
Conjugated polyelectrolyte-based ternary exciton funnels via liposome scaffolds†
Jack
Palmer‡
a,
Carmen J.
Segura
a,
Levi
Matsushima
 a,
Benjamin
Abrams
b,
Hsiau-Wei
Lee
a and
Alexander L.
Ayzner
*a
a,
Benjamin
Abrams
b,
Hsiau-Wei
Lee
a and
Alexander L.
Ayzner
*a
aDepartment of Chemistry and Biochemistry, University of California Santa Cruz, Santa Cruz, CA, USA. E-mail: aayzner@ucsc.edu
bLife Sciences Microscopy Center, University of California Santa Cruz, Santa Cruz, CA, USA
First published on 19th January 2022
Abstract
There is great interest in developing inexpensive, molecular light-harvesting systems capable of efficiently converting photon energy to chemical potential energy. It is highly desirable to do so using self-assembly and in a manner that supports environmentally benign processing. A critical consideration in any such assembly is the ability to absorb a substantial fraction of the solar emission spectrum and to be able to efficiently move excited states through the space to a functional interface. We have previously shown that aqueous inter-conjugated polyelectrolyte (CPE) complexes can act as ultrafast and efficient energy-transfer antennae. Here we demonstrate formation of a hierarchically assembled, aqueous system based on an inter-CPE exciton donor/acceptor network and a lipid vesicle scaffold. Using a model small-molecule organic semiconductor embedded in the vesicle membrane, we form a ternary exciton funnel that is oriented towards the membrane interior. We show that, although energy transfer is efficient, the assembly morphology depends sensitively on preparation conditions and relative ionic stoichiometry. We propose several approaches towards stabilizing such aqueous assemblies. This work highlights a path to formation of an aqueous, panchromatic light-harvesting system, whose functional complexity can be systematically increased with modularity.
Design, System, ApplicationIn this report, we have proposed a way to use soft lipid vesicles to self-assemble three optically complementary light absorbers to form a quasi-panchromatic light-harvesting precursor system in water. Such a design allows a path towards spectrally broad light absorption, directional migration of electronic states, and the potential for introduction of an artificial electron transport chain. As such, this system aims to mimic multiple core functions of natural light-harvesting organelles with tractable structural complexity. We further propose additional design variables that should be manipulated to achieve structural robustness and enhanced stability against large-scale aggregation. The potential application of this construct is formation of inexpensive, light-weight light-harvesting systems via highly environmentally benign processing conditions. |
I. Introduction
The promise of solar light-harvesting – conversion of photon energy to electrical or chemical potential energy – has led to the emergence of many diverse classes of materials capable of executing such an energy conversion process. Materials based on semiconducting organic molecules are highly attractive for this application due to their tunability and relatively inexpensive processing demands. Within this molecular materials class, hierarchical self-assembly is perceived to be a particularly promising route to constructing inexpensive and tunable light-harvesting systems.1–11The beginning stage of the energy conversion process requires efficient light absorption to generate electronic excited states, or excitons. An efficient light-harvesting system should be able to absorb a broad region of the solar emission spectrum; that is, the system should be panchromatic. Molecular semiconductors display well-defined optical absorption bands as opposed to a quasi-continuum. To fulfill the need for panchromatic light harvesting, multiple absorbers with complementary absorption spectra must be co-localized and excitonically coupled. Conversion of excitons to chemical potential energy requires directional exciton migration to a heterointerface to eventually form spatially separated electron/hole pairs. In multichromophoric systems, the process of exciton migration via electronic energy transfer (EET) is particularly critical.12–16 EET helps to ensure that excitons generated far from the heterointerface can still migrate to its vicinity during the excited-state lifetime.
The goal of this work is to elucidate whether such a multichromophoric, soft-matter-based system could be realized using electrostatic self-assembly. This allows us to use water as the assembly medium, which is highly attractive from a processing perspective. We envisioned a charged membranous particle whose outer surface would serve as a structural scaffold for multiple light absorbers. An optically complementary small-molecule organic semiconductor, which would act as the final exciton acceptor, could then be placed within the membrane. Such a hierarchically assembled system could then in principle assist in separating electron/hole pairs across the membrane bilayer and thus serve as a further foundation for an artificial electron-transport chain. The advantage of the membrane as opposed to a nanoparticle is that, in principle, the inner membrane/water interface can be further exploited in future work in the construction of an artificial electron transport chain. A cartoon of such a system is shown in Fig. 1. A criticism of such a system may be that it wastes some energy during sequential energy-transfer steps. However, some wasted energy is a sacrifice that must be accepted in any molecular light-harvesting system that aims to be panchromatic and that utilizes EET to achieve spectrally broad responsivity. We believe the gain of broader spectral light harvesting more than compensates for the energy that is necessarily wasted in such a system.
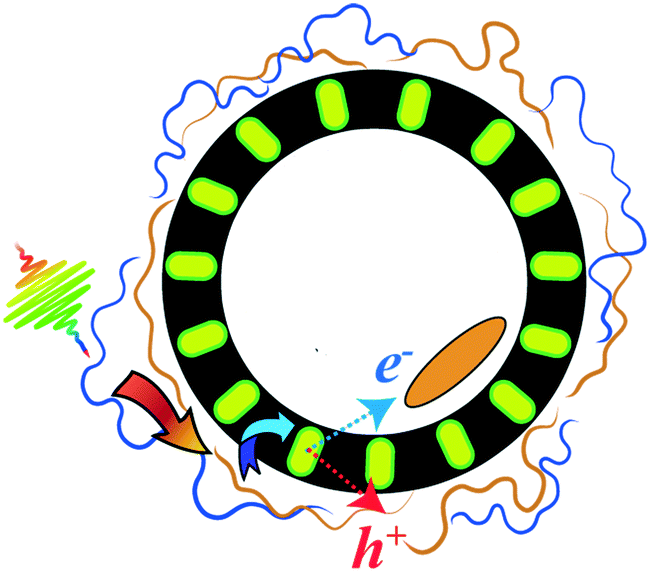 | ||
| Fig. 1 Cartoon of a possible vesicle-based system containing a ternary exciton funnel. The high-energy exciton-donor polymer is shown in blue, and the acceptor polymer is shown in brown. The ternary exciton acceptor is embedded in the membrane interior (shown in yellow), and an electron acceptor (orange oval) is localized at the inner water/membrane interface. Curved arrows signify a hypothetical exciton transfer path. Thin dashed blue and red arrows denote hypothetical electron and hole transfer, respectively, thereby spatially separating the charge carriers. | ||
To form such a system, we chose to work with lipid vesicles (liposomes) which would act as the soft structural scaffold for a proof-of-principle ternary exciton funnel. However, vesicular assemblies can be formed using a variety of soft materials. Some of the biggest challenges in constructing such a soft aqueous system include (i) the stability of the multicomponent assembly, (ii) the need to selectively and asymmetrically partition various organic semiconductors between the different environments and interfaces, and (iii) the demand to manipulate a hierarchy of intermolecular many-body interactions. A reasonable guiding principle is to use a gradient of intermolecular interaction strengths, including electrostatic, hydrophobic and van der Waals.
In this work we progress towards such a system by constructing an excitonically coupled liposome-based assembly from the inside out by first incorporating a ternary exciton acceptor in the membrane interior during liposome assembly. On the outer membrane surface we then electrostatically assemble a bichromophoric excitonic network formed by complexation between two oppositely charged conjugated polyelectrolytes (CPEs).17–19 One CPE absorbs in the blue and forms the outermost, high-photon-energy exciton donor. The complementary CPE simultaneously acts as an exciton acceptor with respect to the higher-energy donor CPE and as a lower-energy exciton donor for the ternary, membrane-embedded acceptor. The exciton funnel is thus directed from the outside in towards the membrane interior.
Our choice to use an inter-CPE network on the outside of the liposome instead of small molecules as the primary absorbers is rooted in the following. First, we showed previously that (i) exciton transfer between electrostatically assembled donor and acceptor CPEs is ultrafast (sub-250 fs),20 and that (ii) the photoluminescence from the acceptor CPE within an inter-CPE complex is not self-quenched.21 In contrast, densely packing small molecules often leads to self-quenching and substantial exciton trapping. This in turn leads to a lowering of the exciton diffusion coefficient and thus is considered undesirable, particularly when EET proceeds via a Förster-type mechanism.13,22 Second, polyelectrolytes are readily amenable to layer-by-layer construction on the surface of colloidal particles, as shown previously using non-conjugated polyelectrolytes.23 Third, the advantage of using a highly charged ion such as a CPE as the primary light absorber leads to the possibility of overcharging the ionic liposome surface due to the electrostatic correlation energy.24–26 The significance of this possibility is that a relatively large density of CPE chromophores may be packed on the liposomal surface at a stoichiometry that reverses the net assembly charge and thus keeps it colloidally stable. Doing so ensures that a single assembly has a maximal absorption coefficient. In contrast, the membrane-intercalated ternary acceptor must be a small molecule to conform to the mean thickness of the lipid bilayer membrane.
Prior work has already demonstrated that single CPEs interact with lipid vesicles.27–29 CPE nanoparticles have also been used to augment the operation of natural light-harvesting systens.30,31 Finally, it has been demonstrated that EET can be efficient between a single CPE and a small molecule within the membrane.27 These observations provide a natural starting point upon which this work is built.
We find that exciton transfer within the CPE network on the liposome surface is highly efficient, leading to a quenching efficiency of the high-energy CPE donor of 95%. We also show that CPE excitons are harvested by the membrane-bound ternary acceptor, albeit with a diminished quantum efficiency compared to EET between the two CPEs. The overall assembly structure is mildly altered in the presence of one outer CPE and the membrane-based small molecule. However, we observe a large change in structure when the inter-CPE network is completed with a secondary CPE, leading to aggregates and generally more complex particles. This work demonstrates that soft self-assembled vesicles hold promise as structural scaffolds for panchromatic CPE-based exciton funnels, but there remains a substantial need to achieve structural robustness. We propose that judicious sidechain engineering is needed to arrest large microstructural changes when the inter-CPE network is adsorbed. We also discuss how the membrane interior may be modified to limit structural rearrangements of the ternary acceptor.
II. Experimental methods
2.1 Materials
The cationic poly(fluorene-alt-phenylene) CPE derivative (PFPI) (MW = 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da; polydispersity = 1.2) was obtained from Solaris Chem, Inc. The anionic regiorandom polythiophene-based CPE (raPTAK) (MW = 8000 Da; polydispersity = 1.8) was obtained from Rieke Metals. Tris(hydroxymethyl)aminomethane (Tris) was obtained from Sigma Aldrich. Sodium chloride (NaCl) was obtained from Sigma Aldrich. The cationic lipid 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt), 18:1 TAP (DOTAP) was purchased from Avanti Polar Lipids (Alabaster, AL). These materials were used as received.
000 Da; polydispersity = 1.2) was obtained from Solaris Chem, Inc. The anionic regiorandom polythiophene-based CPE (raPTAK) (MW = 8000 Da; polydispersity = 1.8) was obtained from Rieke Metals. Tris(hydroxymethyl)aminomethane (Tris) was obtained from Sigma Aldrich. Sodium chloride (NaCl) was obtained from Sigma Aldrich. The cationic lipid 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt), 18:1 TAP (DOTAP) was purchased from Avanti Polar Lipids (Alabaster, AL). These materials were used as received.
Copper(II) 2,3,9,10,16,17,23,24-octakis(octyloxy)-29H,31H-phthalocyanine (oCuPc) was obtained from Sigma-Aldrich (95% nominal purity) and purified via column chromatography using 2 column volumes (CV) of EtOAc, 2 CV MeOH, then eluted with a 73:24:3 mixture of CHCl3:MeOH:triethylamine. Purity was verified via TLC and UV-vis spectroscopy.
2.2 Sample preparation
With the raPTAK/DOTAP mole ratio fixed, we evaluated the following conditions: (1) introducing PFPI at room temperature, (2) introducing PFPI at 70 °C, and quickly cooling the sample in an ice water bath after a 5 min heating period, and (3) heating for 2 hours at 70 °C and slowly cooling to room temperature. Microscopy images showing the resulting assembly morphologies are displayed in Fig. S1.† We found that method (1) led to large particle colonies without a well-defined shape, method (2) led to relatively well-defined particles, and method (3) formed a kind of intermediate between methods (1) and (2), forming colonies composed of more defined particles than method (1). Clearly the method used to prepare the raPTAK/PFPI network depends strongly on preparation conditions. Based on these results, to make the investigation tractable, we chose to focus on assemblies prepared via method (2).
III. Results
Attempts to largely span the visible spectrum with the combined 3-pigment absorption spectrum and to enable directional EET will require that the ternary exciton acceptor absorb on the red side of the visible spectrum. Because of this, we chose to use a lipophilic phthalocyanine derivative as the membrane-embedded acceptor. The two CPEs were chosen to be optically complementary with each other and with the phthalocyanine derivative. Fig. 2a shows the chemical structures of the exciton donor and acceptor CPEs, PFPI and raPTAK, respectively, the lipid DOTAP, and phthalocyanine derivative oCuPc. Fig. 2b shows peak-normalized optical density (OD) and photoluminescence (PL) spectra of the two CPEs and oCuPc. There is clear spectral overlap between PFPI PL and raPTAK OD, and between raPTAK PL and oCuPc OD. Therefore, EET is thermodynamically allowed from PFPI to raPTAK and from raPTAK to oCuPc. There is also mild spectral overlap between PFPI PL and oCuPc OD, leading to the possibility of direct EET from PFPI to oCuPc. The totality of the three OD spectra demonstrates that, if assembled together, a quasi-panchromatic system would be formed. Below, we characterize the structure and the steady-state photophysics in the “inside-out” construction of the ternary exciton funnel templated by the cationic lipid DOTAP. We begin by showing incorporation of the hydrophobic oCuPc into liposomes at the lipid assembly stage. We then go on to characterize the structures formed by complexing raPTAK with both bare and oCuPc-loaded liposomes. Although the parameter space that determines the efficiency of CPE binding to liposomes is in principle vast, below we limit ourselves to a subset of reasonable parameters. We then evaluate the raPTAK → oCuPc EET efficiency. Next, we demonstrate the formation of the inter-CPE network on the surface of lipid particles. Finally, we explore the colocalization of the three light-absorbing components and characterize the EET efficiency in the ternary exciton funnel.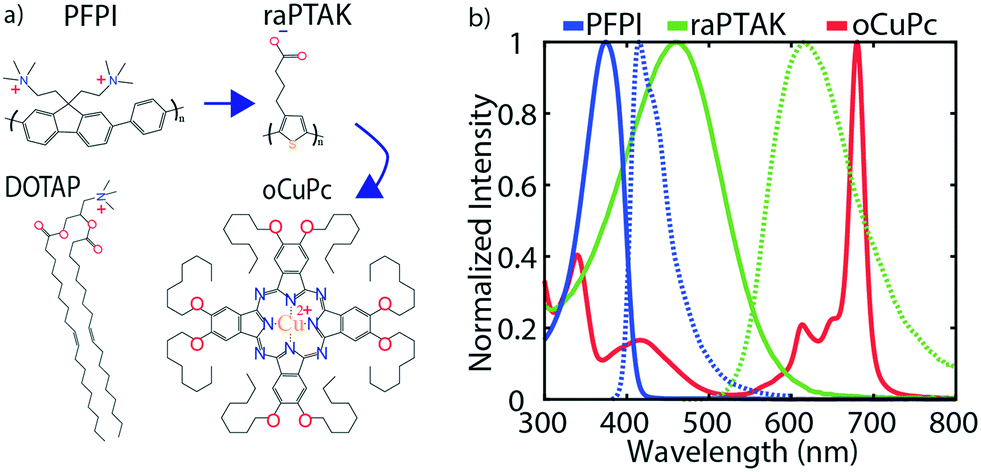 | ||
| Fig. 2 (a) Chemical structures of anionic CPE raPTAK, cationic CPE PFPI, cationic lipid DOTAP, and neutral phthalocyanine derivative oCuPc. (b) Peak-normalized optical density (solid) and photoluminescence (dashed) spectra for the organic semiconductors shown in (a). Blue arrows show the path of exciton flow. | ||
3.1 Forming oCuPc-embedded liposomes
To begin, oCuPc and DOTAP were studied in the absence of CPEs to gain insight into how to best prepare oCuPc-containing liposomes. We found that after hydration of oCuPc:DOTAP cake films, sequential extrusion through a 200 nm filter resulted in the solution losing nearly all oCuPc, as judged by the OD loss (Fig. S2†). Fig. 3 shows that in unextruded samples oCuPc incorporates into the lipid membrane, and that liposomes retain their spherical shapes. This was determined via the weak-yet-measurable emission from the S2 state of oCuPc following excitation of the Soret band (∼490 nm). We could not detect emission from S1 following excitation of the Q band (∼790 nm). | ||
| Fig. 3 oCuPc:DOTAP liposomes with 5% oCuPc loading. Samples were not extruded. a) Transmission image. b) PL image. The weak PL signal is due to emission from the S2 state of oCuPc. | ||
3.2 raPTAK/liposome complex formation
Our prior work had shown that inter-CPE binding may be characterized by a significant thermal activation barrier.32 Thus, we first aimed to determine whether raPTAK binding to the liposome surface was similarly temperature-dependent. We first prepared raPTAK/liposome complexes at 70% raPTAK by mole relative to DOTAP based on the total anionic raPTAK charge concentration (1 charge per monomer). To help interrogate the influence of raPTAK adsorption on liposome structure, liposomes were first extruded to narrow the initial size distribution prior to introducing raPTAK. We note that this was impossible to do with oCuPc, since the nonpolar oCuPc had to be introduced at the liposome formation stage, and extrusion led to loss of oCuPc. Fig. 4 shows representative confocal microscopy images of raPTAK/DOTAP samples as a function of preparation temperature. The room-temperature transmission image displayed in Fig. 4a shows the presence of relatively small liposomes and a few large liposomes with sizes larger than the pore size of the extrusion filter. Comparing the transmission image to the corresponding PL image (Fig. 4d) shows that PL signal is dominated by a few very bright liposomes, which set the contrast maximum. Nevertheless, we find that the great majority of liposomes are fluorescent. Additional evidence for efficient raPTAK binding to liposomes comes from diffusion-ordered NMR (DOSY-NMR) spectroscopy (Fig. S3†).33 | ||
| Fig. 4 Transmission (top row) and PL (bottom row) images of raPTAK/DOTAP samples prepared at RT, 35 °C, and 70 °C (a/d, b/e, c/f respectively). The PL image color temperature is proportional to the relative PL intensity within a given image. | ||
Raising the temperature to 35 °C (Fig. 4b and e) leads to increased aggregation. We observed that liposome samples formed at 35 °C were unstable against precipitation shortly after preparation. In contrast, Fig. 4c and f show that liposomes prepared at 70 °C give spherical, non-aggregated and fluorescent liposomes. We also observe formation of a fraction of liposomes with diameters between 5 and 10 μm. Unlike samples prepared at 35 °C, 70 °C samples were stable for at least several days. Evidently, the interaction between DOTAP liposomes and raPTAK at elevated temperatures appears to promote vesicle fusion.
Binding to the liposome surface is accompanied by changes to the basic photophysical properties of raPTAK. Fig. 5 shows that the normalized optical density (OD) of raPTAK undergoes a blueshift from a λmax of ∼460 nm to ∼430 nm. This is likely primarily due to a decrease in the number density of π-stacking contacts between chromophores on the same raPTAK chain or between different chains when going from the dissolved to the adsorbed state. This interpretation is consistent with the substantial increase in raPTAK PL intensity upon binding (Fig. 5b).34
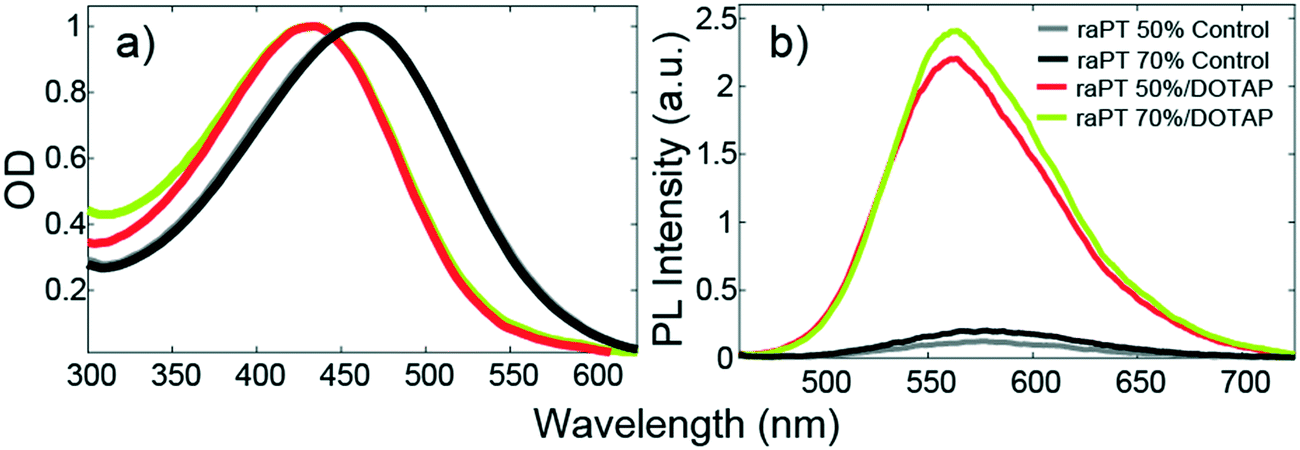 | ||
| Fig. 5 Steady-state photophysics of raPTAK/DOTAP liposome complexes. (a) OD and (b) PL spectra normalized by OD at λmax. The percentage in the legend refers to raPTAK ionic charges relative to those of the lipids. Controls are isolated pristine raPTAK solutions. Binding to DOTAP liposomes leads to a blue shift in raPTAK OD and a concomitant increase in its PL intensity relative to control samples. | ||
To complement the characterization of particle structure on the microscale with that of the nanoscale, we performed small-angle X-ray scattering (SAXS) measurements. Fig. 6 shows the scattering intensity as a function of the length of the scattering vector Q for a pure extruded liposome solution (dashed blue line), as well as raPTAK/liposome complexes created at room temperature (solid lines). The liposome control sample shows multiple fringes, which is consistent with spherical particles and a relatively small polydispersity. Binding to raPTAK leads to an increase in intensity at low Q compared to the pure liposome solution and a loss of low-Q fringes. However, all samples show a characteristic peak at Q ∼ 0.15 Å−1, which corresponds to a real-space distance of 2π/Q ∼ 42 Å.35 This spacing corresponds to the thickness of the lipid bilayer membrane.
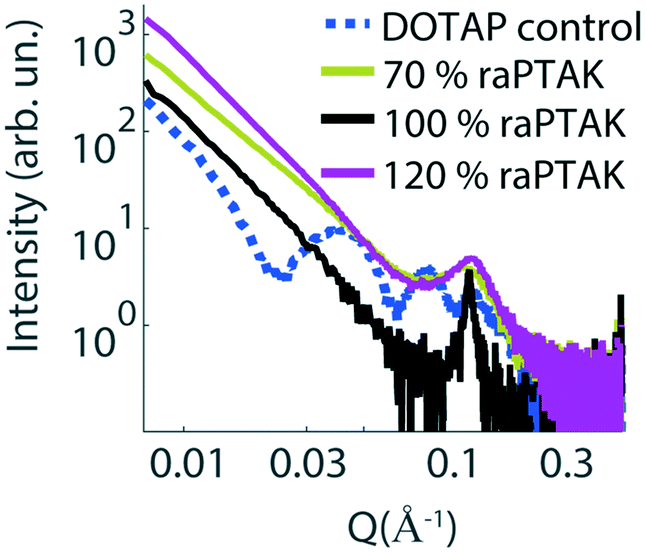 | ||
| Fig. 6 SAXS intensities of raPTAK/liposome complexes as a function of raPTAK % relative to lipids prepared at room temperature. The dashed blue line shows the pure DOTAP liposome control, whereas solid lines correspond to various raPTAK coverages in raPTAK/liposome complexes. | ||
3.3 Formation and EET of raPTAK/oCuPc:DOTAP assemblies
Fig. 7 shows representative confocal transmission and PL microscopy images of samples prepared with 50% raPTAK and 5% oCuPc formed at room temperature and at 70 °C. oCuPc:DOTAP liposomes were not extruded prior to introducing raPTAK, as was the case for all assemblies containing oCuPc discussed below. Even at room temperature, the shape of raPTAK/oCuPc:DOTAP particles begins to depart from sphericity that we observed with raPTAK/DOTAP and oCuPc:DOTAP liposome controls. Thus, colocalization of raPTAK and oCuPc leads to morphological changes. However, the majority of raPTAK/oCuPc:DOTAP particles maintain reasonably well-defined shapes. Fig. 7b and d demonstrates that raPTAK/oCuPc:DOTAP samples prepared at 70 °C displayed a much larger degree of aggregation. Thus, samples for photophysical measurements were prepared at room temperature. | ||
| Fig. 7 Transmission (top row) and PL (bottom row) images of raPTAK/oCuPc:DOTAP samples prepared at RT and 70 °C (a/c and b/d respectively). The oCuPc loading was 5%. | ||
Fig. 8a shows OD spectra of raPTAK/oCuPc:DOTAP complexes as a function of oCuPc mole fraction relative to DOTAP. Q-Band OD of oCuPc (600–800 nm) increases monotonically with oCuPc mole fraction. At 10%, there is a slight reshuffling of oscillator strength above 700 nm, suggesting a subtle change in the structural organization of oCuPc. As shown in Fig. S4,† the OD spectrum shape of raPTAK appears to be approximately independent of oCuPc concentration over the investigated range. We chose to limit the ceiling oCuPc composition to 10% due to solubility concerns at higher concentrations.
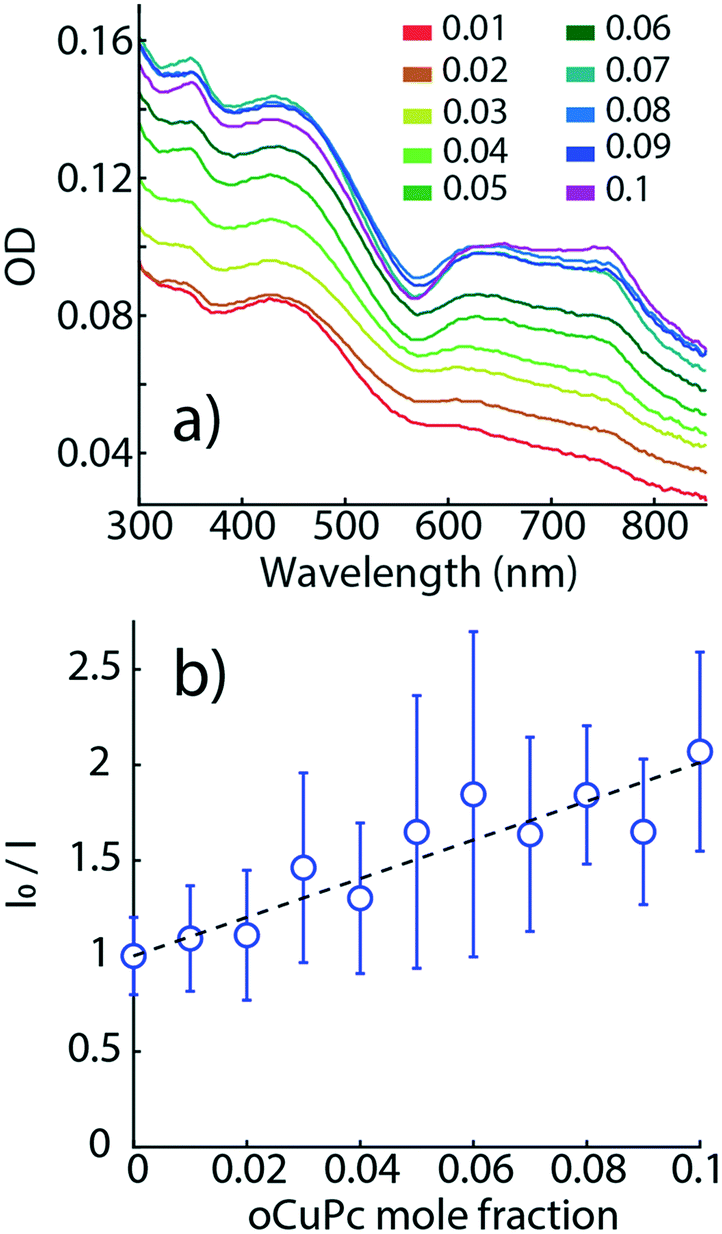 | ||
| Fig. 8 (a) OD of raPTAK/oCuPc:DOTAP liposome complexes as a function of oCuPc mole fraction relative to lipids. (b) Stern–Volmer plot of integrated raPTAK PL, which shows progressive quenching of raPTAK PL with increasing [oCuPc]. | ||
Once the preparation of raPTAK/oCuPc:DOTAP liposomes was established, we went on to investigate whether EET occurred between the surface-bound raPTAK and the membrane-embedded oCuPc. Here, raPTAK acts as the exciton donor with respect to oCuPc. oCuPc is non-emissive from its S1 state, which is expected to be populated via EET from relaxed exciton states of raPTAK. Thus, we used the quenching of raPTAK PL as a measure of EET.36
Fig. 8b shows a Stern–Volmer plot for raPTAK, which shows the ratio of the spectrally integrated raPTAK PL intensity between unquenched (0% oCuPc) and quenched assemblies as a function of oCuPc mole fraction. The raPTAK PL intensity is a monotonically decreasing function of oCuPc concentration, leading to a positive linear slope and a Stern–Volmer constant of ∼5000 M−1. By 10% oCuPc loading, raPTAK emission is quenched by a factor of ∼2 relative to the control sample without oCuPc. Since we find that the raPTAK absorption spectrum is effectively unchanged with increasing oCuPc mole fraction, we attribute this quenching to EET from raPTAK to oCuPc. The EET efficiency ε in terms of donor PL can be written as36
 | (1) |
To further characterize EET between raPTAK and oCuPc, we first calculated the spectral overlap integral J given by
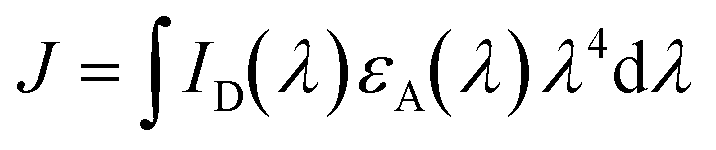 | (2) |
| R0 = 0.02108(κ2ΦDn−4J)1/6 | (3) |
3.4 Assembling an inter-CPE donor/acceptor network on the liposome surface
Using the procedure detailed in the Experimental section, we show in Fig. S5† that we find excellent colocalization of raPTAK and PFPI, which was determined by separately measuring PL intensity in the polymers' distinct emission windows.32 Two questions immediately arise: (A) do dissolved CPE chains coexist with adsorbed CPEs states? (B) Does the introduction of PFPI lead to desorption of raPTAK from the liposome surface, thereby forming dissolved PFPI/raPTAK complexes? We turned to NMR spectroscopy to answer these questions. Fig. 9a and b show representative 1H NMR spectra of raPTAK and PFPI, respectively, in isolated solution at the same concentrations as in the complex. The separation of the aromatic region of the PFPI and raPTAK is convenient in that it allows for straightforward discrimination of the two CPEs. Compared to pure CPE solutions, Fig. 9c shows the PFPI/raPTAK complex in the absence of liposomes shows strong broadening of the raPTAK aromatic resonances and a decrease in the PFPI peak. This is consistent with our prior dynamic light scattering and X-ray scattering measurements on such complexes, which showed formation of inter-CPE networks that were larger than the isolated coil sizes and thus diffused slowly.20Fig. 9d displays the NMR spectrum for the full PFPI/raPTAK/liposome assembly. The spectrum clearly shows that, due to the low tumbling rate of the liposome complex, the raPTAK peak has broadened away into the baseline as before. The PFPI peak is concomitantly strongly diminished both relative to isolated PFPI and the PFPI/raPTAK complex on its own. We thus conclude that at the relative compositions used in this work, (A) effectively all CPE chains are associated with lipid assemblies, and (B) introduction of PFPI does not lead to desorption of raPTAK from the liposome surface. The latter is also consistent with PL microscopy images shown in the ESI† (Fig. S5).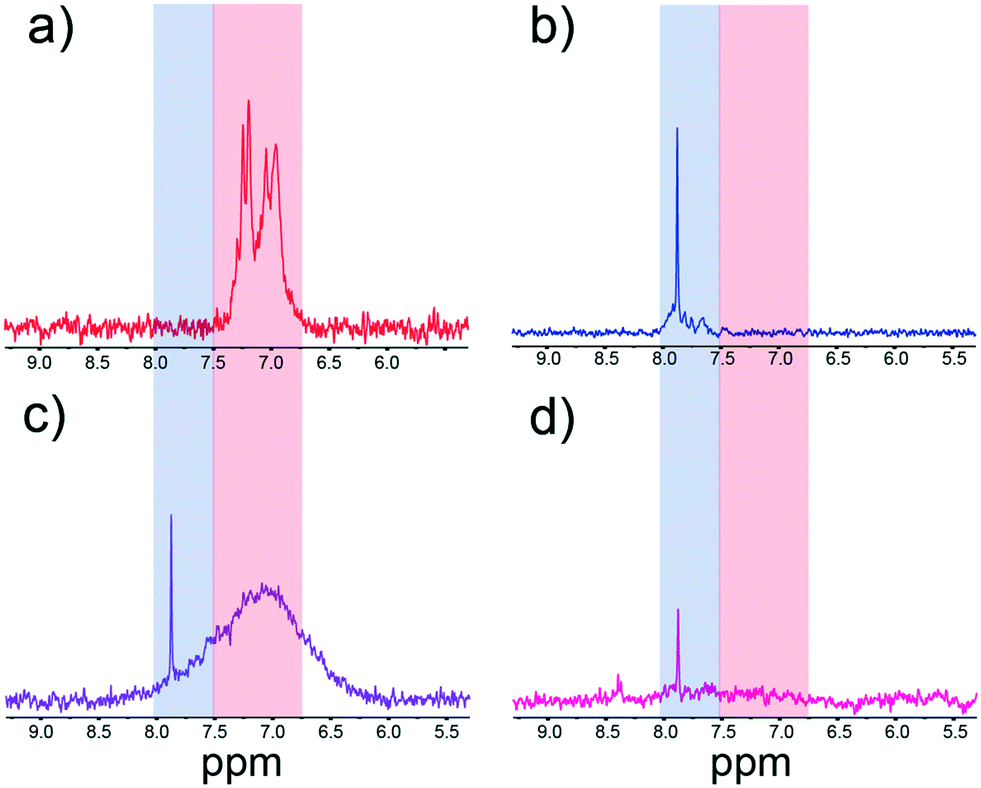 | ||
| Fig. 9 1H NMR spectra of (a) isolated raPTAK solution, (b) isolated PFPI solution, (c) PFPI/raPTAK complex solution, and (d) PFPI/raPTAK/liposome complex solution. The blue and red boxes largely correspond to PFPI and raPTAK aromatic resonance regions, respectively. | ||
We found that PFPI PL was ∼95% quenched when it was introduced to raPTAK/DOTAP complex solutions relative to isolated PPFI at the same concentration (Fig. S6†). Furthermore, we observed that the PL excitation (PLE) spectrum of raPTAK shows a clear contribution from the region that corresponds to maximal PFPI absorption (Fig. S7a and c†). Taken together, this is clear indication of efficient EET from PFPI to raPTAK within the complex assembly, which is consistent with our prior results.
3.5 Formation of the ternary exciton funnel
To complete the ternary exciton funnel, PFPI was added at room temperature to raPTAK/oCuPc:DOTAP assemblies at 10% and 25% charge fractions relative to PTAK charges, while raPTAK was fixed at 50% relative to DOTAP. Samples were then stirred at room temperature for 2 hours prior to performing measurements. Fig. 10a shows PL spectra for PFPI/raPTAK/oCuPcC/DOTAP assemblies as a function of oCuPc mole fraction, excited near the PFPI OD maximum. The functional form of a conjugated polymer PL spectrum encodes structural information about the chromophore ensemble. Fig. 10a shows that the short-wavelength 0–0 vibronic peak of the raPTAK spectrum has a larger magnitude than its longer-wavelength 0–1 shoulder. Such a ratio is consistent with the situation where the magnitude of the intrachain (through-bond) electronic coupling dominates the interchain (through-space) coupling. Therefore, adsorbed raPTAK chains on the liposomal surface are relatively extended and not significantly π-stacked with proximal chains. | ||
| Fig. 10 (a) PL spectra of PFPI/raPTAK/oCuPc:DOTAP complexes as a function of oCuPc mole fraction. (b) PLE spectra of the same complexes with the PL wavelength fixed at 650 nm, corresponding to raPTAK emission. Samples in (a) and (b) contained 25% PFPI relative to raPTAK, which was at 50% relative to DOTAP. Similar results are shown for 10% PFPI in the ESI† (Fig. S8). (c) Stern–Volmer plot of raPTAK PL as a function of oCuPc mole fraction for 10% PFPI relative to raPTAK. (d) same as (c) but for 25% PFPI. PL integration windows are indicated by colored bars in (a). | ||
Interestingly, the 0–0/0–1 PL intensity ratio of PFPI is reversed, suggesting that most PFPI chains adsorbed onto raPTAK are coiled. However, such a conclusion is likely to be misleading, because PFPI PL remains dramatically quenched relative to its isolated state. The small residual PFPI PL may thus not be representative of its statistical chromophore ensemble.
The residual PPFI emission intensity decreased slightly as the oCuPc mole fraction was increased, likely indicating a small amount of EET directly between PFPI and oCuPc. At the same time, PLE spectra of raPTAK shown in Fig. 10b clearly show a large contribution to raPTAK PL from excitation wavelengths associated with PFPI absorption. Consistent with strong quenching of PFPI PL, this shows that EET between raPTAK and PFPI continues to be highly efficient. At the same time, the relative magnitudes of both the raPTAK PL (Fig. 10a) and PLE (Fig. 10b) decrease monotonically with oCuPc mole fraction.
However, Stern–Volmer plots of raPTAK emission as a function of oCuPc mole fraction show that quenching of raPTAK PL by oCuPc appears to be less effective in the presence of PFPI. Fig. 10c shows the raPTAK Stern–Volmer plot of the ternary assembly when the PPFI charge mole fraction is 10% relative to raPTAK. Interestingly, the Stern–Volmer plot appears to deviate from linearity and displays an upward curvature, suggesting that a polynomial may be a more accurate representation of the data. Such a 2nd-order polynomial form may be expected within a simple model where the PL quenching proceeds via two mechanisms: static complex formation and collisional quenching. We were unable to fit the data to such a model to a satisfactory degree. Thus, we chose to impose a linear fit to allow for a quantitative comparison. This yields a mean Stern–Volmer constant of ∼3600 M−1, which is ∼28% smaller than its value in the absence of PFPI. The Stern–Volmer constant obtained from the linear fit for 25% PFPI (Fig. 10d) is ∼4100 M−1, which is ∼18% smaller than its value in the absence of PFPI.
To further investigate the possibility of simultaneous static and collisional quenching, we performed time-resolved PL measurements, shown in Fig. S13 of the ESI.† We selectively excited (430 nm) and detected (530 nm) PL signal due to raPTAK only. We then deconvolved PL decays from the instrument response function; the resulting average lifetimes are listed in Table S1.† Compared to raPTAK/DOTAP control liposomes, we find that introduction of oCuPc at both 5% and 10% leads to a decrease in the average PL lifetime relative to the control: 0.078 ns and 0.088 ns, respectively, compared to 0.101 ns for the control. Together with the steady-state data, this implies that the majority of raPTAK PL quenching by oCuPc occurs on a time scale that is short compared to the ∼120 ps width of the instrument response function in the time-resolved measurement. Although the average lifetime is dominated by the short lifetime component, Fig. S13† shows that there is a decrease in the contribution that the long component (>0.5 ns) makes to the decay upon introduction of oCuPc. We believe that quenching on this time scale is likely to be associated with collisional quenching and is thus an indication that collisional quenching makes a relatively small contribution to the quenching mechanism.
Upon introduction of 10% PFPI relative to raPTAK, overall we find that quenching on the 100 s of ps time scale is further decreased. This is consistent with the smaller degree of steady-state raPTAK PL quenching by oCuPc observed upon addition of PFPI. However, the decrease in the magnitude of quenching is smaller in the time-resolved PL measurement compared to the steady-state one. This again points to the fact that a significant fraction of EET from raPTAK to oCuPc occurs on an ultrafast time scale and is thus not accounted for in the time-resolved PL measurement. Interestingly, the quenching of the long-time component is also decreased, which is consistent with a larger proportion of static quenching seen in the steady-state measurement.
Having demonstrated the simultaneous quenching of PFPI PL by raPTAK and of raPTAK PL by oCuPc, we went on to characterize the microstructure of the ternary assemblies. Fig. 11 shows representative confocal microscopy images of PFPI/raPTAK/oCuPc:DOTAP complexes with 10% PFPI as a function of oCuPc mole fraction. We find that introduction of PFPI leads to a substantial change in morphology relative to raPTAK/oCuPc:DOTAP particles, resulting in particles with ill-defined shapes coexisting with large aggregates. The PL images show apparent colocalization of PFPI and raPTAK intensities. Although in principle patches of isolated polymers could exist on the nanoscale, the fact that PFPI PL is strongly quenched indicates that the two CPEs are in fact largely colocalized.
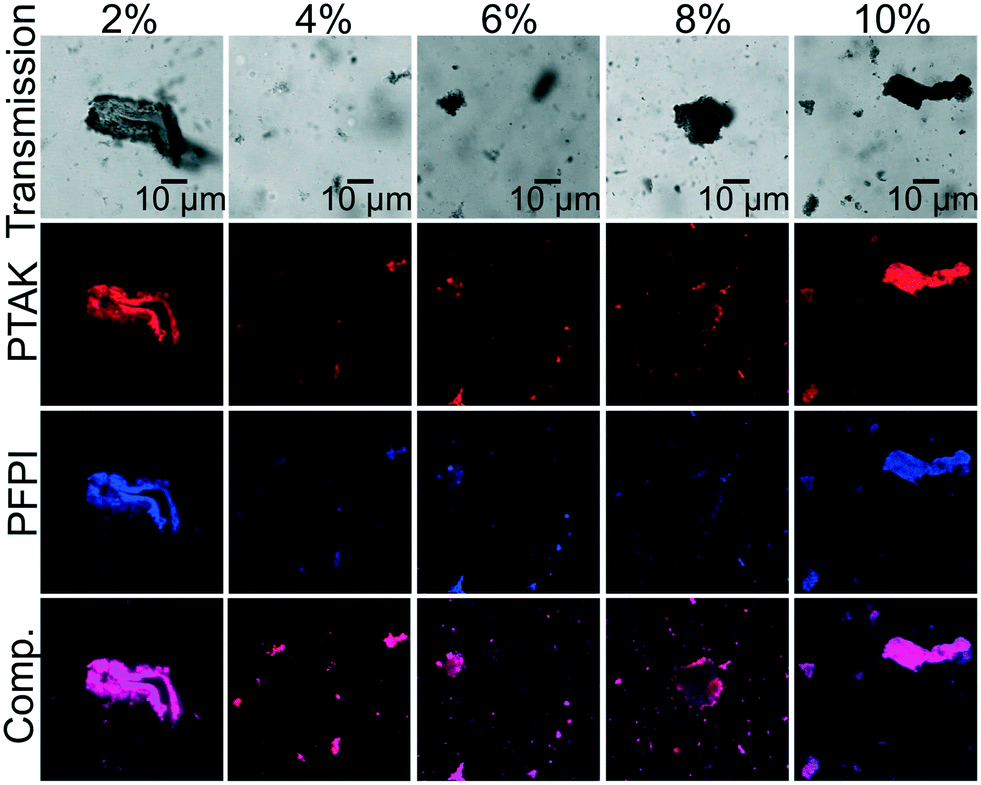 | ||
| Fig. 11 Representative confocal microscopy images of PFPI/raPTAK/oCuPcC:DOTAP complexes as a function of oCuPc mole fraction, labeled at the top of each image. Samples were prepared with a 5/1 raPTAK/PFPI molar charge ratio. From top to bottom, the rows correspond to transmission images, raPTAK PL, PFPI PL, and the composite overlay of raPTAK and PFPI PL. | ||
IV. Discussion
Before discussing our findings, we first summarize the primary takeaways of our results: (1) lipophilic phthalocyanines can be incorporated into DOTAP liposomes, but further manipulation of liposomes such as via extrusion can lead to loss of the pigment from the membrane. (2) The binding of a single oppositely charged CPE to DOTAP liposomes forms well-defined particles with a size distribution that depends on the binding temperature. (3) Adsorption of a CPE to DOTAP liposomes with embedded phthalocyanines can give rise to EET from the CPE to the phthalocyanine but also leads to a modification of the soft particle morphology. (4) An oppositely charged, donor/acceptor inter-CPE complex network can be readily adsorbed onto lipid particles, but the presence of the second outer-most CPE can lead to large morphological changes and large-scale aggregation, particularly in the presence of the ternary membrane-imbedded exciton acceptor.In this work, we endeavored to form a ternary exciton funnel that was quasi-panchromatic across the visible spectrum and that exhibited directional EET oriented from the outside in. This necessitated that the ternary exciton acceptor was membrane-embedded. The fact that we were unable to extrude liposomes without losing oCuPc suggests that the membrane acceptor/liposome assembly must be further stabilized. A major question that must be addressed in follow-on work is, how can the chemical structure of the ternary acceptor be altered to satisfy the dual constraints of (A) strong absorption on the red side of the visible spectrum, and (B) molecular size and geometry that does not lead to membrane destabilization?
The fact that we observed spherical raPTAK/DOTAP assemblies but nonspherical raPTAK/oCuPc:DOTAP particles suggests that binding of raPTAK leads to a rearrangement of lipids and oCuPc. This is not entirely surprising, since the melting point of DOTAP is well below room temperature, and thus the hydrophobic interior of the membrane is fluid. It is possible that to minimize the raPTAK/DOTAP binding free energy, oCuPc molecules are pushed away from raPTAK adsorption sites. This is possibly further aided by the disruption of the membrane packing associated with incorporation of pancake-like oCuPc molecules. These factors are likely partially responsible for the limited EET efficiency between raPTAK and oCuPc. It is interesting to once again note the subtle change in the oCuPc OD at a mole fraction of 0.1, which suggests that this mole fraction is close to the onset of oCuPc ordering. This is intriguing because the possibility of forming ordered small-molecule assemblies within the membrane may be further used to increase the EET efficiency. On the other hand, order could lead to greater membrane destabilization.
We believe that if one desires to minimize morphological changes in such assemblies – which may or may not be necessary – a lipid mixture with a higher melting point may be advantageous. For example, one may choose to introduce TAP lipids into the DOTAP matrix at a prescribed mole fraction. TAP is nearly chemically identical to DOTAP, but the hydrophobic tail of TAP is saturated, leading to greater tail ordering than DOTAP. We speculate that lipid ordering would be coupled with the ordering of the small-molecule acceptor. Nevertheless, the fact that we clearly observe quenching of raPTAK excitons by oCuPc is encouraging and leads us to believe that CPE → membrane acceptor EET can be made substantially more efficient, with Stern–Volmer constants well in access of ∼5000 M−1. The raPTAK-to-oCuPc EET efficiency can in principle be further increased either by increasing oCuPc loading or by increasing the density of adsorbed raPTAK chains. In fact, one can imagine the circumstance where the net positive surface charge of the liposome is overcharged by the net negative raPTAK charge, leading to dense raPTAK chain adsorption.
When the inter-CPE network was adsorbed onto the lipid membrane surface, we found that EET from the exciton donor CPE to the acceptor CPE remained highly efficient. We argue that the inter-CPE network is an attractive multi-chromophoric structure both for transferring excitons between donors and acceptors as well as for facile exciton diffusion along the acceptor CPE network. Such exciton diffusion is desirable to maximize the probability that excitons can eventually find the ternary acceptor during the excited-state lifetime. But the price that was paid was a near complete loss of well-defined structure. This was presumably due to bridging of assemblies by PFPI chains, which act as a kind of electrostatic glue. Recent work on layer-by-layer assembly of nonconjugated polyelectrolytes on the surface of DOTAP-containing liposomes also demonstrated formation of patchy, heterogeneous complexes.38 In this work we also found that preparation conditions had a pronounced influence on PFPI/raPTAK/liposome assemblies.
We propose that to retain well-defined particle shapes while adsorbing the inter-CPE network, the chemical structure of the outermost CPE (PFPI in this case) must be modified to lower its bridging capacity. Specifically, in the case of PFPI the phenyl ring in the backbone structure should be derivatized with bulky, polar but nonionic sidechains. We speculate that the nonionic sidechains would then act to restrict contact between ionic charges on proximal vesicles and thus limit formation of large particle colonies.
V. Conclusion
In this report we have demonstrated the formation of a soft, aqueous and hierarchically structured artificial light-harvesting antenna based on a liposome-scaffolded inter-CPE exciton network. We have shown that in such assemblies electronic energy transfer between donor/acceptor CPEs is efficient. We have also shown that a ternary small-molecule exciton acceptor can be incorporated into the liposome membrane interior. This leads to directional exciton flow along the ternary funnel from the outside of the polyelectrolyte corona towards the membrane. We found that the particle morphology was a sensitive function of preparation conditions and the loading of the membrane-bound exciton acceptor. We hypothesize that judicious modification of CPE sidechains can be used to rationally manipulate liposome aggregation and formation of large colonies as well as to improve stability.Nevertheless, it is encouraging that a few mole % of oCuPc was sufficient to quench half of raPTAK excitons. We believe this observation reflects the ability of delocalized CPE excitons to efficiently explore the underlying polymer network that underpins exciton migration. We expect that molecular engineering of the ternary exciton acceptor may significantly increase the EET efficiency compared to our proof-of-principle demonstration. It is also entirely conceivable that by further molecular engineering, the total wasted energy in the exciton cascade can be lowered significantly.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. 1848069. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract DEAC02-76SF00515. Microscopy was performed in the UCSC Life Sciences Microscopy Center RRID: SCR_021135.References
- A. Ajayaghosh, S. J. George and V. K. Praveen, Gelation-Assisted Light Harvesting by Selective Energy Transfer from an Oligo(P-Phenylenevinylene)-Based Self-Assembly to an Organic Dye, Am. Ethnol., 2003, 115, 346–349 Search PubMed.
- N. Karakostas, I. M. Mavridis, K. Seintis, M. Fakis, E. N. Koini, I. D. Petsalakis and G. Pistolis, Highly Efficient and Unidirectional Energy Transfer within a Tightly Self-Assembled Host-Guest Multichromophoric Array, Chem. Commun., 2014, 50, 1362–1365 RSC.
- T. Zhang, C. Spitz, M. Antonietti and C. F. J. Faul, Highly Photoluminescent Polyoxometaloeuropate-Surfactant Complexes by Ionic Self-Assembly, Chem. – Eur. J., 2005, 11, 1001–1009 CrossRef CAS PubMed.
- H. Tamiaki, T. Miyatake, R. Tanikaga, A. R. Holzwarth and K. Schaffner, Self-Assembly of an Artificial Light-Harvesting Antenna: Energy Transfer from a Zinc Chlorin to a Bacteriochlorin in a Supramolecular Aggregate, Angew. Chem., Int. Ed. Engl., 1996, 35, 772–774 CrossRef CAS.
- J.-H. Olivier, J. Barberá, E. Bahaidarah, A. Harriman and R. Ziessel, Self-Assembly of Charged Bodipy Dyes to Form Cassettes That Display Intracomplex Electronic Energy Transfer and Accrete into Liquid Crystals, J. Am. Chem. Soc., 2012, 134, 6100–6103 CrossRef CAS PubMed.
- J. Vogelsang, T. Adachi, J. Brazard, D. A. Vanden Bout and P. F. Barbara, Self-Assembly of Highly Ordered Conjugated Polymer Aggregates with Long-Range Energy transfer, Nat. Mater., 2011, 10, 942–946 CrossRef CAS PubMed.
- R. A. Haycock, A. Yartsev, U. Michelsen, V. Sundström and C. A. Hunter, Self-Assembly of pentameric Porphyrin Light-Harvesting Antennae Complexes, Am. Ethnol., 2000, 112, 3762–3765 Search PubMed.
- X. Li, L. E. Sinks, B. Rybtchinski and M. R. Wasielewski, Ultrafast Aggregate-to-Aggregate Energy Transfer within Self-Assembled Light-Harvesting Columns of Zinc Phthalocyanine Tetrakis(Perylenediimide), J. Am. Chem. Soc., 2004, 126, 10810–10811 CrossRef CAS PubMed.
- R. Ziessel and A. Harriman, Artificial Light-Harvesting Antennae: Electronic Energy Transfer by Way of Molecular Funnels, Chem. Commun., 2011, 47, 611–631 RSC.
- H. A. M. Ardoña, E. R. Draper, F. Citossi, M. Wallace, L. C. Serpell, D. J. Adams and J. D. Tovar, Kinetically Controlled Coassembly of Multichromophoric Peptide Hydrogelators and the Impacts on Energy Transport, J. Am. Chem. Soc., 2017, 139, 8685–8692 CrossRef PubMed.
- A. M. Sanders, T. J. Magnanelli, A. E. Bragg and J. D. Tovar, Photoinduced Electron Transfer within Supramolecular Donor–Acceptor Peptide Nanostructures under Aqueous Conditions, J. Am. Chem. Soc., 2016, 138, 3362–3370 CrossRef CAS PubMed.
- G. D. Scholes, Long-Range Resonance Energy Transfer in Molecular Systems, Annu. Rev. Phys. Chem., 2003, 54, 57–87 CrossRef CAS PubMed.
- D. Beljonne, C. Curutchet, G. D. Scholes and R. J. Silbey, Beyond Förster Resonance Energy Transfer in Biological and Nanoscale Systems, J. Phys. Chem. B, 2009, 113, 6583–6599 CrossRef CAS PubMed.
- E. Collini and G. D. Scholes, Coherent Intrachain Energy Migration in a Conjugated Polymer at Room Temperature, Science, 2009, 323, 369–373 CrossRef CAS PubMed.
- A. Olaya-Castro and G. D. Scholes, Energy Transfer from Förster–Dexter Theory to Quantum Coherent Light-Harvesting, Int. Rev. Phys. Chem., 2011, 30, 49–77 Search PubMed.
- T. Mirkovic, E. E. Ostroumov, J. M. Anna, R. van Grondelle, Govindjee and G. D. Scholes, Light Absorption and Energy Transfer in the Antenna Complexes of Photosynthetic Organisms, Chem. Rev., 2017, 117, 249–293 CrossRef CAS PubMed.
- H. Jiang, P. Taranekar, J. R. Reynolds and K. S. Schanze, Conjugated Polyelectrolytes: Synthesis, Photophysics, and Applications, Angew. Chem., Int. Ed., 2009, 48, 4300–4316 CrossRef CAS PubMed.
- A. Duarte, K.-Y. Pu, B. Liu and G. C. Bazan, Recent Advances in Conjugated Polyelectrolytes for Emerging Optoelectronic Applications, Chem. Mater., 2011, 23, 501–515 CrossRef CAS.
- B. Liu and G. C. Bazan, Conjugated Polyelectrolytes: Fundamentals and Applications, Wiley, 2013 Search PubMed.
- W. R. Hollingsworth, T. Magnanelli, C. Segura, J. Young, A. E. Bragg and A. L. Ayzner, Polyion Charge Ratio Determines Transition between Bright and Dark Excitons in Donor/Acceptor Conjugated Polyelectrolyte Complexes, J. Phys. Chem. C, 2018, 122, 22280–22293 CrossRef CAS.
- W. R. Hollingsworth, C. Segura, J. Balderrama, N. Lopez, P. Schleissner and A. L. Ayzner, Exciton Transfer and Emergent Excitonic States in Oppositely-Charged Conjugated Polyelectrolyte Complexes, J. Phys. Chem. B, 2016, 120, 7767–7774 CrossRef CAS PubMed.
- W. Barford, Beyond Förster Resonance Energy Transfer in Linear Nanoscale Systems, J. Phys. Chem. A, 2010, 114, 11842–11843 CrossRef CAS PubMed.
- M. Coustet, J. Irigoyen, T. A. Garcia, R. A. Murray, G. Romero, M. Susana Cortizo, W. Knoll, O. Azzaroni and S. E. Moya, Layer-by-Layer Assembly of Polymersomes and Polyelectrolytes on Planar Surfaces and Microsized Colloidal Particles, J. Colloid Interface Sci., 2014, 421, 132–140 CrossRef CAS PubMed.
- T. T. Nguyen and B. I. Shklovskii, Overcharging of a Macroion by an Oppositely Charged Polyelectrolyte, Phys. A, 2001, 293, 324–338 CrossRef CAS.
- A. Y. Grosberg, T. T. Nguyen and B. I. Shklovskii, Colloquium: The Physics of Charge Inversion in Chemical and Biological Systems, Rev. Mod. Phys., 2002, 74, 329–345 CrossRef CAS.
- T. T. Nguyen and B. I. Shklovskii, Complexation of a Polyelectrolyte with Oppositely Charged Spherical Macroions: Giant Inversion of Charge, J. Chem. Phys., 2001, 114, 5905–5916 CrossRef CAS.
- C. F. Calver, K. S. Schanze and G. Cosa, Biomimetic Light-Harvesting Antenna Based on the Self-Assembly of Conjugated Polyelectrolytes Embedded within Lipid Membranes, ACS Nano, 2016, 10, 10598–10605 CrossRef CAS PubMed.
- J. E. Houston, M. Kraft, I. Mooney, A. E. Terry, U. Scherf and R. C. Evans, Charge-Mediated Localization of Conjugated Polythiophenes in Zwitterionic Model Cell Membranes, Langmuir, 2016, 32, 8141–8153 CrossRef CAS PubMed.
- A. T. Ngo and G. Cosa, Assembly of Zwitterionic Phospholipid/Conjugated Polyelectrolyte Complexes: Structure and Photophysical Properties, Langmuir, 2010, 26, 6746–6754 CrossRef CAS PubMed.
- Z. Wang, D. Gao, Y. Zhan and C. Xing, Enhancing the Light Coverage of Photosynthetic Bacteria to Augment Photosynthesis by Conjugated Polymer Nanoparticles, ACS Appl. Bio Mater., 2020, 3, 3423–3429 CrossRef CAS PubMed.
- Y. Wang, S. Li, L. Liu, F. Lv and S. Wang, Conjugated Polymer Nanoparticles to Augment Photosynthesis of Chloroplasts, Angew. Chem., Int. Ed., 2017, 56, 5308–5311 CrossRef CAS PubMed.
- W. R. Hollingsworth, V. Williams and A. L. Ayzner, Semiconducting Eggs and Ladders: Understanding Exciton Landscape Formation in Aqueous Π-Conjugated Inter-Polyelectrolyte Complexes, Macromolecules, 2020, 53, 2724–2734 CrossRef.
- P. Groves, Diffusion Ordered Spectroscopy (Dosy) as Applied to Polymers, Polym. Chem., 2017, 8, 6700–6708 RSC.
- F. C. Spano and C. Silva, H- and J-Aggregate Behavior in Polymeric Semiconductors, Annu. Rev. Phys. Chem., 2014, 65, 477–500 CrossRef CAS PubMed.
- O. Glatter and O. Kratky, Small Angle X-Ray Scattering, Academic Press, 1982 Search PubMed.
- R. M. Clegg, [18] Fluorescence Resonance Energy Transfer and Nucleic Acids, in Methods in Enzymology, Academic Press, 1992, vol. 211, pp. 353–388 Search PubMed.
- S. E. Braslavsky, E. Fron, H. B. Rodríguez, E. S. Román, G. D. Scholes, G. Schweitzer, B. Valeur and J. Wirz, Pitfalls and Limitations in the Practical Use of Förster's Theory of Resonance Energy Transfer, Photochem. Photobiol. Sci., 2008, 7, 1444–1448 CrossRef CAS PubMed.
- Y. Kashcooli, K. Park, A. Bose, M. Greenfield and G. D. Bothun, Patchy Layersomes Formed by Layer-by-Layer Coating of Liposomes with Strong Biopolyelectrolytes, Biomacromolecules, 2016, 17, 3838–3844 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional optical absorption and photoluminescence spectra of liposome complexes, photoluminescence microscopy images of diluted solutions, and small-angle X-ray scattering. See DOI: 10.1039/d1me00139f |
| ‡ Current address: Nanoengineering Department, University of California San Diego, San Diego, CA, USA. |
| This journal is © The Royal Society of Chemistry 2022 |