
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHemilabile MIC^N ligands allow oxidant-free Au(I)/Au(III) arylation-lactonization of γ-alkenoic acids†
Pau
Font
 a,
Hugo
Valdés
*a,
Gregorio
Guisado-Barrios
*b and
Xavi
Ribas
*a
a,
Hugo
Valdés
*a,
Gregorio
Guisado-Barrios
*b and
Xavi
Ribas
*a
aInstitut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Campus de Montilivi, Girona E-17003, Catalonia, Spain. E-mail: hugo.valdes@udg.edu; xavi.ribas@udg.edu
bDepartamento de Química Inorgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH), Universidad de Zaragoza-CSIC, Zaragoza 50009, Spain. E-mail: gguisado@unizar.es
First published on 22nd July 2022
Abstract
Oxidant-free Au-catalyzed reactions are emerging as a new synthetic tool for innovative organic transformations. Oxidant-free Au-catalyzed reactions are emerging as a new synthetic tool for innovative organic transformations. Still, a deeper mechanistic understanding is needed for a rational design of these processes. Here we describe the synthesis of two Au(I) complexes bearing bidentated hemilabile MIC^N ligands, [AuI(MIC^N)Cl], and their ability to stabilize square-planar Au(III) species (MIC = mesoionic carbene). The presence of the hemilabile N-ligand contributed to stabilize the ensuing Au(III) species acting as a five-membered ring chelate upon its coordination to the metal center. The Au(III) complexes can be obtained either by using external oxidants or, alternatively, by means of feasible oxidative addition with strained biphenylene Csp2–Csp2 bonds as well as with aryl iodides. Based on the fundamental knowledge gained on the redox properties on these Au(I)/Au(III) systems, we successfully develop a novel Au(I)-catalytic procedure for the synthesis of γ-substituted γ-butyrolactones through the arylation-lactonization reaction of the corresponding γ-alkenoic acid. The oxidative addition of the aryl iodide, which in turn is allowed by the hemilabile nature of the MIC^N ligand, is an essential step for this transformation.
Introduction
Gold catalysis has been dominated by the Lewis acidity of gold towards activation of alkenes and alkynes for nucleophilic attacks.1–6 On the other hand, redox Au(I)/Au(III) catalysis has been less explored and mainly achieved in the presence of sacrificial external oxidants,7–17 especially due to the reluctance of Au(I) to undergo oxidative addition.18–21 In this regard, several strategies have been explored to overcome this limitation,22–46 among which the chelation-assisted strategy in pre-designed ligands30–34 or the oxidative addition using strained molecules such as biphenylene stand out.35 Bourissou pioneered in 2014 the intramolecular oxidative addition of Csp2–X bonds (X = Br, I) at Au(I) by utilizing rigid 8-halonaphthyl phosphine model substrates, which suitably place the Csp2–X bond close to the Au(I) atom upon coordination to the phosphine moiety. In this manner, facile stabilization of the (P,C)-cyclometalated Au(III) products is obtained via oxidative addition.32 In the same line, Hashmi and Hermange used a similar strategy taking advantage of pendant pyridine groups in monophosphine ligands to stabilize Au(III) systems upon reaction of Au(I) complexes with aryldiazonium salts.47,48 In these examples, the subtle change from linear to square-planar geometry is crucial for the facile reactivity of Au(I) species towards a key oxidative addition process. Noteworthy, P^N MeDalphos ligand showed great performance at promoting the oxidative addition of aryl halides at Au(I), as well as at stabilizing Au(III) intermediates, which paved the way to Au(I)/Au(III) catalytic transformations using the (MeDalphos)AuCl catalyst (Scheme 1a).41 For instance, in 2019 Bourissou and coworkers reported the regioselective Au(I)/Au(III)-catalyzed C3 arylation of indoles, which is rarely attained by other transition metals (Scheme 1a),49 additionally they described the Au(I)/Au(III) catalyzed C3 allylation of indoles using allyl alcohols or allyl acetates.50 Significantly, two independent reports by the groups of Patil51 and Bourissou52 disclosed the Csp2–N cross-coupling reactions of aryl iodides with amines mediated by the MeDalphos-enabled Au(I)/Au(III) catalysis (Scheme 1a). In 2020, Bourissou and Patil groups independently showed that the oxidative addition of aryl iodides and π-activation of olefins can be merged by the Au(I)/Au(III) platform using (MeDalphos)AuCl as catalyst, which involves the oxy- and aminoarylation reactions of alkenols and alkenamines (Scheme 1a).53,54 The 1,2-heteroarylation of alkenes was also achieved using external alcohols and amines.54–56 Interestingly, (MeDalphos)AuCl catalyst has proven to be a key factor in the development of other transformations such as 1,2-diarylation of alkenes,57 or trifluoromethylthiolation and trifluoromethyl-selenolation of organohalides.58 Very recently, Shi and Patil groups developed new chiral-hemilabile P^N-ligands to access enantioselective Au(I)/Au(III) catalysis.59,60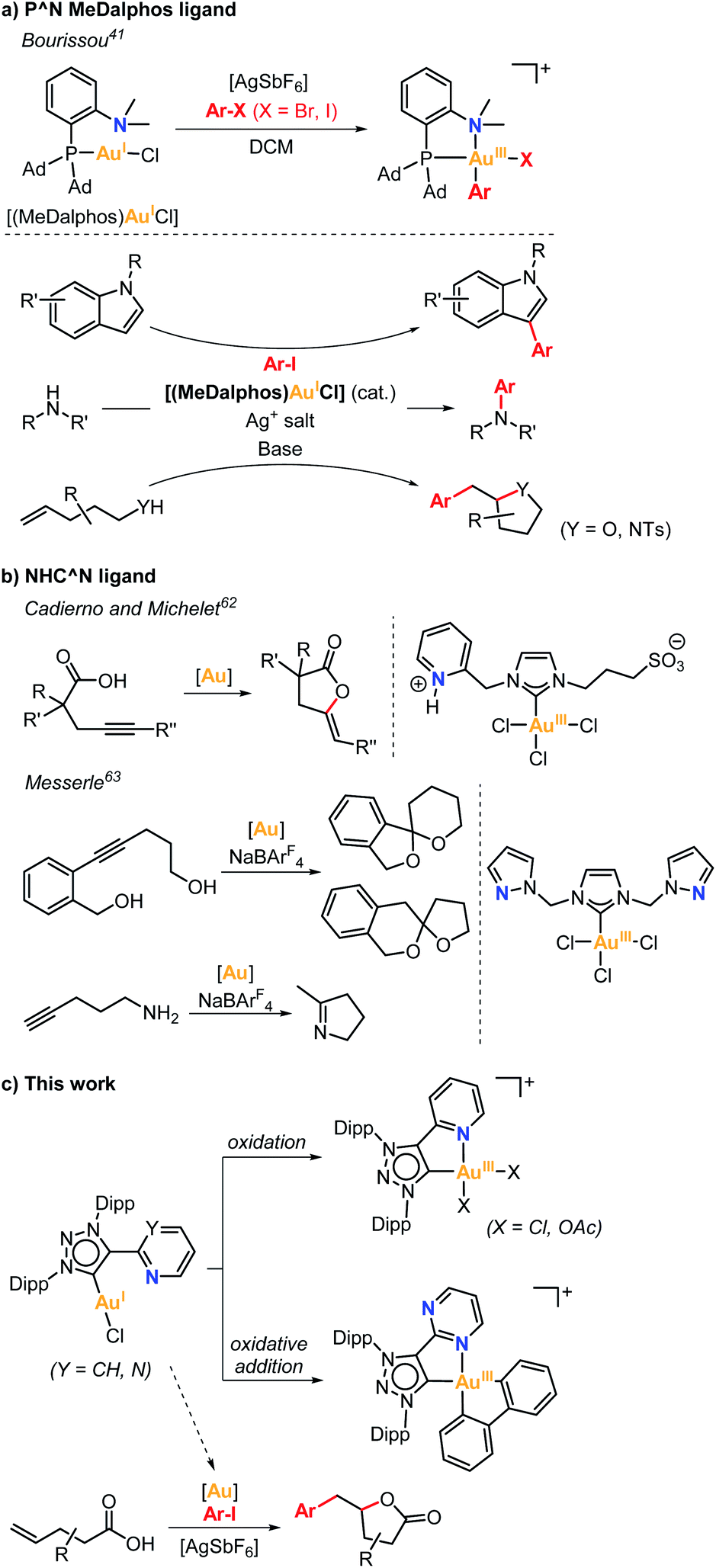 | ||
| Scheme 1 (a) Oxidative addition of aryl halides to a Au(I) complex bearing the hemilabile P^N MeDalphos ligand, and selected examples of its catalytic activity. (b) Selected examples of gold complexes bearing hemilabile NHC^N ligands. (c) Ability of hemilabile MIC^N ligands to stabilize Au(III) complexes obtained from Au(I) via oxidation or via oxidative addition, and application of the (MIC^N)Au(I) complexes to the arylation-lactonization reaction of γ-alkenoic acids. | ||
In contrast to the well-known P^N ligands, the use of bidentate C^N or C^O ligands61 remains scarcely explored, and the few representative examples are depicted in Scheme 1b. Cadierno and Michelet studied the catalytic activity of a (NHC^N)Au(III) complex containing a N-heterocyclic carbene ligand (NHC) with a pyridinium side arm in the cyclization of γ-alkynoic acids (Scheme 1b),62 while Messerle and coworkers explored the dihydroalkoxylation and hydroamination catalyzed by a Au(III) complex bearing a NHC ligand with two pendant pyrazole arms (Scheme 1b).63 Alternatively, Bertrand and coworkers showed that a hemilabile bidentate cyclic (alkyl)(amino)carbene (CAAC) Au(I) complex undergoes oxidative addition of biphenylene.40
More recently, Bourissou and coworkers reported the reactivity of a Au(I) complex containing a N3-alkylated mesoionic 1,2,3-triazol-5-ylidene bearing a pendant pyridine group.64 However, the complex did not undergo oxidative addition towards iodobenzene, thus precluding its exploitation in oxidant-free reactivity. In fact, the formation of a dimeric Au(I) species was observed instead of the formation of the oxidative addition Au(III) product. On the other hand, the Au(I) complex was reactive towards a strong oxidant such as PhICl2, forming a mononuclear square-planar Au(III) species. The latter experiment demonstrated that mesoionic carbene ligands bearing N-based hemilabile groups (MIC^N) can potentially be suitable platforms to stabilize Au(III) species.
Based on the abovementioned, we herein explore the reactivity of two new (MIC^N)Au(I) complexes bearing hemilabile pyridine (py) or pyrimidine (pym) pendant groups, their ability to stabilize (MIC^N)Au(III) species and their performance in oxidant-free oxidative addition of aryl halides and strained C–C bonds. After proving the successful ability to sustain oxidant-free Au(I)/Au(III) redox processes, we further extended its application to arylation-lactonization reactions of γ-alkenoic acids (Scheme 1c).
Results and discussion
Synthesis and characterization of (MIC^N)Au(I) complexes
Initially, two 1,2,3-triazolium ligand salt precursors (MIC^N) bearing 2,6-diisopropylphenyl substituents (Dipp) at the outer nitrogens of the azolium ring and either a pyridine (1a) or a pyrimidine (1b) side arm were prepared by following an adapted method reported in the literature (Fig. 1a).65 We reacted 1,3-bis(2,6-diisopropylphenyl)triaz-1-ene with 2-ethynylpyridine or 2-ethynylpyrimidine in the presence of tert-butyl hypochlorite (tBuOCl) and anhydrous potassium hexafluorophosphate (KPF6), affording the desired products in high and moderate yield (99% for 1a and 67% for 1b). In order to obtain the corresponding (MIC^N)Au(I) complexes, we attempted the Au(I) coordination through a transmetalation reaction to Ag(I) species. For this purpose, we reacted the corresponding triazolium salts with silver oxide (Ag2O) in the presence of cesium carbonate (Cs2CO3) and potassium chloride (KCl) as halide source. Then, the addition of dimethylsulfide gold(I) chloride [AuCl(SMe2)] immediately produced the precipitation of the silver halide. After purification, the desired Au(I) complexes 2a and 2b were obtained in 32% and 35% yield, respectively. The 1H NMR spectra of the complexes showed the absence of the acidic proton of the triazolium salts, indicating that the coordination to gold had occurred. The 13C{1H} NMR spectrum of 2a showed the characteristic signal of the metalated carbon at 162.1 ppm, while in the case of the pyrimidine derivative 2b, such signal was slightly shifted downfield, appearing at 165.3 ppm. Furthermore, the molecular structure of both Au(I) complexes 2a and 2b was unambiguously determined by X-ray diffraction analyses (Fig. 1b). Both complexes are isostructural, showing the mesoionic carbene ligand coordinated to an Au(I) atom, and a chlorine ligand completing the linear coordination sphere around the metal (C–Au–Cl angle is 174.31(18)° for 2a and 179.30(9)° for 2b). Noticeably, no coordination of the hemilabile pyridine or pyrimidine moieties at Au(I) was observed. | ||
| Fig. 1 (a) Synthesis of Au(I) complexes 2a and 2b. (b) Crystal structures of 2a and 2b (ellipsoids set at 50% probability and H atoms removed for clarity). Selected bond distances (Å): for 2a, Au1–C3 1.982(6), Au1–Cl2 2.2854(17); for 2b, Au1–C3 2.006(4), Au1–Cl2 2.2728(16). | ||
Then, Au(I) complexes 2a and 2b were reacted with silver hexafluoroantimonate (AgSbF6) in CD2Cl2 at room temperature (Fig. 2a), seeking the engagement of the pendant pyrimidine or pyridine groups in the coordination to Au(I). The 1H NMR spectra of the corresponding complexes showed some changes in the aromatic and aliphatic region. In particular, the signals of the pyridine or pyrimidine fragments were shifted due to gold coordination. The most significant change was observed in the 13C{1H} NMR spectra. The signal of the carbenic carbon was shifted from 162.1 to 157.2 ppm in the case of the pyridine derivative (3a), and from 165.3 to 161.3 ppm in the case of the pyrimidine derivative (3b).
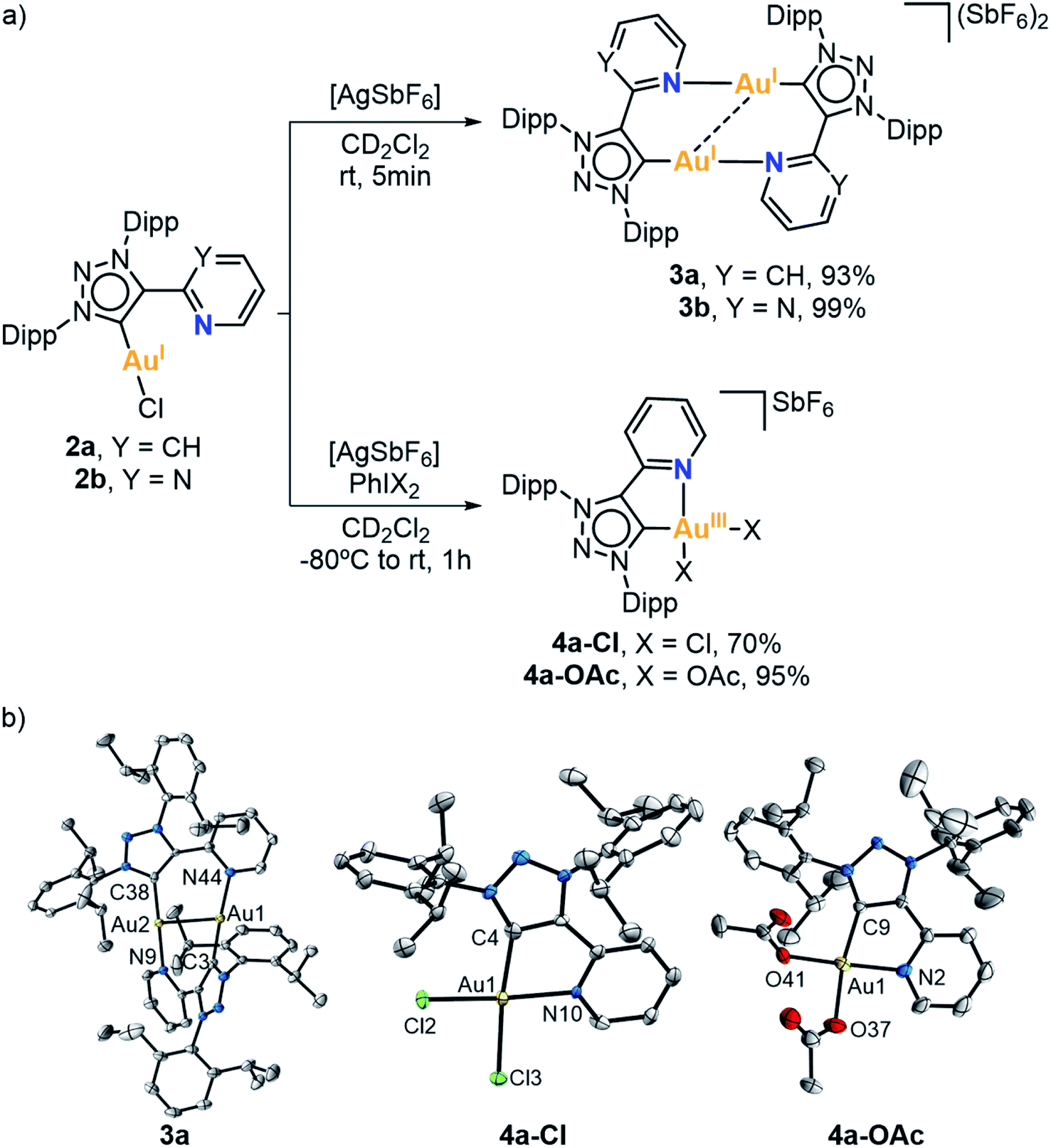 | ||
| Fig. 2 (a) Reactivity of Au(I) complexes 2a and 2b towards halogen scavengers and strong oxidants. (b) Crystal structures of 3a, 4a-Cl and 4a-OAc (ellipsoids set at 50% probability, H atoms and SbF6− anions removed for clarity). Selected bond distances (Å): for 3a, Au1–Au2 2.8212(4), Au1–N44 2.078(6), Au1–C3 1.988(8), Au2–N9 2.069(6), Au2–C38 1.990(8); for 4a-Cl, Au1–C4 2.017(8), Au1–N10 2.089(6), Au1–Cl2 2.268(2), Au1–Cl3 2.302(2); for 4a-OAc, Au1–C9 1.991(11), Au1–N2 2.052(11), Au1–O37 2.032(10), Au1–O41 1.998(9). | ||
X-ray diffraction analysis of complex 3a showed a head-to-tail dimeric Au(I) species where each ligand is bridging two metals (Fig. 2). The Au(I) centers adopted a linear dicoordinate arrangement, showing an aurophilic interaction (Au⋯Au length of 2.8212(4) Å). Analogously to Bourissou's (MIC^N)Au(I) complex,64 the formation of a dimeric Au(I) species was favored rather than a mononuclear Au(I) having both the MIC and py or pym pendant groups coordinated.
Reactivity of (MIC^N)Au(I) complexes with external oxidants and towards oxidative addition
In order to promote the coordination of the side arms of our MIC^N ligands, we attempted the 2 e− chemical oxidation of the Au(I) complexes to mononuclear Au(III). To that end, we studied the reactivity of complex 2a against a 2 e− oxidant, namely PhIX2 (X = Cl or OAc) (Fig. 2a). The reaction was performed at low temperature, from −80 °C to rt, in the presence of AgSbF6 as halogen scavenger. Gratifyingly, the oxidation of Au(I) to Au(III) took place, with the concomitant coordination of the pendant N-moiety to gold. The 13C{1H} NMR spectra of the resulting complexes 4a-Cl and 4a-OAc showed the characteristic signal of the carbenic carbon at higher field when compared to the Au(I) analogue 2a (147.3 ppm for 4a-Cl and 137.1 ppm for 4a-OAcvs. 162.1 ppm for 2a). The molecular structures of complexes 4a-Cl and 4a-OAc were elucidated by X-ray diffraction analyses (Fig. 2b). Both complexes showed a similar arrangement, i.e. the bidentate MIC^N ligand coordinated to Au(III), and two chlorides or acetates completing its tetracoordinated Au(III) center, which featured a slightly distorted square planar geometry. The Ccarbene–Au(III) lengths were slightly longer than those found in the Au(I) analogue (2.017(8) Å for 4a-Cl and 1.991(11) Å for 4a-OAcvs. 1.982(6) Å for 2a).At this point, we had demonstrated that these MIC^N ligands are indeed suitable platforms to stabilize Au(III) species. Thus, we were encouraged to explore the possibility of obtaining Au(III) complexes via oxidative addition. With this aim, we first attempted the oxidative addition of complex 2b towards the strained Csp2–Csp2 bond of biphenylene, by mixing equimolar amount of 2b and biphenylene in dichloromethane, in the presence of a halide scavenger, from −80 °C to room temperature for 10 minutes (Fig. 3a). Unfortunately, we did not observe the desired Au(III) center with a biphenyl moiety but, interestingly, crystallization of the reaction mixture revealed the co-crystallization of the dimeric complex 3b with intact biphenylene (3b·biphenylene) (Fig. 3b). Even after 5 hours at 50 °C, the outcome of the reaction remained the same. To our delight, when the reaction was heated at 90 °C for 2 hours in 1,2-dichloroethane, the Au(I) center underwent oxidative addition of the strained biphenylene Csp2–Csp2 bond, forming the expected Au(III) complex 5b (Fig. 3a). Notably, the presence of a chloride source (such as KCl or tetrabutylammonium chloride) displaced the hemilabile pyrimidine, affording a neutral Au(III) species, 6b. By adding AgSbF6 as halide scavenger, we could reversibly form complex 5b, also reinforcing the hemilabile character of the pyrimidine moiety. Indeed, the NMR spectra of 5b showed a dynamic behaviour of the pyrimidine pendant arm, even at 248 K, as indicated by 1H,1H-NOESY experiment (Fig. S49†). Both the cationic 5b and the neutral 6b Au(III) species were characterized by X-ray diffraction analyses (Fig. 3b), confirming the weak coordination of the pyrimidine moiety with a long Au(III)–Npyrimidine bond of 2.254 Å for 5b. The carbene resonance signals were downfield shifted to 182.8 and 180.1 ppm for 5b and 6b, respectively.
 | ||
| Fig. 3 (a) Study of the reaction conditions for the oxidative addition of a strained Csp2–Csp2 bond to complex 2b. (b) Crystal structures of 3b·biphenylene, 5b and 6b (ellipsoids set at 50% probability, H atoms removed for clarity; in the case of 3b·biphenylene the SbF6− anions have been removed and the biphenylene molecule is painted in green for clarity). Selected bond distances (Å): for 3b·biphenylene, Au1–Au1′ 2.8768(10), Au1–N2 2.105(8), Au1–C9 2.027(10); for 5b, Au1–C2 2.108(5), Au1–N8 2.254(5), Au1–C37 2.066(5), Au1–C44 2.019(5); for 6b, Au1–C3 2.080(6), Au1–Cl2 2.376(3), Au1–C38 2.072(7), Au1–C49 2.057(7). | ||
We then explored the possibility of Au(I) complexes 2a and 2b to undergo oxidative addition of Csp2–I bonds (Fig. 4a). For this purpose, we reacted the Au(I) complexes with para-substituted iodoaryls (R = OMe, Me, F) in the presence of AgSbF6, in 1,2-dichloroethane and heating at 120 °C overnight. Unexpectedly, no Au-containing compounds were isolated, but on the contrary, the 1H NMR and mass spectra were consistent with the full conversion to three triazolium salts (8a-R, 9a, and 1a when 2a was used, and 8b-R, 9b and 1b when 2b was used; R = OMe, Me, F) (see yields in Fig. 4a). When using 2a with 4-iodoanisole, the triazolium salts 8a-OMe and 9a were unambiguously characterized by X-ray diffraction. Both species co-crystallized, and the single crystal X-ray analysis revealed the presence of 8a-OMe and 9a in a 0.89![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.11 ratio (Fig. 4b). Likewise, when 2b was reacted with 4-iodoanisole, the major product was 8b-OMe with a 65% NMR yield, whereas 9b and 1b were both obtained in 16% NMR yield. In this case, 8b-OMe could be isolated and characterized by 1H NMR, HRMS and X-ray diffraction (Fig. 4b). The formation of such compounds suggested that gold(I) compounds underwent oxidative addition of the aryl iodide followed by a fast reductive elimination of the mesoionic carbene ligand either with the aryl or the iodide moiety coordinated in cis, respectively (Fig. 4a). Interestingly, products 8a-R/8b-R were obtained in higher yields than 9a/9b, indicating that after the oxidative addition the major product is the one with the aryl ligand in cis (cis-7a-I or cis-7b-I), probably due to the trans effect of the NHC ligand. By an alternative route,47 the analogous complex cis-7a-Cl was synthesized through the oxidative addition of an aryldiazonium salt promoted by blue LED irradiation (λ = 447 nm). This methodology has the advantage to occur at 25 °C, allowing the isolation and fully characterization of the Au(III) species cis-7a-Cl (see XRD in Fig. 4b). When cis-7a-Cl was heated at 120 °C overnight in 1,2-DCE, product 8a-OMe was formed in 96% yield. This reaction supports that the formation of 8a-R/8b-R and 9a/9b from Au(I) complexes 2a/2b proceeds via an oxidative addition/reductive elimination pathway. We hypothesize that the hemilabile character of the nitrogen atom and the fast decomposition of Au(III) at high temperatures promote the reductive elimination process, which has only been observed, to our knowledge, for a 1,2,3-triazol-5-ylidene based palladium complex.66–68 Nevertheless, our observations sharply contrasted with the lack of reactivity described by Bourissou for a similar (MIC^N)Au(I) system with aryl iodides.64
:0.11 ratio (Fig. 4b). Likewise, when 2b was reacted with 4-iodoanisole, the major product was 8b-OMe with a 65% NMR yield, whereas 9b and 1b were both obtained in 16% NMR yield. In this case, 8b-OMe could be isolated and characterized by 1H NMR, HRMS and X-ray diffraction (Fig. 4b). The formation of such compounds suggested that gold(I) compounds underwent oxidative addition of the aryl iodide followed by a fast reductive elimination of the mesoionic carbene ligand either with the aryl or the iodide moiety coordinated in cis, respectively (Fig. 4a). Interestingly, products 8a-R/8b-R were obtained in higher yields than 9a/9b, indicating that after the oxidative addition the major product is the one with the aryl ligand in cis (cis-7a-I or cis-7b-I), probably due to the trans effect of the NHC ligand. By an alternative route,47 the analogous complex cis-7a-Cl was synthesized through the oxidative addition of an aryldiazonium salt promoted by blue LED irradiation (λ = 447 nm). This methodology has the advantage to occur at 25 °C, allowing the isolation and fully characterization of the Au(III) species cis-7a-Cl (see XRD in Fig. 4b). When cis-7a-Cl was heated at 120 °C overnight in 1,2-DCE, product 8a-OMe was formed in 96% yield. This reaction supports that the formation of 8a-R/8b-R and 9a/9b from Au(I) complexes 2a/2b proceeds via an oxidative addition/reductive elimination pathway. We hypothesize that the hemilabile character of the nitrogen atom and the fast decomposition of Au(III) at high temperatures promote the reductive elimination process, which has only been observed, to our knowledge, for a 1,2,3-triazol-5-ylidene based palladium complex.66–68 Nevertheless, our observations sharply contrasted with the lack of reactivity described by Bourissou for a similar (MIC^N)Au(I) system with aryl iodides.64
 | ||
| Fig. 4 (a) Reactivity towards aryl iodides and decomposition route of Au(I) species 2a and 2b, and synthesis of cis-7a-Cl by oxidative addition of an aryldiazonium salt. NMR yield of the reactions calculated using 1,3,5-trimethoxybenzene as internal standard. (b) Crystal structures of cis-7a-Cl, 8a-OMe, 9a and 8b-OMe (ellipsoids set at 50% probability, H atoms removed for clarity; for cis-7a-Cl, BF4− anion is removed for clarity; for 8a-OMe and 9a, SbF6− anion is not shown for clarity). Selected bond distances for cis-7a-Cl (Å): Au1–C10 2.009(7), Au1–N3 2.154(7), Au1–C38 2.006(8), Au1–Cl2 2.303(2). | ||
Arylation-lactonization of γ-alkenoic acids
Based on the successful catalytic application of the P^N hemilabile ligand strategy, we envisioned that our hemilabile (MIC^N)Au(I) complexes may also mediate a cascade reaction involving the key oxidative addition step. Recently, Bourissou and coworkers described the coupling/cyclization reaction of aryl iodides with alkenol and alkenamine compounds catalyzed by a hemilabile P^N MeDalPhos/Au(I) system.53 Additionally, in 2017, Shi and coworkers reported a system that afforded coupling/cyclization products upon reacting aryldiazonium salts with either alkenamines, alkenols or alkenoic acids, using [(Ph3P)AuCl] under photo-free conditions.69 Thus, we envisioned that our MIC^N/Au(I) system should afford γ-benzyl-γ-butyrolactone products when reacting aryl iodides with γ-alkenoic acids, via coupling/lactonization, provided the oxidative addition step takes place (see reaction scheme embedded in Table 1). Our first attempts consisted in a stoichiometric reaction between 4-iodoanisole, 4-pentenoic acid, complex 2b, K3PO4, and AgSbF6 at 80 °C. When the reactions were carried out in the presence of an excess of base or AgSbF6, we did not observe any conversion, probably due to the rapid decomposition of the Au(I) complex. We also found that by using chlorinated solvents such as 1,2-dichloroethane, the conversion reached up to 49% (Table S3†). We determined by 1H NMR analysis that the catalyst decomposes under the reaction conditions, forming the triazolium salt 1b. Interestingly, the conversion was significantly higher using 2,2,2-trifluoroethanol (TFE) (up to 92%) instead of 1,2-dichloroethane (Table 1 and S3†). When the reaction was carried out at 100 °C, the conversion was quantitative, but the yield was very similar to that found at 80 °C (67% yield at 100 °C vs. 63% yield at 80 °C, entries 1–2 in Table 1), and we also observed the formation of the triazolium salt 8b-OMe. This was a strong indication that complex 2b underwent an oxidative addition of the Csp2–I bond, but rapid reductive elimination to form the coupling product 8b-OMe occurred.|
|
|||||
|---|---|---|---|---|---|
| Entry | [Ag] (eq.) | [Au] mol% | K3PO4 (eq.) | Solvent | Conversion% (yield%) |
| a Reaction conditions: [Ar–I] = 0.08 M, [4-pentenoic acid] = 0.08 M, 80 °C, 16 h. V = 0.55 – 1.40 mL (1H NMR yield obtained with 1,3,5-trimethoxybenzene as internal standard). b [Ar–I] = 0.02 M, [4-pentenoic acid] = 0.1 M, V = 1 mL. c T = 100 °C. d Reaction time = 24 h. | |||||
| 1b | 2.8 | 100 | 1.0 | TFE | 92(63) |
| 2b,c | 2.6 | 100 | 1.2 | TFE | >99(67) |
| 3d | 2.2 | 70 | 1.2 | TFE | 81(65) |
| 4d | 2.0 | 40 | 1.1 | TFE | 79(44) |
| 5b | 1.5 | 10 | 1.0 | TFE | 51(28) |
| 6 | 1.5 | 10 | 0.5 | TFE | 51(24) |
| 7d | 1.5 | 10 | 1.1 | TFE | 54(15) |
| 8d | 1.1 | 1 | 0.5 | TFE | 38(1.6) |
| 9 | 1.5 | 10 | 1.0 | HFIP | 56(29) |
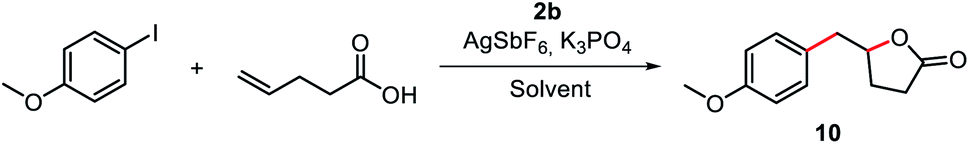
With these results in hand, we explored the possibility to perform the catalytic version of the arylation-lactonization of γ-alkenoic acids. Thus, we carried out the reaction decreasing the loading of 2b, first using substoichiometric 70 mol% of the gold complex. Product 10 was obtained in 65% yield (entry 3), so effectively a quantitative transformation was achieved with respect to Au loading. By using 40 mol% of Au(I) complex the yield was 44%, reaching about 1 TON (entry 4). Interestingly, using a lower amount of 2b (10 mol%) the yield was around 28% (∼3 TON, entry 5). Similarly, by using hexafluoro-2-propanol (HFIP) the yield was 29% (∼3 TON, entry 9); therefore, the best optimized results were obtained with fluorinated alcohols.
To exclude the possibility of oxidizing Au(I) to Au(III) in the presence of Ag(I), complex 2a (1 eq.) was reacted with AgSbF6 (15 eq.) in HFIP at 80 °C for 16 hours. Quantitative formation of the Au(I) dimer 3a was observed by NMR, confirming the role of silver as halide scavenger (see Scheme S12†).
The scope of the reaction was examined with different aryl halides and γ-alkenoic acids (Fig. 5). The arylation-lactonization using iodobenzene and complex 2b reached 50% yield (∼5 TON) in product 11. As expected, by increasing the catalyst loading to 20 mol%, the yield of 11 increased up to 87%, thus maintaining the turnover number (∼4–5 TON). Gratifyingly, the best yields were reached using 4-iodotoluene, obtaining product 12 in 60% yield using 2a (∼6 TON) and 81% yield using 2b (∼8 TON). However, the presence of the strong electron-withdrawing group CF3 in the para-position of the iodoaryl produced product 13 in a lower yield (∼13%). Following this trend,36,38 1-iodo-4-nitrobenzene produced a more dramatic drop of the yield of the corresponding product 14 (1%).
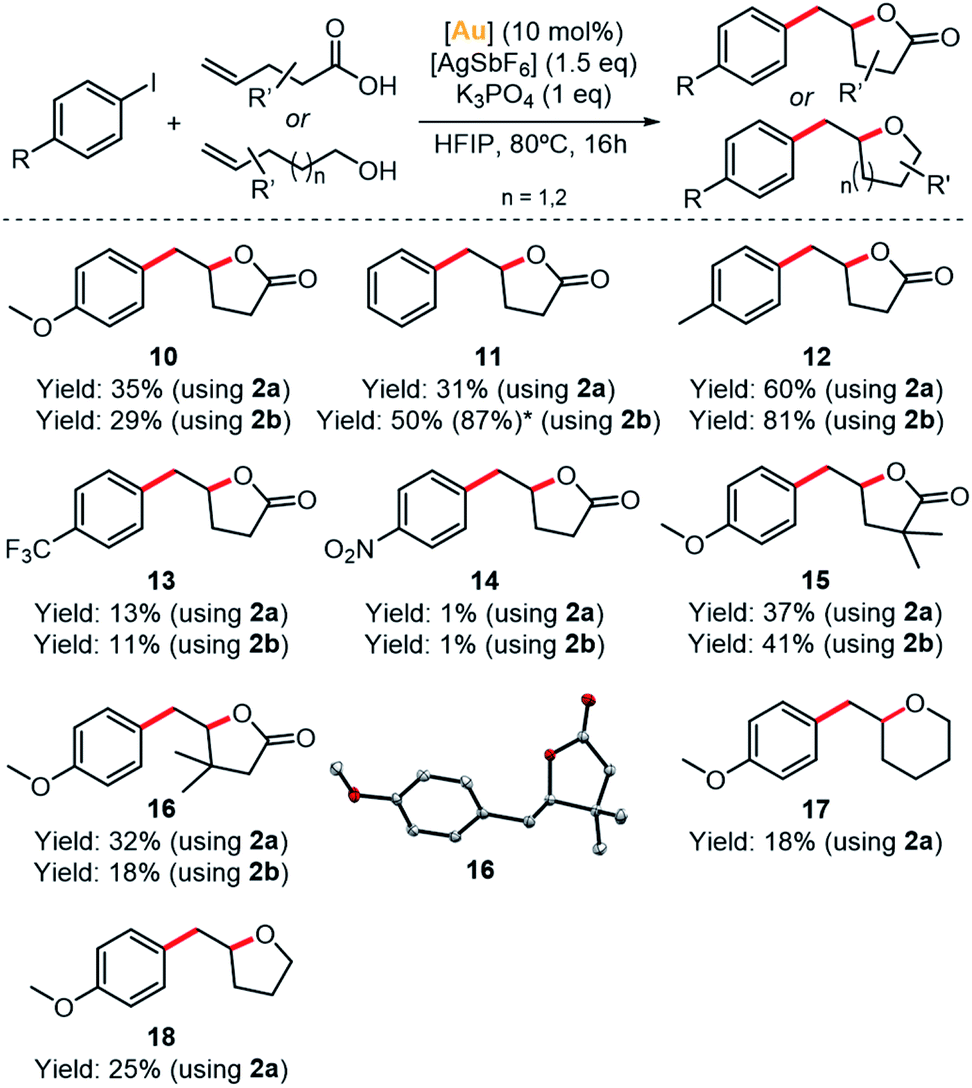 | ||
| Fig. 5 Scope of the arylation-lactonization of alkenoic acids (10–16) and oxyarylation of alkenes (17–18). *NMR yield using 20% of [Au] is shown in parenthesis. Crystal structure of 16 is shown (ellipsoids set at 50% probability, H atoms removed for clarity). | ||
Interestingly, the γ-alkenoic acid with α-methyl groups reacted smoothly, yielding 37% of 15 using 2a and 41% of 15 using 2b. Also, complexes 2a and 2b performed very similar, except in the case of using iodobenzene and 4-pentenoic acid, yet complex 2b is almost two-fold more active than 2a (31% vs. 50% yield of compound 11, for 2a and 2b, respectively). The molecular structure of compound 16 was determined by X-ray diffraction analysis and revealed the formation of the (R)-enantiomer by spontaneous resolution.
Finally, in order to broaden the scope of these (MIC^N)Au(I) complexes, we tested complex 2a in the oxyarylation of alkenols and 1,2-diarylaltion of alkenes under the same catalytic conditions (Scheme S11†). The oxyarylation products 17 and 18 were obtained in low but significant yields (18–25%, ∼2 TON) (Fig. 5 and Table S6†). On the contrary, the 1,2-diarylation reaction was unsuccessful.
Based on the fundamental understanding of the redox behaviour of complexes 2a and 2b, and the previously reported gold-catalyzed processes combining aryl halide oxidative addition and π-activation of alkenes,53 a mechanistic proposal is depicted in Fig. 6. First, the oxidative addition of the Csp2–I bond into a cationic gold(I) species occurs, affording a cationic arylgold(III) species. Then, the silver salt abstracts the iodide from this Au(III) species to generate a vacant site that can be occupied by the π-coordination of the olefin. Subsequent intramolecular γ-lactonization is enabled by the presence of base and the π-activation of the alkene produced by the gold center, to yield an alkylaryl gold(III) intermediate. The latter is proposed to undergo a reductive elimination step that affords the γ-benzyl-γ-butyrolactone product and closes the catalytic cycle by regenerating the initial catalytically active hemilabile [AuI(MIC^N)]+ species.
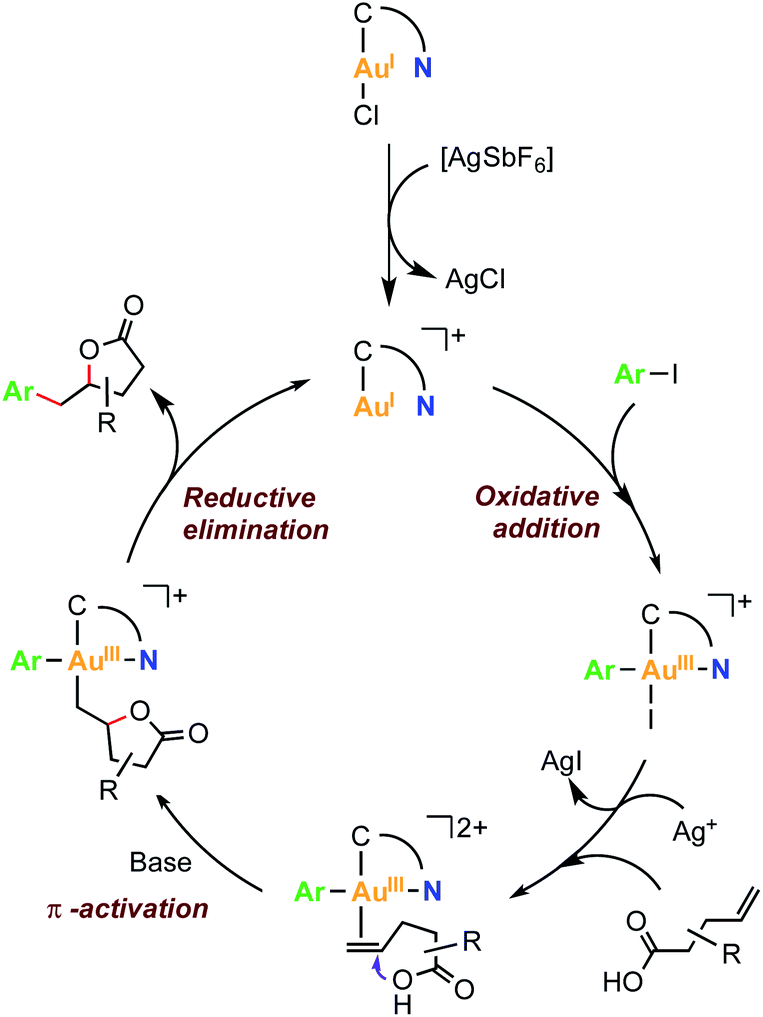 | ||
| Fig. 6 Proposed reaction mechanism for the Au(I)/Au(III)-mediated arylation-lactonization of alkenoic acids. | ||
To gain mechanistic insight, the stoichiometric reaction of cis-7a-Cl with AgSbF6 (1.2 eq.), 4-pentenoic acid (1.3 eq.) and K3PO4 (1.0 eq.) was carried out in HFIP at 80 °C. After 16 hours, γ-benzyl-γ-butyrolactone 10 was obtained in 73% yield (Scheme S13†). Therefore, it is plausible to propose the formation of an arylgold(III) intermediate formed by an oxidative addition reaction of the Csp2–I bond to Au(I).
Conclusions
We synthesized two Au(I) complexes bearing bidentate hemilabile MIC^N ligands, [AuI(MIC^N)Cl], 2a and 2b, and explored their redox reactivity. The formation of the Au(III) complexes 4a-Cl and 4a-OAc was achieved by using 2 e− oxidants of the type PhIX2 (X = Cl, OAc), demonstrating a suitable design of the MIC^N ligand to stabilize Au(III) centers. Next, we showed that effective oxidative addition of a strained Csp2–Csp2 bond was obtained when reacting 2b with biphenylene in the presence of a halide scavenger and heating at 90 °C. In this case, complex 5b was obtained as the desired ensuing Au(III) species.On the other hand, the oxidative addition of aryl iodides to Au(I) required a temperature of 120 °C, and under these conditions the reductive elimination reaction of Au(III) species is also favored, yielding a series of triazolium salts (8a-R/8b-R, 9a/9b and 1a/1b). Although the expected Au(III) complexes were not directly detected, we independently synthesized the analogous Au(III) complex cis-7a-Cl, which after heating produced the same triazolium product (8a-OMe), thus supporting the oxidative addition/reductive elimination pathway for aryl iodides.
Finally, we combined all the redox knowledge gained with this system to develop a novel reaction for the synthesis of γ-benzyl-γ-butyrolactones upon arylation-lactonization of γ-alkenoic acids. The reaction was catalyzed by 2a and 2b (10 mol% catalyst loading), obtaining up to 8 TON. The catalytic activity shown by MIC^N/Au system is significant because it shows that readily available hemilabile C^N ligands are a real alternative to hemilabile P^N ligands, where MeDalphos system stands out among catalytic examples of oxidant-free oxidative addition of aryl halides allowed by the presence of pendant N-groups. Current efforts are devoted to extending the application of these hemilabile [AuI(MIC^N)Cl] complexes to novel catalytic transformations.
Data availability
Data for this work, including experimental procedures, NMR spectra and crystallographic data, are provide in the ESI.†Author contributions
P. F., H. V. and G. G.-B. performed the experiments. All authors discussed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from MINECO of Spain for projects CTQ2016-77989-P, PID2019-104498GB-I00, RTI2018-098903-J-100 and PID2021-122900NB-I00 (financed by MICIN/AEI/10.13039/ 501100011033/ FEDER “Una manera de hacer Europa”), and for a FPU PhD grant to P. F. We also thank the Generalitat de Catalunya for project 2017SGR264 to X. R. and for a Beatriu de Pinós contract to H. V. (Beatriu de Pinós, 2019-BP-0080). X. R. is grateful for an ICREA Acadèmia award. G. G.-B gratefully acknowledges (RYC2019-026693-I/AEI/10.13039/501100011033) “El Fondo Social Europeo invierte en tu futuro’. Gobierno de Aragón/FEDER, UE (GA/FEDER, Reactividad y catálisis en química inorgánica, Group E50_20D). The authors would like to acknowledge the ‘Servicio General de Apoyo a la Investigación-SAI, Universidad de Zaragoza’, and the STR-UdG for technical support.Notes and references
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed.
- A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 CrossRef CAS PubMed.
- A. Fürstner and P. W. Davies, Angew. Chem., Int. Ed., 2007, 46, 3410–3449 CrossRef PubMed.
- J. H. Teles, S. Brode and M. Chabanas, Angew. Chem., Int. Ed., 1998, 37, 1415–1418 CrossRef CAS PubMed.
- Y. Fukuda and K. Utimoto, J. Org. Chem., 1991, 56, 3729–3731 CrossRef CAS.
- A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef CAS.
- M. N. Hopkinson, A. D. Gee and V. Gouverneur, Chem. – Eur. J., 2011, 17, 8248–8262 CrossRef CAS PubMed.
- A. Nijamudheen and A. Datta, Chem. – Eur. J., 2020, 26, 1442–1487 CrossRef CAS PubMed.
- A. Kar, N. Mangu, H. M. Kaiser, M. Beller and M. K. Tse, Chem. Commun., 2008, 386–388 RSC.
- L. T. Ball, M. Green, G. C. Lloyd-Jones and C. A. Russell, Org. Lett., 2010, 12, 4724–4727 CrossRef CAS PubMed.
- L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Chem. – Eur. J., 2012, 18, 2931–2937 CrossRef CAS PubMed.
- G. Zhang, L. Cui, Y. Wang and L. Zhang, J. Am. Chem. Soc., 2010, 132, 1474–1475 CrossRef CAS PubMed.
- T. de Haro and C. Nevado, J. Am. Chem. Soc., 2010, 132, 1512–1513 CrossRef CAS PubMed.
- T. Ball Liam, C. Lloyd-Jones Guy and A. Russell Christopher, Science, 2012, 337, 1644–1648 CrossRef CAS PubMed.
- L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, J. Am. Chem. Soc., 2014, 136, 254–264 CrossRef CAS PubMed.
- X. C. Cambeiro, N. Ahlsten and I. Larrosa, J. Am. Chem. Soc., 2015, 137, 15636–15639 CrossRef CAS PubMed.
- M. Hofer, A. Genoux, R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1021–1025 CrossRef CAS PubMed.
- M. Livendahl, P. Espinet and A. M. Echavarren, Platinum Met. Rev., 2011, 55, 212–214 CrossRef.
- T. Lauterbach, M. Livendahl, A. Rosellón, P. Espinet and A. M. Echavarren, Org. Lett., 2010, 12, 3006–3009 CrossRef CAS PubMed.
- M. Livendahl, C. Goehry, F. Maseras and A. M. Echavarren, Chem. Commun., 2014, 50, 1533–1536 RSC.
- D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395–403 CrossRef CAS PubMed.
- M. Joost, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 15022–15045 CrossRef CAS PubMed.
- B. Huang, M. Hu and F. D. Toste, Trends Chem., 2020, 2, 707–720 CrossRef CAS PubMed.
- P. Font and X. Ribas, Eur. J. Inorg. Chem., 2021, 2021, 2556–2569 CrossRef CAS.
- M. N. Hopkinson, A. Tlahuext-Aca and F. Glorius, Acc. Chem. Res., 2016, 49, 2261–2272 CrossRef CAS PubMed.
- S. Banerjee, V. W. Bhoyare and N. T. Patil, Chem. Commun., 2020, 56, 2677–2690 RSC.
- M. O. Akram, S. Banerjee, S. S. Saswade, V. Bedi and N. T. Patil, Chem. Commun., 2018, 54, 11069–11083 RSC.
- J. Miró and C. del Pozo, Chem. Rev., 2016, 116, 11924–11966 CrossRef PubMed.
- V. W. Bhoyare, A. G. Tathe, A. Das, C. C. Chintawar and N. T. Patil, Chem. Soc. Rev., 2021, 50, 10422–10450 RSC.
- N. Lassauque, P. Gualco, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2013, 135, 13827–13834 CrossRef CAS PubMed.
- P. Gualco, S. Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2011, 50, 8320–8324 CrossRef CAS PubMed.
- J. Guenther, S. Mallet-Ladeira, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 1778–1781 CrossRef CAS PubMed.
- J. Serra, T. Parella and X. Ribas, Chem. Sci., 2017, 8, 946–952 RSC.
- H. Beucher, J. Schörgenhumer, E. Merino and C. Nevado, Chem. Sci., 2021, 12, 15084–15089 RSC.
- C.-Y. Wu, T. Horibe, C. B. Jacobsen and F. D. Toste, Nature, 2015, 517, 449–454 CrossRef CAS PubMed.
- M. Joost, A. Zeineddine, L. Estévez, S. Mallet−Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 14654–14657 CrossRef CAS PubMed.
- M. Joost, L. Estévez, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 5236–5240 CrossRef CAS PubMed.
- M. J. Harper, C. J. Arthur, J. Crosby, E. J. Emmett, R. L. Falconer, A. J. Fensham-Smith, P. J. Gates, T. Leman, J. E. McGrady, J. F. Bower and C. A. Russell, J. Am. Chem. Soc., 2018, 140, 4440–4445 CrossRef CAS PubMed.
- J. A. Cadge, H. A. Sparkes, J. F. Bower and C. A. Russell, Angew. Chem., Int. Ed., 2020, 59, 6617–6621 CrossRef CAS PubMed.
- J. Chu, D. Munz, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 7884–7887 CrossRef CAS PubMed.
- A. Zeineddine, L. Estévez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Nat. Commun., 2017, 8, 565 CrossRef PubMed.
- J. Rodriguez, A. Tabey, S. Mallet-Ladeira and D. Bourissou, Chem. Sci., 2021, 12, 7706–7712 RSC.
- D. Mendoza-Espinosa, D. Rendon-Nava, A. Alvarez-Hernandez, D. Angeles-Beltran, G. E. Negron-Silva and O. R. Suarez-Castillo, Chem.–Asian J., 2017, 12, 203–207 CrossRef CAS PubMed.
- G. Kleinhans, M. M. Hansmann, G. Guisado-Barrios, D. C. Liles, G. Bertrand and D. I. Bezuidenhout, J. Am. Chem. Soc., 2016, 138, 15873–15876 CrossRef CAS PubMed.
- G. Kleinhans, A. K. Chan, M. Y. Leung, D. C. Liles, M. A. Fernandes, V. W. Yam, I. Fernandez and D. I. Bezuidenhout, Chem. – Eur. J., 2020, 26, 6993–6998 CrossRef CAS PubMed.
- R. P. Herrera and M. C. Gimeno, Chem. Rev., 2021, 121, 8311–8363 CrossRef CAS PubMed.
- L. Huang, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem. Commun., 2016, 52, 6435–6438 RSC.
- A. Tabey, M. Berlande, P. Hermange and E. Fouquet, Chem. Commun., 2018, 54, 12867–12870 RSC.
- J. Rodriguez, A. Zeineddine, E. D. Sosa Carrizo, K. Miqueu, N. Saffon-Merceron, A. Amgoune and D. Bourissou, Chem. Sci., 2019, 10, 7183–7192 RSC.
- J. Rodriguez, D. Vesseur, A. Tabey, S. Mallet-Ladeira, K. Miqueu and D. Bourissou, ACS Catal., 2022, 12, 993–1003 CrossRef CAS.
- M. O. Akram, A. Das, I. Chakrabarty and N. T. Patil, Org. Lett., 2019, 21, 8101–8105 CrossRef CAS PubMed.
- J. Rodriguez, N. Adet, N. Saffon-Merceron and D. Bourissou, Chem. Commun., 2020, 56, 94–97 RSC.
- M. Rigoulet, O. Thillaye du Boullay, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2020, 59, 16625–16630 CrossRef CAS PubMed.
- A. G. Tathe, C. C. Chintawar, V. W. Bhoyare and N. T. Patil, Chem. Commun., 2020, 56, 9304–9307 RSC.
- S. Zhang, C. Wang, X. Ye and X. Shi, Angew. Chem., Int. Ed., 2020, 59, 20470–20474 CrossRef CAS PubMed.
- A. G. Tathe, Urvashi, A. K. Yadav, C. C. Chintawar and N. T. Patil, ACS Catal., 2021, 11, 4576–4582 CrossRef CAS.
- C. C. Chintawar, A. K. Yadav and N. T. Patil, Angew. Chem., Int. Ed., 2020, 59, 11808–11813 CrossRef CAS PubMed.
- S. R. Mudshinge, Y. Yang, B. Xu, G. B. Hammond and Z. Lu, Angew. Chem., Int. Ed., 2022, 61, e202115687 CrossRef CAS PubMed.
- X. Ye, C. Wang, S. Zhang, Q. Tang, L. Wojtas, M. Li and X. Shi, Chem. – Eur. J., 2022, e202201018 CAS.
- C. C. Chintawar, V. W. Bhoyare, M. V. Mane and N. T. Patil, J. Am. Chem. Soc., 2022, 144, 7089–7095 CrossRef CAS PubMed.
- T. Shibata, R. Nagai, S. Okazaki, S. Nishibe and M. Ito, Bull. Chem. Soc. Jpn., 2022, 95, 700–706 CrossRef CAS.
- E. Tomás-Mendivil, P. Y. Toullec, J. Borge, S. Conejero, V. Michelet and V. Cadierno, ACS Catal., 2013, 3, 3086–3098 CrossRef.
- A. G. Nair, R. T. McBurney, M. R. D. Gatus, S. C. Binding and B. A. Messerle, Inorg. Chem., 2017, 56, 12067–12075 CrossRef CAS PubMed.
- M. Navarro, A. Tabey, G. Szalóki, S. Mallet-Ladeira and D. Bourissou, Organometallics, 2021, 40, 1571–1576 CrossRef CAS.
- G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed.
- J. Lorkowski, P. Żak, M. Kubicki, C. Pietraszuk, D. Jędrzkiewicz and J. Ejfler, New J. Chem., 2018, 42, 10134–10141 RSC.
- K. J. Cavell and A. T. Normand, in N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, Dordrecht, 2010, vol. 32, pp. 299–314 Search PubMed.
- C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS.
- B. Dong, H. Peng, S. E. Motika and X. Shi, Chem. – Eur. J., 2017, 23, 11093–11099 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For materials, instrumentation, experimental procedures and spectroscopic characterization of all compounds. CCDC 2163608 (4a-OAc), 2163609 (2a), 2163610 (2b), 2163611 (4a-Cl), 2163612 (8a-OMe), 2163613 (3b·biphenylene), 2163614 (5b), 2163615 (6b), 2163616 (8b-OMe), 2163617 (3a), 2163877 (16) and 2176932 (cis-7a-Cl) contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01966c |
| This journal is © The Royal Society of Chemistry 2022 |