
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom stretchable and healable to self-healing semiconducting polymers: design and their TFT devices
Livy
Laysandra
,
Andreas
Njotoprajitno
,
Suhendro Purbo
Prakoso
 and
Yu-Cheng
Chiu
*
and
Yu-Cheng
Chiu
*
Department of Chemical Engineering, National Taiwan University of Science and Technology, No. 43, Sec. 4, Keelung Rd., Da'an Dist., Taipei City 10607, Taiwan. E-mail: ycchiu@mail.ntust.edu.tw
First published on 12th July 2022
Abstract
Flexible thin-film transistor (TFT) devices are essential elements of future flexible electronics owing to their promising applications in various fields ranging from lighting and display, sensors, communication, and smart textiles to medical devices. Currently, these state-of-the-art electronic devices are not only struggling from “brittle” to “stretchable”, but are also expected to advance to “autonomous self-healable”. However, to integrate these features into flexible and stretchable devices, the first requirement to consider is compatibility between their mechanical and electrical properties given that the self-healing ability is associated with mechanical compliance but is incompatible with existing electronic technologies. Therefore, the first step closer to achieve is due to the recent progress in the field of stretchable semiconducting polymers, which thus far consists of two strategies: (1) structural engineering of conjugated polymers and (2) physical blending of conjugated polymers in soft insulating polymers. Together with the emphasis on additional functionalization in novel modified-semiconducting materials to boost large mechanical deformation without damaging their electronic properties, structural engineering can also provide targeted features (i.e., self-healing ability). Finally, we highlight the chemical aspects of self-healing materials (from non-covalent to reversible covalent-based mechanisms) that have been successfully integrated in TFT devices.
 Livy Laysandra | Livy Laysandra received her MSc Degree from National Taiwan University of Science and Technology in 2020 under the guidance of Prof. Yu-Cheng Chiu. Currently, she is pursuing her doctoral degree at the same university. Her current research focuses on designing elastic and self-healing polymers as dielectric, semiconductor, and luminescent layers in thin-film form. |
 Andreas Njotoprajitno | Andreas Njotoprajitno joined Professor Yu-Cheng Chiu's lab at the Department of Chemical Engineering of National Taiwan University of Science and Technology (NTUST) in the spring of 2021 and achieved his BSc in the summer of 2022. His research focuses on the characterization of elastic and self-healing semiconducting polymers. |
 Suhendro Purbo Prakoso | Suhendro Purbo Prakoso received his PhD degree from Taiwan International Graduate Program on Sustainable Chemical Science & Technology (TIGP-SCST) in 2021 under the guidance of Prof. Chien-Lung Wang (Department of Applied Chemistry, National Yang Ming Chiao Tung University) and Prof. Yu-Tai Tao (Institute of Chemistry, Academia Sinica). Currently, he works as a postdoctoral fellow in Prof. Yu-Cheng Chiu’s group. His current research interests involve the low-voltage, tunnelling-effect of photoassisted transistor memory devices and novel photosensitive charge storage materials. |
 Yu-Cheng Chiu | Yu-Cheng Chiu joined the Department of Chemical Engineering at National Taiwan University of Science and Technology (Taiwan Tech) as a tenure-track Assistant Professor since August 2017. Prior to joining the faculty, Yu-Cheng was a postdoc in the Zhenan Bao research group at Stanford University, where he was devoted to research on intrinsically stretchable/healable semiconducting polymers and high-performance OFET by solution shearing technique. Before moving to Stanford, he received his PhD degree under the supervision from Prof. Wen-Chang Chen in December 2012 from the Chem. E. at National Taiwan University, and then stayed in the same group for his first post-doctoral stint until 2014. Currently, his major interests are the elastic and self-healing semiconducting materials, soft organic devices including transistor and transistor memory, and morphology characterization for polymer materials. |
1. Introduction
Since the discovery of polythiophenes and related conductive organic polymers by Heeger, MacDiarmid, and Shirakawa (Nobel Prize in Chemistry 2000),1–3 numerous conjugated polymers have been developed as a new class of organic semiconductors, setting out to renew the semiconducting industry, which has been dominated by their inorganic counterparts such as traditional silicon-based materials over the last 50 years.4 The fruit of this effort is reflected in the electronics revolution from heavy and cumbersome to compact and mobile and rigid to flexible, and is expected to proceed to stretchable, ultimately changing the fundamental interactions between humans and technology. Thus far, striking progress has been achieved in various flexible electronic device applications such as soft robots,5 e-skin sensors,6–8 flexible devices,9–12 wearable electronics,13 and energy-storage devices.14,15 However, these near-magnificence devices tend to fail in long-term use due to repeated wear and tear, accidental scratching, and mechanical fracture during deformation in their practical applications.16 Consequently, researchers have sought to integrate healing capabilities to achieve a substantial improvement in device lifetime and increased reliability, maintainability, and durability.However, the major challenge in the development of these polymers is maintaining good electrical and mechanical properties simultaneously. The extended π-conjugation of the polymer backbone is essential to yield good electronic properties, but the mechanical properties tend to be quite poor due to its high rigidity. Therefore, it is necessary to explore a much broader approach to fabricate semiconducting polymers having excellent mechanical properties without sacrificing their charge carrier mobility, especially for thin-film transistor (TFT) device application.
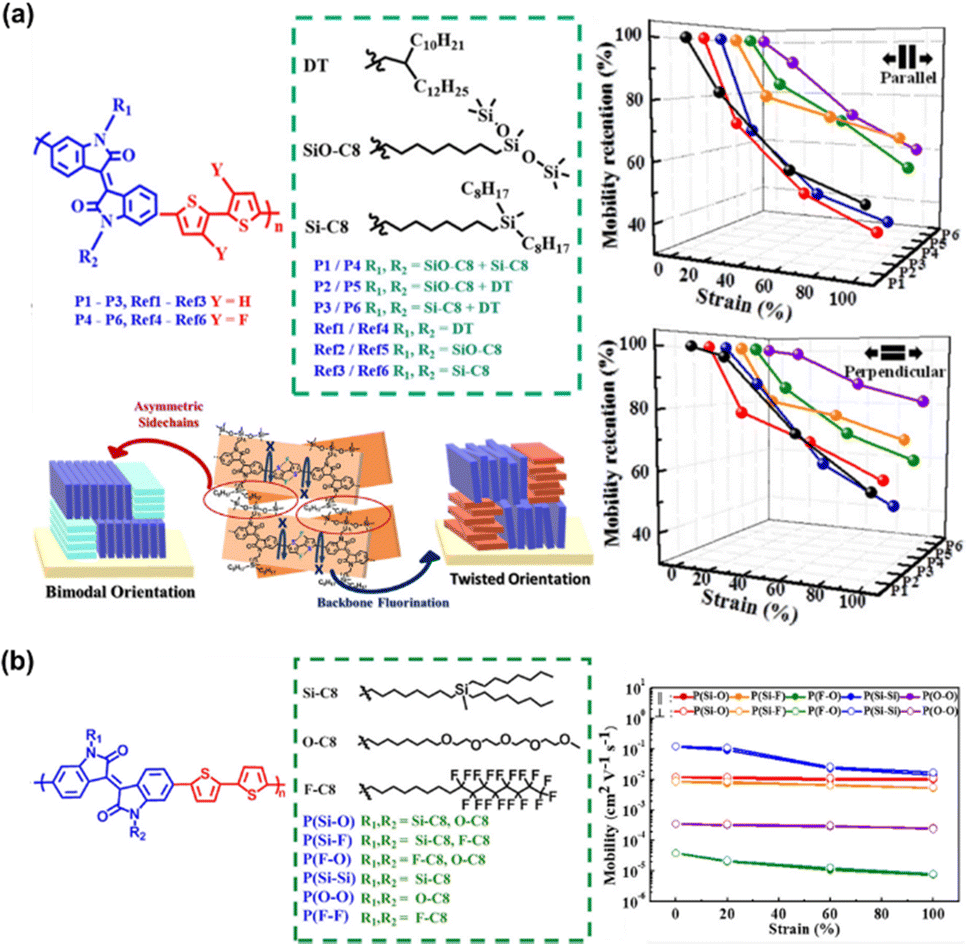
In this progress report, we comprehensively review the various approaches employed by different research groups to develop stretchable semiconducting polymers with emphasis on the design and manipulation of semiconducting polymers toward deformable stretchable electronics, a field which has grown significantly in the past few years.17,18 We also provide an update on the current strategies developed toward intrinsically stretchable semiconducting materials as a starting point, among which two main strategies for diverse structural modifications have been adopted, as follows: (1) backbone engineering and (2) side-chain engineering, with each strategy having several unique approaches, as depicted in Fig. 1a. Then, highlighting the strong impact in the hydrogen bonding units incorporated in the conjugated polymer, intramolecular conformational locks can be established, resulting in increased backbone planarity, enhanced π-delocalization and field-effect mobility (μFETh).19–22 This elegant strategy also creates new properties and functions, one of which is self-healing ability (Fig. 1b).23–25 The exceptionally successful building blocks that will be discussed in this review are exemplified by a range of different conjugated materials, including thiophene, diketopyrrolopyrrole (DPP), and isoindigo (IDG), containing polymers.26–30 Furthermore, to outweigh the extra complexity introduced in modifying structural polymers, the simplicity of the physical blending of conjugated polymers in an elastomeric matrix for TFTs has been exploited (Fig. 1c).31–33 Therefore, surveying the advances from extrinsically stretchable semiconducting polymer blends to self-healing capability is also necessary. We anticipate that the integration of these approaches will be crucial for the development of future-generation stretchable and self-healable polymer semiconductors.
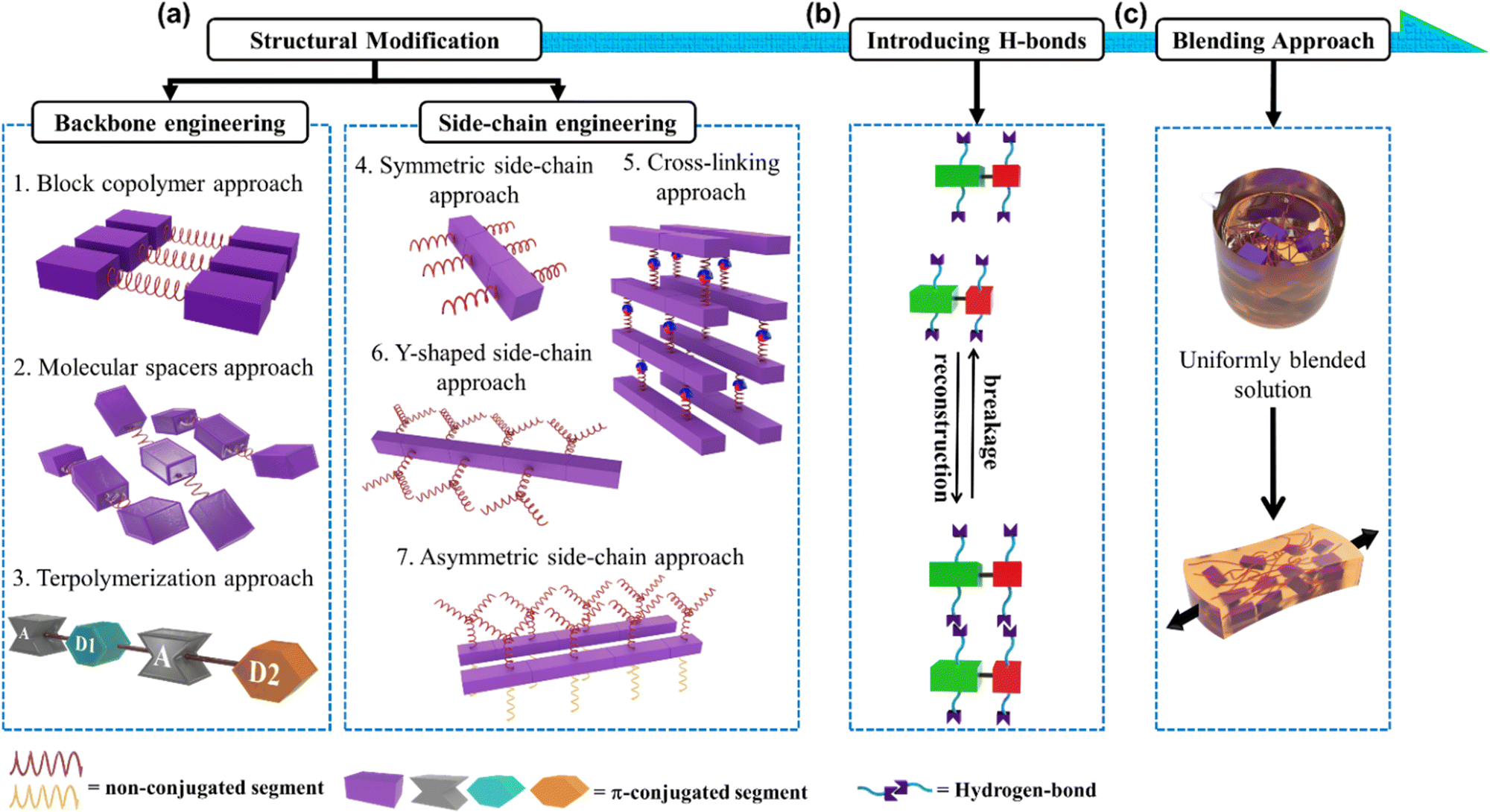 | ||
| Fig. 1 Illustration of designing novel semiconducting materials to promote mechanical and electrical properties through: (a) structural engineering, which consists of two main strategies, and further divided into seven approaches; (b) functionalizing the side-chain using hydrogen bonding units; and (c) simple blending process of π-conjugated polymer with elastomer matrix, generating autonomous self-healing ability. | ||
2. Stretchable semiconducting polymers
Inspired by the overwhelming feature of elastomeric materials, many efforts have been devoted to replicating the mechanical properties of elastomers by integrating conjugated polymers in TFT applications to attain sufficiently high elasticity for stable charge carrier transport under severe external stress.34 Considering that the elastic properties of non-conjugated materials are influenced by the chemical nature of the monomer, branching, conformation, packing structure, extent of crosslinking, and chain length of macromolecules, exploitation of similar attributes in electrically active polymers bears great potential to achieve similar or even better mechanical properties.35 The success of this approach can tremendously promote the potential of flexible devices, including the possibility of using low-cost fabrication methods over large-area substrates. In particular, Prof. Bao at Stanford University contributed greatly to the innovative development of this emerging field by utilizing the semiconducting polymer poly(3-hexylthiophene (P3HT)) to yield high-performance transistors.36–40 Subsequently, flexible electronic devices began to develop rapidly. Several examples are presented and discussed in the next section, with emphasis on the details of the interrelation between the physical characteristics of the conjugated polymer structure with the mechanical-electronical compliance in TFT devices.2.1 Fundamental properties of polythiophene derivatives
P3HT has emerged as a second-generation conjugated polymer that has successfully improved the performance of organic TFT devices.4 Starting from the dedication of scientists to analyze and characterize the structural properties of P3HT, it was revealed that one of the most compelling properties of P3HT is its ability to form highly ordered π-stacks and side-chain lamella stacks.39,41,42 Transmission electron microscopy (TEM) has been used to demonstrate the structural evolution from chain-extended crystals to an architecture consisting of interlinked crystalline lamellae and amorphous regions at a weight-average molecular weight (Mw) of around 25–35 kg mol−1.41,42 Furthermore, the long-range packing behavior and morphology of a P3HT film was observed via atomic force microscopy (AFM), providing information about its assembly mechanism and the aligning effects of various deposition methods. Obviously, chain folding, which leads to lamellar crystalline units, was discovered in classical systems such as polyolefins and poly(ethylene oxides)43–45 and visualized in longer-chain P3HT molecules by scanning tunnelling microscopy (STM) in early 2000 by Mena-Osteriz et al.46 In brief, the well-controlled Mw P3HT chains can fold and may possess sufficient connections (or tie chains) between ordered domains, resulting in higher charge mobility compared with low-Mw P3HT.Focusing on the typical semicrystalline P3HT morphology composed of anisotropic crystalline regions bridged by tie chains (Fig. 2a),47,48 the possibility of intrinsic stretchability can be expected. As the first step in investigating the strong correlation between the strain-alignment and charge mobility of P3HT thin films, O’Connor et al. proposed an innovative strategy to align the crystalline phase in P3HT, which was achieved by laminating a P3HT spin-coated film on a stretchable poly(dimethylsiloxane) (PDMS) substrate, and then stretching to various extents.49 Upon stretching, the crystallites in the P3HT films were reoriented with the polymer backbones aligned in the direction of the applied strain. The mobility of P3HT increased in the applied strain direction and decreased in the perpendicular direction. This strain-induced alignment transformed the predominant edge-on oriented (100) polymer crystallites to face-on orientation (010) crystallites (Fig. 2b). Alternatively, 100% biaxially stretched films showed an increase in facial alignment, but the charge mobility anisotropy was not significant.50 These results suggest that mechanical strain on the micrometer scale can produce changes in texture, anisotropy in elastic modulus, and alter charge transport. Furthermore, molecular engineering seeks to attain the integration of different features by focusing on the structure–property relationship at the molecular level such as molecular weight (Mw), regioregularity (RRe), and other considerations related to solution processing and fabrication techniques. In the following section, we specifically discuss the influence of these crucial parameters on charge transport and mechanical properties.
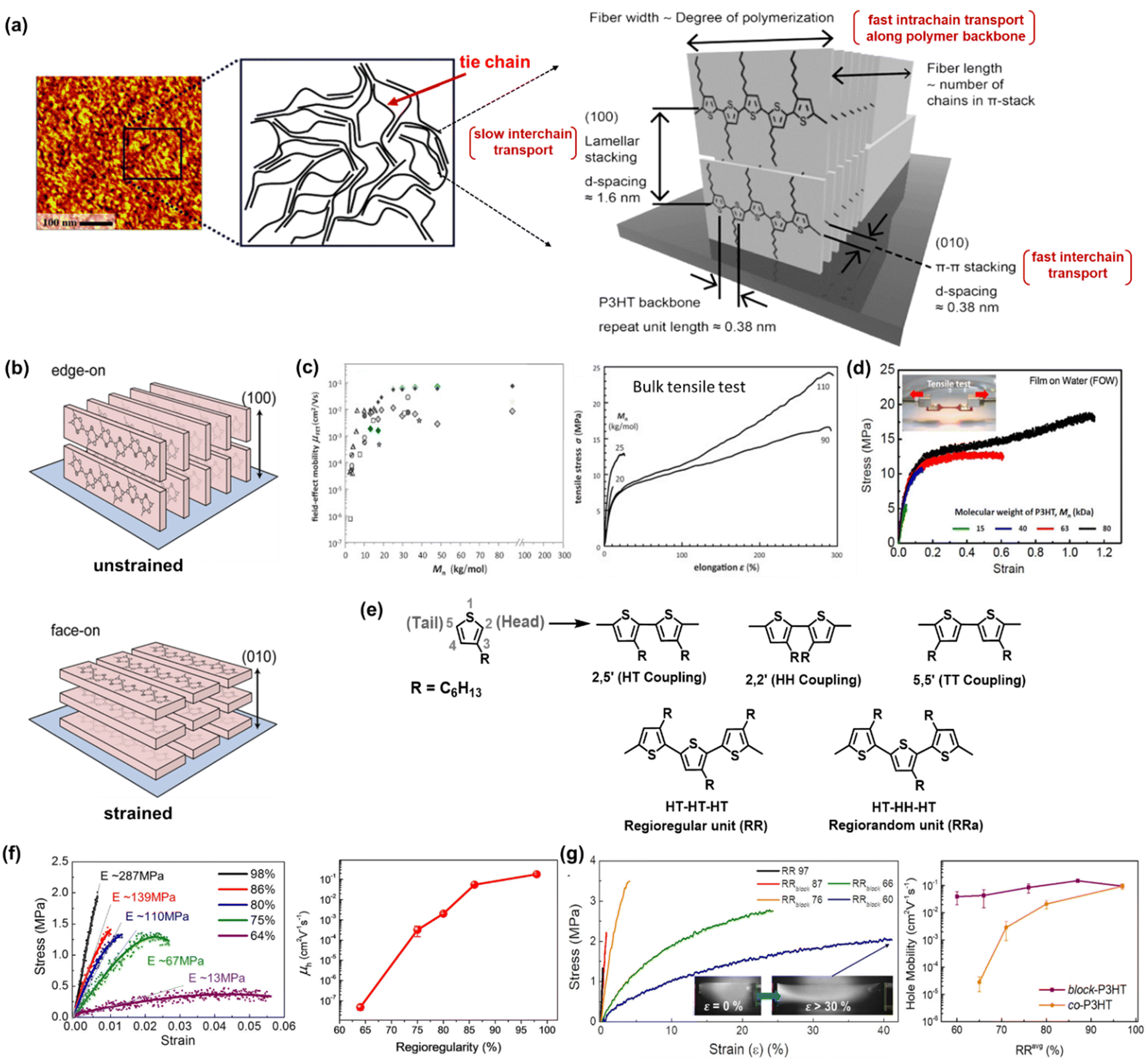 | ||
| Fig. 2 Structure engineering of polythiophene derivatives. (a) Atomic force microscopy (AFM) image of P3HT film (left), schematic of molecular structure consisting of anisotropic crystalline region bridged by amorphous regions (middle), and crystallographic molecular structures of the semicrystalline P3HT packing behavior showing directions of π-stacking and lamellar-stacking (right). (left and middle)47 Copyright: 2005, the American Chemical Society. (right)48 Copyright: 2017, the American Chemical Society. (b) Illustration of the P3HT backbone stacking orientation before and after strain. The polymer crystallites reorient to align in the direction of the applied strain, transforming the edge-on crystallites into face-on crystallites.50 Copyright: 2013, the American Chemical Society. (c) Hole mobilities and stress–strain curves of P3HT tapes as a function of Mn.52 Copyright: 2013, Elsevier. (d) stress–strain curves and tensile moduli of P3HT thin films at different Mn obtained by film-on-water (FOW) technique.53 Copyright 2017, the American Chemical Society. (e) Schematic showing three possible coupling modes and chemical structure of regioregularity (RRe) and regiorandom (RRa) of P3HT. (f) stress–strain curves and hole mobilities of P3HT films with respect to the degree of regioregularity Mn obtained by the FOW technique.58 Copyright: 2015, the American Chemical Society. (g) Schematic illustration of block-P3HTs with increasing lengths of RRa-blocks added for mechanically robust and high-performance TFTs, the stress–strain curves of block-P3HTs (the inserted picture of the RRblock 60 film shows that no damage occurred even after stretching above 30% strain), and hole mobility values as a function of RRe-block-P3HTs.59 Copyright: 2019, the American Chemical Society. | ||
Considering the disadvantages of using a homopolymer with randomly coupled HH defects, in the case with a limited extent of ordered domains, the same research team continued the development of mechanically robust and high-performance TFT using a P3HT regioblock copolymerization strategy (block-P3HTs).59 The authors prepared a series of block-P3HTs consisting of RRe and RRa P3HTs with various Mn values of RRa blocks (from 3 to 33 kg mol−1) and a fixed Mn of RRe blocks (11–12 kg mol−1) via a catalyst-transfer polycondensation strategy (Fig. 2g). Briefly, block-P3HT thin films with longer RRa blocks enhanced the mechanical ductility due to the fact that the amorphous moiety in regioblock copolymers promotes the formation of an entangled and/or folded structure. For example, the elongation at break of the thin films was remarkably enhanced by 100-fold (i.e., from 0.3% to 30.2%) as the Mn of the RRa block increased. Amazingly, all the block-P3HT possessed a similar high μFETh of 1.5 × 10−1 cm2 V−1 s−1 to that of the high RRe P3HT homopolymer. The discovery of the mechanical ductility of block-P3HTs, which can be dramatically enhanced without sacrificing the electrical properties, becomes ones of the promising candidate strategies to fabricate high-performance soft electronics, considering the trade-off between electronic properties and mechanical flexibility often encountered by scientists.
In the preparation of polymers, it is necessary to consider parameters such as the film preparation method (ultrasonication, spin-coating, dip-coating, drop-casting, melt processing, and blade coating) given that they can influence their morphology, regioregularity, and change in Mn in comparison to conjugated polymers, which are structurally different, to fairly assess their structural contribution.38,71,72 Another example by Choi et al. showed that ultrasonication led to the formation of P3HT nanowires with increased mobility, film crystallinity, and decreased crack onset strains.73 Therefore, ultrasonication plays an important role in breaking down the polymer chain, enhancement in aggregation, and local order for inter-and intramolecular interactions. The mechanical behavior could be observed in the tensile test of melt-processed P3HT and spin-coated P3HT, as shown in Fig. 2c and d, respectively. The optimal maximum strain value of the melt-processed high Mn P3HT reached 300%, whereas the spin-coated film was only stretchable up to 100%. The variation in film thickness and its morphology may also cause a significant difference in critical strain.
2.2 Conjugated polymer engineering
Another notable aspect to promote the mechanical deformability and charge-transporting capability of stretchable semiconductor polymers arises from the diverse structural modification of conjugated polymers. This type of approach was highlighted by the finding of O'Connor et al. when comparing RRe P3HT and PBTTT.60 The correlation between μFETh and elastic moduli was displayed with moderately crystalline P3HT and highly crystalline PBTTT, exhibiting low and high μFETh, respectively, while their elastic moduli showed a proportional trend with μFETh. As has been discussed previously (see Fig. 2a), P3HT adopts an edge-on type lamellar packing motif of polymer backbones bridged by alkyl chain regions. The π-stacking direction in a parallel way to the substrate enables efficient 2D charge transport favorable for TFTs. In contrast, PBTTT more easily forms highly ordered films due to the existence of many crystalline domains with larger crystalline sizes and fewer defects. This superiority in terms of μFETh (8 × 10−2 cm2 V−1 s−1) and elastic modulus (E = 0.9 GPa) of PBTTT over P3HT (μFETh = 6 × 10−2 cm2 V−1 s−1 and E = 0.25 GPa) is due to its higher crystalline morphology. Furthermore, the high crystallinity is also responsible for the low crack onset strain for PBTTT (<2.5%) compared to P3HT (>150%). These results illustrate that long-range order is preferable to increase the mobility, but inevitably stiffens and embrittles the polythiophene semiconductor films.Considering the high crystallinity and amorphous nature of conjugated polymers, it is assumed that their electrical-mechanical properties are mutually exclusive. However, through the continuous efforts and exploration by researchers, it was discovered that conjugated polymers based on isoindigo (IDG), naphthalene diimides (NDI), and diketopyrrolopyrrole (DPP)26–30 exhibit impressive charge mobilities of over 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cm2 V−1 s−1 although their long-range order is relatively poorer than that of P3HT and PBTTT. These observations have led to a new way of generating conjugated polymer thin films for effective charge mobility, thus facilitating the optimum intra- and intermolecular charge transport at the device length scale, improving the interconnectedness between aggregated domains, and reducing the amount of disorder in the conjugated segments (making rigid backbones) seem to be the key rather than inducing high crystallinity.74–76 Hence, two main strategies of diverse structural modifications have been adopted, i.e., backbone engineering and side-chain engineering (Fig. 1a). Backbone engineering involves the introduction of conjugated and non-conjugated units into the main chain of a polymer, which is generally classified into three approaches, mainly block copolymerization, molecular spacer, and terpolymerization. Meanwhile, side chain engineering introduces non-conjugated units in the side chain of a conjugated polymer, which can also be classified into four different approaches, including symmetric/asymmetric side-chain, cross-linking, and Y-shaped side-chain. Furthermore, it was revealed that the incorporation of hydrogen bonding moieties in these novel conjugated polymer designs is particularly effective in tailoring the mechanical deformability and maintaining or even boosting the charge transport values in TFTs.19–22 Interestingly, the combination of dynamic H-bonds in organic semiconductors has created new properties and functions such as self-healing ability which will be explained in Section 3.23–25
cm2 V−1 s−1 although their long-range order is relatively poorer than that of P3HT and PBTTT. These observations have led to a new way of generating conjugated polymer thin films for effective charge mobility, thus facilitating the optimum intra- and intermolecular charge transport at the device length scale, improving the interconnectedness between aggregated domains, and reducing the amount of disorder in the conjugated segments (making rigid backbones) seem to be the key rather than inducing high crystallinity.74–76 Hence, two main strategies of diverse structural modifications have been adopted, i.e., backbone engineering and side-chain engineering (Fig. 1a). Backbone engineering involves the introduction of conjugated and non-conjugated units into the main chain of a polymer, which is generally classified into three approaches, mainly block copolymerization, molecular spacer, and terpolymerization. Meanwhile, side chain engineering introduces non-conjugated units in the side chain of a conjugated polymer, which can also be classified into four different approaches, including symmetric/asymmetric side-chain, cross-linking, and Y-shaped side-chain. Furthermore, it was revealed that the incorporation of hydrogen bonding moieties in these novel conjugated polymer designs is particularly effective in tailoring the mechanical deformability and maintaining or even boosting the charge transport values in TFTs.19–22 Interestingly, the combination of dynamic H-bonds in organic semiconductors has created new properties and functions such as self-healing ability which will be explained in Section 3.23–25
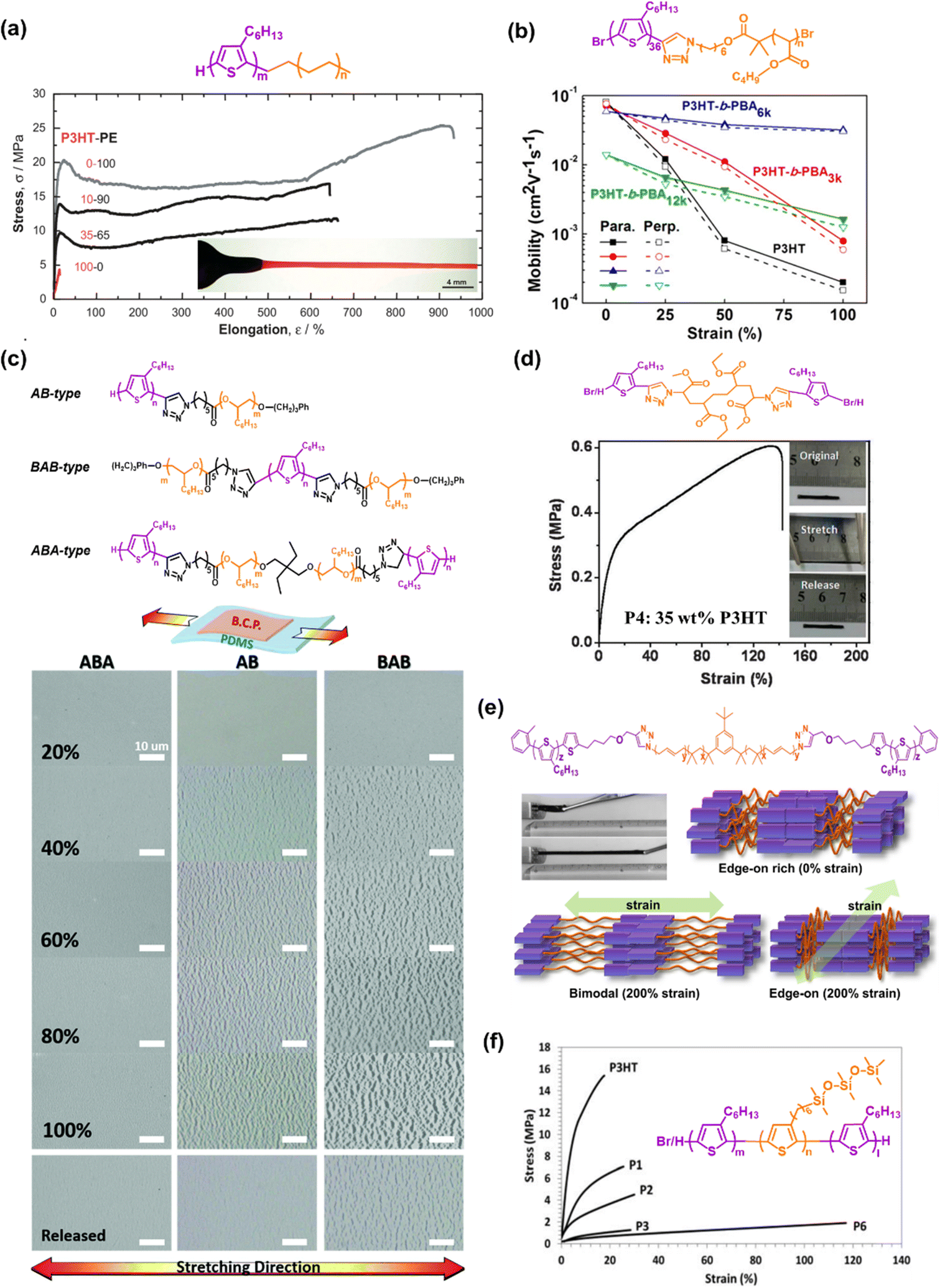 | ||
| Fig. 3 Intrinsically stretchable semiconducting polymers developed based on the block copolymer approach. The chemical structures and mechanical and/or electrical properties of: (a) diblock copolymers with different weight ratios of P3HT and PE. The inset of the bottom image is an optical image of a dog-bone-shaped 35–65 wt% P3HT-PE under 600% stretching.77 Copyright: 2007, Wiley-VCH. (b) P3HT-b-PBA rod-coil diblock copolymers.79 Copyright: 2017, the American Chemical Society. (c) AB-type, BAB-type, and ABA-type of P3HT-b-POO block copolymers.80 Copyright: 2019, The Royal Society of Chemistry. (d) Triblock copolymer P3HT-b-PMA-b-P3HT.81 Copyright: 2015, The Royal Society of Chemistry. (e) P3HT-b-PIB-b-P3HT with a photograph of the triblock copolymer film before and after stretching (upper left) and illustration of the model for the deformation of the microphase separated structure and crystalline orientation of the triblock copolymer thin film (bottom).82 Copyright: 2015, the American Chemical Society. (f) P3HT-b-P3SiHT-b-P3HT, where the stress–strain profile composed of the homopolymer P3HT (Mn = 20 kg mol−1), diblock copolymer P3HT-b-P3SiHT (P1–P3 where PI, P2, and P3 containing 73, 60, and 53 wt% P3HT, respectively), and triblock copolymer P3HT-b-P3SiHT-b-P3HT containing 22 wt% P3HT (P6).83 Copyright: 2018, Wiley-VCH. | ||
As another example of more advanced molecular backbone engineering, Peng et al. (2015) first proposed the concept of utilizing hard-soft-hard (ABA-type) triblock copolymers based on the principle of architectural thermoplastic elastomers (TPEs), leading to the formation of intrinsically stretchable semiconductor TPEs (Fig. 3d).81 The authors succeeded in synthesizing ABA triblock copolymers consisting of crystalline P3HT as A and amorphous polymethacrylate (PMA) block as B. The novel ABA triblock copolymer demonstrated a high Young's modulus of 6 MPa, stretchability of 140%, and μFETh of 1.7 × 10−4 cm2 V−1 s−1 despite containing 65 wt% PMA. Inspired by the semiconductor TPE design, Higashihara et al. (2019) used a similar approach to prepare a novel intrinsically stretchable ABA-type triblock copolymer composed of P3HT and polyisobutylene (PIB) segments (P3HT-b-PIB-b-P3HT) (Fig. 3e). Due to the excellent elastomeric properties of the PIB segments,82 this polymer exhibited an extremely low elastic modulus of 0.00113GPa and a much improved crack onset strain value of 300%, as determined via the tensile test in bulk films, with the μFETh value of 3.0 × 10−3 cm2 V−1 s−1 despite containing 70wt% insulating PIB. Given the fact that these polymers possessed insulating and non-semiconducting segments, it is not surprising that the mobility of the triblock copolymers decreased as the soft-material content increased.
Recognizing this weakness, Miyane et al. (2018) fabricated fully conjugated ABA triblock polymers by utilizing a regioregular polythiophene derivative bearing a trisiloxane group in the side chains (P3SiHT) as the soft segment.83 The merits of the introduction the P3SiHT moiety in conjugated polymers have been further investigated.84–87 Indeed, a P3SiHT film displayed a remarkable change in characteristics such as thermal behavior, chemical stability, mechanical property, and miscibility in solvents. Especially in terms of mechanical behavior, it exhibited 2-orders of magnitude smaller Young's modulus and 20 times higher elongation at break than RRe P3HT.87 Benefiting from the features of P3SiHT, all the block copolymers exhibited a lower tensile modulus and greater elongation as the P3SiHT content increased, which are typical characteristics of a plastic-to-rubber transition. As illustrated in Fig. 3f, P3HT-b-P3SiHT-b-P3HT with the highest P3SiHT content of 78 wt% delivered a low elastic modulus of 0.00335GPa and improved elongation at break of 118%. Interestingly, the resulting μh of the ABA triblock copolymer in the TFTs were unexpectedly comparable or even higher than that of the P3HT homopolymer. P3HT-b-P3SiHT-b-P3HT with the lowest P3HT content of 22% exhibited an even higher average mobility (5.14 × 10−3cm2 V−1 s−1) than the P3HT homopolymer (1.46 × 10−3cm2 V−1 s−1). Among the prepared copolymers, P3HT-b-P3SiHT-b-P3HT with 59 wt% P3HT obtained an excellent average μFETh value of 1.06 × 10−2 cm2 V−1 s−1. Overall, by leveraging its ability to assemble well-defined structures on the nanoscale, the block copolymer approach plays an important role in efficiently optimizing the mechanical properties of materials. This approach offers a way to avoid complex structural engineering for the fabrication of devices, but the development of stretchable materials suitable for electronic devices is a great challenge. Thus far, the low μFETh with P3HT-based modification still cannot satisfy the theoretical intrinsic intracrystalline mobility of RRe P3HT, which can reach over 1 cm2 V−1 s−1.88–90 Thus, it seems that the block copolymer approach still needs to be further developed to actualize the application of high-performance devices in terms of both electrical-mechanical properties.
× 10−4cm2 V−1 s−1, which was comparable to that of its fully conjugated counterpart (P3HT-DPP-10%) of 9.29 × 10−4cm2 V−1 s−1. Interestingly, although the hole mobility decreased with an increase in spacer composition, there is still an increment in hole mobility with respect to the length of the spacer carbon chain. Through the film-on-elastomer measurement, it was revealed that several modified polymers could be stretched beyond 80% strain, and particularly for 10% T-8-T and 10% T-10-T, they could achieve the lowest elastic modulus of 0.14 GPa and 0.15 GPa, respectively. This approach opens an avenue for researchers to overcome the most common obstacle of the tradeoff between mechanical and electronic performance.
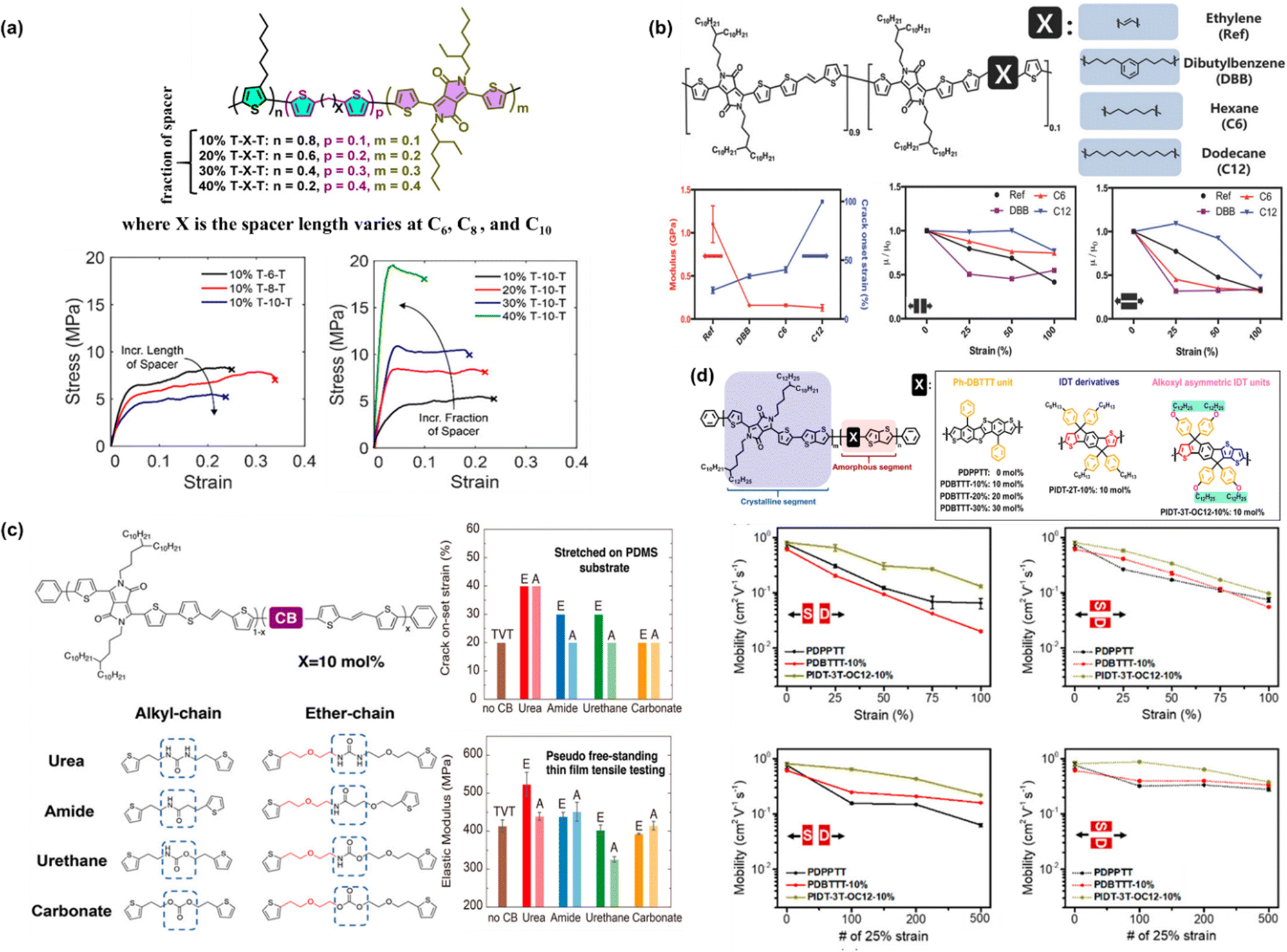 | ||
| Fig. 4 (a) Chemical structure of the designed DPP-based conjugated polymer backbone and the effect of the conjugation-break spacer length and spacer composition on its mechanical and electrical properties. Representative stress–strain curves of modified P3HT-DPP polymer films obtained using the FOW technique.92 Copyright: 2018, the American Chemical Society. (b) Effects of chain breakers on elastic modulus and mobility under strain in DPP-TVT polymers.93 Copyright: 2018, Wiley-VCH. (c) Summary of the crack on-set and elastic modulus measurements of DPP-TVT polymers as a function of the H-bonding conjugation breaker effect.94 Copyright: 2020, the American Chemical Society. (d) Effect of conjugated rigid fused-rings with optimized bulky side groups in the conjugated polymer backbone.95 Copyright: 2021, the American Chemical Society. | ||
Mun et al. investigated the influence of flexible conjugation breakers in the DPP/thienylene vinylene (TVT) polymer semiconductor backbone, in which three different chain breakers were used, i.e., ethylene, dibutylbenzene (DBB), hexane (C6), and dodecane (C12).93 Upon the incorporation of 10% molar ratio chain breaker, the non-conjugated segments could “tune” the mechanical properties, while maintaining high mobility of higher than 1 cm2 V−1 s−1, as shown in Fig. 4b. Remarkably, the C12-DPP/TVT polymer thin films exhibited a largely improved crack onset strain as high as 100% strain and a low modulus of 0.13 GPa, which could maintain moderate mobility (>0.36 cm2 V−1 s−1) under 100% strain even after a hundred cycles of 50% strain. Compared to DPP/TVT-based polymer films containing other conjugation breakers, C12-DPP/TVT was revealed to exhibit the best balanced performance in terms of both mechanical and electrical properties. Moreover, the effect of the three chain breakers on all the polymer derivatives was still more ductile with a lower elastic modulus compared to the reference polymer using ethylene as the chain breaker. It should be noted that the obtained higher stretchability of the C12-DPP/TVT thin films originates from their lower crystallinity, as confirmed by Tg measurements and X-ray diffraction. To further understand the effects of the H-bonding strength, Zheng et al. studied its influence on the mechanical and electrical properties of polymer semiconductor thin films using different types of H-bonding units (urea > amide > urethane > carbonate) and linker flexibilities (ether chain (E) > alkyl chain (A)) (Fig. 4c).94 Basically, H-bonding units were incorporated as CBS into a DPP D–A conjugated polymer backbone. It was observed that E-urea and A-urea with stronger H-bonding (self-association constant of >0.7) and flexible linkers having a higher tendency for denser packing could induce higher polymer chain aggregation and crystallinity in thin films, resulting in an improvement in the modulus and crack on-set strain and less degradation in mobility during stretching. It has to be emphasized that the E-urea/E-amide-derived polymers exhibited the highest charge transport mobilities due to their high crystallinity and intense H-bonding.
The same concept is applicable to other material systems, another example of which is the introduction two distinct classes of rigid fused-ring systems in a poly(thieno[3,2-b]thiophene-diketopyrrolopyrrole) (PDPPTT) backbone. The rigid fused-ring systems investigated by researchers, namely, benzene-substituted dibenzothiopheno[6,5-b:6′,5′-f]thieno[3,2-b]thiophene (Ph-DBTTT) and indacenodithiophene (IDT) systems.95 In fully stretchable transistors, as shown in Fig. 4d, the PIDT-3T-OC12-10% polymer showed a mobility of 0.27 cm2 V−1 s−1 at 75% strain and maintained its mobility after being subjected to hundreds of stretching-releasing cycles at 25% strain. Compared to the reference polymer PDPPTT, the device showed an abrupt drop in mobility when stretched either parallel to the strain direction (‖) or perpendicular to the strain direction (⊥) from 0% to 75% strain, in which the initial mobility of 0.76 cm2 V−1 s−1 decreased to μFETh‖ = 7 × 10−2 cm2 V−1 s−1 and μFETh⊥ = 1.1 × 10−2 cm2 V−1 s−1 at 75% strain. After 500 cycles at 25% strain, the tested devices all showed a low μFETh‖ = 6 × 10−2 cm2 V−1 s−1 in the parallel stretching direction due to crack formation at 25% strain. These results underscore the intimate correlation between chemical structures, mechanical properties, and charge carrier mobility of polymer semiconductors.
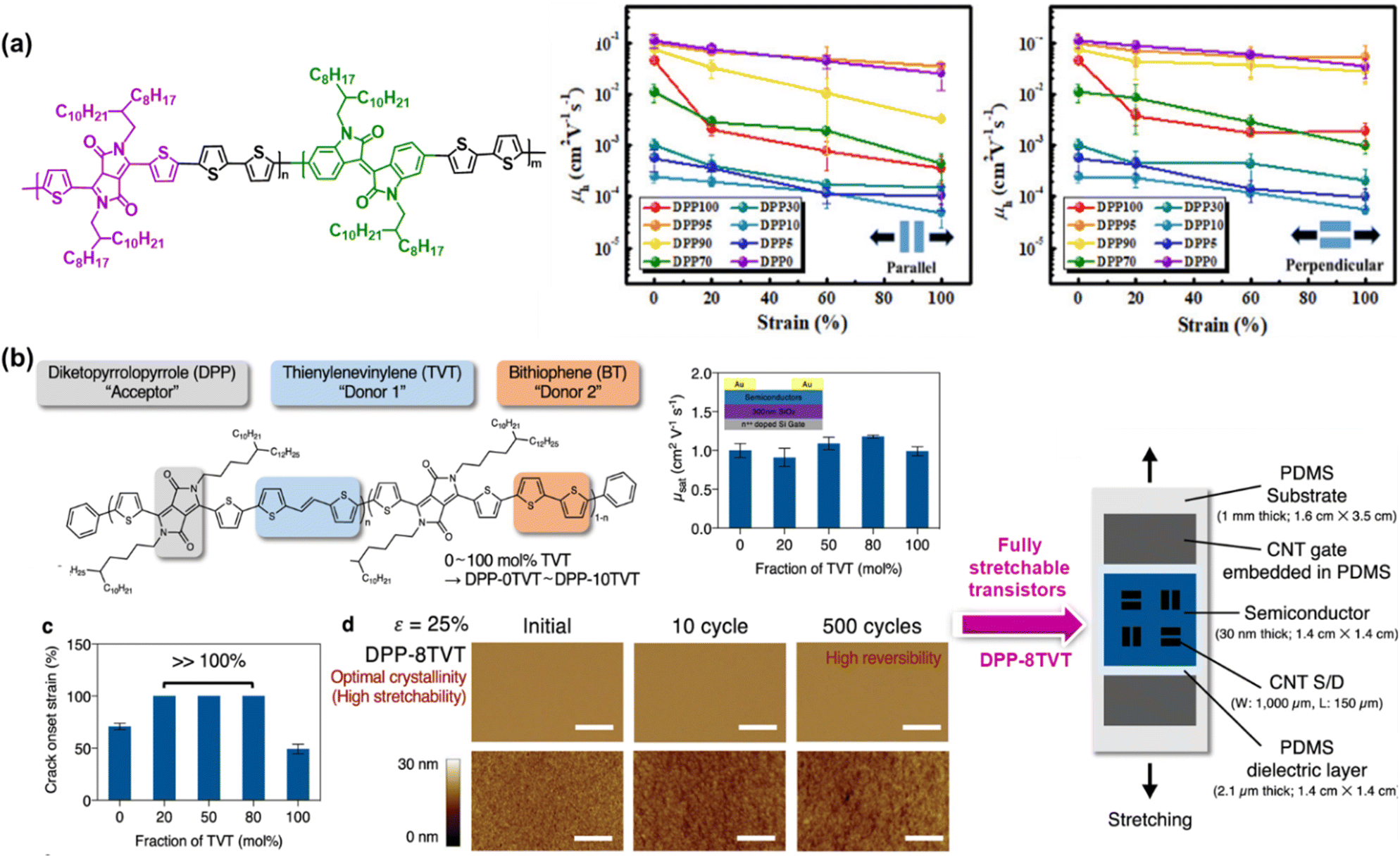 | ||
| Fig. 5 Intrinsically stretchable semiconducting polymer developed based on the ternary copolymer approach. Chemical structure and mechanical and electrical performances of: (a) random terpolymers based on the diketopyrrolopyrrole-bithiophene (DPP-2T) backbone incorporating different amounts of isoindigo (IID) unit.97 Copyright: 2020, the American Chemical Society. (b) Advances in DPP-based terpolymer semiconductors with two different fractions of TVT and BT conjugated co-monomers from observing simple semiconductor layers to fully stretchable transistors.98 Copyright: 2021, Springer Nature. | ||
Recently, Mun et al. proposed a promising strategy for to obtain intrinsically stretchable semiconducting polymers with high mobility and mechanical reversibility against repeated mechanical stress.98 Specifically, a ternary copolymer composed of the DPP unit as a p-type acceptor, TVT unit as donor 1, and bithiophene (BT) unit as donor 2 was prepared based on various concentrations of two types of constituting co-monomers, which were labelled as DPP-0TVT (0 mol% TVT and 100 mol% of BT) to DPP-10TVT (100 mol% TVT and 0 mol% of BT). As depicted in Fig. 5b, the designed terpolymers with a TVT fraction in the range of 20–80 mol% were found to be stretchable to >100% strain without the formation of cracks. In terms of the mechanical reversibility test, DPP-8TVT remained free from mechanical damage despite 500 continuous cycles of 25% strain, while retaining high charge carrier mobility even after repeated strain. Regardless of the TVT fraction, all the terpolymers showed mobility values of >1 cm2 V−1 s−1, which are comparable to or even greater than that of the regular copolymers with only one type of co-monomer units. As the most prominent sample, the DPP-8TVT film was chosen to fabricate fully stretchable transistor arrays, providing tremendous results such as high robustness and improved fracture resistance, as indicated by the slight degradation in mobility under various mechanical deformations (stretching, twisting, and poking) and a negligible decrease in mobility even after 1000 times deformation under 25% strain, respectively. These satisfactory results enable the design of simple polymers that effectively mitigate the long-standing trade-off between mobility and stretchability. Fascinatingly, this molecular design concept is obviously generalizable, and thus allows the construction of stretchable p-type and n-type terpolymer semiconductors by merely utilizing various combinations of electron donors and acceptors molecules. It was hypothesized that the introduction of two different types of fully conjugated co-monomers causes structural randomness in the backbone, which hinders the formation of larger crystalline domains. Substantially, this controlled ordering at different length scales can improve the fracture resistance upon strain without sacrificing the charge transport.
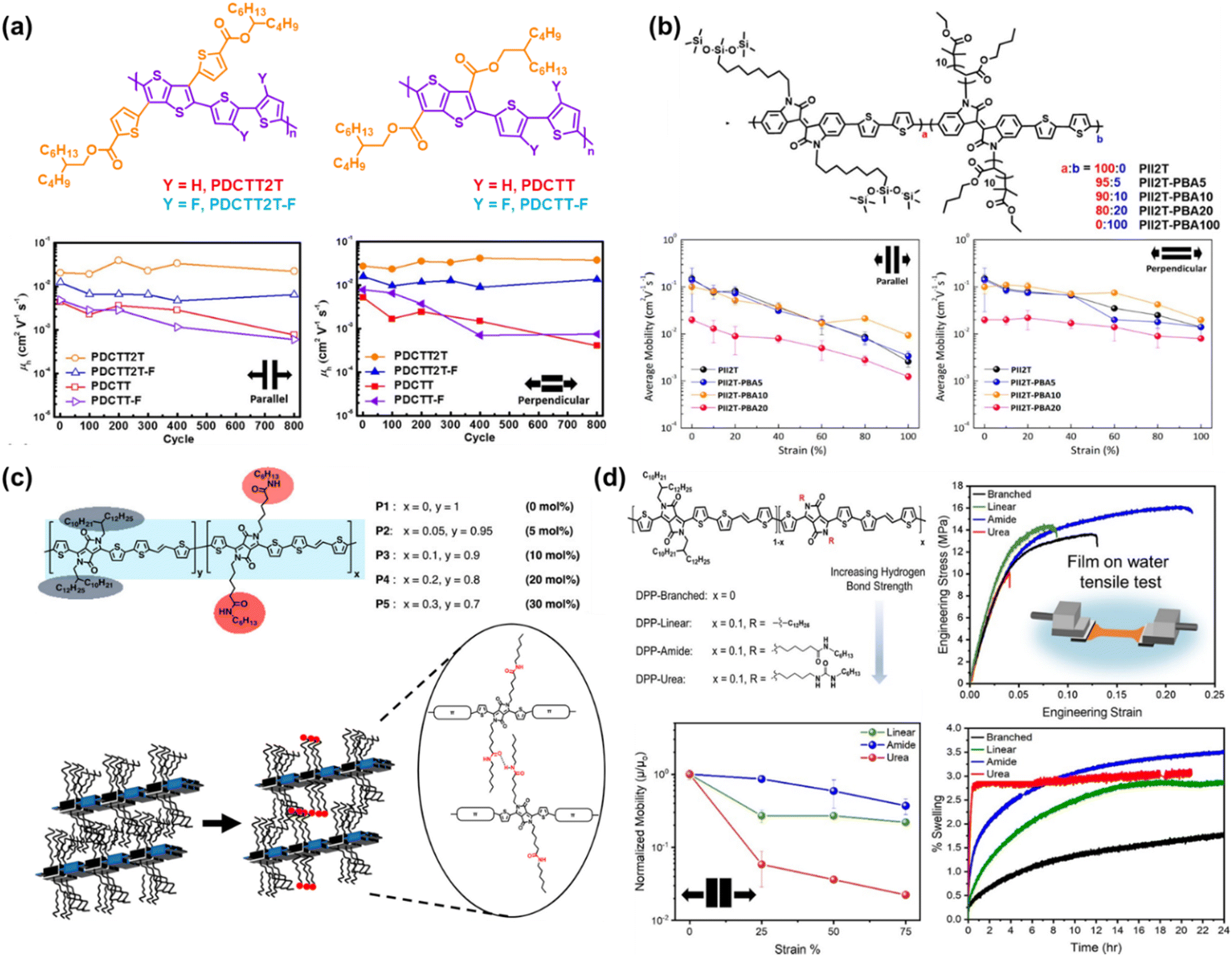 | ||
| Fig. 6 Side chain engineering based on the grafting approach. (a) Charge mobility-stretchability relationship and chemical structure of PDCTT2T and PDCTT2T-F compared to their non-biaxial counterparts of PDCTT and PDCTT-F, respectively.101 Copyright: 2021, the American Chemical Society. (b) Influence of the amount of PBA in mol percentage on the PII2T side chain described by charge mobility-stretchability data.103 Copyright: 2017, the American Chemical Society. (c) Structure of DPP-based conjugated polymers incorporating amide-containing side chains and schematic representation of intermolecular hydrogen bonds formed between adjacent amide-containing side chains.22 Copyright: 2018, the American Chemical Society.(d) Hydrogen bond strength described in chemical structures of DPP-TVT-based conjugated polymers and its influence on swelling, mechanical, and electrical properties as a function of maximum strain properties.21 Copyright: 2022, the American Chemical Society. | ||
Alternatively, the hydrogen bonds located in the side chain also played a role in enhancing the mechanical and charge transport performance and facilitating the morphological organization. Ocheje et al.22 incorporated a range of amide compositions (5% to 30%) in DPP-based polymers as a direct approach to promote the intermolecular hydrogen bonding moieties between polymer chains and tune the solid-state morphology (Fig. 6c). Upon the addition of 5 mol% amide side chain, there was an enhancement in the charge mobilities to 2.46 cm2 V−1 s−1, but the further addition of 30 mol% amide gradually decreased the mobility to 0.59 cm2 V−1 s−1. Besides hydrogen bonding, the increase in mobility can be partially influenced by the noncovalent interaction in the conjugated side chain. In comparison, Galuska et al.21 investigated the incorporation of 10% linear alkyl, amide functional, or urea functional side chains in DPP-TVT based statistical copolymers to assess the influence of hydrogen bonding on the mechanical properties of thin films (Fig. 6d). All the polymers demonstrated a similar elastic modulus with the average modulus following the order of DPP-linear > DPP-branched > DPP-urea > DPP-amide. This study showed that the hydrogen bond strength in the side chain is not linearly correlated with ductility, where DPP-urea possessed the highest hydrogen bonding strength but poor ductility due to its crystalline nature in comparison with DPP-amide, which was the best at tolerating deformation due to the disruption in crystalline packing in water. The embrittlement of DPP-urea was further confirmed in the swelling test, where rapid swelling behavior was observed initially but quickly plateaued due to the greater crystallinity. A similar phenomenon was observed in the measurement of the respective charge transport mobility, where the charge transport of DPP-amide decayed by only 14% from 0.199 cm2 V−1 s−1 after being strained by 25% in the parallel stretching direction. Considering that the typical maximum strain experienced by human skin is 33%, amide functionalization may be a suitable synthetic strategy to achieve tolerable strain for wearable devices, while maintaining charge transport.
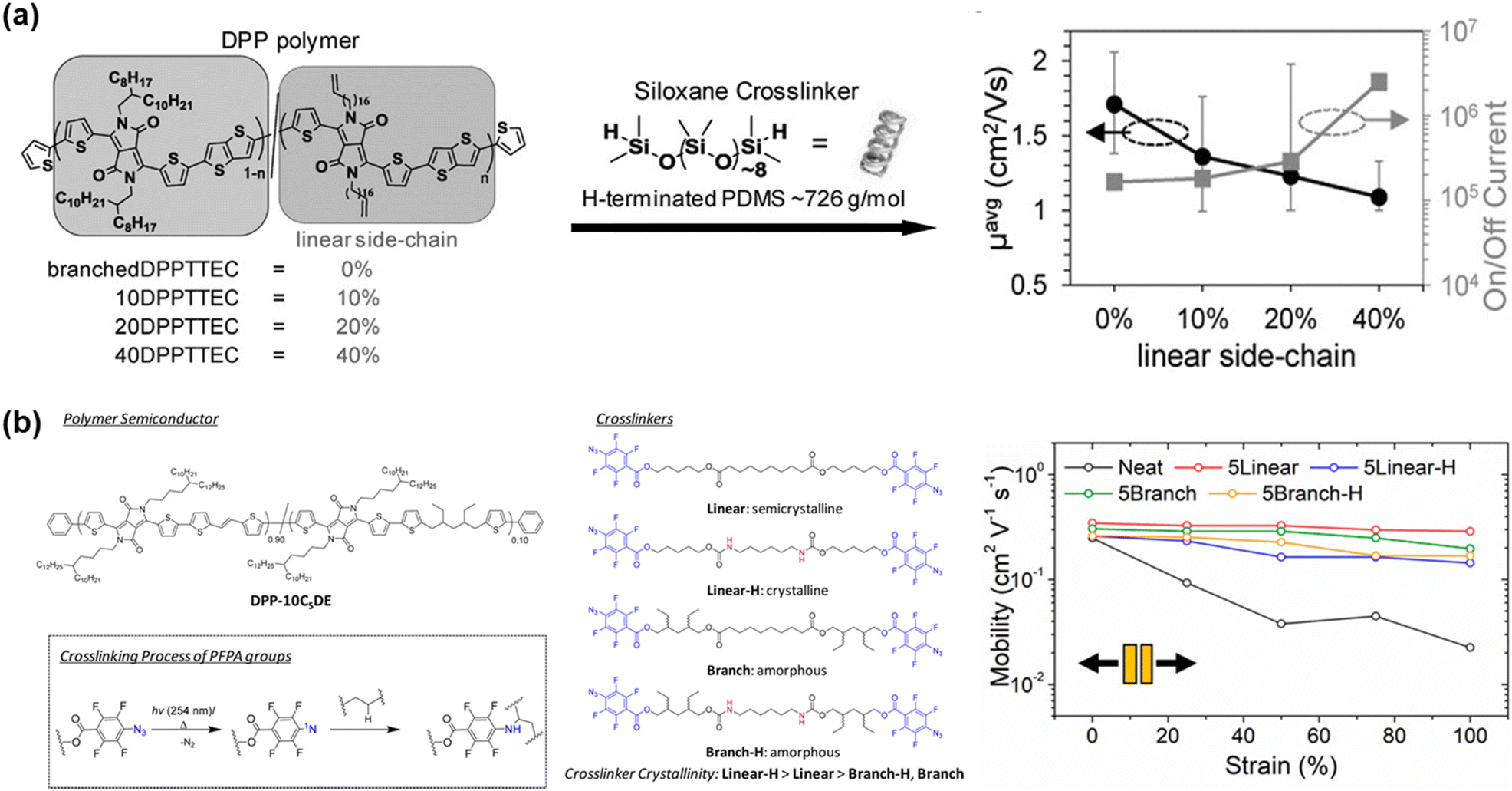 | ||
| Fig. 7 Side chain engineering based on crosslinking approach. Chemical structure and the retention of mobility properties of the polymers in the stretched state. (a) DPP-based random co-polymer containing cross-linkable linear side-chains and linear H-terminated PDMS crosslinker.104 Copyright 2016, Wiley-VCH. (b) Introduction four types of cross-linkers in DPP-based polymer.109 Copyright: 2019, the American Chemical Society. | ||
The embedding of a cross-linked segment in the side chain conjugated polymer has been demonstrated to be an ideal approach because a high crosslinking density can be easily tuned and disruption of the charge transport is minimized. Thus, the same group investigated the importance of crosslinker crystallinity with respect to morphology, mechanical, and electrical properties of polymers by inserting urethane groups and tertiary carbon atoms into the alkyl backbone of perfluorophenyl azide-based cross-linkers.109 Similar studies have also been reported.94,110,111 Here, branched cross-linkers with tertiary carbon are capable of forming an evenly cross-linked network in the polymer blend, stemming from their excellent miscibility, and show a 4-fold increase in fracture strain.109 Furthermore, a stable μFETh of 0.2 cm2 V−1 s−1 was achieved under 100% strain, and a stable μFETh of 0.1 cm2 V−1 s−1 after 2000 stretching-releasing cycles of 25% on fully stretchable organic field-effect transistors. The charge mobility–tensile performance of each specimen in this study is presented in Fig. 7b. Another factor to be considered is the effect of fluorination on the mechanical and electrical properties of conjugated polymers, which was also evaluated by Ocheje et al.112 Their work demonstrated the relationship between a series of fluorination and/or alkylation with benzothiadiazole-based polymer in the solid-state. The crack onset strains of the prepared polymers ranged from as low as 10% for the polymer containing only a fluorinated backbone to as high as 30% elongation for the polymer containing an alkoxy-appended backbone. Ultimately, fluorination embrittled the conjugated polymer thin films, whereas alkylation softened them.
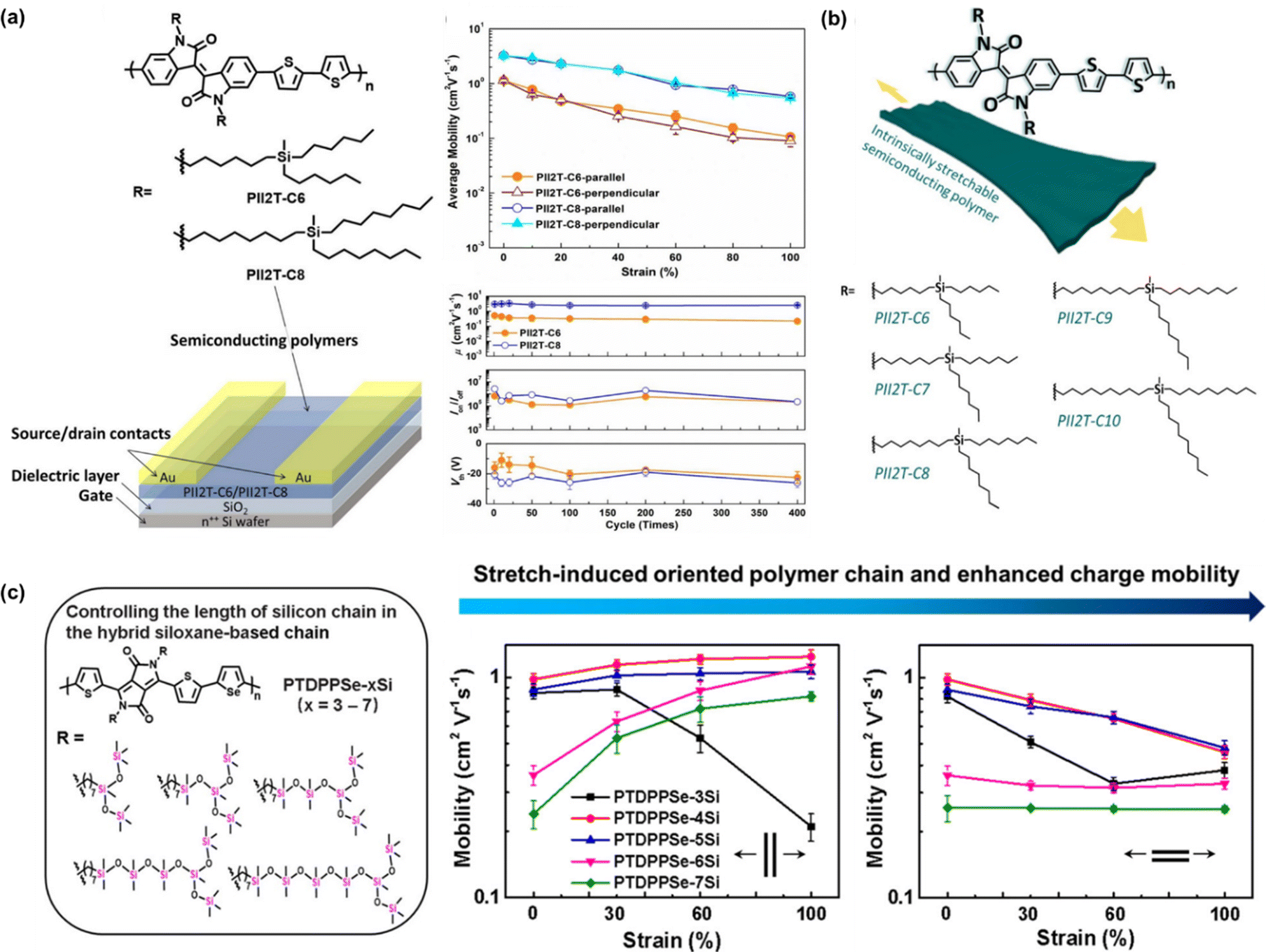 | ||
| Fig. 8 Modulating the mechanical-electrical properties of conjugated polymers via side chain engineering based on y-shaped approach. (a) Average field-effect mobility stretched in the parallel and perpendicular directions and the mechanical-mobility resistance of the of the PII2T-C6 and PII2T-C8 thin films under repeated 400 stretching/releasing cycles with a maximum strain of 60% controlled parallel to the strain direction.113 Copyright: 2016, the American Chemical Society. (b) Chemical structures of PII2T-based polymers with manipulated length of carbosilane side chains, charge mobility distribution in both parallel and perpendicular directions of PII2T with carbon position ranging from C6 to C10 and normalized mobility in the parallel direction of the studied polymer thin films under strain.114 Copyright: 2019, the American Chemical Society. (c) PTDPPSe-based conjugated polymer bearing different lengths of silicon chain.119 Copyright: 2022, the American Chemical Society. | ||
The effects of the carbon spacer present in the branch was investigated further by Chiang et al. (Fig. 8b).114 This group managed to improve the findings based on the work by Wu et al. by further repositioning the carbona of the side chain from 6 (PII2T-C6) to 10 carbons (PII2T-C10) to optimize the charge-transport behavior and the mechanical properties of semiconducting polymer thin films. By increasing the side chain length, the thin film modulus could be further reduced to 250 MPa with an almost unchanged μFETh upon stretching of 1 cm2 V−1 s−1 at 100% strain in both the parallel and perpendicular directions. In contrast, PII2T-C8 showed a higher initial mobility but it notably decreased upon stretching (0.7 cm2 V−1 s−1 at 100%). The initial mobility of PII2T-C10 was not as high as that of PII2T-C8 due to the disturbance of its backbone organization. Additionally, the length of the carbon side chain and the odd-even effect of the carbon position becomes a factor. Cracks could be seen in the PII2T-C6 and PII2T-C7 thin films under 100% strain but were significantly less in PII2T-C10. Polymers with even-numbered carbosilane side chains (C6, C8, and C10) possess smaller π–π stacking than the side chains with an odd carbon number (C7 and C9), and thus a higher mechanical and electrical performance can be achieved in even-numbered side chains.
Departing from the commonly used Y-shaped alkyl side chains, hybrid siloxane-based side chains are more easily deformed owing to their long bond lengths and large bond angles, together with low internal torsional potential barriers.115–117 Mei et al. demonstrated the effectiveness of novel siloxane-terminated solubilizing groups as a side chain in an isoindigo-based conjugated polymer (PII2T-Si) for promoting the field-effect mobility by inducing a denser π–π spacing and a larger crystalline coherence length.28 The hole mobility of PII2T-Si was 2.48 cm2 V−1 s−1, which is 4 times greater than that of the reference polymer with a branched alkyl side chain (PII2T-Ref). This was explained by the polymer packing, where the new polymer exhibited a π–π stacking distance of 3.58 Å, while PII2T-Ref showed a distance of 3.76 Å. Starting from this discovery, other researchers tried to modify the conjugated polymer Y-shaped side chain by incorporating hybrid siloxane-based side chains.87,118 In the latest study by Ding et al., they introduced a novel side chain engineering strategy to realize stretch-induced enhancement of the molecular orientation and charge transport in donor–acceptor conjugated polymers.119 Specifically, five conjugated polymers with a controlled hybrid siloxane-based side chain length were grafted on the backbone of poly-diketo-pyrrolopyrrole-selenophene: PTDPPSe-xSi (x = 3–7). Considering the superior flexibility of silicon chains compared carbon chains, a series of polymers showed a progressively lower elastic modulus and increased fracture strain by increasing the silicon chain length, whereas the charge mobility could be enhanced with an appropriate increase in the silicon chain length (Fig. 8c). Interestingly, the charge mobilities parallel to the stretching direction for PTDPPSe-4Si, PTDPPSe-5Si, and PTDPPSe-6Si were all above 1 cm2 V−1 s−1 at 100% strain, which were higher than that in their unstretched states. This enhancement in charge mobility is attributed to the excellent ductility and high strain-induced alignment of the polymer chains. Different from most other studies on controlling the carbon chain length in hybrid siloxane-based chains, this technique provides an alternative for manipulating the silicon chain length in hybrid siloxane-based chains.
 | ||
| Fig. 9 Tuning the electrical-mechanical properties of conjugated polymers via asymmetric side-chain engineering. (a) Asymmetrical side-chain engineering of IID-based polymers structure; illustration of the effects of the asymmetric side-chain design and backbone fluorination on chain packing; and the mobility retention of these polymers with different strains and with the stretching force parallel and perpendicular to the charge-transporting direction.122 Copyright: 2019, the American Chemical Society. (b) Structure of isoindigo-bithiophene (II2T)-based conjugated polymers with three types of side chains and the orthogonal hole mobilities values of the stretched polymer films with strain ranging from 0 to 100%.123 Copyright: 2021, the American Chemical Society. | ||
With the exception of the block copolymer approach, seven approaches for manipulating semiconducting polymer structures have been successful in providing satisfactory load mobility (μFETh > 1 cm2 V−1 s−1) even under 100% mechanical stretching. However, the modified semiconducting materials still have a high elastic modulus (>0.1 GPa), indicating the inadequate deformability and ductility of thin films. Based on the range of limitations of this review, the block copolymer, CBS, and symmetric side-chain approaches can achieve an elastic modulus close to or lower than 0.1 GPa. To make the comparison much more convenient, in Table 1, we also compare the performances of intrinsically stretchable semiconducting polymers.
| Design Method | Materials | Engineering details | Average mobility (cm2 V−1 s−1) | Mechanical properties | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Before | After | Stretchability (% strain) | Elastic modulus (GPa) | Durability | |||||
| Parallel | Perpendicular | ||||||||
| a Abbreviations: COS = crack onset strain; ref-polymer = DPP-based random copolymers incorporated with 10 mol% ethylene as the conjugation breaker; G1 = polymer with DPP-thiophenevinylenethiophene (PDPPTVT) backbone with side chains consisting of methyl(8-bromooctyl)dioctylsilane (SiC8); G2 = PDPPTVT-SiC8-5% of poly(acryl amide) (PAM); G3 = PDPPTVT-SiC8-9% PAM; G4 = PDPPTVT-SiC8-16% PAM; G5 = polymer with DPP-selenophene (PDPPSe) backbone with side chains consisting of methyl(8-bromooctyl)dioctylsilane (SiC8) and 5% of poly(acryl amide) (PAM); 5Linear = neat with 5 wt% semi-crystalline perfluorophenyl azide-based cross-linker blend; 5LinearH = neat with 5 wt% highly crystalline perfluorophenyl azide-based cross-linker blend; 5Branch = neat with 5 wt% amorphous perfluorophenyl azide-based cross-linker blend (Tg = −44.4 °C); 5Branch-H = neat with 5 wt% amorphous perfluorophenyl azide-based cross-linker blend (Tg = −21.9 °C); P1 = SiOC8 and Si-C8 side chain engineered in isoindigo-bithiophene (PII2T) backbone; P2 = DT/SiO-C8 side chain engineered in isoindigo-bithiophene (PII2T) backbone; P3 = DT/Si-C8 side chain engineered in isoindigo-bithiophene (PII2T) backbone; P4 = SiOC8 and Si-C8 side chain engineered in isoindigo-difluorobithiophene (PII2TF) backbone; P5 = DT/SiO-C8 side chain engineered in isoindigo-difluorobithiophene (PII2TF) backbone; P6 = DT/Si-C8 side chain engineered in isoindigo-difluorobithiophene (PII2TF) backbone; P(Si–O) = carbosilane/oligoether side chains engineered in isoindigo-bithiophene (II2T)-based polymer; P(Si–F) = carbosilane/semi-fluorinated side chains engineered in isoindigo-bithiophene (II2T)-based polymer; and P(F–O) = semi-fluorinated/oligoether side chains engineered in isoindigo-bithiophene (II2T)-based polymer. b Measured by film-on-water tensile test. c Measured by film-on-elastomer tensile test. d Measured by buckling-based metrology. e Thermally annealed at 150 °C for 30 min under nitrogen. | |||||||||
| Block copolymer approach | P3HT | Diblock copolymers | 1 × 10−2 | — | — | 13 | 0.028 | — | 77 |
| P3HT35-PE65 | 5 × 10−2 | — | — | 660 | 0.24 | — | |||
| P3HT | Diblock copolymers | 0.079 | 2.2 × 10−4 at 100% strain | 1.1 × 10−4 at 100% strain | — | 0.93d | — | 79 | |
| P3HT-b-PBA3ka | 0.073 | 7.9 × 10−4 at 100% strain | 6.9 × 10−4 at 100% strain | — | 0.63d | — | |||
| P3HT-b-PBA6ka | 0.061 | 0.025 at 100% strain | 0.025 at 100% strain | — | ∼0.35d | μ FETh‖ and μFETh⊥ ≥0.01 cm2 V−1 s−1 under 100% strain for 1000 cycles | |||
| P3HT-b-PBA12ka | 0.014 | 2.1 × 10−3 at 100% strain | 1.9 × 10−3 at 100% strain | — | 0.19d | — | |||
| P3HT-b-PMA-b-P3HT | ABA triblock copolymer | 1.7 × 10−4 | — | — | 140d | 0.006d | — | 81 | |
| P3HT-b-P3SiHTa | Diblock copolymer | 5.61 × 10−3 | — | — | 25.6 | 0.056b | — | 83 | |
| P3HT-b-P3SiHT-b-P3HT | ABA triblock copolymer | 5.14 × 10−3 | — | — | 118 | 0.003 | — | ||
| Molecular spacer approach | P3HTT-DPP-10a | Fully conjugated semi-random | 9.29 × 10−4 | — | — | COSa: 10 | 0.32c | — | 92 |
| 10% T-10-Ta | Semi-random polymers with CBS | 2.53 × 10−4 | — | — | COS: >80 | 0.13b | — | ||
| 0.15c | |||||||||
| 20% T-10-T | 1.06 × 10−4 | — | — | COS: >80 | 0.32b | — | |||
| 0.52c | |||||||||
| 30% T-10-T | 3.55 × 10−4 | — | — | COS: >80 | 0.33b | — | |||
| 0.60c | |||||||||
| Ref-polymera | Modified chain length and rigidity of CBS | 2.1 | 0.9 at 100% strain | 0.7 at 100% strain | COS: 25c | 1.1c | μ FETh‖ = ∼0.6 and μFETh⊥ = ∼0.08 cm2 V−1 s−1 under 50% strain for 100 cycles | 93 | |
| C12-DPP/TVT | 1.3 | 1.0 at 100% strain | 0.6 at 100% strain | COS: 100c | 0.13c | μ FETh‖ = ∼0.36 and μFETh⊥ = ∼0.8 cm2 V−1 s−1 under 50% strain for 100 cycles | |||
| PDPPTT | Introducing conjugated rigid fused-rings with optimized bulky side groups | 0.76 | 0.07 at 75% strain | 0.11 at 75% strain | COS: 20c | — | μ FETh‖ = 0.06 and μFETh⊥ = ∼0.4 cm2 V−1 s−1 under 25% strain for 500 cycles | 95 | |
| PDBTTT-10% | 0.7 | 0.03 at 75% strain | 0.06 at 75% strain | COS: 40c | — | μ FETh‖ = 0.3 and μFETh⊥ = ∼0.5 cm2 V−1 s−1 under 25% strain for 500 cycles | |||
| PID-3T-OC12-10%a | 0.9 | 0.27 at 75% strain | 0.2 at 75% strain | COS: 60c | — | μ FETh‖ = 0.22 and μFETh⊥ = ∼0.38 cm2 V−1 s−1 under 25% strain for 500 cycles | |||
| Terpolymerization approach | G1a | Introducing PAM-coupled alkyl side chains into the polymer backbone | 0.52 | 0.030 at 100% strain | 0.048 at 100% strain | 0.85 | — | 97 | |
| G2a | 0.26 | 0.038 at 100% strain | 0.052 at 100% strain | 0.80 | μ FETh‖ = 3 × 10−3 and μFETh⊥ = 2 × 10−4 cm2 V−1 s−1 under 60% strain for 400 cycles | ||||
| G3a | 0.38 | 0.10 at 100% strain | 0.12 at 100% strain | 0.76 | μ FETh‖ = 9 × 10−3 and μFETh⊥ = 4 × 10−3 cm2 V−1 s−1 under 60% strain for 400 cycles | ||||
| G4a | 0.15 | 0.011 at 100% strain | 0.031 at 100% strain | 0.71 | — | ||||
| G5a | 0.21 | 0.021 at 100% strain | 0.043 at 100% strain | 0.36 | μ FETh‖ = 0.02 and μFETh⊥ = 3 × 10−3 cm2 V−1 s−1 under 60% strain for 400 cycles | ||||
| DPP-8TVT | A–D–A–D′ type | >1 | >1 at 100% strain | >1 at 100% strain | COS: ≫ 100 | 0.65 | 500 stretching/releasing cycles at 25% strain | 98 | |
| Symmetric side-chain approach | PDCTT2T | Biaxially extended conjugated side chain | 0.06 | 2 × 10−3 at 100% strain | 4 × 10−3 at 100% strain | — | — | μ FETh‖ = 0.02 and μFETh⊥ = 0.03 cm2 V−1 s−1 under 60% strain for 800 cycles | 101 |
| PDCTT2T-F | 0.02 | 2.5 × 10−4 at 100% strain | 2.5 × 10−3 at 100% strain | — | — | μ FETh‖ = 6 × 10−3 and μFETh⊥ = 1.5 × 10−3 cm2 V−1 s−1 under 60% strain for 800 cycles | |||
| PII2T | Introducing soft side chain | 0.15 | 1.4 × 10−3 at 100% strain | 2.6 × 10−4 at 100% strain | COS: <10d | 0.80d | — | 103 | |
| PII2T-PBA5 | 0.14 | 1.4 × 10−3 at 100% strain | 3.4 × 10−4 at 100% strain | COS: ∼20d | 0.63d | — | |||
| PII2T-PBA10 | 0.10 | 0.02 at 100% strain | 9.4 × 10−4 at 100% strain | COS: ∼40d | 0.57d | μ FETh‖ = 8 × 10−3 and μFETh⊥ = 0.01 cm2 V−1 s−1 under 60% strain for 400 cycles | |||
| PII2T-PBA20 | 0.021 | 8.1 × 10−3 at 100% strain | 1.2 × 10−4 at 100% strain | COS: 60d | 0.12d | — | |||
| 20DPPTTEC | Crosslinked with PDMS oligomer | 1.37 | — | — | COS: >150 | 0.28d | Stable μFETh⊥ ≈ 0.4 cm2 V−1 s−1 under 20% strain for 500 cycles | 104 | |
| DPP-10C5DE | Tuning of the cross-linker's crystallinity | 0.25 | 0.02 at 100% strain | 0.02 at 100% strain | Pristine COS: 25b | Pristine: 0.225b | μ FETh‖ = 0.01 and μFETh⊥ = 0.03 cm2 V−1 s−1 under 25% strain for 2000 cycles | 109 | |
| Annealede COS: 25b | Annealede: 0.225b | ||||||||
| 5Lineara | 0.34 | 0.29 at 100% strain | 0.02 at 100% strain | Pristine COSa: 50b | Pristine: 0.205b | μ FETh‖ = 0.01 and μFETh⊥ = 0.06 cm2 V−1 s−1 under 25% strain for 2000 cycles | |||
| Annealede COSa: 100b | Annealede: 0.234b | ||||||||
| 5Linear-Ha | 0.26 | 0.14 at 100% strain | 0.05 at 100% strain | Pristine COSa: 50b | Pristine: 0.234b | μ FETh‖ = 0.05 and μFETh⊥ = 0.03 cm2 V−1 s−1 under 25% strain for 2000 cycles | |||
| Annealede COSa: 75b | Annealede: 0.230b | ||||||||
| 5Brancha | 0.30 | 0.20 at 100% strain | 0.03 at 100% strain | Pristine COSa: 75b | Pristine: 0.236b | μ FETh‖ = 0.03 and μFETh⊥ = 0.12 cm2 V−1 s−1 under 25% strain for 2000 cycles | |||
| Annealede COSa: 100b | Annealede: 0.204b | ||||||||
| 5Branch-Ha | 0.26 | 0.17 at 100% strain | 0.04 at 100% strain | Pristine COSa: 75b | Pristine: 0.2b | μ FETh‖ = 0.03 and μFETh⊥ = 0.11 cm2 V−1 s−1 under 25% strain for 2000 cycles | |||
| Annealede COSa: >100b | Annealede: 0.215b | ||||||||
| Y-Shape approach | PII2T-C6 | Incorporation of branched alkyl side chain | 1.14 | 0.11 at 100% strain | 0.09 at 100% strain | >100 | 0.430c | μ FETh‖ = 0.2 cm2 V−1 s−1 under 60% strain for 400 cycles | 113 |
| PII2T-C8 | 3.24 | 0.58 at 100% strain | 0.54 at 100% strain | >100 | 0.270c | μ FETh‖ ≥1cm2 V−1 s−1 under 60% strain for 400 cycles | |||
| PII2T-C6 | Incorporation of branched alkyl side chain | 2.48 | 0.2 at 100% strain | 0.09 at 100% strain | — | 0.565d | — | 114 | |
| PII2T-C7 | 2.06 | 0.1 at 100% strain | 0.09 at 100% strain | — | 0.465d | — | |||
| PII2T-C8 | 4.67 | 0.7 at 100% strain | 0.7 at 100% strain | — | 0.375d | — | |||
| PII2T-C9 | 0.92 | 0.15 at 100% strain | 0.09at 100% strain | — | 0.280d | — | |||
| PII2T-C10 | 2.23 | 2.0 at 100% strain | 2.2 at 100% strain | — | 0.250d | μ FETh‖ and μFETh⊥ = ∼1 cm2 V−1 s−1 under 60% strain for 500 cycles | |||
| PTDPPSe-3Si | Grafting hybrid siloxane-based side chains | 0.85 | 0.21 at 100% strain | 0.38 at 100% strain | — | 0.378b | — | 119 | |
| PTDPPSe-4Si | 0.98 | 1.24 at 100% strain | 0.46 at 100% strain | — | 0.264b | — | |||
| PTDPPSe-5Si | 0.88 | 1.08 at 100% strain | 0.48 at 100% strain | — | 0.186b | — | |||
| PTDPPSe-6Si | 0.36 | 1.12 at 100% strain | 0.33 at 100% strain | — | 0.129b | — | |||
| PTDPPSe-7Si | 0.24 | 0.82 at 100% strain | 0.25 at 100% strain | — | 0.087b | — | |||
| Asymmetric side-chain approach | P1a | Inserting asymmetric side chains | 0.33 | 0.16 at 100% strain | 0.18 at 100% strain | — | 0.55 | — | 122 |
| P2a | 0.54 | 0.21 at 100% strain | 0.32 at 100% strain | — | 0.55 | — | |||
| P3a | 0.60 | 0.24 at 100% strain | 0.29 at 100% strain | — | 0.51 | μ FETh‖ = 0.15 and μFETh⊥ = 0.2 cm2 V−1 s−1 under 60% strain for 400 cycles | |||
| P4a | 0.21 | 0.14 at 100% strain | 0.12 at 100% strain | — | 0.56 | — | |||
| P5a | 0.22 | 0.12 at 100% strain | 0.14 at 100% strain | — | 0.67 | — | |||
| P6a | 0.26 | 0.16 at 100% strain | 0.21 at 100% strain | — | 0.71 | μ FETh‖ = 0.18 and μFETh⊥ = 0.2 cm2 V−1 s−1 under 60% strain for 400 cycles | |||
| P(Si–O)a | Inserting asymmetric side chains | 0.012 | 9.8 × 10−3 at 100% strain | 0.011 at 100% strain | >80 | 0.45 | Almost unchanged μFETh under 60% strain for 1000 cycles | 123 | |
| P(Si–F)a | 8.7 × 10−3 | 5.3 × 10−3 at 100% strain | 5.3 × 10−3 at 100% strain | ∼60 | 2.09 | — | |||
| P(F–O)a | 3.8 × 10−5 | 7.6 × 10−6 at 100% strain | 8.0 × 10−6 at 100% strain | ∼20 | 2.79 | — | |||
2.3 Stretchable semiconducting polymer blends
Compared with the elegantly structured semiconductor polymers modified through complicated synthetic procedures, blending semiconducting polymers with other conjugated polymers, small molecules, or soft-building block polymers is presented as the most favorable method because sometimes it offers an economic, simpler, or better alternative. Introducing an appropriate secondary component via physical blending leads to polymer dilution, which changes the morphology, cohesion and viscosity, among other physical characteristics, ultimately impacting the electromechanical properties of thin film polymers. However, a dilemma in this method in achieving deformability, typically resulting in a deterioration in the charge transport, and vice versa.124 This is because if only the simple blending method is employed without promoting any suitable morphology, there may be a significant reduction in charge carrier mobility due to the charge transport impediment between the semiconducting domains by the insulating component. Thus, to solve this issue, researchers considered the influence of the processing approach,73,125–128 thickness,129 and nanoconfinement32,130,131 on improving the stretchability of high-mobility polymer blends. As expected, these considerations led to the successful production of blended semiconducting polymers, which can actually effortlessly alter and even optimize the mechanical ductility, mobility/molecular ordering, and stability of conjugated polymer-FETs substantially, together with newly emerging polymer blend characteristics.In the simplest case, the widely studied semiconducting blend composed of 0.75 wt% P3HT:poly(dimethylsiloxane) (PDMS), where P3HT is prepared via “pre-deposition processing” became more resistance to external strain and exhibited an improved electrical performance up to 2 times that of the non-PDMS-containing sample and 44 times greater than that of the untreated P3HT (Fig. 10a).73 Meanwhile, no charge transport behavior was observed for pristine P3HT:PDMS blends. The use of the “pre-deposition processing” technique is known to facilitate enhanced P3HT alignment/ordering/crystallinity with a concomitant improvement in charge carrier mobility.132,133 As analyzed by UV-vis absorption, the samples were red-shifted following the sequence of pristine P3HT, ultrasonicated P3HT/PDMS, and poor solvent/ultrasonicated P3HT/PDMS, implying that agglomeration was induced by the stronger molecular interaction between the two blend components as a result of their improved crystallinity. This result was further supported by the narrower full width at half-maximum (FWHM) of the XRD peaks. The poor miscibility between PDMS and P3HT was also observed via Hansen solubility parameter analysis, leading to the formation of dendritic aggregates by P3HT. However, this research is still in its infancy given that the performance of thin-film transistors upon stretching has not been evaluated to date.
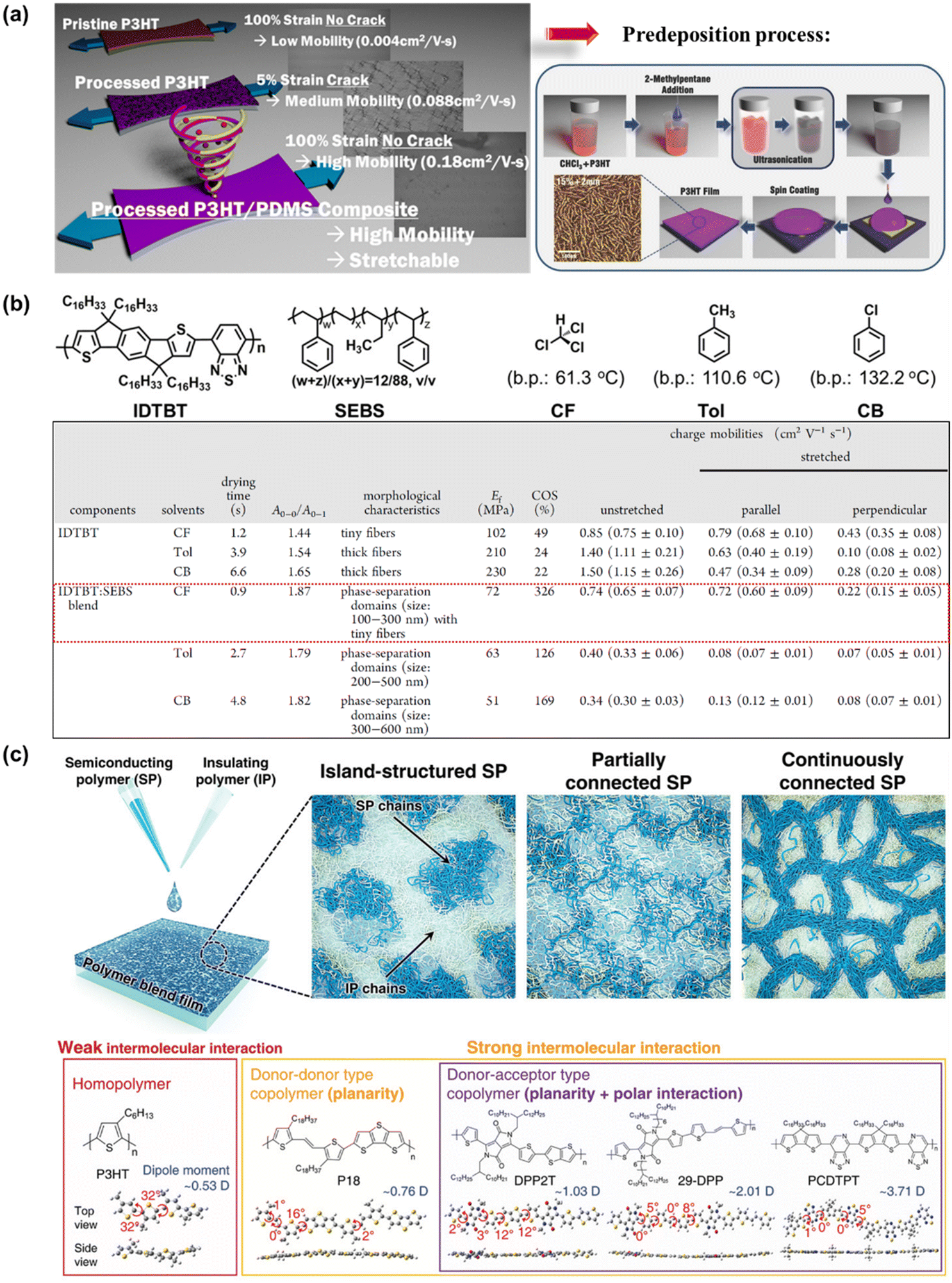 | ||
| Fig. 10 (a) Schematic depicting the processed P3HT:PDMS, affording a blended semiconducting film that exhibits superior ductility and notable mobility versus the single-component polymer semiconductor counterpart.73 Copyright: 2016, the American Chemical Society. Schematic illustration of the pre-deposition process.133 Copyright: 2015, Wiley. (b) Molecular structures of the conjugated polymer IDTBT, elastomer SEBS, and the effect of different deposition treatments on the electrical and mechanical properties of IDTBT and IDTBT:SEBS blend.136 Copyright: 2022, the American Chemical Society. (c) Representative structural morphologies of SP:IP blend and the DFT-optimized molecular models.140 Copyright: 2021, Wiley. | ||
Given that this versatile approach has no limitations on the material chosen, Zhang et al. proposed the fabrication of transparent and elastic semiconductor films by blending only a small amount (less than 1 wt%) of either p-type or n-type commercial semiconductors in PDMS as the transparent stretchable insulating component.134 P3HT and poly[2,5-(2-octyldodecyl)-3,6-diketopyrrolopyrrole-alt-5,5-(2,5di(thien-2-yl)thieno [3,2-b]thiophene)] (DPP-DTT) were used as p-type semiconducting polymers, whereas poly(2,5-bis(2-octyldodecyl)-3,6-di(pyridin-2-yl)-pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione-alt-2,2′-bithiophene) (DPPDPyBT) was used as an electron transport (n-channel) semiconducting polymer. With appropriate semiconductor weight fractions, it was observed that the mobility of 0.49 wt% P3HT/PDMS, 0.83 wt% DPP-DTT/PDMS, and 0.62 wt% DPPDPyBT/PDMS was 0.23, 1.75, and 1.34 cm2 V−1 s−1, respectively. All of them exhibited an enhancement by ∼10-fold, 2-fold, and 3-fold in mobility compared to the control samples (100 wt%) of P3HT, DPP-DTT, and DPPDPyBT, respectively. The electrical-mechanical performance of semiconductor/insulating polymer blend films under 100% strain parallel to the charge transport direction was evaluated via the delamination–stretching–relamination process,33,135 resulting in a slight decrease in charge carrier mobility, while the transfer curve was unaffected. Owing to its good transparency, biocompatible, low cost, and ease of fabrication, commercial PDMS is often selected by researchers as an insulating polymer in these types of binary blend systems. The detailed mechanical and electrical behavior of semiconducting polymer-blended-PDMS films are further summarized in Table 2.
| Semiconductors | Mobility (cm2 V−1 s−1) | Mechanical properties | Ref. | |||||
|---|---|---|---|---|---|---|---|---|
| Type | Material | f semi (wt%) | Original | Parallel at 100% strain | Perpendicular at 100% strain | Crack onset strain (%) | Elongation break (%) | |
| p | P3HT | 100 | ∼2.3 × 10−3 | ∼1 × 10−5 | ∼2.2 × 10−4 | 25 | 50 | 187 |
| p | P3HT/PDMS | 5 | ∼1.8 × 10−3 | ∼1.2 × 10−4 | ∼2.3 × 10−5 | >100 | >100 | |
| p | P3HT/PDMS | 0.49 | 0.17 ± 0.03 | 0.15 ± 0.01 | — | — | 182.1 ± 8.6 | 134 |
| p | P3HT | 100 | 0.02 ± 0.01 | 0.006 ± 0.001 | — | 32.6 ± 4.8 | — | |
| p | DPP-DTT/PDMS | 0.83 | 1.53 ± 0.22 | 1.35 ± 0.04 | — | — | 140.2 ± 10.2 | |
| p | DPP-DTT | 100 | 0.81 ± 0.11 | 0.18 ± 0.01 | — | 20.7 ± 5.2 | — | |
| n | DPPDPyBT/PDMS | 0.62 | 1.25 ± 0.09 | 1.08 ± 0.02 | — | — | 133.5 ± 7.6 | |
| n | DPPDPyBT | 100 | 0.43 ± 0.14 | 0.12 ± 0.01 | — | 22.5 ± 4.5 | — | |
| p | Homo-P3HT NW | 100 | (1.30 ± 0.4) × 10−2 | (1.70 ± 0.9) × 10−5 | — | 50 | 10 | 135 |
| p | P3HT NW/PDMS | 10 | (7.85 ± 2.8) × 10−3 | (3.74 ± 0.7) × 10−4 | — | — | — | |
| p | Pristine P3HT | 0.75 | 0.004 ± 0.0005 | — | — | >120 | — | 73 |
| p | Processed P3HT | 0.75 | 0.18 ± 0.03 | — | — | > 100 | — | |
| p | Processed P3HT/PDMS composite | 0.75 | 0.11 ± 0.016 | — | — | > 100 | — | |
Liu et al. investigated the influence of molecular aggregation on the film morphology of conjugated polymer poly(indacenodithiophene-co-benzothiadiazole) (IDTBT)-blended-elastomer polystyrene-block-poly(ethylene-ran-butylene)-block-polystyrene (SEBS) at much broader parameter choices, including two types of high-boiling-point (bp) solvents such as toluene (Tol) and chlorobenzene (CB) and fast solvent evaporation from low-bp chloroform (CF) (Fig. 10b).136 When a solvent with high-bp was utilized as the processing solvent (Tol and CB), phase separation of the IDTBT:SEBS blend occurred, which resulted in large isolated IDTBT domains, deterring the charge transport efficiency and mechanical deformation of the films. In contrast, the utilization of low-bp such as CF could effectively prevent the large-scale phase separation and promote the formation of a fiber network through the nanoconfinement effect in blend films. This effect enables us to achieve a stretchable film with a low elastic modulus of 72 MPa, large crack at a strain value of 326%, and negligible loss of mobility (μFETh‖ = 0.72 cm2 V−1 s−1) at 100% strain. It is envisaged that the molecular aggregation dynamics plays a critical role in the agglomeration and phase separation in conjugated polymer:elastomer blends, which offers an alternative strategy to manipulate the film morphology for a synergistic improvement in the mechanical and electrical properties of stretchable conjugated polymer films.
Nanoconfinement as the basis for the synergistic effect in semiconducting polymer (SP):insulating polymer (IP) blend systems is defined as the confinement of a material in a nanoscale region, which can cause changes in the physical properties of polymers due to interfacial and finite-size effects.33 Several studies have reported that this confinement can result in a reduction in mechanical properties, glass transition temperature,137 and elastic modulus,138 increased crystalline ordering,139 and better charge transport mobility.131 Therefore, clarifying the confinement effect of SPs and devising an effective strategy are necessary to design polymer blend composites with multi-aptitudes. Park et al. systematically investigated the confinement effect depending on the type of intermolecular interaction of SPs (such as P3HT, P18, DPP2T, 29-DPP, and PCDPTP) in the IP of polystyrene (PS) (Fig. 10c).140 Based on the correlation between the morphological and charge transport properties on various types of SP:IP blend films, it was found that the 29-DPP-type SP exhibited an improvement in field-effect hole mobilities by 3–4 times (maximum μFETh approaching 10 cm2 V−1 s−1 for 60DPP:40PS blended type). The combination of temperature-dependent charge carrier mobility analysis and grazing incident-wide angle X-ray spectroscopy (GIWAXS) indicated that the nanostructure of confined SPs exhibited a straight edge-on stacking structure in the blends. According to the above discussion, the capabilities of the blending method to aptly tune the electrical and mechanical properties in an easy and facile manner, coupled with its unsophisticated processing parameters (e.g., blending ratio, molecular weight, and solvent), have created new avenues for many researchers to develop innovative optically transparent, stretchable, and self-healable semiconducting polymer systems for use in TFT devices.
3. Advancement from non-autonomous healing toward autonomous self-healing semiconducting polymers
According to the numerous strategic options previously reported, developing intrinsically stretchable semiconducting polymers through the insertion of bulky soft side chains or via the utilization of conjugation breaking units seems to be the preferred approach to generate materials with an increased amorphous content, reduced crystallinity, and improved resistance to mechanical failure, while maintaining good charge transport properties. As reported by Mei et al., CBS can successfully reduce the backbone rigidity, and coupled with the fact that the incorporated CBS is capable of forming weak and reversible intra- and inter-chain bonds, the resulting polymer can be stretched excellently.26 Furthermore, Zheng et al. realized that the incorporation of H-bonding units in DPP-based conjugated polymers is a compelling manner to regulate their crystalline behaviors and mechanical properties.94 Undoubtedly, these bonds becomes the most common and accessible types of supramolecular interactions to be implemented in π-conjugated polymeric systems. This type of non-covalent interaction is spontaneous, highly dynamic, and can be achieved through a wide variety of functional groups (amide, urea, urethane, carbonate, etc.), implying the versatility of the noncovalent interaction-based synthetic method. Molecular self-healing is another innovative property achieved through the utilization of dynamic H-bonds.Although the stretchability of polymers has been enhanced, damage due to the forces of stretching, extrusion, and twisting during cumulative use can still occur after long-term repeated operations. By mimicking the self-healing functionality of human skin, artificial self-healing in new smart materials has become a broad concept that covers a wide variety of properties.141 This phenomenon is also often referred to as self-mending or self-repairing characteristics, which is defined as the ability of a material to partially or fully regain its original material properties upon damage. This particular property possesses the ability to be easily damaged and autonomously recover, which can be found in materials containing dynamic covalent or non-covalent bonds.142,143 Hydrogen bonds, π–π interactions, metal-ligand bonds, ionic bonds, host–guest complexes, and hydrophobic interactions are the interactions found in non-covalent bonds,144–151 while Schiff base interactions, Diels–Alder interactions, and borate-ester and disulfide bonds are included in the common dynamic covalent bonds.152–156 In addition to the bond interactions, the physical molecular diffusion also plays a crucial part in determining the healing rate and characteristics of the damaged polymer surface.157–160 The diffusion parameters include the chemical characteristics, the chain length, polydispersity indices, and the mechanical properties of the polymer. In some cases, a non-autonomous healing initiating agent and thermal treatment may be needed to trigger self-healing, which are readily available in the form of capsules released upon cracking of the host materials.161,162
Considering two important features, i.e., the diffusion of polymer chains and the reformation of broken dynamic bonds,160 a low glass transition temperature, which promotes polymer chain movement and abundant sites for dynamic interactions, is an implicit requirement in typical intrinsically self-healing semiconducting polymer systems. Since 2001, the Web of Science has listed more than 6000 publications covering the field of self-healing materials;163 however, only a few studies have reported the successful integration of stretchable conjugated polymers bearing healing capability in TFT devices.23,24,164–166 This limitation is due to the challenges of fabricating self-healing semiconductor layers, while maintaining good electrical properties. Given that the semiconducting layer for most devices is typically very thin, i.e., < 100 nm, it is difficult to align the damaged areas properly for full healing. This case also applies to the development of stretchable and self-healing materials based on conductive polymers (e.g., poly3,4-ethylene dioxythiophene (PEDOT), polypyrrole (PPy), and polyaniline (PANI)) for feasible applications based on organic electrochemical transistors devices (e.g., bioelectronics and wearable electronics). Other interesting topics related to conducting polymers not covered in this section can be found in recently published reports and reviews.167–173
To the best of our knowledge, Bao's group was the first to successfully fabricate magnificent intrinsically stretchable and self-healable semiconducting polymers in TFT devices by utilizing hydrogen-bonding moieties,23 wherein the nano-size cracks formed by external mechanical stress were found to heal with the assistance of solvent vapour and thermal annealing. The design concept applied was through a backbone engineering approach by synthesizing and incorporating a non-covalent crosslinking moiety of 2,3-pyridine dicarboxamide (PDCA) as a conjugation-break spacer in DPP-based conjugated polymers, namely P(DPP-TVT-X).23 The design rationale for this intrinsically stretchable polymer was as follows: (1) the H-bonds between the non-conjugated PDCA moieties are moderate, which facilitate stress-energy dissipation during stretching, thus allowing the breaking/reforming of hydrogen bonds when strain is applied; (2) the PDCA moiety containing two amide groups markedly lowers the tensile modulus of the resulting polymer and (3) the charge transport mobility can be well-sustained due to the small loading fraction of PDCA moieties. The combination of the polymer with a small amount of 10 mol% PDCA moiety (P3) could reduce the elastic modulus from 1 GPa to 0.25 GPa without sacrificing the charge mobility compared to the pristine p(DPP-TVT)(P1). As shown in Fig. 11a, the μFETh of the P3 thin-film reached up to 1.3 cm2 V−1 s−1, and remarkably it could be maintained as high as 1.12 cm2 V−1 s−1 even with 100% elongation after a hundred cycles along the direction perpendicular to the strain. On the contrary, pristine P1 failed to maintain its mobility level under 100% applied strain, resulting in a rapid decline to <10−2 cm2 V−1 s−1. A reference conjugated polymer of P(DPP-TVT-10 mol% Me-PDCA) (P7) with the exclusion of amide-based H-bonding induced by the additional Me-PDCA chain breaker was studied. The distinct influence in mechanical behavior (25% of crack formation strain) of P7 indicates the important contribution of dynamic bonding as a sacrificial bond in the amorphous domain of these systems. Moreover, the mobility of the damaged P3 could be almost fully recovered from 2.4 × 10−3 cm2 V−1 s−1 to 1.13 cm2 V−1 s−1 using a solvent/thermal healing treatment to promote the polymer chain movement. The existence of healing ability in P3 was also proven by AFM characterization, showing that the nano-cracks became less noticeable after thermal treatment.
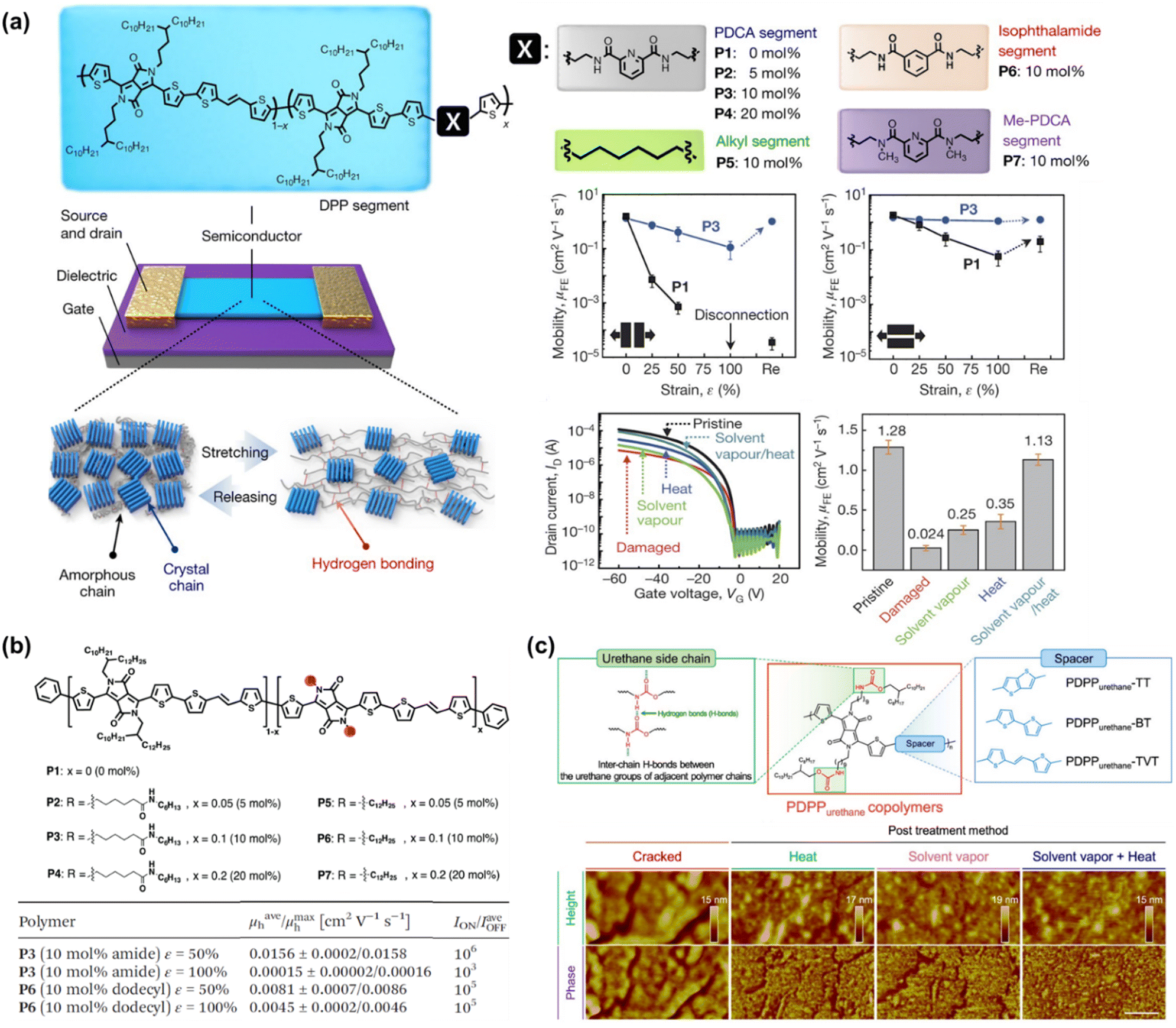 | ||
| Fig. 11 Modulation and influence of the self-healing properties of conjugated polymers based on dynamic hydrogen bonding. (a) DPP-based polymer with various non-conjugated segments.23 Copyright: 2016, Springer Nature. (b) Amide-containing alkyl chains in DPP-based conjugated polymers.164 Copyright: 2018, The Royal Society of Chemistry. (c) Series of DPP-based “alternating” copolymers containing urethane side chains with different donor counterpart co-monomers, denoted as PDPPurethane-TT, -BT, and -TVT.24 Copyright: 2021, Wiley-VCH. | ||
Besides the PDCA unit, other amine-containing outermost side chains were also developed and investigated. Ocheje et al. confirmed the strong influence between the crystallinity, electrical, and mechanical performances of DPP-TVT derivatives with amide-containing alkyl side chains (Fig. 11b).164 Indeed, the inclusion of 10% amide-containing alkyl side chains (P3) significantly promoted their stretching ability up to 75%, reduced their elastic modulus from 203 MPa to 115 MPa, and facilitated molecular alignment after stretching. Upon stretched the polymer at 50% toward 100% strain elongation, the polymer suffered from a 2-fold loss in charge mobility, but P3 showed better charge mobility when stretched at 50% strain in comparison to the reference polymer P6, leading to mobility values one order of magnitude higher for P3 than P6. Taking advantage of the H-bond moiety by side-chain engineering, these polymer thin films can heal through proper treatment. For example, solvent vapour (chlorobenzene, 40 °C for 10 min) and thermal annealing (150 °C for 30 min) were needed to promote the chain mobility and molecular rearrangements. Owing to the easy optimization of the performances of TFTs through typical molecular engineering implemented with OH-bonds, the same group probed the impact of the noncovalent interaction orientation (intramolecular and intermolecular H-bonding) on the physical, optoelectronic, and solid-state properties of novel DPP-conjugated polymers.20 Through a series of detailed characterizations including optical spectroscopy, grazing incidence wide-angle X-ray scattering, and solution small-angle neutron scattering, it was concluded that intramolecular hydrogen bonds were generated in the materials with improved crystallinity and higher effective conjugation in the solid-state.
Similarly, Lee et al. synthesized a series of DPP-based “alternating” copolymers containing urethane side chains with different donor co-monomer counterparts, denoted as PDPPurethane-TT, -BT, and -TVT.24 This system featured a well-defined alternating D–A backbone and long-branched urethane side chains capable of providing structural regularity with moderate H-bonding and sufficient solubility. The PDPPurethane derivative spacer exhibited the maximum μFETh of 2.58 × 10−2, 3.46 × 10−2, and 3.90 × 10−2 cm2 V−1 s−1 for PDPPurethane-TT, -BT, and -TVT, respectively. The PDPPurethane-TVT thin films provided the highest mechanical stability, maintaining their electrical and molecular packing properties up to 100% strain. The typical tensile moduli of DPP-based copolymers are reported to range from 0.1 to 1.0 GPa, whereas PDPPurethane-TVT showed much a lower tensile modulus of 0.048 GPa, implying its superior film deformability and ductility. Owing to its superior electrical and mechanical behavior, the healing feature of PDPPurethane-TVT based on various post-treatments (i.e., heat, solvent vapor, and solvent vapor + heat) was further investigated via AFM analysis. The combination of thermal and solvent vapor provided the most effective healing treatment, as evidenced by the AFM image in Fig. 11c, showing the smooth surface of the thin film after healing. In the case of mild heating and solvent vapor treatment, there were still apparent cracks observed in the AFM image, inferring the poor healing ability, which can plausibly be due to the highly crystalline nature and high glass transition temperature of DPP-based polymers.174
Fundamentally different from the reported IP/SP blends, in which macroscopic phase segregation is often involved, Zhao et al. proposed a general strategy to fabricate self-healing melt-processable semiconductors (Fig. 12a).175 The complementary semiconducting polymer blends were composed of flexible DPP-C5 conjugation-break spacer as a matrix polymer due to its low melting temperature (138 °C) and a fully conjugated polymer (DPP-C0) as a tie chain. After the blend of 95 wt% DPP-C5 and 5 wt% DPP-C0 was melt-processed, the resultant average electrical mobility was reported to be 0.4 cm2 V−1 s−1 and the current on/off ratios were higher than 105 A. With the same combined weight ratio of blended semiconducting polymer, a self-healing test under a melt-processed bottom-gate bottom-contact TFT device was fabricated. It was reported that the initial mobility was 0.29 cm2 V−1 s−1 at room temperature, and when the semiconducting layer was fully separated into two parts, the source-drain current dropped to ≈10−12 A, a level similar to the off current level of the device. The semiconducting layer was partially healed at 160 °C for 2 min, indicating rapid healing ability. Then, it only took 30 min to completely heal and the recovered charge mobility was 0.26 cm2 V−1 s−1. The authors also succeeded in employing this method for functional semiconductor polymers, where most high-performance semiconducting polymers cannot be liquefied to flow. This, this method overcome the problems associated with the solution processing method.
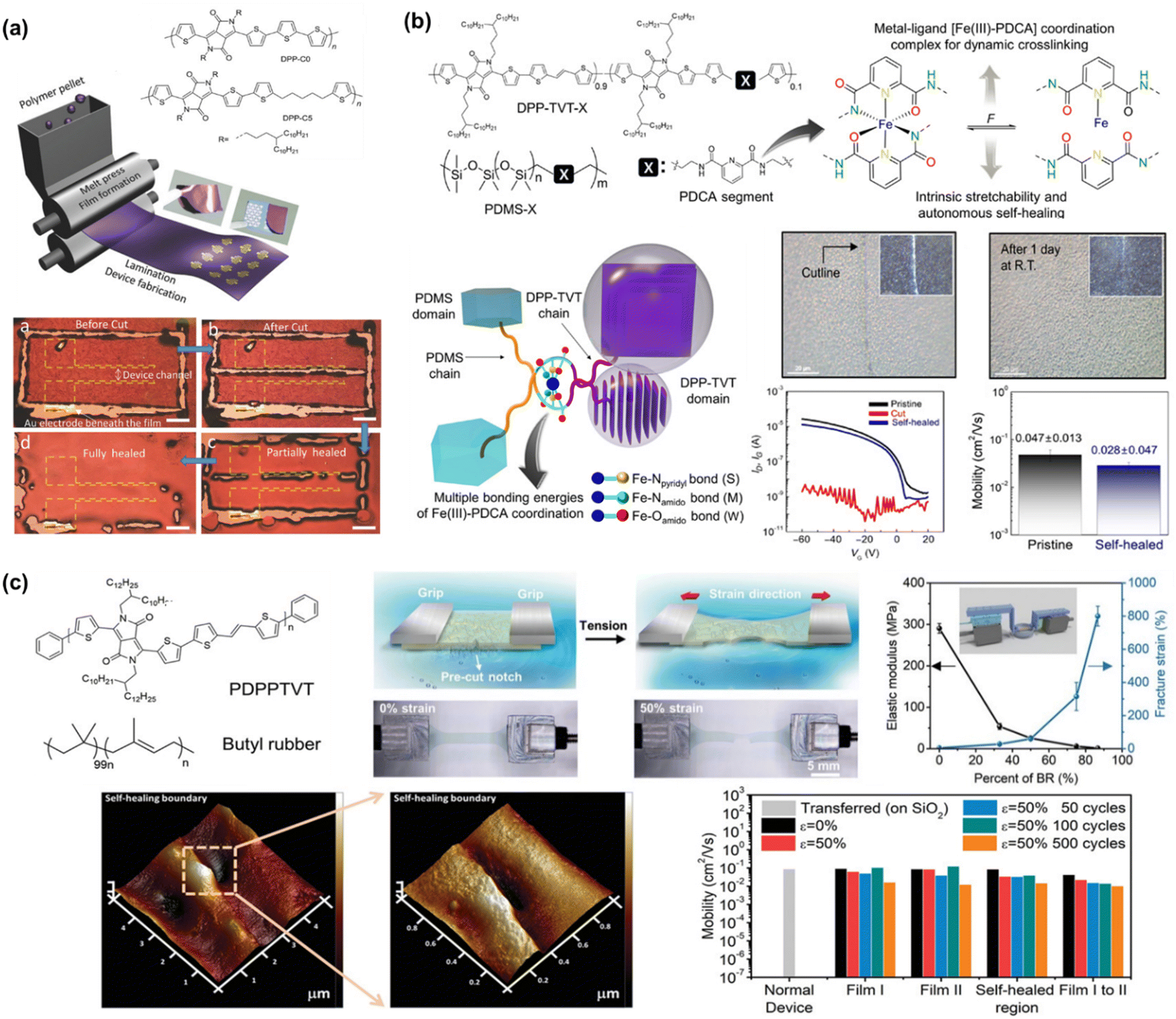 | ||
| Fig. 12 Stretchable and self-healable semiconducting polymer via blending strategy. (a) Materials, fabrication procedure, and TFT characterization of melt-processing of complementary self-healable semiconducting polymer blends.175 Copyright: 2017, Wiley. (b) Illustration and the mechanical-electrical properties of Fe(III)-PDCA ligand bonding of PDMS-PDCA and DPP-TVT-PDCA in the blend film.31 Copyright: 2019, Science Advances. (c) 3D-schematic illustration, optical images of notched film before and after tensile deformation on water surface, and the self-healing test as a function on electrical performances of 2:3 PDPPTVT/BR film.166 Copyright: 2020, Wiley. | ||
Inspired by previous work on self-healing and highly stretchable elastomers via advanced metal-ligand dynamic interactions,165 Oh et al. fabricated a skin-like semiconducting film through physical blending of a polymer semiconductor and a self-healable elastomer, both of which were dynamically cross-linked by metal coordination (Fig. 12b).31 In particular, poly(3,6-di(thiophen-2-yl)diketopyrrolo[3,4-c]pyrrole-1,4-dione-alt-1,2-dithienylethene) with 10 mol% 2,6-pyridinedicarboxamine moieties (DPP-TVT-PDCA) was utilized as the semiconducting material due to its great charge carrier mobility (>1 cm2 V−1 s−1),23 and poly(dimethylsiloxane-alt-2,6-pyridinedicarbozamine) with an Fe(III) ion (PDMS-PDCA-Fe elastomer) as the insulating, stretchable, and healable polymer.165 The multiple dynamic coordinate covalent bonds with three different bonding strengths (Fe–Npyridyl, strong; Fe–Namido, medium; and Fe–Oamido, weak) facilitated the dynamic cross-linking for high stretchability (fracture strain >1300%) and the material could be fully self-healed at room temperature within 1 day. Furthermore, the values of the field-effect mobility evaluated in a bottom-contact bottom-gate-structured TFT on a healed semiconducting film was recovered from the initial mobility value before fracture of 4.7 × 10−3 cm2 V−1 s−1 to 2.8 × 10−3 cm2 V−1 s−1. These results indicate that the practical application of self-healable TFT devices at room temperature without any external stimuli is one-step closer to being achieved, where this behavior is called “autonomous self-healing”. Furthermore, this semiconducting layer has been successfully implemented in the innovative development of fully stretchable active-matrix sensor-based e-skin.
Recently, Zhang et al. also used physical blending to manufacture new self-healing semiconducting composites, but the mechanism of self-healing was different from previous reports.166 Taking advantage of butyl rubber (BR) as the elastomer with outstanding elasticity (a low persistence length of 5.2 Å), strong adhesion, and excellent barrier properties to both oxygen and water, together with PDPPTVT as a p-type donor–acceptor semiconducting polymer with high charge transport mobility,174 these two well-known materials were blended. A free-standing tensile tester was used to evaluate the mechanical and self-healing behavior in various blend ratios between PDPPTVT and BR (Fig. 12c). As the weight fraction of BR increased to ∼90%, the semiconducting polymer composite showed a significant drop in elastic modulus to 1 MPa and great improvement in fracture strain exceeding 800%. Given that the BR polymer endows tackiness and fast segmental motion to low Tg BR polymer chains at room temperature,174,176 the polymer chains that reconstructed during contact led to autonomous self-healing within a few seconds at room temperature, and the film could be stretched up to 150% of its original length after healing. As is the case with electronic performance, the charge mobility was maintained on the same order of magnitude, even after 500 stretch-release cycles at 50% strain. Additionally, this new composite exhibited outstanding tear-resistant properties, which were attributed to the highly entangled BR network being able to redistribute stress, thus preventing crack propagation and increasing the durability. From a mechanical perspective, this material successfully displayed the lowest modulus and the highest deformability compared to other polymer designs.33,77,81,104,177–181 Overall, these tear-resistant and room temperature self-healable semiconducting polymers feature special functionalities, which can facilitate future developments for wearable applications.
4. Perspectives
As has been elucidated from the previous sections, the presence of self-healing ability in semiconducting polymers is mainly derived from the dynamic covalent and non-covalent interaction found in a variety of bond formations.142,143 However, this perspective focuses on the stretchable and self-healable semiconducting polymers that are scarce and less well-exploited in TFTs due to the limitations and challenges associated with this emerging technology. Recent advances in the performance of conjugated polymers through molecular design or physical blending have successfully exhibited field-effect mobilities >1 cm2 V−1 s−1,23,166 similar to the electrical performance of amorphous silicon thin-film transistors.182–184 Furthermore, after integrating self-healable semiconducting polymers in TFT devices, they can withstand extreme mechanical behavior. However, to actualize a complete set of self-healable TFTs, three self-healable components need to be developed concurrently, i.e., semiconductors, dielectrics, and three-terminal electrodes. Compared to the fabrication of self-healable semiconducting polymers, the development of other self-healable components in TFTs is simpler and has met the standards for implementation in TFTs to be integrated into sensors, memory devices, and drive circuits for displays.106,149,165,185,186 For example, in 2016, Bao and co-workers introduced the design of electrically stable and self-healable dielectric layers based on metal-ligand coordination interaction in a PDMS matrix.149 The supramolecular non-covalent bond contained in this interaction allows impeccable self-healing property for potential applications in wearable sensors and electronic skins. Through the proper selection of counter anions, TFTs with hysteresis-free transfer characteristics can be generated. Self-healable conductive materials composed of a 3D percolation network of carbon nanotubes (CNTs) or silver nanowires (AgNWs) embedded in a self-healable polymer matrix were used as source, drain, and gate components to fabricate a fully stretchable and self-healable multi-functional electronic skin system.185 This unique property was achieved by taking advantage of the low glass transition temperature and dynamic hydrogen bonding properties of an elegant elastomeric matrix,160 where the broken conductive network present in the non-covalently bonded hydrogen moieties could be autonomously recovered from disconnection during the self-healing process.Continuing the discussion in Section 3, the same group succeeded in fabricating (DPP-TVT-PDCA)-blended-(PDMS-PDCA-Fe elastomer) as a stretchable and self-healable semiconducting polymer film for an active-matrix strain-sensing array.31 The Fe(III)-PDCA coordination complex inside the polymer blend was comprised of 3 types of bonding forces (Fe–Npyridyl, strong; Fe–Namido, medium; and Fe–Oamido, weak), which could facilitate the crosslinking process for self-healing capability based on non-covalent interaction. Fig. 13 shows that a fully stretchable 5 × 5 active-matrix sensor-based e-skin was capable of detecting pressure-induced deformation, with the simultaneous visualization of the applied strain. The active area of each pixel has a channel width of 1 mm and channel length of 150 μm. All devices showed good uniformity with a maximum mobility of 0.11 cm2 V−1 s−1 and an average mobility of 0.076 ± 0.019 cm2 V−1 s−1. It needs to be emphasized that the authors inserted a self-healing semiconducting film between stretchable sandwich layers. Thus, to construct skin-like sensory devices and verifiably operate within a medically safe voltage range, integration with self-healable conductors and especially high-k dielectric materials still needs to be developed.
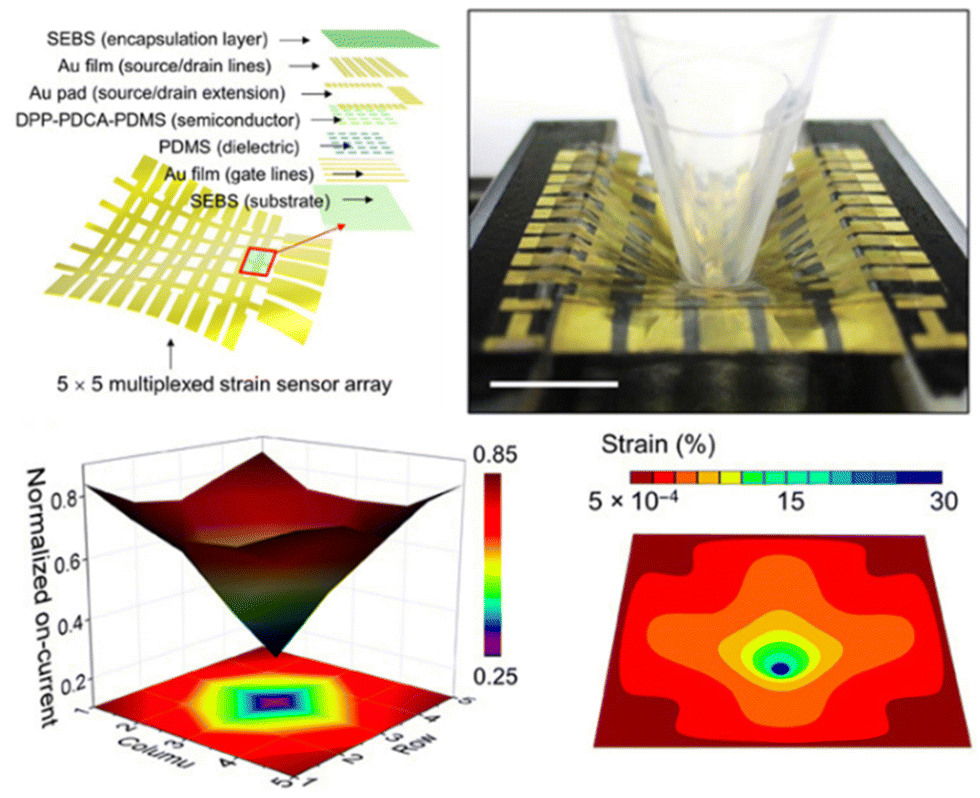 | ||
| Fig. 13 Architecture, photograph, normalized on-current, and simulation results of strain applied by poking with a plastic bar to the fully stretchable active-matrix TFT array containing self-healable semiconducting polymer.31 Copyright: 2019, Science Advances. | ||
Despite the very promising advances in the manufacture of autonomous self-healable semiconducting polymers, their implementation in real applications or commercial products is still in its infancy. To date, the application of self-healing materials in devices is still at the proof-of-concept stage. Besides the possible mismatch between self-healing ability and electrical mobility, the application of self-healable semiconducting polymers in TFT devices is currently limited due to their tedious fabrication process, which may be incompatible with existing electronic technologies established in the silicon industry. Technically, how to make them comparable in size so they can fit in the architecture of boards, especially on ultra-thin semiconductor films at the micro- and nano-scale needs to be considered. Therefore, scalable and reliable fabrication processes such as patterning or printing should be developed for the mass production of self-healing devices and electronics. It should be noted that the self-healing process remains relatively slow and the utilization of self-healing has thus far been limited to small-area damage (nano crack), and thus self-healing capabilities at damage scales larger than layer thickness, which are rapid, repeatable, and autonomous should be explored further. Lastly, the current electronic properties are not yet comparable between non-healable electronic devices and healable functional electronic devices. Thus, further perseverance in the field of new design concepts for self-healable semiconducting polymers is required.
5. Conclusions
This review presented an update on the current status and developments of stretchable to self-healing semiconducting polymers in TFT devices. In the early stages of research, the engineering strategy focused on the fundamental morphological properties of the semiconducting polymer itself such as MW and RRe, and thus other considerations during the preparation of thin films was thoroughly reviewed. As an example, polythiophene derivatives having a high MW possess a superior synergistic relationship between mobility-stretchability properties. An enhanced chain entanglement effect and greater connectivity between crystallites in P3HT and self-aggregation of polymer chains are the advantages of utilizing high MW polymers. The main part of this report, before moving on to autonomous healing properties integrated in TFT devices, elaborated and discussed the seven main approaches and additional function compounds used to overcome the compromise between electrical-mechanical properties. Based on the range of limitations of this review, only the molecular spacer and symmetric side-chain approaches could achieve a field-effect mobility of >1 cm2 V−1 s−1 even in the stretched state up to 100%, together with a lower elastic modulus (<0.1 GPa), indicating excellent deformability and ductility to promote tolerance toward their electrical functionalities under repeated mechanical loading-unloading cycles over long-term usage. The complementary part of this report linked the chosen approaches for designing novel intrinsically stretchable semiconducting polymers with partial or full healing ability implemented with the polymers used in device technologies. Hence, the structural diversity of D–A type polymers has been well selected for the more innovative design of non-covalent interaction-induced self-healing capability. Given that most intrinsically self-healable semiconducting polymers are constructed via complex engineering strategies, the conjugated polymer blending approach was introduced as a modest method with fruitful advantages. This method results in autonomous self-healing capability and a remarkable enhancement in mechanical adherence for high-mobility polymer semiconductors in an easy and facile manner.Author contributions
Y.-C. C. conceived the main concept of the review article. L. L. organized the overall manuscript and wrote the introduction, and the parts relating to fundamental properties of polythiophene derivatives, backbone-engineering approaches, the advances from non-autonomous to autonomous self-healing semiconducting polymers, perspectives and conclusions in the manuscript. A. N. wrote the parts relating to the solution processing and fabrication effect, structural engineering of polymer side chains, and preparing the summary table. S. P. P. organized the overall manuscript. The manuscript was written through contributions from all the authors. All the authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y.-C. C. is thankful for financial support from the Ministry of Science and Technology in Taiwan (MOST 110-2221-E-011-009).Notes and references
- T. Ito, H. Shirakawa and S. Ikeda, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 1943–1950 CrossRef CAS.
- A. J. Heeger, J. Phys. Chem. B, 2001, 105, 8476–8491 CrossRef.
- H. Shirakawa, E. J. Louis, A. G. Macdiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 578–580 RSC.
- R. M. Pankow and B. C. Thompson, Polymer, 2020, 207, 122874 CrossRef CAS.
- E. J. Markvicka, M. D. Bartlett, X. Huang and C. Majidi, Nat. Mater., 2018, 17, 618–624 CrossRef CAS PubMed.
- Y. Liu, M. Pharr and G. A. Salvatore, ACS Nano, 2017, 11, 9614–9635 CrossRef CAS PubMed.
- S. Wang, J. Y. Oh, J. Xu, H. Tran and Z. Bao, Acc. Chem. Res., 2018, 51, 1033–1045 CrossRef CAS PubMed.
- I. You, D. G. Mackanic, N. Matsuhisa, J. Kang, J. Kwon, L. Beker, J. Mun, W. Suh, T. Y. Kim, Z. Bao and U. Jeong, Science, 2020, 370, 961–965 CrossRef CAS PubMed.
- T. Huynh, P. Sonar and H. Haick, Adv. Mater., 2017, 29, 1604973 CrossRef PubMed.
- Z. Zhang, W. Wang, Y. Jiang, Y. X. Wang, Y. Wu, J. C. Lai, S. Niu, C. Xu, C. C. Shih, C. Wang, H. Yan, L. Galuska, N. Prine, H. C. Wu, D. Zhong, G. Chen, N. Matsuhisa, Y. Zheng, Z. Yu, Y. Wang, R. Dauskardt, X. Gu, J. B. H. Tok and Z. Bao, Nature, 2022, 603, 624–630 CrossRef CAS PubMed.
- H. Tien, Y. Huang, Y. Chiu, Y.-H. Cheng, C.-C. Chueh and W.-Y. Lee, J. Mater. Chem. C, 2021, 9, 2660–2684 RSC.
- Y. W. Huang, Y. C. Lin, H. C. Yen, C. K. Chen, W. Y. Lee, W. C. Chen and C. C. Chueh, Chem. Mater., 2020, 32, 7370–7382 CrossRef CAS.
- T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379–385 CrossRef CAS PubMed.
- S. Lage-rivera, A. Ares-pernas and M. Abad, Int. J. Energy Res., 2022, 1–24 Search PubMed.
- D. H. Cho, K. G. Cho, S. An, M. S. Kim, H. W. Oh, J. Yeo, W. C. Yoo, K. Hong, M. Kim and K. H. Lee, Energy Storage Mater., 2022, 45, 323–331 CrossRef.
- J. Kang, J. B. Tok and Z. Bao, Nat. Electron., 2019, 2, 144–150 CrossRef.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- S. Holliday, J. E. Donaghey and I. McCulloch, Chem. Mater., 2014, 26, 647–663 CrossRef CAS.
- A. Gasperini, G. J. N. Wang, F. Molina-Lopez, H. C. Wu, J. Lopez, J. Xu, S. Luo, D. Zhou, G. Xue, J. B. H. Tok and Z. Bao, Macromolecules, 2019, 52, 2476–2486 CrossRef CAS.
- M. U. Ocheje, R. B. Goodman, K. T. Lu, Y. Wang, L. A. Galuska, L. Soullard, Z. Cao, S. Zhang, M. Yadiki, X. Gu, Y. C. Chiu and S. Rondeau-Gagné, Chem. Mater., 2021, 33, 8267–8277 CrossRef CAS.
- L. A. Galuska, M. U. Ocheje, Z. C. Ahmad, S. Rondeau-Gagné and X. Gu, Chem. Mater., 2022, 34, 2259–2267 CrossRef CAS.
- M. U. Ocheje, B. P. Charron, Y. Cheng, C. Chuang, A. Soldera, Y. Chiu and S. Rondeau-gagne, Macromolecules, 2018, 51, 1336–1344 CrossRef CAS.
- J. Y. Oh, S. Rondeau-Gagné, Y. C. Chiu, A. Chortos, F. Lissel, G. J. N. Wang, B. C. Schroeder, T. Kurosawa, J. Lopez, T. Katsumata, J. Xu, C. Zhu, X. Gu, W. G. Bae, Y. Kim, L. Jin, J. W. Chung, J. B. H. Tok and Z. Bao, Nature, 2016, 539, 411–415 CrossRef CAS PubMed.
- M. Y. Lee, S. Dharmapurikar, S. J. Lee, Y. Cho, C. Yang and J. H. Oh, Chem. Mater., 2020, 32, 1914–1924 CrossRef CAS.
- X. Yu, C. Li, C. Gao, X. Zhang, G. Zhang and D. Zhang, SmartMat, 2021, 2, 347–366 CrossRef.
- Y. Zhao, X. Zhao, Y. Zang, C. Di, Y. Diao and J. Mei, Macromolecules, 2015, 48, 2048–2053 CrossRef CAS.
- H. J. Cheon, T. K. An and Y. H. Kim, Macromol. Res., 2022, 30, 71–84 CrossRef CAS.
- J. Mei, D. H. Kim, A. L. Ayzner, M. F. Toney and Z. Bao, J. Am. Chem. Soc., 2011, 133, 20130–20133 CrossRef CAS PubMed.
- I. Kang, H. Yun, D. S. Chung, S. Kwon and Y. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS PubMed.
- C. Lu, W. Y. Lee, X. Gu, J. Xu, H. H. Chou, H. Yan, Y. C. Chiu, M. He, J. R. Matthews, W. Niu, J. B. H. Tok, M. F. Toney, W. C. Chen and Z. Bao, Adv. Electron. Mater., 2017, 3, 1600311 CrossRef.
- J. Y. Oh, D. Son, T. Katsumata, Y. Lee, Y. Kim, J. Lopez, H. C. Wu, J. Kang, J. Park, X. Gu, J. Mun, N. G. J. Wang, Y. Yin, W. Cai, Y. Yun, J. B. H. Tok and Z. Bao, Sci. Adv., 2019, 5, eaav3097 CrossRef CAS PubMed.
- S. Nikzad, H. Wu, J. Kim, C. M. Mahoney, J. R. Matthews, W. Niu, Y. Li, H. Wang, W. Chen, M. F. Toney, M. He and Z. Bao, Chem. Mater., 2020, 32, 897–905 CrossRef CAS.
- J. Xu, S. Wang, G. N. Wang, C. Zhu, S. Luo, L. Jin, X. Gu, S. Chen, V. R. Feig, J. W. F. To, S. Rondeau-gagné, J. Park, B. C. Schroeder, C. Lu, J. Y. Oh, Y. Wang, Y. Kim, H. Yan, R. Sinclair, D. Zhou, G. Xue, B. Murmann, C. Linder, W. Cai, J. B. H. Tok, J. W. Chung and Z. Bao, Science, 2017, 64, 59–64 CrossRef PubMed.
- K. Sim, Z. Rao, F. Ershad and C. Yu, Adv. Mater., 2019, 32, 1902417 CrossRef PubMed.
- Y. Zheng, S. Zhang, B. Tok and Z. Bao, J. Am. Chem. Soc., 2022, 144, 4699–4715 CrossRef CAS PubMed.
- Z. Bao, A. Dodabalapur and A. J. Lovinger, Appl. Phys. Lett., 1996, 69, 4108 CrossRef CAS.
- K. R. Amundson, B. J. Sapjeta, A. J. Lovinger and Z. Bao, Thin Solid Films, 2002, 414, 143–149 CrossRef CAS.
- B. H. Yang, T. J. Shin, L. Yang, K. Cho, C. Y. Ryu and Z. Bao, Adv. Energy Mater., 2005, 15, 671–676 Search PubMed.
- H. Yang, T. A. E. J. O. O. Shin, Z. Bao and C. Y. Ryu, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 1303–1312 CrossRef CAS.
- A. Chortos, J. Lim, J. W. F. To, M. Vosgueritchian, T. J. Dusseault, T. Kim, S. Hwang and Z. Bao, Adv. Mater., 2014, 26, 4253 CrossRef CAS PubMed.
- B. M. Brinkmann and P. Rannou, Adv. Funct. Mater., 2007, 17, 101–108 CrossRef.
- M. Brinkmann, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1218–1233 CrossRef CAS.
- C. P. Buckley and A. J. Kovacs, Colloid Polym. Sci., 1976, 254, 695–715 CrossRef CAS.
- E. Chen, A. J. Jing, X. Weng, P. Huang, S. Lee, S. Z. D. Cheng, B. S. Hsiao and F. Yeh, Polymer, 2003, 44, 6051–6058 CrossRef CAS.
- S. J. Organ, G. Ungar and A. Keller, Macromolecules, 1989, 22, 1995–2000 CrossRef CAS.
- E. Mena-osteritz, A. Meyer, B. M. W. Langeveld-voss, R. A. J. Janssen, E. W. Meijer and P. Bäuerle, Angew. Chem., Int. Ed., 2000, 39, 2679–2684 CrossRef PubMed.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu, J. M. J. Fréchet and M. F. Toney, Macromolecules, 2005, 38, 3312–3319 CrossRef CAS.
- N. E. Persson, P. H. Chu, M. McBride, M. Grover and E. Reichmanis, Acc. Chem. Res., 2017, 50, 932–942 CrossRef CAS PubMed.
- B. O’Connor, R. J. Kline, B. R. Conrad, L. J. Richter, D. Gundlach, M. F. Toney and D. M. DeLongchamp, Adv. Funct. Mater., 2011, 21, 3697–3705 CrossRef.
- D. Gargi, R. J. Kline, D. M. Delongchamp, D. A. Fischer, M. F. Toney and B. T. O’Connor, J. Phys. Chem. C, 2013, 117, 17421–17428 CrossRef CAS.
- R. J. Kline, M. D. McGehee, E. N. Kadnikova, J. Liu and J. M. J. Frechet, Adv. Mater., 2003, 15, 1519–1522 CrossRef CAS.
- F. Peter, V. Koch, J. Rivnay, S. Foster, C. Müller, J. M. Downing, E. Buchaca-domingo, P. Westacott, L. Yu, M. Yuan, M. Baklar, Z. Fei, C. Luscombe, M. A. Mclachlan, M. Heeney, G. Rumbles, C. Silva, A. Salleo, J. Nelson, P. Smith and N. Stingelin, Prog. Polym. Sci., 2013, 38, 1978–1989 CrossRef.
- D. Rodriquez, J. Kim, S. E. Root, Z. Fei, P. Bou, M. Heeney, T. Kim and D. J. Lipomi, Appl. Mater. Interfaces, 2017, 9, 8855–8862 CrossRef CAS PubMed.
- Y. Kim, S. Cook, S. M. Tuladhar, S. A. Choulis, J. Nelson, J. R. Durrant, D. D. C. Bradley, M. Giles, I. Mcculloch, C. Ha and M. Ree, Nat. Mater., 2006, 5, 197–203 CrossRef CAS.
- C. H. Woo, B. C. Thompson, B. J. Kim, M. F. Toney and J. M. J. Fre, J. Am. Chem. Soc., 2008, 130, 16324–16329 CrossRef CAS PubMed.
- H. Sirringhaus, N. Tessler and R. H. Friend, Science, 1998, 280, 1741–1744 CrossRef CAS PubMed.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS.
- J. Kim, J. Kim, W. Lee, H. Yu, H. J. Kim, I. Song, M. Shin, J. H. Oh, U. Jeong, T. Kim and B. J. Kim, Macromolecules, 2015, 48, 4339–4346 CrossRef CAS.
- H. Park, B. S. Ma, J. Kim, Y. Kim, H. J. Kim, D. Kim, H. Yun, J. Han, F. S. Kim, T. Kim and B. J. Kim, Macromolecules, 2019, 52, 7721–7730 CrossRef CAS.
- B. O’Connor, E. P. Chan, C. Chan, B. R. Conrad, L. J. Richter, R. J. Kline, M. Heeney, I. McCulloch, C. L. Soles and D. M. DeLongchamp, ACS Nano, 2010, 4, 7538–7544 CrossRef PubMed.
- B. C. Schroeder, Y. Chiu, X. Gu, Y. Zhou, J. Xu, J. Lopez, C. Lu, M. F. Toney and Z. Bao, Adv. Electron. Mater., 2016, 2, 1600104 CrossRef.
- Y. D. Park, J. K. Park, J. H. Seo, J. D. Yuen, W. H. Lee, K. Cho and G. C. Bazan, Adv. Energy Mater., 2011, 1, 63–67 CrossRef CAS.
- C. Mcdowell, M. Abdelsamie, M. F. Toney and G. C. Bazan, Adv. Mater., 2018, 1707114, 1–30 Search PubMed.
- N. E. Jackson, K. L. Kohlstedt, B. M. Savoie, O. De Cruz, G. C. Schatz, L. X. Chen and M. A. Ratner, J. Am. Chem. Soc., 2015, 137, 6254–6262 CrossRef CAS PubMed.
- W. Lee, G. Giri, Y. Diao, C. J. Tassone, J. R. Matthews, M. L. Sorensen, S. C. B. Mannsfeld, W. Chen, H. H. Fong, J. B. Tok, M. F. Toney, M. He and Z. Bao, Adv. Funct. Mater., 2014, 24, 3524–3534 CrossRef CAS.
- K. Zhao, X. Yu, R. Li, A. Amassian and Y. Han, J. Mater. Chem. C, 2015, 3, 9842–9848 RSC.
- H. Hu, K. Zhao, N. Fernandes, P. Boufflet, J. H. Bannock, L. Yu, J. C. de Mello, N. Stingelin, M. Heeney, E. P. Giannelis and A. Amassian, J. Mater. Chem. C, 2015, 3, 7394–7404 RSC.
- L. Janasz, D. Chlebosz, M. Gradzka, W. Zajaczkowski, T. Marszalek, K. Mullen, J. Ulanski, A. Kiersnowski and W. Pisula, J. Mater. Chem. C, 2016, 4, 11488–11498 RSC.
- J. Y. Oh, M. Shin, T. Il Lee, W. S. Jang, Y. Min, J. Myoung, H. K. Baik and U. Jeong, Macromolecules, 2012, 45, 7504–7513 CrossRef CAS.
- H. Liu, C. H. Reccius and H. G. Craighead, Appl. Phys. Lett., 2005, 87, 253106 CrossRef.
- Y. Yuan, G. Giri, A. L. Ayzner, A. P. Zoombelt, S. C. B. Mannsfeld, J. Chen, D. Nordlund, M. F. Toney, J. Huang and Z. Bao, Nat. Commun., 2014, 5, 3005 CrossRef PubMed.
- J. Chen, H. Hsieh, Y. Chiu, W. Lee, C. Hung, C. Chueh and W. Chen, J. Mater. Chem. C, 2020, 8, 873–882 RSC.
- D. Choi, H. Kim, N. Persson, P. Chu, M. Chang, J. Kang, S. Graham and E. Reichmanis, Chem. Mater., 2016, 28, 1196–1204 CrossRef CAS.
- R. Noriega, J. Rivnay, K. Vandewal, F. P. V. Koch, N. Stingelin, P. Smith, M. F. Toney and A. Salleo, Nat. Mater., 2013, 12, 1038–1044 CrossRef CAS PubMed.
- J. S. Ross, S. Wu, H. Yu, N. J. Ghimire, A. M. Jones, G. Aivazian, J. Yan, D. G. Mandrus, D. Xiao, W. Yao and X. Xu, Nat. Commun., 2013, 4, 1474 CrossRef PubMed.
- D. Venkateshvaran, M. Nikolka, A. Sadhanala, V. Lemaur, M. Zelazny, M. Kepa, M. Hurhangee, A. J. Kronemeijer, V. Pecunia, I. Nasrallah, I. Romanov, K. Broch, I. McCulloch, D. Emin, Y. Olivier, J. Cornil, D. Beljonne and H. Sirringhaus, Nature, 2014, 515, 384–388 CrossRef CAS PubMed.
- C. Müller, S. Goffri, D. W. Breiby, J. W. Andreasen, H. D. Chanzy, R. A. J. Janssen, M. M. Nielsen, C. P. Radano, H. Sirringhaus, P. Smith and N. Stingelin-Stutzmann, Adv. Funct. Mater., 2007, 17, 2674–2679 CrossRef.
- J. Chang, B. Sun, D. W. Breiby, M. M. Nielsen, T. I. Sölling, M. Giles, I. Mcculloch and H. Sirringhaus, Chem. Mater., 2004, 16, 4772–4776 CrossRef CAS.
- J.-T. Wang, S. Takshima, H.-C. Wu, C.-C. Shih, T. Isono, T. Kakuchi, T. Satoh and W.-C. Chen, Macromolecules, 2017, 50, 1442–1452 CrossRef CAS.
- Y.-C. Chiang, S. Kobayashi, T. Isono, C.-C. Shih, T. Shingu, C.-C. Hung, H.-C. Hsieh, S.-H. Tung, T. Satoh and W.-C. Chen, Polym. Chem., 2019, 10, 5452–5464 RSC.
- R. Peng, B. Pang, D. Hu, M. Chen, G. Zhang, X. Wang, H. Lu, K. Cho and L. Qiu, J. Mater. Chem. C, 2015, 3, 3599–3606 RSC.
- T. Higashihara, S. Fukuta, Y. Ochiai, T. Sekine, K. Chino, T. Koganezawa and I. Osaka, ACS Appl. Polym. Mater., 2019, 1, 315–320 CrossRef CAS.
- S. Miyane, H. Wen, W. Chen and T. Higashihara, Polym. Chem., 2018, 56, 1787–1794 CrossRef CAS.
- M. Funahashi and A. Sonoda, Org. Electron., 2012, 13, 1633–1640 CrossRef CAS.
- J. Lee, A. Han, J. Kim, Y. Kim, J. H. Oh and C. Yang, J. Am. Chem. Soc., 2012, 134, 20713–20721 CrossRef CAS PubMed.
- K. Fujita, Y. Sumino, K. Ide, S. Tamba, K. Shono, J. Shen, T. Nishino, A. Mori and T. Yasuda, Macromolecular, 2016, 49, 1259–1269 CrossRef CAS.
- J. Shen, K. Fujita, T. Matsumoto, C. Hongo, M. Misaki, K. Ishida, A. Mori and T. Nishino, Macromol. Chem. Phys., 2017, 218, 1700197 CrossRef.
- A. Salleo, T. W. Chen and A. R. Völkel, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 115311 CrossRef.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. Mcculloch and T. D. Anthopoulos, Adv. Mater., 2018, 1801079, 1–33 Search PubMed.
- Y. Guan, A. Thukral, S. Zhang, K. Sim, X. Wang, Y. Zhang, Z. R. Faheem Ershad5, F. Pan, P. Wang, J. Xiao and C. Yu, Sci. Adv., 2020, 6, eabb3656 CrossRef CAS PubMed.
- S. Savagatrup, X. Zhao, E. Chan, J. Mei and D. J. Lipomi, Macromol. Rapid Commun., 2016, 37, 1623–1628 CrossRef CAS PubMed.
- E. L. Melenbrink, K. M. Hilby, M. A. Alkhadra, S. Samal, D. J. Lipomi and B. C. Thompson, Appl. Mater. Interfaces, 2018, 10, 32426–32434 CrossRef CAS PubMed.
- J. Mun, G. N. Wang, J. Y. Oh, T. Katsumata, F. L. Lee, J. Kang, H. Wu, F. Lissel, S. Rondeau-gagné and J. B. Tok, Adv. Funct. Mater., 2018, 28, 1804222 CrossRef.
- Y. Zheng, M. Ashizawa, S. Zhang, J. Kang, S. Nikzad, Z. Yu, Y. Ochiai, C. Wu, H. Tran, J. Mun, Y. Zheng, J. B. Tok, X. Gu and Z. Bao, Chem. Mater., 2020, 32, 5700–5714 CrossRef CAS.
- D. Liu, J. Mun, G. Chen, N. J. Schuster, W. Wang, Y. Zheng, S. Nikzad, J. Lai, Y. Wu, D. Zhong, Y. Lin, Y. Lei, Y. Chen, S. Gam, J. W. Chung, Y. Yun, B. Tok and Z. Bao, J. Am. Chem. Soc., 2021, 143, 11679–11689 CrossRef CAS PubMed.
- K. Shi, W. Zhang, D. Gao, S. Zhang, Z. Lin, Y. Zou, L. Wang and G. Yu, Adv. Mater., 2018, 30, 1705286 CrossRef PubMed.
- Y. C. Lin, Y. W. Huang, C. C. Hung, Y. C. Chiang, C. K. Chen, L. C. Hsu, C. C. Chueh and W. C. Chen, ACS Appl. Mater. Interfaces, 2020, 12, 50648–50659 CrossRef CAS PubMed.
- J. Mun, Y. Ochiai, W. Wang, Y. Zheng, Y. Zheng, H. Wu, N. Matsuhisa, T. Higashihara, J. B. Tok, Y. Yun and Z. Bao, Nat. Commun., 2021, 12, 3572 CrossRef CAS PubMed.
- A. D. Printz, S. Savagatrup, D. J. Burke, T. N. Purdy and D. J. Lipomi, RSC Adv., 2014, 4, 13635–13643 RSC.
- S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Macromolecules, 2014, 47, 1981–1992 CrossRef CAS.
- Y. C. Lin, Y. W. Huang, Y. S. Wu, J. S. Li, Y. F. Yang, W. C. Chen and C. C. Chueh, ACS Appl. Polym. Mater., 2021, 3, 1628–1637 CrossRef CAS.
- S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070–4098 CrossRef CAS PubMed.
- H. F. Wen, H. C. Wu, J. Aimi, C. C. Hung, Y. C. Chiang, C. C. Kuo and W. C. Chen, Macromolecules, 2017, 50, 4982–4992 CrossRef CAS.
- G. N. Wang, L. Shaw, J. Xu, T. Kurosawa, B. C. Schroeder, J. Y. Oh, S. J. Benight and Z. Bao, Adv. Funct. Mater., 2016, 26, 7254–7262 CrossRef CAS.
- W. Zhao, T. Cao and J. M. White, Adv. Funct. Mater., 2004, 14, 783–790 CrossRef CAS.
- X. Yan, Z. Liu, Q. Zhang, J. Lopez, H. Wang, H. C. Wu, S. Niu, H. Yan, S. Wang, T. Lei, J. Li, D. Qi, P. Huang, J. Huang, Y. Zhang, Y. Wang, G. Li, J. B. H. Tok, X. Chen and Z. Bao, J. Am. Chem. Soc., 2018, 140, 5280–5289 CrossRef CAS PubMed.
- P. Song and H. Wang, Adv. Mater., 2020, 32, 1901244 CrossRef CAS PubMed.
- H. C. Wu, F. Lissel, G. J. N. Wang, D. M. Koshy, S. Nikzad, H. Yan, J. Xu, S. Luo, N. Matsuhisa, Y. Cheng, F. Wang, B. Ji, D. Li, W. C. Chen, G. Xue and Z. Bao, Adv. Funct. Mater., 2021, 31, 2009201 CrossRef CAS.
- G. J. N. Wang, Y. Zheng, S. Zhang, J. Kang, H. C. Wu, A. Gasperini, H. Zhang, X. Gu and Z. Bao, Chem. Mater., 2019, 31, 6465–6475 CrossRef CAS.
- F. J. Kahle, C. Saller, A. Köhler and P. Strohriegl, Adv. Energy Mater., 2017, 7, 1700306 CrossRef.
- Y. Zheng, G. J. N. Wang, J. Kang, M. Nikolka, H. C. Wu, H. Tran, S. Zhang, H. Yan, H. Chen, P. Y. Yuen, J. Mun, R. H. Dauskardt, I. McCulloch, J. B. H. Tok, X. Gu and Z. Bao, Adv. Funct. Mater., 2019, 29, 1905340 CrossRef CAS.
- M. U. Ocheje, M. Comı, R. Yang, Z. Chen, Y. Liu, N. Yousefi, M. Al-hashimi and S. Rondeau-gagne, J. Mater. Chem. C, 2022, 10, 4236–4246 RSC.
- H. C. Wu, C. C. Hung, C. W. Hong, H. S. Sun, J. T. Wang, G. Yamashita, T. Higashihara and W. C. Chen, Macromolecules, 2016, 49, 8540–8548 CrossRef CAS.
- Y. C. Chiang, H. C. Wu, H. F. Wen, C. C. Hung, C. W. Hong, C. C. Kuo, T. Higashihara and W. C. Chen, Macromolecules, 2019, 52, 4393–4404 CrossRef.
- S. W. Sides, J. Curro, G. S. Grest, M. J. Stevens, T. Soddemann, A. Habenschuss and J. D. Londono, Macromolecules, 2002, 35, 6455–6465 CrossRef CAS.
- J. E. Mark, Acc. Chem. Res., 2004, 37, 946–953 CrossRef CAS PubMed.
- N. Kamatham, O. A. Ibraikulov, P. Durand, J. Wang, O. Boyron, B. Heinrich, T. Heiser, P. Lévêque, N. Leclerc and S. Méry, Adv. Funct. Mater., 2020, 31, 2007734 CrossRef.
- Y. Ding, L. Jiang, Y. Du, S. Kim, X. Wang, H. Lu, G. Zhang, K. Cho and L. Qiu, Chem. Commun., 2020, 7, 11867–11870 RSC.
- Y. Ding, Y. Zhu, X. Wang, Y. Wang, S. Zhang, G. Zhang, X. Gu and L. Qiu, Chem. Mater., 2022, 34, 2696–2707 CrossRef CAS.
- G. Xue, X. Zhao, G. Qu, T. Xu, A. Gumyusenge, Z. Zhang, Y. Zhao, Y. Diao, H. Li and J. Mei, ACS Appl. Mater. Interfaces, 2017, 9, 25426–25433 CrossRef CAS PubMed.
- Z. Wang, Z. Liu, L. Ning, M. Xiao, Y. Yi, Z. Cai, A. Sadhanala, G. Zhang, W. Chen, H. Sirringhaus and D. Zhang, Chem. Mater., 2018, 30, 3090–3100 CrossRef CAS.
- Y. C. Lin, F. H. Chen, Y. C. Chiang, C. C. Chueh and W. C. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 34158–34170 CrossRef CAS PubMed.
- H. C. Yen, Y. C. Lin and W. C. Chen, Macromolecules, 2021, 54, 1665–1676 CrossRef CAS.
- A. Babel and S. A. Jenekhe, Macromolecules, 2004, 37, 9835–9840 CrossRef CAS.
- L. Qiu, X. Wang, W. H. Lee, J. A. Lim, J. S. Kim, D. Kwak and K. Cho, Chem. Mater., 2009, 21, 4380–4386 CrossRef CAS.
- J. I. Scott, X. Xue, M. Wang, R. J. Kline, B. C. Ho, D. Dougherty, C. Zhou, G. Bazan and B. T. O’Connor, ACS Appl. Mater. Interfaces, 2016, 8, 14037–14045 CrossRef CAS PubMed.
- N. Onojima, N. Akiyama, Y. Mori, T. Sugai and S. Obata, Org. Electron., 2020, 78, 105597 CrossRef CAS.
- Q. Y. Yan, Y. W. Shia, D. Y. Guo and W. Y. Lee, Macromol. Res., 2020, 28, 660–669 CrossRef CAS.
- S. G. Lee, H. S. Lee, S. Lee, C. W. Kim and W. H. Lee, Org. Electron., 2015, 24, 113–119 CrossRef CAS.
- J. Xu, S. Wang, G. N. Wang, C. Zhu, S. Luo, L. Jin, X. Gu, S. Chen, V. R. Feig, J. W. F. To, S. Rondeau-gagné, J. Park, B. C. Schroeder, C. Lu, J. Y. Oh, Y. Wang, Y. Kim, H. Yan, R. Sinclair, D. Zhou, G. Xue, B. Murmann, C. Linder, W. Cai, J. B. Tok, J. W. Chung and Z. Bao, Science, 2017, 355, 59–64 CrossRef CAS PubMed.
- F. Ge, S. Wei, Z. Liu, G. Wang, X. Wang, G. Zhang, H. Lu, K. Cho and L. Qiu, ACS Appl. Mater. Interfaces, 2018, 10, 9602–9611 CrossRef CAS PubMed.
- A. R. Aiyar, J. Hong, R. Nambiar, D. M. Collard and E. Reichmanis, Adv. Funct. Mater., 2011, 21, 2652–2659 CrossRef CAS.
- D. Choi, M. Chang and E. Reichmanis, Adv. Funct. Mater., 2015, 25, 920–927 CrossRef CAS.
- G. Zhang, M. Mcbride, N. Persson, S. Lee, T. J. Dunn, M. F. Toney, Z. Yuan, Y. Kwon, P. Chu, B. Risteen and E. Reichmanis, Chem. Mater., 2017, 29, 7645–7652 CrossRef CAS.
- E. Song, B. Kang, H. H. Choi, D. H. Sin, H. Lee, W. H. Lee and K. Cho, Adv. Electron. Mater., 2016, 2, 1500250 CrossRef.
- D. Liu, Z. Ding, Y. Wu, S. F. Liu, Y. Han and K. Zhao, Macromolecules, 2022, 55, 297–308 CrossRef CAS.
- C. J. Ellison and J. M. Torkelson, Nat. Mater., 2003, 2, 695–700 CrossRef CAS PubMed.
- C. M. Stafford, B. D. Vogt, C. Harrison, D. Julthongpiput and R. Huang, Macromolecules, 2006, 39, 5095–5099 CrossRef CAS.
- J. L. Keddie, R. A. L. Jones and R. A. Cory, Europhys. Lett., 1994, 27, 59–64 CrossRef CAS.
- B. Park, H. Kang, Y. H. Ha, J. Kim, J. Lee, K. Yu, S. Kwon, S. Jang, S. Kim, S. Jeong, S. Hong, S. Byun, S. Kwon, Y. Kim and K. Lee, Adv. Sci., 2021, 8, 2100332 CrossRef CAS PubMed.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- J. Dahlke, S. Zechel, M. D. Hager and U. S. Schubert, Adv. Mater. Interfaces, 2018, 5, 1800051 CrossRef.
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 CrossRef CAS.
- L. Laysandra, C. H. Chuang, S. Kobayashi, A. N. Au-Duong, Y. H. Cheng, Y. T. Li, M. M. Mburu, T. Isono, T. Satoh and Y. C. Chiu, ACS Appl. Polym. Mater., 2020, 2, 5432–5443 CrossRef CAS.
- F. Song, Z. Li, P. Jia, M. Zhang, C. Bo, G. Feng, L. Hu and Y. Zhou, J. Mater. Chem. A, 2019, 7, 13400–13410 RSC.
- S. C. Cummings, O. J. Dodo, A. C. Hull, B. Zhang, C. P. Myers, J. L. Sparks and D. Konkolewicz, ACS Appl. Polym. Mater., 2020, 2, 1108–1113 CrossRef CAS.
- J. F. Mei, X. Y. Jia, J. C. Lai, Y. Sun, C. H. Li, J. H. Wu, Y. Cao, X. Z. You and Z. Bao, Macromol. Rapid Commun., 2016, 37, 1667–1675 CrossRef CAS PubMed.
- C. H. Li and J. L. Zuo, Adv. Mater., 2020, 32, 1903762 CAS.
- Y. L. Rao, A. Chortos, R. Pfattner, F. Lissel, Y. C. Chiu, V. Feig, J. Xu, T. Kurosawa, X. Gu, C. Wang, M. He, J. W. Chung and Z. Bao, J. Am. Chem. Soc., 2016, 138, 6020–6027 CrossRef CAS PubMed.
- G. Sinawang, M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Chem. Commun., 2020, 56, 4381–4395 RSC.
- Y. Wang, X. Huang and X. Zhang, Nat. Commun., 2021, 12, 1291 CrossRef CAS PubMed.
- Z. Q. Lei, P. Xie, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2015, 3, 19662–19668 RSC.
- C. Mo, L. Xiang and Y. Chen, Macromol. Rapid Commun., 2021, 42, 2100025 CrossRef CAS PubMed.
- C. Ouyang, C. Zhao, W. Li, X. Wu, X. Le, T. Chen, W. Huang, Q. Gao, X. Shan, R. Zhg and W. Zhang, Macromol. Mater. Eng., 2020, 305, 2000089 CrossRef CAS.
- Y. Li, L. Yang, Y. Zeng, Y. Wu, Y. Wei and L. Tao, Chem. Mater., 2019, 31, 5576–5583 CrossRef CAS.
- S. Shan, D. Mai, Y. Lin and A. Zhang, ACS Appl. Polym. Mater., 2021, 3, 5115–5124 CrossRef CAS.
- R. P. Wool and K. M. O’Connor, J. Appl. Phys., 1981, 52, 5953 CrossRef CAS.
- R. P. Wool, Soft Matter, 2008, 4, 400–418 RSC.
- X. Chen, J. Zhu, Y. Luo, J. Chen, X. Ma, D. Bukhvalov, H. Liu, M. Zhang and Z. Luo, Phys. Chem. Chem. Phys., 2020, 22, 17620–17631 RSC.
- J. Kang, D. Son, G. J. N. Wang, Y. Liu, J. Lopez, Y. Kim, J. Y. Oh, T. Katsumata, J. Mun, Y. Lee, L. Jin, J. B. H. Tok and Z. Bao, Adv. Mater., 2018, 30, 1706846 CrossRef PubMed.
- Y. L. Liu and T. W. Chuo, Polym. Chem., 2013, 4, 2194–2205 RSC.
- Y. He, S. Liao, H. Jia, Y. Cao, Z. Wang and Y. Wang, Adv. Mater., 2015, 27, 4622–4627 CrossRef CAS PubMed.
- V. K. Thakur and M. R. Kessler, Polymer, 2015, 69, 369–383 CrossRef CAS.
- M. U. Ocheje, M. Selivanova, S. Zhang, T. H. Van Nguyen, B. P. Charron, C. H. Chuang, Y. H. Cheng, B. Billet, S. Noori, Y. C. Chiu, X. Gu and S. Rondeau-Gagné, Polym. Chem., 2018, 9, 5531–5542 RSC.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. Bao, Nat. Chem., 2016, 8, 618–624 CrossRef CAS PubMed.
- S. Zhang, Y. H. Cheng, L. Galuska, A. Roy, M. Lorenz, B. Chen, S. Luo, Y. T. Li, C. C. Hung, Z. Qian, P. B. J. St. Onge, G. T. Mason, L. Cowen, D. Zhou, S. I. Nazarenko, R. F. Storey, B. C. Schroeder, S. Rondeau-Gagné, Y. C. Chiu and X. Gu, Adv. Funct. Mater., 2020, 30, 2000663 CrossRef CAS.
- S. Zhang and F. Cicoira, Adv. Mater., 2017, 29, 1703098 CrossRef PubMed.
- S. Zhang, Y. Li, G. Tomasello, M. Anthonisen, X. Li, M. Mazzeo, A. Genco, P. Grutter and F. Cicoira, Adv. Electron. Mater., 2019, 5, 1900191 CrossRef.
- Y. Li, S. Zhang, X. Li, V. R. N. Unnava and F. Cicoira, Flex. Print. Electron., 2019, 4, 044004 CrossRef CAS.
- Y. Li, S. Zhang, N. Hamad, K. Kim, L. Liu, M. Lerond and F. Cicoira, Macromol. Biosci., 2020, 20, 2000146 CrossRef CAS PubMed.
- Y. Li, X. Li, S. Zhang, L. Liu, N. Hamad, S. R. Bobbara, D. Pasini and F. Cicoira, Adv. Funct. Mater., 2020, 30, 2002853 CrossRef CAS.
- X. Zhou, A. Rajeev, A. Subramanian, Y. Li, N. Rossetti, G. Natale, G. A. Lodygensky and F. Cicoira, Acta Biomater., 2022, 139, 296–306 CrossRef CAS PubMed.
- Y. Li, X. Zhou, B. Sarkar, N. Gagnon-Lafrenais and F. Cicoira, Adv. Mater., 2022, 34, 2108932 CrossRef CAS PubMed.
- H. Chen, Y. Guo, G. Yu, Y. Zhao, J. Zhang, D. Gao, H. Liu and Y. Liu, Adv. Mater., 2012, 24, 4618–4622 CrossRef CAS PubMed.
- Y. Zhao, X. Zhao, M. Roders, A. Gumyusenge, A. L. Ayzner and J. Mei, Adv. Mater., 2017, 29, 1605056 CrossRef PubMed.
- Z. Qian, Z. Cao, L. Galuska, S. Zhang, J. Xu and X. Gu, Macromol. Chem. Phys., 2019, 220, 1900062 CrossRef.
- Y. Li, W. K. Tatum, J. W. Onorato, S. D. Barajas, Y. Y. Yang and C. K. Luscombe, Polym. Chem., 2017, 8, 5185–5193 RSC.
- H. Wen, H. Wu, J. Aimi, C. Hung, Y. Chiang, C. Kuo and W. Chen, Macromolecules, 2017, 50, 4982–4992 CrossRef CAS.
- M. Shin, J. Y. Oh, K.-E. Byun, Y.-J. Lee, B. Kim, H.-K. Baik, J.-J. Park and U. Jeong, Adv. Mater., 2015, 27, 1255–1261 CrossRef CAS PubMed.
- F. Sugiyama, A. T. Kleinschmidt, L. V. Kayser, M. A. Alkhadra, J. M. Wan, A. S. Chiang, D. Rodriquez, S. E. Root, S. Savagatrup and D. J. Lipomi, Macromolecules, 2018, 51, 5944–5949 CrossRef CAS PubMed.
- H. Wu, C. Hung, C. Hong, H. Sun, J. Wang, G. Yamashita, T. Higashihara and W. Chen, Macromolecules, 2016, 49, 8540–8548 CrossRef CAS.
- M. J. Powell, IEEE Trans. Electron Devices, 1989, 36, 2753–2763 CrossRef CAS.
- C.-S. Yang, L. L. Smith, C. B. Arthur and G. N. Parsons, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2000, 18, 683 CrossRef CAS.
- K. H. Cherenack, A. Z. Kattamis, B. Hekmatshoar, J. C. Sturm and S. Wagner, Mater. Res. Soc. Symp. Proc., 2008, 1066, 10662003 CrossRef.
- D. Son, J. Kang, O. Vardoulis, Y. Kim, N. Matsuhisa, J. Y. Oh, J. W. To, J. Mun, T. Katsumata, Y. Liu, A. F. McGuire, M. Krason, F. Molina-Lopez, J. Ham, U. Kraft, Y. Lee, Y. Yun, J. B. H. Tok and Z. Bao, Nat. Nanotechnol., 2018, 13, 1057–1065 CrossRef CAS PubMed.
- B. C. K. Tee, C. Wang, R. Allen and Z. Bao, Nat. Nanotechnol., 2012, 7, 825–832 CrossRef CAS PubMed.
- J. Zhao, L. Sun, Z. Chu, T. Li, F. Zhang, L. Li and W. Zhang, AIP Adv., 2020, 10, 035020 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |