
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-dimensional heterostructures built from ultrathin CeO2 nanosheet surface-coordinated and confined metal–organic frameworks with enhanced stability and catalytic performance†
Haiyan
An
a,
Yang
Hu
b,
Nan
Song
a,
Tingliang
Mu
a,
Shiqiang
Bai
 a,
Yong
Peng
b,
Liangliang
Liu
*a and
Yu
Tang
*a
a,
Yong
Peng
b,
Liangliang
Liu
*a and
Yu
Tang
*a
aState Key Laboratory of Applied Organic Chemistry, Key Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China. E-mail: liull@lzu.edu.cn; tangyu@lzu.edu.cn
bKey Laboratory of Magnetism and Magnetic Materials of Ministry of Education, School of Physical Science and Technology, Lanzhou University, Lanzhou 730000, China
First published on 14th February 2022
Abstract
Two-dimensional (2D) metal–organic framework (MOF) based heterostructures will be greatly advantageous to enhance catalytic performance because they increase the contact surface and charge transfer. Herein, a novel 2D heterostructure named CeO2@NiFe-MOFs, in which monolayer NiFe-MOFs is coordinated with ceria (CeO2) to improve catalytic and stability performance, is successfully constructed by the strategy of in situ growth on the surface of ultrathin CeO2 nanosheets being functionalized with monolayer carboxylic acid groups. The 2D heterostructure possesses a sandwich structure, where monolayer NiFe-MOFs are coordinated to both the top and bottom surface of CeO2 nanosheets via joining carboxylic acid groups. In particular, CeO2 with robust coordination plays a significant role in the anchoring of carboxylic acid groups and binding strength of heterostructures. The 2D CeO2@NiFe-MOF heterostructure with a joint effect of metal–ligand coordination not only presents good structural stability but also significantly enhances the oxygen evolution reaction (OER) efficiencies in comparison to bare NiFe-MOFs, achieving a current density of 20 mA cm−2 at a low overpotential of 248 mV as well as durability for at least 40 h. Meanwhile, the electronics, optics, band gap energy and local strains of CeO2 decorated with 2D NiFe-MOFs are different to the properties of bare CeO2. Our study on the construction of an ultrathin CeO2 surface-coordinated and confined MOF layer may pave a new way for novel 2D MOF composites/heterostructures or multi-functional 2D CeO2 materials to be used in energy conversion or other fields.
Introduction
The energy crisis and environmental pollution are the most crucial challenges facing humanity today. Electrocatalytic water splitting plays a key role in addressing issues of energy sustainability and environmental protection.1 The development of a high-performance electrocatalyst for the oxygen evolution reaction (OER) with sluggish kinetics is a great challenge in the field. Heterostructures have attracted great attention in the fields of materials, catalysis and energy, because the unique structures generally exhibit enhanced catalytic activity in comparison with other uniformly structured counterparts.2,3 Metal–organic frameworks (MOFs) are a class of porous crystal coordination polymers, which are commonly constructed by the coordination of metal nodes with diversiform organic ligands,4 possessing ultrahigh porosity, enormous surface areas, accessible metal active sites, hybrid composition, good designability and structural diversity. Benefitting from these merits, MOFs have attracted considerable interest in electrocatalytic water splitting.5 However, many bulk MOFs still suffer from poor conductivity, buried active sites and chemical instability caused by the intrinsic shortages of organic ligands. Compared to bulk MOFs, ultrathin two-dimensional (2D) MOFs afford not only an enlarged surface area but also more active sites exposed on the surface, which can be beneficial to the adsorption and activation of reactants and improvement of catalytic performance.6 MOF-based heterostructures can concurrently provide architectural features of MOFs and additional physicochemical properties.7,8 Therefore, to take full advantage of MOFs, ultrathin 2D MOFs and MOF-based heterostructures as efficient electrocatalysts in the field of water splitting and related energy conversion have aroused the attention of researchers.Ultrathin 2D MOFs with thickness on a nanometer scale are beneficial to improving the performance of electrocatalytic water splitting.9,10 Accordingly, two fabrication strategies have been developed to synthesize ultrathin 2D MOFs, including the top-down strategy (exfoliation methods) and bottom-up strategy (direct syntheses from metal and organic precursors).11 The exfoliation method mainly assisted by ultrasound is only applicable to bulk MOFs with intralayer coordinated bonds that can be easily broken, resulting in inhomogeneity, fragmentation and restacking of MOF-nanosheets.12 On the other hand, the bottom-up strategy has proved to be an efficient method to prepare ultrathin 2D MOFs. For example, the ultrathin 2D Co-MOF nanosheets (thickness of ∼2 nm), prepared under hydrothermal conditions using polyvinylpyrrolidone as the surfactant, showed high intrinsic activity and enhanced electrical conductivity.13 The surfactant may block some of the active sites. In order to avoid the introduction of an organic surfactant, recently, Lang and co-workers prepared ultrathin Ni–M-MOFs (M = Fe, Al, Co, Mn, Zn, and Cd) with a thickness of several atomic layers via a one-step hydrothermal method, where the solvent played a crucial role in modulating the formation of 2D MOFs.14 Although many research achievements have been achieved in the preparation of 2D MOFs, developing the ideal technology to simultaneously control the growth of 2D MOFs along the lateral direction only and suppress vertical growth to the nanometer scale still remains challenging.15 2D MOF-based heterostructures can provide the architectural features of nanometer MOFs; moreover, heterostructures can achieve the controllable growth of ultrathin MOFs. Therefore, the novel functional heterostructures, especially those fabricated from 2D MOFs or other 2D materials, have become the current focus of research on electrocatalytic water splitting, due to their increased contact surface and enhanced charge transfer.16,17
Various strategies have been reported to fabricate MOF-based heterostructures, such as spin coating,18 electrochemical deposition,19 layer-by-layer assembly (LBL),20in situ growth21 and so on.22 However, most of the proposed strategies are not suitable to uniquely construct heterostructures by the controllable integration of 2D MOFs and nanomaterials with ultrathin thickness and efficient activities. Therefore, it is urgent to develop a facile and efficient method to synthesize heterostructures of 2D MOFs with 2D nanomaterials. Moreover, the accurate selection of 2D nanomaterials with good properties to support and control the growth of MOFs will lend a hand to improve the activities and stabilities of the heterostructures. Among the abundant 2D nanomaterials,23 ceria (CeO2) is widely used to improve the performance of materials in catalytic conversion processes.24 Its excellent stability and corrosion resistance also result in the structural robustness and durability of materials.25 In particular, ultrathin CeO2 nanosheets display the largest specific surface area, coordination-unsaturated Ce sites and a surface defect-rich structure, which is conducive to catalytic performance.26 As far as we are concerned, CeO2 is an ideal substrate for coordinated functionalization of carboxylic acid groups, which provides a strategy for the design of new CeO2-based materials with tailored multiple functionalities. Considering all the above, we envisage that ultrathin CeO2 would be an ideal nanomaterial to support MOFs. Besides, a Ce-decorated transition metal was reported by Zheng and co-workers,27 and to the best of our knowledge, the heterostructures of ultrathin CeO2 nanosheets with surface-coordinated and confined transition metal-MOFs were poorly understood until fairly recently.
Herein, we report the coordinated functionalization of ultrathin CeO2 nanosheets by attaching oxygen-containing carboxylic functional groups to their surface via an approach of ligand exchange, and then using a straightforward protocol of the coordination between the carboxylic groups and the metal ions to form monolayer MOFs on the surface of CeO2 and construct CeO2@NiFe-MOFs heterostructures. Since the surface of CeO2 can expose coordination-unsaturated Ce atoms, which is beneficial to coordinate with oxygen-containing functional groups, ligand exchange has been adopted to anchor the oxygen-containing functional groups. We demonstrate that the protocol is not only beneficial for the chemical functionalization of CeO2 with tailored oxygen-containing functional groups but also affords well-defined 2D CeO2@NiFe-MOF heterostructures, in which ultrathin CeO2 nanosheets with the largest specific surface area provide a substrate for the dispersion of the 2D NiFe-MOF layer and the thickness of the 2D NiFe-MOFs could be controlled down to nanometers. Therefore, ultrathin 2D heterostructures efficiently increase the contact surface to capture the targeted molecules and will accelerate charge transfer between active NiFe-MOFs and inorganic CeO2. In addition, compared with the properties of bare CeO2, CeO2 decorated with 2D NiFe-MOFs exhibits different properties of electronics, optics, band gap energy and local strain. Meanwhile, 2D CeO2@NiFe-MOF heterostructures assisted by CeO2 nanosheets can be used as an electrocatalyst, exhibiting significantly enhanced performance and long-term stability in the OER.
Results and discussion
Ultrathin heterostructures have been prepared through an in situ growth strategy on the CeO2 surface functionalized by carboxylic acid groups. As shown in Fig. 1a, the ultrathin 2D CeO2@NiFe-MOF heterostructures are prepared via a two-step procedure (ligand exchange and in situ growth). Firstly, terephthalic acid (1,4-BDC) coordinates with the cerium cation on the CeO2 surface, in place of oleic acid (OA) on the surface by two-phase ligand exchange. Thus, ultrathin CeO2 nanosheets were functionalized by monolayer carboxylic acid groups (CeO2-BDC).28 Secondly, CeO2-BDC provides the templates for the crystallization of MOFs, where monolayer NiFe-MOFs are coordinated to both the top and bottom surface of CeO2 nanosheets via joining carboxylic acid groups, obtaining ultrathin 2D CeO2@NiFe-MOF heterostructures with a sandwich structure. In the process of reaction, when CeO2-BDC is suspended in an alkaline solution, the carboxylic acid groups can be polarized in the alkaline solution, and then uniformly adsorb metal ions (e.g. Ni2+, Fe2+) via strong electrostatic adsorption. As a consequence, MOFs crystallize onto the CeO2 surface as films. Undoubtedly, these 2D templated 1,4-BDC tails coordinated with metal ions can create abundant open metal sites, and the 1,4-BDC bridged heterostructures featured 2D layered structures, facilitating the increased interlaminar porosity and largely promoted ion transport.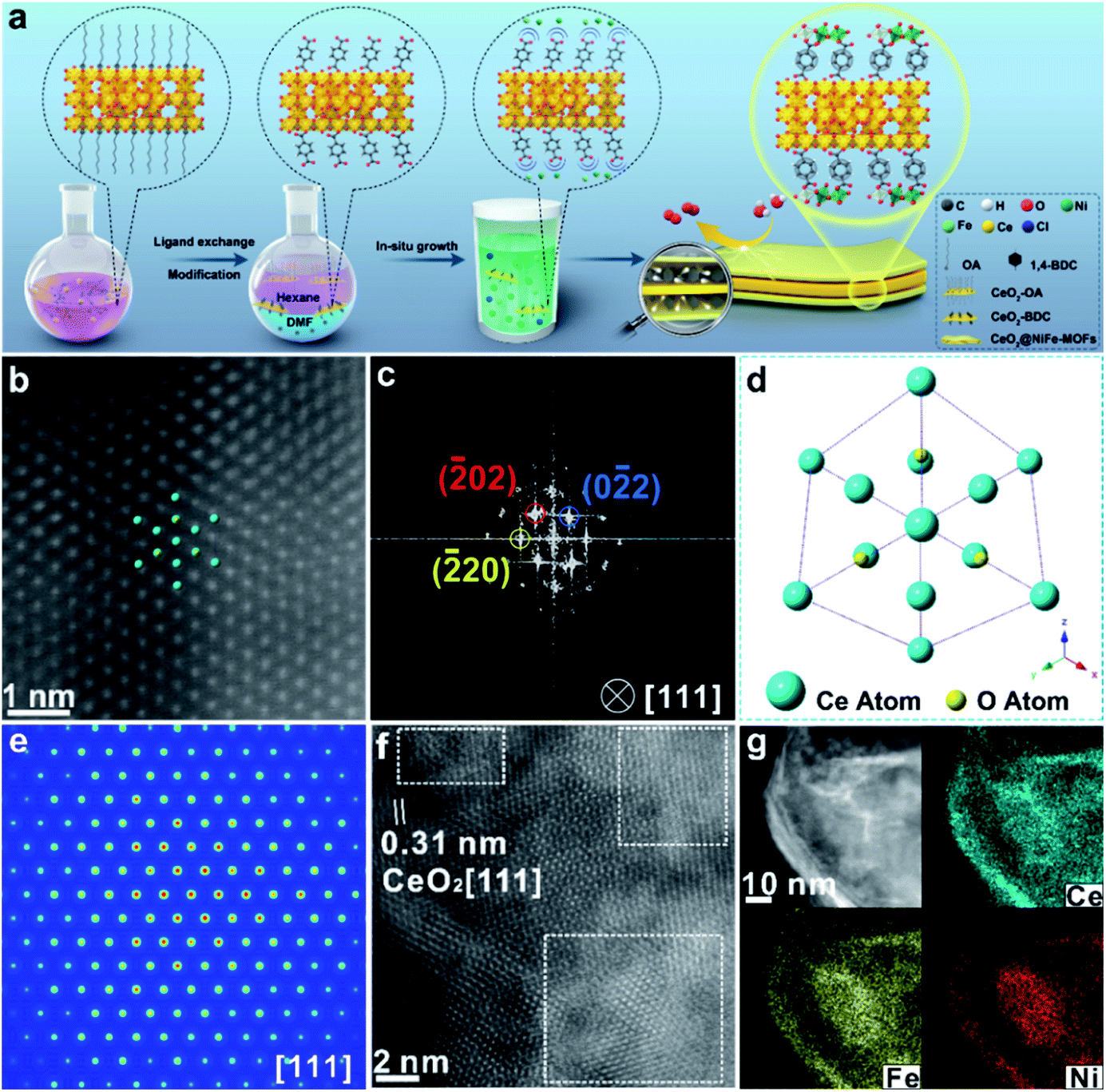 | ||
| Fig. 1 (a) Schematic illustration of the CeO2@NiFe-MOF heterostructure preparation procedure including two steps: two-phase ligand exchange and in situ growth. (b) Atomic HAADF-STEM image of CeO2@NiFe-MOFs along the [111] orientation of CeO2. (c) Corresponding FFT pattern from (b). (d) The projected structural model of CeO2 in CeO2@NiFe-MOFs. (e) FFT-filtered atomic resolution simulation image. (f) Enlarged HR-TEM image of CeO2@NiFe-MOFs from Fig. S10e.† (g) The corresponding elemental mapping images of CeO2@NiFe-MOF heterostructures. | ||
CeO2-OA nanosheets with an ultrathin thickness and organic ligand coordinately functionalized ultrathin CeO2 were prepared according to previous reports (Fig. S1–S3†).29,30 The success of coordinated modification of organic ligands on CeO2 nanosheets can be observed by vibrational spectroscopy (Fig. S4 and S5†).31 In this work, we mainly focus on CeO2-BDC nanosheets. CeO2-OA and CeO2-BDC were investigated by powder X-ray diffraction (PXRD). As shown in Fig. S6,† the shifted diffraction peaks of CeO2-BDC compared to that of pristine CeO2-OA can probably be ascribed to the partial transition from Ce3+ to Ce4+ in the process of substitution. The appearance of a peak at ≈15.38° can be ascribed to the coordinated combination of the Ce-ligand.32 The water contact angle was also measured to further investigate the CeO2 surface (Fig. S7†). The above results elucidate the successful functionalization of carboxylic acid groups onto the CeO2 surface.
The transmission electron microscopy (TEM) image of CeO2-OA shows the characteristic structure of nanosheets with diverse lateral sizes (Fig. S8a†). The nanosheet structure is well preserved in CeO2-BDC nanosheets (Fig. S8b†) with a relatively more disrupted morphology compared to pristine CeO2-OA nanosheets. Morphologies of CeO2-OH and CeO2-PMA (Fig. S9†) are also strikingly different from CeO2-OA, although nanosheets can be observed. The dramatic change in the morphologies of CeO2-BDC, CeO2-OH and CeO2-PMA is mainly caused by coordination modification, which can be regarded as a sign of chemical modification. The TEM images of 2D CeO2@NiFe-MOF heterostructures (Fig. S10a and b†) show an ultrathin sandwich-like structure with multiple stacking faults. High-resolution TEM (HR-TEM) reveals the crystal structure of 2D CeO2@NiFe-MOF heterostructures. As depicted in Fig. S10c,† the lattice spacing of CeO2 is measured to be 0.31 nm corresponding to the [111] interplanar spacing, and the d = 0.21 nm corresponds to the interplanar distance in the [103] plane of NiFe-MOFs.14,33 The corresponding selected area electron diffraction (SAED) pattern (Fig. S10d†) can be indexed to the [111] and [103] lattice planes of CeO2 and NiFe-MOFs, respectively. The high-angle annular dark field aberration corrected scanning TEM (HAADF-STEM) image reveals the fine structure of CeO2 within CeO2@NiFe-MOF heterostructures (Fig. 1b). The fast Fourier transform (FFT) image in Fig. 1c shows the coexistence of two clearly distinct structural domains. Fig. 1d shows the corresponding crystal structure of CeO2 unit cells in CeO2@NiFe-MOF heterostructures taken along the [111] orientation. The FFT-filtered atomic resolution image was further used to visualize the corroboration along the [111] orientation (Fig. 1e), and Fig. S11† shows the corresponding three-dimensional atom intensity profile. 2D NiFe-MOFs are nonuniform on the surface of CeO2 nanosheets, and this may be attributed to the nonuniformity of the monolayer carboxylic acid groups (Fig. 1f). The elemental composition of CeO2@NiFe-MOF heterostructures is identified by HAADF-STEM mapping (Fig. 1g and S10f†), confirming the uniform distribution of Ce, Ni, Fe and O elements throughout the entire nanosheets.
The structure of CeO2@NiFe-MOF heterostructures is further confirmed by the PXRD pattern (Fig. 2a), in which the decrease of crystallinity results from the increased amounts of open metal sites.34 The diffraction peaks of CeO2@NiFe-MOF heterostructures are slightly shifted, revealing structural transformation induced by ligand distortion. Comparing Raman spectra of CeO2@NiFe-MOF heterostructures with those of NiFe-MOFs, the intensity of CeO2@NiFe-MOF heterostructures at 525 cm−1 is found to be considerably lower (Fig. S12†). The atomic force microscopy (AFM) tests are further performed to observe the thickness and surface of CeO2-OA and CeO2@NiFe-MOFs deposited on silicon substrates. As shown in Fig. S13,† compared to CeO2-OA, CeO2@NiFe-MOF heterostructures exhibit a significant change of thickness in the range of 3.8–8 nm and surface morphology. These results verified the successful synthesis of ultrathin CeO2@NiFe-MOF heterostructures. Fig. 2b shows the specific surface area of CeO2@NiFe-MOFs which is estimated to be 53 m2 g−1. Besides, the pore size distribution can also be seen from the inset of Fig. 2b, exhibiting a pore diameter of 2.3 nm, allowing the faster diffusion of ions. The thermogravimetric (TG) analysis also demonstrated good stability of NiFe-MOF nanosheets in CeO2@NiFe-MOFs (Fig. S14†).
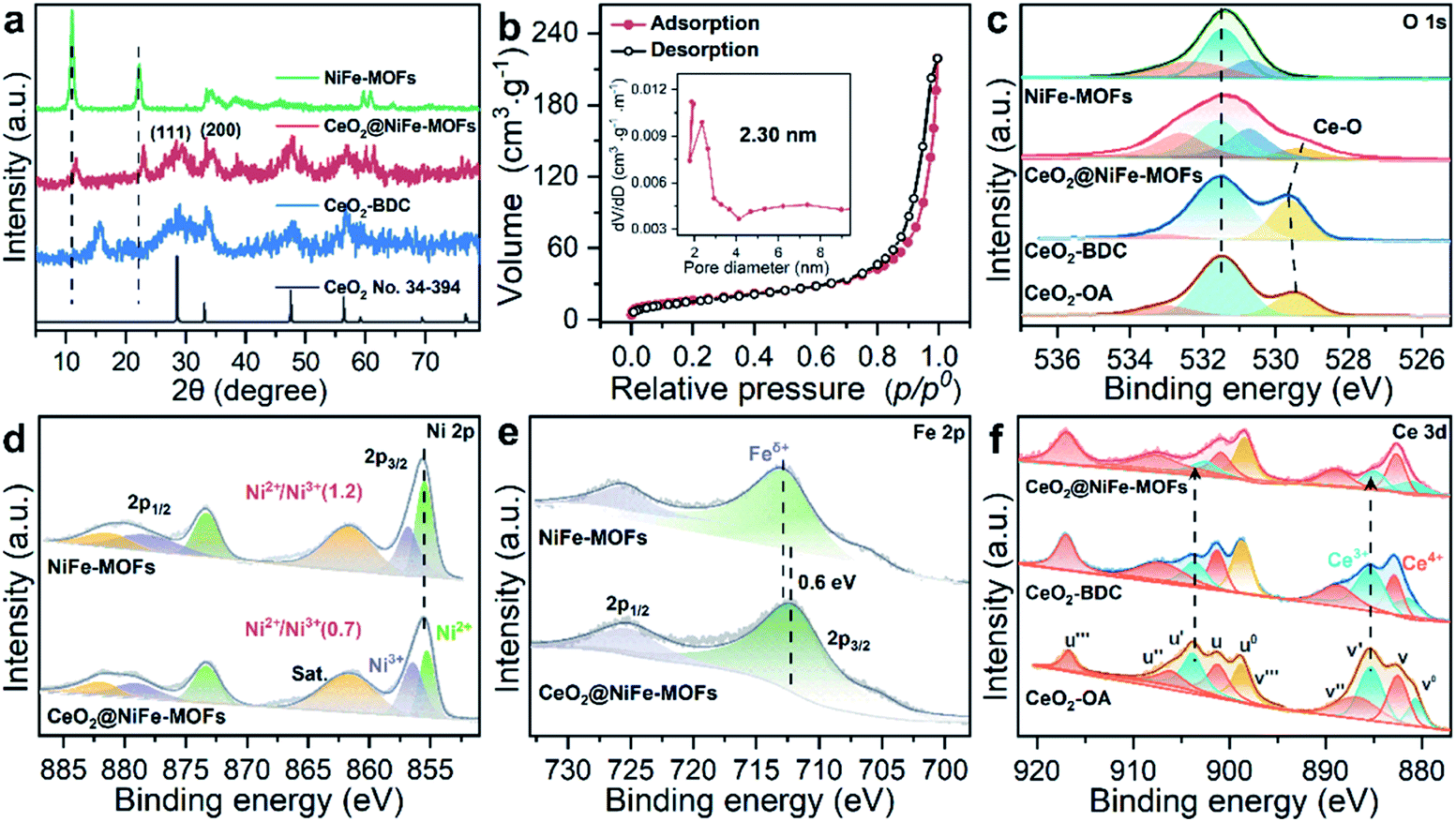 | ||
| Fig. 2 (a) PXRD patterns of CeO2 (no. 34-394), CeO2-BDC, CeO2@NiFe-MOFs and NiFe-MOFs. (b) Nitrogen adsorption/desorption isotherm of CeO2@NiFe-MOFs, pore size distributions (inset). (c–f) XPS spectra of O 1s, Ni 2p, Fe 2p and Ce 3d; Ce 3d XPS spectra reveal the change of valence of Ce caused by coordinated functionalization. | ||
Surface-sensitive X-ray photoelectron spectroscopy (XPS) characterization was carried out to establish the surface elemental composition and valence state, which helps to analyze the intrinsic electronic structure of the prepared materials. As shown in the full XPS spectra of CeO2@NiFe-MOF heterostructures (Fig. S15†), Ce, Ni, Fe, O, and C elements were identified. In the high-resolution O 1s XPS spectra (Fig. 2c), the broad peak at 529 eV can be assigned to the characteristic peak of CeO2;25,35,36 the O 1s of CeO2-BDC (529.6 eV) shifts to higher binding energy compared to that of CeO2-OA (529.3 eV). A much broader peak at around 529.3 eV in CeO2@NiFe-MOFs suggests possible electronic interactions between carboxylic acid groups and CeO2. The ratio of the NiII/NiIII XPS peak in Ni (2p3/2) is used as the reference, and the NiII/NiIII ratio on the surface of CeO2@NiFe-MOFs (0.7) decreases compared with the NiFe-MOFs (1.2) (Fig. 2d), suggesting that the NiII is oxidized when NiFe-MOFs is coordinated with CeO2. The Fe peaks of CeO2@NiFe-MOFs show apparent shifts toward the lower binding energies (Fig. 2e). The Fe3+ sites serve as Lewis acid sites,37 and Ce atoms act as electron-accepting sites, confirming partial electron transfer from Ni to Fe and Ce atoms.38
XPS spectra of Ce 3d are shown in Fig. 2f, the peaks v′′′, v′′, v′ and v0 arise from the ionization of the 3d5/2 electron, the u′′′, u′′, u′, u and u0 peaks arise from the 3d3/2 electron, and the peaks (labeled v0, v′, u0, u′) are coincident with previously reported XPS peaks of Ce3+ of the Ce2O3 compound.39 It is clear that CeO2-OA shows binding energy related to Ce3+, while CeO2-BDC shows decrease of intensity of Ce3+ related peaks, indicating the efficient replacement of OA by 1,4-BDC, which leads to a partial transition from Ce3+ to Ce4+. For CeO2@NiFe-MOFs, the Ce3+ related peaks at 903.8 and 885.5 eV dramatically decrease, showing features related to Ce4+. The comparison of results shows that coordinated functionalization to CeO2-OA may induce gradual oxidation of Ce3+. Therefore, the interactions between CeO2 and organic ligands can be testified according to the electronic charge transfer. To further confirm valence states of Ce ions in CeO2@NiFe-MOFs, the Ce-M edge was measured by STEM-EELS (electron energy loss spectroscopy), which was performed to directly collect the cerium signal. EELS spectra are depicted in Fig. S16,† where the intensity of the M4 edge is higher than that of the M5 edge, which is attributed to Ce4+.40,41 The result of EELS spectral analysis for the valence state of Ce ions is coincident with that of the XPS spectral analysis. The valence of Ce ions is nominally +4 in CeO2@NiFe-MOFs, indicating that reversible charge transfer is easier between the Ce4+ and Ce3+ oxidation stages, which is advantageous to the oxygen evolution reaction.
The lattice distortion in CeO2 can be caused by first dissolution of surface Ce of CeO2 by carboxylic acid and then coordination of carboxylic acid groups onto the CeO2 surface. To further confirm the existence of lattice distortion and surface strains, the HR-TEM lattice images were used to measure the nanoscale spatial distribution of the strains (Fig. 3a–c). The degree of CeO2 surface strains is associated with the extent of dissolution of surface Ce in CeO2, and thus, surface strain is readily adjusted by organic ligands. CeO2 has a standard lattice spacing and thus ensures the degree of strain. Strain mapping measured by geometric phase analysis (GPA) can indicate the existence of legible CeO2 lattice strains upon coordinated modification, resulting from the interactions of CeO2 and organic ligands. The NiFe-MOFs on CeO2 nanosheets are negligible (Fig. 3a), giving CeO2 only slight lattice strain. As shown in Fig. 3b and c, the surface strain of CeO2 revert proportional to dense coverage of NiFe-MOFs, demonstrating that surface strain is caused by dissolution of surface Ce of CeO2. Meanwhile, the FFT diffractogram of the HR-TEM lattice in Fig. 3c also unambiguously identifies the coexistence of CeO2 and NiFe-MOFs (Fig. 3d). The dislocations are clearly observed in Fig. 3e, which indicates the existence of a defect-rich structure.
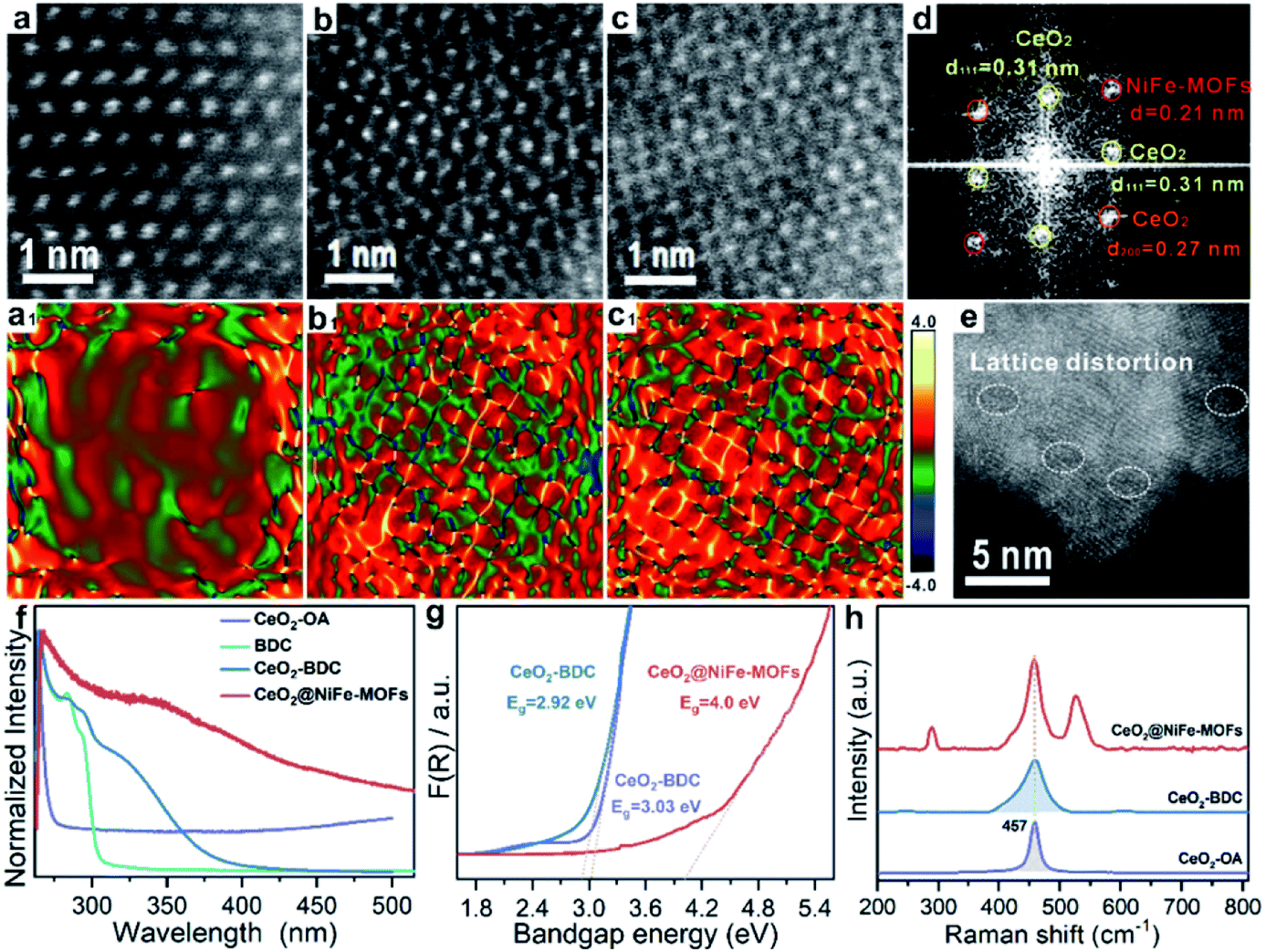 | ||
| Fig. 3 Characterization of lattice strain of CeO2@NiFe-MOFs. (a–c) The HR-TEM images of CeO2@NiFe-MOFs from the local magnification of Fig. S17.† (a1–c1) The corresponding strain mapping in (a–c). (d) FFT diffractogram of the HR-TEM lattice of (c). (e) HR-TEM image of CeO2@NiFe-MOFs. (f) UV-vis absorption spectra of CeO2-OA cyclohexane solution and CeO2-BDC, NiFe-MOFs and CeO2@NiFe-MOFs DMF solution. (g) The band gap energy of CeO2-OA, CeO2-BDC and CeO2@NiFe-MOFs calculated from the UV/Vis diffuse reflectance spectra. (h) The corresponding Raman spectroscopy. | ||
Subsequently, the research is focused on understanding intrinsic interactions of electronic charge transfer, and to then monitor how coordinated modification affects the optical properties of CeO2. The excitation spectra of CeO2-OA display intense bands in the ultra-violet region (Fig. 3f). As expected, the excitonic transition bands of CeO2-BDC were red shifted in comparison to CeO2-OA, and the band of 2D CeO2@NiFe-MOF heterostructures is up-shifted compared to that of CeO2-OA and CeO2-BDC, which could be ascribed to increased scattering effects arising from aggregate and electronic interactions between CeO2 and NiFe-MOFs.42,43 To further elucidate the influence of coordinated functionalization for optical properties of CeO2, UV-vis diffuse reflection spectroscopy was carried out, which showed that band gap energies are 3.03 eV and 2.92 eV for CeO2-OA and CeO2-BDC, respectively (Fig. 3g), indicating that surface coordination can result in fine-tuning of electronic and optical properties of CeO2. The red-shift of band gap energy can be also assigned to the increase of surface strain and defects from CeO2-OA to CeO2-BDC. Conversely, CeO2@NiFe-MOFs exhibit a broad band gap, which can be attributed to the increase of oxygenated groups and indicating the CeO2 surface coordinated with NiFe-MOFs. Different characteristics of CeO2 after coordination have been demonstrated. The Raman spectrum signal of CeO2-BDC (Fig. 3h) is asymmetrically broadened relative to that of pristine CeO2-OA, caused by the increase of the lattice constant.44 Meanwhile, the asymmetrical broadening also indicates the decreasing CeO2 size. The decreasing size proves that carboxylic acids properly dissolve surface Ce of CeO2 and coordinate with CeO2. The tight bonding between CeO2 nanosheets and NiFe-MOF layers is the linchpin. Thus, carboxylic acid groups strongly coordinate with cerium and the tails anchor on the metal ions. Besides, the π-conjugated structure in carboxylic acid groups also provides a platform to facilitate electron transfer between them in the process of catalytic conversion.
The OER is chosen as the primary catalytic model to verify the catalytic performance because NiFe-MOFs have a good water oxidation potential, because Fe incorporation enhances the reducibility of Ni in NiFe-MOFs, thereby potentially increasing the generation of oxygen vacancies and facilitating OER catalytic activity.45,46 Here, we mainly emphasize the importance of coordination of ultrathin CeO2 in promoting catalytic performance. In comparison, control groups and commercial IrO2 catalysts toward the OER were tested under the same conditions of a standard three-electrode measuring system in 1 M KOH solution at room temperature. Control groups were also prepared (Fig. S18–S20†). As revealed by the linear sweep voltammetry (LSV) polarization curves with iR correction (Fig. 4a), CeO2-OA shows very limited OER activity, and CeO2@NiFe-MOFs are testified to have the best activity toward the OER with the lowest overpotential of 248 mV at a current density of 20 mA cm−2, and outperform control groups at the same current density. The detailed comparison of the overpotentials of these samples is presented in Fig. 4b, and the electrocatalytic activity follows this order: CeO2@NiFe-MOFs (248 mV) > CeO2/NiFe-MOFs (280 mV) > NiFe-MOFs (282 mV) > IrO2 (297 mV) > CeO2@Ni-MOFs (352 mV). In addition, OER catalytic activity of CeO2@NiFe-MOFs was also measured at various temperatures to investigate the impact of temperature on catalytic activity (Fig. S21†). According to the experimental results, electrocatalytic activity was positively correlated with temperature, implying feasible application of CeO2@NiFe-MOFs in alkaline solution at various temperatures. Tafel plots obtained from LSV curves were further used to evaluate the OER kinetics of the prepared samples (Fig. 4c). As expected, the Tafel slope of CeO2@NiFe-MOFs is lower than that of the other control groups, indicating that NiFe-MOFs with coordination of CeO2 shows better OER kinetics. Benefiting from the unique electronic configurations of cerium, the coordination of CeO2 may optimize the electronic structure and spatial arrangements of materials.47 In addition, NiFe-MOFs are nonuniformly distributed on the surface of CeO2 nanosheets, which can expose more active sites and enhance catalytic performance. Notably, the summary and comparison of CeO2@NiFe-MOFs and different materials are listed in Table S1.† Comparatively, CeO2@NiFe-MOF heterostructures in this work have relatively better performance and stability.
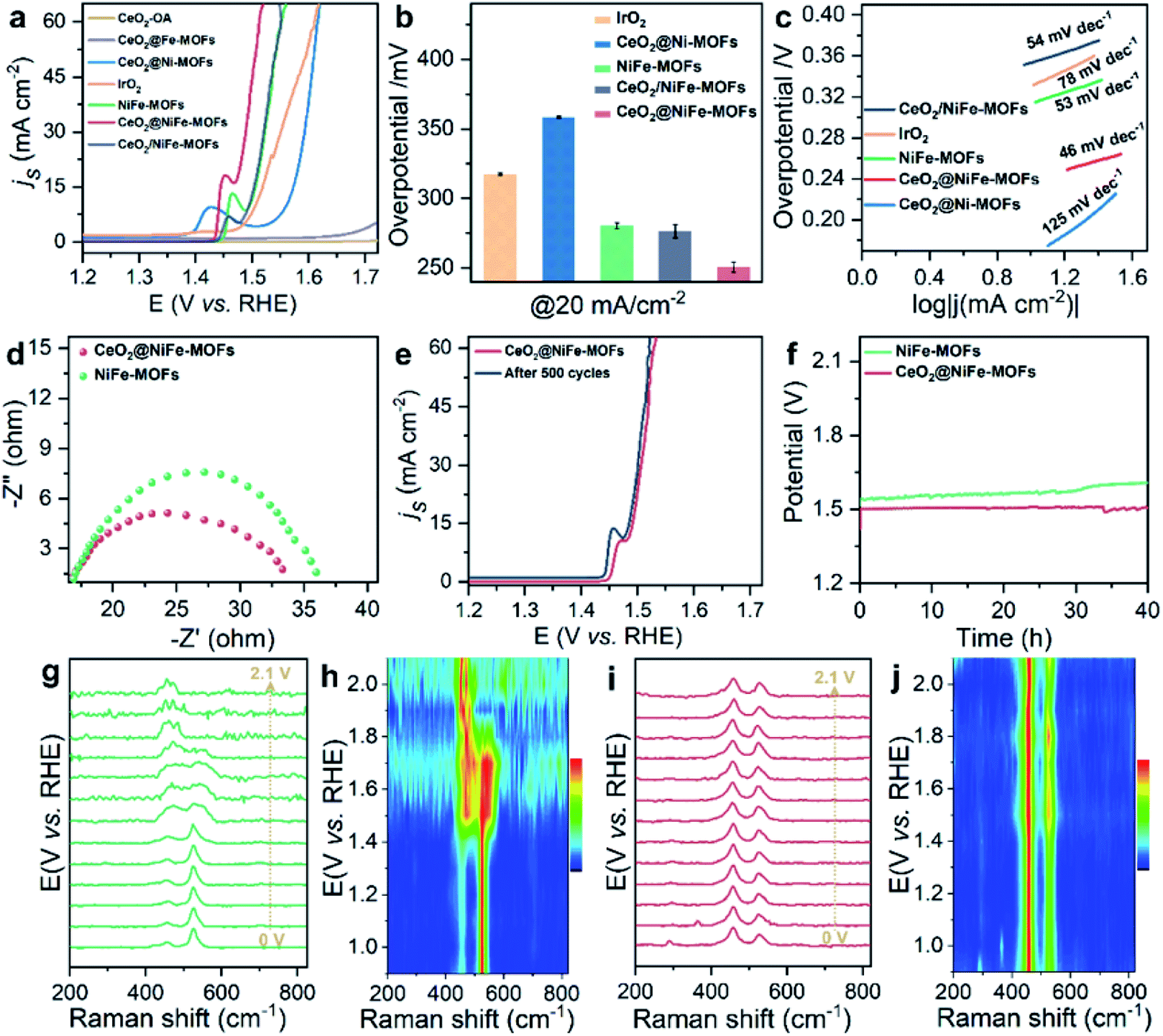 | ||
Fig. 4 (a) OER polarization curves for different electrocatalysts in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) M KOH solution. (b) The corresponding overpotentials to drive 20 mA cm−2. (c) Corresponding Tafel plots. (d) Nyquist plots of NiFe-MOFs and CeO2@NiFe-MOFs recorded at 1.53 V versus RHE. (e) Polarization curves of CeO2@NiFe-MOFs before and after 500 cycles. (f) Chronopotentiometry curves of the NiFe-MOFs and CeO2@NiFe-MOFs at 20 mA cm−2. In situ electrochemical Raman spectra of (g) NiFe-MOFs and (i) CeO2@NiFe-MOFs under different applied potentials. (h and j) Corresponding contour plots of NiFe-MOFs and CeO2@NiFe-MOFs. M KOH solution. (b) The corresponding overpotentials to drive 20 mA cm−2. (c) Corresponding Tafel plots. (d) Nyquist plots of NiFe-MOFs and CeO2@NiFe-MOFs recorded at 1.53 V versus RHE. (e) Polarization curves of CeO2@NiFe-MOFs before and after 500 cycles. (f) Chronopotentiometry curves of the NiFe-MOFs and CeO2@NiFe-MOFs at 20 mA cm−2. In situ electrochemical Raman spectra of (g) NiFe-MOFs and (i) CeO2@NiFe-MOFs under different applied potentials. (h and j) Corresponding contour plots of NiFe-MOFs and CeO2@NiFe-MOFs. | ||
Electrochemical impedance spectroscopy (EIS) was carried out to investigate the charge transfer resistance (Fig. 4d) for CeO2@NiFe-MOFs, and the smaller semi-circular diameter of Nyquist plots indicated that the coordination of CeO2 effectively enhanced the electron transfer rate of NiFe-MOFs, implying that there was charge transfer between CeO2 and MOFs. Long-term electrocatalytic stability is an important challenge for electrocatalysts under alkaline conditions, and especially some unsupported MOF electrocatalysts are prone to degeneration.4 Therefore, it is essential to survey the stability to shed light on the importance of CeO2 coordination. As seen in Fig. 4e, the activity of CeO2@NiFe-MOFs shows almost no decay after 500 cycles. As shown in Fig. 4f, the stability of NiFe-MOFs and CeO2@NiFe-MOFs was evaluated by chronopotentiometric measurement at J = 20 mA cm−2. Obviously, the required potential of CeO2@NiFe-MOFs for 40 h shows almost no change, whereas the potential applied on NiFe-MOFs increases over 40 h. The morphology and structure of CeO2@NiFe-MOFs were again characterized after the stability test, and they were found to roughly preserve the initial morphology and structure (Fig. S22 and S23†). The above results reveal enhanced catalytic activity and structural durability of CeO2@NiFe-MOFs toward the OER. CeO2/NiFe-MOFs, which is an electrostatic composite material, show that CeO2 almost does not improve OER activity. Electrostatically associated materials have moderate binding strengths, and are unstable in solvents, while the coordinated linked conjugates have robust and strong bonding.31,48 Therefore, this further explains that the coordination of CeO2 can promote OER catalytic performance (Fig. S24–S26†).
To further explore electrochemical behavior of the obtained samples, in situ electrochemical Raman spectroscopy measurements were conducted to capture structural evolution under OER conditions. The potential-dependent spectral traces of samples were measured (1 M KOH). Fig. S27a† shows Raman spectra of the O–Ce–O vibration of CeO2-OA at approximately 446 cm−1 in electrolyte, which is down-shifted compared to that of dry CeO2-OA (457 cm−1, Fig. 3h) and the CeO2 fluorite crystal (460 cm−1).49 The Raman signals of CeO2-BDC emerge at approximately 217, 350, 440 and 462 cm−1 (Fig. S27c†), and the Raman signal at 217 cm−1 was previously ascribed to a functionalization related to phonon mode.50 The above results suggest lattice distortions and creation of a fluorite crystal unit of CeO2 indicating that CeO2-OA and CeO2-BDC present excellent stability with increasing applied potential. As for NiFe-MOFs, two main Raman peaks at 456 and 525 cm−1 are correlated with the bending and stretching vibration modes of Ni–O in NiOOH, respectively. NiOOH, acting as potential active sites in electrocatalysis, significantly decreases with increasing applied potential, and the Raman peak at 525 cm−1 disappears at 1.9 V vs. RHE (Fig. 4g and h). As expected, the intensity of Raman peaks centered at 458 and 524 cm−1, originating from CeO2@NiFe-MOFs, changes a little upon the potential increasing from 1.0 to 2.1 V vs. RHE (Fig. 4i and j) demonstrating that materials with CeO2 will give better mechanistic functionalities.
We propose the following electron transfer paths responsible for the observed enhancements in performance over CeO2@NiFe-MOF electrode under the OER conditions (Fig. 5). Monolayer NiFe-MOFs are coordinated to both the top and bottom surface of CeO2 nanosheets via joining carboxylic acid groups, and thus the π-conjugated structure in carboxylic acid groups will facilitate electron transfer from potential NiOOH active species to the CeO2 substrate. Therefore, generating NiIII/IV active species under the process of electrocatalysis then extracting electrons from adsorbate OH* and accomplishing desorption of the formed O2 (Fig. S28†) enhance the OER process, which is testified by the clearly observed enhancement in performance (Fig. 4a). In addition, CeO2 easily undergoes reversible charge transfer between Ce4+ and Ce3+ oxidation stages, which is advantageous for facilitating OER catalysis. On the other hand, oxygen vacancies on CeO2 functioning as effective electron trappers can capture electrons, and this is beneficial for the extraction of electrons from adsorbate OH* and promoting the electrocatalytic reaction. According to previous reports, CeO2[111] has been recognized to present the most stable surface, and CeO2[110] can very easily generate oxygen vacancies.51,52 Nevertheless, CeO2 nanosheets present the most stable surface of CeO2[111] in CeO2@NiFe-MOFs (Fig. S10c†); meanwhile, no typical peak of defect-induced mode at around 592 cm−1 was observed in the Raman spectra (Fig. 3h and 4i), which was attributed to the fact that carboxylic acid functional groups can heal the oxygen vacancies of CeO2. The result indicates that intrinsic properties of CeO2 rather than oxygen vacancies contribute to catalytic activity.
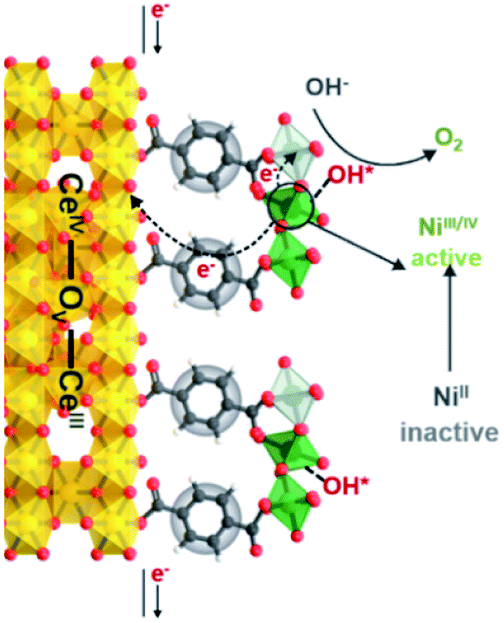 | ||
| Fig. 5 Schematic illustration of possible electron transfer paths occurring in the CeO2@NiFe-MOF heterostructures under electrocatalysis processes for the OER catalysis. | ||
The XPS spectra further confirm electronic behavior of CeO2@NiFe-MOFs before and after OER testing (Fig. 6a–c and S29†). Compared with the original CeO2@NiFe-MOFs, Ni 2p3/2 and 2p1/2 peaks of CeO2@NiFe-MOFs after OER show apparent shifts toward the higher binding energies by 0.4 eV and 0.2 eV, respectively (Fig. 6a) and Fe 2p peaks show shifts toward the lower binding energies by 0.5 eV (Fig. 6b), indicating the reduction of Fe to a lower valence state. This result suggests that there is partial electron transfer from Ni to Fe sites via the bridging O atoms.38 The intensity of the Ce3+ related peak at 903.8 eV also increases (Fig. 6c), demonstrating that the charge transfer occurs from NiOOH active species to the CeO2 substrate. In addition, the NiII/NiIII ratio on the surface of CeO2@NiFe-MOFs after OER increases, and likewise, the consistent reduction of the valence state is also observed from the negative shift in the corresponding electron energy loss spectra (Fig. 6d–f and S30†), indicating the reduction of the valence state to a lower valence state after OER testing. Therefore, the high valence state Ce4+ as electron-accepting sites on the pristine CeO2@NiFe-MOF surface significantly promotes intrinsic electron transfer from NiOOH to CeO2 through π-donation.53 Electron conjugation can improve electron communication between them,54 making a greater amount of coordinated NiOOH electrochemically active and then facilitating the generation of NiIII/IV active species in the CeO2@NiFe-MOF electrode during catalysis.43 This could also be clearly evaluated by EIS analysis. Simultaneously, the oxygen storage capacity generated from the fast reversible variation between Ce3+ and Ce4+ and the surface defect-rich structure of ultrathin CeO2 serves as an oxygen buffer to enhance wettability and release stress from a drastic reaction.55 Though CeO2 is not a good OER catalyst, these results clearly demonstrate that CeO2 could essentially improve activity and stability of electrocatalysis by coordination with particular 2D MOF materials.56 In comparison with other uniformly structured counterparts, 2D heterostructures exhibit greater stability and provide more efficient charge transfer.57,58
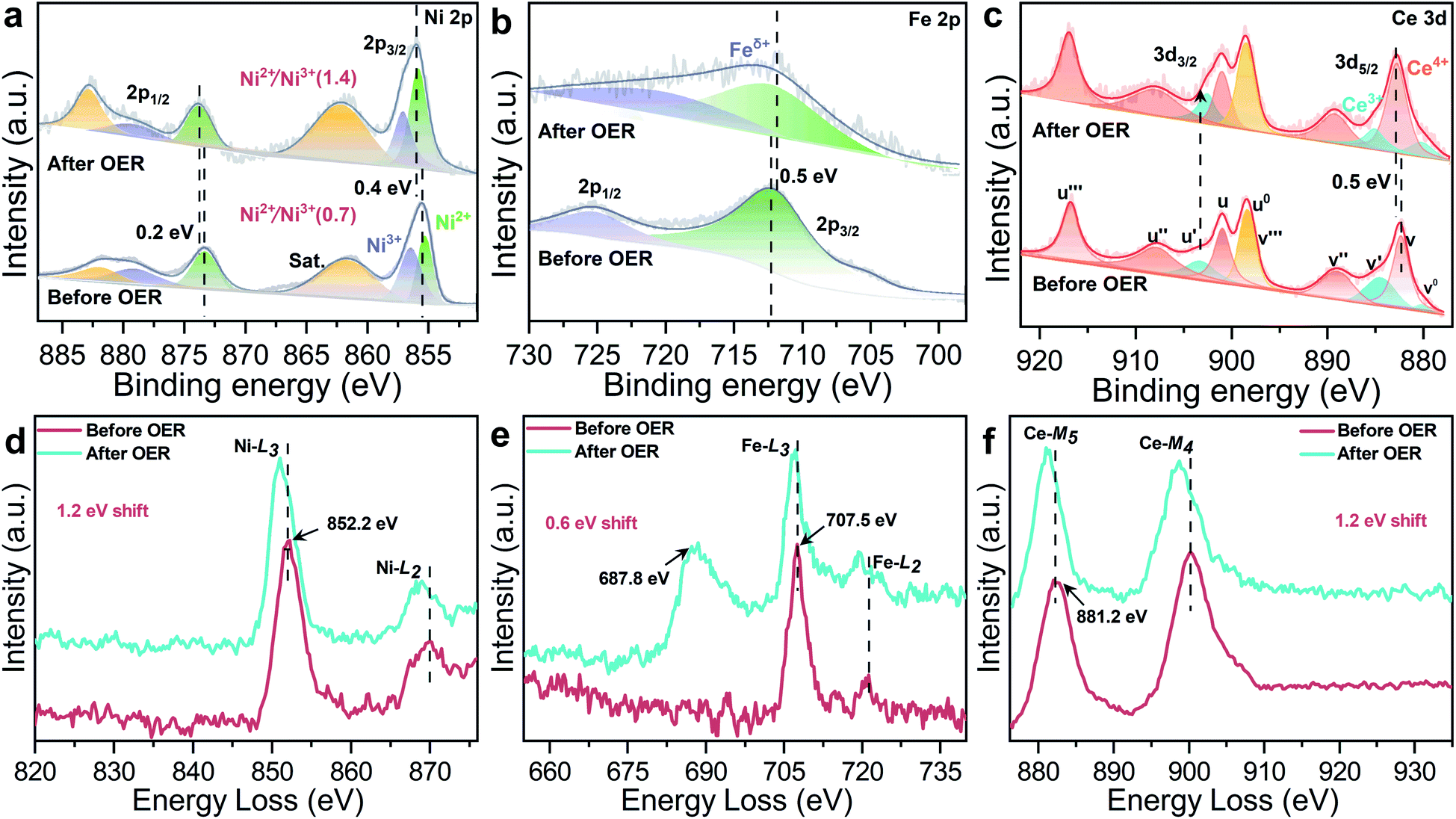 | ||
| Fig. 6 High-resolution XPS spectra of CeO2@NiFe-MOFs before and after the OER: (a) Ni 2p, (b) Fe 2p, and (c) Ce 3d. (d–f) The corresponding EELS spectra of CeO2@NiFe-MOFs before and after the OER: (d) Ni-L, (e) Fe-L, the peak at 687.8 eV is attributed to F-K (Fig. S31†), and (f) Ce-M. | ||
The coordinated CeO2@NiFe-MOF heterostructures as a structural model were constructed, and their electrocatalytic performance was examined by using the OER as a model system. In the designed 2D heterostructures with a sandwich structure, CeO2 not only assisted the coordinately supported and confined MOFs to easily expose more accessible active sites, but also provided a surface defect-rich and unique electronic structure in favor of the catalytic reaction. Meanwhile, the organic–inorganic hybrid heterostructures provide sufficient surface area for the catalytic reaction. The structure and catalytic performance, as well as the key electron transfer in 2D heterostructures were well proved by the characterization studies. Therefore the clear coordination environment of the 2D heterostructures is beneficial to establish the catalytic process and elucidate the catalytic mechanism in the heterostructures. Besides, the clear coordinated combination is not only structurally suitable for 2D CeO2-supported MOFs, but also can be extended to a general strategy for the combination of 2D CeO2 and other molecular layers or precursors, where the interactions between metal oxides and molecular layer require a deeper understanding.59 As we developed this strategy to construct the integrated structure as a superior model system, researchers can be inspired to regulate other hybrid structures for the improvement of catalytic performance by considering the role of CeO2 coordination. Therefore, the hybrid materials of coordinated combination possess superiority as a model system, and high activity of 2D MOF-based or CeO2-based heterostructures can be expected by adjusting interactions between the constituents.
Conclusions
In this work, by employing the means of coordination, we first construct ultrathin 2D CeO2@NiFe-MOF heterostructures. It proves the multiple effects of metal-coordinated interactions in CeO2@NiFe-MOF heterostructures. CeO2 nanosheets decorated with 2D NiFe-MOFs present different electronic and optical properties, as well as local strain. On the other hand, 2D NiFe-MOFs on the surface of the CeO2 nanosheet substrate not only present an excellent OER catalytic activity but also display good long-term stability, achieving a current density of 20 mA cm−2 at a low overpotential of 248 mV as well as long-term durability for at least 40 h. In addition, the ultrathin 2D CeO2 surface can also be functionalized by other organic ligands through this approach. As a consequence, an extensive class of multi-functional 2D CeO2 materials can be predictably synthesized by rationally tailoring structures of organic ligands. It is anticipated that our study will provide a reference for the improvement of the catalytic process by using coordinated heterostructures and open new avenues for the formation of novel 2D MOF composites/heterostructures or multi-functional 2D CeO2 for extensive applications.Data availability
All relevant data is presented in the paper and ESI.†Author contributions
Y. T. and L. L. contributed to valuable guidance and comments and supervised the project. H. A. performed the synthesis, characterizations and electrochemical experiments. Y. H. and Y. P. assisted with the HR-TEM testing and data analysis. The manuscript was written through contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22131007, 21871127, and 21971097), the National Key R&D Program of China (2021YFA1501101), Science and Technological Plan of Gansu Province (20YF3GA012) and the 111 Project (B20027).References
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 4998 CrossRef PubMed.
- Y. Liang, C. Lu, D. Ding, M. Zhao, D. Wang, C. Hu, J. Qiu, G. Xie and Z. Tang, Chem. Sci., 2015, 6, 4103–4108 RSC.
- F. Li, J. Du, X. Li, J. Shen, Y. Wang, Y. Zhu and L. Sun, Adv. Energy Mater., 2018, 8, 1702598 CrossRef.
- J. Du, F. Li and L. Sun, Chem. Soc. Rev., 2021, 50, 2663–2695 RSC.
- Y. Wu, Y. Li, J. Gao and Q. Zhang, SusMat., 2021, 1, 66–87 CrossRef.
- C. Li, K. Wang, J. Li and Q. Zhang, ACS Mater. Lett., 2020, 2, 779–797 CrossRef CAS.
- S. Hou, W. Li, S. Watzele, R. M. Kluge, S. Xue, S. Yin, X. Jiang, M. Döblinger, A. Welle, B. Garlyyev, M. Koch, P. Müller-Buschbaum, C. Wöll, A. S. Bandarenka and R. A. Fischer, Adv. Mater., 2021, 33, 2103218 CrossRef CAS PubMed.
- S. Ahmad, J. Liu, C. Gong, J. Zhao and L. Sun, ACS Appl. Energy Mater., 2018, 1, 249–253 CrossRef CAS.
- S. Zhao, Y. Wang, J. Dong, C.-T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- F. Li, L. Bai, H. Li, Y. Wang, F. Yu and L. Sun, Chem. Commun., 2016, 52, 5753–5756 RSC.
- Y. Li, L. Lin, M. Tu, P. Nian, A. J. Howarth, O. K. Farha, J. Qiu and X. Zhang, Nano Res., 2018, 11, 1850–1860 CrossRef CAS.
- P.-Z. Li, Y. Maeda and Q. Xu, Chem. Commun., 2011, 47, 8436–8438 RSC.
- Y. Xu, B. Li, S. Zheng, P. Wu, J. Zhan, H. Xue, Q. Xu and H. Pang, J. Mater. Chem. A, 2018, 6, 22070–22076 RSC.
- F.-L. Li, P. Wang, X. Huang, D. J. Young, H.-F. Wang, P. Braunstein and J.-P. Lang, Angew. Chem., Int. Ed., 2019, 58, 7051–7056 CrossRef CAS PubMed.
- M. Zhao, Y. Huang, Y. Peng, Z. Huang, Q. Ma and H. Zhang, Chem. Soc. Rev., 2018, 47, 6267–6295 RSC.
- H. Yin and Z. Tang, Chem. Soc. Rev., 2016, 45, 4873–4891 RSC.
- H. Tang, J. Wang, H. Yin, H. Zhao, D. Wang and Z. Tang, Adv. Mater., 2015, 27, 1117–1123 CrossRef CAS PubMed.
- Y. Cheng, X. Wang, C. Jia, Y. Wang, L. Zhai, Q. Wang and D. Zhao, J. Membr. Sci., 2017, 539, 213–223 CrossRef CAS.
- N. Campagnol, T. R. C. Van Assche, M. Li, L. Stappers, M. Dincă, J. F. M. Denayer, K. Binnemans, D. E. De Vos and J. Fransaer, J. Mater. Chem. A, 2016, 4, 3914–3925 RSC.
- W. Li, S. Watzele, H. A. El-Sayed, Y. Liang, G. Kieslich, A. S. Bandarenka, K. Rodewald, B. Rieger and R. A. Fischer, J. Am. Chem. Soc., 2019, 141, 5926–5933 CrossRef CAS PubMed.
- S. Roy, Z. Huang, A. Bhunia, A. Castner, A. K. Gupta, X. Zou and S. Ott, J. Am. Chem. Soc., 2019, 141, 15942–15950 CrossRef CAS PubMed.
- J. Liu and C. Wöll, Chem. Soc. Rev., 2017, 46, 5730–5770 RSC.
- Z. Tang, Chem.–Asian J., 2020, 15, 2240 CrossRef CAS PubMed.
- J. Xia, H. Zhao, B. Huang, L. Xu, M. Luo, J. Wang, F. Luo, Y. Du and C.-H. Yan, Adv. Funct. Mater., 2020, 30, 1908367 CrossRef CAS.
- K. Obata and K. Takanabe, Angew. Chem., Int. Ed., 2018, 57, 1616–1620 CrossRef CAS PubMed.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- Y. Li, X. Zhang and Z. Zheng, CCS Chem., 2021, 3, 2657–2679 Search PubMed.
- B. Ma, M. Blanco, L. Calvillo, L. Chen, G. Chen, T.-C. Lau, G. Dražić, J. Bonin, M. Robert and G. Granozzi, J. Am. Chem. Soc., 2021, 143, 8414–8425 CrossRef CAS PubMed.
- R. Liu, K. Wu, L.-D. Li, L.-D. Sun and C.-H. Yan, Inorg. Chem. Front., 2017, 4, 1182–1186 RSC.
- H. Zhang, J. Jang, W. Liu and D. V. Talapin, ACS Nano, 2014, 8, 7359–7369 CrossRef CAS PubMed.
- R. Canton-Vitoria, T. Scharl, A. Stergiou, A. Cadranel, R. Arenal, D. M. Guldi and N. Tagmatarchis, Angew. Chem., Int. Ed., 2020, 59, 3976–3981 CrossRef CAS PubMed.
- R. Ramachandran, W. Xuan, C. Zhao, X. Leng, D. Sun, D. Luo and F. Wang, RSC Adv., 2018, 8, 3462–3469 RSC.
- Z.-P. Wu, H. Zhang, S. Zuo, Y. Wang, S. L. Zhang, J. Zhang, S.-Q. Zang and X. W. Lou, Adv. Mater., 2021, 33, 2103004 CrossRef CAS PubMed.
- Z. Qiao, Y. Liang, Z. Zhang, D. Mei, Z. Wang, M. D. Guiver and C. Zhong, Adv. Mater., 2020, 32, 2002165 CrossRef CAS PubMed.
- J. Zhang, H. Wong, D. Yu, K. Kakushima and H. Iwai, AIP Adv., 2014, 4, 117117 CrossRef.
- M. C. Spadaro, S. D'Addato, G. Gasperi, F. Benedetti, P. Luches, V. Grillo, G. Bertoni and S. Valeri, Beilstein J. Nanotechnol., 2015, 6, 60–67 CrossRef PubMed.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486–1491 CrossRef CAS PubMed.
- M. Liu, K.-A. Min, B. Han and L. Y. S. Lee, Adv. Energy Mater., 2021, 11, 2101281 CrossRef CAS.
- D. R. Mullins, S. H. Overbury and D. R. Huntley, Surf. Sci., 1998, 409, 307–319 CrossRef CAS.
- J. A. Fortner and E. C. Buck, Appl. Phys. Lett., 1996, 68, 3817–3819 CrossRef CAS.
- T. Manoubi, C. Colliex and P. Rez, J. Electron Spectrosc. Relat. Phenom., 1990, 50, 1–18 CrossRef CAS.
- W.-K. Wang, M.-L. Zheng, W.-Q. Chen, F. Jin, Y.-Y. Cao, Z.-S. Zhao and X.-M. Duan, Langmuir, 2011, 27, 3249–3253 CrossRef CAS PubMed.
- G. Liu, P. Li, G. Zhao, X. Wang, J. Kong, H. Liu, H. Zhang, K. Chang, X. Meng, T. Kako and J. Ye, J. Am. Chem. Soc., 2016, 138, 9128–9136 CrossRef CAS PubMed.
- J. E. Spanier, R. D. Robinson, F. Zhang, S.-W. Chan and I. P. Herman, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 245407 CrossRef.
- Y. Li, M. Lu, Y. Wu, Q. Ji, H. Xu, J. Gao, G. Qian and Q. Zhang, J. Mater. Chem. A, 2020, 8, 18215–18219 RSC.
- C. Kuai, C. Xi, A. Hu, Y. Zhang, Z. Xu, D. Nordlund, C.-J. Sun, C. A. Cadigan, R. M. Richards, L. Li, C.-K. Dong, X.-W. Du and F. Lin, J. Am. Chem. Soc., 2021, 143, 18519–18526 CrossRef CAS PubMed.
- S. Zhang, S. E. Saji, Z. Yin, H. Zhang, Y. Du and C.-H. Yan, Adv. Mater., 2021, 33, 2005988 CrossRef CAS PubMed.
- S. Ma, X. S. Wang, D. Yuan and H. C. Zhou, Angew. Chem., Int. Ed., 2010, 120, 4198–4201 CrossRef.
- X. Tao, R. Long, D. Wu, Y. Hu, G. Qiu, Z. Qi, B. Li, R. Jiang and Y. Xiong, Small, 2020, 16, 2001782 CrossRef CAS PubMed.
- S. Jiménez Sandoval, D. Yang, R. F. Frindt and J. C. Irwin, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 3955–3962 CrossRef PubMed.
- N. Kumari, M. A. Haider, M. Agarwal, N. Sinha and S. Basu, J. Phys. Chem. C, 2016, 120, 16626–16635 CrossRef CAS.
- S. Jiang, R. Zhang, H. Liu, Y. Rao, Y. Yu, S. Chen, Q. Yue, Y. Zhang and Y. Kang, J. Am. Chem. Soc., 2020, 142, 6461–6466 CrossRef CAS PubMed.
- Y. Liu and T.-C. Lau, J. Am. Chem. Soc., 2019, 141, 3755–3766 CrossRef CAS PubMed.
- J. Wang, X. Huang, S. Xi, H. Xu and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 19162–19167 CrossRef CAS PubMed.
- T. Dai, X. Zhang, M. Sun, B. Huang, N. Zhang, P. Da, R. Yang, Z. He, W. Wang, P. Xi and C.-H. Yan, Adv. Mater., 2021, 33, 2102593 CrossRef CAS PubMed.
- X. Shi, B. Cao, J. Liu, J. Zhang and Y. Du, Adv. Mater., 2021, 17, 2005371 CAS.
- H. Yan, Y. Xie, Y. Jiao, A. Wu, C. Tian, X. Zhang, L. Wang and H. Fu, Adv. Mater., 2018, 30, 1704156 CrossRef PubMed.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS PubMed.
- V. Montes-García and P. Samorì, Chem. Sci., 2022, 13, 315–328 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Optical photographs, FT-IR spectra, water contact angles, AFM images, TG analysis, additional PXRD patterns, TEM and HR-TEM images, Raman spectroscopy, XPS spectra, and EELS spectra. See DOI: 10.1039/d2sc00308b |
| This journal is © The Royal Society of Chemistry 2022 |