
Insight into the mechanism of the arylation of arenes via norbornene relay palladation through meta- to para-selectivity†
Shengnan
Liu
,
Qiong
Wang
,
Fang
Huang
 ,
Wenjuan
Wang
,
Chong
Yang
,
Jianbiao
Liu
* and
Dezhan
Chen
*
,
Wenjuan
Wang
,
Chong
Yang
,
Jianbiao
Liu
* and
Dezhan
Chen
*
College of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Functionalized Probes for Chemical Imaging in Universities of Shandong, Institute of Molecular and Nano Science, Shandong Normal University, Jinan 250014, P. R. China. E-mail: jianbiaoliu.thu@sdnu.edu.cn; chendzh@sdnu.edu.cn
First published on 9th November 2021
Abstract
A novel mechanism of the arylation of arenes via norbornene (NBE) relay palladation through meta- to para-selectivity was revealed via density functional theory (DFT) calculations. Our calculated results revealed that the reaction was initiated by a [mono-N-protected amino acid ligand (MPAA)–Pd] complex to activate at first the meta-C–H guided by the directing group (DG), and para-arylation was subsequently achieved by NBE relay palladation from meta- to para-position. Significantly, the palladium/norbornene (Pd/NBE) cooperative catalysis was catalyzed by a Pd–Ag bimetallic complex, which accounted for the experimental fact that no yield detected without Ag. The reaction pathway through para- to meta-selectivity was also investigated, while this pathway was kinetically unfavorable. The results revealed that the initial DG guided C–H site activation was the rate-determining step and played an important role in determining site-selectivity. The primary meta-activation was favorable in energy due to the less ring strain in the cyclic nitrile-coordinated C–H transition states in the meta position. Moreover, the perfect cooperation of a remote directing template and a transient mediator NBE through the alternating association with the Pd center achieved the relay through meta- to para-position. The present results provide a reasonable insight into the para-C–H arylation by the Pd/MPAA/NBE cooperative catalysis in conjunction with a precise DG and Ag(I) additive.
1. Introduction
Undoubtedly, practical and site-selective arene functionalization plays an important role in pharmaceutical, agrichemical and materials research.1 However, the precise site-selective C–H functionalization of aromatic compounds is a fundamental challenge because of the inherently similar electronic and steric properties of the C–H bonds. To achieve the C–H bond functionalization of aromatic compounds at a desired position, directing group (DG) assistance has become an effective strategy.2 This strategy is widely employed for proximal ortho-C–H activation (Scheme 1a),3 but the selective functionalization of remote meta- and para-position is still underdeveloped. Although a few elegant approaches were reported to expand the scope of arene meta-C–H functionalizations (Scheme 1b),4 in contrast, studies of para-selective C–H functionalization5 are significantly limited. Recently, several protocols of para-selective C–H functionalizations were developed by the groups of Itami,6 Chattopadhyay,7 Hiyama8 and others,9 but these protocols were successful only for borylation, olefination,10 alkylation,11 acetoxylation,12 ketonisation13 and cyanation14 reactions. It is urgently needed to develop an alternate method to diversify the scope of para-functionalizations, particularly arylation, because biaryls are basic building blocks of pharmaceuticals and agricultural chemicals.15 Therefore, it is important to selectively introduce an aromatic ring at the para position, which will promote the development of the diaryl synthesis.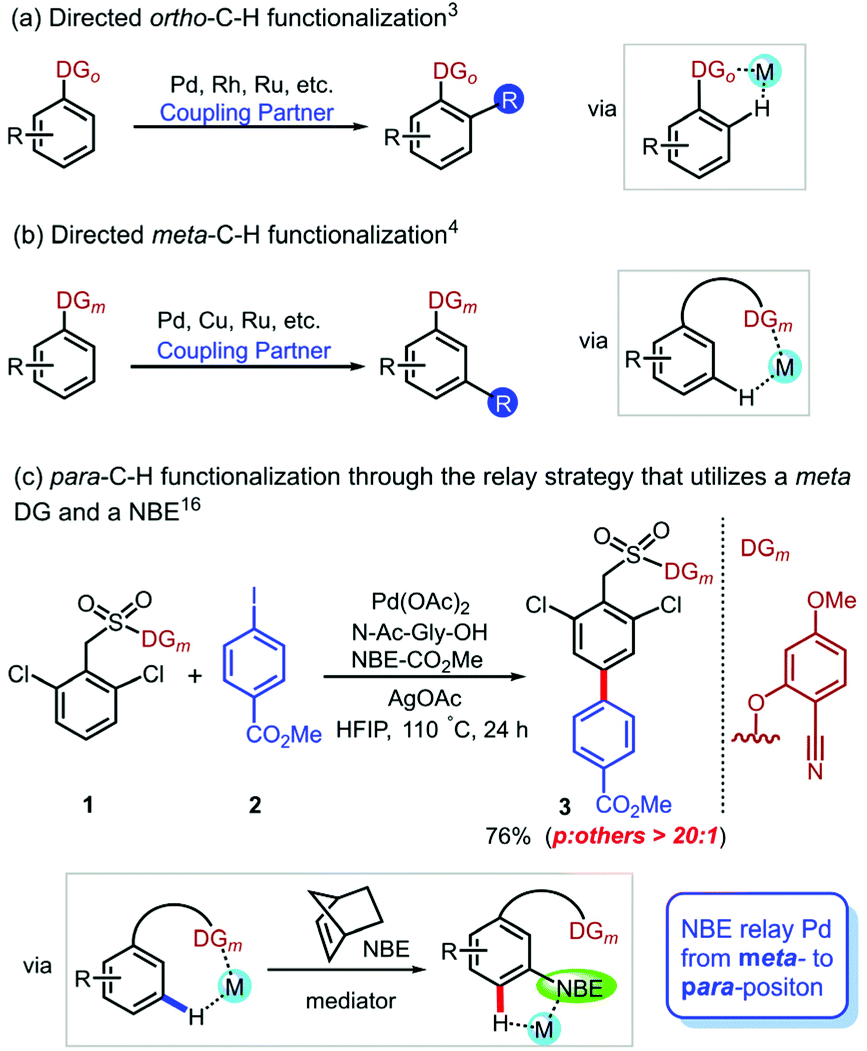 | ||
| Scheme 1 Site-selective C–H functionalization. | ||
In 2020, Maiti et al. developed a para-selective C–H arylation method by a unique combination of a meta-DG and NBE as a transient mediator (Scheme 1c).16 This strategy first implements meta-C–H activation via DG guidance; subsequently, the transient mediator NBE relays Pd to the para-position to achieve precise site-selectivity. The effective ligand was N-acetylglycine (Ac-Gly-OH), which is a mono-N-protected amino acid (MPAA).17 However, the mechanism of how to utilize a transient mediator to combine the remote directing effect with a ligand and palladium relay is still ambiguous. Moreover, the experimental observations showed that Ag(I) was a crucial additive, and the reaction could not proceed without Ag(I).16 How does Ag(I) play an important role in certain Pd-catalyzed C–H functionalization reactions? Investigation into this subject has been scarce and limited to only a few theoretical studies that proposed heterodimeric transition states of C–H activation.18 The mechanism of this novel strategy achieving precise para-C–H arylation and the role of additive, such as AgOAc, are still unknown. Herein, we reported density functional theory (DFT) calculations to explore the mechanism of NBE relay palladation and origins of the site selectivity.
2. Computational methods
All geometry optimizations and single point energy calculations were performed using Gaussian 09.19 The intermediates and transition states were optimized at the B3LYP level20 of DFT. The basis set of SDD21 was employed for Pd, Ag, I and the 6-31G (d,p)22 for other atoms. Vibrational frequencies were calculated to confirm if each optimized structure is a local minimum or a transition state structure. All optimized structures were calculated for single point energy using M06 functional23 and mixed basis set of [6-311++G(d,p)+SDD] at the hexafluoro-2-propanol solvent with parameters “solvent = generic, eps = 16.7, epsinf = 1.63” to simulate by the SMD solvation model.24 In a previous study of Pd-catalyzed C–H activation, the similar level of method was reported to have demonstrated satisfactory performance.18,25The NBO 6.026 program was used to perform the Natural Bond Orbital analysis using the wavefunction obtained from the B3LYP level. The 3D structures were prepared using CYLView.27
3. Results and discussion
We selected the arylation of arene reaction as a representative system to explore the mechanism of the site-selective C–H activation under Pd/MPAA/NBE cooperative catalysis, as shown in Scheme 1c. Based on our calculations and the experimental results, we designed two reaction pathway scheme (Scheme 2) of NBE relay palladation through meta- to para-position and para- to meta-position, respectively. For the pathway of the para-C–H-arylated product pp (left cycle),16 the overall catalytic cycle mainly included the primary meta-C–H activation, NBE insertion, secondary para-C–H activation, oxidative addition and reductive elimination, and then, NBE extrusion and protodemetallation. NBE as a transient mediator achieves the expected palladium relay via migratory insertion. The pathway of meta-arylation pm was derived from the primary para-C–H activation and secondary meta-C–H arylation (right cycle). We also explored the possible side reactions that the active aryl-palladium intermediates are direct meta-functionalized (A to F) and reductive eliminated (cyclobutane G formation from C and D).28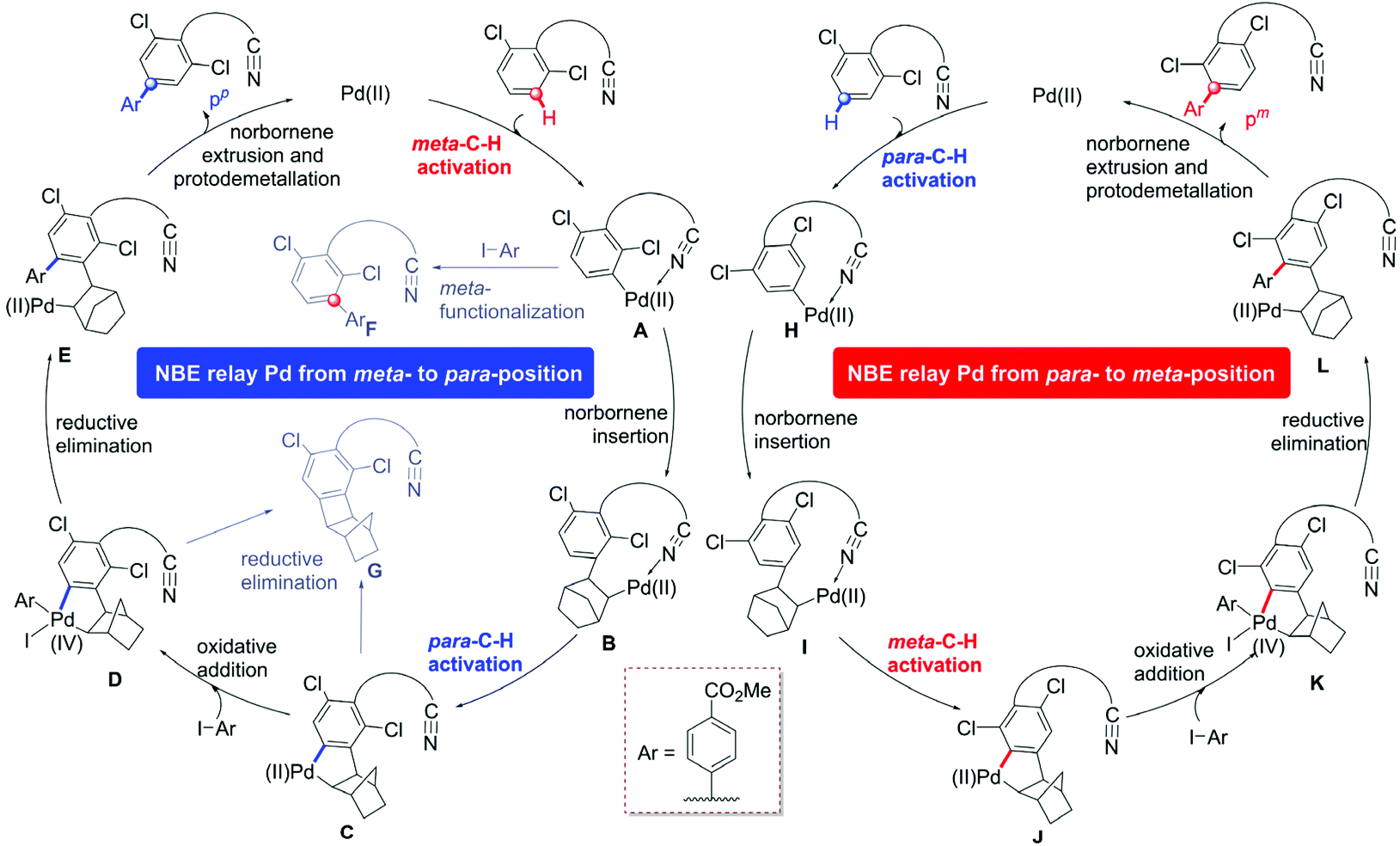 | ||
| Scheme 2 Proposed mechanism for para-C–H arylation (left cycle) and meta-C–H arylation (right cycle) catalyzed by the Pd/MPAA. | ||
3.1 Energy profiles for the NBE relay palladation through meta- to para-position
Fig. 1 depicts the formation of the active catalyst and the CMD pathway of primary C–H activation. Mono-deprotonation of MPAA by one of the acetates leads to monodentate Pd–MPAA complex int0, and then further deprotonation of the MPAA N–H bond by another acetate is highly favored,30 forming a bidentate Pd–MPAA complex int1 coordinated with two neutral acetic acid molecules. Para-Arylation and meta-arylation products are derived from the primary meta-C–H activation and primary para-C–H activation, respectively. We next investigated ortho-, meta- and para-C–H activation steps. The calculation results indicated that the energy barrier for the meta-C–H activation of TS1m was lower than that of the ortho-TS1o and para-TS1p.
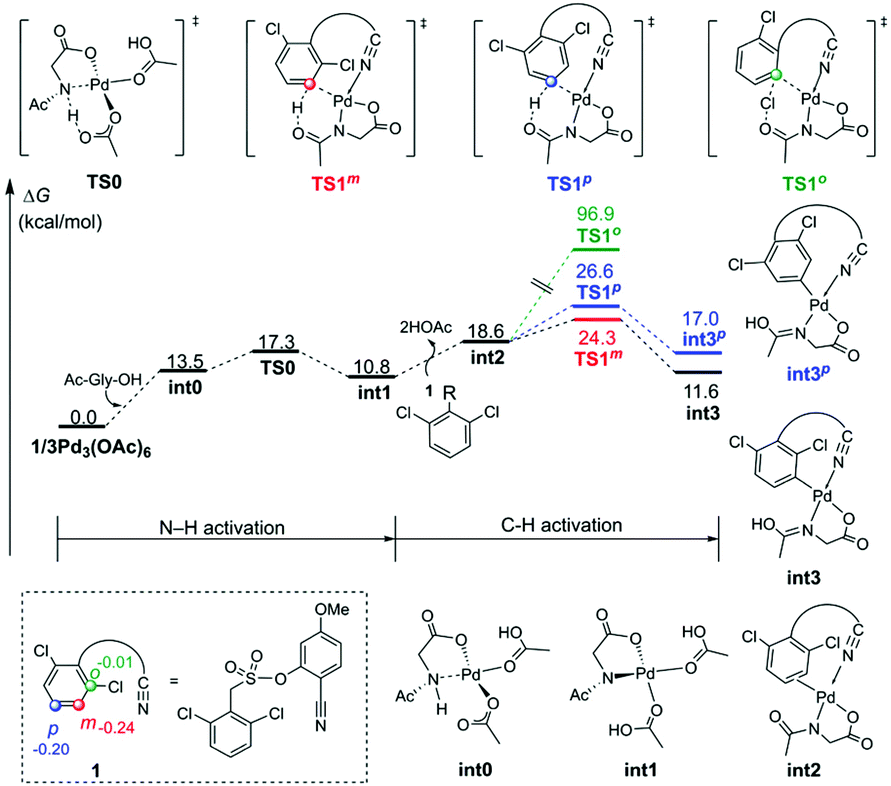 | ||
| Fig. 1 Free energy profiles for the primary C–H activation steps. The selected NPA atomic charges are given in numbers in substrate 1. | ||
The MPAA ligand promoted the primary C–H activation step, and then provided a vacant coordination site for subsequent NBE insertion.35 As shown in Fig. 2, int3 isomerizes to the more stable int4, allowing the competitive direct meta-arylation and NBE insertion. The energy barrier of the direct oxidation addition (TS2) is higher than that of NBE insertion (TS3 and TS4), so the direct meta-functionalization is overridden by the faster NBE coordination to allow the expected Pd relay. Moreover, the MPAA ligand is more beneficial to the stability of the transition state (TS3) than acetate (TS4). In addition, other three kinetically unfavorable pathways of the NBE insertion are given in Fig. S3.†
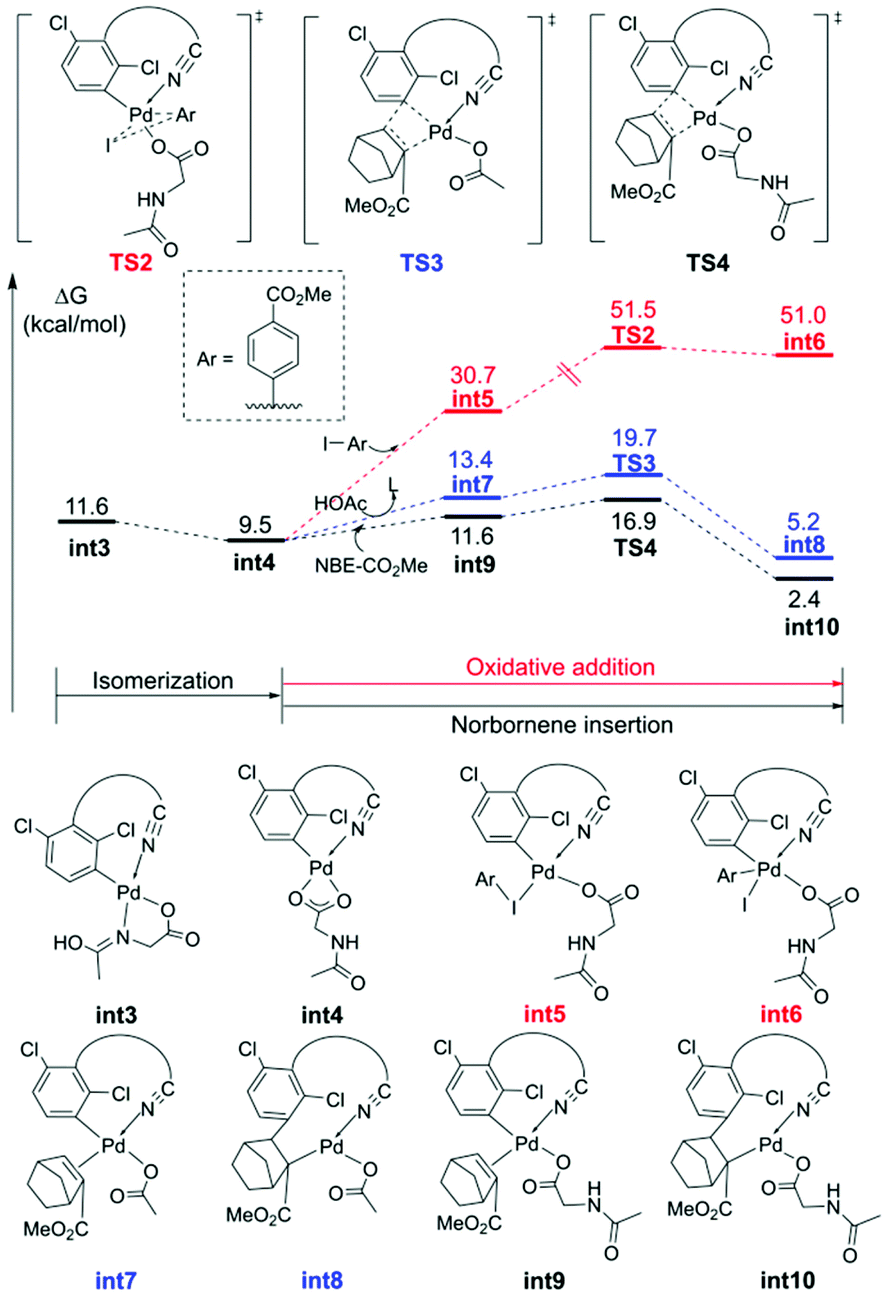 | ||
| Fig. 2 Free energy profiles for the NBE insertion. | ||
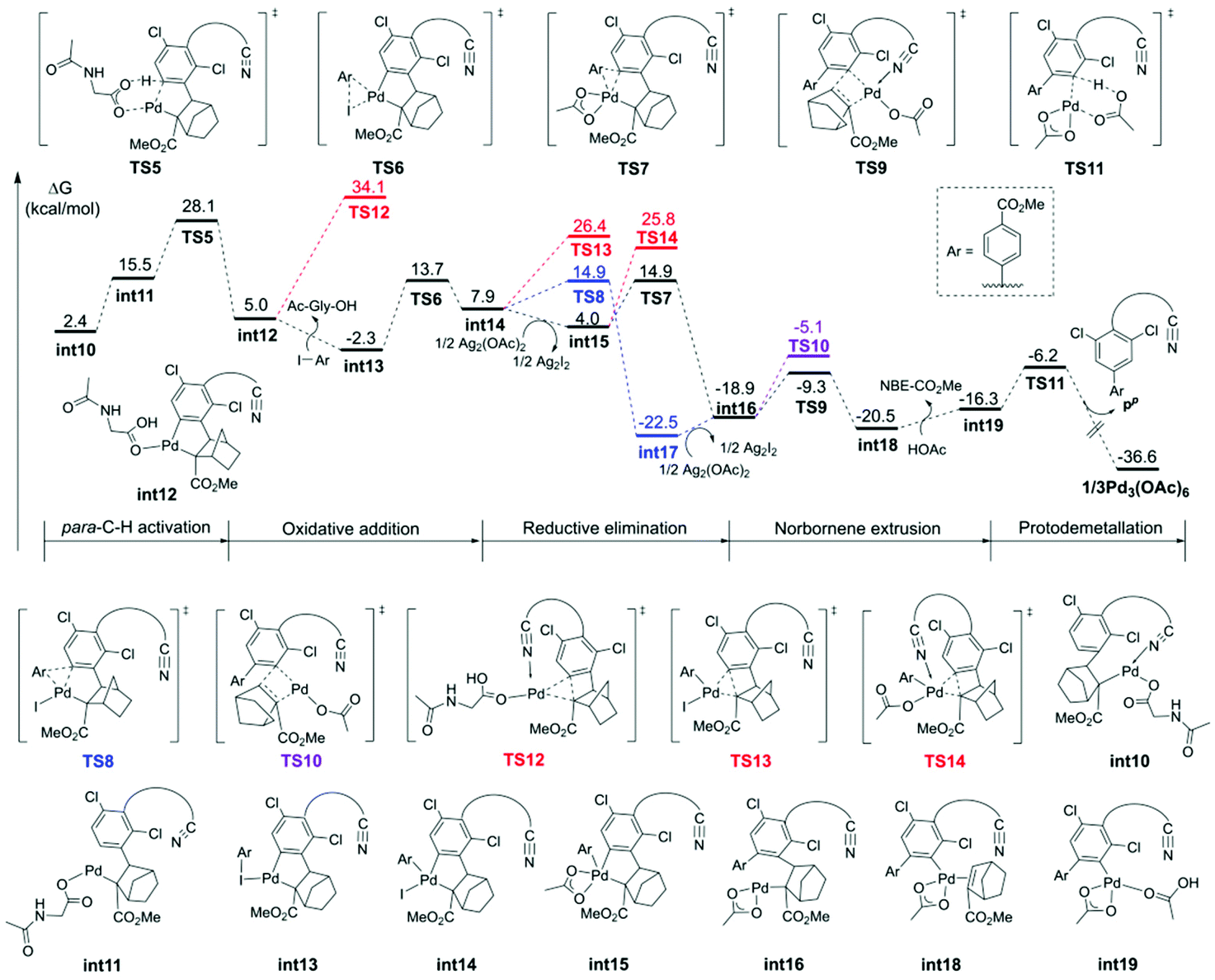 | ||
| Fig. 3 Free energy profiles for the para-C–H arylation catalyzed by the monomeric Pd complex. | ||
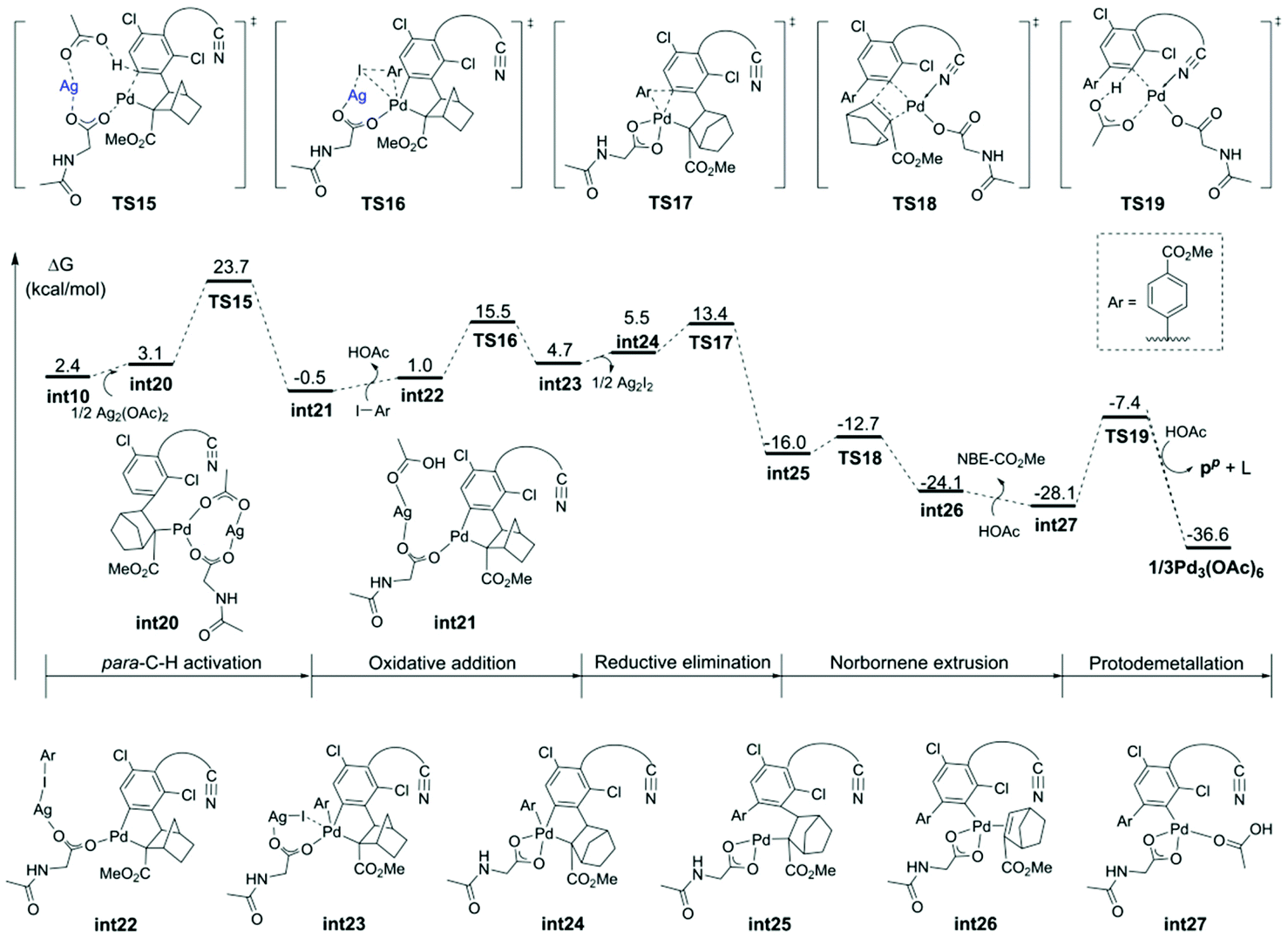 | ||
| Fig. 4 Free energy profiles for the para-C–H arylation catalyzed by the Pd–Ag bimetallic complex. | ||
The calculated free energy barrier of the Pd–Ag bimetallic complex catalyzed para-C–H activation (TS15) is lower than that of the aforementioned monomeric Pd mechanisms (TS5) by 4.4 kcal mol−1 (Fig. 5). It suggested that the Pd–Ag bimetallic complex played a significant role in stabilizing TS15 in a secondary para-C–H activation. We also explored the effect of the tether length on regioselectivity differences. We used an acetic acid to replace silver acetate to calculate the energy barrier of the para-C–H activation step. The result showed that the energy barrier is 31.3 kcal mol−1 (TS15′), which is 10.0 kcal mol−1 higher than that of bimetallic Pd–Ag (TS15), and 5.6 kcal mol−1 higher than monomeric Pd–MPAA (TS5). This suggested that the regioselectivity difference in this reaction did not depend on the tether length. We also calculated the para-C–H activation step with two silver acetates to increase the tether length (Fig. S7†), and it needed to overcome a higher energy barrier (33.4 kcal mol−1). It followed that the bimetallic Pd–Ag played a significant stabilization role in the secondary para-C–H activation.
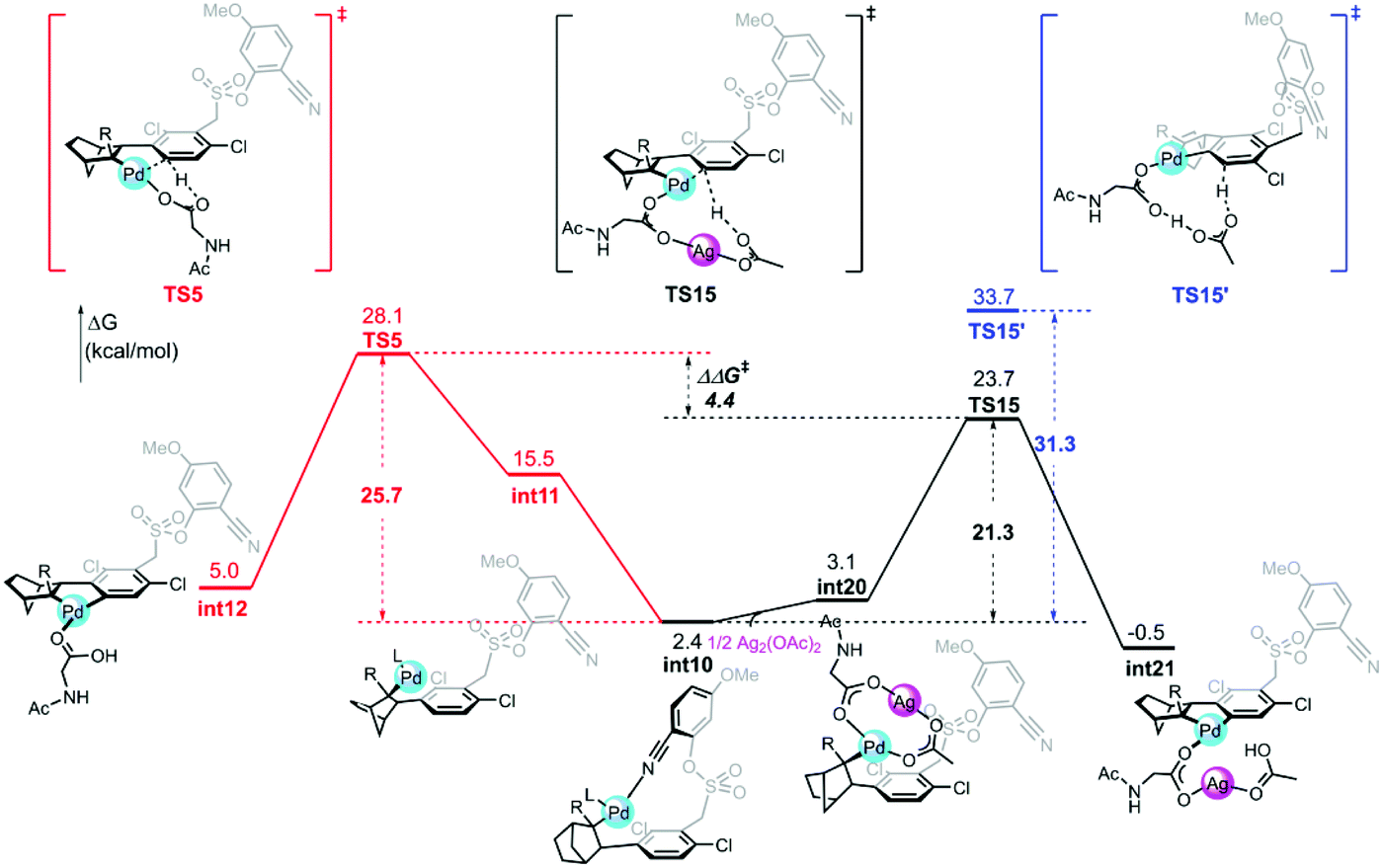 | ||
| Fig. 5 Free energy profiles for the para-C–H activation catalyzed by monomeric Pd and heterodimeric Pd–Ag. | ||
3.2 Origin of site-selectivity for NBE relay Pd through meta- to para-position
The calculated energy profiles of the NBE relay Pd through meta- to para-position to produce para-arylated product are summarized in Fig. 6a. The calculated studies revealed that the reaction was initiated by a monomeric Pd/MPAA complex to achieve a primary meta-C–H activation and a subsequent secondary para-C–H activation catalyzed by a Pd–Ag bimetallic complex (black pathway). In 2019, Yu et al.40 found that the meta-arylated product originates from the initial para- and ortho-C–H activation, followed by subsequent meta-C–H activation in the arylation of the electron-rich arene reaction. Inspired by Yu's report, we further optimized the energy profiles of NBE relay palladation through para- to meta-position (blue pathway). The mechanistic details can be seen in the ESI.† We analyzed free energy pathways for two different meta- and para-arylations based on the Curtin-Hammett scenario41 (Fig. 6b). The results indicated that the competition between the primary C–H activation transition states TS1m and TS1p controlled the site selectivity. The calculated selectivity was ΔΔG‡ = 2.3 kcal mol−1, and the ratio of pp and pm was calculated to be 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. It is in good agreement with the experimental observation of ratio 20:1.16 The present result shows that the site selectivity of the reaction was controlled by kinetics.
:1. It is in good agreement with the experimental observation of ratio 20:1.16 The present result shows that the site selectivity of the reaction was controlled by kinetics.
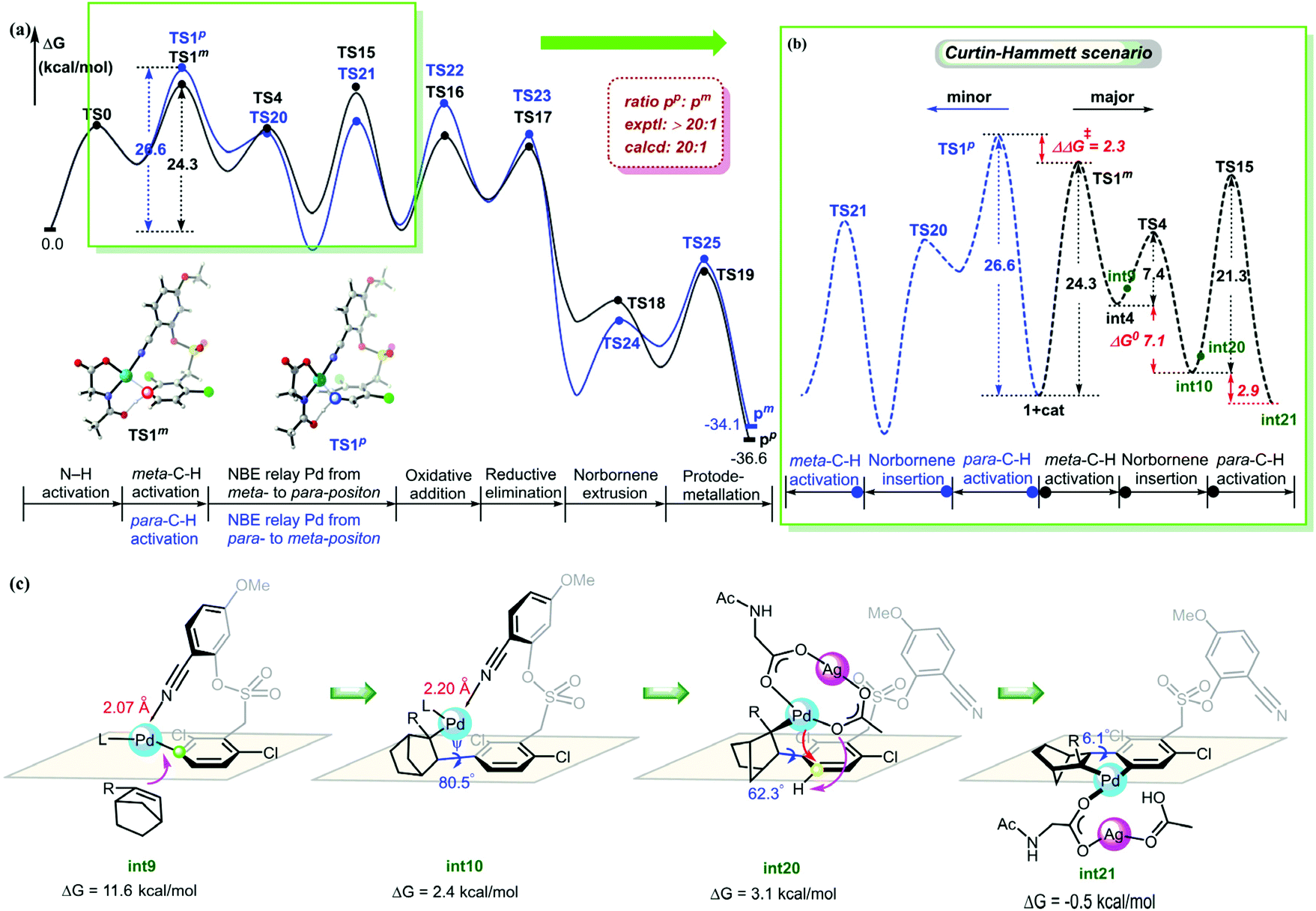 | ||
| Fig. 6 (a) Free energy profiles of the full catalytic cycle of the two pathways. (b) Free energy profiles for the major and minor pathway based on the Curtin-Hammett scenario. (c) Structure conversion of NBE relay Pd from meta- to para-position. | ||
In order to understand the origins of the cooperative mechanism of the transient mediator and remote directing group to achieve palladium relay, we further investigated the structure conversion of the NBE relay Pd from meta- to para-position (Fig. 6c). After Pd accesses meta-position, the norbornene attacks the meta-Pd–Csp2 bond from below the substrate benzene ring (int9). With the rotation of the CNBE–Csp2 bond from 80.5° (int10) to 62.3° (int20), Ag(I) coordinates with Pd/MPAA to get rid of the binding of the directing group, and then, Pd is relayed to the para-position. The Ag(I)-coordinated acetate acts as an internal base to abstract proton (int21). To quantitatively illustrate the NBE relay palladation, we further analyzed the thermodynamics and kinetics for the process of conversion based on the Curtin-Hammett scenario, as illustrated by the Gibbs energy profile in Fig. 6b. Here, as for the single intermediate, ratio [int4]/[int10] was determined by their relative stabilities.42 This means that NBE insertion as the key step can take place spontaneously with exothermic of 7.1 kcal mol−1via a low barrier (7.4 kcal mol−1). The energy released in this step basically compensates for that needed to cross the barrier. It follows that the Pd relay process is driven thermodynamically to achieve the activation from meta- to para-position.
The discussion above shows that the DG-directed Pd activates favorably the meta-position (TS1m) compared to the para-position (TS1p). To further understand how these different factors control the site selectivity, we performed the distortion/interaction analysis43 on these competitively transition states (Fig. 7). The distortion energies of the substrate in TS1p were 3.4 kcal mol−1 higher than that in TS1m, and demonstrated that the distortion of substrate in the C–H activation transition states was a dominant factor (38.5 and 41.9 kcal mol−1 for TS1m and TS1p, respectively). The greater strain in TS1p was due to the significant distortion of the C–S bond from −70.4° to −50.2° compared to substrate 1. However, TS1m barely changed (ψ2 = −69.3°).
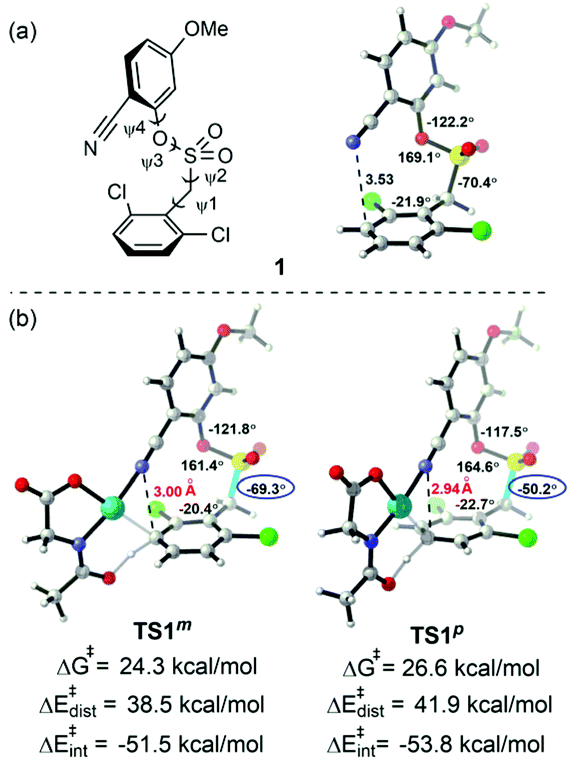 | ||
| Fig. 7 (a) Optimized geometry of the toluene derivative substrate with the nitrile-containing template. (b) Optimized geometries of the meta- and para-C–H activation transition states of Pd/MPAA catalysis. | ||
4. Conclusions
A novel mechanism of the site selective arylation of arenes via Pd/MPAA/NBE cooperative catalysis in conjunction with a precise DG and silver(I) additive was explored using DFT calculations. The results revealed that the reaction was initiated by a Pd/MPAA complex to activate at first the meta-C–H guided by the directing group, and the para-arylation was achieved by the NBE relay palladation from meta- to para-position. The secondary para-C–H activation was catalyzed by a Pd–Ag bimetallic complex, which unraveled the crucial role of the silver additive. We also explored energy pathways through para- to meta-selectivity. The calculation results showed that the DG-assisted primary C–H activation was the rate-determining step for the overall catalytic cycle, and also the key step of determining the site selectivity. The meta-C–H activation was favorable in energy due to the less ring strain in the cyclic nitrile-coordinated C–H activation transition states in meta-position.Notably, we explored in detail the origin of the cooperative mechanism of the transient mediator and remote directing group to achieve the palladium relay. The directing group guided the active monomeric Pd/MPAA catalyst to achieve primary meta-C–H activation. Then, it had to cooperate with a transient mediate NBE to alternate association with the Pd center driven by thermodynamics for the catalytic cycle to proceed. The NBE insertion, as the key step to achieve the expected Pd relay, can take place spontaneously with exothermic of 7.1 kcal mol−1, which can compensate for the energy needed to cross the barrier. The palladium relay process was driven thermodynamically to achieve the activation from meta- to para-position. We expect that this study would have implications for the understanding of C–H functionalization chemistry by norbornene relay palladation and would be helpful to design new reaction systems to access more accurate site selectivity in syntheses.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Shandong Province (ZR2019YQ11).Notes and references
- (a) L. McMurray, F. O'Hara and M. J. Gaunt, Recent Developments in Natural Product Synthesis Using Metal-Catalysed C-H Bond Functionalisation, Chem. Soc. Rev., 2011, 40, 1885–1898 RSC; (b) J. Wencel-Delord and F. Glorius, C-H Bond Activation Enables the Rapid Construction and Late-Stage Diversification of Functional Molecules, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (c) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Recent Applications of C-H Functionalization in Complex Natural Product Synthesis, Chem. Soc. Rev., 2018, 47, 8925–8967 RSC; (d) J. W. Lee, K. N. Lee and M.-Y. Ngai, Synthesis of Tri- and Difluoromethoxylated Compounds by Visible-Light Photoredox Catalysis, Angew. Chem., Int. Ed., 2019, 58, 11171–11180 CrossRef CAS; (e) K. N. Lee and M.-Y. Ngai, Recent Developments in Transition-Metal Photoredox-Catalysed Reactions of Carbonyl Derivatives, Chem. Commun., 2017, 53, 13093–13112 RSC; (f) D. Qian and J. Sun, Recent Progress in Asymmetric Ion-Pairing Catalysis with Ammonium Salts, Chem. – Eur. J., 2019, 25, 3740–3751 CrossRef CAS PubMed; (g) A. J. Metrano and S. J. Miller, Peptide-Based Catalysts Reach the Outer Sphere through Remote Desymmetrization and Atroposelectivity, Acc. Chem. Res., 2019, 52, 199–215 CrossRef CAS PubMed; (h) S. Govaerts, A. Nyuchev and T. Noel, Pushing the Boundaries of C-H Bond Functionalization Chemistry Using Flow Technology, J. Flow Chem., 2020, 10, 13–71 CrossRef CAS.
- (a) K. Murali, L. A. Machado, R. L. Carvalho, L. F. Pedrosa, R. Mukherjee, E. N. da Silva Júnior and D. Maiti, Decoding Directing Groups and their Pivotal Role in C-H Activation, Chem. – Eur. J., 2021, 27, 12453–12508 CrossRef CAS PubMed; (b) S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Toolbox for Distal C-H Bond Functionalizations in Organic Molecules, Chem. Rev., 2021 DOI:10.1021/acs.chemrev.1c00220; (c) G. Meng, N. Y. S. Lam, E. L. Lucas, T. G. Saint-Denis, P. Verma, N. Chekshin and J.-Q. Yu, Achieving Site-Selectivity for C–H Activation Processes Based on Distance and Geometry: A Carpenter's Approach, J. Am. Chem. Soc., 2020, 142, 10571–10591 CrossRef CAS.
- (a) D. A. Colby, R. G. Bergman and J. Ellman, Rhodium-Catalyzed C-C Bond Formation via Heteroatom-Directed C-H Bond Activation, Chem. Rev., 2009, 110, 624–655 CrossRef; (b) H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, Pd(II)-Catalyzed ortho- or meta-C-H Olefination of Phenol Derivatives, J. Am. Chem. Soc., 2013, 135, 7567–7571 CrossRef CAS; (c) C. Wang, H. Chen, Z. Wang, J. Chen and Y. Huang, Rhodium(III)-Catalyzed C-H Activation of Arenes Using a Versatile and Removable Triazene Directing Group, Angew. Chem., Int. Ed., 2012, 51, 7242–7245 CrossRef CAS; (d) C. Wang, H. Sun, Y. Fang and Y. Huang, General and Efficient Synthesis of Indoles through Triazene-Directed C-H Annulation, Angew. Chem., Int. Ed., 2013, 52, 5795–5798 CrossRef CAS; (e) Y. Ogiwara, M. Miyake, T. Kochi and F. Kakiuchi, Syntheses of RuHCl(CO)(PAr3)3 and RuH2(CO)(PAr3)3 Containing Various Triarylphosphines and Their Use for Arylation of Sterically Congested Aromatic C-H Bonds, Organometallics, 2017, 36, 159–164 CrossRef CAS; (f) Q. Wang, F. Huang, L. Jiang, C. Zhang, C. Sun, J. Liu and D. Chen, Comprehensive Mechanistic Insight into Cooperative Lewis Acid/Cp*CoIII-Catalyzed C-H/N-H Activation for the Synthesis of Isoquinolin-3-ones, Inorg. Chem., 2018, 57, 2804–2814 CrossRef CAS; (g) B. Li, K. Seth, B. Niu, L. Pan, H. Yang and H. Ge, Transient-Ligand-Enabled ortho-Arylation of Five-Membered Heterocycles: Facile Access to Mechanochromic Materials, Angew. Chem., Int. Ed., 2018, 57, 3401–3405 CrossRef CAS PubMed; (h) Y.-Y. Xing, J.-B. Liu, Q.-M. Sun, C.-Z. Sun, F. Huang and D.-Z. Chen, A Computational Mechanistic Study of Pd(II)-Catalyzed Enantioselective C(sp3)-H Borylation: Roles of APAO Ligands, J. Org. Chem., 2019, 84, 10690–10700 CrossRef CAS PubMed; (i) A. J. Metrano and S. J. Miller, Peptide-Based Catalysts Reach the Outer Sphere through Remote Desymmetrization and Atroposelectivity, Acc. Chem. Res., 2019, 52, 199–215 CrossRef CAS; (j) H. Kim, R. S. Thombal, H. D. Khanal and Y. R. Lee, Rhodium(III)-Catalyzed Regioselective Distal Ortho C-H Alkenylation of N-Benzyl/Furanylmethylpyrazoles Directed by N-Coordinating Heterocycles, Chem. Commun., 2019, 55, 13402–13405 RSC; (k) S. Chen, M. Wang and X. Jiang, C-H Functionalization Strategies for the Construction of Thioethers, Acta Phys.-Chim. Sin., 2019, 35, 954–967 CAS; (l) R. S. Thombal and Y. R. Lee, Palladium-Catalyzed Direct Oxidative C-H Activation/Annulation for Regioselective Construction of N-Acylindoles, Org. Lett., 2020, 22, 3397–3401 CrossRef CAS.
- (a) R. J. Phipps and M. J. Gaunt, A meta-Selective Copper-Catalyzed C-H Bond Arylation, Science, 2009, 323, 1593–1597 CrossRef CAS; (b) H. A. Duong, R. E. Gilligan, M. L. Cooke, R. J. Phipps and M. J. Gaunt, Copper(II)-Catalyzed meta-Selective Direct Arylation of α-Aryl Carbonyl Compounds, Angew. Chem., Int. Ed., 2011, 50, 463–466 CrossRef CAS; (c) O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, Ruthenium-Catalyzed Meta Sulfonation of 2-Phenylpyridines, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS; (d) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Activation of Remote meta-C-H bonds Assisted by an End-on Template, Nature, 2012, 486, 518–522 CrossRef CAS; (e) G. A. Chotana, M. A. Rak and M. R. Smith, Sterically Directed Functionalization of Aromatic C-H Bonds: Selective Borylation Ortho to Cyano Groups in Arenes and Heterocycles, J. Am. Chem. Soc., 2005, 127, 10539–10544 CrossRef CAS PubMed; (f) N. Hofmann and L. Ackermann, Meta-Selective C-H Bond Alkylation with Secondary Alkyl Halides, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS; (g) Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, A meta-Selective C-H Borylation Directed by a Secondary Interaction Between Ligand and Substrate, Nat. Chem., 2015, 7, 712–717 CrossRef CAS PubMed; (h) M. Bera, S. Agasti, R. Chowdhury, R. Mondal, D. Pal and D. Maiti, Rhodium-Catalyzed meta-C-H Functionalization of Arenes, Angew. Chem., Int. Ed., 2017, 56, 5272–5276 CrossRef CAS PubMed; (i) Y.-Y. Xing, J.-B. Liu, C.-Z. Sun, F. Huang and D.-Z. Chen, The Underlying Factors Controlling the Pd-Catalyzed Site-Selective Alkenylation of Aliphatic Amines, Dalton Trans., 2017, 46, 9430–9439 RSC; (j) S.-D. Li, H. Wang, Y.-X. Weng and G. Li, Carboxy Group as a Remote and Selective Chelating Group for C-H Activation of Arenes, Angew. Chem., 2019, 131, 18673–18678 CrossRef; (k) A. Dey, S. K. Sinha, T. K. Achar and D. Maiti, Accessing Remote meta- and para-C(sp2)−H Bonds with Covalently Attached Directing Groups, Angew. Chem., Int. Ed., 2019, 58, 10820–10843 CrossRef CAS PubMed.
- (a) K. Sun, Y. Li, T. Xiong, J. Zhang and Q. Zhang, Palladium-Catalyzed C-H Aminations of Anilides with N-Fluorobenzenesulfonimide, J. Am. Chem. Soc., 2011, 133, 1694–1697 CrossRef CAS; (b) X. Guo and C.-J. Li, Ruthenium-Catalyzed para-Selective Oxidative Cross-Coupling of Arenes and Cycloalkanes, Org. Lett., 2011, 13, 4977–4979 CrossRef CAS; (c) C.-L. Ciana, R. J. Phipps, J. R. Brandt, F.-M. Meyer and M. J. Gaunt, A Highly para-Selective Copper(II)-Catalyzed Direct Arylation of Aniline and Phenol Derivatives, Angew. Chem., Int. Ed., 2011, 50, 458–462 CrossRef CAS PubMed; (d) L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Gold-Catalyzed Direct Arylation, Science, 2012, 337, 1644–1648 CrossRef CAS; (e) Z. Wu, F. Luo, S. Chen, Z. Li, H. Xiang and X. Zhou, Palladium-Catalyzed para-Selective Arylation of Phenols with Aryl Iodides in Water, Chem. Commun., 2013, 49, 7653–7655 RSC; (f) B. Ma, Z. Chu, B. Huang, Z. Liu, L. Liu and J. Zhang, Highly para–Selective C-H Alkylation of Benzene Derivatives with 2,2,2-Trifluoroethyl α-Aryl-α-Diazoesters, Angew. Chem., Int. Ed., 2017, 56, 2749–2753 CrossRef CAS; (g) M. T. Mihai, B. D. Williams and R. J. Phipps, Para-Selective C-H Borylation of Common Arene Building Blocks Enabled by Ion-Pairing with a Bulky Countercation, J. Am. Chem. Soc., 2019, 141, 15477–15482 CrossRef CAS; (h) T. Adak, J. Schulmeister, M. C. Dietl, M. Rudolph, F. Rominger and A. S. K. Hashmi, Gold-Catalyzed Highly Chemo- and Regioselective C-H Bond Functionalization of Phenols with Haloalkynes, Eur. J. Org. Chem., 2019, 3867–3876 CrossRef CAS; (i) F. de Azambuja, M.-H. Yang, T. Feoktistova, M. Selvaraju, A. C. Brueckner, M. A. Grove, S. Koley, P. H.-Y. Cheong and R. A. Altman, Connecting Remote C-H Bond Functionalization and Decarboxylative Coupling Using Simple Amines, Nat. Chem., 2020, 12, 489–496 CrossRef CAS PubMed; (j) R. L. Carvalho, G. G. Dias, C. L. M. Pereira, P. Ghosh, D. Maiti and E. N. da Silva Júnior, A Catalysis Guide Focusing on C-H Activation Processes, J. Braz. Chem. Soc., 2021, 32, 917–952 CAS.
- B. Haines, Y. Saito, Y. Segawa, K. Itami and D. G. Musaev, Flexible Reaction Pocket on Bulky Diphosphine-Ir Complex Controls Regioselectivity in para-Selective C-H Borylation of Arenes, ACS Catal., 2016, 6, 7536–7546 CrossRef CAS.
- M. E. Hoque, R. Bisht, C. Haldar and B. Chattopadhyay, Noncovalent Interactions in Ir-Catalyzed C-H Activation: L-Shaped Ligand for para-Selective Borylation of Aromatic Esters, J. Am. Chem. Soc., 2017, 139, 7745–7748 CrossRef CAS PubMed.
- Y. Nakao, Y. Yamada, N. Kashihara and T. Hiyama, Selective C-4 Alkylation of Pyridine by Nickel/Lewis Acid Catalysis, J. Am. Chem. Soc., 2010, 132, 13666–13668 CrossRef CAS.
- (a) C.-C. Tsai, W.-C. Shih, C.-Y. Fang, C.-Y. Li, T.-G. Ong and G. A. P. Yap, Bimetallic Nickel Aluminun Mediated para-Selective Alkenylation of Pyridine: Direct Observation of η2,η1-Pyridine Ni(0)-Al(III) Intermediates Prior to C-H Bond Activation, J. Am. Chem. Soc., 2010, 132, 11887–11889 CrossRef CAS PubMed; (b) S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, Para-Selective Alkylation of Benzamides and Aromatic Ketones by Cooperative Nickel/Aluminum Catalysis, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed; (c) L. Yang, K. Semba and Y. Nakao, Para-Selective C-H Borylation of (Hetero)Arenes by Cooperative Iridium/Aluminum Catalysis, Angew. Chem., Int. Ed., 2017, 56, 4853–4857 CrossRef CAS; (d) S. Okumura and Y. Nakao, Para-Selective Alkylation of Sulfonylarenes by Cooperative Nickel/Aluminum Catalysis, Org. Lett., 2017, 19, 584–587 CrossRef CAS PubMed; (e) A. Maji, S. Guin, S. Feng, A. Dahiya, V. K. Singh, P. Liu and D. Maiti, Experimental and Computational Exploration of para-Selective Silylation with a Hydrogen-Bonded Template, Angew. Chem., Int. Ed., 2017, 56, 14903–14907 CrossRef CAS PubMed.
- S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, Remote para-C-H Functionalization of Arenes by a D-Shaped Biphenyl Template-Based Assembly, J. Am. Chem. Soc., 2015, 137, 11888–11891 CrossRef CAS PubMed.
- S. Okumura, S. Tang, T. Saito, K. Semba, S. Sakaki and Y. Nakao, Para-Selective Alkylation of Benzamides and Aromatic Ketones by Cooperative Nickel/Aluminum Catalysis, J. Am. Chem. Soc., 2016, 138, 14699–14704 CrossRef CAS PubMed.
- M. H. Li, M. Shang, H. Xu, X. Wang, H.-X. Dai and J.-Q. Yu, Remote para-C-H Acetoxylation of Electron-Deficient Arenes, Org. Lett., 2019, 21, 540–544 CrossRef CAS.
- A. Maji, A. Dahiya, G. Lu, T. Bhattacharya, M. Brochetta, G. Zanoni, P. Liu and D. Maiti, H-Bonded Reusable Template Assisted para-Selective Ketonisation Using Soft Electrophilic Vinyl ethers, Nat. Commun., 2018, 9, 3582–3592 CrossRef.
- S. Pimparkar, T. Bhattacharya, A. Maji, A. Saha, R. Jayarajan, U. Dutta, G. Lu, D. W. Lupton and D. Maiti, Para-Selective Cyanation of Arenes by H-Bonded Template, Chem. – Eur. J., 2020, 26, 11558–11564 CrossRef CAS.
- (a) M. Simonetti, D. M. Cannas, X. JustBaringo, I. J. Vitorica-Yrezabal and I. Larrosa, Cyclometallated Ruthenium Catalyst Enables Late-Stage Directed Arylation of Pharmaceuticals, Nat. Chem., 2018, 10, 724–731 CrossRef CAS; (b) H. Shi, A. N. Herron, Y. Shao, Q. Shao and J.-Q. Yu, Enantioselective Remote meta-C-H Arylation and Alkylation via a Chiral Transient Mediator, Nature, 2018, 558, 581–585 CrossRef CAS.
- U. Dutta, S. Porey, S. Pimparkar, A. Mandal, J. Grover, A. Koodan and D. Maiti, Para-Selective Arylation of Arenes: A Direct Route to Biaryls by Norbornene Relay Palladation, Angew. Chem., Int. Ed., 2020, 59, 20831–20836 CrossRef CAS.
- B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, PdII-Catalyzed Enantioselective Activation of C(sp2)-H and C(sp3)-H Bonds Using Monoprotected Amino Acids as Chiral Ligands, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 CrossRef CAS.
- (a) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, Palladium-Catalyzed meta-Selective C-H Bond Activation with a Nitrile-Containing Template: Computational Study on Mechanism and Origins of Selectivity, J. Am. Chem. Soc., 2014, 136, 344–355 CrossRef CAS PubMed; (b) T. Bhattacharya, S. Dutta and D. Maiti, Deciphering the Role of Silver in Palladium Catalyzed C-H Functionalizations, ACS Catal., 2021, 11, 9702–9714 CrossRef CAS; (c) W. Liu, Z. Liu, X. Liu and Y. Dang, Mechanism of Pd-Catalysed C(sp3)-H Arylation of Thioethers with Ag(I) Additives, Org. Biomol. Chem., 2021, 19, 6766–6770 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (b) A. D. Becke, Density-Functional Thermochemistry., III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- (a) D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Energy-Adjustedab Initio Pseudopotentials for the Second and Third Row Transition Elements, Theor. Chem. Acc., 1990, 77, 123–144 Search PubMed; (b) L. E. Roy, P. J. Hay and R. L. Martin, Revised Basis Sets for the LANL Effective Core Potentials, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS PubMed.
- (a) S. Huzinaga, Basis Sets for Molecular Calculations, Comput. Phys. Rep., 1985, 2, 281–339 CrossRef; (b) S. Liu, J. Liu, Q. Wang, J. Wang, F. Huang, W. Wang, C. Sun and D. Chen, The Origin of Regioselectivity in Cu-Catalyzed Hydrocarbonylative Coupling of Alkynes with Alkyl Halides, Org. Chem. Front., 2020, 7, 1137–1148 RSC.
- R. Valero, R. Costa, P. R. De, I. Moreira, D. G. Truhlar and F. Illas, Performance of the M06 Family of Exchange-Correlation Functionals for Predicting Magnetic Coupling in Organic and Inorganic Molecules, J. Chem. Phys., 2008, 128, 114103 CrossRef PubMed.
- (a) A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed; (b) J.-B. Liu, X. Wang, A. M. Messinis, X.-J. Liu, R. Kuniyil, D.-Z. Chen and L. Ackermann, Understanding the Unique Reactivity Patterns of Nickel/JoSPOphos Manifold in the Nickel-Catalyzed Enantioselective C-H Cyclization of Imidazoles, Chem. Sci., 2021, 12, 718–729 RSC.
- (a) X. Qi, J. Wang, Z. Dong, G. Dong and P. Liu, Compatibility Score for Rational Electrophile Selection in Pd/NBE Cooperative Catalysis, Chem, 2020, 6, 2810–2825 CrossRef CAS PubMed; (b) X. Ma, X. Zhao, R. Zhu and D. Zhang, Computational Study on Why and How of Nonconventional meta-C-H Arylation of ElectronRich Arenes via Pd/Quinoxaline-Based Ligand/Norbornene Cooperative Catalysis, J. Org. Chem., 2020, 85, 5995–6007 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013, http://nbo6.chem.wisc.edu/ Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, Canada, 2009, http://www.cylview.org Search PubMed.
- (a) X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Ligand-Enabled meta-C-H Activation Using a Transient Mediator, Nature, 2015, 519, 334–338 CrossRef CAS PubMed; (b) J. Wang, R. Li, Z. Dong, P. Liu and G. Dong, Complementary Siteselectivity in Arene Functionalization Enabled by Overcoming the Ortho Constraint in Palladium/Norbornene Catalysis, Nat. Chem., 2018, 10, 866–872 CrossRef CAS PubMed.
- T. Bhattacharya, A. Ghosha and D. Maiti, Hexafluoroisopropanol: the Magical Solvent for Pd-Catalyzed C-H Activation, Chem. Sci., 2021, 12, 3857–3870 RSC.
- G.-J. Cheng, Y.-F. Yang, P. Liu, P. Chen, T.-Y. Sun, G. Li, X. Zhang, K. N. Houk, J.-Q. Yu and Y.-D. Wu, Role of N-Acyl Amino Acid Ligands in Pd(II)-Catalyzed Remote C-H Activation of Tethered Arenes, J. Am. Chem. Soc., 2014, 136, 894–897 CrossRef CAS PubMed.
- (a) B. Bhaskararao, S. Singh, M. Anand, P. Verma, P. Prakash, C. Athira, S. Malakar, H. F. Schaefer and R. B. Sunoj, Is Silver a mere Terminal Oxidant in Palladium Catalyzed C-H Bond Activation Reactions?, Chem. Sci., 2020, 11, 208–216 RSC; (b) W. Feng, T. Wang, D. Liu, X. Wang and Y. Dang, Mechanism of the Palladium-Catalyzed C(sp3)-H Arylation of Aliphatic Amines: Unraveling the Crucial Role of Silver(I) Additives, ACS Catal., 2019, 9, 6672–6680 CrossRef CAS; (c) L. Fang, T. G. Saint-Denis, B. L. Taylor, S. Ahlquist, K. Hong, S. Liu, L. Han, K. N. Houk and J. Q. Yu, Experimental and Computational Development of a Conformationally Flexible Template for the meta-C-H Functionalization of Benzoic Acids, J. Am. Chem. Soc., 2017, 139, 10702–10714 CrossRef CAS; (d) N. Zhao, X. Jin and Y. Dang, Exploring the Pivotal Role of Silver(I) Additives in Palladium-Catalyzed NH2-Directed C(sp3)-H Arylation Reactions, Chin. Chem. Lett., 2021 DOI:10.1016/j.cclet.2021.04.044.
- T. Yang, C. Kong, S. Yang, Z. Yang, S. Yanga and M. Ehara, Reaction Mechanism, Norbornene and Ligand Effects, and Origins of meta-Selectivity of Pd/Norbornene-Catalyzed C-H Activation, Chem. Sci., 2020, 11, 113–125 RSC.
- (a) J. A. Tunge and L. N. Foresee, Mechanistic Studies of Fujiwara Hydroarylation. C-H Activation versus Electrophilic Aromatic Substitution, Organometallics, 2005, 24, 6440–6444 CrossRef CAS; (b) M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, Catalytic Intermolecular Direct Arylation of Perfluorobenzenes, J. Am. Chem. Soc., 2006, 128, 8754–8756 CrossRef CAS PubMed; (c) S. I. Gorelsky, D. Lapointe and K. Fagnou, Analysis of the Concerted Metalation-Deprotonation Mechanism in Palladium-Catalyzed Direct Arylation Across a Broad Range of Aromatic Substrates, J. Am. Chem. Soc., 2008, 130, 10848–10849 CrossRef CAS PubMed.
- (a) S. Winstein and T. G. Traylor, Mechanisms of Reaction of Organomercurials. II. Electrophilic Substitution on Saturated Carbon. Acetolysis of Dialkylmercury Compounds, J. Am. Chem. Soc., 1955, 77, 3747–3752 CrossRef CAS; (b) L. Ackermann, Carboxylate-Assisted Transition-Metal-Catalyzed C-H Bond Functionalizations: Mechanism and Scope, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS; (c) B. Biswas, M. Sugimoto and S. Sakaki, C-H Bond Activation of Benzene and Methane by M(η2-O2CH)2 (M = Pd or Pt). A Theoretical Study, Organometallics, 2000, 19, 3895–3908 CrossRef CAS; (d) W. Guan, F. B. Sayyed, G. Zeng and S. Sakaki, σ-Bond Activation of Small Molecules and Reactions Catalyzed by Transition-Metal Complexes: Theoretical Understanding of Electronic Processes, Inorg. Chem., 2014, 53, 6444–6457 CrossRef CAS PubMed; (e) C. Shan, R. Bai and Y. Lan, Theoretical Advances of Transition Metals Mediated C-H Bonds Cleavage, Acta Phys.-Chim. Sin., 2019, 35, 940–953 CAS.
- H. Shi, Y. Lu, J. Weng, K. L. Bay, X. Chen, K. Tanaka, P. Verma, K. N. Houk and J. Q. Yu, Differentiation and Functionalization of Remote C-H Bonds in Adjacent Positions, Nat. Chem., 2020, 12, 399–404 CrossRef CAS PubMed.
- (a) M. Catellani, F. Frignani and A. Rangoni, A Complex Catalytic Cycle Leading to a Regioselective Synthesis of o,o’-Disubstituted Vinylarenes, Angew. Chem., Int. Ed. Engl., 1997, 36, 119–122 CrossRef CAS; (b) G. Maestri, E. Motti, N. Della Ca’, M. Derat, E. Malacria and M. Catellani, Of the Ortho Effect in Palladium/Norbornene-Catalyzed Reactions: A Theoretical Investigation, J. Am. Chem. Soc., 2011, 133, 8574–8585 CrossRef CAS PubMed.
- M. Catellani and G. P. Chiusoli, Palladium-(II) and -(IV) Complexes as Intermediates in Catalytic C-C Bond-Forming Reactions, J. Organomet. Chem., 1988, 346, C27–C30 CrossRef CAS.
- (a) J. Wang and G. Dong, Palladium/Norbornene Cooperative Catalysis, Chem. Rev., 2019, 119, 7478–7528 CrossRef CAS PubMed; (b) A. R. Dick, J. W. Kampf and M. S. Sanford, Unusually Stable Palladium(IV) Complexes: Detailed Mechanistic Investigation of C-O Bond-Forming Reductive Elimination, J. Am. Chem. Soc., 2005, 127, 12790–12791 CrossRef CAS PubMed.
- (a) J. Vicente, A. Arcas, F. Julia-Hernandez and D. Bautista, Synthesis of a Palladium(IV) Complex by Oxidative Addition of an Aryl Halide to Palladium(II) and Its Use as Precatalyst in a C-C Coupling Reaction, Angew. Chem., Int. Ed., 2011, 50, 6896–6899 CrossRef CAS PubMed; (b) Y. Dang, S. Qu, J. W. Nelson, H. D. Pham, Z.-X. Wang and X. Wang, The Mechanism of a Ligand-Promoted C(sp3)-H Activation and Arylation Reaction via Palladium Catalysis: Theoretical Demonstration of a Pd(II)/Pd(IV) Redox Manifold, J. Am. Chem. Soc., 2015, 137, 2006–2014 CrossRef CAS PubMed; (c) Y.-F. Yang, X. Hong, G. Chen, J.-Q. Yu and K. N. Houk, The Origins of Dramatic Differences in Five-Membered vs Six-Membered Chelation of Pd(II) on Efficiency of C(sp3)-H Bond Activation, J. Am. Chem. Soc., 2017, 139, 8514–8521 CrossRef CAS PubMed.
- L. Y. Liu, J. X. Qiao, K. S. Yeung, W. R. Ewing and J. Q. Yu, Meta C-H Arylation of Electron-Rich Arenes: Reversing the Conventional Site Selectivity, J. Am. Chem. Soc., 2019, 141, 14870–14877 CrossRef CAS PubMed.
- (a) S.-Q. Zhang, B. L. H. Taylor, C.-L. Ji, Y. Gao, M. R. Harris, L. E. Hanna, E. R. Jarvo, K. N. Houk and X. Hong, Mechanism and Origins of Ligand-Controlled Stereoselectivity of Ni–Catalyzed Suzuki-Miyaura Coupling with Benzylic Esters: A Computational Study, J. Am. Chem. Soc., 2017, 139, 12994–13005 CrossRef CAS PubMed; (b) T. Földes, A. Madarász, A. Révész, Z. Dobi, S. Varga, A. Hamza, P. R. Nagy, P. M. Pihko and I. Pápai, Stereocontrol in Diphenylprolinol Silyl Ether Catalyzed Michael Additions: Steric Shielding or Curtin-Hammett Scenario?, J. Am. Chem. Soc., 2017, 139, 17052–17063 CrossRef PubMed.
- Q. Peng, F. Duarte and R. S. Paton, Computing Organic Stereoselectivity - from Concepts to Quantitative Calculations and Predictions, Chem. Soc. Rev., 2016, 45, 6093–6107 RSC.
- (a) D. H. Ess and K. N. Houk, Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity, J. Am. Chem. Soc., 2007, 129, 10646–10647 CrossRef CAS PubMed; (b) F. M. Bickelhaupt and K. N. Houk, Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model, Angew. Chem., Int. Ed., 2017, 56, 2–19 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1qo01500a |
| This journal is © the Partner Organisations 2022 |