
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeveloping Pd(II) based amphiphilic polymeric nanoparticles for pro-drug activation in complex media†
Anjana
Sathyan
 a,
Stephen
Croke
b,
Ana M.
Pérez-López‡
b,
Bas F. M.
de Waal
a,
Asier
Unciti-Broceta
b and
Anja R. A.
Palmans
*a
a,
Stephen
Croke
b,
Ana M.
Pérez-López‡
b,
Bas F. M.
de Waal
a,
Asier
Unciti-Broceta
b and
Anja R. A.
Palmans
*a
aLaboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, P.O box 513, 5600 MB Eindhoven, The Netherlands. E-mail: a.palmans@tue.nl
bEdinburgh Cancer Research, Institute of Genetics and Cancer, University of Edinburgh, Crewe Road South, Edinburgh, EH4 2XR, UK
First published on 19th September 2022
Abstract
Novel approaches to targeted cancer therapy that combine improved efficacy of current chemotherapies while minimising side effects are highly sought after. The development of single-chain polymeric nanoparticles (SCPNs) as bio-orthogonal catalysts for targeted site-specific pro-drug activation is a promising avenue to achieve this. Currently, the application of SCPNs as bio-orthogonal catalysts is in its early stages due to reduced performance when increasing the medium's complexity. Herein, we present a systematic approach to identify the various aspects of SCPN-based catalytic systems, to improve their efficiency in future in vitro/in vivo studies. We developed amphiphilic polymers with a polyacrylamide backbone and functionalised with the Pd(II)-binding ligands triphenylphosphine and bipyridine. The resulting polymers collapse into small-sized nanoparticles (5–6 nm) with an inner hydrophobic domain that comprises the Pd(II) catalyst. We systematically evaluated the effect of polymer microstructure, ligand–metal complex, and substrate hydrophobicity on the catalytic activity of the nanoparticles for depropargylation reactions in water, PBS or DMEM. The results show that the catalytic activity of nanoparticles is primarily impacted by the ligand–metal complex while polymer microstructure has a minor influence. Moreover, the rate of reaction is increased for hydrophobic substrates. In addition, Pd(II) leaching studies confirmed little to no loss of Pd(II) from the hydrophobic interior which can reduce off-target toxicities in future applications. Careful deconstruction of the catalytic system revealed that covalent attachment of the ligand to the polymer backbone is necessary to retain its catalytic activity in cell culture medium while not in water. Finally, we activated anti-cancer pro-drugs based on 5-FU, paclitaxel, and doxorubicin using the best-performing catalytic SCPNs. We found that the rate of pro-drug activation in water was accelerated efficiently by catalytic SCPNs, whereas in cell culture medium the results depended on the type of protecting group and hydrophobicity of the prodrug. We believe our findings will aid in the development of suitable catalytic systems and pro-drugs for future in vivo applications.
Design, System, ApplicationTransition-metal-mediated bio-orthogonal reactions for synthesising drugs in situ that target tumour cells can help to develop side-effect-free cancer therapies. Challenges arise from insolubility of efficient catalysts or prodrugs in water, catalyst deactivation or sequestration by proteins in cells, biocompatibility, and the need for high activity at low substrate concentrations. Our approach to tackling these involves designing a catalytic system where the catalyst ligand is covalently attached to an amphiphilic polymer that folds around the metal–ligand complex into polymeric nanoparticles. The hydrophobic domain inside these polymeric nanoparticles ensures catalyst and substrate solubilisation and accumulation, resulting in high local concentrations and fast reactions. We report here on amphiphilic polymer-based designs with different ligands that bind to Pd(II) and tested the activation of propargyl-protected pro-dyes and pro-drugs in aqueous media of increasing complexity. We developed a systematic approach to determine factors that affect the performance of catalytic nanoparticles such as substrate hydrophobicity, nature of ligands, and microstructure of the polymers, which helped to find the best catalytic system based on stability, activity, and accessibility depending on the medium. The ability of our designed system to activate the several prodrugs motivates further improvements and development of catalytic nanoparticles for application in cancer therapies. |
Introduction
Nature showcases a myriad of chemical reactions performed with utmost efficacy and specificity despite the complexity of cells, with the help of macromolecular biological catalysts called enzymes. Their unrivalled efficiency and selectivity have inspired many chemists to explore and push the boundaries of chemical transformations. In this respect, chemical reactions that do not interact nor interfere with native biochemical processes, bio-orthogonal reactions, have attracted a great deal of interest.1–7 Initially utilised to understand the molecular details of biological processes, bio-orthogonal reactions were diversified to perform in situ synthesis in cells.8 With the advances in bio-orthogonal chemistry, reactions using various transition metal catalysts such as Pd(II)/(0), Ru, Au, or Cu in living cells became promising for biomedical applications.9–18 Among them, Pd is known for both cross-coupling and bond cleavage reactions in cellular media,19–21 and has been explored for protein modification or activation,22,23 cell surface remodeling,24,25 DNA modification26 and pro-drug activation.4,27 Pd(0)/Pd(II) can perform C–O bond cleavage of propargylic or allylic carbamates, ethers, amines, or carbonates in cellular media.7,11,12,28–33 Hence, it displays therapeutic potential exemplified by its ability to activate pro-drugs or pro-dyes in a controlled manner with minimal toxicity and high specificity. Bradley and co-workers first reported intracellular de-allylation and Suzuki–Miyaura coupling reactions using Pd(0) entrapped polystyrene microspheres of 0.5 μm in diameter.12 Later, Pd-mediated depropargylation reactions were reported by Chen and coworkers using discrete Pd(II) complexes for protein activation which proved to function better than deallylation reactions.22 Parallel studies performed by Unciti-Broceta and coworkers with extracellular Pd(0) resins found equivalent results, i.e. Pd-mediated depropargylation reactions are faster than deallylations, enabling the activation of the propargyl-protected anticancer drug 5-fluorouracil34 and propargyloxycarbonyl-protected gemcitabine38 in cell cultures. They also highlighted the compatibility of these catalysts in vivo by locally activating a pro-dye in zebrafish.34 Weissleder and coworkers reported in vivo pro-drug activation of doxorubicin and monomethyl auristatin E by Pd-based nanoparticles thereby inhibiting the growth of solid tumours in mice models opening exciting opportunities to expand in vivo palladium chemistry for developing new cancer therapies.9,32 Further developments from Unciti-Broceta and co-workers on Pd-activatable non-toxic pro-drugs from chemotherapeutics such as doxorubicin, 5-fluorouracil, paclitaxel, etc., widened the scope of new therapies exploiting Pd catalysed pro-drug activation.11,34–38The use of heterogeneous Pd catalysts proved to be advantageous and promising over discrete palladium complexes for in vivo pro-drug activation, as it helps overcome the issues of biocompatibility, stability, deactivation, or their sequestration by proteins.39 However, they are often employed as implants near the tumour tissue which may need to be surgically removed after treatment.11 To cope with this issue, metal complexes can be loaded into the hydrophobic domain of polymeric scaffolds to form homogeneous systems such as micelles,40–42 dendrimers,43 polymerosomes,44 star polymers,45 or polymeric nanoparticles.46–49 They offer the possibility of systemic administration and can be localised to tumour tissues by EPR-mediated passive and RGD or NGR-based active targeting.50
Our group has demonstrated that a single amphiphilic polymer with randomly distributed hydrophobic and hydrophilic side-chains folds/collapses in water into a single-chain polymeric nanoparticle (SCPN).46 These nanoparticles comprise a hydrophobic interior, are in the nanometre-size range, and are completely soluble in water. When ligands capable of binding to transition-metal ions are covalently attached, catalytically-active nanoparticles are obtained, with properties akin to those of metalloenzymes.46,47,51–53 The hydrophobic interior creates a microenvironment for substrates and catalysts allowing high local concentrations, which results in fast kinetics of the reactions.53,54 Compared to other polymeric scaffolds, SCPNs have a discrete, small size (5–10 nm), which will benefit tissue permeability and renal clearance.55–57 SCPNs form a versatile, biocompatible platform that allows easy functionalisation and preparation. Further, they can be readily modified with targeting ligands to improve their localisation in tumour sites for in vivo applications.58 Altogether, SCPNs offer advantages over other reported systems such as heterogeneous Pd(0) resins, offering the possibility of systemic administration, targeted delivery, better penetration into tumour micro-environment and renal clearance. Zimmerman and co-workers reported promising applications of copper and ruthenium-containing SCPNs that perform enzyme-like click reactions or allyl carbamate cleavage in cellular media.48,49,59 Pd(II)-based SCPNs are a logical extension for in vivo prodrug activation owing to their low toxicity but have been less explored in complex media.51
We previously evaluated the potential of a first-generation Pd(II)-based SCPN in the depropargylation of a protected rhodamine dye in the cytosol and lysosomal compartments of HeLa cells. Our work revealed that while deprotection was feasible, a boost in the activity and enhancement of the stability of the catalytic system is required to make the system amenable for in vivo pro-drug activation.51 In addition, we found that the polymer's microstructure affected the size and shape of the formed SCPNs and optimised the hydrophobic/hydrophilic balance to attain well-defined, compartmentalised systems with a structured hydrophobic interior.60 Using the optimised polymer design, we recently observed that the biocompatibility of SCPNs without metals incorporated is excellent and that SCPNs retain their folded, compartmentalised structure in complex media and in the cytoplasm of a variety of cell types.61 Thus, a profound understanding on how different aspects of the catalytic system affect its efficiency when increasing the complexity of the medium is required to make the step to in vivo applications.
We present here our systematic approach to increase the efficiency of Pd(II)-based polymeric nanoparticles for catalysing depropargylation reactions in cellular media by tuning the different aspects of the catalytic system. We focus on depropargylation reactions as they are reported to be faster than deallylation reactions34 and various propargyl-protected pro-drugs of clinically used chemotherapeutics are already reported to be non-toxic to cells.11,34–38 Moreover, depropargylation is cleaner and activates the pro-drug/dye without any toxic side products.30 Hereto, we use our optimised polymer microstructure and investigate (1) the effect of the metal–ligand combination, (2) the effect of the hydrophobicity of the substrate, (3) the effect of the polymer's microstructure on catalyst's activity and (4) the effect of medium complexity. The best design was then applied in the prodrug activation of the well-established cancer chemotherapeutics paclitaxel (pac), doxorubicin (doc) and 5-fluorouracil (5-FU) in the cell culture medium DMEM. The results indicate that while some prodrugs are activated even in DMEM, a careful balance is required between the substrate and product hydrophobicity. Our bottom-up approach highlights not only the challenges associated with pro-drug activation in complex cellular media but also that a fundamental understanding of all aspects of the applied system is crucial for progress.
Results and discussion
Design and preparation of catalytic polymeric nanoparticles and substrates
The design of the catalytic polymeric nanoparticle system (Fig. 1) is based on our previously studied amphiphilic polymers with a polyacrylamide backbone, grafted Jeffamine M-1000 to ensure water solubility, n-dodecyl groups to induce a hydrophobic collapse, and benzene-1,3,5-tricarboxamide groups for imparting a secondary structure formation in the nanoparticle's interior via hydrogen bonding.60 In addition, selected ligands are covalently attached to the polymer backbone capable of binding Pd(II). Following the work of Mascareñas, we select triphenylphosphine as this affords active, discrete Pd(II) complexes, also inside HeLa cells.62 Incorporating phosphine ligands into amphiphilic polymers improves their compatibility with aqueous media, while extending the substrate scope to more hydrophobic molecules. In addition, bipyridine-based ligands were attached to the polymer backbone for reference as we have observed in previous work that these are capable of pro-dye activation in the presence of HeLa cells, albeit with low activity.51 To obtain an efficient catalytic system, the Pd(II) complex should be well protected inside the hydrophobic pocket and should not leak out into the complex biological environment.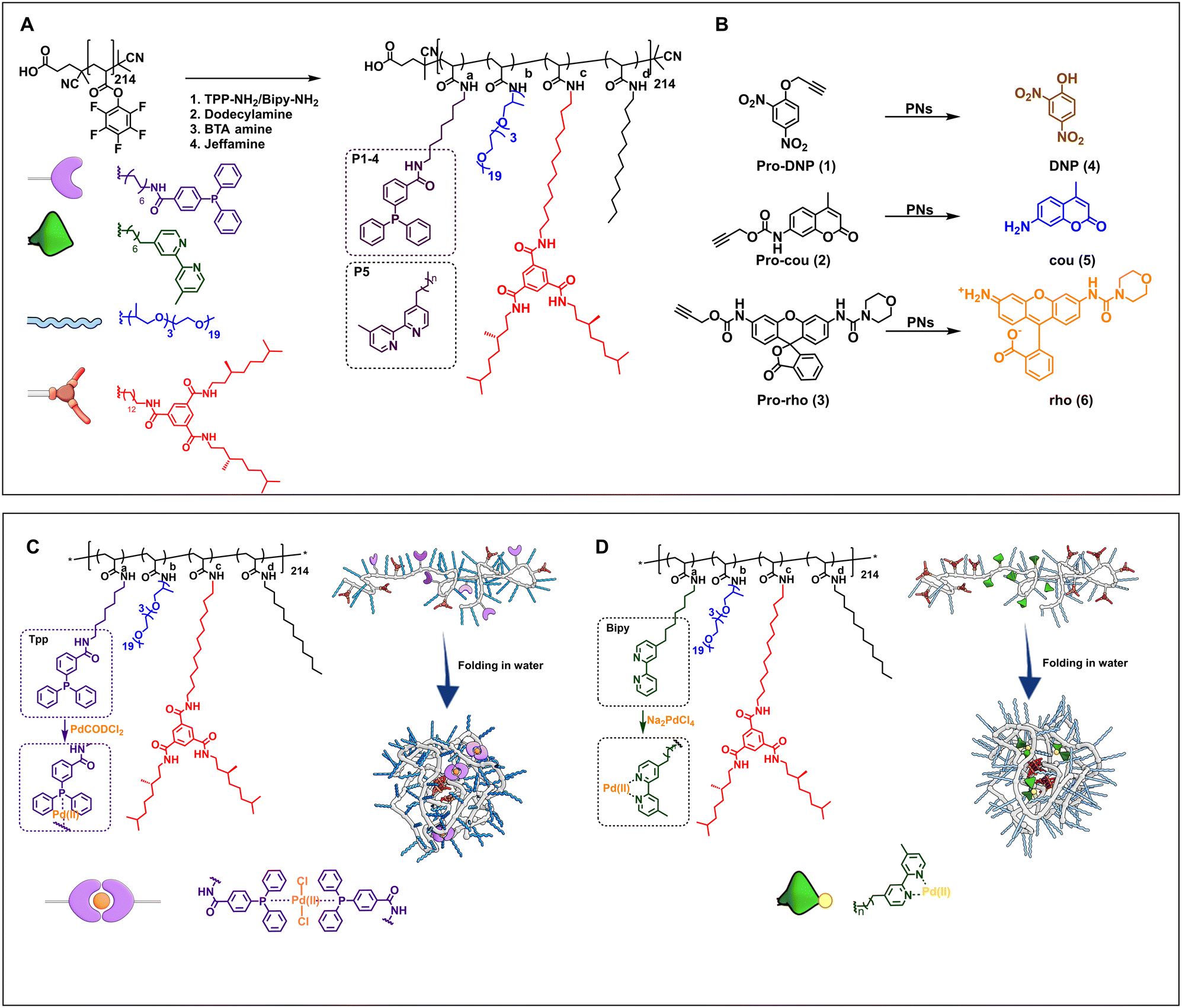 | ||
| Fig. 1 A) Chemical structures of the amphiphilic polymers P1–P6 functionalized from pPFPA in DMF at 50 °C (see ESI† for details). P1–P4 are equipped with triphenylphosphine ligands, P5 with bipyridine ligands, and P6 without any ligands. B) Chemical structures of propargyl protected pro-dyes, and representative depropargylation into the corresponding dye, PNs = polymeric nanoparticles in water/PBS/DMEM. Representation of Pd(II) complexation and folding of C) TPP-functionalised polymers, complexation performed in dry and degassed chloroform at R.T. and D) bipy-functionalised polymers, complexation performed in water at R.T. (see ESI† for details). | ||
Table 1 summarises the details of the composition of the different polymers P1–P6. Amine-functionalised TPP ligand was synthesized starting from 4-(diphenylphosphaneyl)benzoic acid in two steps (see ESI† for details, Section 3.2–3.3), and amine-functionalised bipyridine ligand was synthesised following a reported procedure (see ESI† for details, Section 3.8–3.10).63 All polymers were synthesised starting from the same poly(pentafluorophenylacrylate) homopolymer (pPFPA, DP = 214, Đ = 1.23, Fig. S21 in the ESI†) by a post-functionalisation approach, using previously developed procedures (Fig. S22–44†).46 This post-functionalisation approach allows easy functionalisation and ensures a random distribution of the side groups of interest while keeping the same average degree of polymerisation and molar mass dispersity of the polymer backbone.46,60,64 Polymers P1–P4 comprised triphenylphosphine (TPP) ligands (∼10% in P1–P3, ∼20% in P4), variable amounts of hydrophobic and supramolecular BTA units, and hydrophilic Jeffamine M-1000 (Fig. 1A, Table 1) to vary the microstructure of polymers. P1 contains 10% BTAs. BTAs attached to a polymer backbone assemble via threefold hydrogen-bonding interactions forming M-helical stacks.60 These stacks provide a structured, hydrophobic interior inside the nanoparticles, which is known to enhance the nanoparticle's stability.61P2 contains 5% BTA and 15% dodecyl chains on the backbone, which affords both a structured as well as a compact interior while preventing multichain aggregates due to BTA stacking.60P3 contains 20% dodecyl chains, while P4 has just the hydrophobicity of the ligand to form a hydrophobic domain inside the particles. The simple chemical structures of P3 and P4 highlight the potential of developing easily accessible catalytic polymers for bio-orthogonal catalysis using this approach. P5 is equipped with 10% bipyridine ligand, 5% BTA, 15% dodecyl, and Jeffamine, similar to the polymer reported previously.51 A control polymer P6 without ligands was also prepared to breakdown the complex catalytic system into a simple system which permits physical encapsulation of a Pd(II) complex and study its effect on activity with increasing medium complexity.
| Polymer | a | b | c | d | n | Đ | M n,SEC (kD) | M n,theoretical (kD) |
|---|---|---|---|---|---|---|---|---|
| a–d were determined by 19F NMR. Mn and Đ were measured by SEC.a THF, relative to poly(styrene) standards.b DMF with 10 mM LiBr, relative to poly(ethylene oxide) standards. | ||||||||
| pPFP 214 | 1.23a | 36.7a | 51 | |||||
| P1 | 9 | 80 | 11 | — | — | 1.43b | 46.8b | 183 |
| P2 | 8 | 68 | 4 | 20 | — | 1.18b | 42.2b | 163 |
| P3 | 9 | 76 | — | 15 | — | 1.28b | 62.0b | 158 |
| P4 | 18 | 82 | — | — | — | 1.34b | 55.4b | 178 |
| P5 | 10 | 66 | 5 | 19 | 6 | 1.42b | 46.9b | 179 |
| P6 | — | 76 | — | 24 | — | 1.41b | 57.6b | 164 |
Since P1–P4 are susceptible to oxidation of the TPP ligand, workup and dialysis were performed in degassed solvents. The covalent attachment of the TPP ligands to polymers P1–P4 was indicated by a resonance peak at −5 ppm in the 31P NMR spectrum (Fig. S36†). Next, TPP functionalised polymers were complexed with Pd(II) using PdCODCl2 as the palladium source in degassed chloroform under argon atmosphere and highly diluted conditions to minimise intermolecular crosslinking of the particles.65 We refer to polymer nanoparticles comprising Pd(II) as P@Pd(II). 31P NMR showed that the signal of triphenylphosphine at −5 ppm disappeared, and a new signal downfield between 23–27 ppm formed, confirming the complexation of Pd(II) to TPP (Fig. S45†). We observed minor oxidation of triphenylphosphine in all polymers during complexation as indicated by a small peak at 28 ppm, characteristic of triphenylphosphine oxide.66P1–P4@Pd(II) were dialysed in chloroform to remove most of the unbound Pd(II) salt. Next, the complexed polymers were formulated into nanoparticles by adding water and sonication for 30 min, followed by equilibration for 1 h. In contrast, bipyridine-based polymer P5 does not suffer from sensitivity to oxygen and was first formulated to nanoparticles in water by dissolution, followed by complexation of Pd(II) using the water-soluble Pd(II) precursor Na2PdCl4. The complexation of P5 to Pd(II) was followed by UV-vis spectroscopy, where the characteristic absorption of bipyridine was red-shifted after complexation with Pd(II) (Fig. S46†).51
Dynamic light scattering (DLS) studies of P1–P4@Pd(II) show hydrodynamic radii (RH) of 5–6 nm, well in line with previously reported SCPN systems.51P5@Pd(II), obtained via a slightly different procedure, also showed a RH of 6 nm (Fig. S47†). The sizes of P1–P5 based nanoparticles did not change significantly before and after complexation (Fig. S48†). Due to the presence of a small fraction of aggregates after complexation observed in DLS, we adopted the name polymeric nanoparticles instead of SCPNs in this work.
The negative Cotton effect with a minimum at λ = 225 nm in the CD spectra of polymers P1, P2, and P5 indicated the presence of M-helical BTA aggregates that form threefold hydrogen bonding between the pendant BTA units (Fig. S49†).67,68 The palladium concentration in all nanoparticles was analysed by MP-AES spectroscopy prior to catalysis studies and the results indicated the presence of ∼30–80 Pd(II) ions per particle, meaning that an excess of palladium is trapped in the PNs, which are not able to diffuse out during dialysis (see ESI† for details, Table S1). For all catalytic studies, the total concentration of Pd(II) is kept constant.
The hydrophobicity of the substrates and protecting group plays an essential role during their deprotection by catalytic polymeric nanoparticles, especially in complex media in the presence of competing molecules. The greater the hydrophobicity, the higher is the tendency of substrates to accumulate in hydrophobic reaction space inside the nanoparticles.53 Therefore, we designed and synthesised a set of propargyl-protected, palladium-activatable pro-dyes based on o-dinitrophenol (pro-DNP (1)), coumarin (pro-cou (2)), and rhodamine (pro-rho, (3)) that show different hydrophobicities (for Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values, see Fig. S50†), following reported procedures.24,30,54 Pro-DNP (1) yields DNP (4) upon depropargylation. The reaction is monitored using UV-vis spectroscopy at λ = 400 nm.30 In the case of pro-cou (2), the activation to coumarin (5) is monitored using fluorescence spectroscopy where the uncaged product has an λex,max of 370 nm and λem,max of 440 nm when inside the SCPNs.24 Pro-rho (3) yields the fluorescent rhodamine 110 derivative (6) upon depropargylation, which has an λex,max of 495 nm and λem,max of 520 nm.54
P values, see Fig. S50†), following reported procedures.24,30,54 Pro-DNP (1) yields DNP (4) upon depropargylation. The reaction is monitored using UV-vis spectroscopy at λ = 400 nm.30 In the case of pro-cou (2), the activation to coumarin (5) is monitored using fluorescence spectroscopy where the uncaged product has an λex,max of 370 nm and λem,max of 440 nm when inside the SCPNs.24 Pro-rho (3) yields the fluorescent rhodamine 110 derivative (6) upon depropargylation, which has an λex,max of 495 nm and λem,max of 520 nm.54
Activation of pro-dyes in aqueous solution – influence of polymer microstructure and substrate hydrophobicity
We first check the catalytic performance of newly developed Pd(II) loaded TPP-based polymeric nanoparticles in the depropargylation of O-propargyl and N-propargyloxycarbonyl protected dyes 1–3. Given these are model reactions toward pro-drug activation in cells, the reaction parameters were chosen to fit with the biological environment. Therefore, reactions were performed in aqueous solutions at physiological temperature (37 °C) and at micromolar concentration of substrates, concentrations used for pro-drug administration in vivo.32 Reactions were monitored using fluorescence/UV-vis spectroscopy in real-time and quantified using HPLC-UV (see ESI† for details). The rate of the reaction was first studied using pro-DNP 1 as substrate in water and PBS. P1–P4@Pd(II) nanoparticles were prepared in water or PBS with a concentration of 30 μM Pd(II). To this, a substrate stock solution in DMSO (0.2% in water) was added ([Pro-DNP] = 100 μM). The formation of product DNP was monitored using UV-vis spectroscopy. The kinetic curves in water (Fig. 2B) show saturation after 210 min for P1@Pd(II) and around 700 min for P2–P4@Pd(II). For the free catalyst, PdCODCl2, no saturation is observed, even after 1200 min. In PBS, P1–P3@Pd(II) and PdCODCl2 show faster formation of DNP, whereas P4@Pd(II) showed a similar kinetic profile as in water (Fig. 2C). The results indicate that catalytically active nanoparticles, both in water and PBS, show significantly faster rates compared to free palladium salt PdCODCl2. The kinetic data also imply that the exact microstructure of the polymers P1–P4 has a minor influence on their catalytic activity in water and PBS (Fig. 2B and C).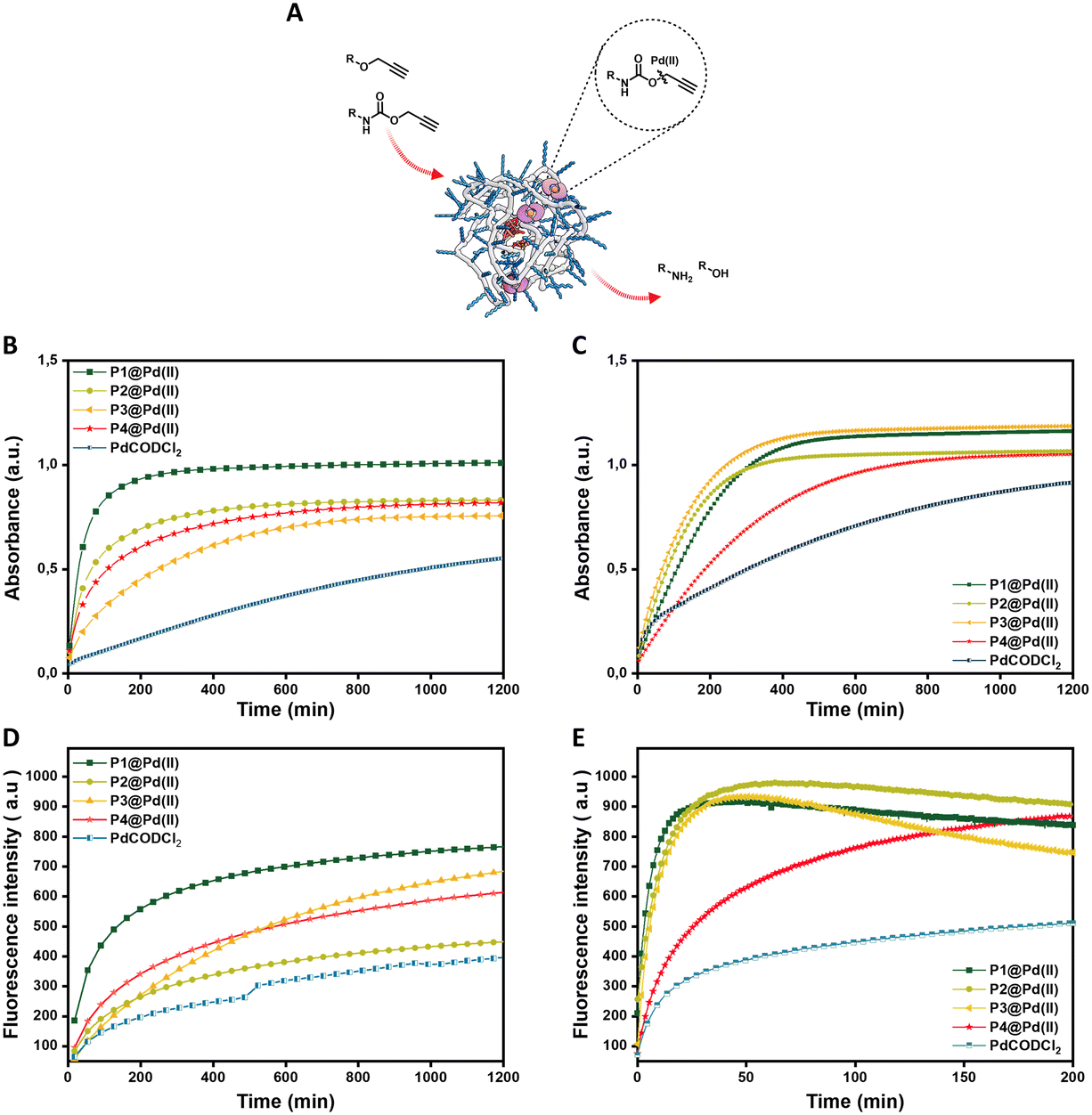 | ||
| Fig. 2 A) Representation of depropargylation by Pd(II) inside hydrophobic cavity of nanoparticles. Activation of pro-DNP 1 (100 μM) to DNP 4 monitored by UV-vis spectroscopy overtime at λ = 400 nm B) in water C) in PBS D) activation of pro-cou 2 (100 μM) to cou 5 monitored by fluorescence spectroscopy over time λex = 370 nm and λem = 420 nm in water E) activation of pro-rho 3 (100 μM) to rho 6 monitored by fluorescence spectroscopy over time λex = 485 nm and λem = 520 nm in water; all reactions were performed at 37 °C by P1–P4@Pd(II) and PdCODCl2; in all cases [Pd(II)] = 30 μM. | ||
The free palladium salt PdCODCl2 shows different behaviour in water and in PBS. In PBS two rate regimes, one fast and one slow, can be observed (Fig. 2C, blue curve). This is similar to what was reported previously for depropargylations by Pd(II) salts in PBS, where it was studied that depropargylation proceeds via two phases, one fast and the other slow.30 The fast phase ends within two turnovers due to product inhibition by the propargylic hydrolytic product, which is followed by a slower reaction phase promoted by Pd(0) nanoparticles formed from Pd(II) in the mixture.30,69 In contrast, P1–P4@Pd(II) nanoparticles catalysed the reaction faster without a slow phase. This could be due to two reasons, a) products formed, which are more hydrophilic than the starting substrate, will have a higher tendency to partition into the aqueous phase, decreasing the chances of product inhibition or b) Pd(0) formed during the cycle is stabilised within the nanoparticles which further allows the continuation of the catalytic cycle.70 The conversion of pro-DNP after 24 h was between 80–90% in the case of the P1–P4@Pd(II), outperforming the PdCODCl2 salt where the conversion was only 55%.
The new polymers were further tested on N-propargyloxycarbonyl protected dyes pro-cou 2 (hydrophilic) and pro-rho 3 (hydrophobic) to assess the effect of substrate hydrophobicity on the rate of the reaction. The deprotection of pro-cou 2 in water (Fig. 2D) proceeds very slow, reaching only 50% even after 24 h. There is no clear trend between the activity of P1–P4@Pd(II) and the free PdCODCl2 salt (Fig. 2D), albeit that P1@Pd(II) seems to be the faster catalyst system. On the other hand, deprotection of pro-rho 3 proceeds very fast, with saturation of the fluorescence increase already after 10 min in the case of P1@Pd(II). The kinetic curves also show that P4@Pd(II), the most hydrophilic nanoparticle with the least hydrophobic content, performed slower compared to others (Fig. 2E), which is also seen for the free PdCODCl2 salt. Also, it is important to note that P4@Pd(II) has ligand incorporation twice compared to P1–P3@Pd(II) resulting in higher Pd(II) loading. As a consequence of keeping the Pd(II) concentration constant, the concentration of nanoparticles P4@Pd(II) in the solution is less, resulting in an overall lesser hydrophobicity to accommodate hydrophobic substrate pro-rho 3.
Quantification of the conversion with HPLC-UV showed quantitative conversion of pro-rho 3 to rho 6 after 3.3 h using P1–P3@Pd(II). The faster conversion of pro-rho 3 by the more hydrophobic nanoparticles P1–P3@Pd(II) suggests that the rate of the deprotection correlates with the hydrophobicity of the substrate, since the rates are significantly slower in the case of pro-DNP 1 and pro-cou 2. The results also show that P1@Pd(II) comprising 10% BTA units outperforms the other nanoparticles in most cases in water. However, the differences between P1–P3@Pd(II) are rather small, indicating that as long as the interior of the nanoparticle is sufficiently hydrophobic to accommodate the substrates, the rate of the reaction is similar. We also observed that the product formed tends to aggregate inside the hydrophobic interior, which was inferred from the quenching of the fluorescence over time. Also, P1@Pd(II) did not show any deactivation after one cycle, as the addition of more substrate pro-rho 3 resulted in the continuation of reaction to reach full conversion (Fig. S51†). Owing to the fast kinetics of activation of pro-rho substrate 3, we select this substrate for subsequent experiments in more complex media.
Role of ligand–metal complex in the catalytic activity of nanoparticles
The choice of ligands attached to the polymer backbone to bind Pd(II) plays an important role in the catalytic system design. The ligands should be labile to allow substrate binding but if they are too labile, nucleophiles in the complex media can deactivate the catalyst faster. Therefore, a fine balance on the lability of ligands is necessary to achieve a high turnover in complex media.69 Here, we compare two ligands, TPP and bipyridine, where TPP is a more labile ligand compared to bipyridine on binding with Pd(II). The labile TPP–Pd(II) complex will allow facile substrate binding and thereby can be more reactive than the stable bipy–Pd(II) complex in water. However, their reactivity in competing environments like in cell culture medium such as DMEM may vary. DMEM medium contains different amino acids such as histidine, cysteine, methionine etc. that are known to complex with Pd(II), with a higher affinity to sulphur-containing amino acids.73 Therefore, these amino acids can interact with Pd(II) displacing the ligands, hence deactivating the catalyst. Here, we studied the effect of TPP and bipyridine on the catalytic activity of polymer nanoparticles in the depropargylation of pro-rho 3 (Fig. 3) in both water and DMEM. P1@Pd(II) and P5@Pd(II) were compared, as well as their Pd(II) precursors PdCODCl2 and Na2PdCl4.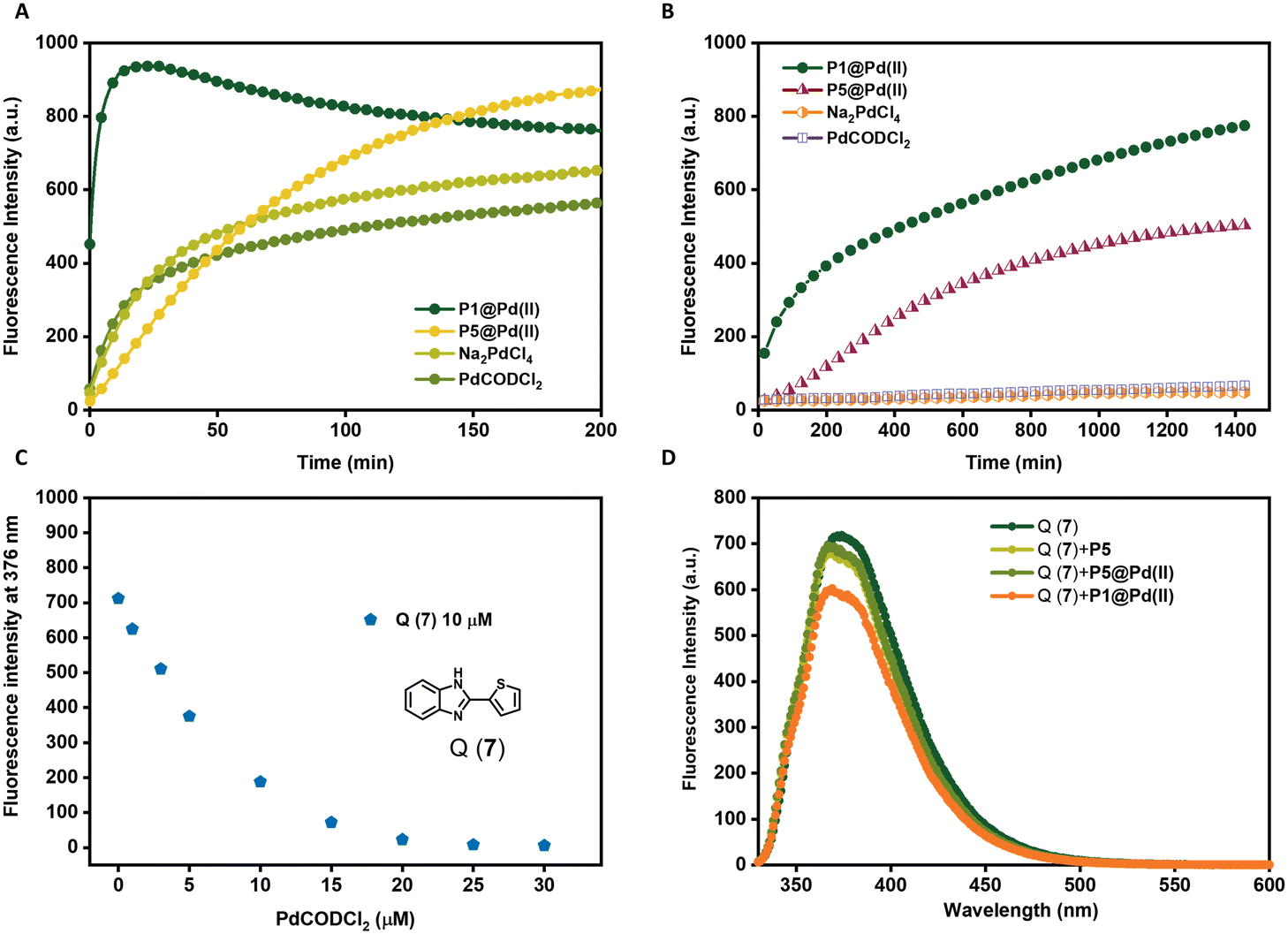 | ||
| Fig. 3 Activation of pro-rho 3 (100 μM) monitored by fluorescence spectroscopy over time λex = 485 nm and λem = 520 nm A) in water, B) in DMEM all reactions were performed at 37 °C by P1@Pd(II), P5@Pd(II), Na2PdCl4 and PdCODCl2, [Pd(II)] = 30 μM in water and [Pd(II)] = 100 μM in DMEM. C) Fluorescent quenching of Q(7) (10 μM) with increasing PdCODCl2 concentration (1–30 μM). D) Pd(II) leaching test, emission of Q(7) at λex = 320 nm in the presence of P1&P5@Pd(II) filtrate solutions [Q(7)] = 10 μM, [P] = 0.25 mg mL−1, [Pd(II)] = 30 μM (before filtration), T = 20 °C, in H2O. | ||
In water, TPP-based P1@Pd(II) performed the depropargylation faster, reaching full conversion in 3.3 h, compared to bipy-based P5@Pd(II) which was slower and did not reach full conversion even after 16 h (Fig. 3A, Table 2). This suggests that the TPP–Pd(II) complex accelerates the depropargylation more efficiently than the bipy–Pd(II) complex. Also in DMEM, TPP-based P1@Pd(II) performed slightly better than bipy-based P5@Pd(II) (Fig. 3B, Table 2). Interestingly the free Pd salts PdCODCl2 and Na2PdCl4 showed a decent activity in water (Fig. 3A) but were fully deactivated in the presence of DMEM (Fig. 3B). This result suggests that TPP and bipy ligands when bound to Pd(II) prevent fast deactivation, and the presence of ligands is essential to retain the catalytic activity of Pd(II) catalysts. However, there is a significant rate decrease when the reactions are conducted in DMEM compared to water.
| Catalyst | Medium* | Pd(II)* mol% | Conversion | |
|---|---|---|---|---|
| a 3.3 h. b 16 h. c 24h. d 48 h. n.d. = not determined as no conversion was observed in DMEM during fluorescence kinetic experiments. Reactions performed at T = 37 °C. *Concentration of Pd(II) and medium of reaction as specified. | ||||
| P1@Pd(II) | Water | 30 | 100%a | |
| DMEM | 100 | 29b | 78%c | |
| P5@Pd(II) | Water | 30 | 56%a | 86%b |
| DMEM | 100 | 28%c | 56%d | |
| PdCODCl 2 | Water | 30 | 49%a | 66%b |
| DMEM | 100 | n.d.c | n.d.d | |
| Na 2 PdCl 4 | Water | 30 | 56%a | 70%b |
| DMEM | 100 | n.d.c | n.d.d | |
To get more insight into this, we designed an experiment to test the leaching of Pd(II) catalysts from the nanoparticles. P1@Pd(II) and P5@Pd(II) in water ([Pd(II)] = 30 μM, [P1/P2] = 0.25 mg mL−1) were centrifuged with centrifugal filters with a molecular weight cutoff of 50 kD, to separate the polymers from the solution. The solution was then tested for the presence of leached-out Pd(II) using an imidazole derivative-based dye Q(7), which exhibits fluorescence quenching in the presence of Pd(II) (Fig. 3C).71
The concentration of Q(7) was fixed at 10 μM while mixing with the filtrate solution (final [Pd(II)] = 25 μM, if there is complete leaching of Pd(II)). Total quenching of Q(7) fluorescence will be observed if 100% Pd(II) is leached out. In the case of P5@Pd(II), there was no significant reduction in the emission of 7 and the result was similar to the control polymer P5 without Pd(II) (Fig. 3D). This indicates that there is no significant leaching of Pd(II) and that the ligand–Pd(II) complex is very stable in P5@Pd(II). However, for P1@Pd(II) there was a slight reduction in fluorescence intensity, which corresponded to ∼4–8% of Pd(II) leaching out. Still, the overall loss of Pd(II) is rather low. We conclude from the leaching experiment that our design of ligand-based nanoparticles ensures good catalyst encapsulation, while limiting its deactivation compared to free Pd(II) catalysts. Also, the activity of P1@Pd(II) in complex media is improved compared to our Pd(II)-SCPNs reported earlier, even at lower catalyst loading (Fig. 3B).51 Likely, the reduction in activity when going from water to DMEM is related to sequestration of Pd(II) by constituents of the DMEM medium.
Complexity of the system vs. catalytic activity in different media
Polymeric nanoparticles perform functions utilising the hydrophobic compartment where both hydrophobic substrates and catalysts can accumulate, accelerating reactions in the aqueous medium. If the catalysts are sufficiently hydrophobic, the catalytic system can be simplified by encapsulation of the catalyst in a simple and easily accessible amphiphilic polymer, which can perform the same function. If such a system remains active in a complex medium, many synthesis steps can potentially be avoided, making the applicability of such nanoparticles more versatile. In order to test this, we designed, evaluated, and compared four systems: a) a simple Pd(II) salt encapsulated in amphiphilic polymers (P6@PdCODCl2); b) phosphine–Pd(II) complex encapsulated in amphiphilic polymers (P6@TPPPd2Cl2); c) phosphine–Pd(II) complex covalently attached to polymer (P1@Pd(II)); and d) the free PdCODCl2 as a reference.The four catalytic systems were compared for their efficiency to catalyze the depropargylation reaction of pro-rho 3 in water and DMEM medium (Fig. 4). In water (Fig. 4A), a steep increase in the fluorescence intensity is observed for all systems except for free PdCODCl2. Although P1@Pd(II) is by far the fastest catalyst, covalent attachment of the catalyst to the polymer backbone is not necessary to achieve conversion in a reasonable time scale. This indicates that the free catalysts either accumulate in the hydrophobic pocket or get trapped inside the polymer microstructure, which then aids in the solubilisation of substrates converting them to products. Remarkably, in DMEM (Fig. 4B), the covalent attachment of the Pd(II) ligand to the polymer backbone as in P1@Pd(II) is crucial to retain catalytic activity. In all other catalysts systems, activity is strongly decreased as in P6@TPPPd2Cl2 or almost completely lost (free PdCODCl2 and P6@PdCODCl2). Thus, encapsulation of the Pd(II) salt does not provide sufficient protection to the catalysts as it is likely not hydrophobic enough to remain inside the hydrophobic reaction pocket (Fig. 4B). The TPP–Pd(II) complex is more hydrophobic, due to which the encapsulated complex performs slightly better than Pd(II) salts in DMEM (Fig. 4B). All in all, our results show that an increase in the complexity of the system, aids the catalyst activity when the reaction is performed in competitive media such as DMEM (Fig. 4C).
 | ||
| Fig. 4 Activation of pro-rho 3 (100 μM) by P1@Pd(II), P6@PdCODCl2, P6@TppPd(II)Cl2 and PdCODCl2 in A) water B) DMEM monitored by fluorescence spectroscopy over time λex = 485 nm and λem = 520 nm; all reactions were performed at 37 °C, [Pd(II)] = 30 μM in water and [Pd(II)] = 100 μM in DMEM. C) Representation of the four catalytic systems with increasing system complexity and their catalytic activity with increasing medium complexity. | ||
Activation of anti-cancer pro-drugs
TPP-based P1@Pd(II) shows promising activity in complex media such as DMEM. Therefore, we selected P1@Pd(II) for evaluating the ability to activate anticancer pro-drugs. Pro-drugs based on 5-fluorouracil, paclitaxel, and doxorubicin (Scheme 1) with different hydrophobicities (for LogP values, see Fig. S50†), and protecting groups were chosen as substrates for the activation to find the best substrate suitable for the nanoparticle-based catalytic system. Masking of these drugs with Pd labile protecting groups reduces their cytotoxicity but on activation converts to corresponding drugs that induce cell death. The activation pathway of all four pro-drugs (Scheme 1) starts with depropargylation mediated by Pd(II). Catalytic reactions were performed with P1@Pd(II) and the results were compared to those obtained by free PdCODCl2 salt. The qualitative conversion was monitored using HPLC-UV/MS.
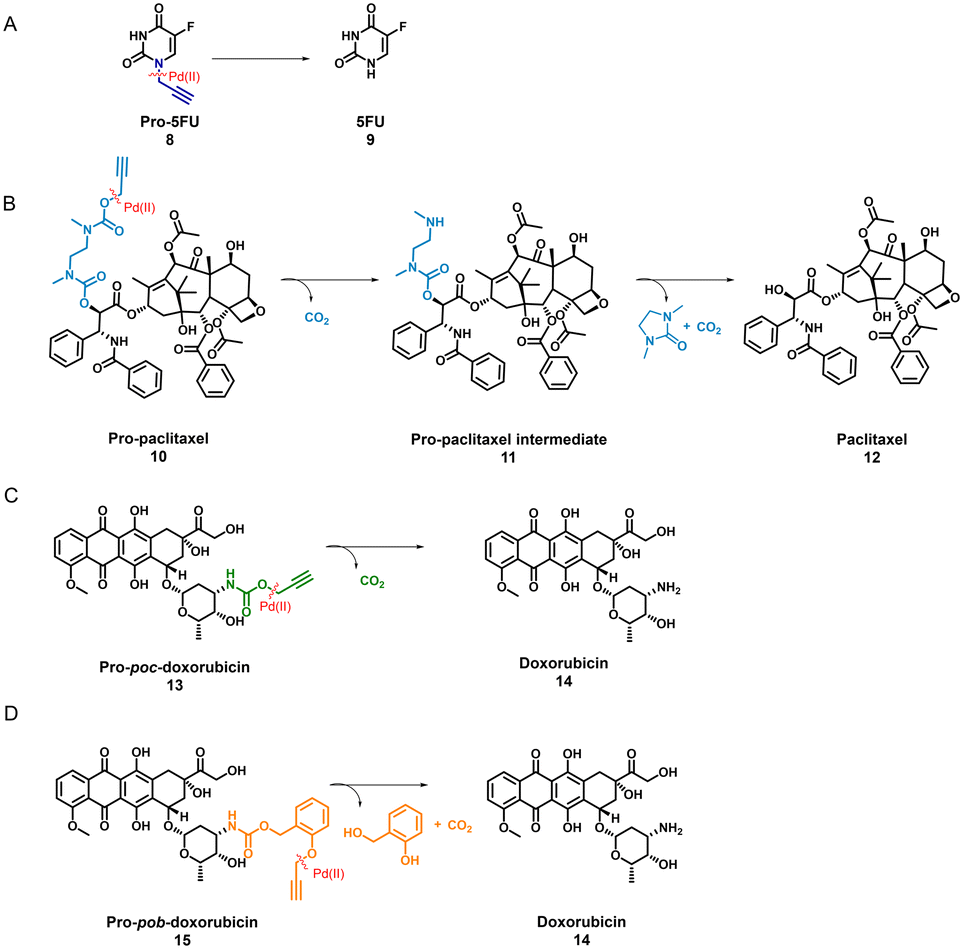 | ||
| Scheme 1 Scheme for pro-drug activation catalysed by Pd(II) catalyst on 4 different anti-cancer pro-drugs (A) pro-5FU to 5-FU (B) pro-paclitaxel to paclitaxel (C) pro-poc-doxorubicin to doxorubicin and (D) pro-pob-doxorubicin to doxorubicin. | ||
Pro-5FU 8 is a very hydrophilic pro-drug of the widely used therapeutic drug 5-FU 9 and is of interest to test if it is compatible with the nanoparticle-based catalytic system. Pro-5FU activation was tested in water, PBS, and DMEM. In water, P1@Pd(II) activates pro-5FU in 16 h when equimolar concentrations of Pd(II) were used. This is significantly faster than when free PdCODCl2 is applied, where no full conversion is observed after 16 h. The same trend was observed when using PBS (Fig. 5A). When the amount of Pd(II) was reduced to 30 mol% in PBS, the reaction progressed slower, and no significant difference between free PdCODCl2 and P1@Pd(II) was observed (Fig. 5B). In DMEM, the trend was difficult to observe by UV/MS detection due to the presence of its components that influence detection. However, pro-5FU activation proceeded at a slow rate, and full conversion was not achieved, not even at an equimolar concentration of Pd(II) (Fig. S55A†). These findings suggest that hydrophilic pro-drugs are indeed suitable for nanoparticle-based catalytic systems where the reaction proceeds, even if slowly, at catalytic amounts of Pd(II), which is promising for future development of targeted cancer therapy where these polymeric nanoparticles can be decorated with tumour-targeting ligands.
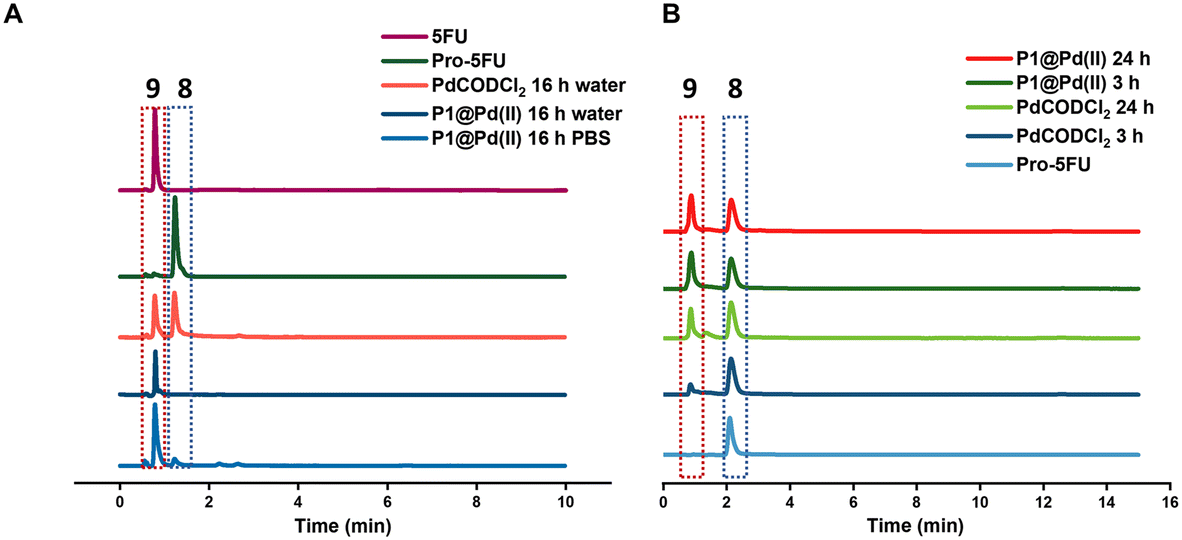 | ||
| Fig. 5 Pro-5FU activation A) in water and PBS [Pd(II)] = 100 μM, [pro-5FU] = 100 μM (HPLC-UV detection 265 nm) B) in PBS(HPLC-MS). [Pd(II)] = 30 μM, [pro-5FU] = 100 μM, all reactions were performed at 37 °C. | ||
In contrast to pro-5FU, pro-paclitaxel is a very large and hydrophobic drug and therefore an interesting substrate to be tested. Due to its high hydrophobicity, the parent drug paclitaxel is usually delivered using nanoplatforms such as micelles, or liposomes for clinical therapy.72 During activation, depropargylation of the terminal propargyl group of pro-paclitaxel can be followed by the disappearance of pro-paclitaxel 10, resulting in an intermediate 11 which should undergo intramolecular cyclisation to form paclitaxel 12. In PBS, complete disappearance of 10 was observed within 9 h in case of reaction catalysed by P1@Pd(II) (30 mol% Pd(II) with respect to substrate) while this was not the case for the PdCODCl2 salt (Fig. 6A and B). This shows that substrate and catalyst accumulation indeed result in good kinetics in the case of P1@Pd(II). However, in both cases, the formed intermediate 11 was stable, eluted earlier, and did not further convert to paclitaxel 12, not even after 48 h (Fig. 6A and B). In DMEM, the PdCODCl2 salt did not activate pro-paclitaxel both at 30 mol% and 100 mol% catalyst concentrations (Fig. 6A and D). In the case of the P1@Pd(II), 11 and 12 were formed in trace amounts within 50 min although full conversion was not achieved even after 48 h (Fig. 6C). The HPLC trace indicates two peaks corresponding to paclitaxel (confirmed by MS) in the presence of polymers which is more obvious in DMEM medium. We hypothesise that the paclitaxel present in the hydrophobic cavity of the nanoparticles and paclitaxel in solution elute at different time points. These results indicate that pro-drugs/activated drugs with high hydrophobicity are stabilised in the hydrophobic pocket and are likely less suitable for designing nanoparticle-based pro-drug activation in cells.
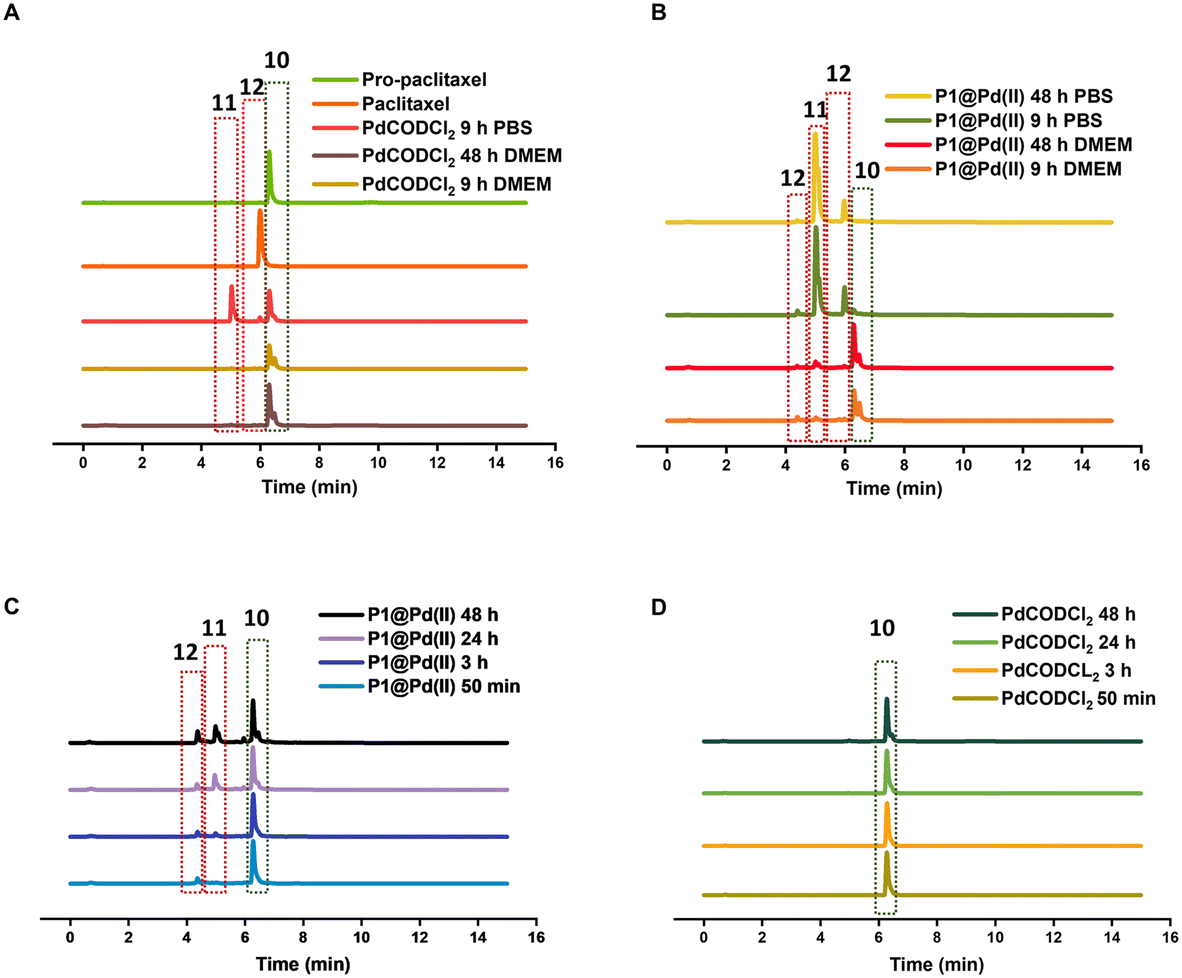 | ||
| Fig. 6 HPLC-MS chromatogram of paclitaxel activation by A) PdCODCl2 in PBS and DMEM B) P1@Pd(II) in PBS and DMEM. [Pd(II)] = 30 μM, [pro-paclitaxel] = 100 μM C) by P1@Pd(II) in DMEM D) by PdCODCl2 in DMEM, [Pd(II)] = 100 μM, [pro-paclitaxel] = 100 μM monitored by HPLC-MS overtime, all reactions were performed at 37 °C. | ||
We also tested pro-poc-doxorubicin 13 and pro-pob-doxorubicin 15 in water. The PdCODCl2 salt activated 13 faster than P1@Pd(II) because complete disappearance of 13 and 15 was observed after 16 h with PdCODCl2 salt while in the case of P1@Pd(II) the pro-drug still remained in the reaction mixture (Fig. S55B and C†). Even though the peaks are not clearly resolved in HPLC chromatogram for the reaction catalysed by P1@Pd(II), substrates 13 and 15 can be detected together with product 14. Easily cleavable propargyl carbamate protecting group and hydrophilicity together makes these prodrugs more prone to activation without the presence of nanoparticles. This shows that our nanoparticle-based catalytic system does not improve the activation of dox-based pro-drugs dramatically. Therefore, careful selection of new pro-drugs with optimum hydrophobicity and stable protecting groups is necessary to further develop a polymeric nanoparticle-based pro-drug activation strategy. A good balance of pro-drug and drug hydrophobicity is required for this system for efficient efflux of drugs to induce cell death.
Conclusions
In conclusion, we have evaluated different aspects of the SCPN-based catalytic system to improve its performance in complex cellular media. Our findings confirmed that the polymer microstructure has only a minor influence on the catalytic activity of nanoparticles when there is enough hydrophobicity in the pocket to accommodate substrates. The most crucial aspect of the design is the selection of the ligand–metal complex, where an optimal balance of stability and activity is required. TPP-based P1@Pd(II) nanoparticles were the most efficient and activated pro-rho to reach full conversion in 3.3 h at 30 mol% catalyst loading in water. Notably, unlike free Pd(II) catalysts, both bipy-based and TPP-based catalytic systems retained their activity in the cell culture medium DMEM, and P1@Pd(II) activated pro-rho reaching 78% conversion in 48 h at 100 mol% catalyst loading. The catalyst loadings used in this work are lower compared to other reports in the field, highlighting the efficiency of our system to perform at low Pd(II) concentrations in complex media. In addition, Pd(II) leaching studies revealed that 4–8% of Pd(II) is lost from the hydrophobic interior in the case of TPP-based system P1@Pd(II), while no leaching was observed in the case of bipy-based P5@Pd(II). This confirmed that our designed nanoparticles are efficient in preventing the leaching of Pd(II), which is critical for in vivo applications as free Pd(II) ions can cause off-target toxicity and decrease the efficiency of catalysts. Careful deconstruction of the catalytic system revealed that the design can be modified to be more synthetically accessible depending on the medium complexity. Physical encapsulation of catalysts in an amphiphilic polymer proved to be sufficient for activity in less complex media such as water and PBS.TPP-based nanoparticles proved to be the best in terms of activity, which was further evaluated for pro-drug activation of pro-5FU, pro-paclitaxel, and pro-doxorubicin in relevant media. The rate of pro-drug activation in water is accelerated efficiently by nanoparticles, conversion in complex media was more sensitive to the protection group and the substrate polarity. The activation of pro-5FU and pro-paclitaxel in PBS by our designed system is as efficient as Pd(0) resins and Pd-nanosheets, respectively, that are already reported to work efficiently near tumour tissue, which highlights the possibility of translating our system for in vivo applications.36,38 The catalytic efficiency of TPP-based nanoparticles in complex media could not be directly compared to other studies, as often these are performed in cells instead of cell culture media. However, hydrophobic drugs such as paclitaxel tend to accumulate in the hydrophobic pocket of nanoparticles, which can affect catalyst performance by product inhibition while also limiting its bioactivity. Overall, our results indicated that hydrophobic substrates in combination with nanoparticles accelerated reaction rates, but a good balance of substrate and product hydrophobicity is required for further improvements and new designs. The ligand-based approach is essential to retain the catalytic activity in a complex medium while screening of new ligand–metal complexes that can withstand nucleophiles in complex media is necessary to further improve the efficiency of nanoparticles. We believe that careful evaluation of the cause of poor catalyst performance is more meaningful than increasing catalyst loading for cellular studies. In short, our findings can aid in the development of more efficient synthetically accessible, stable, and active catalytic nanoparticles, which, when combined with improved substrate design, can greatly increase the potential for in vivo pro-drug activation.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is financed by the European Union's Horizon 2020 research and innovation program under the Marie Sklodowska-Curie Grant Agreement no. 765497 (THERACAT). AMPL and AUB thank EPSRC (EP/N021134/1) for funding. We would like to acknowledge Vishnu Sureshkumar and Alperen Uslu for help with MP-AES measurements, Dr. Gijs ter Huurne for BTA–NH2, and ICMS animation studio for artwork. Xianwen Lou is acknowledged for help with LC–MS.Notes and references
- E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666–676 CrossRef CAS PubMed.
- J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13–21 CrossRef CAS PubMed.
- T. Liang, Z. Chen, H. Li and Z. Gu, Trends Chem., 2022, 4, 157–168 CrossRef CAS.
- W. Wang, X. Zhang, R. Huang, C.-M. Hirschbiegel, H. Wang, Y. Ding and V. M. Rotello, Adv. Drug Delivery Rev., 2021, 176, 113893 CrossRef CAS PubMed.
- M. O. N. van de L'Isle, M. C. Ortega-Liebana and A. Unciti-Broceta, Curr. Opin. Chem. Biol., 2021, 61, 32–42 CrossRef PubMed.
- B. Lozhkin and T. R. Ward, Bioorg. Med. Chem., 2021, 45, 116310 CrossRef CAS PubMed.
- Y. Bai, J. Chen and S. C. Zimmerman, Chem. Soc. Rev., 2018, 47, 1811–1821 RSC.
- C. R. Bertozzi, Acc. Chem. Res., 2011, 44(9), 651–653 CrossRef CAS PubMed.
- M. A. Miller, H. Mikula, G. Luthria, R. Li, S. Kronister, M. Prytyskach, R. H. Kohler, T. Mitchison and R. Weissleder, Nano Lett., 2018, 12, 12814–12826 CAS.
- A. M. Pérez-López, B. Rubio-Ruiz, V. Sebastián, L. Hamilton, C. Adam, T. L. Bray, S. Irusta, P. M. Brennan, G. C. Lloyd-Jones, D. Sieger, J. Santamaría and A. Unciti-Broceta, Angew. Chem., 2017, 129, 12722–12726 CrossRef.
- T. L. Bray, M. Salji, A. Brombin, A. M. Pérez-López, B. Rubio-Ruiz, L. C. A. Galbraith, E. E. Patton, H. Y. Leung and A. Unciti-Broceta, Chem. Sci., 2018, 9, 7354–7361 RSC.
- R. M. Yusop, A. Unciti-Broceta, E. M. V. Johansson, R. M. Sánchez-Martín and M. Bradley, Nat. Chem., 2011, 3, 239–243 CrossRef CAS PubMed.
- C. Vidal, M. Tomás-Gamasa, P. Destito, F. López and J. L. Mascareñas, Nat. Commun., 2018, 9, 1–9 CrossRef CAS.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540 CrossRef PubMed.
- C. Vidal, M. Tomás-Gamasa, A. Gutiérrez-González and J. L. Mascarenas, J. Am. Chem. Soc., 2019, 141, 5125–5129 CrossRef CAS PubMed.
- T. Völker and E. Meggers, Curr. Opin. Chem. Biol., 2015, 25, 48–54 CrossRef PubMed.
- M. Martínez-Calvo and J. L. Mascareñas, Coord. Chem. Rev., 2018, 359, 57–79 CrossRef.
- M. Tomás-Gamasa, M. Martínez-Calvo, J. R. Couceiro and J. L. Mascarenãs, Nat. Commun., 2016, 7, 1–10 Search PubMed.
- J. Li and P. R. Chen, ChemBioChem, 2012, 13, 1728–1731 CrossRef CAS PubMed.
- C. P. Ramil and Q. Lin, Chem. Commun., 2013, 49, 11007–11022 RSC.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes and B. G. Davis, Acc. Chem. Res., 2011, 44, 730–741 CrossRef CAS PubMed.
- J. Wang, B. Cheng, J. Li, Z. Zhang, W. Hong, X. Chen and P. R. Chen, Angew. Chem., Int. Ed., 2015, 54, 5364–5368 CrossRef CAS.
- C. D. Spicer, T. Triemer and B. G. Davis, J. Am. Chem. Soc., 2012, 134, 800–803 CrossRef CAS PubMed.
- L. Lercher, J. F. McGouran, B. M. Kessler, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2013, 52, 10553–10558 CrossRef CAS PubMed.
- J. Li and P. R. Chen, Nat. Chem. Biol., 2016, 12, 129–137 CrossRef CAS PubMed.
- Z. Chen, H. Li, Y. Bian, Z. Wang, G. Chen, X. Zhang, Y. Miao, D. Wen, J. Wang, G. Wan, Y. Zeng, P. Abdou, J. Fang, S. Li, C. J. Sun and Z. Gu, Nat. Nanotechnol., 2021, 16, 933–941 CrossRef CAS PubMed.
- S. V. Chankeshwara, E. Indrigo and M. Bradley, Curr. Opin. Chem. Biol., 2014, 21, 128–135 CrossRef CAS.
- S. E. Coelho, F. S. S. Schneider, D. C. de Oliveira, G. L. Tripodi, M. N. Eberlin, G. F. Caramori, B. de Souza and J. B. Domingos, ACS Catal., 2019, 9, 3792–3799 CrossRef CAS.
- J. Konč, V. Sabatino, E. Jiménez-Moreno, E. Latocheski, L. R. Pérez, J. Day, J. B. Domingos and G. J. L. Bernardes, Angew. Chem., 2022, 61, e2021135 Search PubMed.
- M. A. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 1–13 CrossRef CAS PubMed.
- R. C. Brewster, E. Klemencic and A. G. Jarvis, J. Inorg. Biochem., 2021, 215, 111317 CrossRef CAS PubMed.
- J. T. Weiss, J. C. Dawson, K. G. Macleod, W. Rybski, C. Fraser, C. Torres-Sánchez, E. E. Patton, M. Bradley, N. O. Carragher and A. Unciti-Broceta, Nat. Commun., 2014, 5, 1–9 Search PubMed.
- A. M. Pérez-López, B. Rubio-Ruiz, T. Valero, R. Contreras-Montoya, L. Á. De Cienfuegos, V. Sebastián, J. Santamaría and A. Unciti-Broceta, J. Med. Chem., 2020, 63, 9650–9659 CrossRef PubMed.
- C. Adam, A. M. Pérez-López, L. Hamilton, B. Rubio-Ruiz, T. L. Bray, D. Sieger, P. M. Brennan and A. Unciti-Broceta, Chem. – Eur. J., 2018, 24, 16783–16790 CrossRef CAS PubMed.
- J. T. Weiss, N. O. Carragher and A. Unciti-Broceta, Sci. Rep., 2015, 5, 1–7 Search PubMed.
- J. T. Weiss, J. C. Dawson, C. Fraser, W. Rybski, C. Torres-Sánchez, M. Bradley, E. E. Patton, N. O. Carragher and A. Unciti-Broceta, J. Med. Chem., 2014, 57, 5395–5404 CrossRef CAS PubMed.
- G. Y. Tonga, Y. Jeong, B. Duncan, T. Mizuhara, R. Mout, R. Das, S. T. Kim, Y.-C. C. Yeh, B. Yan, S. Hou and V. M. Rotello, Nat. Chem., 2015, 7, 597–603 CrossRef CAS PubMed.
- S. Tevet, S. S. Wagle, G. Slor and R. J. Amir, Macromolecules, 2021, 54, 11419–11426 CrossRef CAS PubMed.
- M. Cortes-Clerget, N. Akporji, J. Zhou, F. Gao, P. Guo, M. Parmentier, F. Gallou, J. Y. Berthon and B. H. Lipshutz, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- S. Wallace, E. P. Balskus, M. Cortes-Clerget, N. Akporji, J. Zhou, F. Gao, P. Guo, M. Parmentier, F. Gallou, J. Y. Berthon and B. H. Lipshutz, Angew. Chem., Int. Ed., 2016, 55, 6023–6027 CrossRef CAS PubMed.
- C. Deraedt, N. Pinaud and D. Astruc, J. Am. Chem. Soc., 2014, 136, 12092–12098 CrossRef CAS.
- K. T. Kim, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Adv. Mater., 2009, 21, 2787–2791 CrossRef CAS.
- Q. Lu, S. Bai, Z. Chen, N. Zheng, X. Feng and Y. Bai, ACS Mater. Lett., 2020, 2, 89–94 CrossRef CAS.
- Y. Liu, T. Pauloehrl, S. I. Presolski, L. Albertazzi, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 13096–13106 CrossRef CAS PubMed.
- M. Artar, T. Terashima, M. Sawamoto, E. W. Meijer and A. R. A. Palmans, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 12–20 CrossRef CAS.
- J. Chen, J. Wang, Y. Bai, K. Li, E. S. Garcia, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2018, 140, 13695–13702 CrossRef CAS.
- J. Chen, J. Wang, K. Li, Y. Wang, M. Gruebele, A. L. Ferguson and S. C. Zimmerman, J. Am. Chem. Soc., 2019, 141, 9693–9700 CrossRef CAS PubMed.
- S. Kunjachan, R. Pola, F. Gremse, B. Theek, J. Ehling, D. Moeckel, B. Hermanns-Sachweh, M. Pechar, K. Ulbrich, W. E. Hennink, G. Storm, W. Lederle, F. Kiessling and T. Lammers, Nano Lett., 2014, 14, 972–981 CrossRef CAS PubMed.
- Y. Liu, S. Pujals, P. J. M. Stals, T. Paulöhrl, S. I. Presolski, E. W. Meijer, L. Albertazzi and A. R. A. Palmans, J. Am. Chem. Soc., 2018, 140, 3423–3433 CrossRef CAS PubMed.
- T. Terashima, T. Mes, T. F. A. A. de Greef, M. A. J. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742–4745 CrossRef CAS PubMed.
- M. Artar, E. R. J. Souren, T. Terashima, E. W. Meijer and A. R. A. Palmans, ACS Macro Lett., 2015, 4, 1099–1103 CrossRef CAS PubMed.
- Y. Liu, P. Turunen, B. F. M. de Waal, K. G. Blank, A. E. Rowan, A. R. A. Palmans and E. W. Meijer, Mol. Syst. Des. Eng., 2018, 3, 609–618 RSC.
- Z. Li, C. Xiao, T. Yong, Z. Li, L. Gan and X. Yang, Chem. Soc. Rev., 2020, 49, 2273–2290 RSC.
- J. Liu, M. Guo and C. Chen, Adv. Drug Delivery Rev., 2022, 186, 114318 CrossRef CAS PubMed.
- R. van der Meel, E. Sulheim, Y. Shi, F. Kiessling, W. J. M. Mulder and T. Lammers, Nat. Nanotechnol., 2019, 14, 1007–1017 CrossRef CAS PubMed.
- X. Zhang, R. Huang, S. Gopalakrishnan, R. Cao-Milán and V. M. Rotello, Trends Chem., 2019, 1, 90–98 CrossRef CAS PubMed.
- E. S. Garcia, T. M. Xiong, A. Lifschitz and S. C. Zimmerman, Polym. Chem., 2021, 12, 6755–6760 RSC.
- G. M. ter Huurne, L. N. J. de Windt, Y. Liu, E. W. Meijer, I. K. Voets and A. R. A. Palmans, Macromolecules, 2017, 50(21), 8562–8569 CrossRef CAS PubMed.
- L. Deng, L. Albertazzi and A. R. A. Palmans, Biomacromolecules, 2022, 23, 326–338 CrossRef CAS PubMed.
- M. Martínez-Calvo, J. R. Couceiro, P. Destito, J. Rodríguez, J. Mosquera and J. L. Mascareñas, ACS Catal., 2018, 8, 6055–6061 CrossRef PubMed.
- U. Schatzschneider and J. K. Barton, J. Am. Chem. Soc., 2004, 126, 8630–8631 CrossRef CAS PubMed.
- M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41(7), 1569–1575 CrossRef CAS.
- J. Willenbacher, O. Altintas, V. Trouillet, N. Knöfel, M. J. Monteiro, P. W. Roesky and C. Barner-Kowollik, Polym. Chem., 2015, 6, 4358–4365 RSC.
- D. F. Shriver and P. W. Atkins, Inorganic chemistry, Oxford University Press, Oxford, 3rd edn, 1999 Search PubMed.
- M. A. J. Gillissen, T. Terashima, E. W. Meijer, A. R. A. Palmans and I. K. Voets, Macromolecules, 2013, 46, 4120–4125 CrossRef CAS.
- E. Huerta, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Angew. Chem., 2013, 125, 2978–2982 CrossRef.
- E. Latocheski, G. M. Dal Forno, T. M. Ferreira, B. L. Oliveira, G. J. L. Bernardes and J. B. Domingos, Chem. Soc. Rev., 2020, 49, 7710–7729 RSC.
- S. U. Son, Y. Jang, K. Y. Yoon, E. Kang and T. Hyeon, Nano Lett., 2004, 4(6), 1147–1151 CrossRef CAS.
- S. Suresh, N. Bhuvanesh, A. Raman, P. Sugumar, D. Padmanabhan, S. Easwaramoorthi, M. N. Ponnuswamy, S. Kavitha and R. Nandhakumar, J. Photochem. Photobiol., A, 2019, 385, 112092 CrossRef CAS.
- R. J. Mumper, J. A. Mcneill and P. Ma, J. Nanomed. Nanotechnol., 2013, 4, 164 Search PubMed.
- D. B. Hobart, M. A. G. Berg, H. M. Rogers and J. S. Merola, Molecules, 2021, 26, 4331 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2me00173j |
| ‡ AMPL present address: Technische Universität Berlin, Institut für Biotechnologie, Aufgang 17-1, Level 4, Raum 472, Gustav-Meyer-Allee 25, 13355, Berlin, Germany. |
| This journal is © The Royal Society of Chemistry 2022 |