
Harvesting mechanical energy for hydrogen generation by piezoelectric metal–organic frameworks†
Shiyin
Zhao‡
abc,
Maosong
Liu‡
 d,
Yuqiao
Zhang
d,
Zhicheng
Zhao
e,
Qingzhe
Zhang
ef,
Zhenliang
Mu
e,
Yangke
Long
a,
Yinhua
Jiang
d,
Yong
Liu
e,
Jianming
Zhang
d,
Shun
Li
*d,
Xuanjun
Zhang
*b and
Zuotai
Zhang
*a
d,
Yuqiao
Zhang
d,
Zhicheng
Zhao
e,
Qingzhe
Zhang
ef,
Zhenliang
Mu
e,
Yangke
Long
a,
Yinhua
Jiang
d,
Yong
Liu
e,
Jianming
Zhang
d,
Shun
Li
*d,
Xuanjun
Zhang
*b and
Zuotai
Zhang
*a
aSchool of Environmental Science and Engineering, Southern University of Science and Technology, Shenzhen 518055, Guangdong, China. E-mail: zhangzt@sustech.edu.cn
bMOE Frontiers Science Center for Precision Oncology, Faculty of Health Sciences, University of Macau, Macau, SAR 999078, China. E-mail: XuanjunZhang@um.edu.mo
cCollege of Liberal Arts and Science, National University of Defense Technology, Changsha 410073, Hunan, China
dInstitute of Quantum and Sustainable Technology (IQST), School of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, Jiangsu, China. E-mail: shun@ujs.edu.cn
eFoshan (Southern China) Institute for New Materials, Foshan 528200, Guangdong, China
fShandong Key Laboratory of Environmental Processes and Health, School of Environmental Science and Engineering, Shandong University, Qingdao 266237, Shandong, China
First published on 29th April 2022
Abstract
Piezocatalysis, the process of directly converting mechanical energy into chemical energy, has emerged as a promising alternative strategy for green H2 production. Nevertheless, conventional inorganic piezoelectric materials suffer from limited structural tailorability and small surface area, which greatly impedes their mechanically driven catalytic efficiency. Herein, we design and fabricate a novel UiO-66(Zr)-F4 metal–organic framework (MOF) nanosheet for piezocatalytic water splitting, with the highest H2 evolution rate reaching 178.5 μmol g−1 within 5 h under ultrasonic vibration excitation (110 W, 40 kHz), far exceeding that of the original UiO-66 host. A reduced bandgap from 2.78 to 2.43 eV is achieved after introducing a fluorinated ligand. Piezoresponse force microscopy measurements demonstrate a much stronger piezoelectric response for UiO-66(Zr)-F4, which may result from the polarity of the introduced fluorinated ligand. This work highlights the potential of MOF-based porous piezoelectric nanomaterials in harvesting mechanical energy to drive chemical reactions such as water splitting.
New conceptsPiezoelectric nanomaterials can generate charges in response to stress and drive chemical reactions by converting mechanical energy, providing an alternative means for mild mechanochemistry towards various catalytic applications such as water-splitting for H2 generation. Nevertheless, conventional inorganic semiconductors suffer from limited structural tailorability, small surface area, and relatively long transport distance of the generated charge carriers, which impedes their piezocatalytic efficiency. Piezo-/ferroelectric metal–organic frameworks (MOFs) are ideal candidates for piezocatalysis to replace conventional inorganic semiconductor catalysts, owing to their large surface areas, great structural variability, and mild synthetic procedures. Here, for the first time, we design and fabricate UiO-66-F4 MOFs with a strong piezoelectric response by introducing a polar fluorinated ligand into UiO-66 for H2 production from water driven by ultrasonication-induced periodic mechanical vibrations. A high H2 evolution rate of 178.5 μmol g−1 within 5 h is realized under ultrasonic mechanical vibration (110 W, 40 kHz), which is a more than 100 times enhancement compared to the original UiO-66 host. This work not only provides a feasible strategy to regulate the piezoelectricity of UiO-66-type MOFs, but also sheds light on utilizing novel MOF-based porous piezoelectric nanomaterials as efficient piezocatalysts for mechanically driven water splitting. |
Introduction
The energy shortage has been widely recognized as a critical global issue in the past few decades. Hydrogen (H2) is regarded as one of the most ideal clean energy resources to replace fossil fuels in the future, owing to its advantages of high energy density, low cost, and widespread properties.1 In recent years, green H2 production has been realized by converting various forms of renewable energy, such as solar, thermal, and mechanical energy.2–4 In particular, piezocatalysis has been emerging as a promising alternative technology for H2 generation via water-splitting, as it can harvest various waste mechanical energies including vibrations, wind, and water waves from the surrounding environment.5,6 The currently developed piezocatalysts are mainly confined to inorganic semiconductor materials such as metal oxides (e.g., BaTiO3,7 BiFeO3,8 Bi0.5Na0.5TiO39 and AgNbO310), nitrides (e.g., C3N411 and GaN12), and transition metal dichalcogenides (TMDs).13–15. Nevertheless, these inorganic semiconductors suffer from limited structural tailorability, small surface area, and relatively long transport distance of the generated charge carriers, which greatly impedes their piezocatalytic efficiency. Therefore, there is a critical need to develop new material systems toward high efficient piezocatalytic H2 production driven by weak mechanical energy.Metal–organic frameworks (MOFs), a class of crystalline porous materials with high surface area and great structural variability, have aroused broad interests in various fields such as gas adsorption and separation, drug delivery, and catalysis.16–18 In recent years, MOFs have also been considered as promising candidates to replace conventional piezo-/ferroelectric materials because of their unique superiority combining both inorganic bricks with favorable chemical, thermal, and mechanical stabilities, and organic linkers with easily tailored molecular structures.19,20 Moreover, MOFs are usually synthesized by simple and facile methods at relatively low temperatures, which is a great asset when compared with traditional oxide-based piezo-/ferroelectrics. Until now, significant progress has been made in developing polar MOF materials with outstanding piezo-/ferroelectric properties,21–23 showing promising potential for various applications in actuators, sensors, and energy harvesting technologies. Due to their large surface areas, strong piezo-/ferroelectric responses, and mild synthetic procedures, MOFs are ideal candidates to be utilized in the field of piezocatalysis. However, to the best of our knowledge, research work on the piezocatalytic process that harvests mechanical energy for clean energy production (e.g., water splitting for H2 evolution) over MOF-based materials is very rare.24 Therefore, the design and fabrication of novel MOF-based piezocatalysts for high efficient H2 production from water is highly required.
Herein, for the first time, we report UiO-66-type UiO-66(Zr)-F4 MOF nanosheets as piezocatalysts for H2 production from water driven by ultrasonication-induced periodic mechanical vibrations. Although UiO-66 was believed to be piezo-/ferroelectrically inactive previously, very recent works combining X-ray absorption fine structure analysis and ab initio calculations suggested a low symmetric crystal structure [F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3m (216)].25 In addition, nanoscale probing characterizations further proved that the UiO-66 nanocrystals indeed show certain piezo-/ferroelectric responses.26,27 Motivated by these studies, we designed and fabricated a novel UiO-66-F4 nanosheet MOFs with a strong piezoelectric response by introducing a polar fluorinated ligand and applied it in piezocatalytic water splitting. A high H2 evolution rate of 178.5 μmol g−1 within 5 h was realized over the UiO-66-F4 nanosheets under ultrasonic mechanical vibration (110 W, 40 kHz), which is a more than 100 times enhancement compard to the original UiO-66 host. The piezoelectric properties were corroborated by piezoresponse force microscopy (PFM) measurements. Moreover, density functional theory (DFT) calculations were performed to investigate the electronic band structures and thermodynamic reaction mechanisms.
3m (216)].25 In addition, nanoscale probing characterizations further proved that the UiO-66 nanocrystals indeed show certain piezo-/ferroelectric responses.26,27 Motivated by these studies, we designed and fabricated a novel UiO-66-F4 nanosheet MOFs with a strong piezoelectric response by introducing a polar fluorinated ligand and applied it in piezocatalytic water splitting. A high H2 evolution rate of 178.5 μmol g−1 within 5 h was realized over the UiO-66-F4 nanosheets under ultrasonic mechanical vibration (110 W, 40 kHz), which is a more than 100 times enhancement compard to the original UiO-66 host. The piezoelectric properties were corroborated by piezoresponse force microscopy (PFM) measurements. Moreover, density functional theory (DFT) calculations were performed to investigate the electronic band structures and thermodynamic reaction mechanisms.
Results and discussion
A microwave-assisted hydrothermal method was employed to construct the UiO-66(Zr)-F4 MOFs. Experimental details are described in the ESI.† Briefly, a mixed water/HAc (30/20) solution containing ZrCl4 and tetrafluoroterephthalic acid (H2BDC-F4) was introduced into a microwave oven and treated at 100 °C for 4 h. As shown in Fig. 1(a) and (b), the as-obtained powder sample displays a uniform sheet morphology with lateral sizes in the range of about 20 to 150 nm and a thickness less than 20 nm, as confirmed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) characterizations. Distinctly different from previously reported particle-shaped UiO-66-F4,28,29 this is the first report of ultrathin nanosheets with the assistance of microwaves during the hydrothermal synthetic process. Meanwhile, the energy dispersive spectroscopy (EDS) mapping images (Fig. 1(c)–(f)) clearly display the homogenous distribution of Zr, C, O and F elements throughout the nanosheets. The high-resolution TEM (HRTEM) image (Fig. 1(g)) exhibits well-defined lattice fringes, which demonstrates good crystallinity of the as-prepared UiO-66-F4 MOFs. The observed spacings of about 1.28 nm correspond to the (111) crystal plane of UiO-66-type MOFs. Moreover, the integrated differential phase-contrast scanning transmission electron microscopy (iDPC-STEM) image (Fig. 1(h)) clearly shows benzene rings and metal clusters, which perfectly correspond to the crystal structure of UiO-66.25,30,31 For comparison, the UiO-66 host was also synthesized, presenting octahedron-shaped nanocrystals with sizes ranging from around 200 to 900 nm with uniform distribution of Zr, C and O elements (Fig. S1, ESI†). The size distributions of both UiO-66 and UiO-66-F4 MOFs are displayed in Fig. S2 (ESI†). The well-defined selected area electron diffraction (SAED) patterns and lattice fringes prove the single-crystalline nature of the UiO-66 MOFs. It is noteworthy that the lattice fringe of UiO-66 (1.18 nm) is smaller than that of UiO-66-F4, indicating a larger distance and weak coordination between the H2BDC-F4 ligand and the Zr metals. | ||
| Fig. 1 (a) SEM, (b) TEM, (c)–(f) EDS mapping, (g) high-resolution TEM, and (h) STEM iDPC images (inset shows the corresponding structure) of UiO-66-F4. | ||
The crystal structure of the UiO-66-F4 framework is depicted in Fig. 2(a) and Fig. S3 (ESI†). UiO-66(Zr) contains inorganic Zr6O4(OH)4 units and BDC linkers. UiO-66-F4, an isostructural perfluorinated UiO-66, was constructed by ZrCl4 and tetrafluoroterephthalic acid (H2BDC-F4), employing acetic acid as a modulator. The F-BDC2− linker is labile and coordinates weakly to the Zr(IV) metal centers due to the four electron-withdrawing atoms on the benzene ring.32 The X-ray diffraction (XRD) patterns for the UiO-66 and UiO-66-F4 samples are shown in Fig. 2(b). The diffraction peaks of both samples match well with the simulated profile for UiO-66 (fcu), confirming the successful assembly of the crystalline phase of the UiO-66-type MOFs.33 The relatively wide peaks of the UiO-66-F4 indicate inferior crystallinity compared to UiO-66. Consistent with previous reports,34 the peaks of the UiO-66-F4 shifted to lower diffraction angles, which is in good agreement with the lattice variation observed by HRTEM, demonstrating the expansion of the UiO-66 unit cell after introducing a fluorinated ligand.
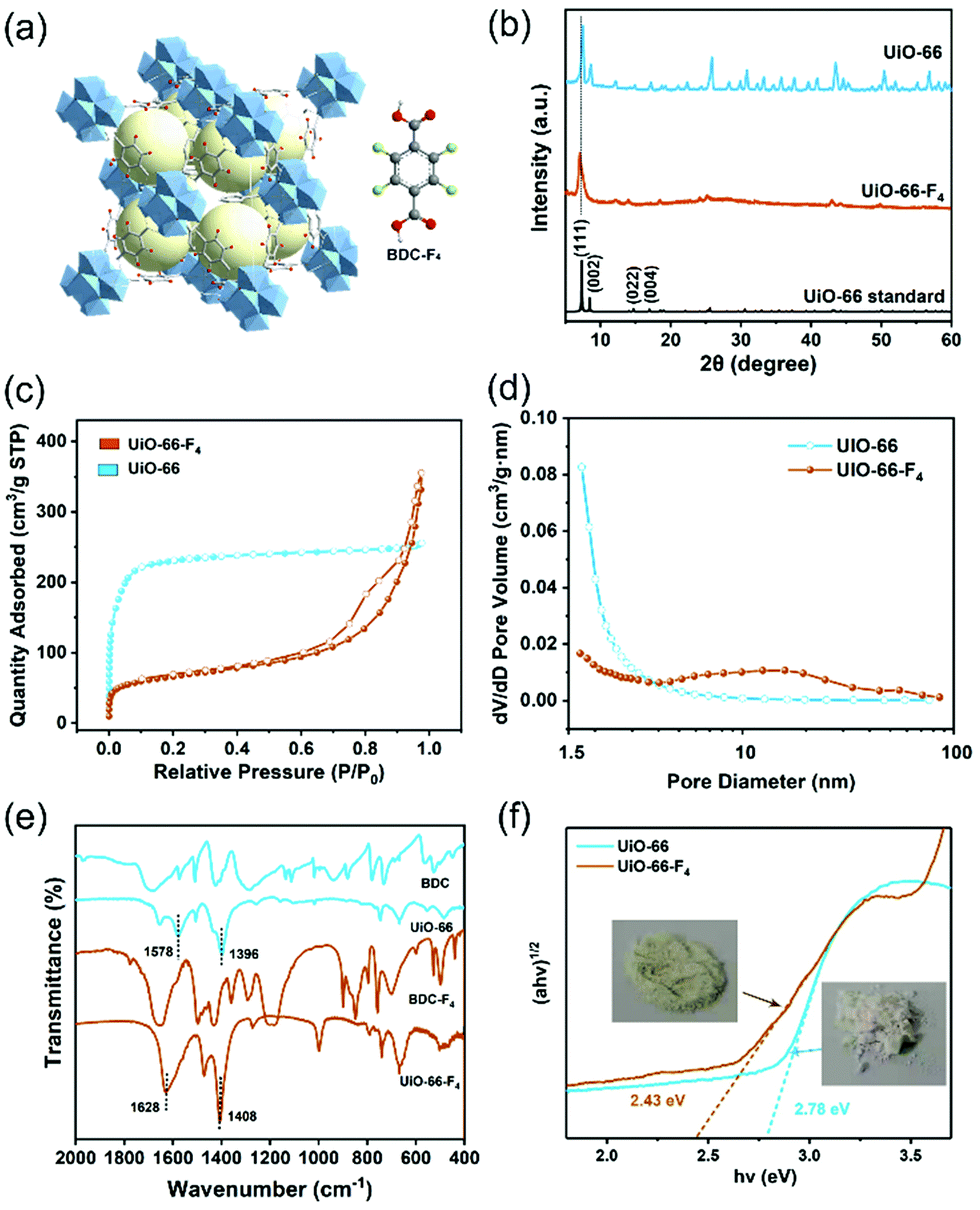 | ||
| Fig. 2 (a) Crystal structure of the UiO-66-F4 framework and H2BDC-F4 linker. (b) XRD patterns, (c) Ar adsorption–desorption isotherms, (d) size distributions, (e) FTIR spectra, and (f) optical band gap calculation using Tauc's equation of UiO-66 and UiO-66-F4. | ||
The surface areas of the two different UiO-66-type MOFs were evaluated by the Brunauer–Emmett–Teller (BET) method from Ar adsorption–desorption isotherms (Fig. 2(c)). The calculated BET surface areas of the UiO-66 and UiO-66-F4 MOFs are 832.8 and 212.0 m2 g−1, respectively. Meanwhile, their porosity was analyzed by the Barrett–Joyner–Halenda (BJH) method (Fig. 2(d)). UiO-66-F4 presents a larger average pore diameter (13.94 nm) and cumulative volume of the pores (0.31 cm3 g−1) (Fig. S4, ESI†) than UiO-66 (3.03 nm and 0.28 cm3 g−1). As reported in previous works on UiO-66-type MOFs, the surface areas and pore structures could be drastically changed after introducing ligands such as OH, COOH, NO2, NH2, Br and F.35,36 This is because of the significant role of the polar properties of different ligands in the crystallization process. The extremely high surface area and porous structure of the as-prepared piezoelectric MOFs overcome the disadvantage of conventional piezoelectric materials as catalysts.
Fourier-transform infrared spectroscopy (FTIR) measurements were carried out to investigate the coordination mode between the metal cluster and ligands. As shown in Fig. 2(e), both UiO-66 and UiO-66-F4 exhibit entirely distinct spectra with the precursor ligands. For UiO-66, two dominant FTIR bands appearing at around 1396 and 1578 cm−1 are attributed to vs and vasy of the carboxyl group, respectively. The difference of Δv = 182 cm−1 indicates that the carboxyl group in the organic linkers mainly adopts a bridging bidentate coordination mode with the Zr nodes. For UiO-66-F4, two dominant FTIR bands appearing at 1408 and 1628 cm−1 are attributed to vs and vasy of the carboxyl group, respectively, and the Δv = 230 cm−1 demonstrates that it adopts a similar coordination mode with UiO-66, according to reported works.37–39 The optimized crystal structures obtained by density functional theory (DFT) calculations also reveal that bidentate is the dominant coordination mode (Fig. S3, ESI†), which strongly supports the FTIR results. The chemical states of the elements and the coordination mode were further characterized using X-ray photoelectron spectroscopy (XPS), as shown in Fig. S5 (ESI†). The Zr 3d XPS spectra for both samples exhibit a spin-orbit doublet that splits into 3d5/2 (182.7 eV) and 3d3/2 (185.1 eV), which are attributed to Zr4+ and Zr8+, respectively, demonstrating the formation of Zr6O4(OH)4(–COO)12 in the frameworks.40,41 The deconvoluted peaks for O 1s at around 531.6 and 529.8 to 530.6 eV are ascribed to the –COO– and lattice O bands, respectively. For UiO-66-F4, the binding energy at 688.7 eV can be assigned to the F 1s orbital.
The color variations of the two MOF samples (inset of Fig. 2(f)) indicate that the electronic band structure around the band gap is significantly modified. The optical band gaps were estimated to be 2.78 and 2.43 eV for the UiO-66 and UiO-66-F4, respectively (Fig. 2(f)), based on UV−Vis diffuse reflectance spectroscopy (DRS). Moreover, the photoluminescence (PL) spectra were also obtained (Fig. S6, ESI†), which show that the excitation peak slightly shifts toward a longer wavelength, demonstrating the band gap reduction of UiO-66-F4. According to the previously reported theoretical and experimental studies, the band gap of UiO-66 can be effectively regulated upon hydrogen substitutions (e.g., NH2 and NO2) on the organic linker.42,43 Our results clearly demonstrate that the introduction of the fluorinated ligand could largely reduce the band gap. To get a deeper understanding of the band structure, electrochemical measurements were carried out using the UiO-66 and UiO-66-F4 MOF-based electrodes. As shown in Fig. 3(a), the UiO-66-F4 shows a much smaller arc radius of the electrochemical impedance curve, indicating a smaller resistance and band gap than that of the UiO-66 host. Mott–Schottky plots were further obtained from the capacitance impedance measurements at three different frequencies (0.5, 1 and 1.5 kHz). As shown in Fig. 3(b), the positive slope of the C−2 curve indicates an n-type semiconductor, and the x-axis intercept occurs at −0.44 V (vs. Ag/AgCl). As a result, the conduction band (CB) was estimated to be at −0.24 eV, which is more negative than the potential of H+/H2 (0 eV). The UiO-66 shows a similar band position (Fig. S7, ESI†). Therefore, both UiO-66 and UiO-66-F4 could fulfill the requirement for hydrogen evolution via water splitting.
 | ||
| Fig. 3 (a) Electrochemical impedance spectra of UiO-66 and UiO-66-F4. (b) Mott–Schottky plots of UiO-66-F4 at different frequencies. DFT calculation of the band structures of (c) UiO-66 and (d) UiO-66-F4. PFM phase and amplitude hysteresis loops of (e) UiO-66 and (f) UiO-66-F4. | ||
To obtain further insight into the band structure, DFT calculations were carried out on the two UiO-66-type MOFs. The results show that both UiO-66 and UiO-66-F4 have a direct energy bandgap. The theoretical band gap values were calculated to be 2.87 eV and 2.38 eV for UiO-66 (Fig. 3(c)) and UiO-66-F4 (Fig. 3(d)), respectively, which is consistent with the UV-Vis spectral analysis. Contrary to the unfunctionalized UiO-66 material, the UiO-66-F4 MOFs possess a gap state within the original band gap. Densities of states (DOS) provide more insight into the origins of distinct bands (Fig. S8, ESI†). The CB mainly corresponds to that of the Zr 3d electrons or to orbitals delocalized over the entire linker, which is rather insensitive to the functional group. This result is in good agreement with the Mott–Schottky electrochemical measurements. In contrast, upon functionalization, the fluorinated ligand forms a bond with the π orbitals of the aromatic ring,43 giving rise to the top levels of the valence band (VB). As a result, a new VB was formed, thus lowering the effective band gap substantially.
The piezoelectricity of the UiO-66 and UiO-66-F4 MOFs was investigated by piezoresponse force microscopy (PFM). As shown in Fig. 3(e) and (f), both samples exhibit well-defined 180° phase-reversal hysteresis, demonstrating a characteristic polarization switching behavior of the as-prepared MOFs. Furthermore, typical amplitude-voltage butterfly loops were obtained for both samples under a 10 V DC bias field. The larger amplitude displacement of UiO-66-F4 (52.1 mV, Fig. 3(e)) indicates a much stronger piezoelectric response than that of UiO-66 (17.5 mV, Fig. 3(f)). Previous reports disclose that the structure of UiO-66 may not be the highly centrosymmetric Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (225) but a reduced symmetry form.26 Moreover, the polarities of MOFs can be endowed by modifying traditional MOF linkers or introducing novel polar linkers.44 In the present work, the substitution by the F4-BDC2− linker that coordinates weakly to the Zr(IV) metal centers may cause a strong polarity in the UiO-66 type MOF framework. In addition, the strong hydrogen bonding between F and μ3-OH could also lead to a larger polarization of the modified MOFs. Further studies are needed to clarify the origin of the strong polarization in UiO-66-F4 MOFs.
m (225) but a reduced symmetry form.26 Moreover, the polarities of MOFs can be endowed by modifying traditional MOF linkers or introducing novel polar linkers.44 In the present work, the substitution by the F4-BDC2− linker that coordinates weakly to the Zr(IV) metal centers may cause a strong polarity in the UiO-66 type MOF framework. In addition, the strong hydrogen bonding between F and μ3-OH could also lead to a larger polarization of the modified MOFs. Further studies are needed to clarify the origin of the strong polarization in UiO-66-F4 MOFs.
The piezocatalytic water splitting performance of the as-prepared UiO-66–type MOFs was evaluated in a Pyrex reaction cell connected to a closed gas circulation system (Labsolar-6A, Beijing Perfectlight Co., Ltd) using Na2SO3 as the sacrificial agent. An ultrasonic cleaner (Branson 2800) with a maximum power of 110 W and a frequency of 40 kHz was used to periodically apply a local mechanical strain to the MOF nanostructures. The time-dependent H2 evolution curves over different samples are shown in Fig. 4(a). The UiO-66 MOF presents a negligible H2 production amount of merely about 5 μmol within 5 h. In contrast, the UiO-66-F4 sample exhibits a significantly enhanced water splitting performance, with 178.5 μmol g−1 H2 being produced under the same conditions, which is more than 100 times higher than that of the original UiO-66 host. The piezocatalytic H2 production activity of UiO-66-F4 is higher than that of previously reported organolead halide perovskite CH3NH3PbI3,45 GaN nanowires,12 and few-layer TMDs.14 The piezocatalytic activity of the UiO-66-F4 nanosheets was further evaluated under different frequencies of ultrasonication (Fig. S9, ESI†), demonstrating that the catalytic efficiency is highly dependent on the frequency, with the highest H2 generation rate obtained at 45 kHz. In addition, cycling tests were conducted to investigate the stability of the as-prepared MOFs under mechanical vibration (Fig. 4(a)), which shows that the catalytic performance almost remained constant in three cycles. The stable performance can be ascribed to the high crystallinity of the UiO-66 type MOFs. Moreover, the morphology and crystal structure of the UiO-66 nanosheets were perfectly reserved after 15 h piezocatalytic reactions (Fig. S10, ESI†), demonstrating excellent structural stability of the MOF piezocatalysts under ultrasonic vibration. To validate the generated transient radical intermediates from the UiO-66-F4 piezoelectric MOFs under mechanical pressure, electron spin resonance (ESR) trapping experiments were carried out by adding 5,5-dimethyl-1-pyrroline N-oxide (DMPO) in aqueous and methanol solutions, respectively. As shown in Fig. S11 (ESI†), characteristic peaks of a spin adduct of DMPO-˙OH and DMPO-˙ O2− were observed after applying ultrasonic vibration for 10 min, suggesting the generation of positive and negative piezoelectric charges.
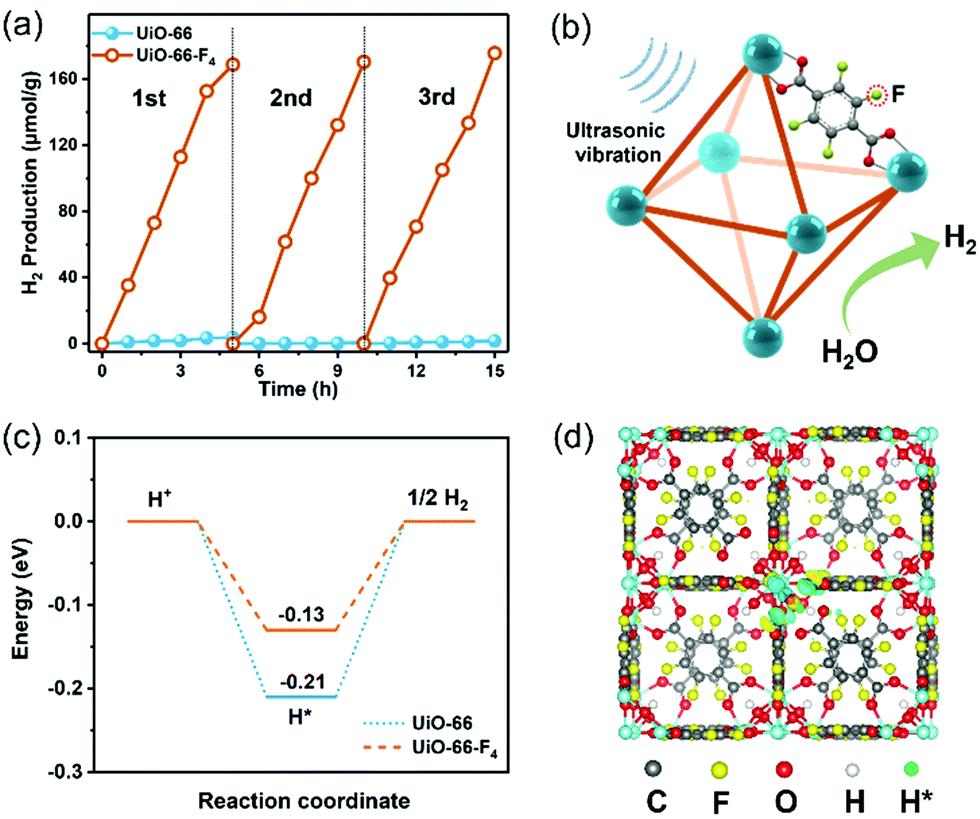 | ||
| Fig. 4 (a) Three cycles of time-dependent H2 evolution curves over UiO-66 and UiO-66-F4 under ultrasonic vibration. (b) Schematic illustration of the mechanism of piezocatalytic H2 production from water over the UiO-66-F4via harvesting vibration energy. (c) Gibbs free energy (ΔGH*) diagram for hydrogen adsorption on UiO-66 and UiO-66-F4. (d) Most stable atomic configurations of hydrogen adsorption on UiO-66-F4. | ||
Based on the above results, the proposed mechanism for piezocatalytic H2 production via water splitting over the UiO-66-type MOFs can be explained as follows (Fig. 4(b)). When the piezoelectric MOF nanocrystals were subjected to mechanical vibration, numerous free charges are excited on the surface of the MOFs. The electrons on the CB could effectively harvest the free H+ to produce H2. Simultaneously, the holes accumulated on the VB will be fulfilled by the sacrificial agents. As discussed earlier, the CB edge for the UiO-66-type MOF is more negative than the potential of H+/H2, thus strongly supporting its continuous and stable H2 production capability. The band gap of the original UiO-66 host was substantially reduced after introducing a fluorinated ligand. Consequently, the excitation of the linker electrons from this newly split-off gap state is much easier to transfer to the CB and drive water-splitting catalytic reactions. Meanwhile, the UiO-66-F4 is found to exhibit much stronger piezoelectricity than the UiO-66 MOF, which is crucial for the significantly enhanced piezocatalytic activity. Herein, to gain a deeper insight into the effects of F atoms on the interfacial H2 evolution reaction, the free energies of H adsorption (ΔGH*) for the UiO-66 and UiO-66-F4 were calculated based on the DFT method (Fig. 4(c) and (d)). For the UiO-66, the ΔGH* value was calculated to be −0.21 eV. In contrast, the ΔGH* value of the UiO-66-F4 catalyst is obviously weakened (−0.13 eV), which facilitates the subsequent desorption of Hads and promotes the rapid interfacial H2-production rate. Our results clearly demonstrate that the introduction of fluorinated ligand into the UiO-66 framework successfully promotes the piezocatalytic performance for water splitting H2 production by simultaneously optimizing the band gap structure and the piezoelectric response. The present work has enriched the candidate material systems for harvesting mechanical energy toward green H2 production via a piezocatalysis process. With the great advantages of large surface areas, high structural variability for modulating piezoelectricity, and mild synthetic procedures, well-designed MOF materials are highly promising to be utilized in the field of piezocatalysis in the future.
Conclusions
In summary, piezocatalytic H2 evolution via water splitting was realized over ultrathin UiO-66-F4 nanosheets. A combined spectroscopy analysis and DFT calculations revealed a reduction in the band gap from 2.78 to 2.43 eV upon hydrogen substitution on the organic linker of the UiO-66 by a fluorinated ligand. Moreover, PFM measurements demonstrated a much stronger piezoresponse for the UiO-66-F4 than the unfunctionalized UiO-66 counterpart. The piezocatalytic H2 yield rate over UiO-66-F4 was 178.5 μmol g−1 under 110 W ultrasonic vibration excitation at the frequency of 40 kHz for 5 h, far exceeding that of the original UiO-66 host. This work not only provides a feasible strategy to regulate the piezoelectricity of the UiO-66 MOFs, but also sheds light on utilizing MOFs as efficient piezocatalysts for water splitting.Author contributions
S. L. conceived and designed the experiments. S. Z., M. L. and Q. Z. conducted the materials synthesis and catalytic measurements. Y. Z., Z. Z. and Y. K. L. performed structural characterizations. Y. L., Z. M., Y. J. and J. Z. analysed the results. S. L., Z. Z. and X. Z. supervised the work. S. L. and S. Z. co-wrote the paper. All authors commented on the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22075126), the Shenzhen Science and Technology Innovation Committee (JCYJ20200109141437586 and JCYJ20180504165648211), the Science and Technology Development Fund, Macau SAR (019/2017/AMJ, 0114/2019/A2 and 085/2020/A2), the Guangdong Basic and Applied Basic Research Foundation (2021A1515111137), and the startup funding for Jinshan Distinguished professor project of Jiangsu University. In addition, we appreciate the technological support from Thermo Fisher Scientific for TEM characterizations http://www.shiyanjia.com.References
- J. K. Dawson, Nature, 1974, 249, 724–726 CrossRef CAS.
- P. Ganguly, M. Harb, Z. Cao, L. Cavallo, A. Breen, S. Dervin, D. D. Dionysiou and S. C. Pillai, ACS Energy Lett., 2019, 4, 1687–1709 CrossRef CAS.
- J. Qi, W. Zhang and R. Cao, Adv. Energy Mater., 2018, 8, 1701620 CrossRef.
- S. Li, Z. Zhao, J. Zhao, Z. Zhang, X. Li and J. Zhang, ACS Appl. Nano Mater., 2020, 3, 1063–1079 CrossRef CAS.
- S. Tu, Y. Guo, Y. Zhang, C. Hu, T. Zhang, T. Ma and H. Huang, Adv. Funct. Mater., 2020, 30, 2005158 CrossRef CAS.
- M. Wang, B. Wang, F. Huang and Z. Lin, Angew. Chem., Int. Ed., 2019, 58, 7526–7536 CrossRef CAS PubMed.
- R. Su, H. A. Hsain, M. Wu, D. Zhang, X. Hu, Z. Wang, X. Wang, F.-t Li, X. Chen, L. Zhu, Y. Yang, Y. Yang, X. Lou and S. J. Pennycook, Angew. Chem., Int. Ed., 2019, 58, 15076–15081 CrossRef CAS PubMed.
- H. You, Z. Wu, L. Zhang, Y. Ying, Y. Liu, L. Fei, X. Chen, Y. Jia, Y. Wang, F. Wang, S. Ju, J. Qiao, C.-H. Lam and H. Huang, Angew. Chem., Int. Ed., 2019, 58, 11779–11784 CrossRef CAS PubMed.
- Z. Zhao, L. Wei, S. Li, L. Zhu, Y. Su, Y. Liu, Y. Bu, Y. Lin, W. Liu and Z. Zhang, J. Mater. Chem. A, 2020, 8, 16238–16245 RSC.
- S. Li, Z. C. Zhao, M. S. Liu, X. B. Liu, W. Huang, S. K. Sun, Y. H. Jiang, Y. Liu, J. M. Zhang and Z. T. Zhang, Nano Energy, 2022, 95, 107031 CrossRef CAS.
- C. Hu, F. Chen, Y. Wang, N. Tian, T. Ma, Y. Zhang and H. Huang, Adv. Mater., 2021, 33, 2101751 CrossRef CAS PubMed.
- M. Zhang, S. Zhao, Z. Zhao, S. Li and F. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 10916–10924 CrossRef CAS PubMed.
- Y. Su, L. Zhang, W. Wang, X. Li, Y. Zhang and D. Shao, J. Mater. Chem. A, 2018, 6, 11909–11915 RSC.
- S. Li, Z. Zhao, D. Yu, J.-Z. Zhao, Y. Su, Y. Liu, Y. Lin, W. Liu, H. Xu and Z. Zhang, Nano Energy, 2019, 66, 104083 CrossRef CAS.
- S. Karmakar, A. Pramanik, A. K. Kole, U. Chatterjee and P. Kumbhakar, J. Hazard. Mater., 2022, 424, 127702 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS.
- W. Zheng and L. Y.-S. Lee, ACS Energy Lett., 2021, 6, 2838–2843 CrossRef CAS.
- W. Zhang and R.-G. Xiong, Chem. Rev., 2012, 112, 1163–1195 CrossRef CAS PubMed.
- K. Asadi and M. A. van der Veen, Eur. J. Inorg. Chem., 2016, 4332–4344 CrossRef CAS.
- M. Mon, J. Ferrando-Soria, M. Verdaguer, C. Train, C. Paillard, B. Dkhil, C. Versace, R. Bruno, D. Armentano and E. Pardo, J. Am. Chem. Soc., 2017, 139, 8098–8101 CrossRef CAS PubMed.
- H.-R. Zhao, D.-P. Li, X.-M. Ren, Y. Song and W.-Q. Jin, J. Am. Chem. Soc., 2010, 132, 18–19 CrossRef CAS PubMed.
- G.-C. Xu, X.-M. Ma, L. Zhang, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2010, 132, 9588–9590 CrossRef CAS PubMed.
- C. Zhang, D. Lei, C. Xie, X. Hang, C. He and H.-L. Jiang, Adv. Mater., 2021, 33, 2106308 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- Y. Sun, J. Gao, Y. Cheng, Y.-W. Zhang and K. Zeng, J. Phys. Chem. C, 2019, 123, 3122–3129 CrossRef CAS.
- Y. Sun, Z. Hu, D. Zhao and K. Zeng, Nanoscale, 2017, 9, 12163–12169 RSC.
- F. Zhang, X. Hu, E. W. Roth, Y. Kim and S. T. Nguyen, Chem. Mater., 2020, 32, 4292–4302 CrossRef CAS.
- M. Campanelli, T. Del Giacco, F. De Angelis, E. Mosconi, M. Taddei, F. Marmottini, R. D’Amato and F. Costantino, ACS Appl. Mater. Interfaces, 2019, 11, 45031–45037 CrossRef CAS PubMed.
- P. Ji, T. Drake, A. Murakami, P. Oliveres, J. H. Skone and W. Lin, J. Am. Chem. Soc., 2018, 140, 10553–10561 CrossRef CAS PubMed.
- S. Øien, D. Wragg, H. Reinsch, S. Svelle, S. Bordiga, C. Lamberti and K. P. Lillerud, Cryst. Growth Des., 2014, 14, 5370–5372 CrossRef.
- M. Kalaj, K. E. Prosser and S. M. Cohen, Dalton Trans., 2020, 49, 8841–8845 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- K. Sini, D. Bourgeois, M. Idouhar, M. Carboni and D. Meyer, New J. Chem., 2018, 42, 17889–17894 RSC.
- R. D’Amato, R. Bondi, I. Moghdad, F. Marmottini, M. J. McPherson, H. Naïli, M. Taddei and F. Costantino, Inorg. Chem., 2021, 60, 14294–14301 CrossRef PubMed.
- Z. Hu, Y. Peng, Z. Kang, Y. Qian and D. Zhao, Inorg. Chem., 2015, 54, 4862–4868 CrossRef CAS.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- X. Zhang, Z. K. Chen and K. P. Loh, J. Am. Chem. Soc., 2009, 131, 7210–7211 CrossRef CAS.
- J. Wei, W. Zhang, W. Pan, C. Li and W. Sun, Environ. Sci.: Nano, 2018, 5, 1441–1453 RSC.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- D. Banerjee, W. Xu, Z. Nie, L. E. Johnson, C. Coghlan, M. L. Sushko, D. Kim, M. J. Schweiger, A. A. Kruger, C. J. Doonan and P. K. Thallapally, Inorg. Chem., 2016, 55, 8241–8243 CrossRef CAS PubMed.
- T. Musho, J. Li and N. Wu, Phys. Chem. Chem. Phys., 2014, 16, 23646–23653 RSC.
- K. Hendrickx, D. E.-P. Vanpoucke, K. Leus, K. Lejaeghere, A. Van Yperen-De Deyne, V. Van Speybroeck, P. Van Der Voort and K. Hemelsoet, Inorg. Chem., 2015, 54, 10701–10710 CrossRef CAS PubMed.
- F. Xue, J. Cao, X. Li, J. Feng, M. Tao and B. Xue, J. Mater. Chem. C, 2021, 9, 7568–7574 RSC.
- M. Wang, Y. Zuo, J. Wang, Y. Wang, X. Shen, B. Qiu, L. Cai, F. Zhou, S. P. Lau and Y. Chai, Adv. Energy Mater., 2019, 9, 1901801 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d1mh01973b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |