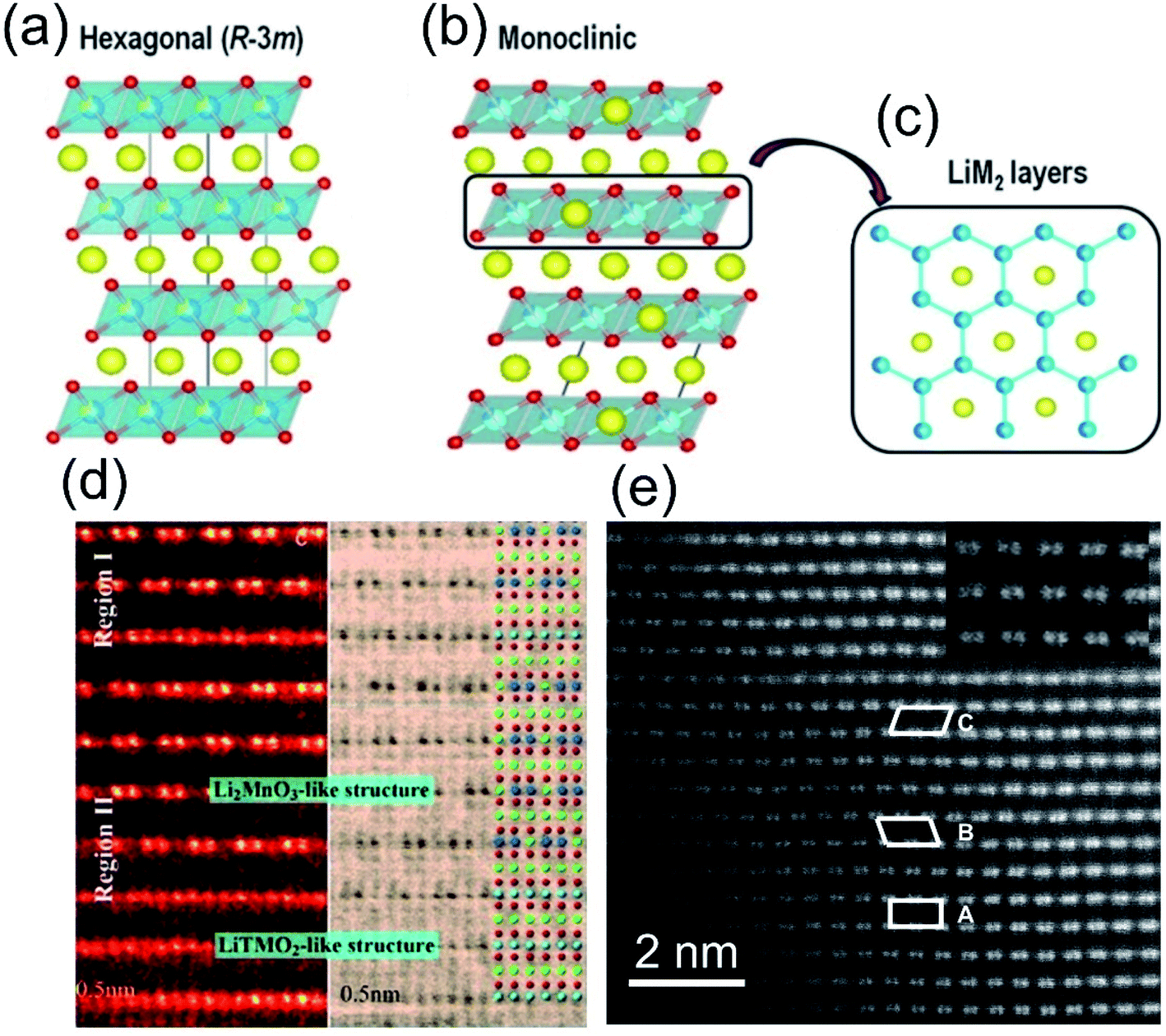 | ||
| Fig. 2 Structural representation of (a) O3-type layered oxides; (b) the overall cell of Li-rich layered oxides described as monoclinic and (c) M/Li ordering within the LiM2 layer leading to a honey-comb pattern. Reproduced with permission from ref. 30. Copyright 2015, the American Chemical Society. (d) HAADF images of the intergrowth two-phase and hetero-interface in the same local region along the [001]rh zone axis direction. Reproduced with permission from ref. 35. Copyright 2013, Wiley-VCH. (e) Aberration-corrected STEM image of a Li[Li0.2Ni0.2Mn0.6]O2 crystal. Reproduced with permission from ref. 36. Copyright 2011, the American Chemical Society. | ||
The unique structure of LLOs is the fundamental reason for triggering the OR reaction. First, the occupation of Li in the TM layer changes the local electron distribution of oxygen and the formation of the Li–O–Li configuration. Since the Li ions residing in the local Li–O–Li configuration are weakly bonded with the adjacent O2p state, the oxygen ions are more inclined to lose electrons in the high-voltage region. The induced oxygen redox behavior supplies extra capacity to the LLOs, but it also leads to irreversible local structural transformation (discussed later). Second, the existence of the LiM6 superstructure also brings about uneven distribution of elements and differences in the local structure. According to the chemical formula of Li2MnO3, the average valence state of the elements in the TM layer is +3. This means that when a +1 valence Li ion exists in the TM layer, in order to keep the average +3 valence state of the TM layer unchanged, two +4 valence Mn are required to be distributed around. But in general, there are at least 4 Mn around Li, so the elements around the superlattice are mostly Li and Mn, while Ni, Co, and Mn elements are mostly distributed in the places without the superlattice.31
In the composite, it is still under debate whether Li2MnO3 and LiTMO2 is solid solution or not. Some of the researches claimed that these two phases are separated in the LLO composite.20,32–34 Yu et al. directly observed a clear two-phase structure through HADDF-STEM characterizations (Fig. 2d).35 In contrast, other researchers observed the uniform solid solution structure of LLOs (Fig. 2e),36 and the experimental results of Bragg peaks were also consistent with Vegard's law described for the solid solution phase.37 It is hard to reach a conclusion, due to the complexity of the LLO structure and the difference in the synthesis process.38,39 Despite the complex composite structure, it is determined that there is a strong synergy between the two-phase structures of Li2MnO3 and LiTMO2, which promotes the redox reaction of the material at high voltages. The excess consumption of lithium ions in the tetrahedral sites in the LiTMO2 structure can be supplemented by the migration of lithium ions in the TM layer of Li2MnO3, thereby alleviating the phase transition at high voltage.40 In turn, LiMO2 can promote the activation process of Li2MnO3.41 Besides, the addition of Ni and Co also suppresses the irreversible migration of Mn and increases the reversibility of the OR process.42
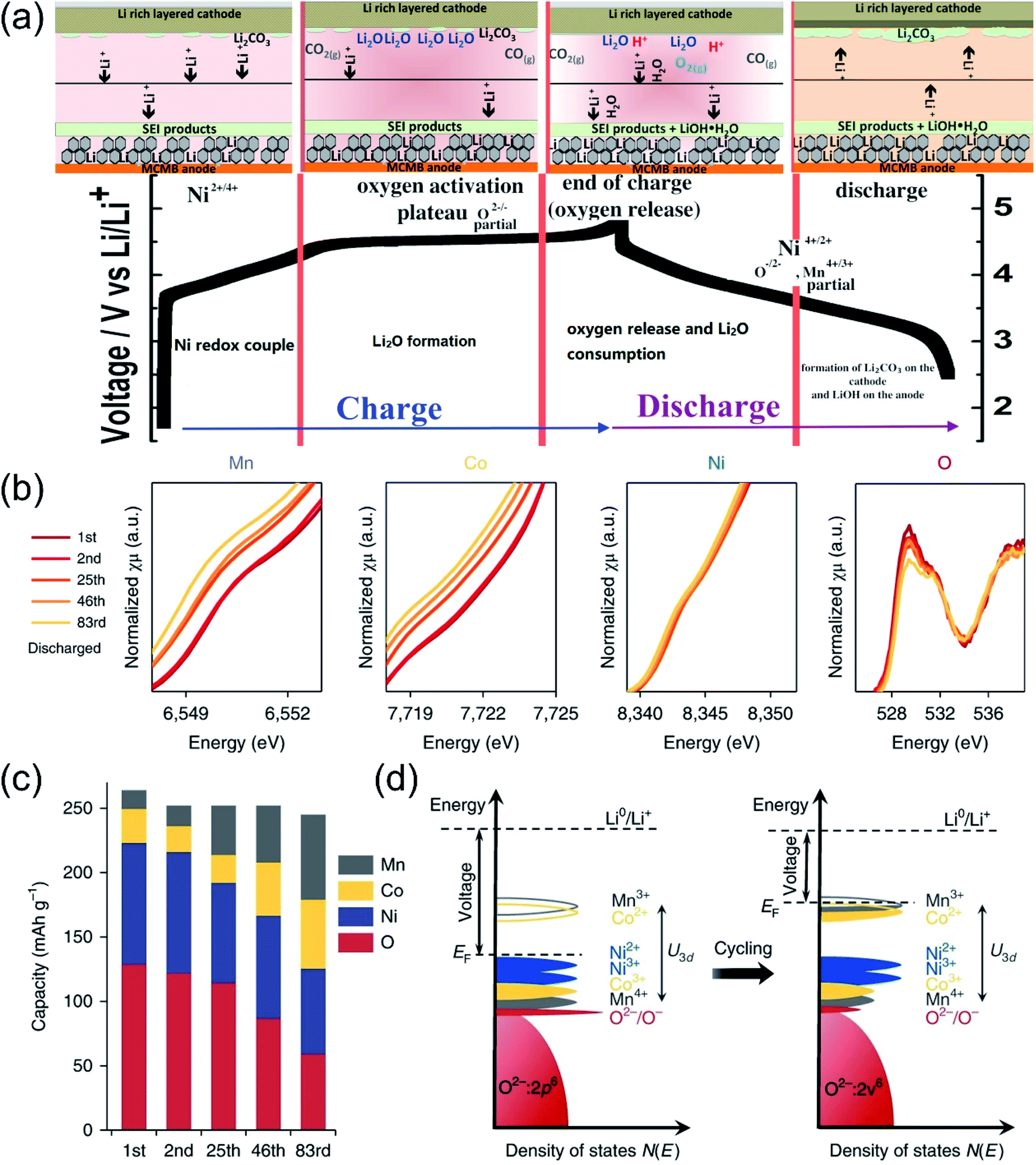 | ||
| Fig. 3 Proposed redox reaction of Li[NixLi(1−2x)/3Mn(2−x)/3]O2 for oxygen activating members during the charging and discharging process. Reproduced with permission from ref. 43. Copyright 2014, the American Chemical Society. (b) K-edge XAS of Mn, Co, Ni and O for Li1.2Ni0.15Co0.1Mn0.55O2 collected after the 1st, 2nd, 25th, 46th and 83rd cycles. For transition metals (Mn, Co and Ni), XAS is collected in the transmission mode; for oxygen, it is collected in the FY mode. (c) The contribution towards the discharge capacity from each element at various cycles. (d) An illustration of the Fermi level being lifted up as a result of electronic structure change. As the voltage is determined by the energy gap between the Fermi level and the Li+/Li0 energy level, it is lowered accordingly. U3d is the on-site coulombic repulsion energy that splits up successive redox potentials. Reproduced with permission from ref. 10. Copyright 2018, Springer Nature. | ||
Hu et al. employed in situ K-edge XAS data of TMs and O to investigate the evolution process of redox couples during cycling based on typical Li1.2Ni0.15Co0.1Mn0.55O2 cathodes.10 As shown in Fig. 3b, with the increase of the cycle numbers, the average valence state of TMs and intensity of the pre-edge peak continued to decrease, indicating the weakening of the hybridization strength between the TMs and oxygen. Besides, semiquantitative analysis of the capacity contributions were carried out and the results are summarized in Fig. 3c. It showed that the capacity contributions of O and Ni gradually decreased as the cycle progressed, while the capacity contribution of Mn and Co increased steadily. It meant that the redox couples were continuously transferred from O/Ni to Mn/Co during the charge–discharge cycles. Fig. 3d illustrates the evolution of the density of states during the cycles. Initially, the Fermi level just lied above the dominant Ni redox couples. Oxygen was gradually lost with each cycle and induced the decrease in the valence state of the TMs. The reduction of Ni induced the reconstruction of the surface to the rock-salt phase.48 Besides, the reduction of Mn and Co also promoted the Fermi level shift to higher positions, resulting in decreased operating voltages. In turn, the reduction of TMs also exacerbated the oxygen loss due to the weakened TM–O covalent bonds.
2.2 Structure–OR coupling mechanism
In order to demystify the structural origin of the OR process, researchers studied the degree of overlap between TM ions and oxygen ion orbits from the perspective of molecular orbital theory, and proposed the anion redox reaction theory.24,49,50 For the layered cathode materials, the redox couple is composed of holes above the Fermi level (EF) and electrons below the EF. The intersections between the d orbitals of the TM and the p orbitals of O will result in the generation of two energy bands: a strong ligand binding state (M–O) and an antibonding state (M–O)* with metallic characteristics.51–53 For traditional cathode materials (not including the anion redox process), the ligand bonding band (M–O) does not provide additional electrons, but only maintains the integrity of the material's crystal structure. The single TM redox process only involves the antibonding band (M–O)* around the Fermi position.24,54–56 However, scientists have also discovered the non-bonded state of oxygen in the band structure of lithium-rich materials. As we know, the energy band of O2− is divided into a pair of 2s bands and three pairs of 2p bands (the bonded state and the anti-bonded state are a pair of each other). Since the 2s energy band is far away from the EF, it can be considered electrochemically inert, while the O 2p energy band with higher energy (closer to the EF) tends to participate in the formation of M–O bonds.52,57 The participation degree of the O 2p energy band is mainly affected by the material structure. For example, in the classic layered oxide LiMO2, the ratio of O/M is 2, and the O electrons of three 2p orbitals are all involved in bonding;22,58 while in the lithium-rich structure Li2MnO3 (O/M is 3), one/third of the lithium ions are present in the TM layer, so in addition to Li–O–M, the oxygen composition environment also has Li–O–Li formation. Since there is no TM orbital available for hybridization, the O 2p orbitals along the Li–O–Li configuration are weakly bonded to the 2s orbitals of lithium.59 Therefore, isolated unhybridized O 2p orbitals will be formed in the Li–O–Li configuration (Fig. 4a–c). Just as the energy level of the t2g state is close to that of the unhybridized dxy/dyz/dxz orbitals of the TM, the energy of this isolated Li–O–Li configuration is very similar to the energy of the unhybridized O 2p orbitals. The energy of the isolated O 2p orbitals is above the bonded state (t1ub, a1gb, egb), but lower than the anti-bonding hybrid state of the TM (eg*, a1g*, t1u*), as shown in Fig. 4d and e.24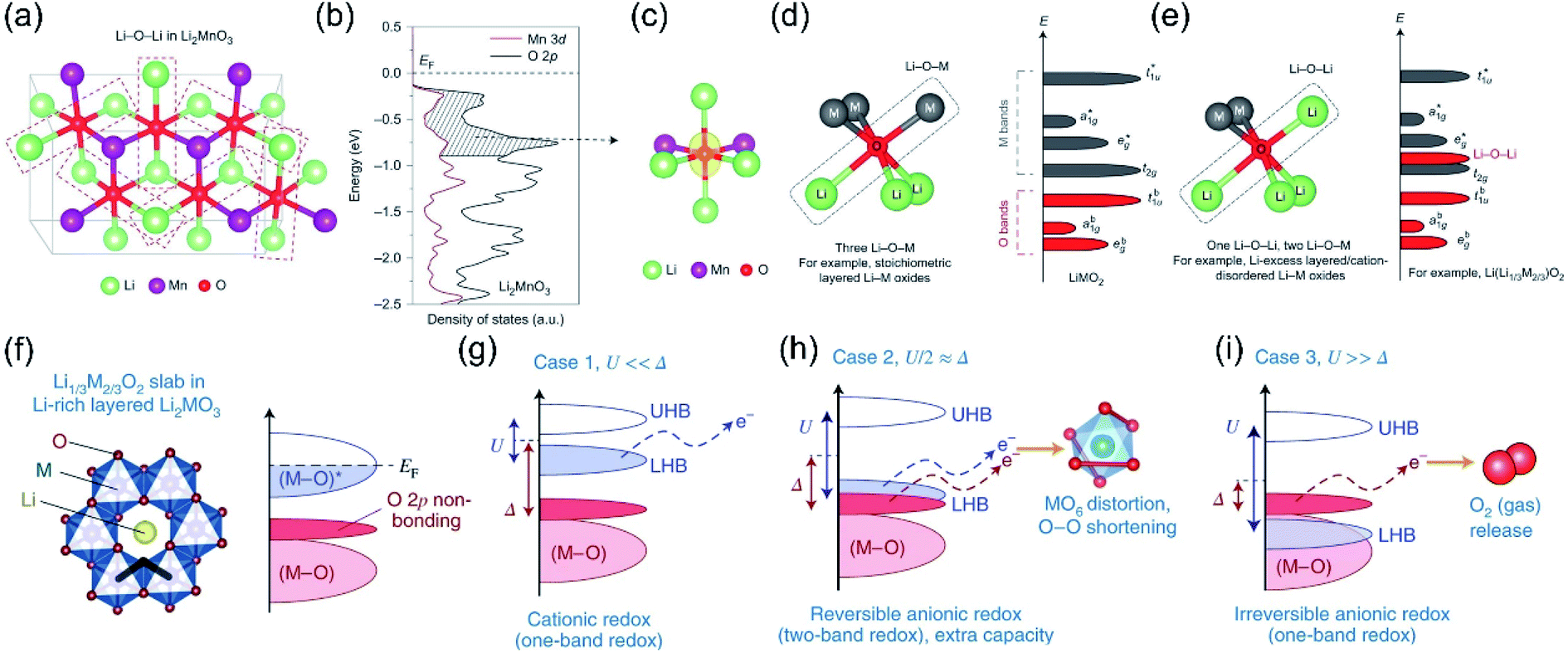 | ||
| Fig. 4 (a) Illustration of Li–O–Li configurations in Li2MnO3. (b) pDOS of the O 2p orbitals (black) and Mn 3d orbitals (red) in Li2MnO3. (c) Isosurface of the charge density (yellow) around oxygen in Li2MnO3, in the energy range of 0 to −0.9 eV. (d) Local atomic coordination around oxygen consisting of three Li–O–M configurations in stoichiometric layered Li metal oxides and band structure. (e) Local atomic coordination around oxygen with one Li–O–Li and two Li–O–M configurations in Li-excess layered or cation-disordered Li–M oxides and the band structure for Li-excess layered Li–M oxides such as Li2MnO3. The Li–O–Li configurations lead to unhybridized O 2p states (Li–O–Li states) whose energies are higher than those of hybridized O 2p states (t1ub, a1gb, egb) and as a result are more easily oxidized. Reproduced with permission from ref. 24. Copyright 2016, Springer Nature. (f) Crystal structures of Li1/3M2/3O2 and the relevant parts of their band structures, thick black lines highlight three M neighbours for each O in LiMO2, compared with only two in the honeycomb-arranged Li2MO3, thus giving rise to O 2p non-bonding states in the latter. Taking Mott–Hubbard splitting into account, the Li2MO3 band structure is further classified under three cases (g–i), depending on the interplay between the d–d Coulomb repulsion term U and the charge transfer term Δ. Reproduced with permission from ref. 8. Copyright 2018, Springer Nature. | ||
It is found that the extent to which nonbonding oxygen participates in the charge compensation is related to the relative position of the antibonding state (M–O)* energy band.60,61 Here, we employ the function of U (d–d Coulomb reciprocity term) and Δ (charge transmission parameter) to express the relative positions of the Hubbard band and O-2p non-bonding energy bands. U characterizes the repulsion of d orbital electrons. The Mott–Hubbard split causes the antibonding state (M–O)* to split into two Hubbard bands. The upper Hubbard band (UHB) is empty, and the lower Hubbard band (LHB) is fully occupied.62,63 The U parameter represents the energy difference between the two Hubbard bands, while Δ is the charge transmission parameter, which is the energy dissimilarity of the bonding state (M–O) and the antibonding state (M–O)*. The value of Δ is related to the difference between the electronegativity of M and O, reflecting the strength (ionicity or covalent) of the M–O bond.60,64 Therefore, according to the relationship between U and Δ, the relative positions of LHB and O-2p nonbonding bands can be expressed as three situations (Fig. 4f).8 Case 1: U ≪ Δ, this situation is a classic single-energy band cation redox reaction. The strong ion M–O bonding makes LHB much higher than the O 2p nonbonding band and closer to the EF, from where the electronic structure changes (Fig. 4g). Case 2: U ≫ Δ, the O-2p nonbonding band is located above the LHB, closer to the EF. When this type of material is charged, electrons will be extracted from the O 2p bonds, and the partially nonbonding O 2p electrons are easily converted into the boosted reactive O2n− groups. Then the O2n− groups will further react with the electrolyte to regain electrons, which are separated from the original coordination environment and released in the form of oxygen gas or carbon dioxide. This lattice oxygen release process is irreversible, which leads to partial irreversibility of the oxygen redox reactions in some lithium-rich materials (Fig. 4i). The process is irreversible, which leads to partial irreversibility of the oxygen redox reactions in lithium-rich materials. Case 3: U/2 ≈ Δ, this special case leads to the overlap of the LHB and the nonbonding O 2p energy bands, which indicates that both energy bands can participate in the charge compensation process and achieve dual-band redox to obtain additional capacity (Fig. 4h).
The extra OR capacity contribution has been generally accepted, which mainly originated from the two redox couples: O2−/O2 or O2−/O2n−. The evolution process of O2−/O2 was usually accompanied by the serious capacity and phase transition problems of lithium-rich materials. Typically, Armstrong et al. conducted in situ differential electrochemical mass spectrometry (DEMS) experiments and found the oxygen release of the Li[Ni0.2Li0.2Mn0.6]O2 material when it was initially charged to 4.5 V.20 Koyama et al. also confirmed that the 4.6 V platform corresponded to the O 2p oxidation process through first-principles calculations.21 Besides, Armstrong et al.65–67 proposed that O 2p bonds in the Li2MnO3 component were oxidized (O2− precipitated from the material lattice) and combined with the liberated Li+, and finally released from the electrode in the form of Li2O. At the same time, the TM on the surface will migrate from the surface to the vacancies of the Li+ in the bulk phase. This process induces the extracted lithium ions to be unable to re-intercalate, causing the first irreversible capacity loss of lithium-rich materials. The research by Quine et al. confirmed that the release of Li2O from the surface of the Li[NixLi(1−2x)/3Mn(2−x)/3]O2 material will induce the generation of TM vacancies in the bulk materials.68 Weill et al. conducted the electron diffraction experiments of lithium-rich materials and demonstrated that the precipitation of oxygen induces the rearrangement of the material structure during the first charge and discharge process. The material changes from a layered O3 type structure to a new layered MO2 type structure.69 As for the charge compensation behavior of the material in the discharge process, Yabuuchi et al. deeply studied it based on Li1.2Mn0.54Ni0.13Co0.13O2 materials through X-ray diffraction, X-ray absorption spectroscopy, secondary ion mass spectrometry, and X-ray photoelectron spectroscopy characterizations.47 They believed that during the discharge process, the Ni4+, Co3+ and Mn4+ reduction reaction happens in the bulk phase of the lithium-rich materials (Fig. 5a), and the O2 reduction reaction occurs on the surface of the material (Fig. 5b). First, Ni4+ and Co3+ are reduced to Ni2+ and Co2+, and then Mn4+ is reduced to Mn3+ when the discharge voltage is lower than 3.5 V. Next, when the discharge voltage is lower than 3.0 V, surface absorbed O2 will be reduced to O2− on the surface of the LLO material. Since the interface is in contact with the electrolyte, a layer of lithium carbonate will be formed on the surface of the electrode material.
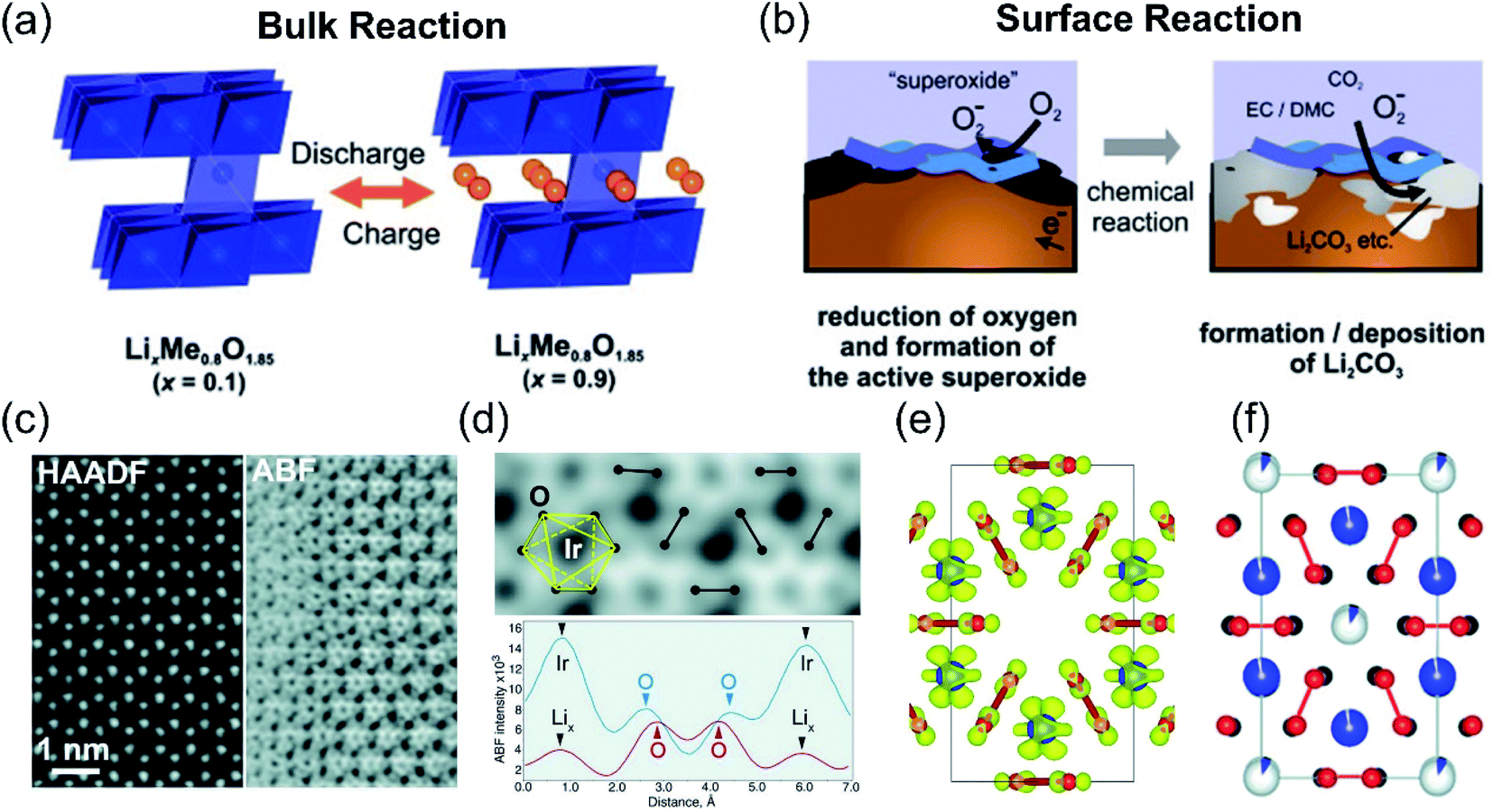 | ||
| Fig. 5 Proposed reaction mechanisms of (a) bulk particles and (b) surface reactions in the LixNi0.13Co0.13Mn0.54O2−δ composite electrodes. Reproduced with permission from ref. 47. Copyright 2011, the American Chemical Society. (c) [001] HAADF-STEM and ABF-STEM images of the charged Li0.5IrO3 sample. (d) Enlarged ABF-STEM image and ABF intensity profiles along the O–O pairs with long (blue) and short (red) projected distances. O–O pairs with short projected distances are marked with dumbbells. (e) [001] projection of the Li0.5IrO3 in the O1 stacking configuration, obtained with DFT calculations. Li atoms are omitted for clarity, oxygen atoms are shown in red, and Ir atoms are in blue. The yellow surfaces are the Fukui orbitals. (f) Structure of the charged Li–Ir–O material, as obtained from neutron powder diffraction. Reproduced with permission from ref. 23. Copyright 2015, AAAS. | ||
In addition to the O2−/O2 redox couple, the evolution of the O2n− peroxide formation was also confirmed by several research experiments. In 2003, Hong et al. found that the weight loss of the lithium-rich electrode during the first charging process was not consistent with the amount of oxygen released.70 Therefore, the entire process cannot be simply explained by the oxygen loss reaction. Subsequently, Koga et al. proposed a new mechanism that the surface and bulk oxygen undergo different redox reactions.71,72 They believed that the surface lattice oxygen is oxidized to molecular oxygen accompanied by the inside TM migration, while the oxygen in the bulk undergoes a reversible redox reaction (2O2−/O22−), In this process, there is no oxygen release and structural transformation. Moreover, Oishi and Han et al. further gave direct evidence for the formation of peroxide-like compounds during the charging process through XAS characterization techniques.73,74 Tarascon et al. used ex situ X-ray photoelectron spectroscopy to study the Li2Ru1−ySnyO3 material system. They found that the charge compensation behavior of lattice oxygen is mainly involved in the formation of peroxo-like O2n− radicals.22 Peroxide-like radicals (O2n−) can exist stably in the materials and can be reversibly converted into O2− during the charge and discharge process, so that the redox reaction of lattice oxygen can be reversibly realized. Moreover, O–O peroxo-like dimers were further directly observed in the bulk phase of Li2IrO3 materials through neutron powder diffraction and spherical aberration transmission electron microscopy (Fig. 5c–f). The experimental results confirm that the peroxy-like (O2n−) charge compensation mechanism not only occurs on the surface, but also occurs in the bulk of the lithium-rich cathode materials.23 In addition, a TM-driven reduction coupling mechanism (RCM) related to the O2n− evolution theory was also proposed to realize the invertible anion redox process. As we know, the gradual extraction of electrons induces the formation of an unstable EF. To avoid this instability, if the metastable oxygen vacancies formed during the oxidation process can be stabilized in the form of O2n− peroxides and are connected to the TM in the form of covalent bonds, then the process will be reversible.75,76 The reversibility of the oxygen redox process could be improved through Jahn-Teller or Peierls distortions. These distortions include O network reorganization and the reduction of symmetry to shorten O–O distances, which is conducive to the stability of TM–O2n− interactions. In general, scientific researchers need to adjust U and Δ by selecting a suitable TM-anion combination to meet the condition of U/2 ≈ Δ, and then achieve the reversible additional capacity.
3. Structural evolution related to the OR reaction
In LLOs, reversible and irreversible anion redox reactions occur simultaneously. The anionic redox reaction in the bulk is generally reversible, which is the origin of the excess discharge specific capacity, while the irreversible OR reaction (especially occurring on the surface) is the key to the instability of the LLO structure, capacity and voltage decay. The OR process is not isolated, it involves the structural evolution of the entire electrode. The OR process needs to be accompanied by TM migration to stabilize the structure. If the back-insertion of the TM ions is thermodynamically unfavorable, the TM ions are permanently trapped at the new site, resulting in a drop in voltage. On the other hand, TM migration also promotes the formation of O vacancies and induces the generation of O2. Meanwhile, the O vacancies also promote irreversible TM migration, allowing TM ions to accumulate on the surface and in the bulk phase, inducing spinel phase transitions. The excessive O vacancy concentration can also lead to crystal shrinkage. The local spinel transformation and lattice shrinkage will diffuse throughout the crystal over long cycles, resulting in particle fracture. These structural evolutions all have a significant impact on the OR process. Therefore, in this section, we introduce several structural evolutions accompanying the OR reaction and explore the relationship of their coupling and mutual promotion.3.1 Transition metal migration
The inevitable migration process of TM ions during charging and discharging in Li-rich materials has been widely accepted. However, the TM ion migration mechanism is complex and affected by various factors such as the OR reaction, lattice O species, O vacancies, and Li+ migration. Therefore, it is difficult to establish a comprehensive understanding of the relationship between TM ion migration and structural dynamic evolutions. Among them, the OR reaction has the most critical impact on TM migration.30,77 Here, we focus on the dynamic coupling relationship between the OR reaction and TM migration, and the effects of the irreversible TM migration on the OR.The intrinsic link between the redox pairs of lattice O and structural evolution has been neglected by researchers for a long time. Therefore, Gent et al. explored the coupling mechanism of the OR reaction and TM migration in detail.9 As shown in Fig. 6a, the redox order of the TM and O is reversed during the charge–discharge process, implying that there is no O 2p orbital participation during the discharge process. This phenomenon can be explained by the structural evolution of the LLO when it is charged above 4.6 V. Hence, they calculated the dynamic changes of the O coordination structure and electronic structure through the TM ion migration model. The migration of TM to the Li layer will induce the oxygen electronic state moving to a higher energy level, resulting in a significant reduction of the nonbonding O 2p electronic state, which illustrates that TM migration and the OR reaction are dynamically coupled (Fig. 6b). Subsequently, in the study based on Li2IrO3 (LIO) lithium-rich model compounds, Hong et al. further elucidated the strong coupling relationship between TM migration and the OR reaction.27 LIO model compounds were generally considered to exhibit a stable OR reaction and reversible structural evolution due to their abundant Li–O–Li structures, whereas in practice, no anionic redox behavior is observed even when 4.6 V is reached. Instead, a highly hybridized Ir–O redox reaction is involved. sXAS and DOS revealed that although LIO formed a 2.5 Å O–O dimer at high voltage, the OR reaction still did not occur due to the highly covalent Ir–O state and low redox potential (Fig. 6c–e). After doping with redox-inert Sn, the formed Li2−xIr1−ySnyO3 (LISO) electrode can stimulate the OR reaction (Fig. 6f). Compared with the former strong covalent behavior of Ir–O, Sn is more likely to migrate to the lithium layer at high voltage, which makes O decoordinate from a single covalent TM ligand and forms SnLi–VSn anti-vacancy defects (Fig. 6g). During the OR reaction of LSIO, this defect pair induces the formation of a metastable 1.8 Å M–O π bond and a 1.4 Å O–O dimer structure. Meanwhile, the Ir![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–O dimers realize ligand–metal electron transfer from O to Ir and facilitate the migration of TM ions. This theory proposes that the O electronic state and structural evolution of LSIR electrodes arise from the synergistic effect of the OR reaction and TM ion migration. Recently, House et al. reported a coupling mechanism of in-plane transition metal migration and O2 formation in the bulk.29 They found that the formation of O2 occurs not only at the surface, but also in the entire bulk phase. Furthermore, density functional theory (DFT) results confirmed that Li+ vacancies in the TM layer and vacancy clusters formed by in-plane TM migration lead to the formation of O2 in the bulk during charging. O2 will be confined in the vacancy clusters and then re-reduced to O2− with the back-insertion of Li+ during discharging. However, the back-intercalated Li+ cannot return to the original position, resulting in the inability of the reduced O2− to coordinate with the TM, further causing the voltage hysteresis.
O and O–O dimers realize ligand–metal electron transfer from O to Ir and facilitate the migration of TM ions. This theory proposes that the O electronic state and structural evolution of LSIR electrodes arise from the synergistic effect of the OR reaction and TM ion migration. Recently, House et al. reported a coupling mechanism of in-plane transition metal migration and O2 formation in the bulk.29 They found that the formation of O2 occurs not only at the surface, but also in the entire bulk phase. Furthermore, density functional theory (DFT) results confirmed that Li+ vacancies in the TM layer and vacancy clusters formed by in-plane TM migration lead to the formation of O2 in the bulk during charging. O2 will be confined in the vacancy clusters and then re-reduced to O2− with the back-insertion of Li+ during discharging. However, the back-intercalated Li+ cannot return to the original position, resulting in the inability of the reduced O2− to coordinate with the TM, further causing the voltage hysteresis.
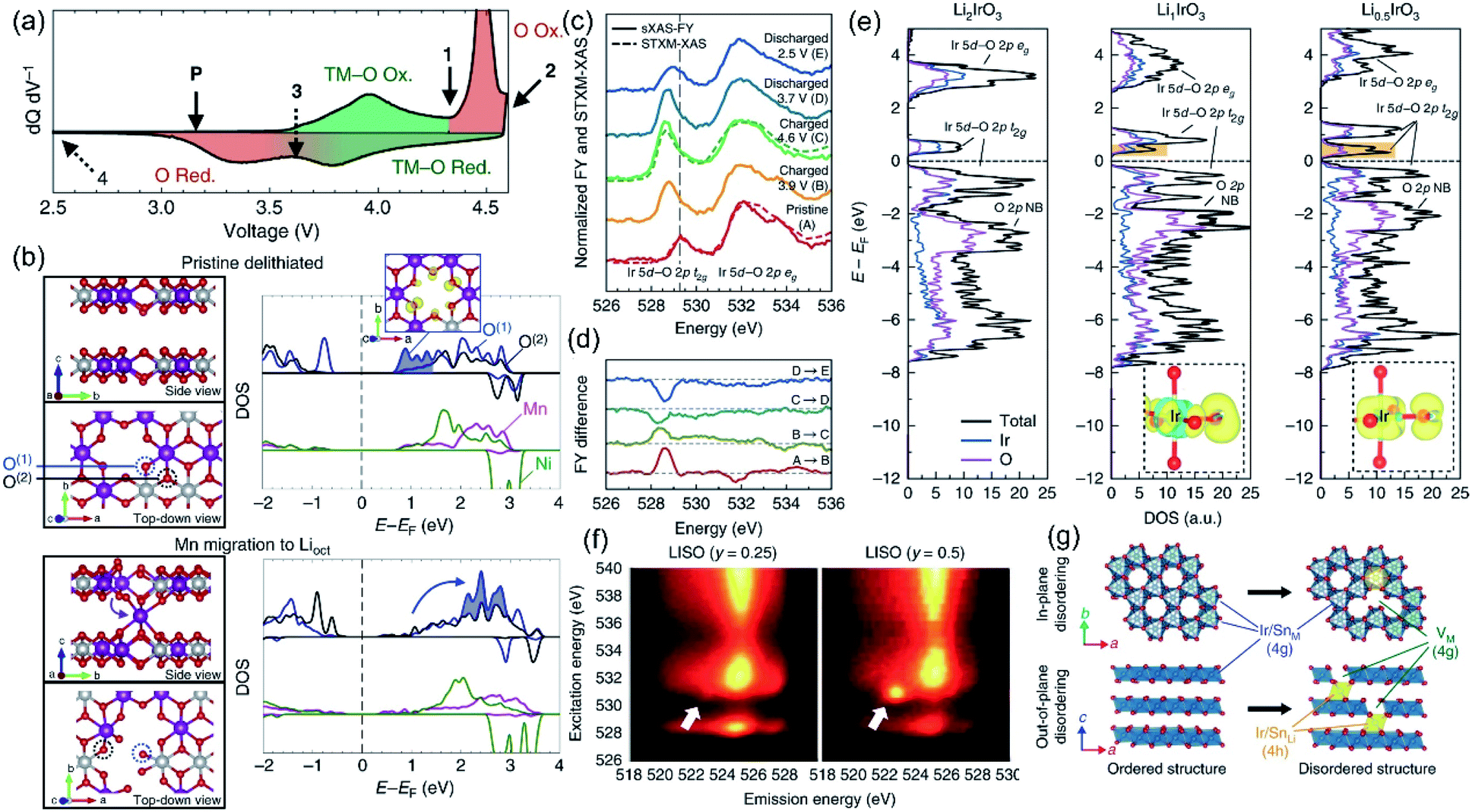 | ||
| Fig. 6 (a) dQ dV−1 of the first cycle showing the voltages at 4.35 V (1), 4.60 V (2), 3.65 V (3), and 2.00 V (4). Regions of the dQ dV−1 are shaded to show the hysteresis in the O redox relative to the TM–O redox. (b) pDOS for the TMs and the two-coordinate (O(1), blue) and three-coordinate (O(2), black) oxygen environments in the pristine delithiated state (top), and after Mn (middle) and Ni (bottom) migration into octahedral sites in the Li layer. The integrated charge density for the lowest unoccupied states in the pristine delithiated structure (blue shaded area of the pristine DOS) is shown in the top right inset. Schematics of each supercell used to generate the pDOS are shown to the left, with the plotted O(1) and O(2) oxygen environments circled. Reproduced with permission from ref. 9. Copyright 2017, Springer Nature. (c) sXAS-FY spectra and STXM-XAS of the O K edge of Li2−xIrO3 at various voltages throughout the first cycle. (d) Difference plot of sXAS obtained from (c) showing the intensity evolution of Ir 5d–O 2p t2g and eg* peaks. (e) Ir- and O-projected density of states of Li2IrO3, Li1IrO3 and Li0.5IrO3 calculated from first principles. Yellow and blue show negative and positive changes in charge density, respectively. Li ions are omitted for clarity. (f) O K-edge RIXS maps of LISO25 and LISO50 charged to 4.60 V showing a localized RIXS feature at 530.7 eV excitation energy and 522.8 eV emission energy. (g) In-plane and out-of-plane disorder quantified by iterative XRD Rietveld refinement. Reproduced with permission from ref. 27. Copyright 2019, Springer Nature. | ||
Several other mechanisms of TM ion migration have also been reported. Using theoretical calculations, Eum et al. revealed that the migration of TM to octahedral sites in the Li layer is thermodynamically favorable during charging, stating that the TM migration process occurs easily.28 Besides, Chen et al. used theoretical calculations to find that O22− and O2− in the lattice would promote the Mn migration to empty Li octahedral sites in the lithium layer.46 In addition, Fell et al. revealed that the formation of oxygen vacancies also has a promotion effect on TM migration.79 In short, these coupling relationships reveal that TM ion migration is a prerequisite for the occurrence of the OR reaction. In other words, the oxygen redox in Li-rich materials must be accompanied by the migration of TM. Therefore, the corresponding modification strategies should not be limited to the inhibition of TM migration, but devoted to the reversible back-intercalation of TMs.28,78
Since TM migration is highly correlated with the OR process, irreversible TM migration will cause permanent structural evolution and lead to an irreversible OR process. It is mainly attributed to the complexity of the TM migration path during cycling. Regardless of whether the TM migrates in-plane or towards the Li layer, the coordination environment of TM ions will be changed during discharging (e.g., Li back-insertion and O loss), which hinders TM ion back-migration.80–82 Hence, the accumulation of irreversibly migrated TM ions will cause O2 release and phase transition, resulting in significant voltage and capacity fading.83 Some reports verified this process. Adrien et al. found that because of the unsaturated TM-O ligands on the surface, the TM ions will migrate into the layer inducing the loss of the lattice O.84 Upon cycling, a densified spinel phase gradually formed on the surface. In addition, some studies reported that the formation of the spinel phase and O2 also occurs in the whole bulk phase. TM ions will permanently migrate and accumulate in the octahedral sites of the Li layer during cycling, resulting in the formation of spinel phases.85 Meanwhile, TM accumulation also forms hole clusters, and the mismatched lattice O is directly reduced to O2 in the hole clusters.29 Once the spinel phase and O2 are formed, the Li-rich layered structure and the honeycomb superstructure are destroyed, implying an irreversible OR process. This transition of the electrode from a high-energy configuration to a low-energy configuration resulted in a sustained capacity and voltage decay.86
3.2 Oxygen defects
The formation of O vacancies is considered to be an important feature of the irreversible OR reaction. O vacancies exacerbate unfavorable processes such as the irreversible OR reaction, TM migration, O release and phase transition. Exploring the O vacancy mechanism is an effective way to realize the reversible OR reaction and alleviate the voltage and capacity fading. Therefore, here we summarize the related reports on the origin of O vacancy formation and its subsequent structural evolution.Yabuuchi et al. found that Li and O-containing species were simultaneously extracted from the lattice throughout the charging process.47 Meanwhile, SXRD and X-ray adsorption near-edge structure (XANES) showed that cation rearrangement and TM ion migration also occurred simultaneously. Afterward, Fell et al. confirmed the formation of O vacancies in the crystals by electron energy loss spectroscopy (EELS) and SXRD, and the O vacancies may further promote cation migration and phase transition.79 Furthermore, they proposed the coupling mechanism of TM ion migration and O vacancy through theoretical calculations: the formation of O vacancies on the surface would lead to an unstable TM–O unsaturated coordination structure; then the unstable TM ions would gradually migrate to the interior of the structure and form the spinel phase.87 In addition, Okamoto et al. found that the content of O vacancies increases proportionally with the extraction amount of Li through DFT calculations, because when the extraction amount of Li is higher, the formation of O vacancies is more favorable thermodynamically.88 However, an excessively high density of O vacancies induces volume shrinkage and severe structural deformation of the crystal. Correspondingly, Lee et al. found that excessive Li and O vacancies can cause rearrangement of TM ions and induce phase transition.89 Besides, Chen et al. used DFT to establish the link between the OR reaction and O holes.46 The results indicated that the unstable O hole structure formed O dimers, which induced the formation of O2 molecules and the migration of Mn. Subsequently, Shim et al. found that O vacancies were related to the formation of O2 by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy dispersive spectroscopy (EDS), and the unstable Mn valence state caused by oxygen vacancies caused irreversible capacity decline.90 Furthermore, Hu et al. believed that O vacancies were responsible for the formation of the pore structure in the particles, which accelerated the voltage decay.10 Subsequently, Yan et al. confirmed this process by STEM and theoretical calculations.52 They found that the OR reaction promoted the formation of O vacancies and the diffusion of oxidized oxygen species, resulting in the gradual diffusion of O vacancies into the bulk phase, which eventually led to the formation of nanopores.
3.3 Evolution of lattice parameters
The charge–discharge process of LLO is accompanied by the TM and oxygen redox, the irreversible lattice oxygen release, and the Li de-intercalation process, resulting in the crystal structure evolution and change in lattice parameters. Especially in the high voltage region (>4.5 V), the OR reaction is inseparably related to the change of the unit cell parameters. In situ X-ray diffraction is a powerful characterization method to reflect the crystal structure evolution and the OR reaction behaviors during the charge/discharge process.91,92 Mohanty et al. explored the structural transformation of the Li1.2Co0.1Mn0.55Ni0.15O2 cathode during cycling through in situ XRD characterization (Fig. 7a).93 During the charging process, in the region of 2.4–4.4 V, the LiMO2 structure mainly contributed to the capacity. During this process, the diffraction peak of the (003) crystal plane shifted to a low angle, accompanied by the Li+ extraction and TM oxidation reactions. When the Li+ was extracted from the Li layer in the crystal structure, the two adjacent O layers generated electrostatic repulsion, which increased the crystal plane spacing and the cell parameter c increased (Fig. 7c and d), while the ionic radius of TM ions decreased due to the oxidation reaction, resulting in the shortening of the TM–O bond and the shrinkage of the a and b axes in the crystal cell.94 In the region of 4.4–4.6 V, the cell parameters a and c were not changed significantly, indicating that there was no change in the valence state of the TM, which proved the redox mechanism of lattice oxygen. In the region of 4.6–4.8 V, the diffraction peak of the (003) crystal plane shifted to a high angle, accompanied by the TM migration and excess Li+ extraction from the TM layer. Simultaneously, the OR reaction and irreversible oxygen release occur, leading to a decrease in electrostatic repulsion between adjacent O layers and c-cell parameters (Fig. 7c and d), while the a-cell parameter was not changed during this process, indicating that no TM redox was involved. During the discharge process, the ionic radius of TM ions increased continuously due to the redox process, increasing the a-parameter. Meanwhile, the re-insertion of Li-ion initially caused the expansion of the c-axis, whereas, with the decrease of the TM valence state, the distance between adjacent oxygen layers became smaller, resulting in a decrease of the c-cell parameter. Besides, Lu et al. attributed the anomalous c-parameter change on the plateau to the redox reaction of lattice oxygen and the extreme lithium-ion extraction.19 With further cycles, the (004) crystal facet of the spinel structure gradually appeared (Fig. 7b), which confirmed the gradual transformation of the layered structure to the spinel phase during the cycling. The coincident formation of the spinel phase led to the voltage decay during the cycling of the cathode material. Similar structural evolutions have also been observed by in situ XRD in other research papers.95,96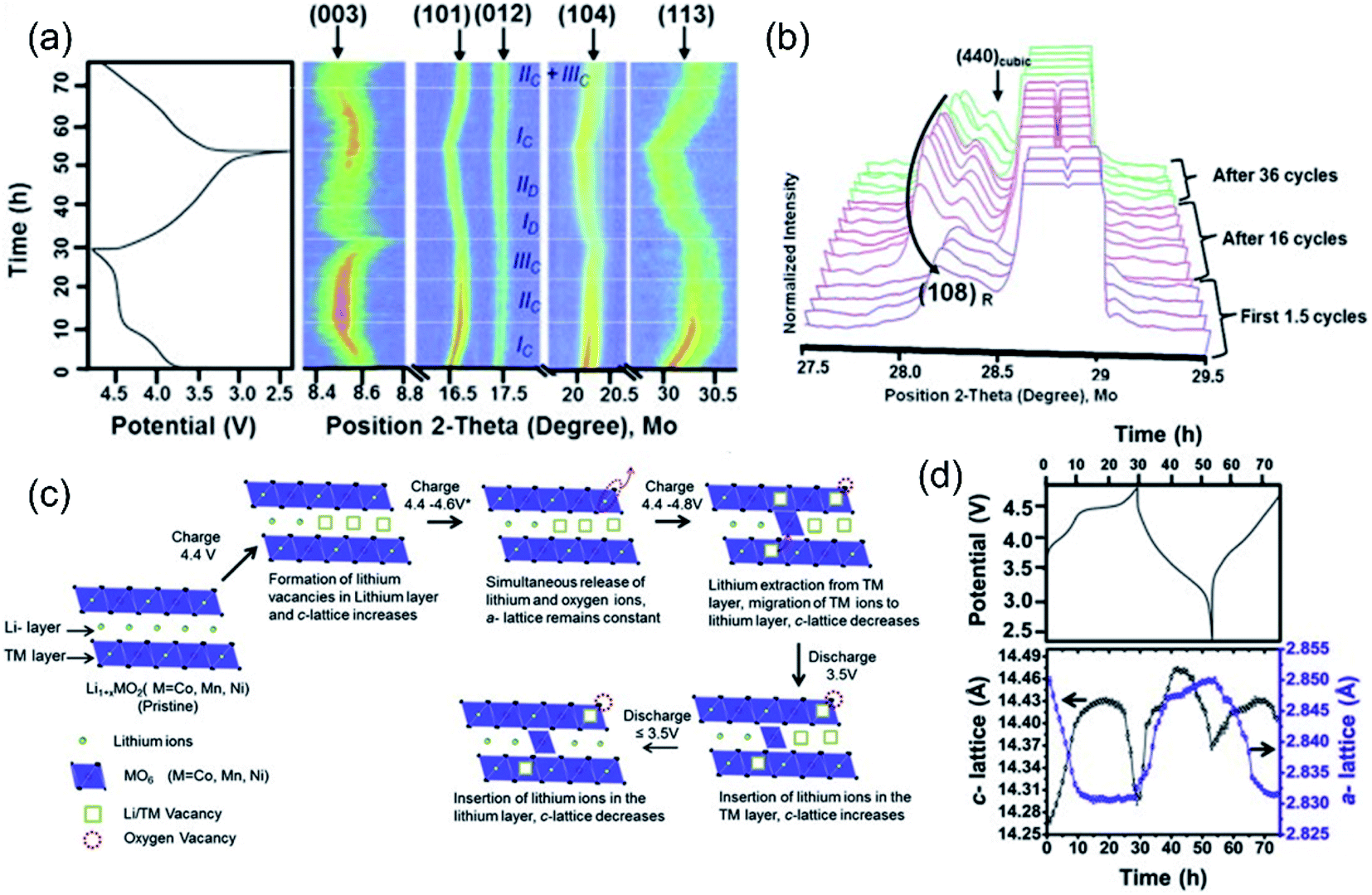 | ||
| Fig. 7 (a) Intensity plots of (003), (101), (012), (104), and (113) peaks along with the electrochemical profile of the cell during the first 1.5 cycles. (b) Change in lattice parameters (with error bars) as a function of the electrochemical charge/discharge profile. (c) Schematic of change in the crystal structure of Li1.2Co0.1Mn0.55Ni0.15O2 in the course of the charge/discharge process. (d) Normalized intensity XRD patterns of the Li1.2Co0.1Mn0.55Ni0.15O2 cathode during the first 1.5 cycles, after 16 cycles and after 36 cycles. The patterns were selected in the region of 3.5–2.4 V discharge. (e) Galvanostatic charge/discharge profiles of the lithium rich NMC cathode. Reproduced with permission from ref. 89. Copyright 2013, Elsevier. | ||
3.4 Phase transformation
The structural phase transformation of LLO due to the TM migration during cycling is closely related to the participation of lattice oxygen redox in charge compensation reactions.4,97,98 The irreversible OR reaction induces the release of lattice oxygen, which weakens the binding force of TMs.84,99 During the charging and delithiation process, the migration energy barrier of manganese ions decreases owing to the continuous formation of lithium and oxygen vacancies, and manganese ions tend to migrate to the lithium layer to form a defect-like spinel structure.52,100 In addition, the release of oxygen will continuously promote the valence reduction of TM and further induce interfacial reactions and surface TM dissolution.101 Meanwhile, the irreversible re-intercalation of TM ions in the Li layer during the discharge process also aggravates the LLO to spinel phase transition.81,102 The synergistic contribution of the above-mentioned inducements makes the TM migration and structural phase transformation more likely to occur.103 Upon cycling, the lattice oxygen release and the TM migration continue to intensify, and the spinel phase will continue to extend from the surface to the interior of the material, resulting in the permanent phase transition from the layered to the spinel phase.84,104 Xiao et al. explored the reaction mechanism between TMs and lattice oxygen through XAS and in situ electrochemical tests. The results confirmed that lattice oxygen participated in the reaction and contributed a large amount of lithium storage capacity. However, the lattice oxygen was unstable and can be extracted at high voltage, causing the unstable TM to migrate from the octahedral site to the nearby tetrahedral Li site, resulting in the generation of the spinel phase.105 Besides, Gu et al. performed STEM imaging on the LLO single particles after cycling, and intuitively observed spinel-like phase regions with different orientations on the surface (Fig. 8a).106 During cycling, the spinel phase was formed due to the gradual migration of TM ions from the TM layer to the Li sites of the Li layer and the movement of Li ions to the tetrahedral sites. The transformation from Rm to the spinel phase tended to form the mosaic spinel grain without disrupting the lattice structure, whereas the transformation of the C2/m phase to the spinel phase involved the removal of lithium ions and oxygen from the lattice, which induced large strains and resulted in lattice fracture of the bulk particles (Fig. 8b). Moreover, Zheng et al. explored the phase transformation process from the layered to the spinel phase through the HAADF-STEM characterization methods.107 They found that the layered material first transformed from the C2/m phase into the Rm phase, and then transformed into the LT-LiCoO2-like defect spinel structure (Fig. 8c). It was also confirmed that the layer–spinel structural transformation starts at the surface of the material and then gradually permeates into the interior structure. During cycling, the LT-LiCoO2-like spinel phase was further converted to a disordered rock-salt [110] structure. In addition, Hong et al. also provided evidence for the transformation of LLO from a layered phase into a spinel-like phase through Raman spectroscopy.45 They proposed that the layered phase to spinel-like phase transition conformed to the solid-phase nucleation growth mechanism, and the spinel-like phase mainly evolved from the surface to the interior. Therefore, it is highly necessary to evolve from surface protection and defense structure to the bulk in lithium-rich materials.108,109
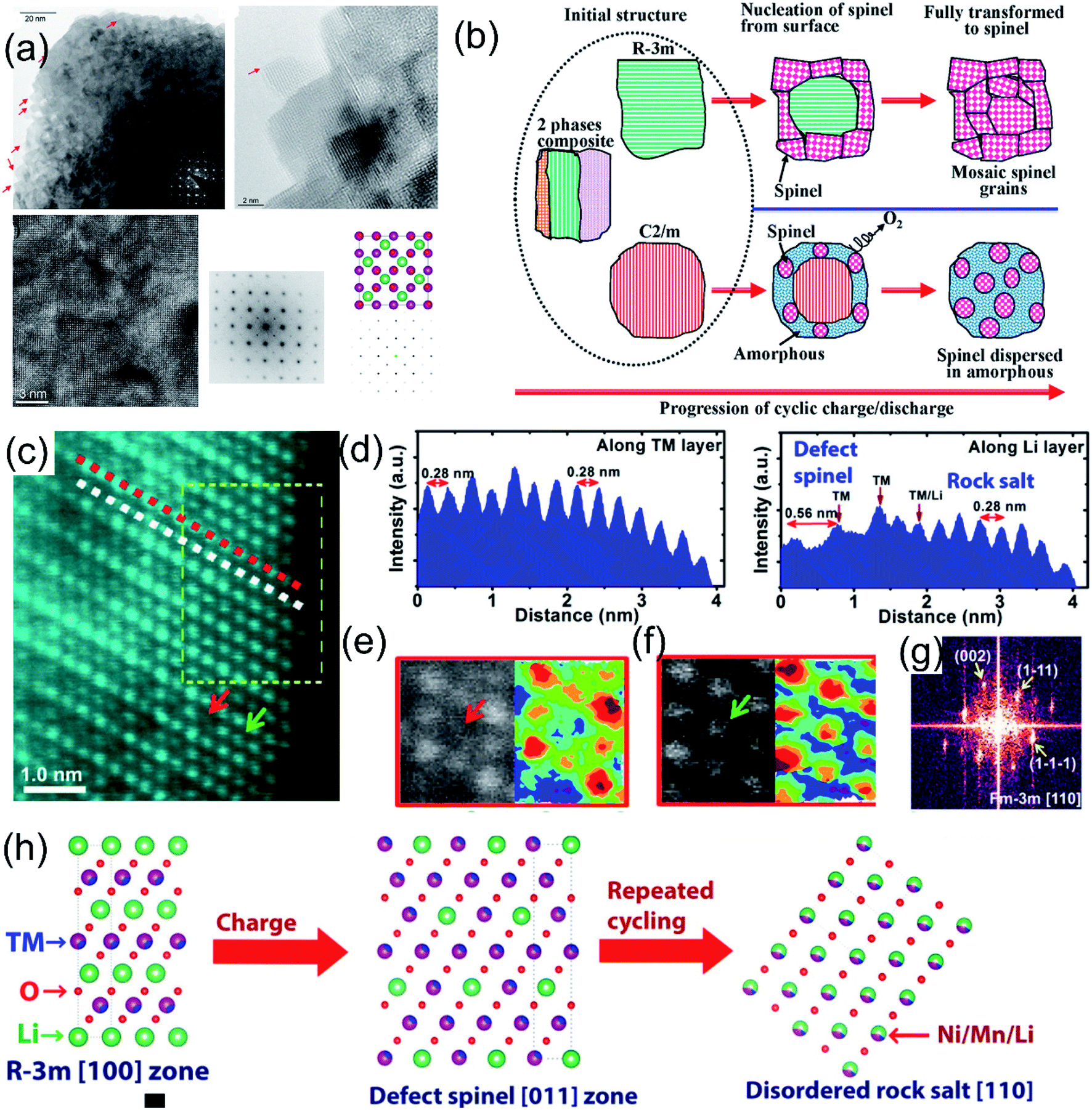 | ||
| Fig. 8 (a) Overview of the nanoparticle with the spinel structure after 300 cycles. TEM image collected from the bulk region also revealed a cubic lattice after 60 cycles; corresponding FFT at [001] zone matched with the atomic model and simulated diffraction pattern of the LiMn2O4 spinel [001] zone axis. (b) Schematic drawing showing that the initial material is composed of three phases: Rm, C2/m, and the nanocomposite of intergrowth of Rm and C2/m. The transition from the Rm and C2/m layered structure to the spinel follows different routes, leading to different structural features of the spinel grains. Reproduced with permission from ref. 103. Copyright 2013, the American Chemical Society. Crystal structure of the LMR material after 10 cycles at C/10 rate. (c) High resolution STEM image. (d) Intensity plot along the red and white dashed lines. (e) Enlarged STEM image showing the structure of the defect spinel structure (LT-LiCoO2 type structure) with empty 16c octahedral sites. (f) Enlarged STEM image showing the structure with filled octahedral sites (disordered rock-salt structure). (g) Fast Fourier transform (FFT) from the region indicated by the yellow dashed rectangle in (c). (h) Atomic models explaining the structural evolution pathway based on the close observation from the structural changes in cycled materials. Reproduced with permission from ref. 107. Copyright 2015, the American Chemical Society. | ||
4. Structural modifications to stabilize the OR reaction
The irreversible OR reaction (especially that occurring on the surface) is closely related to the TM migration, shrinkage of the O–O distance and phase transformation of LLO, which in turn accelerate the oxygen loss, capacity and voltage decay during cycling. To stabilize and enhance the reversibility of the OR reaction, several modification strategies have been explored to improve the electrochemical performance of LLO materials, such as regulating TM–O bonds, heterointerfaces, surface defective engineering, O2 phase construction and so on.4.1 Regulating O-ligand bonds
Atom doping is the most common modification method to regulate O-ligand bonds of LLO materials, which can directly introduce different ions into the crystal lattice of the material and further stabilize its structure and increase the Li+ diffusion rate. The doped element usually has a similar radius to that of the replacement ions, but is effective in building a stronger TM–O bond strength, thereby reducing the irreversible OR reaction and oxygen loss.110,111 Currently, the most widely researched doping methods of LLO materials can be divided into three types: cationic doping, anionic doping and cationic-anionic co-doping.The doped cations will occupy the TM sites or Li sites inside the crystal lattice. Common cations, such as Y3+,112,113 Ta5+,114 Zr4+,115 Sn4+,116 Yb3+,117 and Nb5+,118 tend to occupy the octahedron position of TM sites, thereby forming stronger TM-O bands, increasing the TM transition energy barrier and inhibiting the irreversible oxygen release. Typically, Shin et al. systematically screened cationic doping elements capable of stabilizing O–TM bonds to improve surface O retention through the high-throughput DFT approach.114 In this study, five optimal doping elements (Os, Sb, Ru, Ir, and Ta) were screened out by calculating the doping segregation energy, surface defect formation energy, and thermodynamic stability of surface oxygen. Further experimental results also confirmed that Ta-doping can effectively suppress the oxygen release in the cathode materials and improve their structural stability and electrochemical performance. Besides, Li et al. realized the doping of LLO materials with a large ionic radius element Y3+ by the oxalate co-precipitation method. The large radius Y3+ replaces Mn4+ sites in the layered crystal structure, which expanded the diffusion channels of lithium ions.113 Meantime, undoped Y2O3 that existed in LLO materials effectively retains the lithium and oxygen vacancies on the surface, and Y–O bonds with stronger binding energy ensure the stability of the LLO layered structure during cycling. Using the same scientific research idea, Liu et al. synthesized Nb5+ doped LLO materials with 4–5 TM layers on the surface (Fig. 9a–c). Strong Nb–O bonds formed on the surface were helpful to passivate the surface oxygen and increase the energy barrier for TM migration, which effectively ensured the structural stability of LLO materials (Fig. 9d) and suppressing the phase transition and oxygen loss during cycling.118 Moreover, Bao et al. doped an appropriate amount of Yb3+ in LLO materials by the sol–gel method to effectively enhance its discharge specific capacity, cycling stability, and rate capability.117 In addition, Yu et al. synthesized the Sb-doped LLO nanofibers through an electrospinning process.119 Doping elemental Sb tended to occupy the TM sites and supply the extra electrons to O within the Li2MnO3 phase, which effectively narrowed the bandgap and stabilize the lattice oxygen (Fig. 9g). Hence, the O2 gas evolution, layer-spinel phase transformation and structural degradation were significantly suppressed. Meantime, Sb substitution also expanded the layered phase lattice parameters, thereby enhancing the Li+ diffusion rate and electronic conductivity (Fig. 9e and f). Benefitting from the higher Sb–O bond strength, enhanced kinetics and reformative electronic structure, Sb-doped LLO nanofibers delivered a high reversible discharge capacity (272.8 mA h g−1 at 0.1 C) and excellent capacity retention (86.9% after 200 cycles).
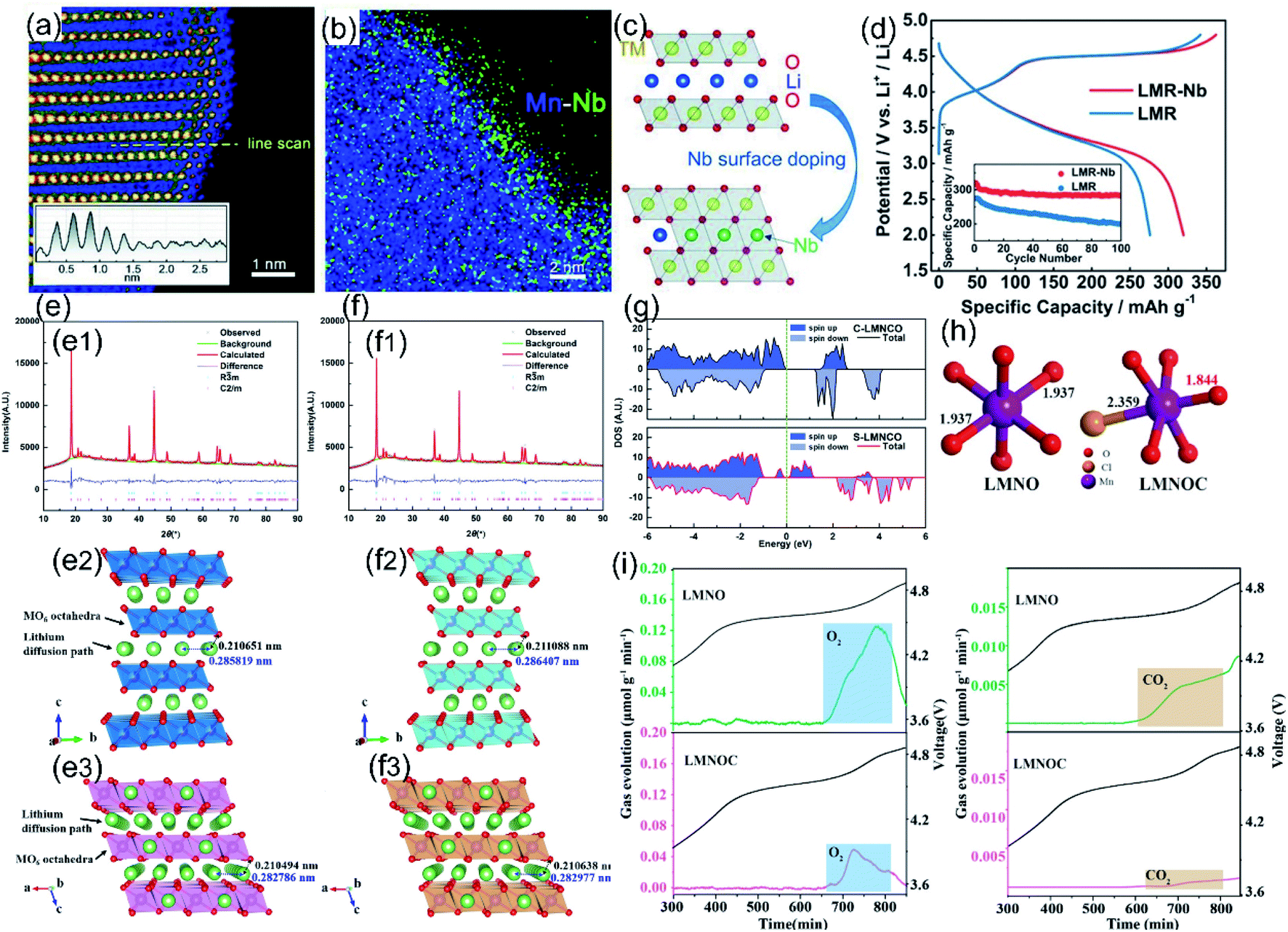 | ||
| Fig. 9 (a) The STEM-HAADF images of the LMR-Nb sample near the surface. (b) EDS mapping of Mn and Nb for the corresponding HAADF image of the surface doping layer. (c) Schematic process of surface doping and the Nb-enhanced surface structure. (d) Comparison of the initial charge/discharge profiles at a rate of 0.1 C between 2.0 and 4.8 V (inset for the cycling performances). Reproduced with permission from ref. 118. Copyright 2018, Wiley-VCH. (e) (f) XRD patterns and the related Rietveld refinement profiles of (e1) C-LMNCO and (f1) S-LMNCO. Structural models of the Rm phase within (e2) C-LMNCO and (f2) S-LMNCO and the C2/m phase within (e3) C-LMNCO and (f3) S-LMNCO. (g) TDOS of pure Li2MnO3 (C-LMNCO) and Sb-doped Li2MnO3. (S-LMNCO). Reproduced with permission from ref. 119. Copyright 2018, the American Chemical Society. (h) Length of TM–O (Cl) bonds of LMNO and LMNOC. (i) DEMS curves of O2 and CO2 for LMNO and LMNOC. Reproduced with permission from ref. 131. Copyright 2021, the American Chemical Society. | ||
Different from the TM site substitution, Na+,120,121 K+,122 Mg2+,123,124 Ti4+,4 doping elements usually occupy the tetrahedral position of the Li sites, thereby creating the pillar effect by stabilizing the Li layer structure.125 In this way, the Li–O–Li configuration could be tuned, thus improving the reversibility of oxygen redox reactivity. Besides, doping atoms with a large ion radius can also increase the interlayer distance of the Li layer, ensuring the rapid extraction and insertion of lithium ions during the charge and discharge process. Hy et al. found that changing the Ni/Mn ratio in the LLO structure could affect the binding ability of TM and O, thereby effectively regulating the degree and reversibility of the OR reaction.126 Increasing the Ni content in LLO was useful in reducing the Li/Ni mixing degree and effectively shortening the oxygen loss plateau length, which could increase the reversibility of the material structure and the cycling stability. Besides, Qing et al. synthesized Na+-surface gradient doped LLO materials by utilizing the diffusion-driven Na+ concentration in the molten state during the calcination process. Owing to the gradient Na+ doping, the lithium layer (003) interplanar spacing and Li+ migration rate in the bulk structure were promoted.127 At the same time, Na acted as a pillar between Li layers, which could inhibit the material phase transition caused by the TMs migration from oxygen octahedral sites (TM layers) to adjacent oxygen tetrahedral sites (Li layers). After Na+ doping, the initial discharge specific capacity and coulombic efficiency are enhanced to 286 mA h g−1 and 87%, respectively. Moreover, Li et al. also synthesized LLO materials by in situ K+ doping, which prevented the formation of the spinel structure during cycling and stabilized the layered structure.122 K+ doping atoms in lithium sites could decrease the formation of Li tri-vacancies, thereby hindering the migration of Mn ions in the TM layer to the tetrahedral position of the Li layer, and effectively suppressing the oxygen loss and phase transformation in the delithiated cathodes.122,128 In addition, Luo et al. provided a new strategy to enhance the capacity and voltage stability simultaneously by bulk Ti-doping and surface Fdm integrated layer construction.4 Several atomic layers of Li ions were substituted by Ti ions on the surface, which effectively alleviates the Mn and Ni ions migration in the bulk. At the same time, the generation of surface Li vacancies and the phase transformation of the surface structure during lithium extraction were also suppressed.
Another strategy to form stronger TM–O bonds is anion doping, which replaces the oxygen sites with other anions or polyanions. F−,129 S2−,130 Cl−,131 and (XmO3m+1)n− (X = P, B, S, As, Mo, W)132–135 are all commonly employed for anion doping to suppress the TM migration, oxygen release and voltage decay. Luo et al. achieved accurate control of the initial coulombic efficiency for LLO materials through an NH4F-assisted hydrothermal treatment strategy.129 Since F substituted the surface unstable oxygen, the oxygen close-packed framework and surface structural stability were significantly improved. Similarly, An et al. synthesized S-doped Li1.2Mn0.6Ni0.2O1.97S0.03 cathode materials; the Li/Ni mixing degree was greatly reduced and the stability of the layered crystal structure was also well maintained. The initial coulombic efficiency and discharge specific capacity of the S-doped cathode material were as high as 96% and 293 mA h g−1, respectively. Even at a high rate of 5C, the discharge capacity remained at 117 mA h g−1.136 Besides, Cl− doping can enhance the covalency of the Mn–O bond (Fig. 9h), and the oxygen released during the charging process was significantly alleviated (Fig. 9i). As a result, the Li+ migration rate and redox reversibility of oxygen on Li, Mn-based cathode materials were enhanced simultaneously.131 Meanwhile, Yan et al. used the DFT calculations to research the effect of Cl− doping on the anion redox reaction of the LLO electrodes.137 It was found that Cl− doping increased the TM–O bond spacing, which in turn reduced the bandgap of the cathode material and effectively decreased the charging potential. This adjustment enabled the contraction of the oxygen loss plateau and reduction of the charging voltage, thereby enhancing the structural and cycling stability. Polyanion groups have strong covalent bonds with TM to form polyhedral structures, hence are commonly used to stabilize the layered structure and prevent the migration of TMs during cycling.132,138 Li et al. found that (BO3)3− doping could reduce the covalency of the M–O bond and the energy of the O 2p orbital, making the anionic redox reaction more stable under high voltage.139 Hence, the (BO3)3− doped sample displayed a significantly improved layered structure, cycling performance, redox potential and thermal stability. In addition, the (PO4)3− doped Li(Li0.17Ni0.20Co0.05Mn0.58)O2 sample was also found to minimize the local structural changes and provide relatively stable energy density during long cycles due to the synergistic function of (PO4)3− with O2−.132
The multi-element co-doping, which has been gradually developed in recent years, could combine the advantages of single-element doping to better improve the electrochemical performance of cathode materials.135,140 Chen et al. synthesized Cd2+ and S2− anion–cation co-doped LLO materials, which exhibited a more attenuated voltage drop during cycling.141 Besides, Liu et al. explored the Na+/F− co-doping method to improve the cycling stability and rate performance of Li1.2Ni0.2Mn0.6O2 cathode materials simultaneously.140 Na substitution was effective in enhancing the layered structural framework and inhibiting the TM transition, while F− doping could greatly improve the electronic and ionic conductivity.
4.2 Surface heterostructure engineering
The lattice oxygen terminated on the surface of an LLO is not fully coordinated, and has a weaker bonding strength than bulk oxygen and is more prone to contact and react with the electrolyte during the charging–discharging process. Therefore, the irreversible OR reaction and structural degradation mainly originate from the surface and gradually expand to the inner region during cycling. Based on this, researchers have explored the surface heterostructure engineering method to change the surface lattice oxygen framework, thereby stabilizing the surface oxygen environment to improve the reversibility of the OR reaction. Some metal oxides such as LiCoO2,142,143 with similar space group to LLOs, spinel structures,144 polyanions,145 or fluorinated146etc. are often used in the surface heterostructure. Similar space groups and the oxygen stacking method ensure the compatibility of the heterojunction with the bulk phase. Besides, the different local structures promote the enhanced Li–O interaction at the interface, which in turn suppresses the structural collapse during cycling. For example, Liu et al. in situ grew a spinel LiCoO2 heterostructure layer with space group Fdm on the surface of Li1.2Ni0.13Co0.13Mn0.54O2 by a wet chemical method (Fig. 10a).142 The surface Fdm spinel phase heterostructure not only achieved lattice compatibility with the host Rm phase LiTMO2 but also significantly changed the ligand orientation on the surface of Li2MnO3 with the C2/m phase. The pair distribution function analysis revealed that the heterostructure induced an octahedral distortion in the Li2MnO3 lattice, corresponding to the shortening of the Mn–O bond and the stretching of the Mn···Mn pair in the C2/m phase (Fig. 10b and c). The computational results further demonstrated that the octahedral distortion modulated the relative positions of the Mn–O* antibonding band and the non-bonding O 2p band (Fig. 10d and e), thereby stimulating the double-band redox reaction to stabilize the OR reaction. As a result, the ligand-orientation-regulated LLO achieved an excellent capacity retention rate of 85.9% after 300 cycles. Similarly, Guo et al. explored a KMnO4 oxidation strategy to construct a 14 nm epitaxial spinel Li4Mn5O12 layer on the surface of LLO.143In situ XRD and electrochemical techniques indicated that the Li4Mn5O12 layer can significantly alleviate the undesired oxygen loss and phase transition due to its lattice stability in the oxygen framework structure. Besides, Hu et al. used hexagonal La0.8Sr0.2Mn3−y (LSM) as a phase-compatible surface protective layer to mitigate the TM migration and capacity degradation of LLO cathodes. As shown in Fig. 10a and f strong Mn–O–M bonding was formed at the heterostructure interface by sharing O, which effectively reduced the Mn dissolution and unwanted phase transition, defect generation and oxygen release.147 Moreover, research on the use of fluoride as an LLO modification layer was carried out subsequently. An AlF3 layer provided F–O bonding on the surface and inhibited the layer–spinel phase transition and oxygen vacancy migration, thus the voltage decay of LLO materials was effectively mitigated.148 Phosphate structures are more stable than oxides and fluorides due to the presence of PO double bonds and often used to improve the cycling stability of LLO materials at high temperatures and voltages. Ma et al. synthesized AlPO4-coated LLO cathodes through the atomic layer deposition (ALD) method; the process of depositing AlPO4 induced the formation of the spinel phase on the surface, which promoted a significant increase in the initial coulombic efficiency of the LLO materials.145 The differential scanning calorimetry (DSC) results also showed that compared with Al2O3, the initial thermal decomposition temperature of the AlPO4-coated material in the charged state was also increased, indicating that the AlPO4 coating can improve the thermal stability of the LLO materials.
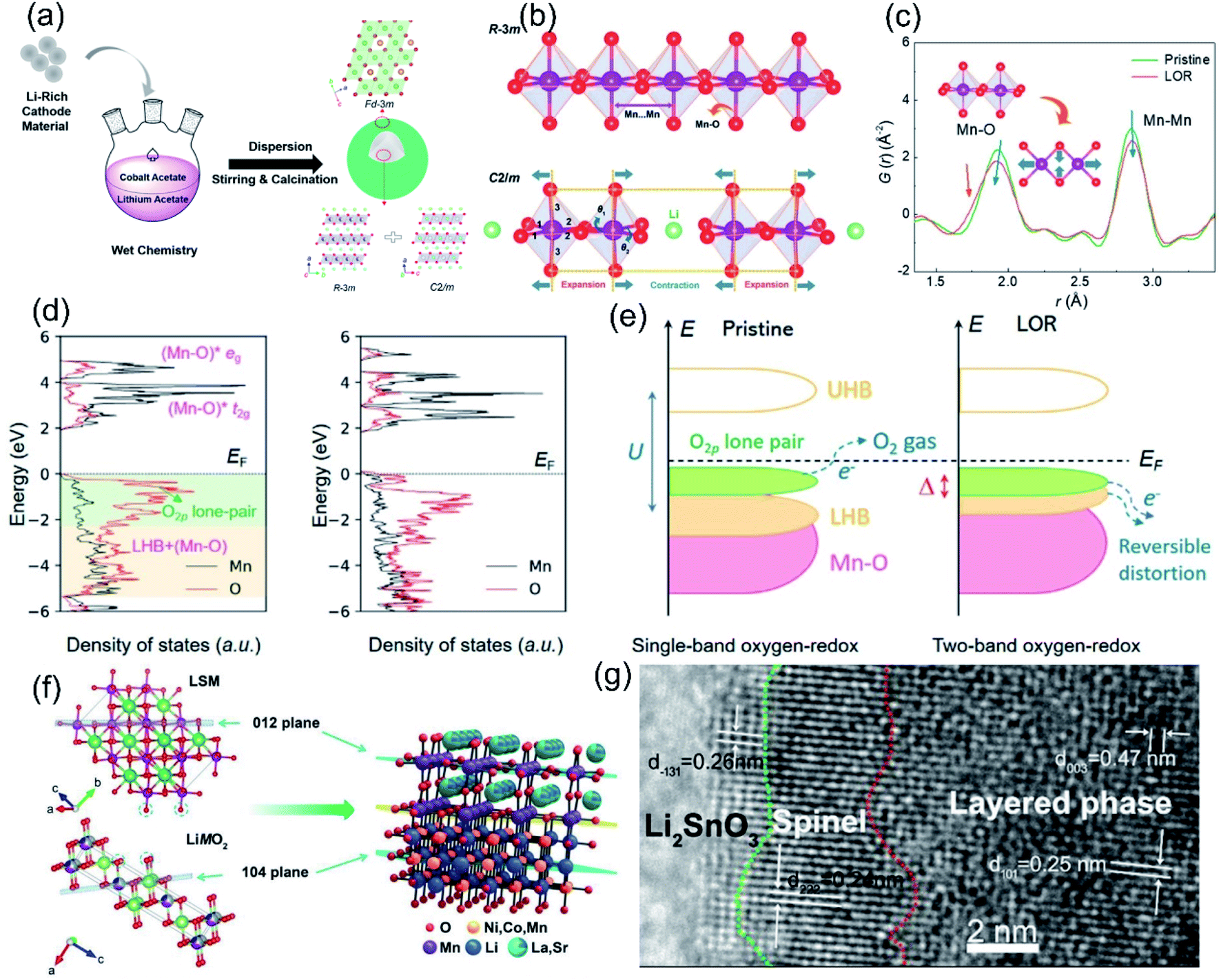 | ||
| Fig. 10 (a) Schematic diagram of the growth of the heterostructured spinel-type LCO outer layer. (b) Schematic of the local ligand orientations for the Rm and C2/m components. (c) Low-r PDF patterns of the pristine and LOR samples. The inset shows the local structural distortion correlated with the PDF peak shifts. (d) pDOS of the Mn and O states for the pristine and LOR samples based on the Li2MnO3 structures extracted from the PDF refinements. (e) Schematic of the single-band oxygen redox process of the pristine sample and the two-band oxygen redox process of the LOR sample. Reproduced with permission from ref. 142. Copyright 2021, Wiley-VCH. (f) The heterostructural interface and bonding structure of the LSM-coated sample. Reproduced with permission from ref. 147. Copyright 2019, Wiley-VCH. (g) HRTEM image of the 1% LSO sample, the structures from the inside to outside are the layered phase, spinel phase, and Li2SnO3 coating layers, respectively. Reproduced with permission from ref. 116. Copyright 2019, Wiley-VCH. | ||
In addition, researchers have also developed a combined lattice doping-surface modification strategy to simultaneously enhance the TM–O bond strength and stabilize the surface oxygen-redox process. For example, Li et al. achieved Li2SnO3 coating and surface Sn4+ doping on Li1.2Mn0.6Ni0.2O2 cathode materials through the in situ lithiation method.116 As revealed by the high-resolution transmission electron microscopy (HRTEM) images in Fig. 10g, the structures from the inside to outside are the layered phase, spinel phase, and Li2SnO3 coating layers, respectively. XAS analysis confirmed that Sn4+ doping increased the Li interlayer spacing and further decreased the Li+ activation energy, which alleviated the problems of electrode polarization and oxygen evolution in the redox reaction. In addition, the surface spinel phase induced by Sn4+ doping, together with the Li2SnO3 layer, not only ensured the Li+ migration rate, but also hindered the side reactions at the interface. Liu et al.149 achieved Mg3(PO4)2 coating and Mg2+ doping on the surface of Li-rich manganese-based cathode materials, and the modified sample displayed a high specific discharge capacity of 180 mA h g−1 after 250 cycles at 60 °C.
In summary, surface heterostructures can effectively enhance the stability of the interface, especially to bind the lattice oxygen on the surface. As for regulation of the TM–O bond, although its improvement in interfacial stability is limited, it can significantly enhance the structural stability of the materials during the charging and discharging process. Combining the above two advantages, comprehensive construction of the lattice structure and surface modification can greatly improve the electrochemical performance of LLO materials. However, their preparation process is complex and still in the laboratory stage, and researchers still need to simplify and explore the joint modification synthesis method.
4.3 Surface defective structure construction
Anion-substituted oxygen ions and cationic substitutions can control the degree of hybridization between O 2p orbitals and TM d/s/p orbitals, while the surface heterostructure engineering is effective in promoting the enhanced Li–O interaction at the interface and stabilizing the oxygen redox. Besides the two above-mentioned methods, researchers found that the creation of lithium or oxygen defects was also effective in controlling the anionic redox reaction.52,150 On the one hand, the nanoscale defect structure on the surface of the LLO produced a pinning effect, which could hinder its dislocation evolution and further phase transition of the layered structure during cycling. On the other hand, the transport of Li+ was also more active in some surface disordered Li-rich material structures, ensuring high reversible capacity at high rates.71,151 Luo et al. constructed oxygen vacancies and F-doping on the surface of LLO materials by NH4F treatment.129 Besides, Qiu et al. constructed a 20 nm oxygen-deficient layer on the surface of LLOs through the CO2 gas–solid interface reaction (GSIR), realizing the regulation of oxygen activity.152 As shown in Fig. 11a, the generation of oxygen vacancies lowered the Li+ migration energy barrier, providing a favorable environment for Li+ diffusion in the bulk structure. Meanwhile, it also hindered the generation of highly reactive oxygen radicals during the electrochemical process, effectively inhibiting the oxygen aggregation and oxygen release. The modified LLO material showed excellent performance, the discharge specific capacity was still higher than 300 mA h·g−1 after 100 cycles, and the voltage was basically not attenuated. Similarly, Erickson et al. used ammonia gas to pretreat LLO materials in a high-temperature environment, which greatly improved the stability and rate performance of the cathode materials.153 It has been confirmed that NH3 at high temperature reduced the valence states of Mn and Co, forming oxygen vacancies and partial spinel structures on the surface, which in turn passivated the surface oxygen and expanded the 3D Li+ diffusion channels. Moreover, Ding et al. designed a new three-in-one surface modification method to improve the capacity and voltage stability of Li1.2Mn0.6Ni0.2O2 simultaneously.97 As shown in Fig. 11b, through the pyrolysis of urea treatment, oxygen vacancies, N-doped carbon nanolayers and a spinel-coherent phase were synchronously built on the LLO surface. Therefore, the release of oxygen is largely suppressed, and the initial coulombic efficiency can be adjusted almost linearly by changing the amount of NH4F. Oxygen vacancies together with the spinel phase could suppress irreversible O2 loss, while the N-doped carbon nanolayer was effective in mitigating the electrolyte side reactions.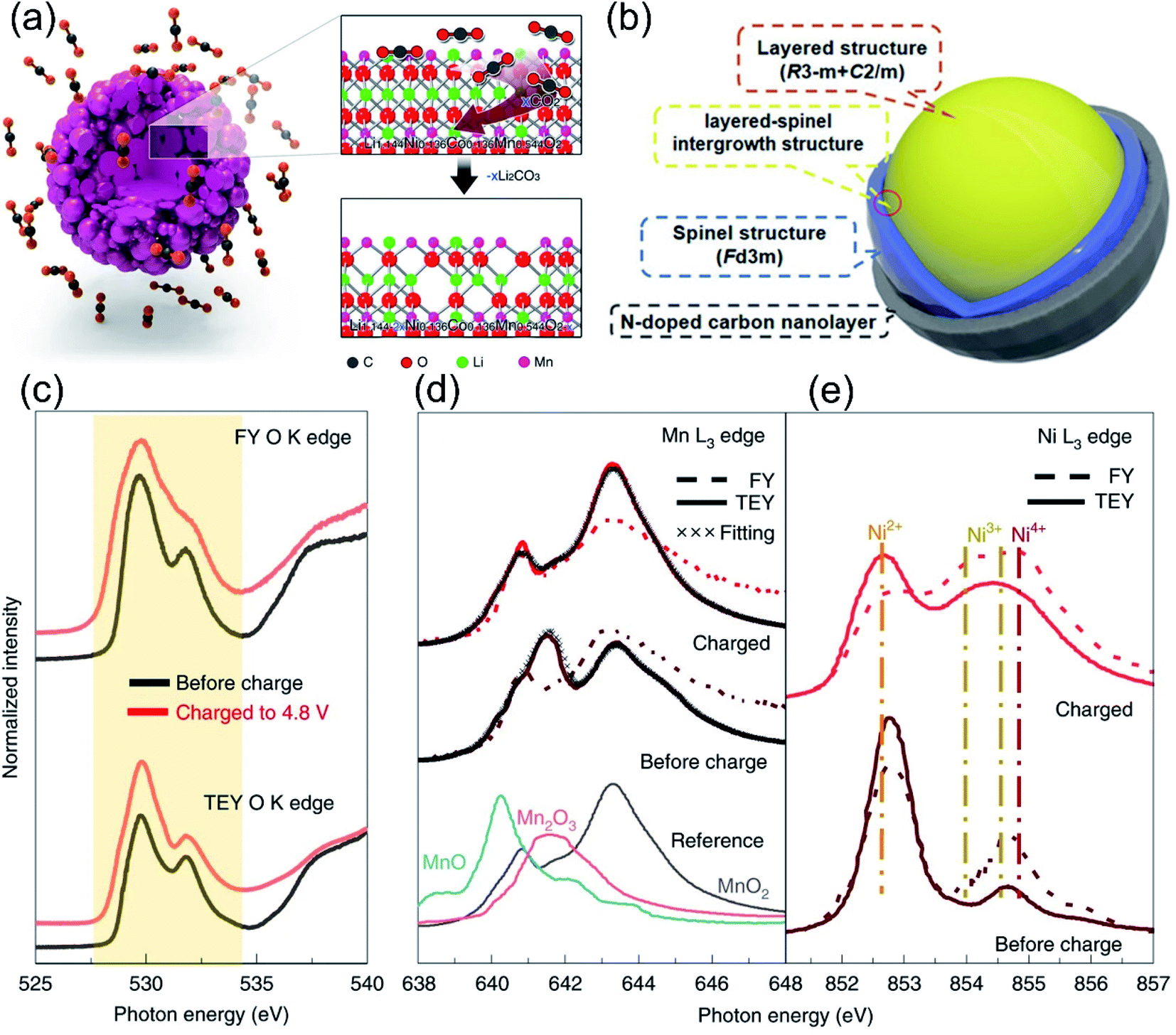 | ||
| Fig. 11 (a) Schematic of the GSIR between Li-rich layered oxides and carbon dioxide. Reproduced with permission from ref. 152. Copyright 2016, Springer Nature. (b) Illustration of the structural components of the M-LMNO sample. Reproduced with permission from ref. 97. Copyright 2020, Wiley-VCH. (c) Normalized sXAS O K edge of G4 from discharged to charged states collected from FY and TEY modes. The normalized intensity was integrated from 527.5 eV to 534.2 eV, as indicated by the light orange background. (d) sXAS Mn L3 edge for discharged and charged states under FY and TEY modes. The TEY Mn L3 edge was fitted by the linear combination of Mn2+, Mn3+ and Mn4+ TEY references. c, sXAS Ni L3 edge for discharged and charged states under FY and TEY modes. (e) Distribution of Mn valence at the surface for discharged and charged states. Reproduced with permission from ref. 156. Copyright 2019, Springer Nature. | ||
In addition to the construction strategies of oxygen vacancies, the modification methods of surface lithium defects and pre-extraction of lithium–oxygen have also been extensively studied. Van et al. found that Li vacancies could induce the transition of Li+ in layered materials from the oxygen dumbbell mode (ODH) with a higher energy barrier to the tetrahedral mode (TSH) with a lower energy barrier.154 Besides, Pimenta et al. found that depletion of lithium would lead to the formation of a spinel phase crystal structure on the surface of the LLO, and the migration of Ni ions from the TM layer to the lithium layer, thereby stabilizing the crystal structure and suppressing lattice oxygen release during cycling.155 Moreover, Zhu et al. pioneered a lithium gradient modification strategy on LLO surfaces to immunize oxygen release through (NH4)6Mo7O24·4H2O-assisted Li2O leaching treatment.156 The Li-poor phase on the surface effectively inhibited the irreversible anionic redox reactions and structural collapse of the surface. In addition, atomic-scale mixing of the bulk and surface regions was achieved, sharing a fully occupied oxygen framework with no grain boundaries, ensuring the preservation of the structure during cycling. Soft X-ray absorption spectroscopy (sXAS) results showed that oxygen in the surface was not involved in the redox process, while the core region contributed significant capacity (Fig. 11c–e). The altered-valence oxygen mobility could enhance the reversibility of the OR reaction and stabilize the Mn valence during cycling. There are also some studies on the leaching part of Li2O in the Li2MnO3 structure through Na2S2O8,157 (NH4)2S2O8,158 (NH4)2SO4,159 N2H4·H2O160,161 and acid treatment162–164 to improve the initial coulombic efficiency and reduce the oxygen loss. For example, Kang et al. pre-extracted part of the lithium-containing oxides in the Li2MnO3 component through HNO3 treatment, which reduced the initial irreversible capacity in the LLO, and the first coulombic efficiency increased to 100%.164
The above studies have proved that the introduction of defects can reduce the degree of surface oxygen ion participation in the electrochemical process by changing the orbital overlap between TM ions and oxygen ions, thereby making the anion redox reaction more reversible. However, the construction process of defects is usually complicated and cumbersome, which is not suitable for mass production. Therefore, simplifying the modification process and synergizing with other modification methods are the research directions for future defective structure construction.
4.4 O2 phase construction
The irreversible TM migration in conventional Li-rich materials significantly exacerbates the process of irreversible OR reaction and the release of lattice oxygen. Unfortunately, as previously mentioned, the dynamic coupling relationship between the OR reaction and the TMLi–VTM defect pair has revealed that the migration process of TMs is inevitable due to the inherent structural properties of LLO. However, common modification strategies (such as anion doping,133 cation doping,119 and the construction of surface heterostructures143) are mainly aimed at alleviating the migration of TMs, which are unable to fundamentally solve the reversible back-intercalation of TMs. In recent years, researchers have developed novel O2-phase Li-rich materials through a Li–Na ion-exchange strategy, inspired by the high reversibility of the OR reaction and TM migration of sodium-ion cathodes.55,56 Emu et al. prepared the O2 phase Lix(Li0.2Ni0.2Mn0.6)O2 (O2-LLO) material, achieving the reversible migration of TM ions, and systematically revealed the TM migration process in O3 and O2 phases.28 As shown in Fig. 12a, the C2/m-type O3-LLO structure consists of LiO6 octahedra (Lioct) and TMO6 octahedra (TMoct), in which the stacking arrangement of oxygen is ABCABC type, a typical O3 structure. It is not difficult to see that Lioct is coplanar with tetrahedral vacancies (Vtet) and shares edge with TMoct. Corresponding DFT results confirmed that during the charging process, the TM ions in the TMoct can easily migrate into the adjacent Vtet. Furthermore, the TM ions in the Vtet are thermodynamically favorable and even permanently migrate into the empty Lioct with the extraction of Li ions. During the discharge process, the mixed arrangement of TM ions and Li ions in the Li layer inevitably leads to the complexity of the return path of TM ions. During cycles, more and more TM ions are trapped in the Li layer, resulting in permanent voltage decay and even spinel phase transition.7,165 Hence, they believe that the fundamental factor of voltage decay in Li-rich materials is not the TM ion migration, but the irreversibility of TM ions in the Li layer returning to the TM layer. Based on this, the O2-LLO was designed to build an energy barrier between the Vtet and Lioct sites to prevent the permanent retention of TM ions in the Li layer and realize the reversible back-intercalation of TM ions. As shown in Fig. 12b, the oxygen stacking mode in O2-LLO (space group: p63mc) is ABCBA. The O2 phase is characterized by a special Lioct sharing structure: the Lioct of the O2 phase is coplanar with one TMoct. Therefore, the TM ions in the Lioct (or Vtet) will generate a strong electrostatic repulsion with the coplanar TMoct due to the short TM–TM spacing. Hence, TM ions in Lioct (or Vtet) are more likely to migrate back to the TMoct. Subsequently, the authors observed the reversible migration process of TM ions by HADDF imaging. Fig. 12c shows the HAADF image of the O2 phase when fully charged, in which bright spots representing TM ions appear in the Li layer, illustrating the migration of TM ions to the Li layer, while in the fully discharged HAADF image (Fig. 12d), the TM ions in the Li layer completely disappeared, proving that they have reversibly migrated back to the TM layer. Furthermore, theoretical calculations confirmed that the migration of TM ions in the Li layer of the O2 phase needs to overcome a higher energy barrier compared to the O3 phase (Fig. 12e and f). Therefore, during the discharge process, the TM ions in the O2 phase can easily migrate back to the TM layer from the intermediate positions. Ultimately, O2-LLO showed excellent cycling performance with negligible voltage drop due to this unique thermodynamic barrier (Fig. 12g).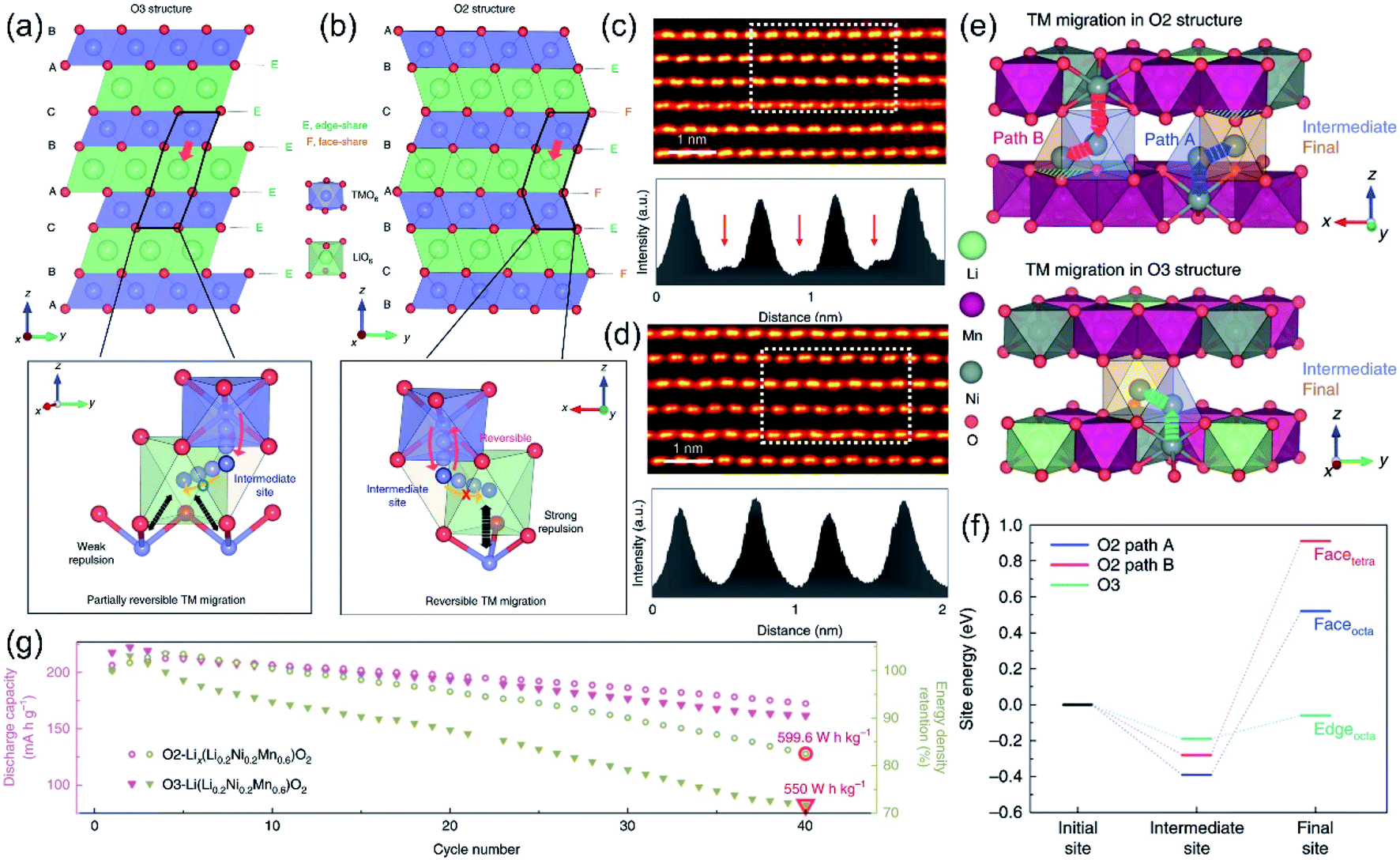 | ||
| Fig. 12 Schematic illustrations of the crystal structures of O3-type (a) and O2-type (b) lithium layered oxides. The figures below show the TM migration paths on a magnified scale. Red and yellow arrows indicate the interlayer and the intra-layer TM migrations, respectively. And black arrows represent the electrostatic repulsion in the TM layer. (c and d) HAADF-STEM images for 4.8 V charged (c) and 2.0 V discharged (d) O2-LLNMOs. The graphs below are the HAADF signal profiles of the regions enclosed by the dotted lines in the STEM images. (e) TM migration paths from the initial to the intermediate to the final Li sites. (f) Relative site energies of intermediate and final sites calculated along the migration paths of TM ions. (g) Comparison of discharge capacity and energy density retention in O2- and O3-LLNMOs. Reproduced with permission from ref. 28. Copyright 2020, Springer Nature. | ||
This special structure in the O2 phase has attracted the great attention of many researchers.5,78 Zuo et al. synthesized a single-layer O2-phase Li2MnO3 superstructure with an ultra-high reversible capacity of 400 mA h g−1, which showed good charge–discharge efficiency and stable voltage after 50 cycles.166 Meanwhile, Cui et al. adopted a synergistic strategy of O2 phase LLO and fluorinated electrolytes to reduce O2 release, which achieved a coulombic efficiency of nearly 100%.167 The O2-LLO guaranteed the reversible back-intercalation of the TM, while the fluorinated electrolyte was effective in growing a protective interface on the material surface in situ during the charge–discharge process. The combined strategy effectively stabilized the reversible TM migration and OR reaction, thereby suppressing the structural transition upon cycling. Moreover, Cao et al. compared two synthesized methods of the O2 phase, chemical and electrochemical ion-exchange strategies.5 The results elucidated that the chemically synthesized O2-LLO involved a more stably reversible oxygen redox, which effectively suppressed the phase transition and lattice O loss. Overall, the construction of the O2-phase LLO structure is a promising modification strategy to fundamentally achieve reversible TM ion migration from the bulk structure, thereby suppressing the irreversible OR reaction and lattice O loss, and finally stabilizing the voltage and capacity.
4.5 Summary of the structural modification strategies
In conclusion, we summarize the structural modification strategies to address the irreversible OR issues of LLOs and provide a discussion on its advantages and disadvantages (Fig. 13). Regulating O-ligand bonds involves mainly introducing different ions to adjust the electronic and crystal structure, thereby effectively forming stronger TM-O bonds. It can enhance the stability of the lattice structure and suppress the irreversible phase transitions. Surface heterostructure design can restrain the generation of surface oxygen defect and reducing the oxygen release, which is necessary to prevent the structural degradation from the surface to the interior. Surface defective construction is effective to stabilize the lattice structural evolution during the charge and discharge process, especially in the high voltage region. Surface Li defect can compensate for the drastic lattice evolution caused by the extreme extraction of Li ions under high voltages, while the surface O defect can suppress the irreversible OR process and drastic lattice shrinkage. O2 phase construction emerging in recent years can achieve the reversible back-intercalation of the TMs, thereby suppress the irreversible OR reaction and voltage decay. However, these above-mentioned modification methods all have some general disadvantages. Modification treatment is difficult to control and achieve uniformity, while the resulting local ligand improvement is difficult to fully protect the entire LLO grain. Besides, the modification process is often complex and cumbersome, which hinders its commercial production.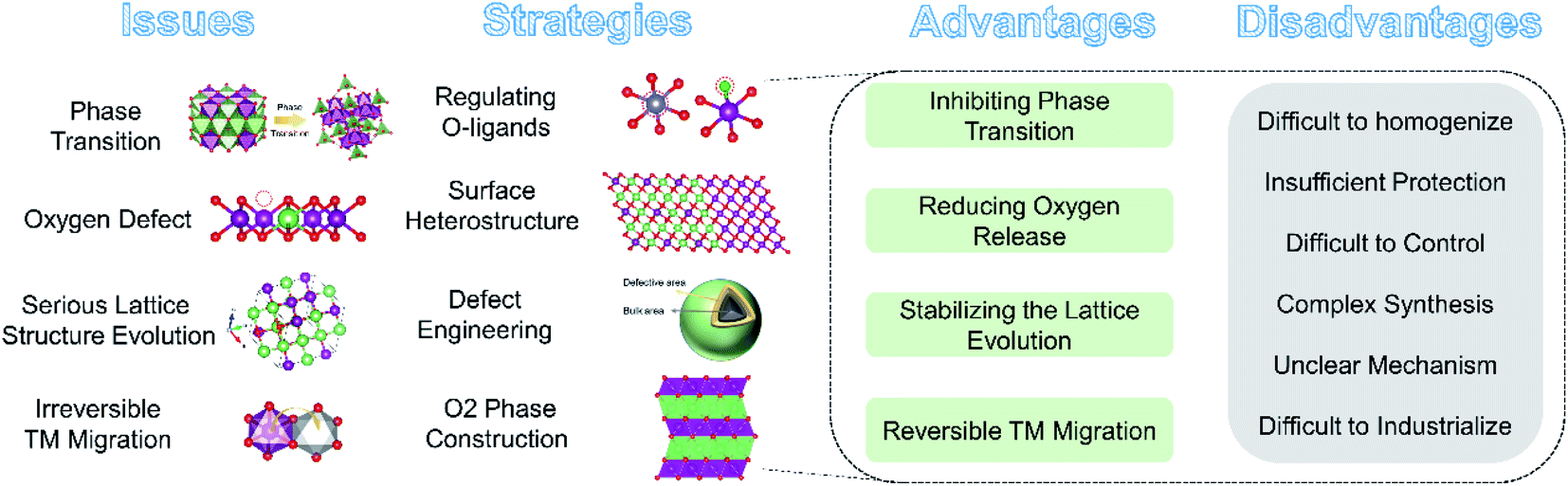 | ||
| Fig. 13 Summary of the structural modification strategies to address the irreversible OR issues of LLOs and their corresponding advantages and disadvantages. | ||
5. Conclusion and perspectives
The development of lithium-rich cathode materials with high energy density has always been a hot spot in the field of LIB research. In this paper, the structure, OR mechanism, OR structural evolution and corresponding stabilized OR reaction strategies of Li-rich materials are reviewed. Starting from the unique structure of Li-rich materials, the advantages and disadvantages of anionic redox mechanisms are systematically discussed. On the one hand, the unique anionic redox reaction in the bulk phase is the source of the ultra-high discharge specific capacity of Li-rich materials. On the other hand, the irreversible anionic redox reaction especially on the surface is also the factor of structural instability, which causes surface oxygen release and structural rearrangement, resulting in serious irreversible capacity loss, and subsequent cycling and voltage decrease. To enhance the reversibility of the OR reaction, many modification methods, such as regulating TM–O bonds, heterointerfaces, surface defective engineering, O2 phase construction, etc., have been extensively studied to adjust the degree of orbital overlap between TM ions and O2− or change the crystal structure, and the oxygen environment of LLO, thereby realizing the reversible anionic-redox chemistry. In this paper, from the perspective of molecular orbital theory, the complexity of the crystal structure and charge–discharge mechanism is explained, which is beneficial to guide the modification design of crystal oxygen structure, and then to improve the electrochemical performance of LLO materials in a targeted manner.It is worth mentioning that the research on LLO is still lacking in the following aspects, which limit its commercial development: (1) the dynamic coupling relationship between the OR reaction, TM ion migration and electronic structure during charge and discharge still lacks a more systematic understanding; (2) the oxygen loss mechanism, lattice O structure change, and the relationship of the O loss path are not clear enough in the irreversible OR reaction; (3) there is a lack of more efficient synthesis or modification strategies to suppress the decay of voltage and capacity; (4) the structural evolution of anionic redox reactions during long cycles still needs to be explored. In response to the above challenges, we make the following prospects. (1) Using in situ and atomic-resolution characterization techniques to explore the evolution of atomic structures coupled with OR reactions during cycling. (2) Limited by characterization techniques, theoretical calculations are powerful tools to analyze the relationship between the changes in the LLO atomic structure, electronic structure and the OR reaction evolution process in static and dynamic processes. Meanwhile, high-throughput and machine learning techniques can effectively extract structural features to screen out high-performance Li-rich materials. (3) O2-phase Li-rich materials are a promising development direction because of their unique structure through the reversible TM ion migration to achieve a reversible OR reaction, thereby realizing the high voltage and capacity stability. (4) It is difficult to comprehensively solve the problems caused by the irreversible OR reaction with a single modification strategy. Therefore, the multi-level hybrid modification method of atom-surface-crystal is an effective strategy to realize the reversible OR reaction. Overall, exploring the mechanism and high reversibility of the OR reaction in Li-rich materials during long-term cycling still requires continuous efforts by researchers and further innovation in characterization techniques. We believe that with the increasingly clear reaction mechanism and the continuous innovation of large-scale preparation and synthesis methods, Li-rich layered oxide cathode materials will gradually meet the commercial demand for high-energy density Li-ion batteries in the future.
Conflicts of interest
There are no conflicts to declare.List of abbreviations
| AM | Alkali metal |
| ALD | Atomic layer deposition |
| DOS | Density of states |
| DEMS | Differential electrochemical mass spectrometry |
| DFT | Density functional theory |
| DSC | Differential scanning calorimetry |
| EDS | Energy dispersive spectroscopy |
| EELS | Electron energy loss spectroscopy |
| E F | Fermi level |
| EV | Electric-vehicle |
| GSIR | Gas–solid interface reaction |
| HAADF-STEM | High-angle annular dark-field scanning transmission electron microscopy |
| HRTEM | High-resolution transmission electron microscopy |
| LIBs | Lithium-ion batteries |
| LIO | Li2IrO3 |
| Lioct | LiO6 octahedra |
| LISO | Li2−xIr1−ySnyO3 |
| LHB | Lower Hubbard band |
| LLO | Layered Li-rich oxide |
| LSM | La0.8Sr0.2Mn3−y |
| ODH | Oxygen dumbbell mode |
| OR | Oxygen redox |
| RCM | Reduction coupling mechanism |
| SIMS | Secondary ion mass spectrometry |
| STEM | Scanning transmission electron microscopy |
| STXM- XAS | Scanning X-ray transmission microscopy-X-ray absorption spectroscopy |
| SXRD | Synchrotron X-ray diffraction |
| sXAS | Soft X-ray absorption spectroscopy |
| TM | Transition metal |
| TMoct | TMO6 octahedra |
| TSH | Tetrahedral mode |
| UHB | Upper Hubbard band |
| Vtet | Tetrahedral vacancies |
| XAS | X-ray absorption spectroscopy |
| XANES | X-ray adsorption near-edge structure |
Acknowledgements
This study was supported by the National Key R&D Program of China (2020YFA0406203), the Shenzhen Science and Technology Innovation Commission (JCYJ20180507181806316, JCYJ20200109105618137), the ECS scheme (CityU 21307019), and the Shenzhen Research Institute, City University of Hong Kong.References
- M. Li and J. Lu, Science, 2020, 367, 979–980 CrossRef CAS PubMed.
- S. L. Cui, M. Y. Gao, G. R. Li and X. P. Gao, Adv. Energy Mater., 2021, 12, 2003885 CrossRef.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- D. Luo, J. Cui, B. Zhang, J. Fan, P. Liu, X. Ding, H. Xie, Z. Zhang, J. Guo, F. Pan and Z. Lin, Adv. Funct. Mater., 2021, 31, 2009310 CrossRef CAS.
- X. Cao, H. Li, Y. Qiao, M. Jia, P. He, J. Cabana and H. Zhou, Energy Storage, 2021, 38, 1–8 CrossRef.
- M. S. Whittingham, Chem. Rev., 2014, 114, 11414–11443 CrossRef CAS PubMed.
- Z. Li, Y. Li, M. Zhang, Z. W. Yin, L. Yin, S. Xu, C. Zuo, R. Qi, H. Xue, J. Hu, B. Cao, M. Chu, W. Zhao, Y. Ren, L. Xie, G. Ren and F. Pan, Adv. Energy Mater., 2021, 11, 2101962 CrossRef CAS.
- G. Assat and J.-M. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- W. E. Gent, K. Lim, Y. Liang, Q. Li, T. Barnes, S.-J. Ahn, K. H. Stone, M. McIntire, J. Hong, J. H. Song, Y. Li, A. Mehta, S. Ermon, T. Tyliszczak, D. Kilcoyne, D. Vine, J.-H. Park, S.-K. Doo, M. F. Toney, W. Yang, D. Prendergast and W. C. Chueh, Nat. Commun., 2017, 8, 2091 CrossRef PubMed.
- E. Hu, X. Yu, R. Lin, X. Bi, J. Lu, S. Bak, K.-W. Nam, H. L. Xin, C. Jaye, D. A. Fischer, K. Amine and X.-Q. Yang, Nat. Energy, 2018, 3, 690–698 CrossRef CAS.
- W. Zuo, M. Luo, X. Liu, J. Wu, H. Liu, J. Li, M. Winter, R. Fu, W. Yang and Y. Yang, Energy Environ. Sci., 2020, 13, 4450–4497 RSC.
- J. Liu, J. Wang, Y. Ni, K. Zhang, F. Cheng and J. Chen, Mater. Today, 2021, 43, 132–165 CrossRef CAS.
- J. Yang, Y. Niu, X. Wang and M. Xu, Inorg. Chem. Front., 2021, 8, 4300–4312 RSC.
- Y. Y. Hwang, J. H. Han, S. H. Park, J. E. Jung, N. K. Lee and Y. J. Lee, Nanotechnology, 2022, 33, 182003 CrossRef PubMed.
- H. Zhu, Y. Huang, J. Ren, B. Zhang, Y. Ke, A. K. Jen, Q. Zhang, X. L. Wang and Q. Liu, Adv. Sci., 2021, 8, 2003534 CrossRef CAS PubMed.
- H. Zhu, Y. Huang, H. Zhu, L. Wang, S. Lan, X. Xia and Q. Liu, Small Methods, 2019, 4, 1900223 CrossRef.
- S. Hu, A. S. Pillai, G. Liang, W. K. Pang, H. Wang, Q. Li and Z. Guo, Electrochem. Energy Rev., 2019, 2, 277–311 CrossRef CAS.
- G. Ceder, Y. M. Chiang, D. R. Sadoway, M. K. Aydinol, Y. I. Jang and B. Huang, Nature, 1998, 392, 694–696 CrossRef CAS.
- Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A815–A822 CrossRef CAS.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S.-H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed.
- Y. Koyama, I. Tanaka, M. Nagao and R. Kanno, J. Power Sources, 2009, 189, 798–801 CrossRef CAS.
- M. Sathiya, G. Rousse, K. Ramesha, C. P. Laisa, H. Vezin, M. T. Sougrati, M. L. Doublet, D. Foix, D. Gonbeau, W. Walker, A. S. Prakash, M. Ben Hassine, L. Dupont and J. M. Tarascon, Nat. Mater., 2013, 12, 827–835 CrossRef CAS PubMed.
- E. McCalla, A. M. Abakumov, M. Saubanère, D. Foix, E. J. Berg, G. Rousse, M.-L. Doublet, D. Gonbeau, P. Novák, G. Van Tendeloo, R. Dominko and J.-M. Tarascon, Science, 2015, 350, 1516–1521 CrossRef CAS PubMed.
- D.-H. Seo, J. Lee, A. Urban, R. Malik, S. Kang and G. Ceder, Nat. Chem., 2016, 8, 692–697 CrossRef CAS PubMed.
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y.-S. Liu, K. Edström, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684–691 CrossRef CAS PubMed.
- Y. Xie, M. Saubanère and M. L. Doublet, Energy Environ. Sci., 2017, 10, 266–274 RSC.
- J. Hong, W. E. Gent, P. Xiao, K. Lim, D.-H. Seo, J. Wu, P. M. Csernica, C. J. Takacs, D. Nordlund, C.-J. Sun, K. H. Stone, D. Passarello, W. Yang, D. Prendergast, G. Ceder, M. F. Toney and W. C. Chueh, Nat. Mater., 2019, 18, 256–265 CrossRef CAS PubMed.
- D. Eum, B. Kim, S. J. Kim, H. Park, J. Wu, S. P. Cho, G. Yoon, M. H. Lee, S. K. Jung, W. Yang, W. M. Seong, K. Ku, O. Tamwattana, S. K. Park, I. Hwang and K. Kang, Nat. Mater., 2020, 19, 419–427 CrossRef CAS PubMed.
- R. A. House, G. J. Rees, M. A. Pérez-Osorio, J.-J. Marie, E. Boivin, A. W. Robertson, A. Nag, M. Garcia-Fernandez, K.-J. Zhou and P. G. Bruce, Nat. Energy, 2020, 5, 777–785 CrossRef CAS.
- P. Rozier and J. M. Tarascon, J. Electrochem. Soc., 2015, 162, A2490–A2499 CrossRef CAS.
- J. Xu, D. H. Lee, R. J. Clément, X. Yu, M. Leskes, A. J. Pell, G. Pintacuda, X.-Q. Yang, C. P. Grey and Y. S. Meng, Chem. Mater., 2014, 26, 1260–1269 CrossRef CAS.
- Y. S. Meng, G. Ceder, C. P. Grey, W. S. Yoon, M. Jiang, J. Bréger and Y. Shao-Horn, Chem. Mater., 2005, 17, 2386–2394 CrossRef CAS.
- J. Bréger, M. Jiang, N. Dupré, Y. S. Meng, Y. Shao-Horn, G. Ceder and C. P. Grey, J. Solid State Chem., 2005, 178, 2575–2585 CrossRef.
- J. Bareño, M. Balasubramanian, S. H. Kang, J. G. Wen, C. H. Lei, S. V. Pol, I. Petrov and D. P. Abraham, Chem. Mater., 2011, 23, 2039–2050 CrossRef.
- H. Yu, R. Ishikawa, Y. G. So, N. Shibata, T. Kudo, H. Zhou and Y. Ikuhara, Angew. Chem., Int. Ed., 2013, 52, 5969–5973 CrossRef CAS PubMed.
- K. A. Jarvis, Z. Deng, L. F. Allard, A. Manthiram and P. J. Ferreira, Chem. Mater., 2011, 23, 3614–3621 CrossRef CAS.
- Z. Lu, Z. Chen and J. R. Dahn, Chem. Mater., 2003, 15, 3214–3220 CrossRef CAS.
- E. McCalla, C. M. Lowartz, C. R. Brown and J. R. Dahn, Chem. Mater., 2013, 25, 912–918 CrossRef CAS.
- E. McCalla, A. W. Rowe, R. Shunmugasundaram and J. R. Dahn, Chem. Mater., 2013, 25, 989–999 CrossRef CAS.
- J. Choi and A. Manthiram, Electrochem. Solid-State, 2004, 7, A365 CrossRef CAS.
- A. D. Robertson and P. G. Bruce, Chem. Mater., 2003, 15, 1984–1992 CrossRef CAS.
- J. Yang and Y. Xia, ACS Appl. Mater. Interfaces, 2016, 8, 1297–1308 CrossRef CAS PubMed.
- S. Hy, F. Felix, J. Rick, W.-N. Su and B. J. Hwang, J. Am. Chem. Soc., 2014, 136, 999–1007 CrossRef CAS PubMed.
- H. Zhu, Y. Tang, K. M. Wiaderek, O. J. Borkiewicz, Y. Ren, J. Zhang, J. Ren, L. Fan, C. C. Li, D. Li, X.-L. Wang and Q. Liu, Nano Lett., 2021, 21, 9997–10005 CrossRef CAS PubMed.
- J. Hong, D.-H. Seo, S.-W. Kim, H. Gwon, S.-T. Oh and K. Kang, J. Mater. Chem., 2010, 20 RSC.
- H. Chen and M. S. Islam, Chem. Mater., 2016, 28, 6656–6663 CrossRef CAS.
- N. Yabuuchi, K. Yoshii, S.-T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef CAS PubMed.
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 CrossRef PubMed.
- B. Li and D. Xia, Adv. Mater., 2017, 29, 1701054 CrossRef PubMed.
- A. Grimaud, W. T. Hong, Y. Shao-Horn and J. M. Tarascon, Nat. Mater., 2016, 15, 121–126 CrossRef CAS PubMed.
- L. A. Montoro, M. Abbate, E. C. Almeida and J. M. Rosolen, Chem. Phys. Lett., 1999, 309, 14–18 CrossRef CAS.
- P. Yan, J. Zheng, Z.-K. Tang, A. Devaraj, G. Chen, K. Amine, J.-G. Zhang, L.-M. Liu and C. Wang, Nat. Nanotechnol., 2019, 14, 602–608 CrossRef CAS PubMed.
- J. Chen, W. Deng, X. Gao, S. Yin, L. Yang, H. Liu, G. Zou, H. Hou and X. Ji, ACS Nano, 2021, 15, 6061–6104 CrossRef CAS PubMed.
- K. Luo, M. R. Roberts, N. Guerrini, N. Tapia-Ruiz, R. Hao, F. Massel, D. M. Pickup, S. Ramos, Y.-S. Liu, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, J. Am. Chem. Soc., 2016, 138, 11211–11218 CrossRef CAS PubMed.
- M. Ben Yahia, J. Vergnet, M. Saubanère and M.-L. Doublet, Nat. Mater., 2019, 18, 496–502 CrossRef CAS PubMed.
- X. Rong, J. Liu, E. Hu, Y. Liu, Y. Wang, J. Wu, X. Yu, K. Page, Y.-S. Hu, W. Yang, H. Li, X.-Q. Yang, L. Chen and X. Huang, Joule, 2018, 2, 125–140 CrossRef CAS.
- K. Luo, M. R. Roberts, R. Hao, N. Guerrini, D. M. Pickup, Y.-S. Liu, K. Edström, J. Guo, A. V. Chadwick, L. C. Duda and P. G. Bruce, Nat. Chem., 2016, 8, 684–691 CrossRef CAS PubMed.
- M. Okubo and A. Yamada, ACS Appl. Mater. Interfaces, 2017, 9, 36463–36472 CrossRef CAS PubMed.
- P. E. Pearce, G. Assat, A. Iadecola, F. Fauth, R. Dedryvère, A. Abakumov, G. Rousse and J.-M. Tarascon, J. Phys. Chem. C, 2020, 124, 2771–2781 CrossRef CAS.
- U.-H. Kim, L.-Y. Kuo, P. Kaghazchi, C. S. Yoon and Y.-K. Sun, ACS Energy Lett., 2019, 4, 576–582 CrossRef CAS.
- Y. Yu, P. Karayaylali, S. H. Nowak, L. Giordano, M. Gauthier, W. Hong, R. Kou, Q. Li, J. Vinson, T. Kroll, D. Sokaras, C.-J. Sun, N. Charles, F. Maglia, R. Jung and Y. Shao-Horn, Chem. Mater., 2019, 31, 7864–7876 CrossRef CAS PubMed.
- S. Kim, M. Aykol, V. I. Hegde, Z. Lu, S. Kirklin, J. R. Croy, M. M. Thackeray and C. Wolverton, Energy Environ. Sci., 2017, 10, 2201–2211 RSC.
- E. McCalla, M. T. Sougrati, G. Rousse, E. J. Berg, A. Abakumov, N. Recham, K. Ramesha, M. Sathiya, R. Dominko, G. Van Tendeloo, P. Novák and J.-M. Tarascon, J. Am. Chem. Soc., 2015, 137, 4804–4814 CrossRef CAS PubMed.
- M. Yoon, Y. Dong, Y. Yoo, S. Myeong, J. Hwang, J. Kim, S. H. Choi, J. Sung, S. J. Kang, J. Li and J. Cho, Adv. Funct. Mater., 2019, 30, 1907903 CrossRef.
- A. R. Armstrong and P. G. Bruce, Electrochem. Solid-State, 2004, 7, A1 CrossRef CAS.
- A. D. Robertson and P. G. Bruce, Electrochem. Solid-State, 2004, 7, A294 CrossRef CAS.
- A. R. Armstrong, A. D. Robertson and P. G. Bruce, J. Power Sources, 2005, 146, 275–280 CrossRef CAS.
- T. E. Quine, M. J. Duncan, A. R. Armstrong, A. D. Robertson and P. G. Bruce, J. Mater. Chem., 2000, 10, 2838–2841 RSC.
- F. Weill, N. Tran, N. Martin, L. Croguennec and C. Delmas, Electrochem. Solid-State, 2007, 10, A194 CrossRef CAS.
- Y.-S. Hong, Y. J. Park, X. Wu, K. S. Ryu and S. H. Chang, Electrochem. Solid-State, 2003, 6, A166 CrossRef CAS.
- H. Koga, L. Croguennec, M. Ménétrier, K. Douhil, S. Belin, L. Bourgeois, E. Suard, F. Weill and C. Delmas, J. Electrochem. Soc., 2013, 160, A786–A792 CrossRef CAS.
- H. Koga, L. Croguennec, M. Ménétrier, P. Mannessiez, F. Weill and C. Delmas, J. Power Sources, 2013, 236, 250–258 CrossRef CAS.
- M. Oishi, C. Yogi, I. Watanabe, T. Ohta, Y. Orikasa, Y. Uchimoto and Z. Ogumi, J. Power Sources, 2015, 276, 89–94 CrossRef CAS.
- S. Han, Y. Xia, Z. Wei, B. Qiu, L. Pan, Q. Gu, Z. Liu and Z. Guo, J. Mater. Chem. A, 2015, 3, 11930–11939 RSC.
- G. Assat, A. Iadecola, C. Delacourt, R. Dedryvère and J.-M. Tarascon, Chem. Mater., 2017, 29, 9714–9724 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- S. Chong, Y. Chen, W. Yan, S. Guo, Q. Tan, Y. Wu, T. Jiang and Y. Liu, J. Power Sources, 2016, 332, 230–239 CrossRef CAS.
- X. Cao, Y. Qiao, M. Jia, P. He and H. Zhou, Adv. Energy Mater., 2021, 12 Search PubMed.
- C. R. Fell, D. Qian, K. J. Carroll, M. Chi, J. L. Jones and Y. S. Meng, Chem. Mater., 2013, 25, 1621–1629 CrossRef CAS.
- N. Tran, L. Croguennec, M. Ménétrier, F. Weill, P. Biensan, C. Jordy and C. Delmas, Chem. Mater., 2008, 20, 4815–4825 CrossRef CAS.
- J. R. Croy, M. Balasubramanian, K. G. Gallagher and A. K. Burrell, Acc. Chem. Res., 2015, 48, 2813–2821 CrossRef CAS PubMed.
- D. Mohanty, A. Huq, E. A. Payzant, A. S. Sefat, J. Li, D. P. Abraham, D. L. Wood and C. Daniel, Chem. Mater., 2013, 25, 4064–4070 CrossRef CAS.
- M. Sathiya, A. M. Abakumov, D. Foix, G. Rousse, K. Ramesha, M. Saubanère, M. L. Doublet, H. Vezin, C. P. Laisa, A. S. Prakash, D. Gonbeau, G. VanTendeloo and J. M. Tarascon, Nat. Mater., 2015, 14, 230–238 CrossRef CAS PubMed.
- A. Boulineau, L. Simonin, J.-F. Colin, C. Bourbon and S. Patoux, Nano Lett., 2013, 13, 3857–3863 CrossRef CAS PubMed.
- D. Mohanty, J. Li, D. P. Abraham, A. Huq, E. A. Payzant, D. L. Wood and C. Daniel, Chem. Mater., 2014, 26, 6272–6280 CrossRef CAS.
- S. G. Rinaldo, K. G. Gallagher, B. R. Long, J. R. Croy, M. Bettge, D. P. Abraham, J. Bareño and D. W. Dees, J. Electrochem. Soc., 2015, 162, A897–A904 CrossRef CAS.
- D. Qian, B. Xu, M. Chi and Y. S. Meng, Phys. Chem. Chem. Phys., 2014, 16, 14665–14668 RSC.
- Y. Okamoto, J. Electrochem. Soc., 2011, 159, A152–A157 CrossRef.
- E. Lee, R. Koritala, D. J. Miller and C. S. Johnson, J. Electrochem. Soc., 2014, 162, A322–A329 CrossRef.
- H. C. Shim, D. Kim, D. Shin, S. Hyun, C.-S. Woo, T. Yu and J.-P. Ahn, Phys. Chem. Chem. Phys., 2017, 19, 1268–1275 RSC.
- W. Zhang, Y. Sun, H. Deng, J. Ma, Y. Zeng, Z. Zhu, Z. Lv, H. Xia, X. Ge, S. Cao, Y. Xiao, S. Xi, Y. Du, A. Cao and X. Chen, Adv. Mater., 2020, 32, 2000496 CrossRef CAS PubMed.
- J. L. Shi, J. N. Zhang, M. He, X. D. Zhang, Y. X. Yin, H. Li, Y. G. Guo, L. Gu and L. J. Wan, ACS Appl. Mater. Interfaces, 2016, 8, 20138–20146 CrossRef CAS PubMed.
- D. Mohanty, S. Kalnaus, R. A. Meisner, K. J. Rhodes, J. Li, E. A. Payzant, D. L. Wood and C. Daniel, J. Power Sources, 2013, 229, 239–248 CrossRef CAS.
- C. S. Johnson, N. Li, C. Lefief, J. T. Vaughey and M. M. Thackeray, Chem. Mater., 2008, 20, 6095–6106 CrossRef CAS.
- C. R. Fell, M. Chi, Y. S. Meng and J. L. Jones, Solid State Ionics, 2012, 207, 44–49 CrossRef CAS.
- V. A. Godbole, J.-F. o. Colin and P. Novák, J. Electrochem. Soc., 2011, 158, A1005–A1010 CrossRef CAS.
- X. Ding, D. Luo, J. Cui, H. Xie, Q. Ren and Z. Lin, Angew. Chem., Int. Ed., 2020, 59, 7778–7782 CrossRef CAS PubMed.
- J. Gao, J. Kim and A. Manthiram, Electrochem. Commun., 2009, 11, 84–86 CrossRef CAS.
- H. Yu, Y.-G. So, Y. Ren, T. Wu, G. Guo, R. Xiao, J. Lu, H. Li, Y. Yang, H. Zhou, R. Wang, K. Amine and Y. Ikuhara, J. Am. Chem. Soc., 2018, 140, 15279–15289 CrossRef CAS PubMed.
- J. Ma, Y.-N. Zhou, Y. Gao, X. Yu, Q. Kong, L. Gu, Z. Wang, X.-Q. Yang and L. Chen, Chem. Mater., 2014, 26, 3256–3262 CrossRef CAS.
- C. Jacob, J. Jian, Q. Su, S. Verkhoturov, R. Guillemette and H. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 2433–2438 CrossRef CAS PubMed.
- Y. Sun, H.-W. Lee, Z. W. Seh, N. Liu, J. Sun, Y. Li and Y. Cui, Nat. Energy, 2016, 1, 15008 CrossRef CAS.
- Y. Gao, X. Wang, J. Ma, Z. Wang and L. Chen, Chem. Mater., 2015, 27, 3456–3461 CrossRef CAS.
- X. Zhang, I. Belharouak, L. Li, Y. Lei, J. W. Elam, A. Nie, X. Chen, R. S. Yassar and R. L. Axelbaum, Adv. Energy Mater., 2013, 3, 1299–1307 CrossRef CAS.
- B. Xiao and X. Sun, Adv. Energy Mater., 2018, 8, 1802057 CrossRef.
- M. Gu, I. Belharouak, J. Zheng, H. Wu, J. Xiao, A. Genc, K. Amine, S. Thevuthasan, D. R. Baer, J.-G. Zhang, N. D. Browning, J. Liu and C. Wang, ACS Nano, 2013, 7, 760–767 CrossRef CAS PubMed.
- J. Zheng, P. Xu, M. Gu, J. Xiao, N. D. Browning, P. Yan, C. Wang and J.-G. Zhang, Chem. Mater., 2015, 27, 1381–1390 CrossRef CAS.
- W. Zuo, M. Luo, X. Liu, J. Wu, H. Liu, J. Li, M. Winter, R. Fu, W. Yang and Y. Yang, Energy Environ. Sci., 2020, 13, 4450–4497 RSC.
- Y. Lei, J. Ni, Z. Hu, Z. Wang, F. Gui, B. Li, P. Ming, C. Zhang, Y. Elias, D. Aurbach and Q. Xiao, Adv. Energy Mater., 2020, 10, 2002506 CrossRef CAS.
- P. K. Nayak, J. Grinblat, M. Levi, E. Levi, S. Kim, J. W. Choi and D. Aurbach, Adv. Energy Mater., 2016, 6, 1502398 CrossRef.
- X. Su, X. Wang, H. Chen, Z. Yu, J. Qi, S. Tao, W. Chu and L. Song, Chin. J. Chem., 2017, 35, 1853–1860 CrossRef CAS.
- S. Kang, H. Qin, Y. Fang, X. Li and Y. Wang, Electrochim. Acta, 2014, 144, 22–30 CrossRef CAS.
- N. Li, R. An, Y. Su, F. Wu, L. Bao, L. Chen, Y. Zheng, H. Shou and S. Chen, J. Mater. Chem. A, 2013, 1, 9760–9767 RSC.
- Y. Shin, W. H. Kan, M. Aykol, J. K. Papp, B. D. McCloskey, G. Chen and K. A. Persson, Nat. Commun., 2018, 9, 4597 CrossRef PubMed.
- Z. He, Z. Wang, H. Chen, Z. Huang, X. Li, H. Guo and R. Wang, J. Power Sources, 2015, 299, 334–341 CrossRef CAS.
- Q. Li, D. Zhou, L. Zhang, D. Ning, Z. Chen, Z. Xu, R. Gao, X. Liu, D. Xie, G. Schumacher and X. Liu, Adv. Funct. Mater., 2019, 29, 1806706 CrossRef.
- L. Bao, Z. Yang, L. Chen, Y. Su, Y. Lu, W. Li, F. Yuan, J. Dong, Y. Fang, Z. Ji, C. Shi and W. Feng, ChemSusChem, 2019, 12, 2294–2301 CrossRef CAS PubMed.
- S. Liu, Z. Liu, X. Shen, W. Li, Y. Gao, M. N. Banis, M. Li, K. Chen, L. Zhu, R. Yu, Z. Wang, X. Sun, G. Lu, Q. Kong, X. Bai and L. Chen, Adv. Energy Mater., 2018, 8, 1802105 CrossRef.
- R. Yu, Z. Zhang, S. Jamil, J. Chen, X. Zhang, X. Wang, Z. Yang, H. Shu and X. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 16561–16571 CrossRef CAS PubMed.
- W. He, P. Liu, B. Qu, Z. Zheng, H. Zheng, P. Deng, P. Li, S. Li, H. Huang, L. Wang, Q. Xie and D. L. Peng, Adv. Sci., 2019, 6, 1802114 CrossRef PubMed.
- S. Chen, Z. Chen, M. Xia, C. Cao and Y. Luo, ACS Appl. Energy Mater., 2018, 1, 4065–4074 CrossRef CAS.
- Q. Li, G. Li, C. Fu, D. Luo, J. Fan and L. Li, ACS Appl. Mater. Interfaces, 2014, 6, 10330–10341 CrossRef CAS PubMed.
- X. Jin, Q. Xu, H. Liu, X. Yuan and Y. Xia, Electrochim. Acta, 2014, 136, 19–26 CrossRef CAS.
- D. Wang, Y. Huang, Z. Huo and L. Chen, Electrochim. Acta, 2013, 107, 461–466 CrossRef CAS.
- S. Sharifi-Asl, J. Lu, K. Amine and R. Shahbazian-Yassar, Adv. Energy Mater., 2019, 9, 1900551 CrossRef.
- S. Hy, J.-H. Cheng, J.-Y. Liu, C.-J. Pan, J. Rick, J.-F. Lee, J.-M. Chen and B. J. Hwang, Chem. Mater., 2014, 26, 6919–6927 CrossRef CAS.
- R.-P. Qing, J.-L. Shi, D.-D. Xiao, X.-D. Zhang, Y.-X. Yin, Y.-B. Zhai, L. Gu and Y.-G. Guo, Adv. Energy Mater., 2016, 6, 1501914 CrossRef.
- W. Cho, S. Myeong, N. Kim, S. Lee, Y. Kim, M. Kim, S. J. Kang, N. Park, P. Oh and J. Cho, Adv. Mater., 2017, 29, 1605578 CrossRef PubMed.
- D. Luo, X. Ding, J. Fan, Z. Zhang, P. Liu, X. Yang, J. Guo, S. Sun and Z. Lin, Angew. Chem., Int. Ed., 2020, 59, 23061–23066 CrossRef CAS PubMed.
- Z. Sun, L. Xu, C. Dong, H. Zhang, M. Zhang, Y. Ma, Y. Liu, Z. Li, Y. Zhou, Y. Han and Y. Chen, Nano Energy, 2019, 63, 103887 CrossRef CAS.
- T. Wang, C. Zhang, S. Li, X. Shen, L. Zhou, Q. Huang, C. Liang, Z. Wang, X. Wang and W. Wei, ACS Appl. Mater. Interfaces, 2021, 13, 12159–12168 CrossRef CAS PubMed.
- H. Z. Zhang, Q. Q. Qiao, G. R. Li and X. P. Gao, J. Mater. Chem. A, 2014, 2, 7454–7460 RSC.
- H.-Z. Zhang, F. Li, G.-L. Pan, G.-R. Li and X.-P. Gao, J. Electrochem. Soc., 2015, 162, A1899–A1904 CrossRef CAS.
- Y. Zhao, J. Liu, S. Wang, R. Ji, Q. Xia, Z. Ding, W. Wei, Y. Liu, P. Wang and D. G. Ivey, Adv. Funct. Mater., 2016, 26, 4760–4767 CrossRef CAS.
- Y. Yu, Z. Yang, J. Zhong, Y. Liu, J. Li, X. Wang and F. Kang, ACS Appl. Mater. Interfaces, 2020, 12, 13996–14004 CrossRef CAS PubMed.
- J. An, L. Shi, G. Chen, M. Li, H. Liu, S. Yuan, S. Chen and D. Zhang, J. Mater. Chem. A, 2017, 5, 19738–19744 RSC.
- H. Yan, B. Li, Z. Yu, W. Chu and D. Xia, J. Phys. Chem. C, 2017, 121, 7155–7163 CrossRef CAS.
- Z. Gong and Y. Yang, Energy Environ. Sci., 2011, 4, 3223–3242 RSC.
- B. Li, H. Yan, J. Ma, P. Yu, D. Xia, W. Huang, W. Chu and Z. Wu, Adv. Funct. Mater., 2014, 24, 5112–5118 CrossRef CAS.
- D. Liu, X. Fan, Z. Li, T. Liu, M. Sun, C. Qian, M. Ling, Y. Liu and C. Liang, Nano Energy, 2019, 58, 786–796 CrossRef CAS.
- G. Chen, J. An, Y. Meng, C. Yuan, B. Matthews, F. Dou, L. Shi, Y. Zhou, P. Song, G. Wu and D. Zhang, Nano Energy, 2019, 57, 157–165 CrossRef CAS.
- Y. Liu, Z. He, H. Zhu, Y. Ren, Y. Zhu, Y. Huang, D. Liang, S. Dou, J. Xu, C.-J. Sun, X.-Li Wang, Y. Deng, Q. Yuan, X. Liu, J. Wu, Y. Chen and Q. Liu, Adv. Energy Mater., 2021, 11, 2003479 CrossRef CAS.
- X. D. Zhang, J. L. Shi, J. Y. Liang, Y. X. Yin, J. N. Zhang, X. Q. Yu and Y. G. Guo, Adv. Mater., 2018, 30, 1801751 CrossRef PubMed.
- L. Dai, N. Li, L. Chen, Y. Su, C. M. Chen, F. Su, L. Bao, S. Chen and F. Wu, Chin. J. Chem., 2021, 39, 345–352 CrossRef CAS.
- J. Ma, B. Li, L. An, H. Wei, X. Wang, P. Yu and D. Xia, J. Power Sources, 2015, 277, 393–402 CrossRef CAS.
- T. Zhao, L. Li, R. Chen, H. Wu, X. Zhang, S. Chen, M. Xie, F. Wu, J. Lu and K. Amine, Nano Energy, 2015, 15, 164–176 CrossRef CAS.
- S. Hu, Y. Li, Y. Chen, J. Peng, T. Zhou, W. K. Pang, C. Didier, V. K. Peterson, H. Wang, Q. Li and Z. Guo, Adv. Energy Mater., 2019, 9, 1901795 CrossRef.
- J. Zheng, M. Gu, J. Xiao, B. J. Polzin, P. Yan, X. Chen, C. Wang and J.-G. Zhang, Chem. Mater., 2014, 26, 6320–6327 CrossRef CAS.
- W. Liu, P. Oh, X. Liu, S. Myeong, W. Cho and J. Cho, Adv. Energy Mater., 2015, 5, 1500274 CrossRef.
- E. Zhao, Q. Li, F. Meng, J. Liu, J. Wang, L. He, Z. Jiang, Q. Zhang, X. Yu, L. Gu, W. Yang, H. Li, F. Wang and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 4323–4327 CrossRef CAS PubMed.
- H. Guo, Z. Wei, K. Jia, B. Qiu, C. Yin, F. Meng, Q. Zhang, L. Gu, S. Han, Y. Liu, H. Zhao, W. Jiang, H. Cui, Y. Xia and Z. Liu, Energy Storage, 2019, 16, 220–227 CrossRef.
- B. Qiu, M. Zhang, L. Wu, J. Wang, Y. Xia, D. Qian, H. Liu, S. Hy, Y. Chen, K. An, Y. Zhu, Z. Liu and Y. S. Meng, Nat. Commun., 2016, 7, 12108 CrossRef CAS PubMed.
- E. M. Erickson, H. Sclar, F. Schipper, J. Liu, R. Tian, C. Ghanty, L. Burstein, N. Leifer, J. Grinblat, M. Talianker, J. Y. Shin, J. K. Lampert, B. Markovsky, A. I. Frenkel and D. Aurbach, Adv. Energy Mater., 2017, 7, 1700708 CrossRef.
- A. Van der Ven and G. Ceder, J. Power Sources, 2001, 97, 529–531 CrossRef.
- V. Pimenta, M. Sathiya, D. Batuk, A. M. Abakumov, D. Giaume, S. Cassaignon, D. Larcher and J.-M. Tarascon, Chem. Mater., 2017, 29, 9923–9936 CrossRef CAS.
- Z. Zhu, D. Yu, Y. Yang, C. Su, Y. Huang, Y. Dong, I. Waluyo, B. Wang, A. Hunt, X. Yao, J. Lee, W. Xue and J. Li, Nat. Energy, 2019, 4, 1049–1058 CrossRef CAS.
- J. Zheng, S. Deng, Z. Shi, H. Xu, H. Xu, Y. Deng, Z. Zhang and G. Chen, J. Power Sources, 2013, 221, 108–113 CrossRef CAS.
- C. Yang, Q. Zhang, W. Ding, J. Zang, M. Lei, M. Zheng and Q. Dong, J. Mater. Chem. A, 2015, 3, 7554–7559 RSC.
- D. Y. W. Yu, K. Yanagida and H. Nakamura, J. Electrochem. Soc., 2010, 157, A1177–A1182 CrossRef CAS.
- C.-H. Shen, L. Huang, Z. Lin, S.-Y. Shen, Q. Wang, H. Su, F. Fu and X.-M. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 13271–13279 CrossRef CAS PubMed.
- J. Zhang, Z. Lei, J. Wang, Y. NuLi and J. Yang, ACS Appl. Mater. Interfaces, 2015, 7, 15821–15829 CrossRef CAS PubMed.
- K. Karthikeyan, K. W. Nam, E. Y. Hu, X. Q. Yang and Y. S. Lee, Bull. Korean Chem. Soc., 2013, 34, 1995–2000 CrossRef CAS.
- Y. Paik, C. P. Grey, C. S. Johnson, J.-S. Kim and M. M. Thackeray, Chem. Mater., 2002, 14, 5109–5115 CrossRef CAS.
- S. H. Kang, C. S. Johnson, J. T. Vaughey, K. Amine and M. M. Thackeray, J. Electrochem. Soc., 2006, 153, A1186–A1192 CrossRef CAS.
- Y. Li, S. Xu, W. Zhao, Z. Chen, Z. Chen, S. Li, J. Hu, B. Cao, J. Li, S. Zheng, Z. Chen, T. Zhang, M. Zhang and F. Pan, Energy Storage, 2021, 45, 422–431 CrossRef.
- Y. Zuo, B. Li, N. Jiang, W. Chu, H. Zhang, R. Zou and D. Xia, Adv. Mater., 2018, 30, 1707255 CrossRef PubMed.
- C. Cui, X. Fan, X. Zhou, J. Chen, Q. Wang, L. Ma, C. Yang, E. Hu, X. Q. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 8918–8927 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally: Zijia Yin and He Zhu. |
| This journal is © The Royal Society of Chemistry 2022 |