
Reactive oxygen species on transition metal-based catalysts for sustainable environmental applications
Yonglin
Wen†
ab,
Jiawei
Yan†
ab,
Bixia
Yang
ab,
Zanyong
Zhuang
 *ab and
Yan
Yu
*ab
*ab and
Yan
Yu
*ab
aCollege of Materials Science and Engineering, Fuzhou University, New Campus, Minhou, Fujian 350108, China. E-mail: zyzhuang@fzu.edu.cn; yuyan@fzu.edu.cn
bKey Laboratory of Advanced Materials Technologies, Fuzhou University, Fuzhou 350108, China
First published on 18th May 2022
Abstract
Tailoring reactive oxygen species (ROS) in advanced oxidation processes (AOPs) in a controlled manner is essential in chemical synthesis and environmental applications. In this paper, we begin with an overview of four ROS, and summarize methods and various precursors (e.g., O2, H2O2, and persulfate) to generate particular ROS. We then examine the use of the ROS in the degradation of pollutants and in the synthesis of value-added chemicals. We highlight the use and mechanism response of transition metal catalysts, generally developed with defect engineering, in advancing AOPs. We conclude with an outlook of current challenges and future perspectives of applying ROS in AOPs. We anticipate this review to inspire researchers to develop green, safe, and efficient ROS systems for sustainable environmental applications.
 Yonglin Wen | Yonglin Wen received his BSc in materials science and engineering from Fuzhou University. He is currently pursuing his PhD degree at Fuzhou University under the supervision of Prof. Yu and Prof. Zhuang. His research focuses on the design of Mn-based catalysts for heterogeneous oxidation catalysis and biomass conversion. |
 Jiawei Yan | Jiawei Yan received his BSc in materials science and engineering from Fujian Agriculture and Forestry University. He is currently pursuing his PhD degree at Fuzhou University under the supervision of Prof. Zhuang and Prof. Yu. His research focuses on the design of transition metal-based materials deployed for advanced oxidation processes. |
 Zanyong Zhuang | Zanyong Zhuang received his BS degree (2006) in chemistry from Xiamen University, and PhD degree (2011) from the Fujian Institute of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS). Currently, he is a full professor at Fuzhou University. His research interests mainly focus on the rational design of transition metal-based catalysts for energy and environmental applications, including advanced oxidation reactions and CO2 reduction reactions. |
 Yan Yu | Yan Yu received her BS, MS, and PhD degrees from Fuzhou University. She was a postdoctoral fellow from 2010–2013 at the Fujian Institue of Research on the Structure of Matter (FJIRSM), Chinese Academy of Sciences (CAS). Currently, she is a full professor at Fuzhou University. Her research interests include environmental remediation, water purification, ecological materials, and photocatalytic CO2 reduction, and H2 production. |
1 Introduction
The growing consumption of petrochemical products is quickly depleting petroleum and fossil resources worldwide and discharging organic pollutants into the environment. Efficient green techniques are needed to reduce pollution and to produce value-added chemicals from renewable feedstock.1–4 Advanced oxidation processes (AOPs) mediated by reactive oxygen species (ROS) have been demonstrated in the past few decades as the underpinning of many essential applications in environmental protection and sustainable development.5,6A key issue in developing suitable AOPs is finding the desired ROS with appropriate reactivity and selectivity.7 Fenton chemistry has been used extensively since 1894 to create the hydroxyl radical (HO˙), a powerful radical that can degrade toxic organic substrates thoroughly to CO2 and H2O. However, HO˙ has a short half-life and cannot be sustained in multi-step processes, and continuous addition of its precursor (e.g. H2O2) is required to produce a sufficient amount of HO˙ to ensure effective degradation of pollutants.8 Moreover, although it has long been used for the thorough degradation of recalcitrant organic contaminants to CO2 and H2O, it has limited applications in chemical synthesis or biomass conversion, as its high reactivity may cause undesired over-oxidation and degradation.9 Alternatively, researchers found that the superoxide radical (O2˙−) and singlet oxygen (1O2) can also engage in AOPs, both of which have a longer half-life than HO˙ as well as a more moderate oxidation potential suitable for controlled oxidation. In addition to these ROS generated over the catalysts, the lattice oxygen (OL) of transition metal oxides (TMO) can serve as a moderate ROS in heterogeneous AOPs to directly oxidize organic matter, and it has allowed successful oxidation reactions based on thermocatalysis, photocatalysis, electrocatalysis, etc.10,11 Using OL in chemical synthesis and biomass conversion is advantageous in that it can selectively oxidize the organic substrate and avoid over-oxidation.
Heterogeneous transition metal-based catalysts (e.g., oxides,12 hydroxides,13,14 oxyhydroxides,15 and sulfides16) are readily used in AOPs. Their advantages include earth abundance, high stability, low cost, etc.17 AOPs using heterogeneous catalysts manipulate the geometrical and/or electronic states of the precursors (e.g., H2O2, O2, PMS, PDS, and TMO) to create ROS on the catalyst surfaces. Hence, surface and defect engineering has emerged in recent years as a popular and appealing technique to customize the catalyst and more effectively activate the percursors.18–21 There are substantial developments in the rational design of deficient transition metal-based materials deployed for advancing AOP strategies.22–26 Anionic vacancies (e.g., oxygen, sulfur, and nitrogen) introduced to transition metal-based catalysts as point defects always have a pronounced effect on the physical and chemical properties of the catalyst.27,28 Engineering point defects creates new opportunities to afford advanced catalysts that can efficiently and selectively create ROS (HO˙, O2˙−, 1O2, OL) from the precursors to thus carry out the AOP as intended.29
Understanding the reactivity and the selective generation of distinct ROS and identifying the mechanism response of deficient catalysts in AOPs are essential for the rational design of potent catalysts with fine-tuned oxidation behavior.30 In this review, we firstly summarize the types and redox properties of ROS. We then systematically discuss the generation of ROS based on the precursor used and the methods to detect and identify the ROS. We also discuss defect engineering techniques that modify the catalyst surface and enable thorough or selective oxidation of organic matter. We next examine oxidation reactions that can be accomplished with the ROS. We finish with a brief comment on the current challenges and future developments of ROS in the burgeoning research area of AOPs.
2 Types of reactive oxygen species
2.1 Hydroxyl radical (HO˙)
The hydroxy radical is highly reactive and has demonstrated widespread use as a nonselective reagent in AOPs for wastewater treatment, as it can initiate complex chain oxidation reactions to convert organic matter to oxidized products or ultimately to CO2 and H2O. In acidic media, HO˙ has an extraordinarily high redox potential (2.8 V versus the normal hydrogen electrode, NHE) which is only second to that of fluorine.31 It can attack most organic pollutants with a high rate constant of 106 to 109 M−1 s−1, which is about 103 to 109 times faster than that of molecular O2 (1–103 M−1 s−1).32 However, the reactivity of HO˙ is pH-dependent according to the Nernst equation,33 and its standard electrode potential drops to 1.55 V vs. NHE in basic media.34 The HO˙ in aqueous solution has a half-life ranging from picoseconds to nanoseconds, and its self-diffusion coefficient in water is estimated to be 2.8 × 10−5 cm2 s−1. Hence, the diffusion distance of HO˙ is a few molecular diameters away from where the HO˙ is produced.35,36Mechanistically, the oxidation of organic matter by HO˙ may involve radical adduct formation, hydrogen abstraction, and electron transfer,37 and is highly dependent on the types of functional groups in the organic substrate.38 Because of its electrophilic nature, HO˙ normally prefers to react with electron-rich sites such as C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN bonds in the organic substrate rather than the CO bonds whose carbon atom is electron-deficient.39 The C–H bonds of saturated aliphatic compounds that have relatively small dissociation energy can experience hydrogen abstraction.40
C and CN bonds in the organic substrate rather than the CO bonds whose carbon atom is electron-deficient.39 The C–H bonds of saturated aliphatic compounds that have relatively small dissociation energy can experience hydrogen abstraction.40
2.2 Superoxide radical (O2˙−)
The superoxide radical, first proposed in 1934 as the radical anion in the potassium salt,41 is a relatively small univalent anion with an O–O bond distance between that of O2 and peroxide.42 The ground-state triplet molecular oxygen (O2) has two electrons occupying the separated π* orbitals of parallel spins (Fig. 1). When the ground-state O2 participates in the redox reaction, the reductants need to offer a pair of electrons of the same spin states, so that the electrons from reductants can fit into the vacant spaces in the π* orbitals of oxygen.33 Due to this limitation, the ground-state O2 is normally of weak oxidizing capacity via the 2-electron transfer process. O2˙− can be formed by one-electron reduction of O2, or by filling the two π* orbitals of O2 with an electron followed by protonation (Fig. 1). The homonuclear diatomic radical O2˙− has a slightly lower redox potential (2.4 V vs. RHE) than HO˙,43 and has demonstrated critical roles in chemical, biological, and environmental applications. Note that (somewhat perplexingly) O2˙− also has a reducing potential of E0 (O2/O2˙−) = −0.33 V and can be oxidized by a ferricenium ion to give singlet oxygen (1O2),44 because it has an extra electron in one of its π* antibonding orbitals compared to ground state dioxygen (3O2). The redox potential of E0 (O2/O2˙−) does no change with pH when the pH value is >4.8. At pH value <4.8, O2˙− will be protonated at pH < 4.8 which creates the perhydroxyl radical (˙O2H) of reduction potential E0 (O2, H+/˙O2H) = −0.046 V vs. NHE.31,33 The half-life of O2˙− can reach up to 51–422 s at pH = 2–10,45 much longer than those of HO˙ and 1O2.46 It means O2˙− can be prepared and work under alkaline conditions. The diffusion distance of O2˙− has been estimated to be about 35 μm, assuming a general diffusion coefficient (about 105 cm2 s−1) of small molecules.47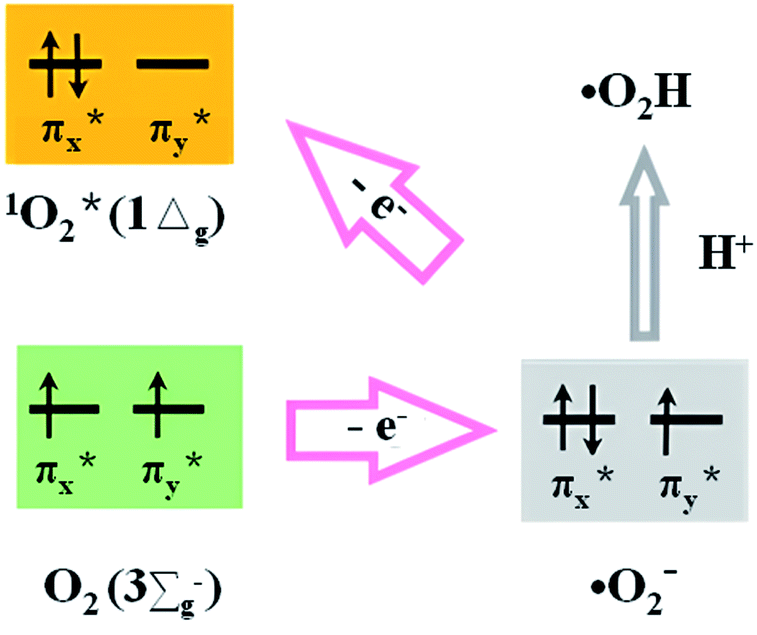 | ||
| Fig. 1 Molecular π* orbitals for O2, 1O2, and O2˙−.33 | ||
However HO˙ is electrophilic, and O2˙− can be both nucleophilic and electrophilic. The half-life of O2˙− is up to 51–422 s at pH = 2–10,45 which is longer than that of HO˙ and 1O2.46 Hence, O2˙− can be prepared and stabilized in strongly basic aqueous solutions. It is readily used to scavenge organic pollutants, such as treating pollutants in underground water under anoxic conditions,33,48,49 and can serve as a major ROS for the degradation of organic compounds such as bisphenol A (BPA) and dyes.50,51 However, HO˙ is highly active, and nonselective and tends to react with organic solvents; O2˙− can be used for the partial or selective oxidation of substrates in organic solvents.33,52–54
The reaction of organic matter with O2˙− may involve radical adduct formation, hydrogen abstraction, and electron transfer. Transferable hydrogen atoms on the substrates can be readily removed by O2˙− (e.g., dihydrophenazine to phenazine radical, N-methylhydrophenazine to N-methylphenazine radical).55 When O2˙− reacts with organic compounds via hydrogen abstraction, it generates carbon-based radicals that can further combine with oxygen to form peroxy intermediates that will decompose further. The nucleophilic substitution reaction of O2˙− was first reported in 1970,56,57 and O2˙− can react with alkyl halides via nucleophilic substitution to break the C–Cl bond to give dechlorinated products.58 Besides, O2˙− is found to also react with α-keto, α-hydroxy, and α-halo carbonyl compounds via nucleophilic addition at the carbonyl carbon, and the ensuing oxidative cleavage gives carboxylic acids.59 As a nucleophile, O2˙− can attack the carbonyl carbon of esters to yield carboxylic acid anions and alcohols, and the carbonyl carbon of acyl halides to yield diacyl peroxides.42 Interestingly, O2˙− may also act as a reducing agent in one-electron transfer reductions, and reactions involving electron transfer from O2˙− to organic substrates (e.g., nitroblue tetrazolium) have been exploited to detect or quantify O2˙−.33
2.3 Singlet oxygen (1O2)
There have been reports on using 1O2 in the synthesis of chemicals and natural products since the 1980s.60 Energy transfer to O2 could create singlet oxygen (1O2). 1O2 has paired electrons of opposite spins (Fig. 1), manifesting higher reactivity compared with the ground-state O2. As the unstable, excited state of molecular oxygen, 1O2 has a lower redox potential (0.81 V vs. NHE) than O2˙− and HO˙. The paired electrons in the external orbital of 1O2 have antiparallel spins.61 The reaction rate of 1O2 with organic compounds generally ranges in 104 to 107 M−1 s−1. The half-life of 1O2 (1Δg state) is several tens of milliseconds in air but only as short as 3 μs in water.33 A growing body of work has suggested that the redox potential of 1O2 does not change significantly with the pH value, so that the 1O2 can work in a wide pH range and exhibit considerable tolerance to environmental interference. Note that the half-life of 1O2 (1Δg state) is several tens of milliseconds in air but only as short as 3 μs in water.33 This very short half-life and the short diffusion distance (∼220 nm) restrict the reactivity of 1O2 to the proximity of where it is formed.62 As a result, it is less effective to rapidly remove organic pollutants in an aqueous environment by 1O2.58,63Nevertheless, 1O2 can be exploited as a moderate oxidant for selective or partial oxidations in chemical synthesis,43 such as the selective oxidation of various organic substrates including amines, alcohols, olefins, and sulfides.31,64–66 Studies have shown that 1O2 can oxidize unsaturated organic compounds by electrophilic attack and electron abstraction. For instance, the electrophilic attack of 1O2 to dioxin and furan gives endoperoxide intermediates,67 and the photosensitized oxygenation of phenol proceeds via hydrogen abstraction by 1O2.68 Because 1O2 is an excited state of O2, it may be deactivated to the original stable 3O2 without undergoing chemical reactions or electron transfer.
2.4 Surface lattice oxygen (O2−x)
The lattice oxygen (OL) of transition metal oxides (TMO) can directly oxidize organic matter through the Mars–van Krevelen (MvK) redox cycle.69,70 In the MvK mechanism, the substrate is firstly oxidized by the OL in the oxides, leaving behind an oxygen vacancy (OV) on the TMO surface that will be subsequently oxidized by O2, i.e., the stoichiometric oxidant, to regenerate OL. The reactivity of OL depends strongly on the formation of OVs from the oxides, the local coordination environment of the OL in oxides, and the strength of the M–O bond, and is sensitive to defects (or impurities), the composition, the surface, the interface, etc.71,72 Normally TMO with transition metals of high valence state exhibit superior OL activity. However reactions based on HO˙, O2˙−, and 1O2 normally proceed at room temperature and the reactivity weakens at elevated temperatures;73,74 OL can serve as an appealing oxidation reagent at elevated temperatures.Because OL is nucleophilic and has a lower redox potential, the oxidation of hydrocarbons with OL can give products in which all C–C bonds are retained (partial oxidation), which is in clear contrast to the electrophilic oxidation (e.g., by HO˙) that cleaves all C–C bonds.75 Thus, OL is readily used for the selective oxidation of biomass for sustainable development, as it can avoid undesired oxidative degradation. The use of OL is also key in soot combustion, the oxidation of CO and NOx, and the thermal oxidation of pollutants such as volatile organic compounds (VOC).76–78
3 Generation of reactive oxygen species
Manipulating the geometrical and electron states of various precursors (H2O2, O2, persulfates, and TMO) is key to generating ROS. In recent years, engineering point defects (e.g., anionic vacancies) on crystals (e.g., TMO) has emerged as an effective strategy to create catalytically active surfaces that help generate ROS. The deficient structure can strongly adsorb and activate the precursor oxidants (e.g., H2O2, O2, PMS, and PDS) by elongating the chemical bond (e.g., the O–O bond in H2O2).79 The electrons conducted from the bulk to the surface of the crystal may be trapped in the vacancies to create electron-rich regions80 as additional carriers for surface reactions to generate ROS.81 For catalytic reactions using OL in oxides as the ROS, the defect structure of oxides can help create OL of high reactivity by increasing the OL mobility and/or by promoting OL regeneration.(1) Activation of H2O2: hydrogen peroxide (H2O2) is a unique, versatile, green oxidizing agent for AOPs, as it has a high active oxygen content and gives H2O as the only reduction product. However, due to its low redox potential (1.77 eV), it does not readily oxidize organic compounds on its own,82 and Fenton chemistry is exploited to activate the H2O2 to generate ROS. The classic, well-known homogeneous Fenton reaction system based on Fe2+/H2O2 suffers from several drawbacks that impede large-scale applications, such as a narrow working pH range (mostly acidic), generation of iron-containing sludge, and limited activity.83 These hurdles prompted efforts to investigate the heterogeneous activation of H2O2 with transition metal-based catalysts, with which the type and amount of ROS can be regulated through distinct surface reactions. Although the decomposition of H2O2 on the catalyst surface is always spontaneous in 25–286 °C,84 the activation energy (20.93–96.30 kJ mol−1) and the rate of ROS generation depend heavily on the surface structure of the catalysts.85,86
(2) Activation of persulfates: peroxymonosulfate (PMS and HSO5−) and peroxydisulfate (PDS and S2O82−) are relatively strong oxidizers with redox potentials of 1.82 and 2.01 V, respectively. AOPs with persulfates were introduced for groundwater remediation in the late 1990s, because methods to activate persulfates are more abundant and the availability of persulfate salts reduces storage and transportation costs.87 Additionally, PMS and PDS attain a higher radical yield, and the treatment efficiency is less dependent on the operational parameters (e.g., pH, initial peroxide loading, and background constituents). Moreover, the activation of PMS/PDS on transition metal-based catalysts is usually carried out under alkali conditions.88 Hence, persulfate AOPs are good substitutes of Fenton chemistry with H2O2. The energy of the O–O bond in PMS is estimated to be in 140–213.3 kJ mol−1, and the activation requires adequate energy input. Appropriate activation of PMS and PDS to give ROS is imperative to ensure the desired oxidation performance, since PMS and PDS react directly with the organic contaminants only at a low rate.89 They can be activated with metal catalysts,90 carbon nanotubes,91 biochars,92 TMO,93etc.
(3) Activation of O2: among all oxidants, oxygen is the most environmentally friendly, inexpensive, and accessible.94 Due to the spin-flip restriction, molecular O2 is normally at the ground triplet state (3O2) and cannot directly engage in aerobic oxidation.95 The activation of molecular O2 into ROS (HO˙, O2˙−, and 1O2) is normally a fundamental step in aerobic catalytic oxidation reactions.96,97 However, the OO bond of O2 has a high bond energy (498 kJ mol−1) that must be overcome to initiate O2-mediated oxidation reactions. Transition metal-based catalysts (e.g., transition metal complexes,98 TMO,99 and transition metal sulfides100) critically improve the reactivity of O2 by effectively adsorbing the O2 molecules and weakening the OO bond of O2 through efficient charge transfer, and photocatalysis is frequently used to promote electron transfer in the activation of O2.101
(4) Lattice oxygen: metal oxides are the carrier of OL and they maintain the lattice structure throughout the reaction. The MvK mechanism of OL-mediating oxidation is well supported by isotope labelling, as using 18O2 as the stoichiometric oxidant will install 18O in the lattice of the metal oxides.102 Heating is generally required to activate OL and regenerate OL from O2, which can increase cost and make the reaction procedure tedious. The reactivity and regeneration of OL can be tuned by regulating the strength of the M–O bond (M: metal) and the chemical states of the metal, to enable stronger oxidation performance at a lower temperature.103 Anionic vacancies on the metal oxides change the electronic state and the surface structure, and they can improve the adsorption of O2, reduce the energy of the M–O bond, modify the electron density distribution, change the ion transport in the crystal, and expedite the migration of OL.104 Doping metal oxides with heteroatoms is an appealing method to create such vacancies.
3.1 Hydroxyl radical
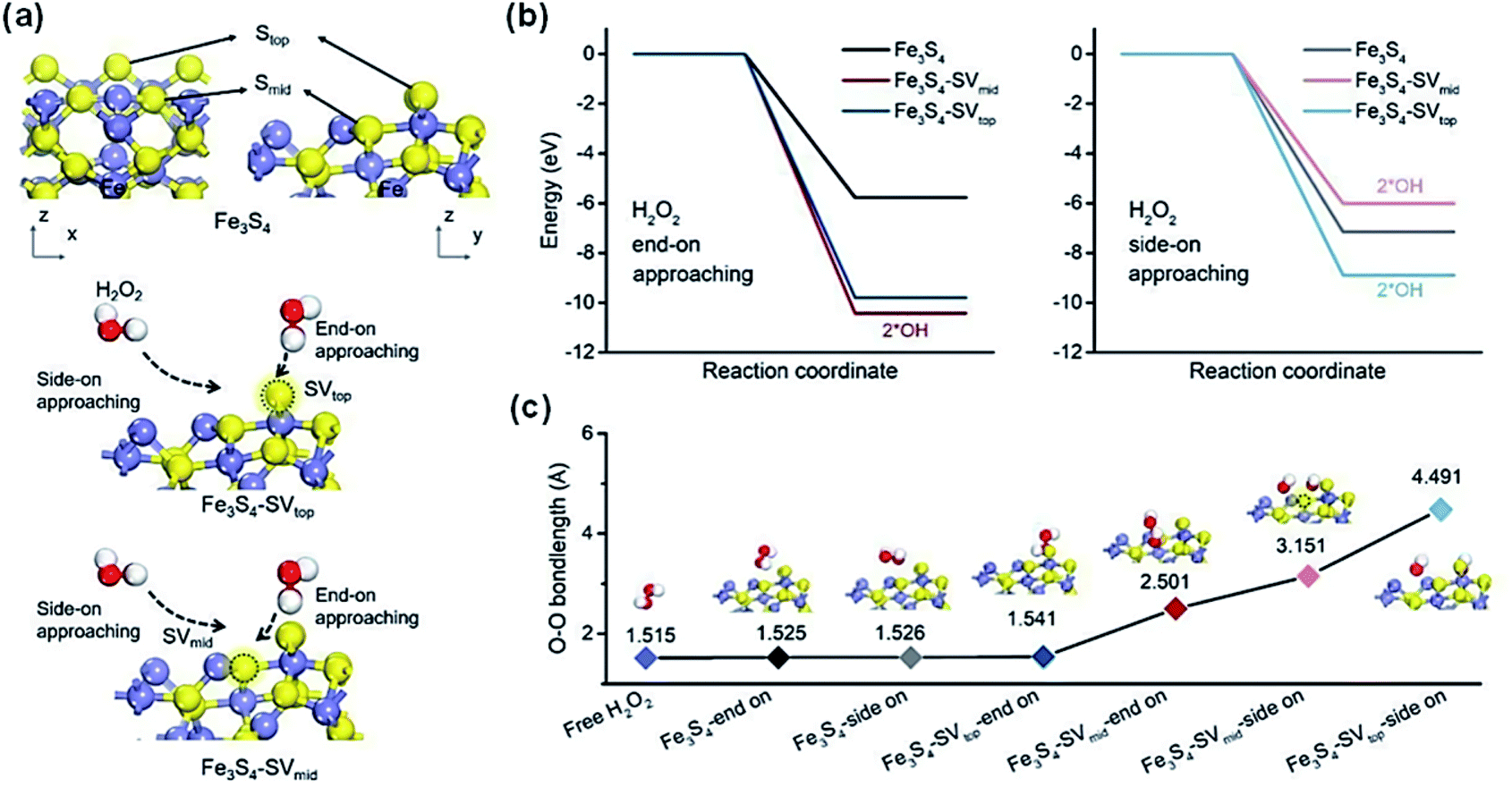 | ||
| Fig. 2 (a) Models of Fe3S4 and SVs-rich Fe3S4 with exposure of the (011) surface. (b) Energy diagrams of H2O2 adsorption on SVs-rich Fe3S4via end-on and side-on attachment models. (c) The O–O bond length of adsorbed H2O2 in different adsorption configurations. Adapted with permission from ref. 110 Copyright (2021) Elsevier. | ||
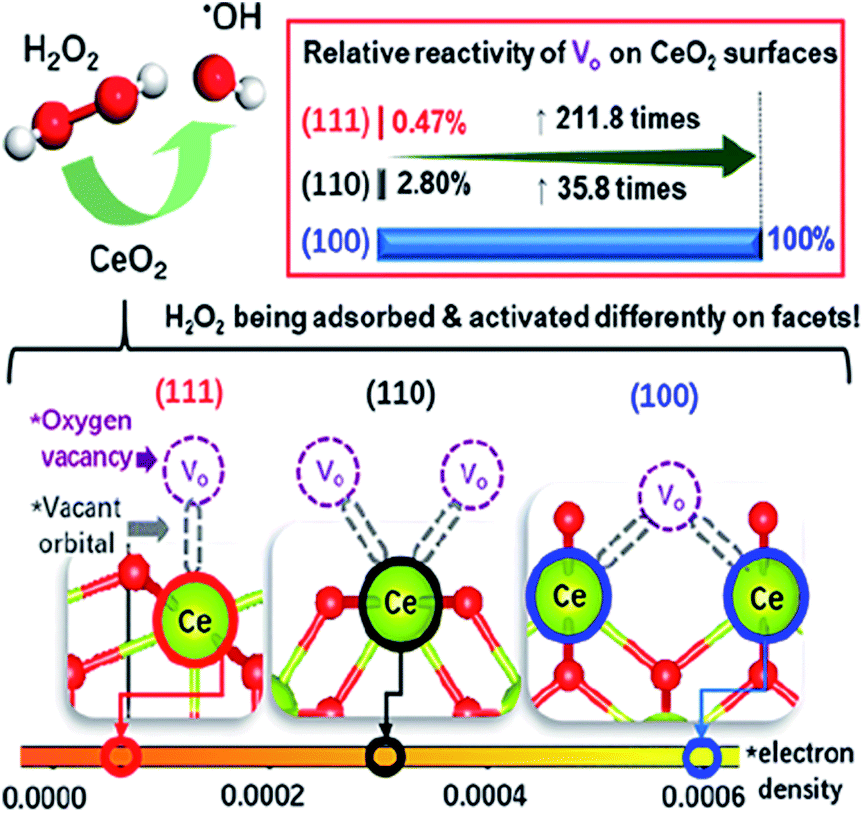 | ||
| Fig. 3 Schematic illustration of the synergy effect of Ce and OVs on the CeO2 surface for the efficient activation of H2O2 into HO˙. Adapted with permission from ref. 118 Copyright (2020) American Chemical Society. | ||
O bond to generate HO˙ when more electrons are transferred to O2.121 Setvin et al. used scanning tunnelling microscopy to pull the vacancies of an anatase crystal to its surface and proved that the surface OVs of TiO2 can serve as electron donation sites to activate the adsorbed O2.122 In an example that is beyond the use of transition metals, Zhao et al. reported that the O2 adsorbed on the surface of Pt SA/MgO with OVs can dissociate more easily to generate HO˙.123 The presence of OVs notably decreases the adsorption energy of O2 (Eads = −3.25 eV), elongates the OO bond (from 1.363 to 1.552 Å), and decreases the energy barrier of activating O2 (down to 2.48 eV). The O2 molecule adsorbed on the surface of Pt SA/MgO gives two adsorbed oxygen atoms [denoted as *O] after the OO bond is broken, and a further reaction between *O and adsorbed H2O gives HO˙ (i.e., O2 → 2*O, *O + H2O → 2HO˙).
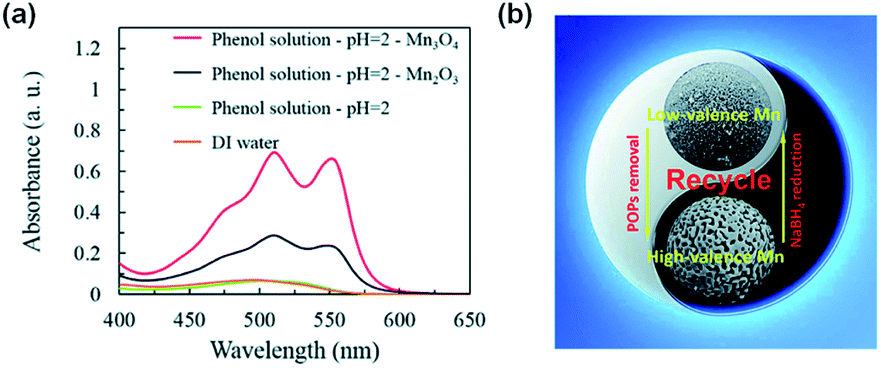 | ||
| Fig. 4 (a) Detection of H2O2 by activation of O2 over Mn3O4 and Mn2O3, respectively. (b) Schematic illustration of the recycling strategy for Mn3O4 and Mn2O3 on O2 activation to generate H2O2 that decomposes into HO˙. Adapted from ref. 120 with permission from The Royal Society of Chemistry. | ||
| Mn+ + HSO5˙− → M(n+1)+ + SO4˙− + OH− | (1) |
| Mn+ + S2O82− → M(n+1)+ + SO4˙− + SO42− | (2) |
| SO4˙− + H2O → HO˙ + SO42− + H+ | (3) |
| SO4˙− + OH− → HO˙ + SO42− | (4) |
3.2 Superoxide radical
 | ||
| Fig. 5 Side view of (a) H2O2 (b) initial configuration of two hydroxyl groups, and (c) superoxide radicals adsorbed on the TiO2 predicted by the GGA-TS method. (d) The decomposition mechanism of H2O2 into superoxide radicals on TiO2 NSs. Adapted with permission from ref. 132 Copyright (2017) American Chemical Society. | ||
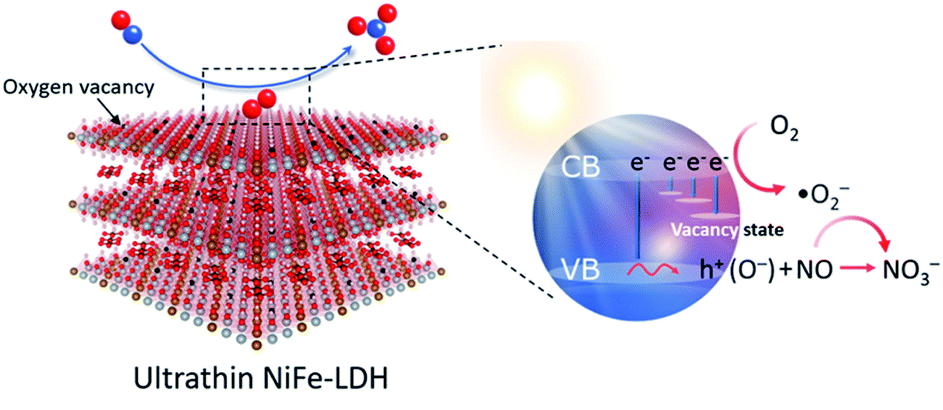 | ||
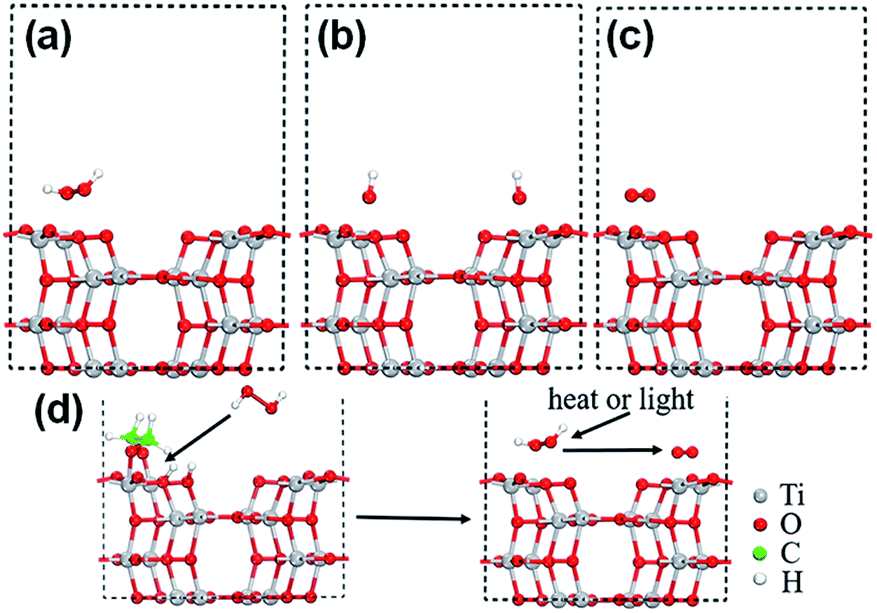
| Fig. 6 Schematic illustration of activation of O2 into O2˙− on NiFe-LDH with an OV for the selective photocatalytic oxidation of NO. Adapted with permission from ref. 140 Copyright (2022) American Chemical Society. | ||
In addition to OVs, S or N vacancies on the catalyst surface have also been exploited to tune the band structure and charge carrier transfer efficiency for O2 activation on photocatalysts. For example, N vacancies can create electron-accumulated centers to activate O2 over the Pt/C3N4 catalyst (Fig. 7b).146 The length of the OO bond is 1.496 Å for the activated O2 over the N-doped catalyst but 1.260–1.365 Å in the control (Fig. 7a). The SVs on In2S3 nanosheets can activate O2 into O2˙−via enhanced electron transfer under visible light irradiation.147 Theoretical calculations reveal that in the presence of the SVs, the density of states (DOS) of In2S3 is significantly enhanced at the valence band maximum (Fig. 7c and d). Consequently, electrons can be photoexcited to the conduction band and transferred to the defective surface more easily, and the defective In2S3 shows stronger interactions with O2 than pristine In2S3 (Fig. 7e and f).
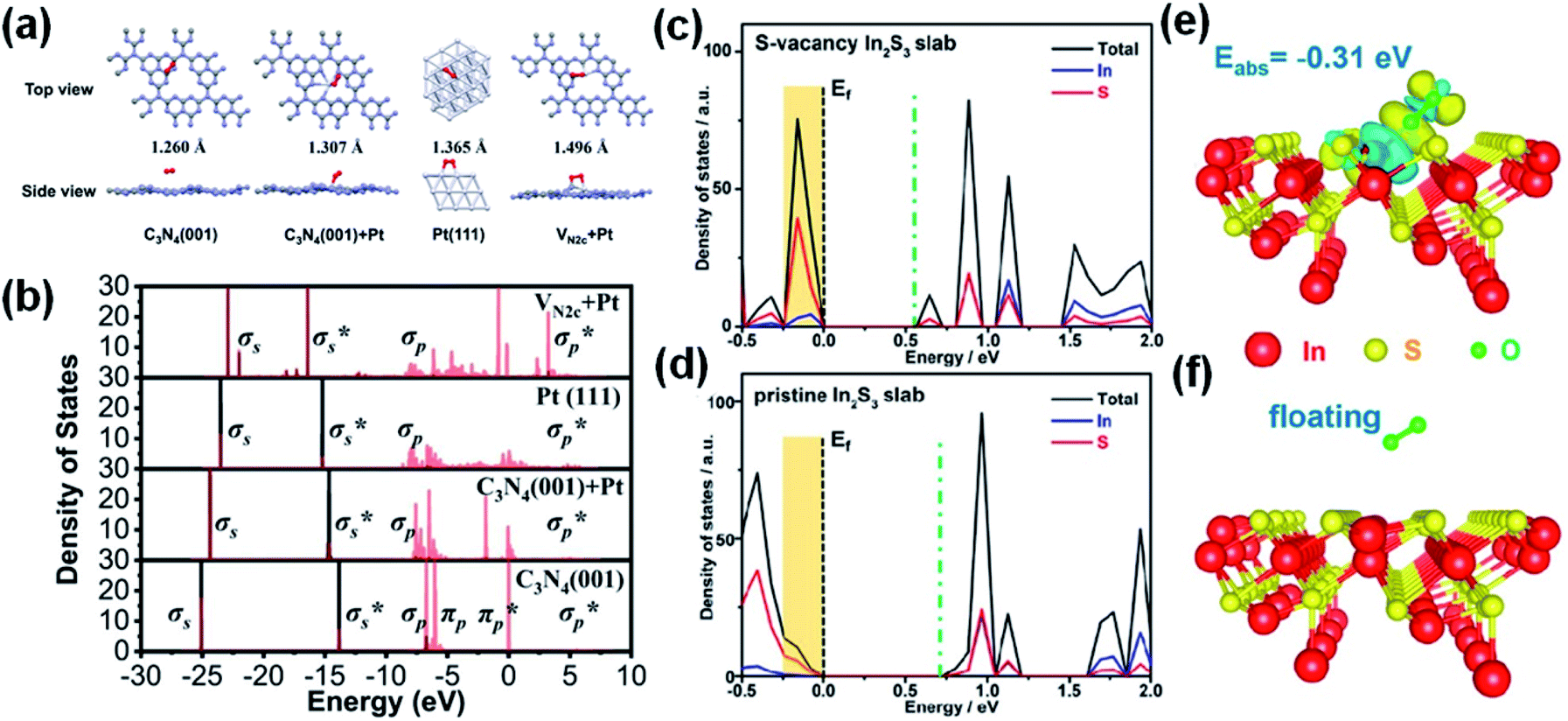 | ||
| Fig. 7 (a) The structures of O2 adsorbed on pure C3N4 (001), C3N4 (001) + Pt, Pt (111), and VN2c + Pt (gray, purple, white, and red represent C, N, Pt, and O). (b) Partial density of states (PDOS) of the adsorbed O2 (black and red lines represent the O 2s and 2p orbitals, respectively). Adapted with permission from ref. 146 Copyright (2021) Springer Nature Group. Calculated DOS of (c) S-vacancy and (d) pristine In2S3 slabs (Ef = Fermi level; orange shading indicates the increased DOS around the VBM). (e) The adsorption of O2 at the S vacancy of the deficient In2S3 slab. (f) O2 floating on the pristine In2S3 slab. Adapted with permission from ref. 147 Copyright (2022) American Chemical Society. | ||
While the vacancies can directly inject localized electrons into O2 to elongate the OO bond and activate O2, the geometry of the vacancies may tune the pathway of ROS generation by changing the band structure of catalysts. There are two different crystallographic positions of the oxygen atoms in the [BiO]22+ layer of (BiO)2CO3 (referred to below as BOC), and Rao et al. constructed two kinds of OVs (i.e., OVs1 and OVs2) in BOC by breaking Bi1-O1 or Bi2-O2 from two distinct lattice oxygens (O1 and O2) binding to the Bi atoms (Fig. 8a–c).148 Both OVs1 and OVs2 extend the lifetime of photogenerated charge carriers in their respective photocatalytic systems and promote the efficient separation of carriers (Fig. 8d and e). While both OVs1 and OVs2 enhance light absorption in the visible region by introducing an intermediate level in the band gap, only OVs1 can enhance light absorption in the infrared region (Fig. 8f). The photogenerated charge carriers in OVs1 react with the adsorbed O2 and H2O to form O2˙− and HO˙, respectively. In contrast, the observed redshift indicates that OVs2 decreases the optical bandgap energy of BOC, and HO˙ cannot be generated on the catalyst surface with OVs2.
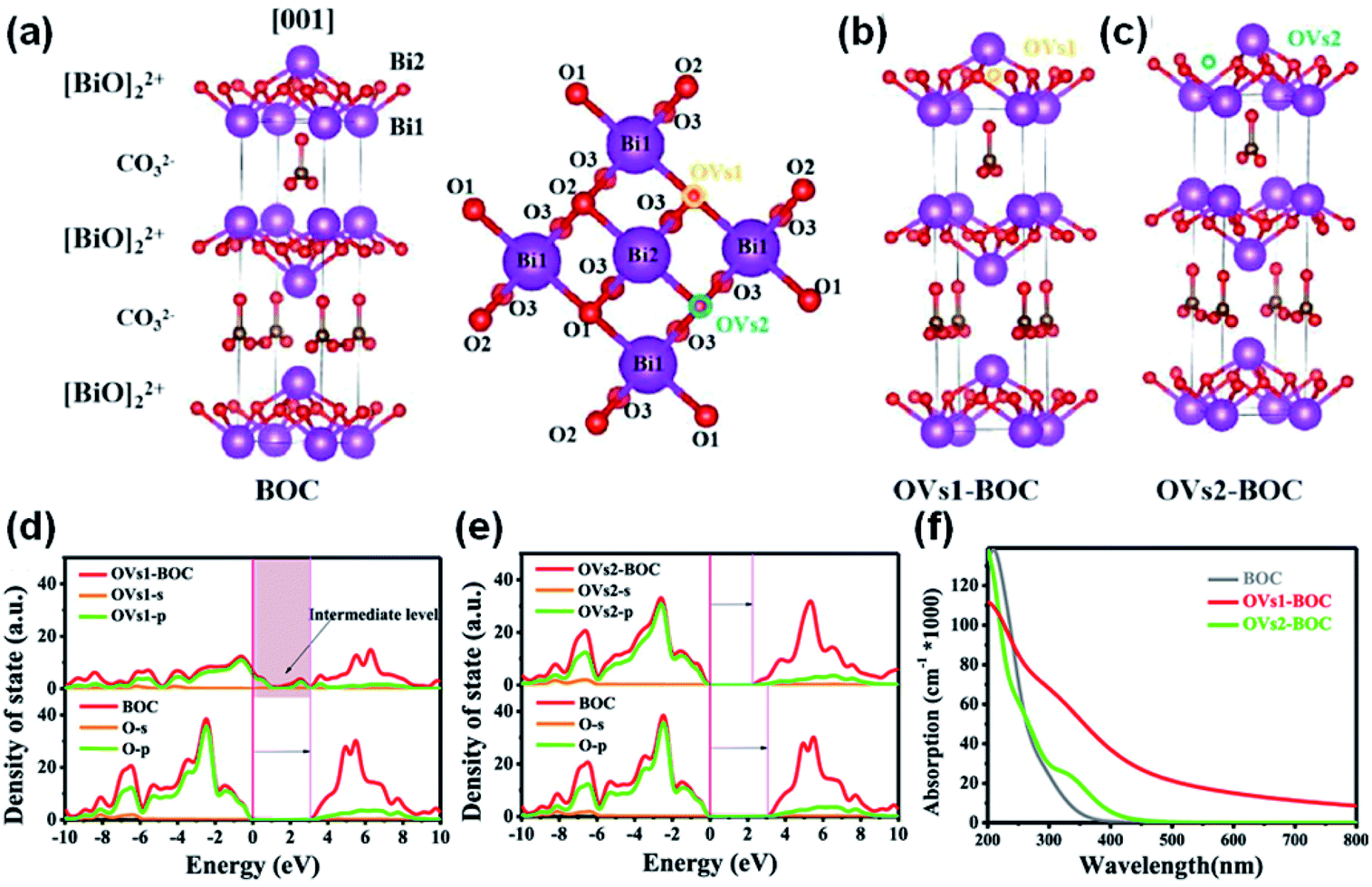 | ||
| Fig. 8 The structural models of (a) BOC, (b) OVs1-BOC, and (c) OVs2-BOC, respectively. Contradistinction of DOS/PDOS (Fermi levels are set to 0 eV) between (d) BOC and OVs1-BOC and (e) BOC and OVs2-BOC. (f) Calculated optical absorption of BOC, OVs1-BOC, and OVs2-BOC. Adapted with permission from ref. 148 Copyright (2021) American Chemical Society. | ||
| HSO5− + H2O → HSO4− + H2O2 | (5) |
| SO52− + H2O → SO42− + H2O2 | (6) |
| H2O2 + HO˙ → H2O + H+ + O2˙− | (7) |
| S2O82− + H2O → SO52− + 2H+ + SO42− | (8) |
| S2O82− + HO2− → SO4˙− + H+ + O2˙− + SO42− | (9) |
3.3 Singlet oxygen
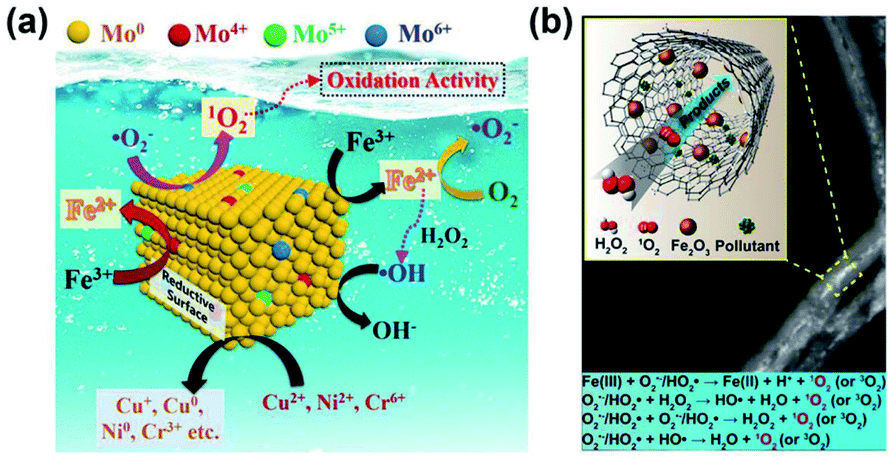 | ||
| Fig. 9 (a) Schematic illustration of Mo catalytic oxidation of O2˙− to 1O2. Adapted with permission from ref. 43 Copyright (2021) American Chemical Society. (b) Schematic illustration of the generation of 1O2 over Fe2O3@FCNT-H/H2O2. Adapted with permission from ref. 151 Copyright (2019) National Academy of Sciences. | ||
O bond length and the O2 adsorption energy are 1.51 Å and 0.5 meV for O2 on pristine Bi2O3, but 1.23 Å and 1.66 meV for O2 on OVs-Bi2O3 (Fig. 10a and b). The O2 adsorbed on OVs-Bi2O3 can be activated to the excited state (i.e., 1O2) because both the π*↑ and π*↓ states of O2 are occupied (Fig. 10e and f).158 Upon light irradiation, a faster generation of 1O2 occurs over OVs-Bi2O3 compared with pristine Bi2O3, and OVs-Bi2O3 displays stronger capability on O2 activation and thus a higher 1O2 generation rate (Fig. 10c and d). However 1O2 is the only ROS over OVs-Bi2O3; the OVs-free Bi2O3 produces other ROS (e.g., H2O2 and HO˙) along with 1O2.158 Ji et al. ultrasonically treated commercial CoS2, and the SVs in the resulting deficient CoS2−x can tune the redox reaction on the CoS2−x surface to release 1O2 in a sustained manner.159 As abundant electrons are confined in the vacant electron orbitals of the SVs, O2 can capture electrons from the SVs to form O2˙− through a one-electron process (eqn (10)) or form H2O2 through a two-electron process (eqn (11)). O2˙− can be quickly oxidized to 1O2 by the highly oxidative Co4+ or Co3+ exposed on the S deficient surface (eqn (12)).| O2 + e− (defect) → O2˙− | (10) |
| O2 + 2e− (defect) → H2O2 | (11) |
| O2˙− + Co4+ (or Co3+) → 1O2 + Co2+ | (12) |
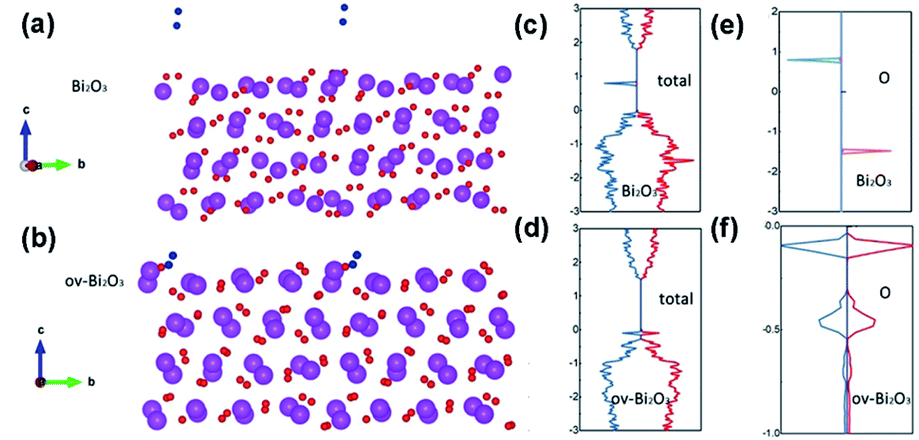 | ||
| Fig. 10 Comparison of O2 adsorption on the (120) surface of (a) pristine Bi2O3 and (b) OVs-Bi2O3 (O2, blue; O atom of Bi2O3, red; Bi, purple); the total DOS of (c) Bi2O3 and (d) OVs-Bi2O3 with O2; and partial DOS plot for O2 absorbed on (e) Bi2O3 and (f) OVs-Bi2O3 surface; the up-spin states are represented by red curves and the down-spin states by blue curves. Adapted with permission from ref. 158 Copyright (2020) Wiley-VCH Verlag GmbH & Co. KGaA. Adapted with permission from ref. 159 Copyright (2021) Wiley-VCH Verlag GmbH & Co. KGaA. | ||
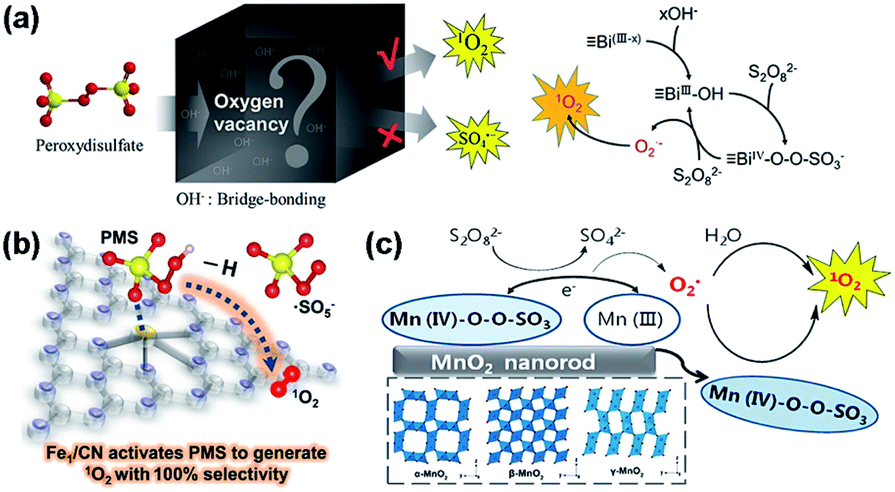 | ||
| Fig. 11 (a) Proposed mechanism of OVs-mediated activation of peroxydisulfate to create 1O2. Adapted with permission from ref. 168 Copyright (2021) American Chemical Society. (b) Schematic illustration of how the Fe single-atom catalyst (Fe1/CN) activates PMS to generate 1O2. Adapted with permission from ref. 160 Copyright (2021) Wiley-VCH Verlag GmbH & Co. KGaA. (c) Activation of PDS over manganese oxides. Adapted with permission from ref. 169 Copyright (2018) American Chemical Society. | ||
Bu et al. activated PDS to generate 1O2 on the deficient surface of BiOBr.168 The OVs of BiOBr interact with hydroxyl ions (HO−) to form BiIII–OH species, which are the major active sites for the adsorption and activation of PDS. The PDS adsorbed at the BiIII–OH sites produces the metastable BiIV–O(−1)–O(−1)–SO3− intermediate, which reacts with another PDS to release O2˙− when the BiIV–O bond breaks. In this process, the O atom bonded to BiIV acts as the electron donor, and BiIV acts as the electron acceptor. Finally, Bi(IV−x)–O(−1)–O(−1)–SO3− reacts with O2˙− to produce 1O2 under alkaline conditions (Fig. 11a).
Many studies have suggested the direct oxidation of O2˙− as a common route to generate 1O2,43,169,170 and the oxidation of O2˙− to 1O2 requires an oxidant with a redox potential of at least 0.34 V vs. NHE. Hu et al. revealed that the OV concentration of Co3O4 impacts its efficiency in activating PMS to give 1O2.171 The PMS decomposition mainly depends on the adsorption energy of PMS on the Co3O4 surface, the cleavage of the O–O bond in the PMS molecule, and the electron transfer between PMS and Co3O4. The direct oxidation of O2˙− to give 1O2 may account for the generation of 1O2 in view of the thermodynamics, i.e., E0 (Co3+/Co2+) = 1.93 V and E0 (O2˙−/1O2) = −0.34 V. Prior studies have also shown that the oxidization of O2˙− to 1O2 is thermodynamically feasible with CuIII and MnIV (Fig. 11c),169,170 as E0 (CuIII/CuII) = 2.3 V vs. NHE and E0 (MnIV/MnIII) = 0.95 V vs. NHE. The reduction of Mo6+ by O2˙− during the Fenton reaction can also give 1O2, i.e., Mo6+ + O2˙− → 1O2 + Mo4+.43
3.4 Lattice oxygen in TMO: reactivity and regeneration
Just like HO˙, O2˙− and 1O2, the OL of TMO itself can also serve as the ROS to participate in AOPs. Since OL already exists stably in TMO, it is not newly generated from a precursor, but it needs to be constantly replenished in the oxidation reaction as described in the MvK mechanism. TMO with highly reactive OL generally have low OV formation energy that arises from a weak M–O bond and strong oxygen migration ability in the TMO lattice.104 Hayashi et al. found that β- and λ-MnO2 are good catalysts for the oxidation of hydroxymethyl furfural (HMF).172 Compared with α- and γ-MnO2, β- and λ-MnO2 have lower OV formation energies due to their distinct crystal structures, and for both α- and γ-MnO2 the OV formation energies are higher at the planar oxygen sites than at the bent oxygen sites (Fig. 12a).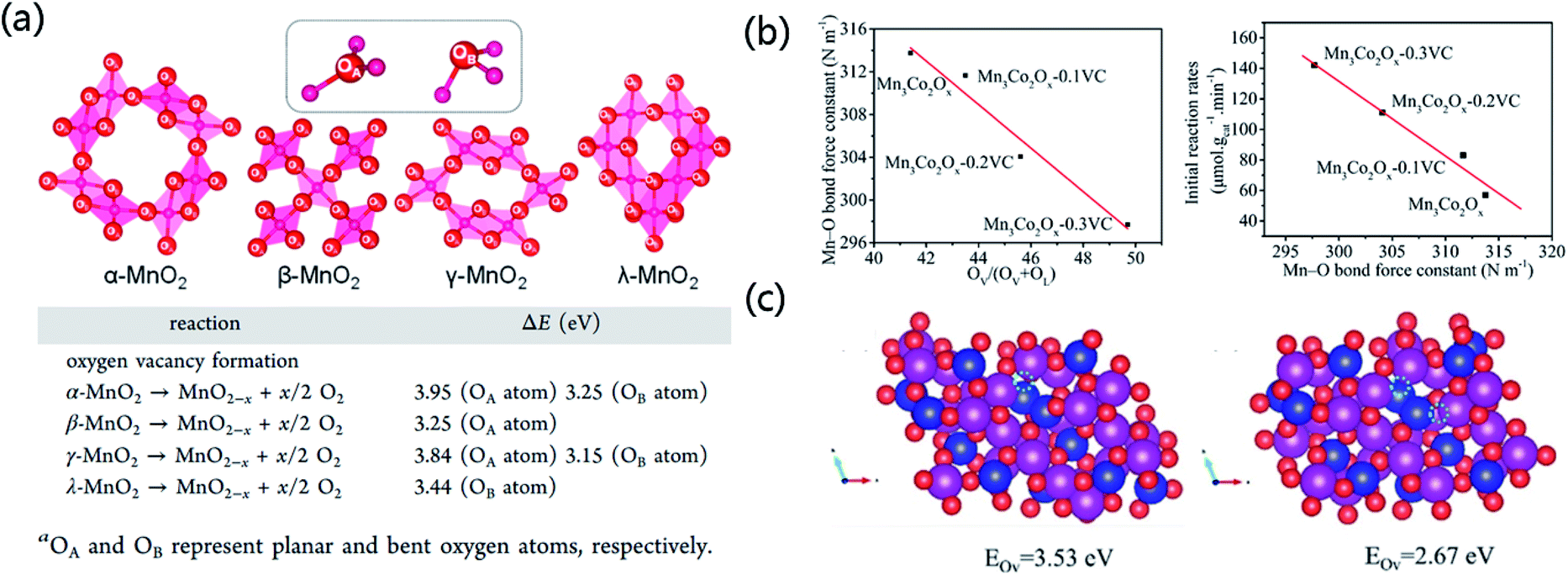 | ||
| Fig. 12 (a) Calculated OV formation energies of MnO2 of distinct phase structures. Adapted with permission from ref. 172 Copyright (2019) American Chemical Society. (b) Relationship between the Mn–O bond force constant and the relative OV content and the initial reaction rates of various Mn3Co2Ox–zVC catalysts. (c) Top views of the (111) plane of the Mn–Co spinel oxide with one OV and two neighboring OVs. Adapted with permission from ref. 173 Copyright (2021) American Chemical Society. | ||
The OVs of deficient TMO can regulate the coordination structure of the TMO and alter the reactivity of OL.103 Liu et al. presented a facile and green vitamin C-assisted solid-state grinding method to synthesize mesoporous Mn–Co spinel oxides with a well-defined OV concentration.173 The Mn–Co oxides with a higher OV concentration have a weaker Mn–O bond and higher OL reactivity (Fig. 12b). According to DFT calculations and in situ FT-IR studies, the removal energy of OL decreases significantly from 3.53 to 2.67 eV when there is an adjacent OV (Fig. 12c). That is, the OL becomes more active when OVs exist in its surrounding. The analyses of Raman spectroscopy, H2-TPR, and O2-TPD show that the Mn3Co2Ox–0.3VC catalyst with a higher OV concentration has stronger OL reactivity than Mn3Co2Ox.
Doping is a common strategy to enhance the reactivity of OL, as doping TMO with cations (e.g., Li+, Mg2+, Ca2+, Sr2+, Al3+, Zn2+, and Ln3+) can decrease the OV formation energy. Low-valence dopants may create OVs and enhance the reactivity of OL.27 Xu et al. prepared Li+-doped NiO which has remarkably enhanced OL reactivity than pristine NiO.174 The OV formation energy is 3.32 eV for pure NiO but only 2.12 eV for Li+-doped NiO. Schlexer et al. introduced Au to TiO2 to capture the excess electrons associated with the vacancies formed at the Au/TiO2 perimeter to stabilize the vacancies.175 The OV formation energy at the Au/TiO2 perimeter is reduced by 1.32 eV with respect to the Au-free surface. Dostagir et al. prepared a catalyst with isolated Co atoms in ZrO2 and demonstrated with DFT calculations that the OV formation energy of ZrO2 is decreased by Co doping to between −1.45 and −1.17 eV from 2.70–3.20 eV.176 That is, OV formation is endothermic on pure ZrO2 but becomes exothermic after Co doping. OV sites are created near Co atoms due to mismatch in the oxidation states of Co2+ and Zr4+ and thus facilitate the need for charge balance.
Doping may also enhance the OL reactivity by inducing structural distortion in the oxides. Zhao et al. found that the Raman peaks of Ni-doped Co3O4 shift to lower frequencies and become broader because lattice distortion weakens the Co–O bond.177 Consequently, more OVs are created in the oxide, the OL has stronger mobility, and the active surface OL become more abundant. Zhang et al. showed that the [FeO6] octahedral distortion in La1−xCexFeO3 promotes the mobility and reactivity of OL,178 suggesting that it is feasible to tailor the reactivity of OL in perovskites by modulating the distortion of BO6 in ABO3 (Fig. 13a).
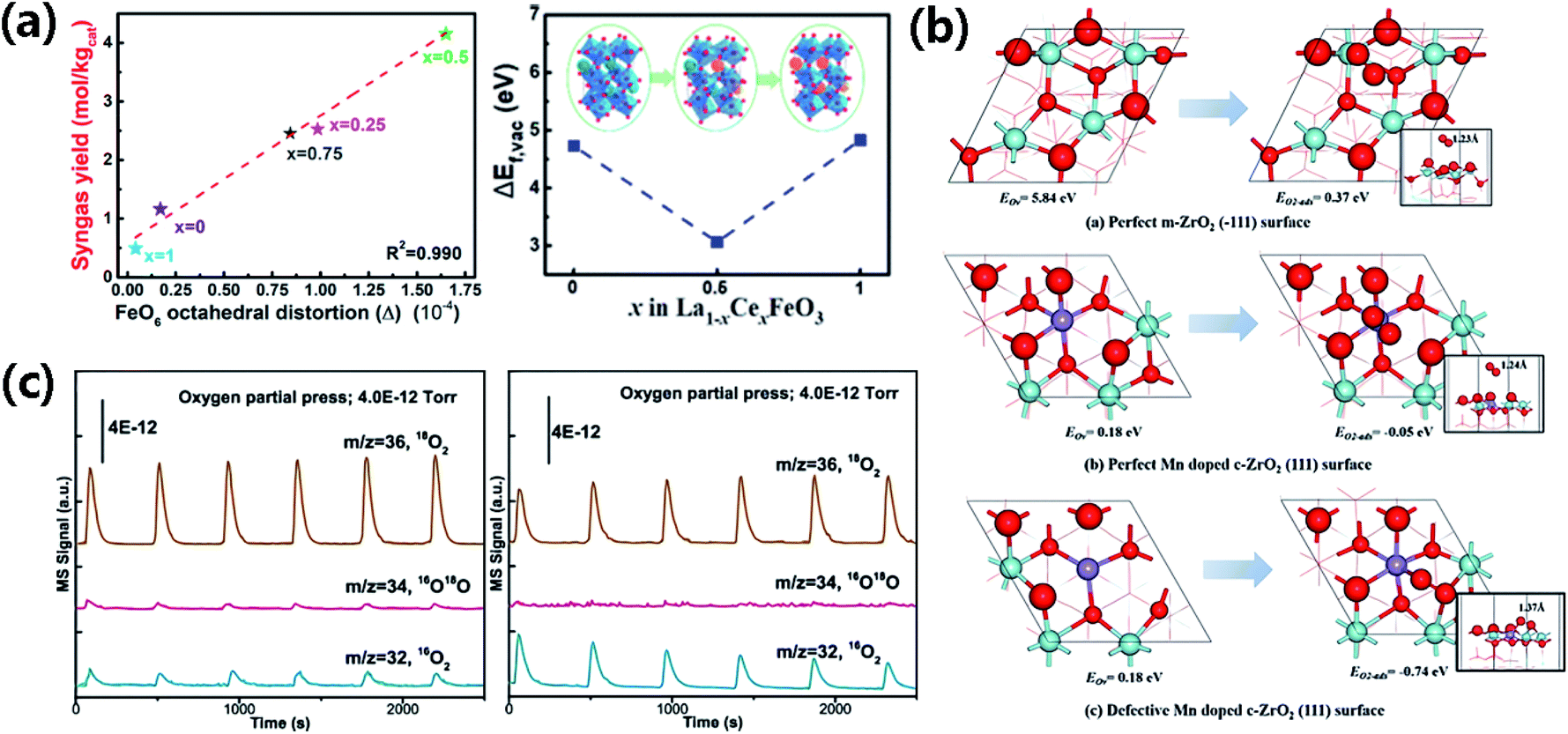 | ||
| Fig. 13 (a) The OV concentration and possible reaction mechanism of POM-CO2 splitting over La1−xCexFeO3 (x = 0, 0.5, 1). Adapted with permission from ref. 178 Copyright (2020) American Chemical Society. (b) OV formation energy (EOv) and adsorption energy of O2 (EO2-ads) over distinct Zr sites. Adapted with permission from ref. 179 Copyright (2018) American Chemical Society. (c) Isotopic 18O2 exchange analysis of Bi2Zr2O7 at 300 °C and 400 °C. Adapted with permission from ref. 102 Copyright (2021) American Chemical Society. | ||
The efficient regeneration of OL can also benefit from OVs. OVs can enhance the adsorption of O2 and the storage of O22− on the TMO surface in the MvK process.180 Lu et al. incorporated Fe into CuO–CeO2 to create the Fe–O–Ce structure with rich OVs and measured in situ the oxygen storage capacity (OSC) of the oxides by the CO pulse technique.181 The OVs readily trap the O2 in the gas phase, which ensures a durable release of subsurface lattice oxygen to supply reactive OL. The initial CuO–CeO2 catalyst has a low OSC value of 46.4 μmol CO2 per g, whereas the Fe-doped CuO–CeO2 has a considerably higher OSC value of 94.8 μmol CO2 per g thanks to the greater number of OVs introduced by Fe doping. Similarly, Wang et al. assembled ultrathin and vacancy-rich CoAl-LDH nanosheets prepared from exfoliating bulk CoAl-LDH with graphite oxide (GO) to obtain the CoAl-ELDH/GO hybrid catalyst that possesses abundant newly generated OVs.182 The OVs promote oxygen adsorption and storage over the deficient CoAl-ELDH/GO, which has larger O2-TPD peaks than the corresponding bulk CoAl-LDH. Yang et al. showed with DFT calculations that O2 can be strongly adsorbed and activated on the defective Mn-doped c-ZrO2 (111) surface with OVs.179 The OVs elongate the OO bond from 1.24 Å on the perfect Mn-doped c-ZrO2 (111) surface to 1.37 Å on the defective surface. The OV formation energy and the adsorption energy of O2 are 5.84 eV and 0.37 eV on the perfect m-ZrO2 (−111) surface, 0.18 eV and −0.05 eV on the perfect Mn-doped c-ZrO2 (111) surface, and down to 0.18 eV and −0.74 eV on the defective Mn-doped c-ZrO2 (111) surface, respectively (Fig. 13b).
Generally, there are two possible pathways (i.e., R1 and R2 mechanism) for the gas phase O2 to oxidize the OVs and replenish OL. In the R1 mechanism, the exchange of oxygen species occurs at one surface vacant site. In the R2 mechanism, two adjacent surface vacant sites are needed to adsorb and decompose the gaseous O2. Feng et al. ran isotopic 18O2 tracing experiments and demonstrated that the adsorption and activation of gas phase O2 on Bi2B2O7 mainly follow an R2 mechanism that requires two adjacent surface vacancies (Fig. 13c).102
3.5 Tailoring the reactive oxygen species generation
PMS, PDS and H2O2 are strong oxidants but normally have low reaction rates with most organic pollutants. Catalysts bearing transition metal ions (e.g., Fe, Mn, Co, Ni, V, Ce, etc.) are promising activators for these oxidants to give powerful ROS.183 Transition metal catalysts can provide a diversity of metals with tunable valence states to afford high redox properties, and an appropriate d-band center to balance the catalyst-adsorbate binding strength, as well as controllable surface and electronic structures to promote light absorption and/or adsorbate adsorption, all crucial for tailoring and boosting the generation of ROS. Note that the catalysts containing different types of transition metals are frequently found to give the same type of ROS.16,184–186 Hence, it should not just be the type of transition metal ions controlling the selectivity and efficiency of ROS generation.187 Recent studies have shown that modulation of the geometrical and electronic structures of catalysts is a key to probe how to customize the ROS production.Tuning the geometrical structure of catalysts can change the binding effect of catalysts and oxidants and monitor the ROS generation. For instance, transition metal based catalysts commonly drive the dissociation of PMS and PDS into SO4˙− and HO˙.169 It has been reported that the catalysts of CoN4 structure prefer to adsorb the O atom connected to S in the PMS, and the system produces a mixture of ROS including SO4˙−, HO˙, O2˙− and 1O2.188,189 In comparison, when Co is coordinated with N to form a CoN[2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) +2] structure, CoN[2+2] can bind the O from –OH in the PMS, while the loss of the H atom of –OH creates a SO5˙− intermediate.188,189 Since SO5˙− has a high reaction rate (∼2 × 108 M−1 s−1) and low activation energy (7.4 ± 2.4 kcal mol−1), it can rapidly transfer into 1O2viaeqn (13)–(15).188,189 Intriguingly, on the deficient surface of BiOBr the PDS can be activated into 1O2.168 The OVs of BiOBr interact with HO− to form BiIII–OH species and produce the BiIV–O(−1)–O(−1)–SO3− intermediate, which reacts with another PDS to release O2˙−, while Bi(IV−x)–O(−1)–O(−1)–SO3− reacts with O2˙− to produce 1O2 under alkaline conditions (Fig. 11a).
+2] structure, CoN[2+2] can bind the O from –OH in the PMS, while the loss of the H atom of –OH creates a SO5˙− intermediate.188,189 Since SO5˙− has a high reaction rate (∼2 × 108 M−1 s−1) and low activation energy (7.4 ± 2.4 kcal mol−1), it can rapidly transfer into 1O2viaeqn (13)–(15).188,189 Intriguingly, on the deficient surface of BiOBr the PDS can be activated into 1O2.168 The OVs of BiOBr interact with HO− to form BiIII–OH species and produce the BiIV–O(−1)–O(−1)–SO3− intermediate, which reacts with another PDS to release O2˙−, while Bi(IV−x)–O(−1)–O(−1)–SO3− reacts with O2˙− to produce 1O2 under alkaline conditions (Fig. 11a).
| HSO5− → SO5˙− + H+ + e− | (13) |
| SO5˙− + SO5˙− → S2O82− + 1O2 | (14) |
| SO5˙− + SO5˙− → 2SO42− + 1O2 | (15) |
Tuning the electronic structure of photocatalysts can boost the generation and transfer of electrons for catalytic reactions. Wu et al. employed OVs to mediate 1O2 generation in the Fe–Co LDH/PMS system, and found that PMS tends to donate one electron to the OV and is thus activated to form 1O2.190 When oxidation reactions involve photocatalytic reactions, a key to tailor ROS generation is by tuning the band and electronic structure of photocatalysts. Introducing point defects (e.g. OVs) could create additional defect levels of photocatalysts, rendering a downshift of the conduction band (CB) minimum, and/or an upshift of the valence band (VB) maximum.143,144 With an appropriate band structure, the electrons in the defect levels could transfer to the CB band through inter-band excitation and be captured by O2 that generates O2˙−,191 while upshift of the VB band may help suppress the oxidation of H2O to HO˙. For instance, CdS and ZnIn2S4 catalysts have appropriate conduction and valence bands for O2˙− generation while avoiding HO˙ generation.192 The presence of OVs could introduce intermediate energy states, leading to a narrowed band gap and enhanced charge carrier separation efficiency,193,194 and helps extend the light response to visible or even near infrared light,195,196 Wang et al. showed that OV-rich sulfur-doped BiOBr has a narrowed band gap responsible for the visible light and can generate O2˙− and HO˙ efficiently for 4-chlorophenol degradation.197
To date, it remains an open question to establish robust principles to develop effective transition-metal-based catalysts for fine control over ROS generation. The roles and mechanism responses of different metal groups in distinct reaction pathways await to be disclosed, by performing more systematical investigations under the assistance of operando spectroscopic studies and theoretical calculations.
4 Detection of reactive oxygen species
4.1 Hydroxyl radical
:2:2:1.200
4.2 Superoxide radical
:1:1:1.207 As the reaction rate of DMPO with O2˙− is much slower than that with HO˙ in aqueous solution, and DMPO-O2˙− is more stable than DMPO-HO˙ in methanol,208 it is preferred to perform the EPR measurements of O2˙− in methanol solvent.
4.3 Singlet oxygen
:1:1 triplet signal.214 For the detection of 1O2, the use of 4-oxo-tetramethylpiperidine,215 and 4-hydroxy-tetramethylpiperidine216 has also been reported.
4.4 Quenching technique
When two or more ROS are generated over the catalyst, a quenching experiment can be employed to identity the dominant ROS in the AOP system. Since many alcohols (e.g., isopropanol (IPA),221 mannitol,222t-butyl alcohol (TBA)223 and methanol224) can efficiently react with HO˙, these substances could be good candidate reagents to quench HO˙. It is worth noting that methanol can also work as the scavenger for quenching SO4˙−,225 while TBA is only effective for quenching OH˙ but not for SO4˙−.226Benzoquinone (BQ),227,228 ascorbic acid,228 superoxide dismutase (SOD)229 and trichloromethane230 can be used to remove O2˙− in oxidation reactions. For instance, in many cases, O2˙− may serve as an intermediate for the generation of other ROS (e.g., 1O2). To find out whether O2˙− is the intermediate of 1O2 or the dominant ROS for pollutant degradation, Yi43 and Zhang189 introduced BQ as a quenching agent of O2˙−, and showed that O2˙− served as an intermediate to form 1O2, the active species in their studies.
Useful quenching agents for 1O2 include NaN3,231 furfur alcohol (FFA),232L-tryptophan233etc. However, since FFA or NaN3 can also react with free radicals,234 it is infeasible to use FFA/NaN3 as a scavenger when the oxidation system contains both the radical and non-radical ROS. In addition, NaN3 can react with PMS rapidly, and hence, it cannot apply to the PMS-involved system. Besides the quenching technique, sometimes, the solvent exchange method that replaces H2O by D2O can help identity the generation of 1O2, as the lifetime of 1O2 in D2O is about ∼55 μs, more than 10 times that (∼4.2 μs) in H2O.235,236
5 Reactions of reactive oxygen species
5.1 Hydroxyl radical
C bonds) by abstracting hydrogen atoms.240 Because of its electrophilic nature, HO˙ prefers to react with the CC and CN bonds in the organic substrate rather than the CO bond whose carbon atom is electron-deficient.39 Nevertheless, it can react with aliphatic carboxylic acids to create highly reactive carbon-centered radicals via abstraction of the hydrogen atom, and react with aromatic carboxyl acids via hydroxylation.
The surface defects of TMO can effectively enhance the oxidation performance of HO˙. For instance, the Co–Cu LDH with abundant OVs can boost the dissociation of H2O2 to generate a sufficient amount of HO˙ for the degradation of anthraquinones.241 The highest COD and TOC removal values (89.9% and 71.3% respectively) are achieved with an initial H2O2/TOC ratio of 75 and an initial pH of 6.8. Zhang et al. examined a ceria-based heterogeneous Fenton-like catalyst.242 Compared with pure iron oxide and ceria, Fe-doped CeO2 with abundant OVs (FeCeOx) degrades rhodamine B (RhB) at a higher efficiency (98%) over a wider working pH range (3.0–9.0). The surface HO˙ is the predominant ROS to oxidize RhB. Annealing the FeCeOx in an oxygen atmosphere eliminates the OVs, and the annealed FeCeOx with fewer OVs has poorer performance in activating H2O2 and degrading the target pollutant. Zhan et al. prepared OV-rich Co–ZnO microparticles (OV–CoZnO MPs) as a Fenton-like catalyst.119 The electron-rich OV sites on the catalyst are the key active sites responsible for the capture and reduction of H2O2 to generate HO˙ to degrade organic pollutants (e.g., BPA, PHT, 2,4-D, phenol, and methylene blue) over a wide pH range (4.5–9.5). In addition, the electron-deficient Co3+ sites of the catalyst appear to actively adsorb the pollutant, and the organic pollutant adsorbed on Co3+ acts as an electron donor to further accelerate the degradation process. The OV-CoZnO MPs/H2O2 system with dual reaction centers can degrade pollutants about ∼17 times faster than the ZnO/H2O2 system that has only one reaction center.
Li et al. performed quantitative analysis and reported an interesting case that shows the selectivity of HO˙ in the degradation of pollutants (Fig. 14e and f).243 The OVs of the BiOCl catalyst demonstrate an electron donor nature and serve as the “Fenton-catalytic” center to dissociate H2O2 to generate HO˙, but it occurs in a surface confinement pathway. The generated HO˙ quickly diffuses away from the (001) surface of BiOCl, because the steric hindrance due to the high density of O atoms on the (001) surface prevents the binding of newly generated HO˙ (Fig. 14a–d). However, free HO˙ prefers to oxidize dissolved pollutants (e.g., benzoic acid, benzene, and phenol) in the solution (Fig. 14g); the nascent HO˙ on the (010) surface of BiOCl prefers to bond and stay on the catalyst surface to react with strongly adsorbed pollutants (e.g., formic acid, rhodamine B, and methyl orange) (Fig. 14g).
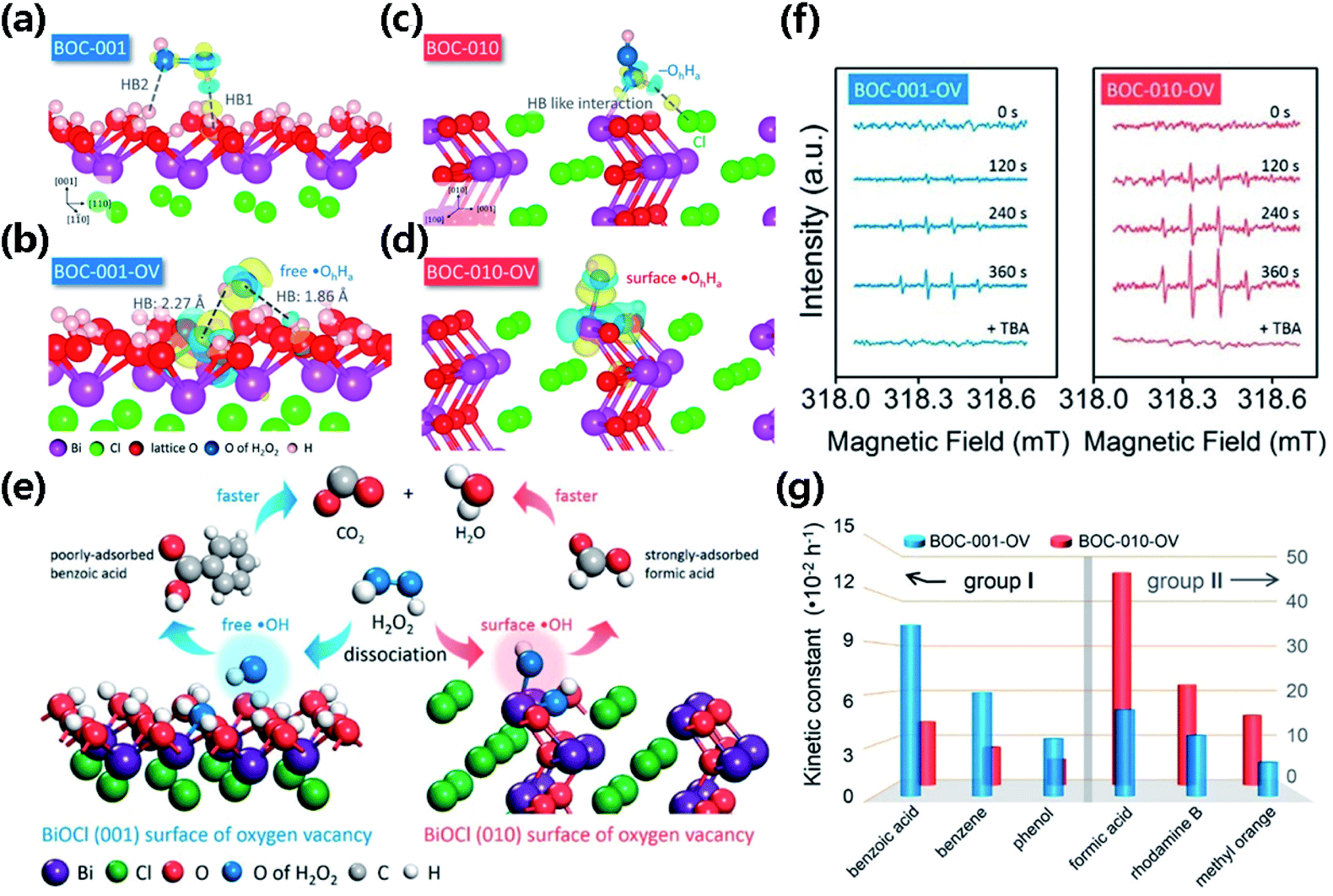 | ||
| Fig. 14 Schematic illustration of H2O2 dissociation on (a) BOC-001, (b) BOC-010, (c) BOC-001-OV and (d) BOC-010-OV, respectively. (e) Schematic illustration of H2O2 dissociation on different surfaces of BiOCl with OVs for scavenging benzoic acid and formic acid, respectively. (f) EPR spectra of spin-reactive HO˙ radicals generated over BOC-001-OV and BOC-010-OV in the presence of H2O2, respectively. (g) Reaction kinetic constants of BOC-001-OV and BOC-010-OV for the removal of group I and group II pollutants. Adapted with permission from ref. 243 Copyright (2017) American Chemical Society. | ||
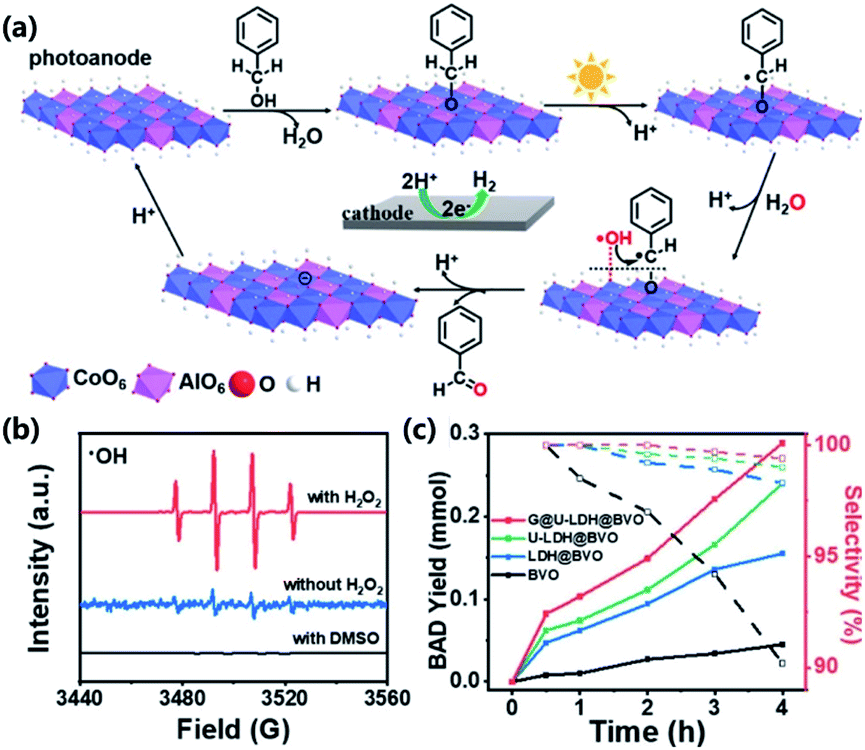 | ||
| Fig. 15 (a) Mechanism of selective benzyl alcohol (BA) activation coupled with PEC water oxidation. (b) ESR spectra of HO˙ generated over the graphene@ultrathin-CoAl-LDH@BiVO4 under different conditions (H2O2, 30%, 0.1 mM; DMSO, 0.5 mM). (c) Benzaldehyde yield (solid squares and lines) and selectivity (empty squares and dashed lines) at different reaction times in the PEC oxidation of BA under illumination (AM 1.5G, 100 mW cm−2) at 1.2 V vs. RHE. Solution: 2 mmol BA in PBS (pH = 7, 0.1 M, 10 mL) at 10 mV s−1 scan rate. Adapted with permission from ref. 246 Copyright (2020) American Chemical Society. | ||
5.2 Superoxide radical
As a free radical, O2˙− can work as both an electron transfer agent and an electron acceptor.253 Xiao et al. studied how the unpaired electrons fit into the resonance π* molecular orbitals in O2˙− and found that the HOMO and LUMO of O2˙− are degenerated.254 Thus, O2˙− is both electrophilic and nucleophilic, and can work as either a reducing agent or an oxidizing agent.255,256 Because the carboxyl group is weakly electron-withdrawing and the hydroxyl group is electron-donating, the efficiency of degradation by O2˙− ranks as hydroquinone < p-hydroxybenzoic acid < p-nitrophenol, which is the exact opposite order of their electron cloud density.251
Defect structures on the catalyst can help generate O2˙− for AOP. In the degradation of ciprofloxacin over W-doped CeO2, W is beneficial for the formation of surface OVs, and the deficient surface effectively adsorbs and activates O2 to O2˙− through a one electron transfer process.257 The 2% W/CeO2 can degrade ciprofloxacin in a wide pH range with excellent efficiency. In some cases, O2˙− and other species in the reaction medium react with the substrate concurrently. For instance, in the oxidation of BPA over AgFe1−xNixO2, the dominant ROS are O2˙− and 1O2, rather than the traditional SO4˙− or HO˙.166 Analogously, the MnO2-mediated degradation of anthracene is mediated by both the non-radical oxidation by Mn3+ and the radical-based oxidation by O2˙−.258
It is worth remarking that the geometry of O2˙− on the catalyst surface may affect the outcome of the oxidation reaction. Li et al. engineered deficient BiOCl that can oxidize NO completely into nitrate under visible light irradiation with >99% selectivity (Fig. 16a).247 When the reaction is carried out at ambient temperature, the carefully crafted OVs on the prototypical (001) surface of BiOCl preferentially give O2˙− in a side-on bridging mode and suppress the thermodynamically stable terminal end-on geometry that will be predominant at an elevated temperature (Fig. 16b–e). This newly developed O2˙−-mediated technique thus avoids the partial oxidation of NO to NO2.
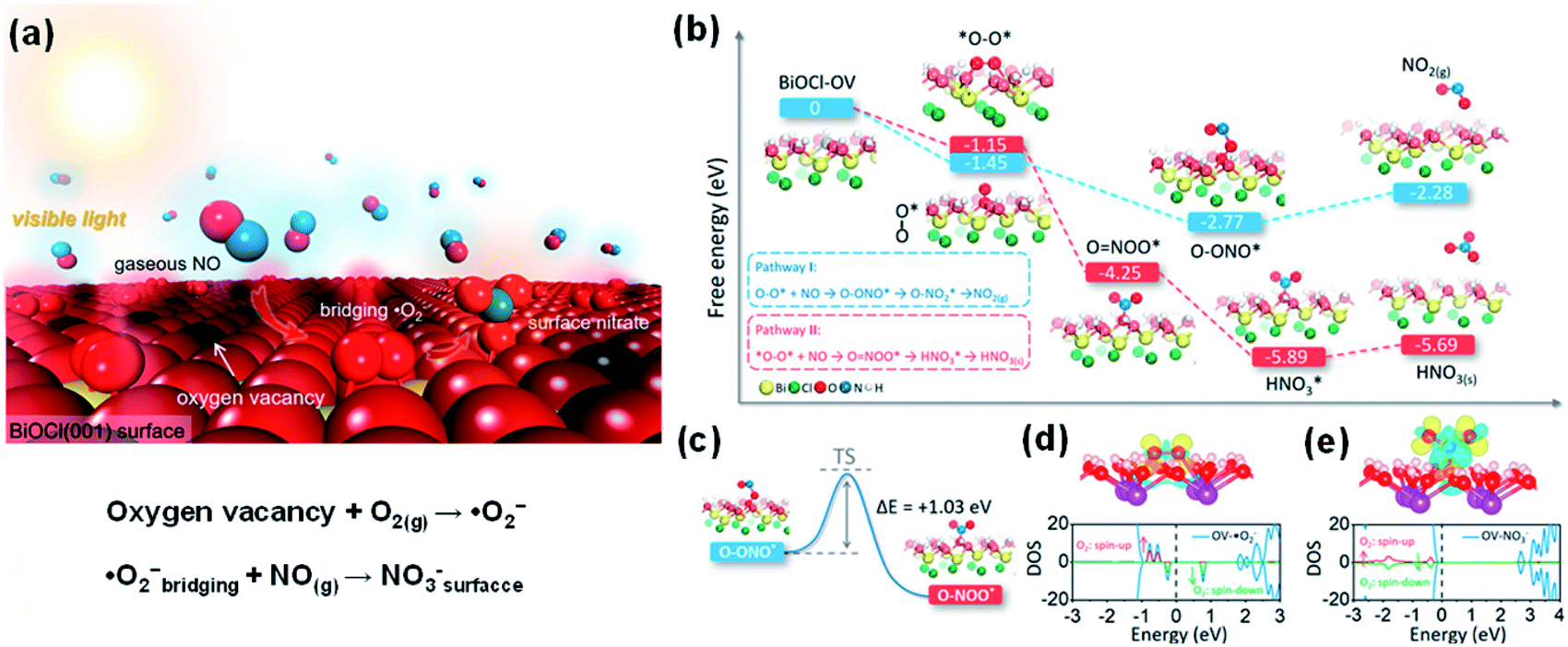 | ||
| Fig. 16 (a) Schematic illustration of photocatalytic NO removal by BiOCl-OV. (b) Free energy change against the reaction coordinate for the oxidation of NO by O2˙− on the BiOCl (001) surface in different geometries. (c) Geometric transition from peroxynitrite to nitrate. TS represents the transition state. Charge density difference and O2 partial DOS of the BiOCl (001) surface adsorbed with (d) O2 and (e) nitrate. The yellow and blue isosurfaces with an isovalue of 0.005 au represent charge accumulation and depletion in space. The vertical dashed line in the DOS shows the VBM. Adapted with permission from ref. 247 Copyright (2018) American Chemical Society. | ||
Enhanced generation of O2˙− for selective photooxidation can also be accomplished on the defective surface with SVs. Sun et al. carried out a photocatalytic selective oxidation to convert alcohols to aldehydes with high efficiency and selectivity using defective In2S3 nanosheets (Fig. 17a and b).147 The SVs-rich-In2S3 activates O2 into O2˙−via electron transfer, and the aldehyde selectivity is ≥98% for all alcohol substrates when SVs-rich In2S3 is used. For the oxidation of benzyl alcohol, the conversion rate of the substrate under ambient conditions is only 30% with SVs-poor In2S3 but 71% with SVs-rich In2S3 (Fig. 17c).
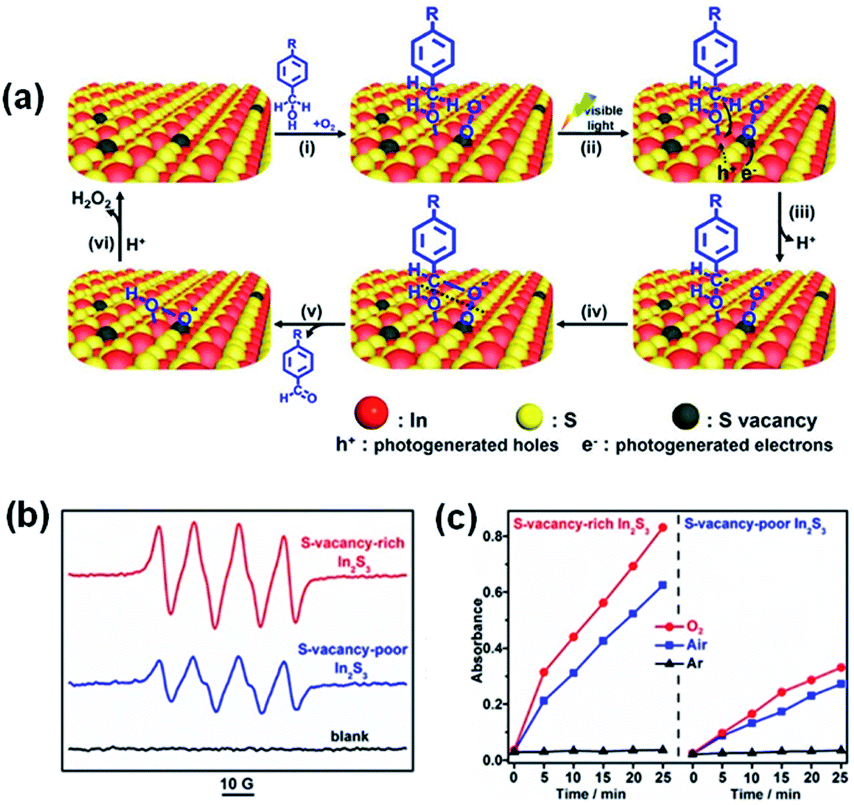 | ||
| Fig. 17 (a) Proposed mechanism of the photooxidation of benzyl alcohol to benzaldehyde. (b) ESR spectra of SV-rich and SV-poor In2S3 in the presence of DMPO. (c) The dramatically different oxidation rates of the indicator molecule 3,3′,5,5′-tetramethylbenzidine in various gas environments (oxygen, air, and nitrogen) indicate that the ROS evolved from O2. Adapted with permission from ref. 147 Copyright (2019) American Chemical Society. | ||
The selective oxidation of primary amines to imines is essential to synthesize dyes, fragrances, fungicides, pharmaceuticals, and agrochemicals.265–268 Imines are normally synthesized in industry by the direct oxidation of amines or by the condensation of amines with aldehydes, which can require using stoichiometric amounts of harmful or hazardous reagents. Khampuanbut et al. utilized a WO3/BiOBr photocatalyst for oxidizing amines to imine, for which O2˙− is the main ROS and the OVs on WO3/BiOBr promote the interfacial charge transfer.269 Carrying out the reaction at room temperature under an O2 atmosphere with visible light irradiation converts nearly 74.1% amines into imines within 4 h. Shi et al. used the phenol–TiO2 complex as an efficient photocatalyst for the selective oxidation of amines to imines with the aid of O2 under visible light irradiation.270 Both primary and secondary amines can be oxidized to imines at >80% conversion rate in 30 min, and O2˙− again proves to be the main ROS.
5.3 Singlet oxygen
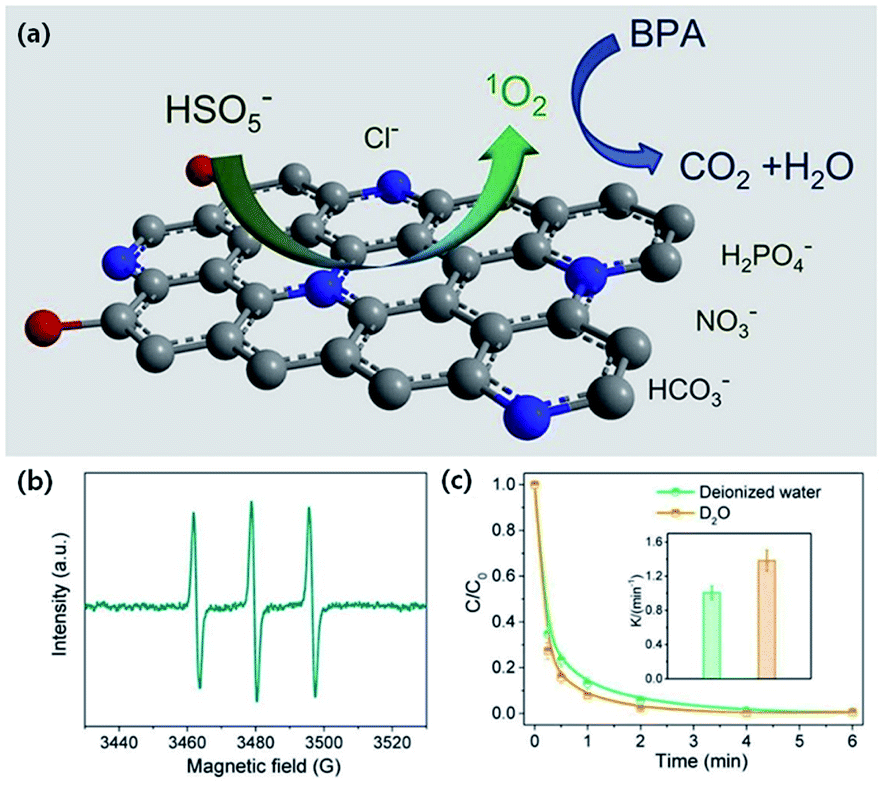 | ||
| Fig. 18 (a) Schematic illustration of BPA degradation. (b) EPR spectrum of PMS activation. (c) Effect of D2O on BPA removal efficiency. Adapted with permission from ref. 275 Copyright (2019) Elsevier. | ||
Zhao et al. showed that 1O2 plays a dominant role in the removal of BPA catalyzed by deficient ZnCoOx.278 Bu et al. reported the OVs-mediated activation of PDS with BiOBr to degrade BPA, for which 1O2 is the main ROS under alkaline conditions.168 The removal ratio of TOC reaches 57.3% after 15 min in the reaction system (pH = 11.5), suggesting that BPA can be effectively mineralized into CO2 and H2O by 1O2. Li et al. developed La1.15FeO3 with rich surface OVs.279 The catalyst has a high Fenton activity (0.0402 min−1) for activating H2O2 to oxidize methyl orange, and the 1O2 formed at the surface OVs is the main ROS. Gao et al. found that the piperazinyl, oxazinyl, and carboxylic substituents of ofloxacin are readily attacked by the 1O2 generated from PMS with perovskites.167 Both the amount of OVs of the perovskites and the degradation efficiency of ofloxacin fall in the order of LaFeO3 < LaZnO3 < LaMnO3 < LaNiO3.
Chen et al. developed a metal–organic framework based on Pt nanocrystals and porphyrin, denoted as Pt/PCN-224(M), to selectively oxidize aromatic alcohols to the corresponding aldehydes with O2 (Fig. 19a).280 Benzyl alcohol is oxidized with 1 atm O2 at ambient temperature (16 °C) in water under visible light irradiation and the reaction is complete within 50 min. The 100% aldehyde selectivity can be attributed to 1O2 as the main ROS, and extending the reaction time does not produce benzoic acid (Fig. 19b and c). Li et al. demonstrated the enhanced photocatalytic generation of 1O2 with Mn–NCs@SiO2–Pt and the selective oxidation of primary benzylic alcohols to aldehydes.283 The energy transfer between the Mn-doped nanocrystals and molecular oxygen is made efficient thanks to the long half-life of the Mn excited state (in the order of milliseconds). Selective oxidization of glycerol to dihydroxyacetone (DHA) is a high-revenue chemical that has wide range of applications, e.g., as a sun tanning agent in the cosmetics industry and as a precursor of pharmaceuticals or a building block for fine chemical synthesis, and can combine with lactic acid for producing biodegradable polymers.284 Zhao et al. developed the Bi/Bi3.64Mo0.36O6.55 heterostructure for the photocatalytic selective oxidization of glycerol.285 The presence of OVs in Bi/Bi3.64Mo0.36O6.55 promotes the mass production of 1O2 as the ROS for the selective oxidation, and glycerol is oxidized in water to 1,3-dihydroxyacetone at ambient temperature (25 °C) with high selectivity (97–99%). Selective oxidation of organic sulfides has also sparked increasing attention as the resultant sulfoxides are key intermediates for the synthesis of agrochemicals, and pharmaceuticals as well as other valuable fine chemicals.286 Wang et al. demonstrated the highly efficient and selective photo-oxidation of phenyl methyl sulfide to phenyl methyl sulfoxide catalyzed by OVs-Bi2O3.158 The OVs in OVs-Bi2O3 can activate O2 into 1O2 in the dark, and the generation of 1O2 is accelerated over both OVs-Bi2O3 and Bi2O3 when there is light irradiation. OVs-Bi2O3 has a stronger capability for the selective oxidation of phenyl methyl sulfide to phenyl methyl sulfoxide owing to its higher 1O2 generation rate.
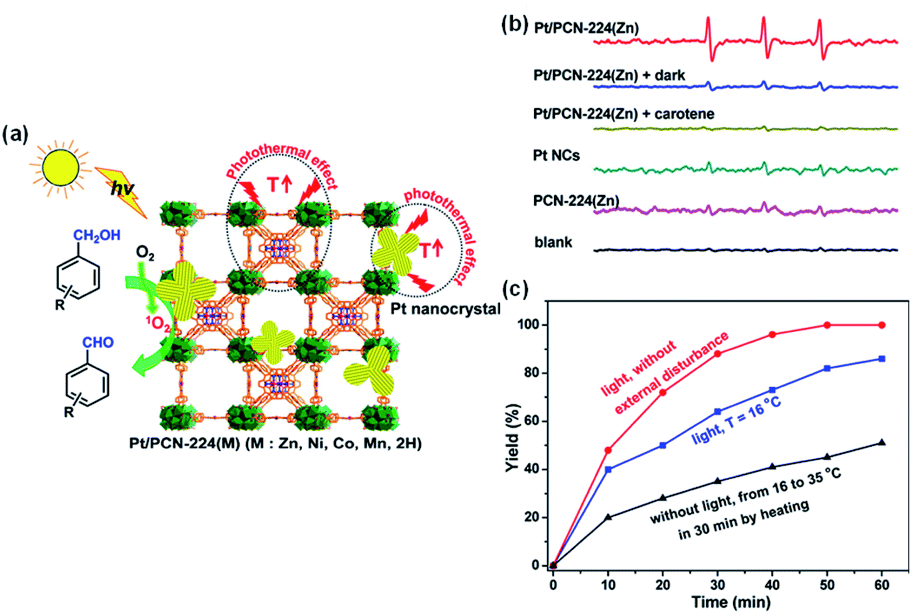 | ||
| Fig. 19 (a) Schematic illustration showing the singlet oxygen-engaged selective oxidation of alcohols over Pt/PCN-224(M) using molecular oxygen under visible-light irradiation. (b) ESR spectra of the samples after mixing 4-oxo-TMP with PCN-224(Zn), Pt NCs, and Pt/PCN-224(Zn) in the absence or presence of carotene under visible-light irradiation or in the dark. (c) Oxidation yield of benzyl alcohol vs. the reaction time over Pt/PCN-224(Zn) in the presence or absence of light irradiation or external heating with other reaction parameters remaining identical. Adapted with permission from ref. 280 Copyright (2017) American Chemical Society. | ||
In addition, the 1O2-mediated oxidation of HMF, a key biomass-derived intermediate, can give value-added products such as 2,5-furandicarboxylic acid (FDCA), which is an important monomer substitute for petrochemically-derived terephthalic acid to make polyethylene furanoate and other polymers (Fig. 20a).287,288 Current technology for the oxidation of HMF to FDCA is costly as it normally needs to be run with noble metal catalysts at elevated temperatures (80–150 °C), thus needing high energy consumption and high oxygen pressure.289 Xu et al. created a photocatalyst of cobalt thioporphyrazine (CoPz) dispersed on g-C3N4 for the oxidation of HMF to FDCA under simulated sunlight (Fig. 20b and c).287 The reaction is performed at ambient temperature and air pressure, and the FDCA yield is 96.1%.
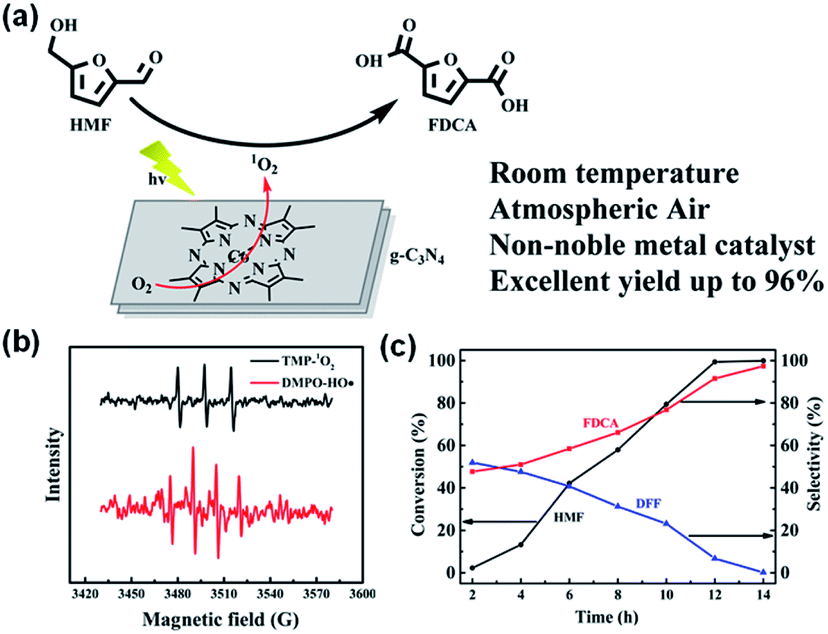 | ||
| Fig. 20 (a) Possible mechanism of the photocatalytic oxidation of HMF with the CoPz/g-C3N4 catalyst. (b) EPR signals of the DMPO-HO˙ adduct in water in the presence of bulk g-C3N4 and the TMP-1O2 adduct in water in the presence of CoPz/g-C3N4 in the photocatalysis process. (c) Photocatalytic performance. Adapted with permission from ref. 287 Copyright (2017) American Chemical Society. | ||
5.4 Surface lattice oxygen
Catalytic oxidation is the most appealing pathway to process effluent streams with dilute VOC (<0.5 vol%).290,291 Conventional catalysts required for the oxidation of HCHO292 normally contain noble metals (e.g., Pt, Au, Pd, and Ag) that are costly and poorly available, and cheap and thermally stable TMO catalysts are excellent alternatives although their efficiency may be somewhat lower. Zha et al. synthesized Co3O4 nanowires with abundant OVs grown in situ on the Ni foam (denoted as r-Co3O4 NW@Ni foam) as a stable catalyst for the highly efficient oxidation of HCHO to formate (Fig. 21a), and used the OVs-free Co3O4 NW@Ni foam and Co3O4 nanoparticles (Co3O4 NPs) to compare and characterize the catalytic performance.293 The T10% (i.e., temperature at which HCHO conversion reaches 10%) of the catalysts ranks as r-Co3O4 NW@Ni foam (75 °C) < Co3O4 NW@Ni foam (100 °C) < Co3O4 NP (132 °C). According to in situ DRIFTs studies, HCHO and formate are slightly more easily adsorbed and formed on the OVs-free Co3O4 NW@Ni foam without O2, but the formate species are more active on the surface of the r-Co3O4 NW@Ni foam (Fig. 21b). Further theoretical studies reveal that the abundant surface OVs reduce the adsorption energy of O2, and r-Co3O4 NW@Ni can thus store more ROS and have a higher catalytic activity in HCHO oxidation (Fig. 21c). In another example, Ma et al. used MOF-derived Co3O4 with different shapes for the catalytic oxidation of o-xylene and studied how the surface twofold-coordinate lattice oxygen (O2f) creates higher catalyst reactivity (Fig. 21d).294 The rod-like Co3O4-R has a lower T90% (i.e., temperature at which o-xylene conversion reaches at 90%) of 270 °C than the spherical Co3O4–S (about 290 °C). According to the OV formation energy and the adsorption energy of O2, OVs are more easily formed and O2 is more easily replenished on the Co3O4-R surface (Fig. 21e and f).
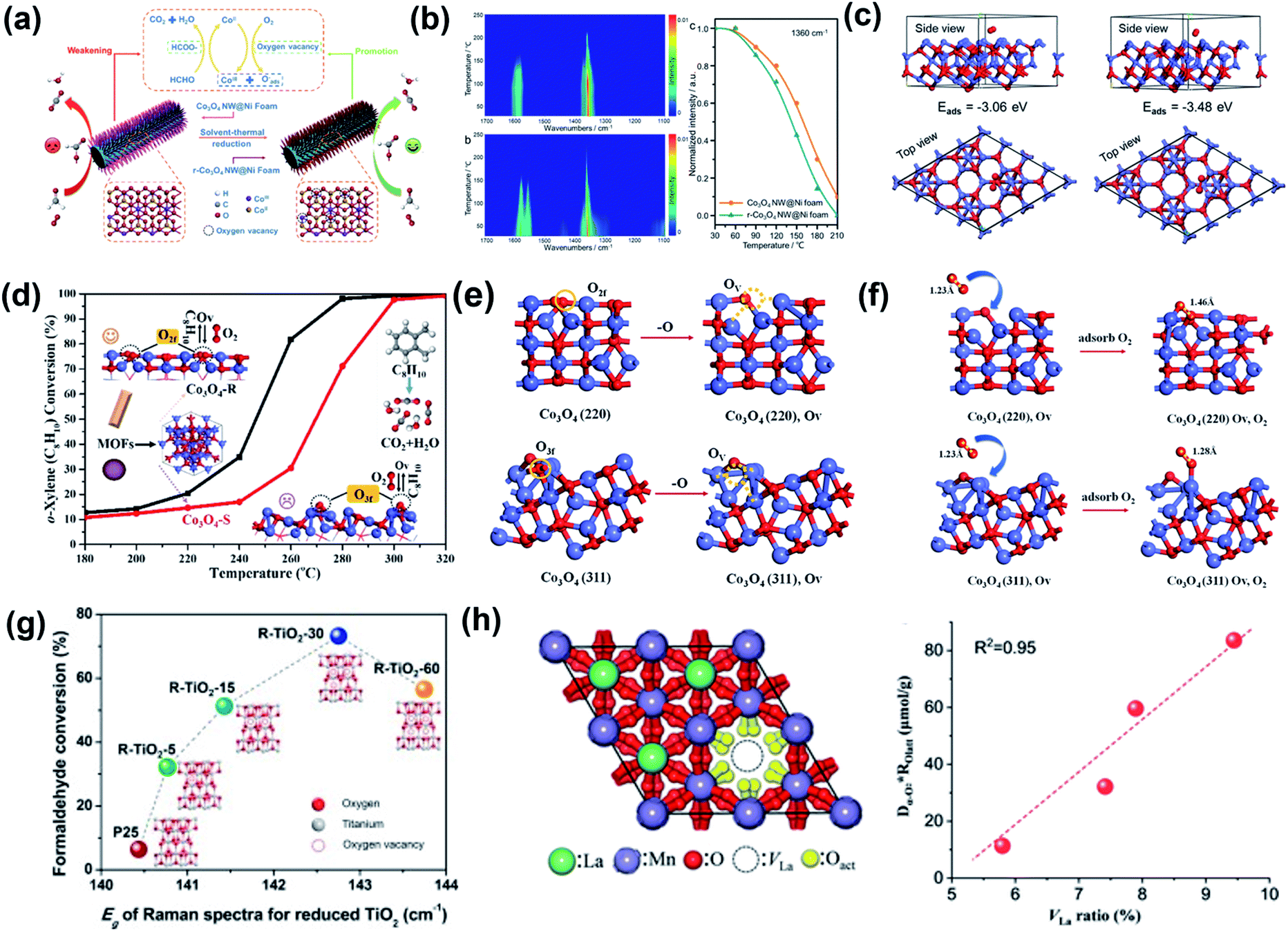 | ||
| Fig. 21 (a) Schematic illustration of the catalytic oxidation of HCHO over r-Co3O4 NW@Ni with OVs; (b) In situ DRIFTs after HCHO adsorption over Co3O4 NW@Ni and r-Co3O4 NW@Ni catalysts as a function of temperature. (c) O2 molecule adsorbed on the {111} surface of Co3O4 NW@Ni foam and r-Co3O4 NW@Ni catalysts. Adapted with permission from ref. 293 Copyright (2020) American Chemical Society. (d) Catalytic oxidation activities of o-xylene over MOF-derived Co3O4 with different shapes. (e) OV formation over the (220) and (311) surfaces of Co3O4, respectively; (f) side view of the adsorption configuration of O2 over the (220) and (311) surfaces of Co3O4 with OVs. The Co and O atoms are in blue and red, respectively. Adapted with permission from ref. 294 Copyright (2021) American Chemical Society. (g) Formaldehyde conversion over reduced TiO2 of different OV concentrations. Adapted with permission from ref. 296 Copyright (2019) Elsevier. (h) Activation of surface lattice oxygen induced by La vacancies and the amount of released surface Olatt (Dα-O2 ROlatt) as a function of VLa ratio. Adapted with permission from ref. 297 Copyright (2021) Wiley-VCH Verlag GmbH & Co. KGaA. | ||
Wang et al. performed catalytic HCHO oxidation with three kinds of Co3O4 with different OV concentrations and demonstrated the dependence of the OL reactivity on the OV concentration.295 The reaction rate constant (k) is 0.14 min−1 for the Co3O4 nanobelts with a high OV concentration, which is about 20 times that of Co3O4 nanoplates (0.0071 min−1). However, excessive OVs may also impair reactivity. For example, He et al. found that the catalytic activity of reduced TiO2 for HCHO oxidation ranks as R-TiO2-30 > R-TiO2-60 > R-TiO2-15 > R-TiO2-5 (Fig. 21g).296 In the catalytic reaction, O2 is adsorbed on the surface of the reduced TiO2, accepts electrons, and is transformed into active OL to degrade HCHO. However, excessive OVs can reduce the adsorbed O2 and trap electrons, and the formation of ROS from O2 then becomes less effective.
Dong et al. developed a CoMn2O4 catalyst that has a lower activation energy (35.5 kJ mol−1) for the oxidation of toluene than other metal oxides (Co3O4, MnOx, and Co3O4/MnOx).293 The obtained CoMn2O4 has a large surface area, rich cationic vacancy, and high mobility of oxygen species, all of which contribute to the high catalytic activity. According to in situ temperature-programmed experiments, the surface OL induces the catalytic reaction, then gaseous O2 is moved to the bulk phase lattice, and finally the bulk oxygen species is migrated to the surface to form more surface OL species at 200–250 °C. Toluene oxidation occurs via the benzyl alcohol-benzoate-anhydride-acetate reaction pathway over spinel CoMn2O4, and the conversion of the surface anhydride is the rate-determining step, especially at 200–210 °C.
For perovskites of the ABO3 general formula, modulation of A-site defects can create effective oxidation catalysts that can be used in clean air applications. Liu et al. regulated the La vacancies (VLa) in LaMnO3.15 by simply introducing urea in the traditional citrate-based synthesis to examine the relationship between the creation of VLa and the activation of surface OL (Fig. 21h).297 The LaMnO3.15 catalyst with optimized VLa achieves toluene oxidation at above 220°C which is 10–25 times faster than the LaMnO3.15 prepared from the traditional urea-free citric acid sol–gel method. The easier charge transfer from Mn to O in the MnO6 octahedron activates the surface OL and enhances the redox properties of the LaMnO3.15 perovskite, thus enabling superior performance.
Conversely, OL is also used in various cases for the thorough oxidation of alcohols to carboxylic acids (RCH2OH → RCOOH). The aerobic oxidation of biomass to carboxylic acids under mild conditions is environmentally and economically advantageous to generate value-added chemicals from renewable feedstocks. Jin et al. found that Co-doped Mg3Al(OH)y(CO3)z can promote the aerobic oxidation of glycerol into tartronic acid,305 which is a high value-added chemical that has been widely used as a pharmaceutical and anti-corrosive protective agent, as well as for the monomer of biopolymers.306 The complete conversion of glycerol is achieved with 64% selectivity of tartronic acid under mild conditions (70 °C, 0.1 MPa O2). Hayashi et al. used MnO2 for the aerobic oxidation of HMF and obtained FDCA in 95% yield under mild conditions (3 eq. NaHCO3, 10 bar O2, 120 °C, 24 h).307 Han et al. prepared a mixed oxide MnOx-CeO2 (Mn/Ce = 6) catalyst via co-precipitation for the aerobic oxidation of HMF.308 After 15 h at 110 °C in the presence of KHCO3, the catalyst gives 91% FDCA yield and 98% HMF conversion. Zhang et al. prepared a series of Mn–Co binary oxides for the catalytic oxidation of HMF to FDCA and found that the MnCo2O4 catalyst is significantly better than Mn3O4, Co3O4, and other Mn–Co binary oxides with different Mn/Co molar ratios.309 The MnCo2O4 catalyst has a Mn/Co molar ratio of 0.5, and gives 99.5% HMF conversion and 70.9% FDCA yield when the reaction is carried out at 100 °C under 10 bar O2 with KHCO3 for 24 h.
Reducing the OV formation energy increases the OL reactivity. Hayashi et al. found that compared with α- and γ-MnO2, β- and λ-MnO2 have lower OV formation energies due to their distinct crystal structures, and the OV formation energies of α- and γ-MnO2 are higher both at the planar oxygen sites and at the bent oxygen sites.172 The initial reaction rate per surface area of the catalysts for the aerobic oxidation of HMF to FDCA decreases in the order of β-MnO2 (16.4 μmol h−1 m−2) > λ-MnO2 (12.2 μmol h−1 m−2) > α-MnO2 (7.6 μmol h−1 m−2) ≈ γ-MnO2 (7.4 μmol h−1 m−2) > δ-MnO2 (5.3 μmol h−1 m−2), which agrees well with what is expected from computational results. Wan et al. found that the high OV concentration of CuMn2O4 promotes the mobility and adsorption of oxygen species in the oxidation of HMF to FDCA.310 Liu et al. demonstrated with DFT calculations that an increasing OV concentration boosts the OL reactivity of manganese oxide by reducing the OV formation energy, and also strengthens the adsorption and activation of O2 to regenerate OL by significantly cutting down the O2 adsorption energy, ultimately increasing the catalytic activity for the oxidation of HMF.184
Regulating the coordination environment of the metal also affects the activity of OL. The coordination structures affect the M–O bond strength, and thu results in different mobilities of the lattice oxygen. From the Rietveld refinement of the X-ray diffractogram, Sabaté et al. determined that the interplanar spacing of the (100) plane of pristine KMn8O16 increased from 7.1 to 7.7 Å after doping with Ru.299,311 With the lattice expansion and the elongation of the Mn–O bond, the Ru-doped KMn8O16 can more easily lose its OL on the surface, thus allowing selective oxidation of alcohols to aldehydes.
Ishikawa et al. oxidized acrolein to acrylic acid with up to 98% selectivity by using orthorhombic Mo–V–Cu oxide as the catalyst.312 The location of Cu under the oxygen defect site moderates the oxygen activation and prevents side reactions such as C–C scission to form COx and acetic acid, thus improving the selectivity for acrylic acid.313 The dispersion of noble metals (Au, Pt, Pd, Ru and Ag) can also regulate the coordination environment of TMO and generate OL with high reactivity thanks to the enhanced electronic metal-support interactions. Li et al. reported that the OL activated at the interfacial sites in Au1/CeO2 has lower OV formation energy (0.36 eV) and is more selective in oxidizing the alcohol substrate compared with the OL in Au NPs/CeO2.314
Various studies have demonstrated reactions in which OL is the ROS directly responsible for the oxidation, and the OVs on the catalyst surface commonly expedite the dissociation of the O–H bond in the alcohol substrate.315,316 Wang et al. used ultrathin CoAl-ELDH to catalyze the oxidation of benzyl alcohol to benzaldehyde.182 The adsorption of benzyl alcohol is spontaneous and exothermic on the catalyst surface. The main active sites are OVs and VCo–Co–OHδ−, where alcohol can be captured through linkage via its –OH group and the –OH group can then be activated and oxidized by the OL (Fig. 22). Dai et al. found that the OL-mediated selective photo-oxidation of alcohols over Bi24O31Br10(OH)δ starts with the photo-dehydrogenation of alcohols to form adsorbed hydrogen species on the surface of the photocatalyst.301 The OL of Bi24O31Br10(OH)δ then oxidizes the alcohol to form aldehydes and OVs, and the OVs are subsequently healed by mild oxidants (O2 or benzoquinone) to complete the photocatalytic cycle.
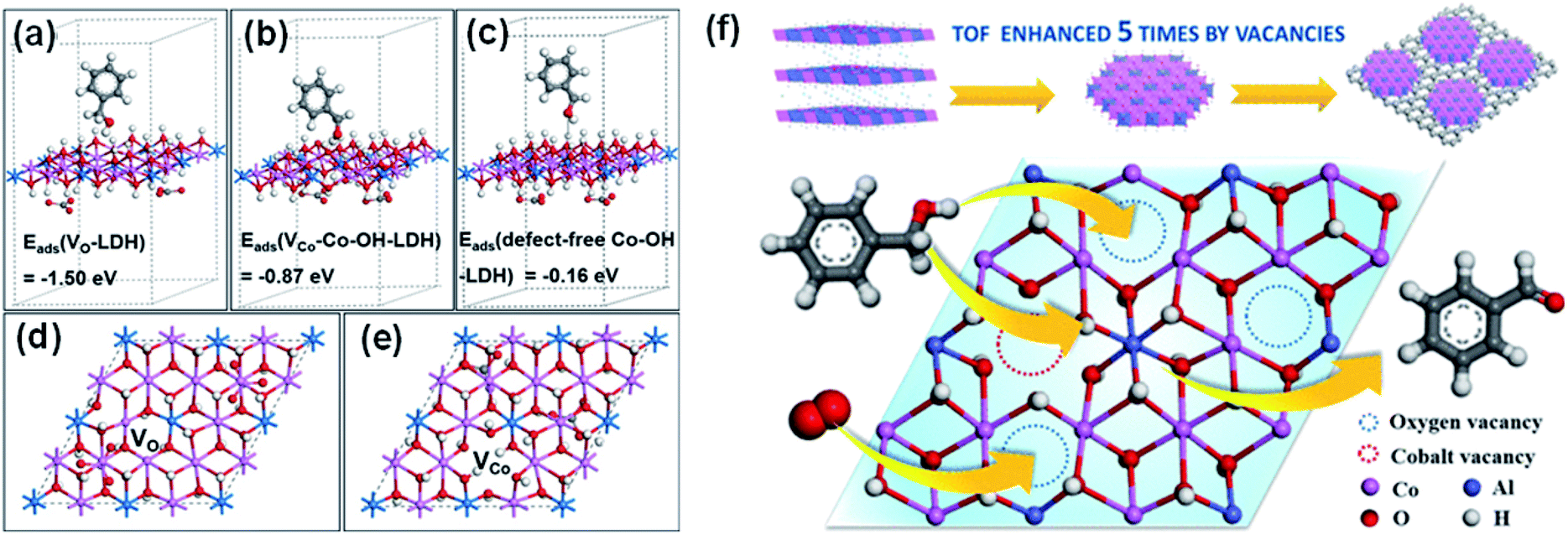 | ||
| Fig. 22 DFT-calculated conformation of adsorption states and adsorption energies of a benzyl alcohol molecule on (a) an OV, (b) a VCo–Co–OHδ− site, and (c) defect-free Co–OH. Top views of the optimized structures of (d) an OV and (e) a Co vacancy. (f) Schematic diagram of Co-containing LDH for the aerobic oxidation of alcohols. Reproduced with permission. Adapted with permission from ref. 182 Copyright (2018) American Chemical Society. | ||
While both 1O2 and OL allow selective oxidations, oxidations mediated by OL are typically carried out under conditions such as high temperature and high humidity. Catalysts may collapse or agglomerate to a certain extent in these harsh environments, which will decrease the specific surface area and the number of exposed active sites. As a result, it may not be possible to maintain high catalytic efficiency and the catalyst may become deactivated. In contrast, 1O2-mediated AOP are more favorable for selective oxidations; its catalytic conditions are milder, thus avoiding the agglomeration, collapse, and thus the deactivation of catalysts.
6 Challenges and perspectives
Various knowledge gaps remain in developing novel AOP and exploring the controllable reactivity of ROS. For example, although defect engineering has proven fruitful in accessing ROS for the desired AOP, some routes are sluggish and give interfering reactive species that do not contribute to the intended chemical transformation. To accomplish the rational design of catalysts and reactions, more understanding is needed regarding how the ROS contribute to the AOP and affect the selectivity and reactivity. To this end, in situ characterization techniques (e.g., Raman, FTIR, and EPR) may be helpful for the on-line detection of ROS, and monitoring the transition states of the ROS, as well as verifying the interactions of the ROS with the organic substrate and oxidation pathways.Controversy still exists regarding how the structures of catalysts, particularly the ubiquitous point defects, participate in the AOP. While in many cases OVs engage directly as active defect sites to accelerate the catalytic reaction,305 at times the intrinsic properties of the catalyst altered by OVs are no less important.118 The selective generation of ROS toward the specified oxidation reaction is still a growing research area, and various questions remain to be answered. For example, given a certain TMO catalyst, what kind of selectivity should be expected? What is the mechanistic aspect that can account for differences in the catalyst's behavior? How do researchers exercise control in designing catalysts? More research is needed on how the surface structure of catalysts regulate the generation and reactivity of ROS for intended reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported primarily by the National Key Research and Development Program of China (2020YFA0710303) and the National Natural Science Foundation of China (No. U1905215, 51772053 and 52072076). We thank Jay Wang for suggestions in writing.Notes and references
- C. Xu, E. Paone, D. Rodriguez-Padron, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- H. Luo, J. Barrio, N. Sunny, A. Li, L. Steier, N. Shah, I. E. L. Stephens and M. M. Titirici, Adv. Energy Mater., 2021, 11, 2101180–2101231 CrossRef CAS.
- Q. Chen, D. Yang, L. Yu, X. Jing and Y. Chen, Mater. Horiz., 2020, 7, 317–337 RSC.
- Y. Shang, X. Xu, B. Gao, S. Wang and X. Duan, Chem. Soc. Rev., 2021, 50, 5281–5322 RSC.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. R. Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- H. Dong, G. Wei, D. Yin and X. Guan, J. Hazard. Mater., 2020, 384, 121497 CrossRef CAS PubMed.
- L. Luo, T. Zhang, M. Wang, R. Yun and X. Xiang, ChemSusChem, 2020, 13, 5173–5184 CrossRef CAS PubMed.
- S. Wu, P. Wang, J. Qin, Y. Pei and Y. Wang, Adv. Funct. Mater., 2021, 31, 2102160 CrossRef CAS.
- J. L. Wang, X. C. Dai, H. L. Wang, H. L. Liu, J. Rabeah, A. Brückner, F. Shi, M. Gong and X. J. Yang, Nat. Commun., 2021, 12, 6840 CrossRef CAS PubMed.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS PubMed.
- B. H. Ko, B. Hasa, H. Shin, Y. Zhao and F. Jiao, J. Am. Chem. Soc., 2022, 144, 1258–1266 CrossRef CAS PubMed.
- K. Wang, C. Han, Z. Shao, J. Qiu, S. Wang and S. Liu, Adv. Funct. Mater., 2021, 31, 2102089 CrossRef CAS.
- Z. Z. Yang, C. Zhang, G. M. Zeng, X. F. Tan, H. Wang, D. l. Huang, K. H. Yang, J. J. Wei, C. Ma and K. Nie, J. Mater. Chem. A, 2020, 8, 4141–4173 RSC.
- Q. Zhang, D. He, X. Li, W. Feng, C. Lyu and Y. Zhang, J. Hazard. Mater., 2020, 384, 121350 CrossRef CAS PubMed.
- R. Pelalak, Z. Heidari, R. Alizadeh, E. Ghareshabani, N. Nasseh, A. Marjani, A. B. Albadarin and S. Shirazian, J. Hazard. Mater., 2021, 411, 125074 CrossRef CAS PubMed.
- Y. Sun, R. Li, C. Song, H. Zhang, Y. Cheng, A. Nie, H. Li, D. D. Dionysiou, J. Qian and B. Pan, Appl. Catal., B, 2021, 298, 120537 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Soc. Rev., 2017, 46, 816–854 RSC.
- J. Wang and S. Wang, Chem. Eng. J., 2021, 411, 128392 CrossRef CAS.
- S. O. Ganiyu, S. Sable and M. Gamal El Din, Chem. Eng. J., 2022, 429, 132492 CrossRef CAS.
- Y. Ding, L. Fu, X. Peng, M. Lei, C. Wang and J. Jiang, Chem. Eng. J., 2022, 427, 131776 CrossRef CAS.
- J. Ye, G. Zhuang, Y. Wen, J. Wei, J. Chen, Z. Zhuang and Y. Yu, Chem. Eng. J., 2019, 372, 260–268 CrossRef CAS.
- Y. Zhu, X. Liu, S. Jin, H. Chen, W. Lee, M. Liu and Y. Chen, J. Mater. Chem. A, 2019, 7, 5875–5897 RSC.
- S. Wu, G. Zhuang, J. Wei, Z. Zhuang and Y. Yu, J. Mater. Chem. A, 2018, 6, 18234–18241 RSC.
- J. Azadmanjiri, V. K. Srivastava, P. Kumar, J. Wang and A. Yu, J. Mater. Chem. A, 2018, 6, 13509–13537 RSC.
- C. Xie, D. Yan, W. Chen, Y. Zou, R. Chen, S. Zang, Y. Wang, X. Yao and S. Wang, Mater. Today, 2019, 31, 47–68 CrossRef CAS.
- S. Guo, Y. Jiang, L. Li, X. Huang, Z. Zhuang and Y. Yu, J. Mater. Chem. A, 2018, 6, 4167–4178 RSC.
- G. Zhuang, Y. Chen, Z. Zhuang, Y. Yu and J. Yu, Sci. China Mater., 2020, 63, 2089–2118 CrossRef CAS.
- M. Ai, J. W. Zhang, Y. W. Wu, L. Pan, C. Shi and J. J. Zou, Chem.–Asian J., 2020, 15, 3599 CrossRef CAS PubMed.
- J. Miao, X. Duan, J. Li, J. Dai, B. Liu, S. Wang, W. Zhou and Z. Shao, Chem. Eng. J., 2019, 355, 721–730 CrossRef CAS.
- Q. Lin and Y. Deng, Environ. Sci. Technol., 2021, 55, 15010–15012 CrossRef CAS PubMed.
- L. Xiong and J. Tang, Adv. Energy Mater., 2021, 11, 2003216 CrossRef CAS.
- J. Hoigné and H. Bader, Water Res., 1983, 17, 185–194 CrossRef.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- W. D. Oh, Z. Dong and T. T. Lim, Appl. Catal., B, 2016, 194, 169–201 CrossRef CAS.
- W. A. Pryor, Annu. Rev. Phytopathol., 1986, 48, 657–667 CAS.
- E. Codorniu-Hernández and P. G. Kusalik, J. Am. Chem. Soc., 2012, 134, 532–538 CrossRef PubMed.
- J. L. Wang and L. J. Xu, Crit. Rev. Environ. Sci. Technol., 2012, 42, 251–325 CrossRef CAS.
- S. Luo, Z. Wei, D. D. Dionysiou, R. Spinney, W. P. Hu, L. Chai, Z. Yang, T. Ye and R. Xiao, Chem. Eng. J., 2017, 327, 1056–1065 CrossRef CAS.
- L. Wojnarovits and E. Takacs, Radiat. Phys. Chem., 2014, 96, 120–134 CrossRef CAS.
- D. Minakata, K. Li, P. Westerhoff and J. Crittenden, Environ. Sci. Technol., 2009, 43, 6220–6227 CrossRef CAS PubMed.
- E. W. Neuman, J. Chem. Phys., 1934, 2, 31–33 CrossRef CAS.
- D. T. Sawyer and J. S. Valentine, Acc. Chem. Res., 1981, 14, 393–400 CrossRef CAS.
- Q. Yi, J. Ji, B. Shen, C. Dong, J. Liu, J. Zhang and M. Xing, Environ. Sci. Technol., 2019, 53, 9725–9733 CrossRef CAS PubMed.
- E. A. Mayeda and A. J. Bard, J. Am. Chem. Soc., 1973, 95, 6223–6226 CrossRef CAS.
- C. Marques, J. M. F. Ferreira, E. Andronescu, D. Ficai, M. Sonmez and A. Ficai, Int. J. Nanomed., 2014, 9, 2713–2725 Search PubMed.
- L. De Trizio and L. Manna, Chem. Rev., 2016, 116, 10852–10887 CrossRef CAS PubMed.
- S. Korshunov and A. Imlay James, J. Bacteriol., 2006, 188, 6326–6334 CrossRef CAS PubMed.
- J. Ma, Z. Wei, R. Spinney, D. D. Dionysiou and R. Xiao, Environ. Sci.: Water Res. Technol., 2021, 7, 1966–1970 RSC.
- Y. Pan, H. Xu, M. Chen, K. Wu, Y. Zhang and D. Long, ACS Catal., 2021, 11, 5974–5983 CrossRef CAS.
- C. Pan, L. Fu, F. Lide, Y. Ding, C. Wang, J. Huang and S. Wang, Chem. Eng. J., 2022, 431, 133957 CrossRef CAS.
- B. Cai, J. F. Feng, Q. Y. Peng, H. F. Zhao, Y. C. Miao and H. Pan, J. Hazard. Mater., 2020, 392, 122279 CrossRef CAS PubMed.
- M. Q. Yang, N. Zhang and Y. J. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 1156–1164 CrossRef CAS PubMed.
- C. Zhang, F. R. F. Fan and A. J. Bard, J. Am. Chem. Soc., 2009, 131, 177–181 CrossRef CAS PubMed.
- X. Ma, X. Li, J. Zhou, Y. Wang and X. Lang, Chem. Eng. J., 2021, 426, 131418 CrossRef CAS.
- E. J. Nanni and D. T. Sawyer, J. Am. Chem. Soc., 1980, 102, 7591–7593 CrossRef CAS.
- R. Dietz, A. E. J. Forno, B. E. Larcombe and M. E. Peover, J. Chem. Soc. B, 1970, 816–820 RSC.
- M. V. Merritt and D. T. Sawyer, J. Org. Chem., 1970, 35, 2157–2159 CrossRef CAS.
- J. Wang and S. Wang, Chem. Eng. J., 2020, 401, 126158 CrossRef CAS.
- J. S. Filippo, C. I. Chern and J. S. Valentine, J. Org. Chem., 1976, 41, 1077–1078 CrossRef PubMed.
- A. A. Ghogare and A. Greer, Chem. Rev., 2016, 116, 9994–10034 CrossRef CAS PubMed.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- R. W. Redmond and I. E. Kochevar, Photochem. Photobiol., 2006, 82, 1178–1186 CrossRef CAS PubMed.
- L. Chen, S. Wang, Z. Yang, J. Qian and B. Pan, Appl. Catal., B, 2021, 292, 120193 CrossRef CAS.
- J. Lin, Q. Dai, H. Zhao, H. Cao, T. Wang, G. Wang and C. Chen, Environ. Sci. Technol., 2021, 55, 8683–8690 CrossRef CAS PubMed.
- X. Liu, A. Mazel, S. Marschner, Z. Fu, M. Muth, F. Kirschhöfer, G. Brenner-Weiss, S. Bräse, S. Diring and F. Odobel, ACS Appl. Mater. Interfaces, 2021, 13, 57768–57773 CrossRef CAS PubMed.
- R. Ma, N. Liu, T. T. Lin, T. Zhao, S. L. Huang and G. Y. Yang, J. Mater. Chem. A, 2020, 8, 8548–8553 RSC.
- N. Zeinali, I. Oluwoye, M. Altarawneh and B. Z. Dlugogorski, Ecotoxicol. Environ. Saf., 2019, 184, 109605 CrossRef CAS PubMed.
- T. Matsuura, N. Yoshimura, A. Nishinaga and I. Saito, Tetrahedron, 1972, 28, 4933–4938 CrossRef CAS.
- Y. Liu, W. Shu, K. Chen, Z. Peng and W. Chen, ACS Catal., 2012, 2, 2557–2565 CrossRef CAS.
- Y. Li, C. Wang, H. Zheng, F. Wan, F. Yu, X. Zhang and Y. Liu, Appl. Surf. Sci., 2017, 391, 654–661 CrossRef CAS.
- W. Li, J. Shi, K. H. L. Zhang and J. L. MacManus-Driscoll, Mater. Horiz., 2020, 7, 2832–2859 RSC.
- S. Huang, T. Ouyang, B. F. Zheng, M. Dan and Z. Q. Liu, Angew. Chem., Int. Ed., 2021, 60, 9546–9552 CrossRef CAS PubMed.
- Y. Li, J. Huang, T. Peng, J. Xu and X. Zhao, ChemCatChem, 2010, 2, 1082–1087 CrossRef CAS.
- L. Ren, M. Mao, Y. Li, L. Lan, Z. Zhang and X. Zhao, Appl. Catal., B, 2016, 198, 303–310 CrossRef CAS.
- S. Albonetti, F. Cavani and F. TrifirÒ, Catal. Rev., 1996, 38, 413–438 CrossRef CAS.
- H. Xu, N. Yan, Z. Qu, W. Liu, J. Mei, W. Huang and S. Zhao, Environ. Sci. Technol., 2017, 51, 8879–8892 CrossRef CAS PubMed.
- C. H. Chung, F. Y. Tu, T. A. Chiu, T. T. Wu and W. Y. Yu, Chem. Lett., 2021, 50, 856–865 CrossRef CAS.
- M. Konsolakis, Appl. Catal., B, 2016, 198, 49–66 CrossRef CAS.
- H. Jin, X. Tian, Y. Nie, Z. Zhou, C. Yang, Y. Li and L. Lu, Environ. Sci. Technol., 2017, 51, 12699–12706 CrossRef CAS PubMed.
- W. Dai, J. Long, L. Yang, S. Zhang, Y. Xu, X. Luo, J. Zou and S. Luo, J. Energy Chem., 2021, 61, 281–289 CrossRef.
- S. Guan, L. Wang, S. M. Xu, D. Yang, G. I. N. Waterhouse, X. Qu and S. Zhou, Chem. Sci., 2019, 10, 2336–2341 RSC.
- Z. Jiang, F. Yang, G. Yang, L. Kong, M. O. Jones, T. Xiao and P. P. Edwards, J. Photochem. Photobiol., A, 2010, 212, 8–13 CrossRef CAS.
- Y. Zhu, R. Zhu, Y. Xi, J. Zhu, G. Zhu and H. He, Appl. Catal., B, 2019, 255, 117739 CrossRef CAS.
- C. M. Lousada, A. J. Johansson, T. Brinck and M. Jonsson, J. Phys. Chem. C, 2012, 116, 9533–9543 CrossRef CAS.
- C. C. Lin, F. R. Smith, N. Ichikawa, T. Baba and M. Itow, Int. J. Chem. Kinet., 1991, 23, 971–987 CrossRef CAS.
- A. Hiroki and J. A. LaVerne, J. Phys. Chem. B, 2005, 109, 3364–3370 CrossRef CAS PubMed.
- J. Lee, U. von Gunten and J. H. Kim, Environ. Sci. Technol., 2020, 54, 3064–3081 CrossRef CAS PubMed.
- R. L. Johnson, P. G. Tratnyek and R. O. B. Johnson, Environ. Sci. Technol., 2008, 42, 9350–9356 CrossRef CAS PubMed.
- J. Wang and S. Wang, Chem. Eng. J., 2018, 334, 1502–1517 CrossRef CAS.
- J. Zhu, S. Wang, H. Li, J. Qian, L. Lv and B. Pan, Water Res., 2021, 202, 117397 CrossRef CAS PubMed.
- W. Ren, L. Xiong, X. Yuan, Z. Yu, H. Zhang, X. Duan and S. Wang, Environ. Sci. Technol., 2019, 53, 14595–14603 CrossRef CAS PubMed.
- S. Cai, Q. Zhang, Z. Wang, S. Hua, D. Ding, T. Cai and R. Zhang, Appl. Catal., B, 2021, 291, 120093 CrossRef CAS.
- J. Hu, H. Dong, J. Qu and Z. Qiang, Water Res., 2017, 112, 1–8 CrossRef CAS PubMed.
- M. M. Montemore, M. A. van Spronsen, R. J. Madix and C. M. Friend, Chem. Rev., 2018, 118, 2816–2862 CrossRef CAS PubMed.
- C. Zhang, Z. Huang, J. Lu, N. Luo and F. Wang, J. Am. Chem. Soc., 2018, 140, 2032–2035 CrossRef CAS PubMed.
- Z. Xie, J. Zhang, Y. Xiao, Y. Xie, W. Zhu, S. Yu, T. Hou, S. Liang and L. Wang, Energy Environ. Sci., 2021, 4, 444–450 CAS.
- L. Wenqian, H. Xu, L. Boda, Z. Bin, L. Ting, H. Ying and M. Jun, Appl. Catal., B, 2021, 305, 121019 Search PubMed.
- H. Zhang, G. P. Hatzis, C. E. Moore, D. A. Dickie, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, J. Am. Chem. Soc., 2019, 141, 9516–9520 CrossRef CAS PubMed.
- Y. Yang, K. Hu, P. Zhang, P. Zhou, X. Duan, H. Sun and S. Wang, Small, 2021, 17, 2100927 CrossRef CAS PubMed.
- Y. Li, H. Dong, L. Li, L. Tang, R. Tian, R. Li, J. Chen, Q. Xie, Z. Jin, J. Xiao, S. Xiao and G. Zeng, Water Res., 2021, 192, 116850 CrossRef CAS PubMed.
- X. Chu, Y. Qu, A. Zada, L. Bai, Z. Li, F. Yang, L. Zhao, G. Zhang, X. Sun, Z. D. Yang and L. Jing, Adv. Sci., 2020, 7, 2001543 CrossRef CAS PubMed.
- X. Feng, J. Xu, X. Xu, S. Zhang, J. Ma, X. Fang and X. Wang, ACS Catal., 2021, 11, 12112–12122 CrossRef CAS.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- H. Arandiyan, S. S. Mofarah, C. C. Sorrell, E. Doustkhah, B. Sajjadi, D. Hao, Y. Wang, H. Sun, B. J. Ni, M. Rezaei, Z. Shao and T. Maschmeyer, Chem. Soc. Rev., 2021, 50, 10116–10211 RSC.
- M. Xing, W. Xu, C. Dong, Y. Bai, J. Zeng, Y. Zhou, J. Zhang and Y. Yin, Chem, 2018, 4, 1359–1372 CAS.
- R. Yang, Y. Fan, R. Ye, Y. Tang, X. Cao, Z. Yin and Z. Zeng, Adv. Mater., 2021, 33, 2004862 CrossRef CAS PubMed.
- N. Thomas, D. D. Dionysiou and S. C. Pillai, J. Hazard. Mater., 2021, 404, 124082 CrossRef CAS PubMed.
- X. Wang, W. Liu, J. Qin and L. Lei, Ind. Eng. Chem. Res., 2020, 59, 2192–2202 CrossRef CAS.
- Q. Wu, H. Yang, L. Kang, Z. Gao and F. Ren, Appl. Catal., B, 2020, 263, 118282 CrossRef CAS.
- W. Jiang, D. D. Dionysiou, M. Kong, Z. Liu, Q. Sui and S. Lyu, Chem. Eng. J., 2020, 380, 122537 CrossRef CAS.
- M. E. Lindsey and M. A. Tarr, Environ. Sci. Technol., 2000, 34, 444–449 CrossRef CAS.
- L. Lyu, L. Zhang, G. He, H. He and C. Hu, J. Mater. Chem. A, 2017, 5, 7153–7164 RSC.
- L. Z. Huang, M. Zhu, Z. Liu, Z. Wang and H. C. B. Hansen, J. Hazard. Mater., 2019, 364, 39–47 CrossRef CAS PubMed.
- J. Y. Qiu, J. H. Chen, B. Y. Xiao, X. X. Li, T. Wan, F. H. Qin, Y. Mi and Z. Y. Huang, Catal. Lett., 2020, 150, 222–233 CrossRef CAS.
- A. Y. Zhang, T. Lin, Y. Y. He and Y. X. Mou, J. Hazard. Mater., 2016, 311, 81–90 CrossRef CAS PubMed.
- B. Gong, C. Ku, H. Q. Yu and P. H. L. Sit, J. Phys. Chem. C, 2021, 125, 8508–8517 CrossRef CAS.
- W. Liu, J. Tian, C. Mao, Z. Wang, J. Liu, R. A. Dahlgren, L. Zhang and X. Wang, Anal. Chim. Acta, 2020, 1127, 246–255 CrossRef PubMed.
- Z. Tan, J. Zhang, Y. C. Chen, J. P. Chou and Y. K. Peng, J. Phys. Chem. Lett., 2020, 11, 5390–5396 CrossRef CAS PubMed.
- S. Zhan, H. Zhang, X. Mi, Y. Zhao, C. Hu and L. Lyu, Environ. Sci. Technol., 2020, 54, 8333–8343 CrossRef CAS PubMed.
- Z. Weng, J. Li, Y. Weng, M. Feng, Z. Zhuang and Y. Yu, J. Mater. Chem. A, 2017, 5, 15650–15660 RSC.
- X. Zhang, T. Li, M. Guan, R. Li, J. Liu, Y. Wang, C. Zhang and C. Fan, Appl. Surf. Sci., 2021, 567, 150828 CrossRef CAS.
- M. Setvin, U. Aschauer, P. Scheiber, Y. F. Li, W. Hou, M. Schmid, A. Selloni and U. Diebold, Science, 2013, 341, 988–991 CrossRef CAS PubMed.
- S. Zhao, Y. Wen, X. Liu, X. Pen, F. Lu, F. Gao, X. Xie, C. Du, H. Yi, D. Kang and X. Tang, Nano Res., 2020, 13, 1544–1551 CrossRef CAS.
- H. Y. Gao, C. H. Huang, L. Mao, B. Shao, J. Shao, Z. Y. Yan, M. Tang and B.-Z. Zhu, Environ. Sci. Technol., 2020, 54, 14046–14056 CrossRef CAS PubMed.
- D. E. Pennington and A. Haim, J. Am. Chem. Soc., 1968, 90, 3700–3704 CrossRef CAS.
- R. O. C. Norman, P. M. Storey and P. R. West, J. Chem. Soc. B, 1970, 1087–1095, 10.1039/J29700001087.
- N. Chen, D. Lee, H. Kang, D. Cha, J. Lee and C. Lee, J. Environ. Chem. Eng., 2022, 10, 107654 CrossRef CAS.
- J. D. Rush and B. H. J. Bielski, J. Phys. Chem., 1985, 89, 5062–5066 CrossRef CAS.
- K. Xu and U. von Gunten, ACS ES&T Engineering, 2021, 1, 1410–1419 Search PubMed.
- Z. Wu, K. Guo, S. Cao, W. Yao and L. Piao, Nano Res., 2020, 13, 551–556 CrossRef CAS.
- E. S. Kumar, S. Venkatesh and M. S. R. Rao, Appl. Phys. Lett., 2010, 96, 232504 CrossRef.
- Z. Wei, D. Liu, W. Wei, X. Chen, Q. Han, W. Yao, X. Ma and Y. Zhu, ACS Appl. Mater. Interfaces, 2017, 9, 15533–15540 CrossRef CAS PubMed.
- T. Mallat and A. Baiker, Chem. Rev., 2004, 104, 3037–3058 CrossRef CAS PubMed.
- T. Matsumoto, M. Ueno, N. Wang and S. Kobayashi, Chem.–Asian J., 2008, 3, 196–214 CrossRef CAS PubMed.
- B. Z. Zhan and A. Thompson, Tetrahedron, 2004, 60, 2917–2935 CrossRef CAS.
- P. Lyu, J. Zhu, C. Han, L. Qiang, L. Zhang, B. Mei, J. He, X. Liu, Z. Bian and H. Li, ACS Appl. Mater. Interfaces, 2021, 13, 2033–2043 CrossRef CAS PubMed.
- H. Shang, M. Li, H. Li, S. Huang, C. Mao, Z. Ai and L. Zhang, Environ. Sci. Technol., 2019, 53, 6444–6453 CrossRef CAS PubMed.
- W. Qin, G. Fang, Y. Wang and D. Zhou, Chem. Eng. J., 2018, 348, 526–534 CrossRef CAS.
- W. Li, X. He, B. Li, B. Zhang, T. Liu, Y. Hu and J. Ma, Appl. Catal., B, 2022, 305, 121019 CrossRef CAS.
- H. Li, H. Zhu, Y. Shi, H. Shang, L. Zhang and J. Wang, Environ. Sci. Technol., 2022, 56, 1771–1779 CrossRef PubMed.
- H. Li, F. Qin, Z. Yang, X. Cui, J. Wang and L. Zhang, J. Am. Chem. Soc., 2017, 139, 3513–3521 CrossRef CAS PubMed.
- J. Li, C. Shu, C. Liu, X. Chen, A. Hu and J. Long, Small, 2020, 16, 2001812 CrossRef CAS PubMed.
- J. Wu, X. Li, W. Shi, P. Ling, Y. Sun, X. Jiao, S. Gao, L. Liang, J. Xu, W. Yan, C. Wang and Y. Xie, Angew. Chem., Int. Ed., 2018, 57, 8719–8723 CrossRef CAS PubMed.
- M. Kim, B. Lee, H. Ju, J. Y. Kim, J. Kim and S. W. Lee, Adv. Mater., 2019, 31, 1903316 CrossRef PubMed.
- Y. Mao, P. Wang, L. Li, Z. Chen, H. Wang, Y. Li and S. Zhan, Angew. Chem., Int. Ed., 2020, 59, 3685–3690 CrossRef CAS PubMed.
- T. Gan, J. Yang, D. Morris, X. Chu, P. Zhang, W. Zhang, Y. Zou, W. Yan, S. H. Wei and G. Liu, Nat. Commun., 2021, 12, 2741 CrossRef CAS PubMed.
- X. Sun, X. Luo, X. Zhang, J. Xie, S. Jin, H. Wang, X. Zheng, X. Wu and Y. Xie, J. Am. Chem. Soc., 2019, 141, 3797–3801 CrossRef CAS PubMed.
- F. Rao, G. Zhu, W. Zhang, Y. Xu, B. Cao, X. Shi, J. Gao, Y. Huang, Y. Huang and M. Hojamberdiev, ACS Catal., 2021, 11, 7735–7749 CrossRef CAS.
- Y. Zhao and H. Wang, Langmuir, 2021, 37, 13969–13975 CrossRef CAS PubMed.
- Y. Yin, R. Lv, X. Li, L. Lv and W. Zhang, Appl. Catal., B, 2021, 299, 120685 CrossRef CAS.
- Z. Yang, J. Qian, A. Yu and B. Pan, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6659–6664 CrossRef CAS PubMed.
- Y. Yin, Y. Ren, J. Lu, W. Zhang, C. Shan, M. Hua, L. Lv and B. Pan, Appl. Catal., B, 2021, 286, 120282 CrossRef.
- X. Li, J. Wang, X. Duan, Y. Li, X. Fan, G. Zhang, F. Zhang and W. Peng, ACS Catal., 2021, 11, 4848–4861 CrossRef CAS.
- B. F. Sels, D. E. De Vos and P. A. Jacobs, J. Am. Chem. Soc., 2007, 129, 6916–6926 CrossRef CAS PubMed.
- X. Li, J. Liu, A. I. Rykov, H. Han, C. Jin, X. Liu and J. Wang, Appl. Catal., B, 2015, 179, 196–205 CrossRef CAS.
- S. Chong, G. Zhang, N. Zhang, Y. Liu, T. Huang and H. Chang, J. Hazard. Mater., 2017, 334, 150–159 CrossRef CAS PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- J. Wang, X. Xu, Y. Liu, Z. Wang, P. Wang, Z. Zheng, H. Cheng, Y. Dai and B. Huang, ChemSusChem, 2020, 13, 3488–3494 CrossRef CAS PubMed.
- J. Ji, Q. Yan, P. Yin, S. Mine, M. Matsuoka and M. Xing, Angew. Chem., Int. Ed., 2021, 60, 2903–2908 CrossRef CAS PubMed.
- W. J. Duan, J. L. He, Z. L. Wei, Z. R. Dai and C. H. Feng, Environ. Sci.: Nano, 2020, 7, 2982–2994 RSC.
- T. Liu, D. Zhang, K. Yin, C. Yang, S. Luo and J. C. Crittenden, Chem. Eng. J., 2020, 388, 124264 CrossRef CAS.
- Q. Ye, J. Wu, P. Wu, S. Rehman, Z. Ahmed and N. Zhu, Chem. Eng. J., 2021, 417, 12911 CrossRef.
- L. Kong, G. Fang, Z. Fang, Y. Zou, F. Zhu, D. Zhou and J. Zhan, Chem. Eng. J., 2021, 416, 128996 CrossRef CAS.
- M. I. Akhtar, F. Junejo, H. I. Naqvi, M. Hamid, Y. Wang, M. Liu, C. Hu, Y. Xin, D. Ma, M. Gao and H. Xie, Chem. Eng. J., 2022, 433, 134048 CrossRef.
- H. Zeng, L. Deng, H. Zhang, C. Zhou and Z. Shi, J. Hazard. Mater., 2020, 400, 123297 CrossRef CAS PubMed.
- Y. Zhao, H. An, G. Dong, J. Feng, T. Wei, Y. Ren and J. Ma, Chem. Eng. J., 2020, 388, 124371 CrossRef CAS.
- P. Gao, X. Tian, Y. Nie, C. Yang, Z. Zhou and Y. Wang, Chem. Eng. J., 2019, 359, 828–839 CrossRef CAS.
- Y. Bu, H. Li, W. Yu, Y. Pan, L. Li, Y. Wang, L. Pu, J. Ding, G. Gao and B. Pan, Environ. Sci. Technol., 2021, 55, 2110–2120 CrossRef CAS PubMed.
- S. Zhu, X. Li, J. Kang, X. Duan and S. Wang, Environ. Sci. Technol., 2019, 53, 307–315 CrossRef CAS PubMed.
- A. Jawad, K. Zhan, H. Wang, A. Shahzad, Z. Zeng, J. Wang, X. Zhou, H. Ullah, Z. Chen and Z. Chen, Environ. Sci. Technol., 2020, 54, 2476–2488 CrossRef CAS PubMed.
- J. Hu, X. Zeng, G. Wang, B. Qian, Y. Liu, X. Hu, B. He, L. Zhang and X. Zhang, Chem. Eng. J., 2020, 400, 125869 CrossRef CAS.
- E. Hayashi, Y. Yamaguchi, K. Kamata, N. Tsunoda, Y. Kumagai, F. Oba and M. Hara, J. Am. Chem. Soc., 2019, 141, 890–900 CrossRef CAS PubMed.
- H. Liu, W. Jia, X. Yu, X. Tang, X. Zeng, Y. Sun, T. Lei, H. Fang, T. Li and L. Lin, ACS Catal., 2021, 11, 7828–7844 CrossRef CAS.
- X. Xu, L. Li, J. Huang, H. Jin, X. Fang, W. Liu, N. Zhang, H. Wang and X. Wang, ACS Catal., 2018, 8, 8033–8045 CrossRef CAS.
- P. Schlexer, D. Widmann, R. J. Behm and G. Pacchioni, ACS Catal., 2018, 8, 6513–6525 CrossRef CAS.
- N. H. M. Dostagir, R. Rattanawan, M. Gao, J. Ota, J. Y. Hasegawa, K. Asakura, A. Fukouka and A. Shrotri, ACS Catal., 2021, 11, 9450–9461 CrossRef CAS.
- M. Zhao, J. Deng, J. Liu, Y. Li, J. Liu, Z. Duan, J. Xiong, Z. Zhao, Y. Wei, W. Song and Y. Sun, ACS Catal., 2019, 9, 7548–7567 CrossRef CAS.
- X. Zhang, C. Pei, X. Chang, S. Chen, R. Liu, Z. J. Zhao, R. Mu and J. Gong, J. Am. Chem. Soc., 2020, 142, 11540–11549 CrossRef CAS PubMed.
- X. Yang, X. Yu, M. Jing, W. Song, J. Liu and M. Ge, ACS Appl. Mater. Interfaces, 2019, 11, 730–739 CrossRef CAS PubMed.
- A. D. Quinonero, E. B. Garcia, S. L. Rodriguez, J. Juan, D. L. Castello, M. G. Melchor, F. C. Herrera, E. Pellegrin, C. Escudero and A. B. Lopez, ACS Catal., 2020, 10, 6532–6545 CrossRef.
- J. Lu, J. Wang, Q. Zou, D. He, L. Zhang, Z. Xu, S. He and Y. Luo, ACS Catal., 2019, 9, 2177–2195 CrossRef CAS.
- Q. Wang, L. Chen, S. Guan, X. Zhang, B. Wang, X. Cao, Z. Yu, Y. He, D. G. Evans, J. Feng and D. Li, ACS Catal., 2018, 8, 3104–3115 CrossRef CAS.
- Q. Gao, G. Wang, Y. Chen, B. Han, K. Xia and C. Zhou, Environ. Sci.: Water Res. Technol., 2021, 7, 1197–1211 RSC.
- W. Sang, Z. Li, M. Huang, X. Wu, D. Li, L. Mei and J. Cui, Chem. Eng. J., 2020, 383, 123057 CrossRef CAS.
- S. Hussain, E. Aneggi and D. Goi, Environ. Chem. Lett., 2021, 19, 2405–2424 CrossRef CAS.
- F. Chen, X. L. Wu, C. Shi, H. Lin, J. Chen, Y. Shi, S. Wang and X. Duan, Adv. Funct. Mater., 2021, 31, 2007877 CrossRef CAS.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- X. Zhao, X. Li, Z. Zhu, W. Hu, H. Zhang, J. Xu, X. Hu, Y. Zhou, M. Xu, H. Zhang and G. Hu, Appl. Catal., B, 2022, 300, 120759 CrossRef CAS.
- L. S. Zhang, X. H. Jiang, Z. A. Zhong, L. Tian, Q. Sun, Y. T. Cui, X. Lu, J. P. Zou and S. L. Luo, Angew. Chem., Int. Ed., 2021, 60, 21751–21755 CrossRef CAS PubMed.
- L. Wu, Z. Sun, Y. Zhen, S. Zhu, C. Yang, J. Lu, Y. Tian, D. Zhong and J. Ma, Environ. Sci. Technol., 2021, 55, 15400–15411 CrossRef CAS PubMed.
- H. Zhao, X. Liu, Y. Dong, Y. Xia and H. Wang, Appl. Catal., B, 2019, 256, 117872 CrossRef CAS.
- C. Y. Toe, C. Tsounis, J. Zhang, H. Masood, D. Gunawan, J. Scott and R. Amal, Environ. Sci. Technol., 2021, 14, 1140–1175 CAS.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., Int. Ed., 2018, 57, 6054–6059 CrossRef CAS PubMed.
- J. Xiang, T. Zhang, R. Cao, M. Lin, B. Yang, Y. Wen, Z. Zhuang and Y. Yu, Sol. RRL, 2021, 5, 2100703 CrossRef CAS.
- M. Yang, T. Xu, X. Jin, Q. Shen and C. Sun, Appl. Surf. Sci., 2022, 581, 152439 CrossRef CAS.
- J. Sahu, S. Kumar, V. S. Vats, P. A. Alvi, B. Dalela, S. Kumar and S. Dalela, J. Lumin., 2022, 243, 118673 CrossRef CAS.
- Q. Wang, Z. Liu, D. Liu, W. Wang, Z. Zhao, F. Cui and G. Li, Chem. Eng. J., 2019, 360, 838–847 CrossRef CAS.
- M. J. Jackson, Proc. Nutr. Soc., 1999, 58, 1001–1006 CrossRef CAS PubMed.
- C. D. Jaeger and A. J. Bard, J. Phys. Chem., 1979, 83, 3146–3152 CrossRef CAS.
- K. Hiramoto, H. Johkoh, K. I. Sako and K. Kikugawa, Free Radical Res. Commun., 1993, 19, 323–332 CrossRef CAS PubMed.
- J. M. Burns, W. J. Cooper, J. L. Ferry, D. W. King, B. P. DiMento, K. McNeill, C. J. Miller, W. L. Miller, B. M. Peake, S. A. Rusak, A. L. Rose and T. D. Waite, Aquat. Sci., 2012, 74, 683–734 CrossRef CAS.
- Y. Jing and B. P. Chaplin, Environ. Sci. Technol., 2017, 51, 2355–2365 CrossRef CAS PubMed.
- C. C. Perry, V. J. Tang, K. M. Konigsfeld, J. A. Aguilera and J. R. Milligan, J. Phys. Chem. B, 2011, 115, 9889–9897 CrossRef CAS PubMed.
- C. Yu, Y. Wu, F. Zeng and S. Wu, J. Mater. Chem. B, 2013, 1, 4152–4159 RSC.
- T. Charbouillot, M. Brigante, G. Mailhot, P. R. Maddigapu, C. Minero and D. Vione, J. Photochem. Photobiol., A, 2011, 222, 70–76 CrossRef CAS.
- M. Náfrádi, L. Farkas, T. Alapi, K. Hernádi, K. Kovács, L. Wojnárovits and E. Takács, Radiat. Phys. Chem., 2020, 170, 108610 CrossRef.
- C. He, Y. Wang, Z. Li, Y. Huang, Y. Liao, D. Xia and S. Lee, Environ. Sci. Technol., 2020, 54, 12771–12783 CrossRef CAS PubMed.
- T. Wu, T. Lin, J. Zhao, H. Hidaka and N. Serpone, Environ. Sci. Technol., 1999, 33, 1379–1387 CrossRef CAS.
- L. Liang, Q. Chang, T. Cai, N. Li, C. Xue, J. Yang and S. Hu, Chem. Eng. J., 2022, 428, 131139 CrossRef CAS.
- M. I. Muniz-Junqueira and V. N. de Paula-Coelho, Int. Immunopharmacol., 2008, 8, 1633–1638 CrossRef CAS PubMed.
- Y. Guo and B. Li, Carbon, 2015, 82, 459–469 CrossRef CAS.
- Y. Kakuma, A. Y. Nosaka and Y. Nosaka, Phys. Chem. Chem. Phys., 2015, 17, 18691–18698 RSC.
- K. K. Mothilal, J. Johnson Inbaraj, R. Gandhidasan and R. Murugesan, J. Photochem. Photobiol., A, 2004, 162, 9–16 CrossRef CAS.
- R. Konaka, E. Kasahara, W. C. Dunlap, Y. Yamamoto, K. C. Chien and M. Inoue, Free Radical Biol. Med., 1999, 27, 294–300 CrossRef CAS PubMed.
- L. R. Barclay, M. C. Basque and M. R. Vinqvist, Can. J. Chem., 2003, 81, 457–467 CrossRef CAS.
- J. A. Rengifo-Herrera, K. Pierzchała, A. Sienkiewicz, L. Forró, J. Kiwi and C. Pulgarin, Appl. Catal., B, 2009, 88, 398–406 CrossRef CAS.
- X. Ragàs, A. Jiménez-Banzo, D. Sánchez-García, X. Batllori and S. Nonell, Chem. Commun., 2009, 2920–2922, 10.1039/B822776D.
- A. M. Asadirad, Z. Erno and N. R. Branda, Chem. Commun., 2013, 49, 5639–5641 RSC.
- H. Wang, X. Yang, W. Shao, S. Chen, J. Xie, X. Zhang, J. Wang and Y. Xie, J. Am. Chem. Soc., 2015, 137, 11376–11382 CrossRef CAS PubMed.
- M. Wozniak, F. Tanfani, E. Bertoli, G. Zolese and J. Antosiewicz, Biochim. Biophys. Acta, Lipids Lipid Metab., 1991, 1082, 94–100 CrossRef CAS.
- Z. C. Redman, J. Wesolowski and P. L. Tomco, Environ. Sci. Technol., 2021, 55, 4974–4983 CrossRef CAS PubMed.
- C. Liang, H. P. Feng, H. Y. Niu, C. G. Niu, J. S. Li, D. W. Huang, L. Zhang, H. Guo, N. Tang and H. Y. Liu, Chem. Eng. J., 2020, 384, 123236 CrossRef CAS.
- Z. Wang, J. Wang, B. Xiong, F. Bai, S. Wang, Y. Wan, L. Zhang, P. Xie and M. R. Wiesner, Environ. Sci. Technol., 2020, 54, 464–475 CrossRef CAS PubMed.
- S. Kang, L. Zhang, C. Yin, Y. Li, L. Cui and Y. Wang, Appl. Catal., B, 2017, 211, 266–274 CrossRef CAS.
- P. Neta, V. Madhavan, H. Zemel and R. W. Fessenden, J. Am. Chem. Soc., 1977, 99, 163–164 CrossRef CAS.
- S. Gligorovski, R. Strekowski, S. Barbati and D. Vione, Chem. Rev., 2015, 115, 13051–13092 CrossRef CAS PubMed.
- K. Bao, S. Zhang, P. Ni, Z. Zhang, K. Zhang, L. Wang, L. X. Sun, W. Mao, Q. Zhou and Y. Qian, Catal. Today, 2020, 340, 311–317 CrossRef CAS.
- L. Hu, Y. Liao, D. Xia, F. Peng, L. Tan, S. Hu, C. Zheng, X. Lu, C. He and D. Shu, Chem. Eng. J., 2020, 385, 123824 CrossRef CAS.
- H. Guo, C. G. Niu, C. Y. Feng, C. Liang, L. Zhang, X. J. Wen, Y. Yang, H. Y. Liu, L. Li and L. S. Lin, Chem. Eng. J., 2020, 385, 123919 CrossRef CAS.
- H. Xu, Q. Ye, J. Zhang, Q. Li, M. Wang, P. Zhou, G. Zhou and Q. Wang, Sci. Total Environ., 2021, 778, 146280 CrossRef CAS PubMed.
- Y. Liu, X. Liu, Y. Zhao and D. D. Dionysiou, Appl. Catal., B, 2017, 213, 74–86 CrossRef CAS.
- A. A. Gorman and M. A. J. Rodgers, Chem. Soc. Rev., 1981, 10, 205–231 RSC.
- X. Chen, W. D. Oh and T. T. Lim, Chem. Eng. J., 2018, 354, 941–976 CrossRef CAS.
- Z. Xiong, Y. Jiang, Z. Wu, G. Yao and B. Lai, Chem. Eng. J., 2021, 421, 127863 CrossRef CAS.
- M. Zhang, R. Luo, C. Wang, W. Zhang, X. Yan, X. Sun, L. Wang and J. Li, J. Mater. Chem. A, 2019, 7, 12547–12555 RSC.
- W. R. Haag and J. Hoigne, Environ. Sci. Technol., 1986, 20, 341–348 CrossRef CAS PubMed.
- M. A. Oturan and J. J. Aaron, Crit. Rev. Environ. Sci. Technol., 2014, 44, 2577–2641 CrossRef CAS.
- X. Ye, Y. Cui, X. Qiu and X. Wang, Appl. Catal., B, 2014, 152, 383–389 CrossRef.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- B. Ervens, S. Gligorovski and H. Herrmann, Phys. Chem. Chem. Phys., 2003, 5, 1811–1824 RSC.
- X. X. Guo, T. T. Hu, B. Meng, Y. Sun and Y. F. Han, Appl. Catal., B, 2020, 260, 118157 CrossRef CAS.
- N. Zhang, E. P. Tsang, J. Chen, Z. Fang and D. Zhao, J. Colloid Interface Sci., 2020, 558, 163–172 CrossRef PubMed.
- H. Li, J. Shang, Z. Yang, W. Shen, Z. Ai and L. Zhang, Environ. Sci. Technol., 2017, 51, 5685–5694 CrossRef CAS PubMed.
- M. T. Bender, Y. C. Lam, S. Hammes-Schiffer and K. S. Choi, J. Am. Chem. Soc., 2020, 142, 21538–21547 CrossRef CAS PubMed.
- Z. Liu, Z. Zhao, W. Jiang, Z. Zhu, Y. Wang, Z. Liu, W. Gu, Y. Yang, Y. Yang, L. Zhang, W. Yao and F. Teng, Appl. Catal., B, 2021, 298, 120517 CrossRef CAS.
- L. Luo, Z. J. Wang, X. Xiang, D. Yan and J. Ye, ACS Catal., 2020, 10, 4906–4913 CrossRef CAS.
- H. Li, H. Shang, X. Cao, Z. Yang, Z. Ai and L. Zhang, Environ. Sci. Technol., 2018, 52, 8659–8665 CrossRef CAS PubMed.
- A. L. Teel, S. A. Khotz and R. J. Watts, J. Adv. Oxid. Technol., 2007, 10, 282–286 CAS.
- R. J. Watts and A. L. Teel, Water Res., 2019, 159, 46–54 CrossRef CAS PubMed.
- M. Ahmad, M. A. Simon, A. Sherrin, M. E. Tuccillo, J. L. Ullman, A. L. Teel and R. J. Watts, Chemosphere, 2011, 84, 855–862 CrossRef CAS PubMed.
- C. Zhang, T. Li, J. Zhang, S. Yan and C. Qin, Appl. Catal., B, 2019, 259, 118030 CrossRef CAS.
- Q. Wang, H. Zhou, X. Liu, T. Li, C. Jiang, W. Song and W. Chen, Environ. Sci.: Nano, 2018, 5, 2864–2875 RSC.
- R. J. Watts, B. C. Bottenberg, T. F. Hess, M. D. Jensen and A. L. Teel, Environ. Sci. Technol., 1999, 33, 3432–3437 CrossRef CAS.
- J. Xiao, Y. Xie, Q. Han, H. Cao, Y. Wang, F. Nawaz and F. Duan, J. Hazard. Mater., 2016, 304, 126–133 CrossRef CAS PubMed.
- J. Jiang, Z. Lu, M. Zhang, J. Duan, W. Zhang, Y. Pan and J. Bai, J. Am. Chem. Soc., 2018, 140, 17825–17829 CrossRef CAS PubMed.
- A. Ishak, K. Dayana, M. H. Mamat, M. F. Malek and M. Rusop, Optik, 2015, 126, 1610–1612 CrossRef CAS.
- Z. Li, W. Yang, L. Xie, Y. Li, Y. Liu, Y. Sun, Y. Bu, X. Mi, S. Zhan and W. Hu, Appl. Surf. Sci., 2021, 549, 149262 CrossRef CAS.
- Z. Wang, H. Jia, T. Zheng, Y. Dai, C. Zhang, X. Guo, T. Wang and L. Zhu, Appl. Catal., B, 2020, 272, 119030 CrossRef CAS.
- Y. H. Kim and B. C. Chung, J. Org. Chem., 1983, 48, 1562–1564 CrossRef CAS.
- X. Xiao, J. Jiang and L. Zhang, Appl. Catal., B, 2013, 142, 487–493 CrossRef.
- A. Yuan, H. Lei, Z. Wang and X. Dong, J. Colloid Interface Sci., 2020, 560, 40–49 CrossRef CAS PubMed.
- S. Ito, Y. Kon, T. Nakashima, D. Hong, H. Konno, D. Ino and K. Sato, Molecules, 2019, 24, 2520 CrossRef CAS PubMed.
- F. Xie, Y. Zhang, X. He, H. Li, X. Qiu, W. Zhou, S. Huo and Z. Tang, J. Mater. Chem. A, 2018, 6, 13236–13243 RSC.
- C. Chen, G. Qiu, T. Wang, Z. Zheng, M. Huang and B. Li, J. Colloid Interface Sci., 2021, 592, 1–12 CrossRef CAS PubMed.
- V. R. Akhmetova, G. R. Khabibullina, E. B. Rakhimova, R. A. Vagapov, R. R. Khairullina, Z. T. Niatshina and N. N. Murzakova, Mol. Diversity, 2010, 14, 463–471 CrossRef CAS PubMed.
- S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626–2704 CrossRef CAS PubMed.
- J. H. Xie, S. F. Zhu and Q. L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed.
- Q. Li, X. Huang, G. Su, M. Zheng, C. Huang, M. Wang, C. Ma and D. Wei, Environ. Sci. Technol., 2018, 52, 13351–13360 CrossRef CAS PubMed.
- A. Khampuanbut, S. Santalelat, A. Pankiew, D. Channei, S. Pornsuwan, K. Faungnawakij, S. Phanichphant and B. Inceesungvorn, J. Colloid Interface Sci., 2020, 560, 213–224 CrossRef CAS PubMed.
- J. L. Shi, H. Hao and X. Lang, Sustainable Energy Fuels, 2019, 3, 488–498 RSC.
- E. J. Corey and W. C. Taylor, J. Am. Chem. Soc., 1964, 86, 3881–3882 CrossRef CAS.
- C. S. Foote and S. Wexler, J. Am. Chem. Soc., 1964, 86, 3880–3881 CrossRef CAS.
- T. Li, L. Ge, X. Peng, W. Wang and W. Zhang, Water Res., 2021, 190, 116777 CrossRef CAS PubMed.
- L. He, Z. W. Jiang, W. Li, C. M. Li, C. Z. Huang and Y. F. Li, ACS Appl. Mater. Interfaces, 2018, 10, 28868–28876 CrossRef CAS PubMed.
- R. Luo, M. Li, C. Wang, M. Zhang, M. A. Nasir Khan, X. Sun, J. Shen, W. Han, L. Wang and J. Li, Water Res., 2019, 148, 416–424 CrossRef CAS PubMed.
- S. Liu, Z. Zhang, F. Huang, Y. Liu, L. Feng, J. Jiang, L. Zhang, F. Qi and C. Liu, Appl. Catal., B, 2021, 286, 119921 CrossRef CAS.
- F. Sun, T. Chen, H. Liu, X. Zou, P. Zhai, Z. Chu, D. Shu, H. Wang and D. Chen, Sci. Total Environ., 2021, 784, 147117 CrossRef CAS PubMed.
- J. Zhao, F. Li, H. Wei, H. Ai, L. Gu, J. Chen, L. Zhang, M. Chi and J. Zhai, Chem. Eng. J., 2021, 409, 128150 CrossRef CAS.
- J. Li, J. Miao, X. Duan, J. Dai, Q. Liu, S. Wang, W. Zhou and Z. Shao, Adv. Funct. Mater., 2018, 28, 1804654 CrossRef.
- Y. X. Chen, J. Y. Gao, Z. W. Huang, M. J. Zhou, J. X. Chen, C. Li, Z. Ma, J. M. Chen and X. F. Tang, Environ. Sci. Technol., 2017, 51, 7084–7090 CrossRef CAS PubMed.
- J. Lee, S. Hong, Y. Mackeyev, C. Lee, E. Chung, L. J. Wilson, J. H. Kim and P. J. J. Alvarez, Environ. Sci. Technol., 2011, 45, 10598–10604 CrossRef CAS PubMed.
- A. Kashyap, E. Ramasamy, V. Ramalingam and M. Pattabiraman, Molecules, 2021, 26, 2673 CrossRef CAS PubMed.
- Z.-J. Li, S. Li, A. H. Davis, E. Hofman, G. Leem and W. Zheng, Nano Res., 2020, 13, 1668–1676 CrossRef CAS.
- P. K. Dikshit, S. K. Padhi and V. S. Moholkar, Bioresour. Technol., 2017, 244, 362–370 CrossRef CAS PubMed.
- S. Zhao, Z. Dai, W. Guo, F. Chen, Y. Liu and R. Chen, Appl. Catal., B, 2019, 244, 206–214 CrossRef CAS.
- H. Li, X. Li, J. Zhou, W. Sheng and X. Lang, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.10.068.
- S. Xu, P. Zhou, Z. Zhang, C. Yang, B. Zhang, K. Deng, S. Bottle and H. Zhu, J. Am. Chem. Soc., 2017, 139, 14775–14782 CrossRef CAS PubMed.
- N. Zheng, X. He, R. Hu, R. Wang, Q. Zhou, Y. Lian and Z. Hu, Appl. Catal., B, 2022, 307, 121157 CrossRef CAS.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- C. He, J. Cheng, X. Zhang, M. Douthwaite, S. Pattisson and Z. Hao, Chem. Rev., 2019, 119, 4471–4568 CrossRef CAS PubMed.
- Y. Li, C. Wang, C. Zhang and H. He, Top. Catal., 2020, 63, 810–816 CrossRef CAS.
- P. Pinto Joseph, G. R. Gladstone and L. Yung Yuk, Science, 1980, 210, 183–185 CrossRef PubMed.
- C. Dong, Z. Qu, Y. Qin, Q. Fu, H. Sun and X. Duan, ACS Catal., 2019, 9, 6698–6710 CrossRef CAS.
- Y. Ma, L. Wang, J. Ma, H. Wang, C. Zhang, H. Deng and H. He, ACS Catal., 2021, 11, 6614–6625 CrossRef CAS.
- Z. Wang, W. Wang, L. Zhang and D. Jiang, Catal. Sci. Technol., 2016, 6, 3845–3853 RSC.
- M. He, J. Ji, B. Liu and H. Huang, Appl. Surf. Sci., 2019, 473, 934–942 CrossRef CAS.
- X. Liu, J. Mi, L. Shi, H. Liu, J. Liu, Y. Ding, J. Shi, M. He, Z. Wang, S. Xiong, Q. Zhang, Y. Liu, Z. S. Wu, J. Chen and J. Li, Angew. Chem., Int. Ed., 2021, 60, 26747–26754 CrossRef CAS PubMed.
- T. Brink Gerd Jan, W. C. E. Arends Isabel and A. Sheldon Roger, Science, 2000, 287, 1636–1639 CrossRef PubMed.
- F. Sabaté, J. L. Jordá, M. J. Sabater and A. Corma, J. Mater. Chem. A, 2020, 8, 3771–3784 RSC.
- Y. Dong, J. Luo, S. Li and C. Liang, Catal. Commun., 2020, 133, 105843 CrossRef.
- Y. Dai, P. Ren, Y. Li, D. Lv, Y. Shen, Y. Li, H. Niemantsverdriet, F. Besenbacher, H. Xiang and W. Hao, Angew. Chem., Int. Ed., 2019, 131, 6331–6336 CrossRef.
- E. Markó István, R. Giles Paul, M. Tsukazaki, M. Brown Stephen and J. Urch Chistopher, Science, 1996, 274, 2044–2046 CrossRef PubMed.
- I. Enache Dan, K. Edwards Jennifer, P. Landon, B. Solsona-Espriu, F. Carley Albert, A. Herzing Andrew, M. Watanabe, J. Kiely Christopher, W. Knight David and J. Hutchings Graham, Science, 2006, 311, 362–365 CrossRef CAS PubMed.
- M. J. Schultz, R. S. Adler, W. Zierkiewicz, T. Privalov and M. S. Sigman, J. Am. Chem. Soc., 2005, 127, 8499–8507 CrossRef CAS PubMed.
- H. Yan, Q. Shen, Y. Sun, S. Zhao, R. Lu, M. Gong, Y. Liu, X. Zhou, X. Jin, X. Feng, X. Chen, D. Chen and C. Yang, ACS Catal., 2021, 11, 6371–6383 CrossRef CAS.
- Y. Wan and J. M. Lee, ACS Catal., 2021, 11, 2524–2560 CrossRef CAS.
- E. Hayashi, T. Komanoya, K. Kamata and M. Hara, ChemSusChem, 2017, 10, 654–658 CrossRef CAS PubMed.
- X. Han, C. Li, X. Liu, Q. Xia and Y. Wang, Green Chem., 2017, 19, 996–1004 RSC.
- S. Zhang, X. Sun, Z. Zheng and L. Zhang, Catal. Commun., 2018, 113, 19–22 CrossRef CAS.
- X. Wan, N. Tang, Q. Xie, S. Zhao, C. Zhou, Y. Dai and Y. Yang, Catal. Sci. Technol., 2021, 11, 1497–1509 RSC.
- F. Sabaté, J. L. Jordà and M. J. Sabater, Catal. Today, 2021 DOI:10.1016/j.cattod.2021.06.033.
- S. Ishikawa, Y. Yamada, C. Qiu, Y. Kawahara, N. Hiyoshi, A. Yoshida and W. Ueda, Chem. Mater., 2019, 31, 1408–1417 CrossRef CAS.
- L. Bui, R. Chakrabarti and A. Bhan, ACS Catal., 2016, 6, 6567–6580 CrossRef CAS.
- T. Li, F. Liu, Y. Tang, L. Li, S. Miao, Y. Su, J. Zhang, J. Huang, H. Sun, M. Haruta, A. Wang, B. Qiao, J. Li and T. Zhang, Angew. Chem., Int. Ed., 2018, 57, 7795–7799 CrossRef CAS PubMed.
- J. S. Chung, R. Miranda and C. O. Bennett, J. Catal., 1988, 114, 398–410 CrossRef CAS.
- S. P. Bates, M. J. Gillan and G. Kresse, J. Phys. Chem. B, 1998, 102, 2017–2026 CrossRef CAS.
Footnote |
| † Y. Wen and J. Yan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |