
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmidosquaramides – a new anion binding motif with pH sensitive anion transport properties†
Luke A.
Marchetti
a,
Tobias
Krämer
 a and
Robert B. P.
Elmes
*ab
a and
Robert B. P.
Elmes
*ab
aDepartment of Chemistry, National University of Ireland Maynooth, Maynooth, Ireland. E-mail: robert.elmes@mu.ie
bSynthesis and Solid State Pharmaceutical Centre (SSPC), Ireland
First published on 9th August 2022
Abstract
Stimuli responsive anion transport is becoming an important aspect of supramolecular anion recognition chemistry. Herein, we report the synthesis of a family of anion receptors that incorporate a new anion binding motif, amidosquaramides. We show using experimental and computational methods that these receptors have pKa values close to physiological pH but also display intramolecular H-bonding interactions that affect anion recognition. Moreover, moderate activity in a Cl−/NO3− exchange assay is observed at physiological pH that can be effectively ‘switched on’ when repeated under acidic conditions. The reported findings provide synthetic methods that can be used for the construction of more complex squaramide based anion receptors and also provide insight into the importance of conformational analysis when considering receptor design.
Introduction
Squaramides, a family of conformationally rigid cyclobutene ring derivatives, are rapidly gaining research interest across diverse areas of the chemical and biological sciences.1 Composed of two carbonyl hydrogen-bond acceptors in close proximity to two NH hydrogen-bond donors, this small molecular scaffold benefits from unique physical and chemical properties that render it extremely useful in the design of anion receptors2,3 and transporters.4,5 This has led to an explosion of research in the area of supramolecular medicinal chemistry where efficient anion transporters have been explored as treatment options for a variety of disease states including anti-cancer agents,6,7 CF treatments,8 and anti-microbial agents.9 The use of squaramides in this regard has been accelerated by their synthetic versatility, strong H-bond donating ability, planar structure and an increase in aromaticity upon guest binding10 – all properties that make squaramides practical for use in a wide variety of medicinal chemistry applications more broadly. However, if the full potential of squaramides are to be realised, further strategies are required to increase their structural diversity while simultaneously increasing their anion binding affinity.One approach that has not been considered in the context of squaramide synthesis is a reversal of reactivity. The large majority of the synthetic literature uses a method developed by Cohen et al.11 where diethylsquarate is employed as an electrophile in a substitution reaction with a primary amine to form an N-aryl or alkyl linkage. A lesser explored route is the exploitation of a squaramide as a nucleophile in a condensation reaction to form an N-amide linkage. While the amine's nucleophilicity is considerably reduced by proximity to two carbonyl groups within the cyclobutene ring, this option should give access to a broader subset of possible receptors. Davis et al. reported such a route where they synthesised N-carbamoyl squaramides and described their self-assembly behaviour, however, anion binding characteristics were not reported.12
Several methods have been used to increase the anion binding affinity of squaramides. Incorporation of an electron withdrawing aromatic substituent can improve H-bond donor ability,13 however, limitations arise where acidity of NHs can be pursued to a level that favours deprotonation by basic anions.14,15 Similarly, inclusion of several squaramide binding sites within a single molecule can give rise to high affinity supramolecular receptors but this can also lead to issues of solubility.16,17
We envisaged that the N-amido linkage previously reported by Davis et al. could be exploited as an anion binding moiety and their synthetic approach could be expanded to produce a more diverse range of anion receptors. Herein, we describe the synthesis of a family of disubstituted squaramides using this approach that allows incorporation of both an N-aryl linkage and an N-amide linkage in the same molecule; so called ‘amidosquaramides’ (Fig. 1). Varying the substituents of these phenyl moieties, allowed us to investigate the effect on the amidosquaramides anion binding affinity in comparison to their parent squaramide. We envisaged that the N-amide linkage would increase the acidity of the squaramide NH, consequently decreasing its pKa, and increasing its anion binding affinity. We have conducted a detailed spectroscopic and computational analysis of their structure, anion recognition ability, and a preliminary analysis of their anionophoric activity.
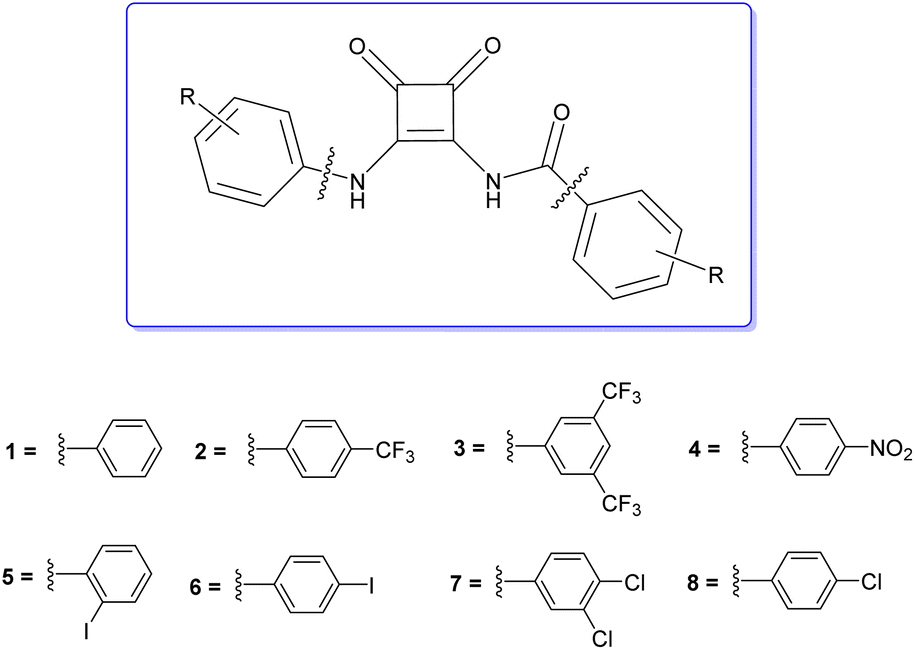 | ||
| Fig. 1 Structure of amidosquaramides. | ||
Results and discussion
Synthesis
Amidosquaramides were accessed through a linear synthetic approach (Scheme 1). Initially, diethyl squarate was obtained using methods adapted by Moore et al.,18 which was then converted to amino-squarate using methods previously reported by Davis et al.12 The N-amide linkage was then formed by coupling the amino-squarate to the relevant benzoic acid derivative using EDCI·HCl. The yield of the coupling reactions varied greatly (25–83%), depending on the strength of the electron-withdrawing groups of the benzoic acid substituents, where the stronger electron-withdrawing groups resulted in lower yields. With the example of 11, the successful formation of the N-amido linkage was indicated in the 1H NMR where the amido-NH signal appeared at 9.88 ppm (see ESI†).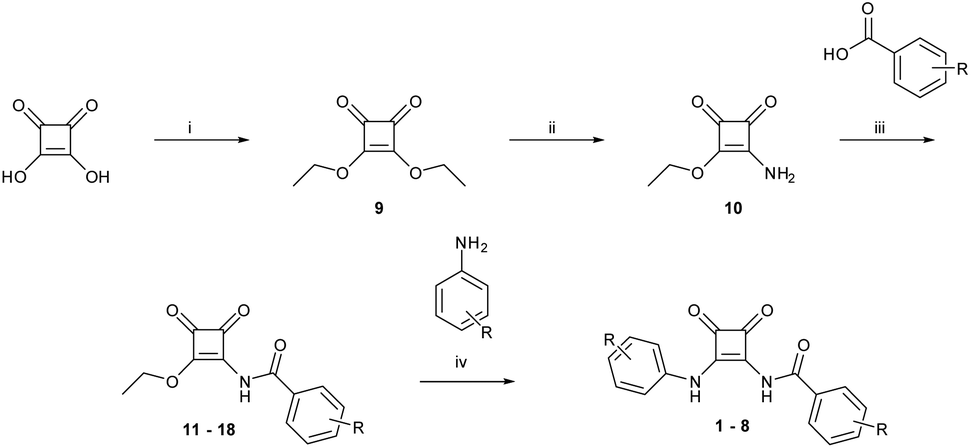 | ||
| Scheme 1 Synthetic pathway to amido-squaramides. Reagents and conditions: (i) triethyl orthoformate, EtOH, 80 °C, 72 h, 91%; (ii) 20% ammonium in ethanol, EtOH, rt, 18 h, 70%; (iii) EDCI·HCl, DMAP, MeCN/DMF (anhydrous), N2, rt −80 °C, 18 h, 10–83%; (iv) Zn(OTf)2, MeCN/DMF, 80 °C, 3–18 h. 10–71%. | ||
The final step of the synthetic pathway was a substitution reaction between the amidosquarate intermediate and the relevant aniline in the presence of zinc trifluoromethanesulfonate, acting as a Lewis-acid catalyst, to yield the target amidosquaramides. The yields of this final step again depended on the electron-withdrawing ability of the aryl substituents, ranging from 10–71%. Synthesis of the target amidosquaramides was evidenced by the presence of a new resonance signal appearing at 11.95 ppm, in the example of 1, attributed to the phenyl-NH proton and the slight downfield shift of the N-amide proton signal to 9.98 ppm (see ESI†). The successful synthesis of compounds 1–8 was confirmed in all cases by 1H and 13C NMR, infrared spectroscopy (IR) and high-resolution mass spectrometry (HRMS) (see ESI† for further details).
pKa determination
pKa is a key parameter when considering the anion recognition properties of charge neutral anion receptors. Thus, to examine the effect the carbonyl has on the pKa of the amido-NH, pH-spectrophotometric titrations were performed to obtain experimental pKa values, using a method described by Schmidtchen et al.19 These titrations were performed in a mixture of DMSO-water (9/1 v/v; in the presence of 0.1 M TBAPF6 at a receptor concentration of 10 μM).During the spectrophotometric titrations, deprotonation events were observed by hypo- and hyperchromism of absorbance bands at various wavelengths. This can be seen in the example of 6 (Fig. 2), where there hypochromism occurs at 284 and 320 nm and hyperchromism at 347 and 407 nm, signalling deprotonation events. The inset of Fig. 2 details the changes of absorbance as a function of pH. A four-parameter sigmoid curve was fitted through these data points using Sigma Plot (Systat Software Inc., Chicago, IL, USA) with the point of inflexion corresponding to the pKa, the results of which are summarised in Table 1. We expected to observe two deprotonation events, one for the amide-NH and the second being the NH of the phenyl substitutent, however, only one deprotonation could be observed for all receptors. This signified that the pKa of only one NH was determined during these experiments.
 | ||
| Fig. 2 Absorption spectra taken over the course of a pH-spectrophotometric titration of 6 (10 μM) in an DMSO–water mixture (9/1 v/v; in presence of 0.1 M TBAPF6) pH 2.52 (blue), pH 12.40 (red). Inset: Comparison plots of absorbance at 284 nm (yellow), 320 nm (grey), 347 nm (orange), and 407 nm (blue) vs. pH. | ||
| Receptor | N-Amide pKa (exp.) | N-Amide pKa (calc.) | N-Phenyl pKa (calc.) |
|---|---|---|---|
| 1 | 8.8 | 8.7 | 12.7 |
| 2 | 7.3 | 7.9 | 11.6 |
| 3 | 6.3 | 6.6 | 10.3 |
| 4 | 7.5 | 7.5 | 10.4 |
| 5 | 8.8 | 8.9 | 12.4 |
| 6 | 8.1 | 9.8 | 11.4 |
| 7 | 7.1 | 7.1 | 9.3 |
| 8 | 8.2 | 7.8 | 11.0 |
We postulated that the pKa of the N-amide NH would be lower than that of the N-phenyl pKa due to the electron-withdrawing effect of the carbonyl. Thus, examining the results of the pH-spectrophotometric titrations, we expected that we had determined the pKa values of the amide-NH as they displayed lower pKa values than those of analogous disubstituted N-phenyl squaramides.20 This was the first piece of evidence suggesting that inclusion of a carbonyl on the squaramide NH increases its acidity.
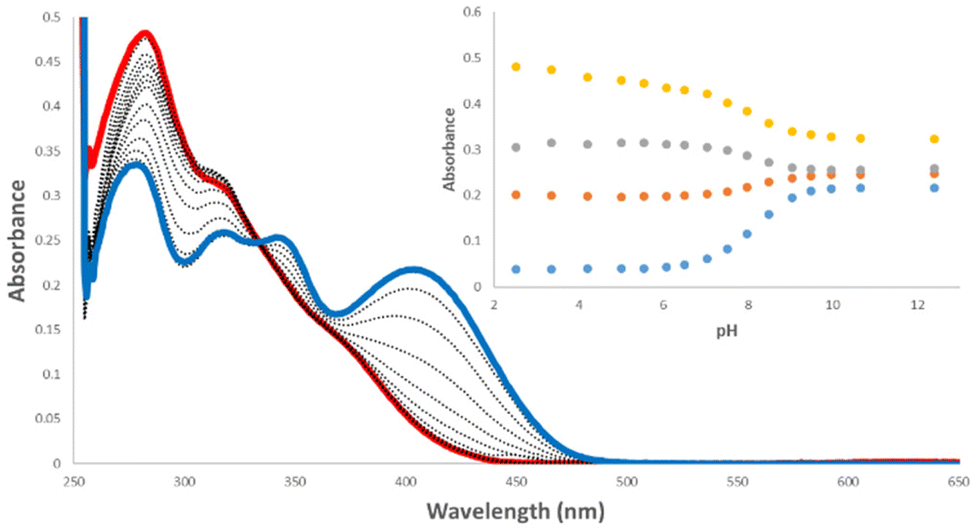
To confirm the validity of our experimental pKa values and to establish an approximate pKa for both the N-phenyl and N-amide NH, quantum chemical calculations were undertaken (full details are given in the ESI†). All receptors and their conjugated bases were subjected to unconstrained geometry optimisations at the M06-2X/6-31+G(d) level of theory, following initial exploration of their conformational space with the conformer–rotamer ensemble sampling tool (crest).21 Thermal and entropic corrections were evaluated within the rigid rotor harmonic oscillator approximation. High-level single point calculations using the DLPNO–CCSD(T) method were performed on the above geometries. From these calculations, the energy-minimised structure of receptor 1 and its derivatives showed that the carbonyl of the amide-linkage participates in an intramolecular hydrogen bond to the NH of the N-phenyl NH (Fig. 3(a)).
 | ||
| Fig. 3 (a) Gibbs Free Energy heat map (in kcal mol−1) of conformational space of 1 illustrating two minima (blue). (b) The two lowest energy conformations of 1, anti/syn (top), anti/anti (bottom). Hydrogens bound to carbon have been omitted. | ||
Reinforcing the finding that the amidosquaramide prefers to maintain a planar conformation stabilised by H-bonding, molecular dynamics simulations of 1 furnish the free energy surface of the system's conformational space as illustrated in the heat map shown in Fig. 3(a). The conformational space is defined by the two torsional angles C–C–N–C(Phenyl) (θ) and C–C–N–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) (φ), i.e. rotations around the bonds attached to the double bond of the cyclobut-3-ene ring of the squaramide. The heat map shows two distinct minima (blue) of the amidosquaramide conformations, one where the amidosquaramide adopts an anti/syn conformation as it participates in intramolecular H-bonding and one where the amidosquaramide adopts an anti/anti conformation.
O) (φ), i.e. rotations around the bonds attached to the double bond of the cyclobut-3-ene ring of the squaramide. The heat map shows two distinct minima (blue) of the amidosquaramide conformations, one where the amidosquaramide adopts an anti/syn conformation as it participates in intramolecular H-bonding and one where the amidosquaramide adopts an anti/anti conformation.
pKa values of all receptors were obtained using a direct method via the thermodynamic cycle depicted in Scheme 2. Solvation free energies in DMSO were obtained using the SMD implicit solvent model and utilising solvent-reoptimized geometries. A proton solvation free energy of −273.30 kcal mol−1 in DMSO was used.22
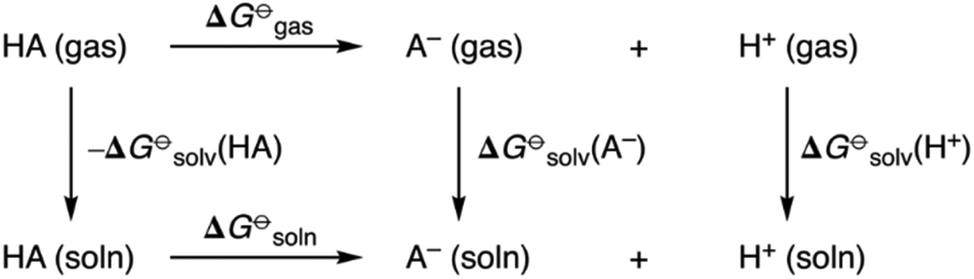 | ||
| Scheme 2 Thermodynamic cycle for quantum chemical prediction of pKa values of amidosquaramide receptors. | ||
The results obtained showed excellent agreement between the experimentally and computational derived pKa values (Table 1). The calculated values of the N-phenyl NH also show alignment with previous reports in the literature from Ni et al.23 Moreover, the observed values correlate with the Hammett constants of the substituents i.e. the pKa of the receptor decreases as the Hammett constant increases.24 For example, receptor 3 bearing two (trifluoro)methyl moieties displayed the lowest pKa while the unsubstituted receptor 1 displayed the highest pKa. The calculations also confirmed that the N-amide linkage decreased the pKa of the amidosquaramide receptors. Although the conditions should have allowed it, we did not observe a 2nd deprotonation event spectroscopically. This suggests that either the 2nd deprotonation is not observed by absorption spectroscopy or the computational values are not accurate. We have previously observed a 2nd deprotonation spectroscopically in a squaramide–anthracene conjugate whereby titration with TBAOH, resulted in stark spectroscopic changes on going from the mono-deprotonated species to the doubly deprotonated derivative.25 While the 2nd pKa for this compound was not calculated it suggests that strongly basic conditions are required to effect a 2nd deprotonation. Moreover, it is known that squaramides are not stable at highly basic pH,26 and this may explain why we were not able to observe a 2nd clear equivalence point.
Given the observed propensity of amidosquaramides to possibly form an intramolecular H-bond, we expected that this may impact on the anion binding ability of this receptor. Indeed, we envisaged that it may cause an additional thermodynamic barrier to anion recognition where the intramolecular interaction would first need to be overcome and a conformational change induced before anion recognition could proceed.
Anion binding studies
Receptors 1–3 were selected to undergo 1H NMR spectroscopic anion screening to assess their interaction in DMSO-d6 (0.5% H2O) with a range of TBA salts of halides and oxoanions (F, Cl, Br, I, NO3, AcO, H2PO42−, SO42−).When treated with basic anions (F−, AcO− and H2PO42−) all receptors underwent deprotonation. This was observed in the 1H NMR spectra with the disappearance of both NH peaks and in some cases, the coalescence of aromatic peaks (see ESI†). In the case of TBAF, the formation of HF2− can be seen by the characteristic peak at 16.15 ppm (see ESI†). Minimal observable changes in the 1H NMR spectra were observed when receptors were treated with 10 molar equivalents of Br−, I− or NO3− indicating that little interaction was taking place. SO42− exhibited quite different behaviour from other anions whereby a broad NH peak at ∼9.5 ppm was observed that was absent with the other anions. Moreover, the aromatic signals were also significantly perturbed suggesting that more complex behaviour is occurring with this anion. When treated with 10 molar equivalents of Cl−, a downfield shift of the amido-NH was observed (Δ ppm ≈1) indicating stronger hydrogen bond formation between this anion and the receptors.
Based on the results of the anion screen, more detailed 1H spectroscopic titrations were performed in DMSO-d6 (0.5% H2O) at 298 K, to determine the binding constants for Cl−. All receptors displayed similar behaviour when treated with TBACl. Sequential additions of Cl− resulted in the NH peak corresponding to the amido-NH (blue) undergoing a downfield shift, while the NH peak corresponding to the N-phenyl (green) displayed a barely discernible shift, as seen with 3 as an example (Fig. 4). Slight shifts in aromatic resonances could also be observed, possibly indicating weak C–H⋯anion interactions.
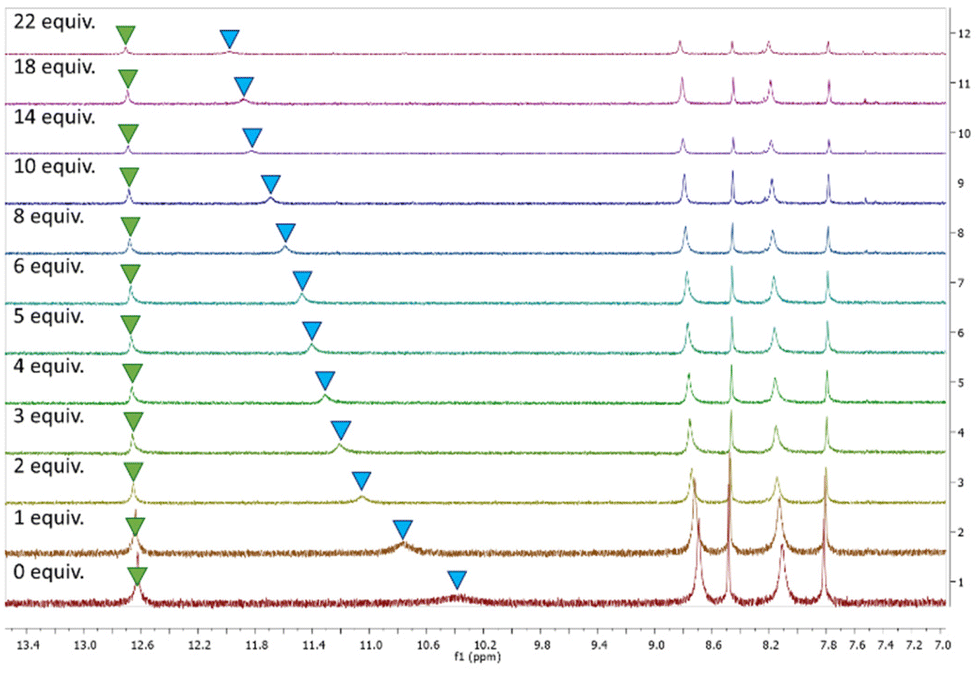 | ||
| Fig. 4 1H NMR stackplot, 7.0–13.5 ppm, of receptor 3 with 0.0–22.0 equiv. TBACl in DMSO-d6/0.5% H2O. | ||
As the results of the geometry optimisation showed the amidosquaramides contain an intramolecular H-bond between the N-phenyl NH and the amide's oxygen atom, it is unsurprising that there was only a slight shift observed for the corresponding signal when treated with TBACl. We postulate that the stability afforded by the intramolecular H-bond results in the amidosquaramide being unwilling to break this intramolecular interaction, resulting in only one NH being available to bind to anionic guests.
Binding constants were determined by plotting the changes in chemical shift of the NH peaks against anion concentration and the resulting plots were analysed using the open access software BindFit27,28 (see ESI† for details), and fitted to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model. The results are summarised in Table 2.
:1 binding model. The results are summarised in Table 2.
:1 binding model
The binding affinity of the amidosquaramides were in the order of 3 > 2 > 1 > 7 > 8 > 4 ≈ 6 > 5. Unsurprisingly, compound 3 displayed the highest binding constant of 123 Ka/M−1 due to the presence of the bis-3,5-(trifluoro)methyl substituents on both aromatic rings. This aligns with previously reported receptors decorated with CF3 moieties displaying the highest binding constants.29 Compound 5 displayed the lowest binding constant of 6 Ka/M−1. We postulate that this is a result of the 2-iodo substituent causing steric hindrance at the binding site, preventing Cl− coming into close proximity with the NH hydrogen bond donor. When anion binding values are correlated with the appropriate Hammett parameter and pKa values (Table 2) no clear trend emerges across the entire series. Some relationship is observed for compounds 1–3 where an increase in the Hammet constant results in a decrease in pKa and an increase in anion binding affinity. However, this trend does not hold for receptors 4–8.
When comparing amidosquaramides to analogous squaramides, ureas, and thioureas (Fig. 5a), we see that while amidosquaramides do not reach the anion binding ability of their analogous squaramides, they do show anion binding abilities greater than that of their urea and thiourea analogues.29 For example, amidosquaramide 3 displays a Ka value for Cl− of 123 M−1, while the binding constants for its analogous squaramide, urea, and thiourea are 643, 88, and 41 Ka/M−1, respectively. We postulate that the anti/syn conformation of the receptors arising from the intramolecular H-bonding was a factor in the relatively low Cl− binding constants, as only one hydrogen bond donor is available for H-bond formation with Cl−, compared to the analogous squaramides where two hydrogen bond donors are available for anion binding (Fig. 5b).
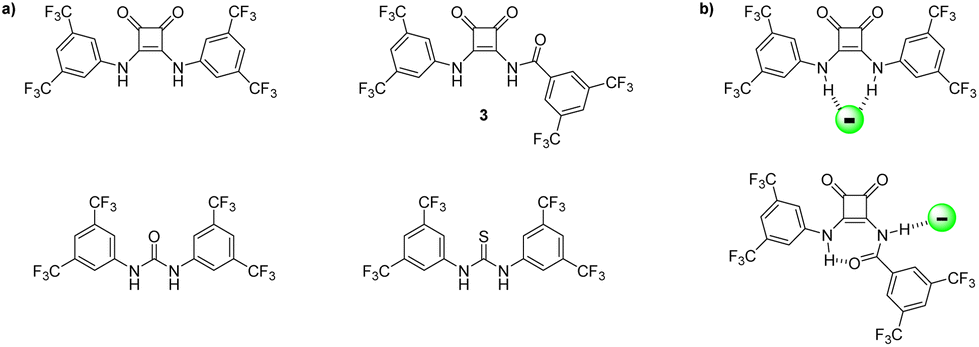 | ||
| Fig. 5 (a) Structure of receptor 3, alongside previously studied analogous squaramides, ureas, and thioureas.29 (b) Comparison of binding modes of squaramide vs. amidosquaramide. | ||
Intramolecular H-bonds that affect anion binding are well known and several examples have been presented where the interactions can either favour the desired conformation30,31 that leads to increased anion binding or result in an undesired conformation that inhibits anion binding.32 For example, Jolliffe et al. recently showed this behaviour for diaminomethyleneindanediones, where addition of SO42− disrupted intramolecular H-bonding and led to enhanced binding. We expected that the intramolecular interaction of the amidosquaramides may be disrupted thereby promoting greater anion binding affinity. While DMSO is already a competitive H-bonding solvent we expected that the addition of acid would further increase the competitiveness and may lead to a disruption of the internal H-bond and, potentially, to a change in conformation. Therefore, we undertook a preliminary 1H NMR study in the presence of camphor sulfonic acid (CSA) where we expected a conformational change may lead to variation in the 1H NMR spectrum. Unfortunately, under our experimental conditions (100 equiv. CSA in a solution of 3 (2.5 mM) in DMSO-d6/0.5% H2O; see ESI†) minimal changes were observed and suggested that no conformational change occurred. The experiment was repeated in the presence of both acetic acid (AcOH) and trifluoroacetic acid (TFA) under the same conditions but in each case, only minor shifts in the NMR were observed. These results suggest that the intramolecular H-bond is stable under very competitive environments and requires a significant amount of energy to ‘reorganise’ into the desired anti/anti conformation.
Anion transport studies
With just moderate Cl− binding affinities, we expected that the amidosquaramides may not exhibit efficient anion transport as we had initially envisaged. Nevertheless, the anionophoric activity of receptors 1–8 were screened using methods reported by Gale et al.33 Briefly, unilamellar vesicles were created using a 7:3 molar ratio of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and cholesterol with an internal environment of a NaCl solution (487 mM), buffered to pH 7.2 using a sodium phosphate buffer (5 mM). The liposomes were then purified by dialysis to suspend the vesicles in an isotonic external solution of NaNO3 solution, buffered to pH 7.20 using a sodium phosphate buffer (5 mM). Using the external solution, the liposomes were diluted to 0.5 mM before each run was performed. The efflux of chloride was measured using a chloride ion selective electrode (ISE). Theamidosquaramides were added to the vesicles as DMSO solutions via micropipette. See ESI† for detailed assay conditions.
Initially, the anionophoric activity of the amidosquaramides were assessed at pH 7.20 at a concentration of 5 mol% (with respect to POPC concentration), repeated in triplicate. The amidosquaramides were added at 0 seconds and the efflux was monitored for 300 seconds before the addition of a solution of Triton X-100 (11 wt% in H2O:DMSO 7:1 v:v) in order to calibrate the 100% efflux value.
The results from the anion transport assays performed at pH 7.20 (Fig. 6a), revealed that the receptors displayed minimal transport at this pH. Receptor 2 demonstrated the most potent anionophoric activity, yet still failed to reach 50% efflux at 5 mol%. 4 and 5 also showed some activity, but to a lesser extent than 2. The anionophoric activity of 2 is likely due to its relatively high binding affinity for Cl− compared to the other receptors as well as its enhanced lipophilicity brought about by the trifluoromethyl substituents. However, while 3 displayed the highest binding constant and contained multiple trifluoromethyl moieties, which are known to increase lipophilicity and anion transport, the receptor was only able to facilitate approximately 20% Cl− efflux.
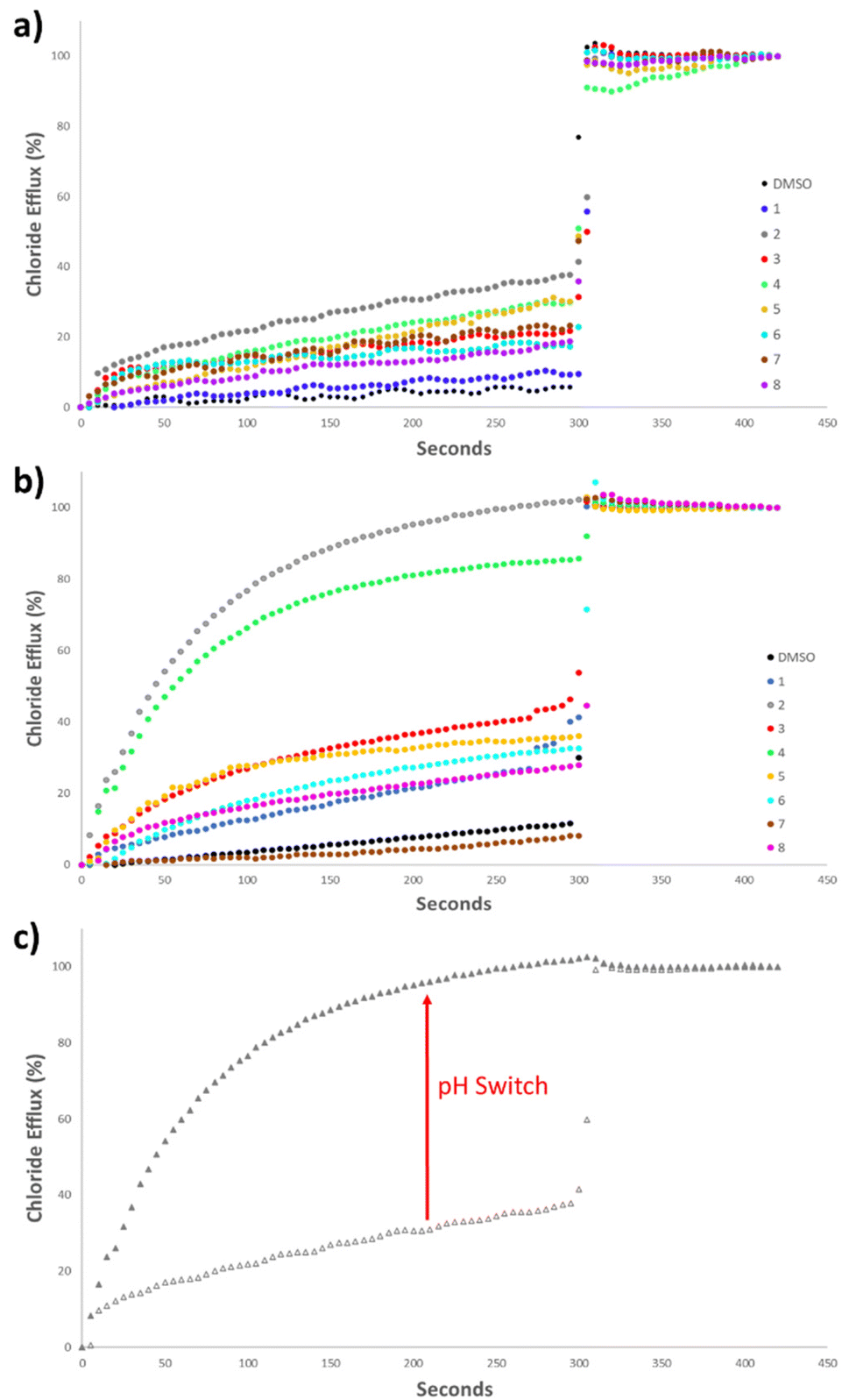 | ||
| Fig. 6 (a) Chloride efflux facilitated by 1–8 (5 mol% receptor to lipid) from unilamellar POPC vesicles loaded with 487 mM NaCl, buffered to pH 7.20 with 5 mM sodium phosphate salts, suspended in 487 mM NaNO3, buffered to pH 7.20 with 5 mM sodium phosphate salts. Each trace represents the average of three trials. (b) Chloride efflux facilitated by 1–8 (5 mol% receptor to lipid) from unilamellar POPC vesicles loaded with 487 mM NaCl, buffered to pH 4.20 with 5 mM citric acid buffer, and suspended in 487 mM NaNO3, buffered to pH 4.20 with 5 mM citric acid buffer. Each trace represents the average of three trials. (c) Chloride efflux facilitated by 2 (5 mol%) at pH 7.2 (empty triangle) and at pH 4.2 (filled triangle). | ||
The poor anion transport ability of the receptors, can be ascribed to a combination of the aforementioned intramolecular H-bonding in addition to their pKa values that fall close to physiological pH. The range of pKa values of the amidosquaramide amide NH were experimentally determined to be between 6.3–8.8. Thus, as the anion transport assays were performed at pH 7.20, a large proportion of the receptors would exist in their deprotonated state. As the amide NH is the only one available to participate in H-bonding, this effectively limits their ability to transport Cl− at neutral pH. Such behaviour has previously been reported for thiosquaramides,20 oxothiosquaramides,34 and phenylthiosemicarbazones.35
Therefore, the anion transport assays were repeated at pH 4.20, using the method described previously, with the solutions buffered to pH 4.20 using a citrate buffer. At acidic pH, a considerable increase was observed in the anionophoric activity of the amidosquaramides (Fig. 6b). 2 facilitated 100% Cl− efflux at 5 mol%, and 4 also demonstrated a stark increase from approximately 30% efflux to approximately 80% efflux. The remaining receptors did not reach 50% efflux of Cl− after 300 seconds, placing 2 and 4 as the most potent anionophores at this pH. Overall, these results established that amidosquaramides can facilitate Cl−/NO3− exchange across a lipid membrane, and display pH-switchable behaviour, where anion transport is ‘switched on’ at acidic pH. Gale and co-workers previously published a QSAR analysis of anion binding and transport where they found that the anion binding abilities of a family of thioureas are dominated by the pKa of the thiourea NH.36 The same work also showed that anion transport ability is mainly dependent on the lipophilicity of the transporter, the size of the transporter and the anion binding affinity. In short, it appears that the factors affecting anion transport are numerous and when dealing with receptors with a pKa close to 7.4, still poorly understood. We postulate that the observed ‘switch’ is either due to the protonation of the receptor at acidic pH or a switch in the conformation of the receptor at the lipid solvent interface (or a combination of both). Further investigation of this phenomenon is ongoing.
Conclusions
In conclusion, we have reported the syntheses of a small family of anion receptors that incorporate a new anion binding motif; amidosquaramides. We have shown, through a combination of experimental and computational methods that these receptors have pKa values close to physiological pH but also display intramolecular H-bonding interactions. Both of these characteristics result in a decrease in binding affinity relative to their squaramide analogues but remain higher than those reported for analogous ureas and thioureas. The entire family of receptors were also screened for chloride transport ability where they showed moderate activity in a Cl−/NO3− exchange assay at physiological pH, however, when experiments were repeated under acidic conditions, a significant increase in transport ability was observed. We suspect that the observed increase under acidic conditions is two-fold; brought about by a reprotonation of acidic NH's as well as a potential disruption of the intramolecular H-bonding (although we were unable to observe this experimentally). We are currently designing receptors containing amidosquaramides with a view to taking advantage of the intramolecular H-bond that could lead to a ‘conformational switch’. Such an advance should provide access to stimuli responsive anion transporters and our detailed investigations will be reported in due course.Experimental
General experimental methods
All chemicals purchased were obtained from commercial suppliers and used without further purification. DCM was distilled over CaH2 and MeCN was dried over 3 Å molecular sieves. Anhydrous DMF was purchased from Sigma Aldrich. TLC was performed on Merck Silica Gel F254 plates and visualised under UV light (λ = 254 nm). Flash chromatography was performed with Merck Silica Gel 60. Compounds were lyophilised on a Labconco Freezone 1 Dry system. NMR spectra were recorded on a Bruker Ascend 500 NMR spectrometer, operated at 500 MHz for 1H NMR analysis and 126 MHz for 13C analysis, both at 293 K. The residual solvent peak was used as an internal standard for DMSO-d6 and (CD3)2CO and TMS for CDCl3. Chemical shifts (δ) were reported in ppm. NMR spectra were processed and stack plots produced using MestReNova 6.0.2 software. Proton and carbon signals were assigned with the aid of 2D NMR experiments (COSY, HSQC and HMBC). Multiplicity is given as s = singlet, bs = broad singlet, d = doublet, brd = broad doublet, dd = doublet of doublets, ddd = doublet of doublet of doublets, t = triplet, q = quartet, m = multiplet as appropriate, and J values are given in Hz. Infrared spectra were obtained via ATR as a solid on a zinc selenide crystal in the region of 4000–400 cm−1 on a PerkinElmer Spectrum 100 FT-IR spectrophotometer. LC-MS was performed on an Agilent Technologies 1200 Series instrument consisting of a G1322A Quaternary pump and a G1314B UV detector (254 nm) coupled to an Advion Expression L Compact Mass spectrometer (ESI) operating in positive mode. High resolution ESI spectra were recorded on an Agilent 6310 LCMS TOF. UV-vis spectra were recorded on a Varian Cary 50 Scan UV-visible spectrophotometer using Cary WinUV software. pH measurements were recorded using a Jenway 350 pH meter. Conductivity measurements were performed using an Orion Star A211 Benchtop pH Meter and a Fisherbrand accumet chloride combination electrode.Author contributions
RE, LM and TK designed the study and wrote the manuscript. RE and TK supervised the study. LM synthesized and characterized the compounds and carried out spectroscopic titrations and anion transport assays. TK conducted computational calculations. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. M. acknowledges Maynooth University for a Hume Scholarship. R. E. and L. M. acknowledges funding from Science Foundation Ireland (SFI), grant number 12/RC/2275/P2, which is co-funded under the European Regional Development Fund. SFI are also acknowledged for the funding of the NMR facility (12/RI/2346/SOF) through the Research Infrastructure Programme and the Advion Compact Mass Spec through the Opportunistic Infrastructure Fund (16/RI/3399). T. K. wishes to acknowledge the DJEI/DES/SFI/HEA Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities and support.Notes and references
- L. A. Marchetti, L. K. Kumawat, N. Mao, J. C. Stephens and R. B. P. Elmes, Chem, 2019, 5, 1398–1485 CAS.
- Y. Li, G.-H. Yang, Y.-Y. Shen, X.-S. Xue, X. Li and J.-P. Cheng, J. Org. Chem., 2017, 82, 8662–8667 CrossRef CAS.
- D. Quiñonero, A. Frontera, G. A. Suñer, J. Morey, A. Costa, P. Ballester and P. M. Deyà, Chem. Phys. Lett., 2000, 326, 247–254 CrossRef.
- L. K. Kumawat, A. A. Abogunrin, M. Kickham, J. Pardeshi, O. Fenelon, M. Schroeder and R. B. P. Elmes, Front. Chem., 2019, 7, 354 CrossRef CAS.
- A. Kerckhoffs and M. J. Langton, Chem. Sci., 2020, 11, 6325–6331 RSC.
- M. Quintana, J. V. Alegre-Requena, E. Marqués-López, R. P. Herrera and G. Triola, MedChemComm, 2016, 7, 550–561 RSC.
- S. Zhang, Y. Wang, W. Xie, E. N. W. Howe, N. Busschaert, A. Sauvat, M. Leduc, L. C. Gomes-da-Silva, G. Chen, I. Martins, X. Deng, L. Maiuri, O. Kepp, T. Soussi, P. A. Gale, N. Zamzami and G. Kroemer, Cell Death Dis., 2019, 10, 242 CrossRef PubMed.
- H. Li, H. Valkenier, A. G. Thorne, C. M. Dias, J. A. Cooper, M. Kieffer, N. Busschaert, P. A. Gale, D. N. Sheppard and A. P. Davis, Chem. Sci., 2019, 10, 9663–9672 RSC.
- E. T. Buurman, M. A. Foulk, N. Gao, V. A. Laganas, D. C. McKinney, D. T. Moustakas, J. A. Rose, A. B. Shapiro and P. R. Fleming, J. Bacteriol., 2012, 194, 5504–5512 CrossRef CAS PubMed.
- D. Quiñonero, R. Prohens, C. Garau, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2002, 351, 115–120 CrossRef.
- S. Cohen and S. G. Cohen, J. Am. Chem. Soc., 1966, 88, 1533–1536 CrossRef CAS.
- A. P. Davis, S. M. Draper, G. Dunne and P. Ashton, Chem. Commun., 1999, 2265–2266, 10.1039/A907179B.
- G. Picci, I. Carreira-Barral, D. Alonso-Carrillo, D. Sanz-González, P. Fernández-López, M. García-Valverde, C. Caltagirone and R. Quesada, Supramol. Chem., 2020, 32, 112–118 CrossRef CAS.
- S. Camiolo, P. A. Gale, M. B. Hursthouse, M. E. Light and A. J. Shi, Chem. Commun., 2002, 758–759, 10.1039/B200980C.
- C. Caltagirone, G. W. Bates, P. A. Gale and M. E. Light, Chem. Commun., 2008, 61–63, 10.1039/B713431B.
- L. Qin, A. Hartley, P. Turner, R. B. P. Elmes and K. A. Jolliffe, Chem. Sci., 2016, 7, 4563–4572 RSC.
- L. Qin, S. J. N. Vervuurt, R. B. P. Elmes, S. N. Berry, N. Proschogo and K. A. Jolliffe, Chem. Sci., 2020, 11, 201–207 RSC.
- H. Liu, C. S. Tomooka and H. W. Moore, Synth. Commun., 1997, 27, 2177–2180 CrossRef CAS.
- F. P. Schmidtchen, Angew. Chem., Int. Ed. Engl., 1977, 16, 720–721 CrossRef.
- N. Busschaert, R. B. P. Elmes, D. D. Czech, X. Wu, I. L. Kirby, E. M. Peck, K. D. Hendzel, S. K. Shaw, B. Chan, B. D. Smith, K. A. Jolliffe and P. A. Gale, Chem. Sci., 2014, 5, 3617–3626 RSC.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- C. P. Kelly, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2007, 111, 408–422 CrossRef CAS.
- X. Ni, X. Li, Z. Wang and J.-P. Cheng, Org. Lett., 2014, 16, 1786–1789 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- R. B. P. Elmes, P. Turner and K. A. Jolliffe, Org. Lett., 2013, 15, 5638–5641 CrossRef CAS PubMed.
- M. Ximenis, E. Bustelo, A. G. Algarra, M. Vega, C. Rotger, M. G. Basallote and A. Costa, J. Org. Chem., 2017, 82, 2160–2170 CrossRef CAS PubMed.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- https://supramolecular.org .
- N. Busschaert, I. L. Kirby, S. Young, S. J. Coles, P. N. Horton, M. E. Light and P. A. Gale, Angew. Chem., Int. Ed., 2012, 51, 4426–4430 CrossRef CAS PubMed.
- K. J. Winstanley and D. K. Smith, J. Org. Chem., 2007, 72, 2803–2815 CrossRef CAS.
- P. V. Santacroce, J. T. Davis, M. E. Light, P. A. Gale, J. C. Iglesias-Sánchez, P. Prados and R. Quesada, J. Am. Chem. Soc., 2007, 129, 1886–1887 CrossRef CAS PubMed.
- J. D. E. Lane, S. N. Berry, W. Lewis, J. Ho and K. A. Jolliffe, J. Org. Chem., 2021, 86, 4957–4964 CrossRef CAS PubMed.
- L. A. Jowett and P. A. Gale, Supramol. Chem., 2019, 31, 297–312 CrossRef CAS.
- R. B. P. Elmes, N. Busschaert, D. D. Czech, P. A. Gale and K. A. Jolliffe, Chem. Commun., 2015, 51, 10107–10110 RSC.
- E. N. W. Howe, N. Busschaert, X. Wu, S. N. Berry, J. Ho, M. E. Light, D. D. Czech, H. A. Klein, J. A. Kitchen and P. A. Gale, J. Am. Chem. Soc., 2016, 138, 8301–8308 CrossRef CAS PubMed.
- N. Busschaert, S. J. Bradberry, M. Wenzel, C. J. E. Haynes, J. R. Hiscock, I. L. Kirby, L. E. Karagiannidis, S. J. Moore, N. J. Wells, J. Herniman, G. J. Langley, P. N. Horton, M. E. Light, I. Marques, P. J. Costa, V. Félix, J. G. Freya and P. A. Gale, Chem. Sci., 2013, 4, 3036–3045 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01176j |
| This journal is © The Royal Society of Chemistry 2022 |