
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lignin alkaline oxidation using reversibly-soluble bases†
Jacob S.
Kruger
 a,
Reagan J.
Dreiling
a,
Daniel G.
Wilcox
a,
Arik J.
Ringsby
a,
Katherine L.
Noon
a,
Camille K.
Amador
a,
David G.
Brandner
a,
Kelsey J.
Ramirez
a,
Stefan J.
Haugen
a,
Bruno C.
Klein
b,
Ryan
Davis
b,
Rebecca J.
Hanes
b,
Renee M.
Happs
a,
Nicholas S.
Cleveland
a,
Earl D.
Christensen
b,
Joel
Miscall
a and
Gregg T.
Beckham
*a
a,
Reagan J.
Dreiling
a,
Daniel G.
Wilcox
a,
Arik J.
Ringsby
a,
Katherine L.
Noon
a,
Camille K.
Amador
a,
David G.
Brandner
a,
Kelsey J.
Ramirez
a,
Stefan J.
Haugen
a,
Bruno C.
Klein
b,
Ryan
Davis
b,
Rebecca J.
Hanes
b,
Renee M.
Happs
a,
Nicholas S.
Cleveland
a,
Earl D.
Christensen
b,
Joel
Miscall
a and
Gregg T.
Beckham
*a
aRenewable Resources and Enabling Sciences Center, National Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, CO 80401, USA. E-mail: Gregg.Beckham@nrel.gov
bCatalytic Carbon Transformation and Scale-Up Center, National Renewable Energy Laboratory, 15013 Denver West Parkway, Golden, CO 80401, USA
First published on 26th October 2022
Abstract
Lignin valorization approaches, which are critical to biorefining, often involve depolymerization to aromatic monomers. Alkaline oxidation has long held promise as a lignin depolymerization strategy, but requires high concentrations of base, typically NaOH, much of which must be neutralized to recover lignin-derived aromatic monomers. This consumption of base and associated waste generation incurs high cost and negative environmental impacts. In this work, we demonstrate that Sr(OH)2 and Ba(OH)2 perform comparably to NaOH in terms of total aromatic monomer yields in the aqueous aerobic alkaline depolymerization of corn stover lignin, and that up to 90% of these reversibly-soluble bases can be recovered via precipitation and filtration. Process modeling suggests that the use of Sr(OH)2 could reduce the cost of alkaline oxidation by 20–60% compared to NaOH, depending on lignin loading. In contrast, the energy required to regenerate the Sr largely offsets potential improvements in sustainability over Na-promoted alkaline oxidation, though the sustainability comparison is likely sensitive to the lignin composition and could be improved by further optimization of the regeneration step.
Introduction
Lignin is a heterogeneous macromolecule consisting of linked phenylpropanoid units and typically comprises 15–30% of lignocellulosic biomass.1,2 Currently, most lignin isolated in biomass processing (e.g., in paper mills) is burned on-site for process heat. However, biorefinery configurations targeting biofuels production from carbohydrates would benefit from conversion of lignin to valuable products to supplement fuel production costs.3–7 In multiple biorefinery concepts, the generation of coproducts from lignin involves depolymerization to aromatic monomers that can be isolated or further upgraded. Many depolymerization approaches have been explored, including oxidative, reductive, solvolytic, acidic, basic, electrochemical, thermochemical, and mechanochemical methods.8–14 Many of the various depolymerization approaches target cleavage of the lignin polymer primarily at β-O-4′ and ester linkages.The different depolymerization approaches also yield very different product distributions.12 In particular, oxidative approaches generate primarily phenolic aldehydes, acids, and ketones, as well as aliphatic acids, which are among the highest-value products available directly from lignin today.9,15 Aromatic aldehydes can be used as flavorings and fragrances,16 intermediates in the synthesis of fine chemicals,17 corrosion inhibitors,18 and monomers for bio-based polymers.19–21 Similarly, some microorganisms are capable of converting aromatic monomers into target products.22–25 These aldehydes and other biologically-available monomers are readily available from oxidative depolymerization.23,26–29
Among oxidative depolymerization strategies, alkaline aerobic oxidation is one of the most studied.30–48 Total monomer yields depend on the type of lignin employed, approaching 30 wt% from hardwood lignin.47 Typical reaction conditions include temperatures of 130–190 °C, reaction times of 10–180 min, oxygen partial pressures of 2–5 bar, and 2 M aqueous NaOH, though other conditions have been explored. Many catalysts have been studied, typically targeting vanillin production.35,40
Although monomer yields of 30 wt% are promising, a major limitation of alkaline aerobic oxidation is the use of 2 M NaOH, which typically represents a large base excess. However, the excess is necessary to mitigate overoxidation of aromatic monomers34,47,49–51 and to promote deprotonation and substitution reactions that are part of the β-O-4′ cleavage mechanism.38,39 While depolymerization requires highly alkaline conditions, recovery of phenolic monomers often requires pH ≤ 7.16 Thus, the excess of base required for high monomer yields is mostly wasted during neutralization and product recovery, which can significantly challenge process economics and sustainability except in certain sulfite pulping configurations. With these configurations, lignin-derived products feature prominently in the biorefining strategy, and the availability of SO2 greatly facilitates product recovery.17,35,52,53
Some researchers have attempted to replace NaOH with less-soluble alternatives. The Ontario Paper Company explored the use of Ca(OH)2 as an alternative base, but achieved relatively low yields of vanillin, even at very high Ca(OH)2 loadings.49 Similarly, Ca(OH)2 and Mg(OH)2 have been explored for biomass pretreatment,54,55 but the lignin was reported to degrade mainly to aliphatic acids. It is important to note that delignification, rather than monomer generation was the primary goal of these studies. Similarly, NH3 can function as an easily-removable base to promote lignin extraction and depolymerization, though a fraction of the NH3 often becomes chemically bonded to the lignin, potentially impacting downstream applications.46,56–60
Solubility profiles of the alkaline earth hydroxides change substantially with increasing molecular weight. While Mg(OH)2 and Ca(OH)2 are relatively insoluble at room temperature and decrease in solubility with increasing temperature, Sr(OH)2 and Ba(OH)2 exhibit an opposite trend,61 wherein their slight solubilities at room temperature increase significantly with increasing temperature. In particular, Ba(OH)2 becomes soluble to ∼2 M at 80 °C, and Sr(OH)2 becomes soluble to ∼2 M at 95 °C.61 We hypothesized that, due to these trends, it would be feasible to obtain the high hydroxide ion concentrations needed for high monomer yields in the alkaline oxidation of lignin at typical reaction temperatures, while maintaining the benefits of a mostly insoluble hydroxide at room temperature. In this way, it could be possible to recover the excess base by filtration. Furthermore, the small amount of residual base in solution should be amenable to neutralization with CO2, precipitating SrCO3 or BaCO3 that could also be filtered and regenerated to the hydroxide, and thus leave a neutral-pH, low-salt monomer solution for separations or further upgrading.16,27,62,63 The envisioned process is illustrated in Fig. 1 and Fig. S1.†
 | ||
| Fig. 1 Envisioned lignin upgrading process using reversibly-soluble alkaline earth metal hydroxides, such as Sr(OH)2 and Ba(OH)2. This process is compared to other lignin upgrading strategies in Fig. S1.† | ||
Towards this concept, in this study we explored the use of alkaline earth hydroxides as reversibly-soluble bases for alkaline oxidation of lignin, including a direct comparison to NaOH, recovery and regeneration of the hydroxides, and techno-economic analysis (TEA) of the envisioned lignin depolymerization process. We also performed a life cycle assessment (LCA) to compare global warming potential (GWP), cumulative energy demand (CED), and eco-toxicity of solubilized lignin from reversibly-soluble bases and from NaOH.
Results and discussion
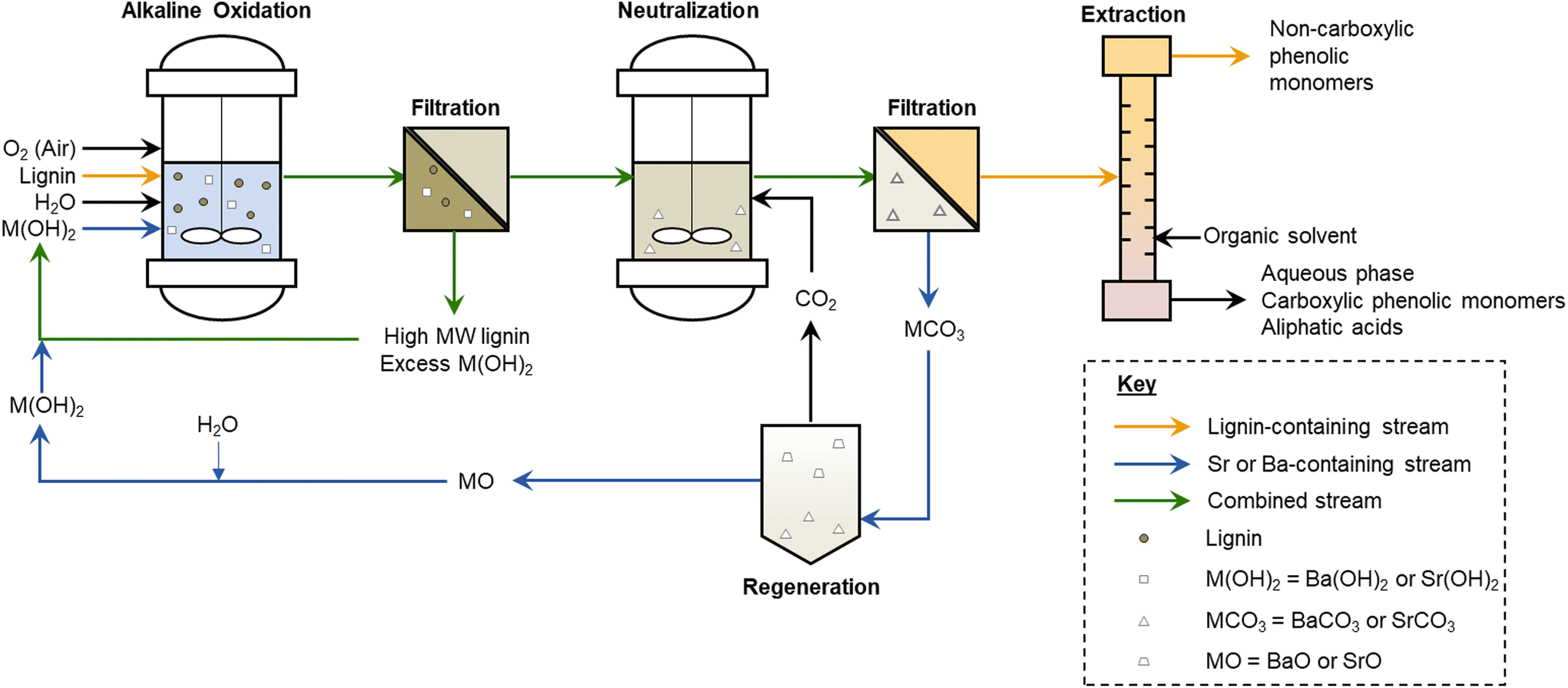
We first compared the performance, measured by aromatic monomer yields, between NaOH, Ba(OH)2, and Sr(OH)2. Initial experiments demonstrated that Ba(OH)2 and Sr(OH)2 perform almost equivalently to NaOH in terms of total monomer yield during alkaline oxidation of native corn stover, as shown in Fig. 2 and Table S1.† Each base generated ∼20 wt% monomer yield from the native lignin. Conversely, Ca(OH)2, which exhibits limited solubility at both room and reaction temperature, produced only 5% monomer yield. We note that hydroxycinnamates are included in the monomer yields because they are valuable aromatic compounds that are released during lignin depolymerization and are counted as lignin in our compositional analysis of the feedstocks, though their origin may not be entirely from the lignin polymer.64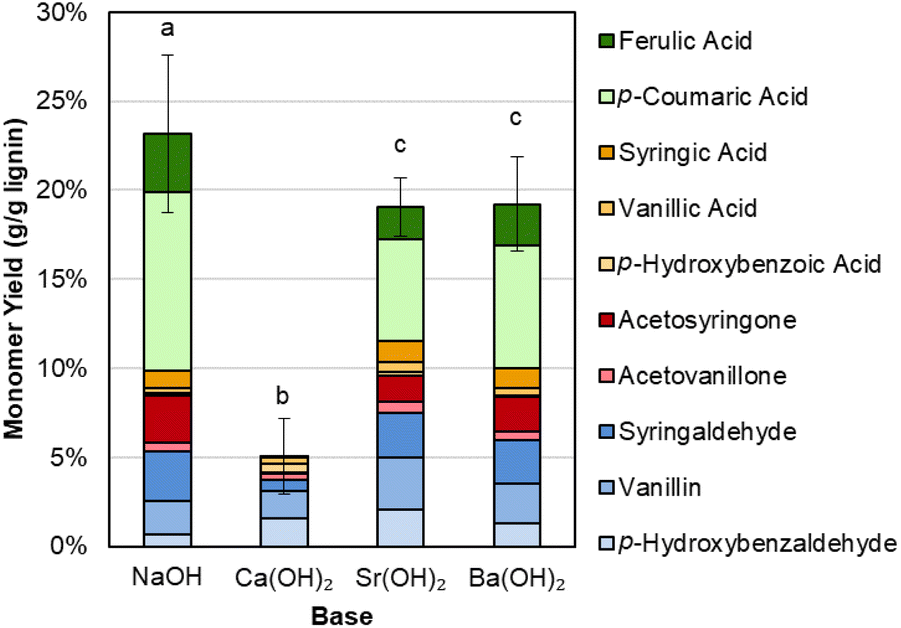 | ||
| Fig. 2 Comparison of monomeric aromatic compound yields from alkaline oxidation of native corn stover. Reaction conditions: 1 g corn stover, 30 mL water, 2 M hydroxide, 175 °C, 10 min reaction time, and air added at reaction temperature to generate 30 bar total pressure (∼22 bar air). Error bars represent the total monomer yield standard deviation of replicate experiments. Letters indicate statistically different means by t-test (α = 0.05). Numerical data for this figure are included in Table S1.† | ||
Interestingly, there is a difference in selectivity between Na, Ba, and Sr hydroxides. NaOH generates a monomer stream with a higher proportion of p-coumaric acid and a lower proportion of aldehydes, while Ba(OH)2 and Sr(OH)2 each yielded less p-coumaric acid and more of the phenolic aldehydes. We further probed this phenomenon by using p-coumaric acid and ferulic acid as starting materials under similar conditions with Sr(OH)2.We determined that Sr(OH)2 is capable of converting the hydroxycinnamates to a greater degree than NaOH, though the yield to the corresponding aldehyde is higher for NaOH (Fig. 3 and Table S2†). It is possible that Sr(OH)2 initially forms insoluble strontium dicoumarate or diferulate salts, which may be less accessible to oxidation. Indeed, while the recovery of p-coumaric acid and ferulic acid is overall higher for NaOH, it decreases after 10 minutes under oxidizing conditions, while the total recovery is only ∼15% even before adding O2, but constant under oxidizing conditions for Sr(OH)2. The higher aldehyde yields also do not appear to be due to some protecting feature of NaOH on the hydroxycinnamates, as recovery of vanillin and p-hydroxybenzaldehyde was the same for both Sr(OH)2 and NaOH at 0 min and 10 min reaction times (Fig. S2 and Table S3†). Finally, Sr(OH)2 produces slightly higher yields to G- and H-type monomers and slightly lower yields to S-type monomers, suggesting that Sr(OH)2 may promote demethoxylation slightly more than NaOH does.
 | ||
| Fig. 3 Recovery of p-coumaric acid and ferulic acid in the presence of NaOH or Sr(OH)2. Reaction conditions: 0.3 g p-coumaric (left four bars) or ferulic acid (right four bars), 30 mL water, 2 M hydroxide, 175 °C. For 10 min reaction times, air was added at reaction temperature to generate 30 bar total pressure (∼22 bar air). Error bars represent the total monomer yield range of duplicate experiments. Numerical data for this figure are included in Table S2.† | ||
Gel Permeation Chromatography (GPC) analysis of the residual solids after reaction also showed similar effects for NaOH, Sr(OH)2, and Ba(OH)2; however, Ca(OH)2 was not as effective for depolymerization, as indicated by the less prominent low-molecular weight features. The GPC traces are shown in Fig. S3.†
While both Sr(OH)2 and Ba(OH)2 appear to be effective and equivalent substitutes for NaOH in the alkaline oxidation of lignin, Sr(OH)2 may be favorable to Ba(OH)2 during base recycle. Specifically, the regeneration of SrCO3 to SrO (which then re-forms the hydroxide on addition of H2O) can occur at 800–900 °C, while the regeneration of BaCO3 to BaO requires temperatures over 1000 °C (Fig. S4†), allowing for less energy consumption in the overall biorefinery when Sr(OH)2 is used. Thus, we focused on Sr(OH)2 for the remainder of the experiments.
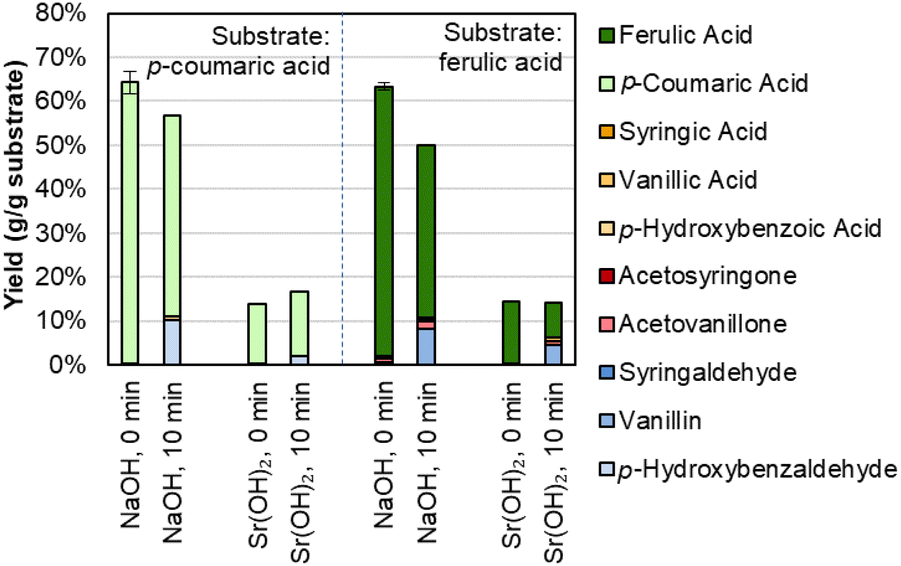
Aromatic monomer yield from native lignin represents a useful measure of overall lignin reactivity, but lignin available in a biorefinery is likely to be isolated from whole biomass. Given the importance of lignin for biorefinery economics, fractionation approaches that maintain lignin in a native-like state are essential. One reported approach to that end is the deacetylation, mechanical refining, and enzymatic hydrolysis (DMR-EH) process.5,65 This process uses a mild alkaline deacetylation followed by milling to enhance enzymatic hydrolysis, and effectively fractionates feedstock lignin into a soluble portion during deacetylation and an insoluble portion recovered in a solid residue after enzymatic hydrolysis. 2D HSQC NMR spectroscopy of the parent stover and isolated lignins shows features corresponding to β-O-4 and ester linkages, confirming the native-like structure of the lignins (Fig. S5†). Monomer yields from these two fractions and the native lignin in the parent corn stover, relative to the lignin content of each substrate, are shown in Fig. 4 and Table S4.†
 | ||
| Fig. 4 Lignin monomer yields from corn stover, deacetylation black liquor, and the residue obtained after deacetylation, mechanical refining, and enzymatic hydrolysis (DMR-EH). Reaction conditions: 1 g corn stover or 0.3 g lignin-rich substrate, 30 mL water, 1 M Sr(OH)2, 175 °C. For 10 min reaction times, air was added at reaction temperature to generate 30 bar total pressure (∼22 bar air). Error bars represent the total monomer yield range of at least duplicate experiments. Letters indicate statistically different means by t-test (α = 0.05), specific to each time point. Numerical data for this figure are included in Table S4.† | ||
The yield from parent corn stover lignin lies between that of the deacetylation black liquor lignin and the DMR-EH residue lignin, highlighting both the low level of deleterious reactions in the lignin polymer that occur in the DMR-EH process, and the ability of DMR-EH processing to fractionate the lignin into a more reactive and a less reactive fraction. For comparison, depolymerization of the DMR-EH residue by reductive catalytic fractionation (RCF), which provides a measure of the maximum monomer yield achievable by cleavage of ester and ether bonds,66 produces a 15.3 wt% monomer yield. Notably, most of the p-coumaric and ferulic acids are solubilized during deacetylation and are present as the free acids in the black liquor. This feature provides an opportunity to separate these monomers prior to oxidation, which may increase the total monomer yield available from biomass in the DMR-EH process.67,68 In the absence of O2, degradation of the hydroxycinnamates is slower and monomers other than the hydroxycinnamates are produced in lower yields (Fig. S6 and Table S5†), consistent with previous reports that these monomers are released under alkaline conditions even without oxygen present.69
As shown in Table S6,† the DMR-EH residue (and the parent corn stover) contains significant carbohydrates. These carbohydrates are mainly cellulose in the DMR-EH residue, but monomeric glucose may also be present from residual hydrolysate. Thus, we were motivated to explore the behavior of these carbohydrates under alkaline oxidation conditions with Sr(OH)2. As shown in Fig. 5 and Table S7,† glucose is primarily converted to lactic acid in both oxidative and nonoxidative conditions, while cellulose remains largely intact. The conversion of glucose to lactic acid likely occurs by retro-aldol reaction.70
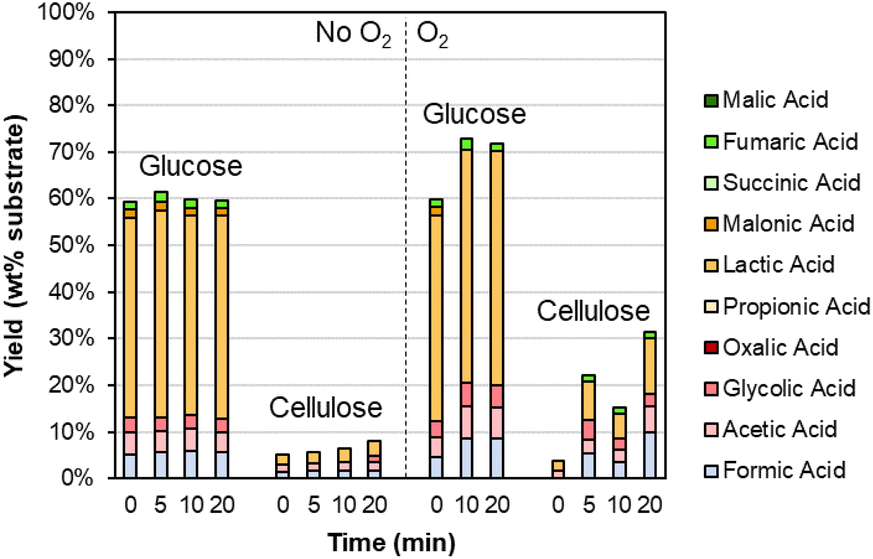 | ||
| Fig. 5 Degradation of carbohydrates during alkaline oxidation of lignin using Sr(OH)2 as a reversibly soluble base. Reaction conditions: 0.3 g carbohydrate, 30 mL water, 1 M Sr(OH)2, 175 °C. For reactions with O2, the reactor was pressurized to 30 bar total pressure with air when reactor reached 175 °C (∼22 bar air). Numerical data for this figure are included in Table S7.† | ||
A critical component of the reversibly-soluble bases concept is the recovery and reuse of the base. After filtration of the excess base, a small amount of base remains in the filtrate. We hypothesized that this base could be recovered simultaneously with neutralization of the filtrate by using CO2 to precipitate SrCO3. Using 35 bar CO2, the neutralization reaction equilibrates within roughly 5 min, during which time the pH of the filtrate decreases from pH 13 to pH 6 (Fig. S7†). Under similar conditions, Ca(OH)2 and Ba(OH)2 also produce a solution of pH 6–6.5, while NaOH produces a solution of pH 3.5. This difference in pH range also has implications in the extraction of monomers. In particular, pH 6–7 is ideal for selective monomer extraction, as it allows recovery of compounds with phenolic groups, which have pKa values in the range of 7.5–9 and are not significantly ionized below pH 7, while leaving compounds with carboxylic acid groups, which have pKa values in the range of 3–4.5 and are ionized, in the aqueous phase. Additionally, biological upgrading is often optimal at pH ∼7 for many microbes studied to date for this purpose.22,24,71,72
In the presence of lignin products, the color of the solution also changes from golden yellow to clear during neutralization, likely indicating some precipitation of oligomeric lignin. This observation is also supported by GPC. However, monomers are not precipitated, as shown in Fig. S8, S9 and Table S8.† That is, monomer yields remain at ∼15% through the entire workup.
The Sr(OH)2 can also be generated from calcination of SrCO3 to SrO, and in situ rehydration of SrO to Sr(OH)2, as shown in Fig. 6 and Table S9.† In contrast, when SrCO3 is used directly or no base is added, monomer yields are significantly lower.
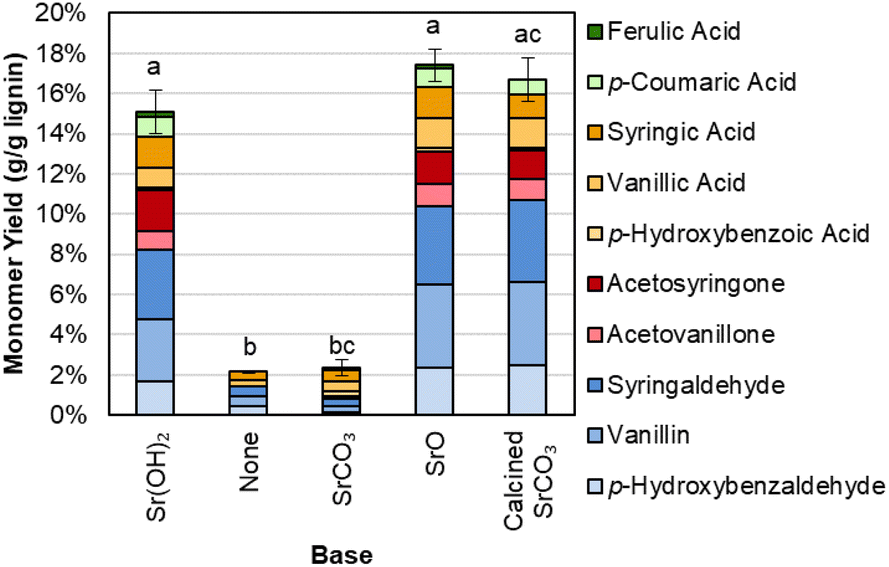 | ||
| Fig. 6 Monomer yields from DMR-EH lignin using Sr(OH)2 from different sources. Reaction conditions: 0.3 g DMR-EH lignin, 30 mL water, 1 M Sr except for biomass-only control, 175 °C, 10 min reaction time. Reactor was pressurized to 30 bar total pressure with air when reactor reached 175 °C (∼22 bar air). Error bars represent the total monomer yield range of at least duplicate experiments. Letters indicate statistically different means by t-test (α = 0.05). Numerical data for this figure are included in Table S9.† | ||
Total Sr recovery exceeds 90% from depolymerized solutions and 98% in the absence of biomass, as shown in Table 1. The difference is likely due to the presence of Sr carboxylate salts, such as strontium acetate, which limit the extent of SrCO3 precipitation. It is important to note that the solid recovered after reaction is primarily Sr(OH)2, but also contains residual biomass solids and SrCO3 formed during the reaction by a reaction of CO2 from overoxidation of the substrate with Sr(OH)2. Under the current experimental conditions, 0.3 g substrate and 7.97 g Sr(OH)2·8H2O are added to the reactor. Thus, the presence of residual substrate could lead to a maximum 7.6% overestimate in the recovery of the Sr(OH)2, but the overestimate is likely lower due to dissolution of the substrate during the reaction. These values are largely consistent with measurement of Sr remaining in solution after each step measured by inductively coupled plasma optical emission spectroscopy (ICP-OES), which showed 14% of the Sr in solution after the initial cooling and 5.9% of the Sr in solution after precipitation with CO2.
| Mode | Recovery as Sr(OH)2 | Recovery as SrCO3 | Total recovery |
|---|---|---|---|
| a Reaction conditions: 0.3 g DMR-EH lignin, 30 mL water, 1 M Sr(OH)2, 175 °C, 10 min reaction time. Reactor was pressurized to 30 bar total pressure with air when reactor reached 175 °C (∼22 bar air). Recovery as SrCO3 calculated after filtering Sr(OH)2 and any residual biomass, and precipitating Sr in solution with 30 bar CO2 at room temperature for 10 min. | |||
| Without biomass | 90.7% | 8.2% | 98.9% |
| With biomassa | 91.7% | 2.7% | 94.5% |
The recovered Sr(OH)2 can also be recycled directly, as shown in Fig. 7 and Table S10.† In the second and third cycles, the monomer yield is slightly increased compared to the first cycle, likely due to the presence of residual lignin in the recycled Sr(OH)2 (similar to the residual lignin signals in GPC, Fig. S3†). This feature, in combination with the slightly higher RCF yield produced from the DMR-EH residue lignin, indicates that the alkaline oxidation conditions employed here may be improved by further optimization. Additionally, there is likely some SrCO3 in the recycled solids, formed by reaction of soluble Sr with CO2 from overoxidation. We note that accumulation of both residual organic solids and SrCO3 can be mitigated by periodic feeding of the post-reaction solids into the regeneration step, where SrCO3 will be converted to SrO and combustion of the organics can provide process heat, lowering the natural gas demand for the regeneration step. Alternatively, the organic solids may potentially be valorized by further functionalization (selective oxidation, sulfonation, or nitration)73 or direct use as, e.g., a concrete plasticizer.74
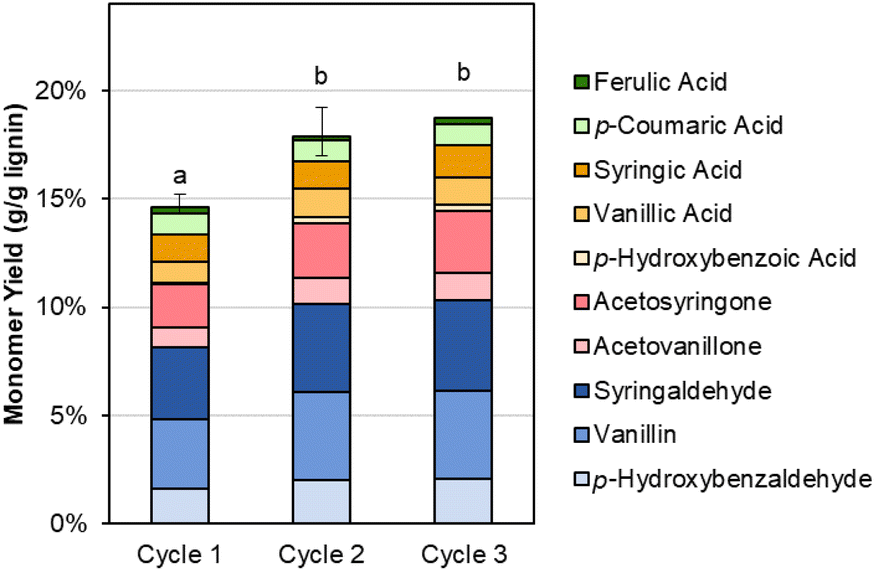 | ||
| Fig. 7 Monomer yields from DMR-EH lignin using virgin (cycle 1) and recycled (cycles 2–3) Sr(OH)2. Reaction conditions: 0.3 g DMR-EH lignin, 30 mL water, 1 M Sr(OH)2, 175 °C, 10 min reaction time. For cycles 2 and 3, the recovered Sr(OH)2 was dried to anhydrous form and loaded directly. The reactor was pressurized to 30 bar total pressure with air when reactor reached 175 °C (∼22 bar air). Error bars represent the total monomer yield range of at least duplicate experiments. Letters indicate statistically different means by t-test (α = 0.05). Numerical data for this figure are included in Table S10.† | ||
While all of the critical components of the reversibly-soluble bases concept have been demonstrated, the concentration of lignin at 10 g L−1 in the reaction conditions is too low for economic viability. Thus, we were interested in the effects of lignin concentration. Substrate concentrations likely need to be at least 100 g L−1 to avoid excess energy consumption for heating and transferring solvent, so we selected this concentration to eliminate one optimization variable. Similarly, previous work has shown that peak monomer yields increase with increasing hydroxide concentration, while concentrations above 2 M can lead to operational issues in the reactor17 and faster monomer degradation.31 We thus selected 2 M hydroxide (1 M Sr(OH)2) as the concentration, and optimized for temperature, time, and pressure using initially a three-factor, three-level design of experiments, analogous to the approach taken by Cao et al.75 As shown in Fig. 8 and Table S11,† at these higher concentrations, optimal conditions of approximately 160 °C, 60 bar air, and 30 min lead to total monomer yields of 4.9 wt%. The optimal conditions likely occur where the rate of monomer generation is maximized while the rate of monomer degradation is minimized. Thus, when the pressure is only 30 bar, the monomer generation rate is too slow relative to monomer degradation, while at 90 bar, monomers are degraded too quickly in our reaction system.
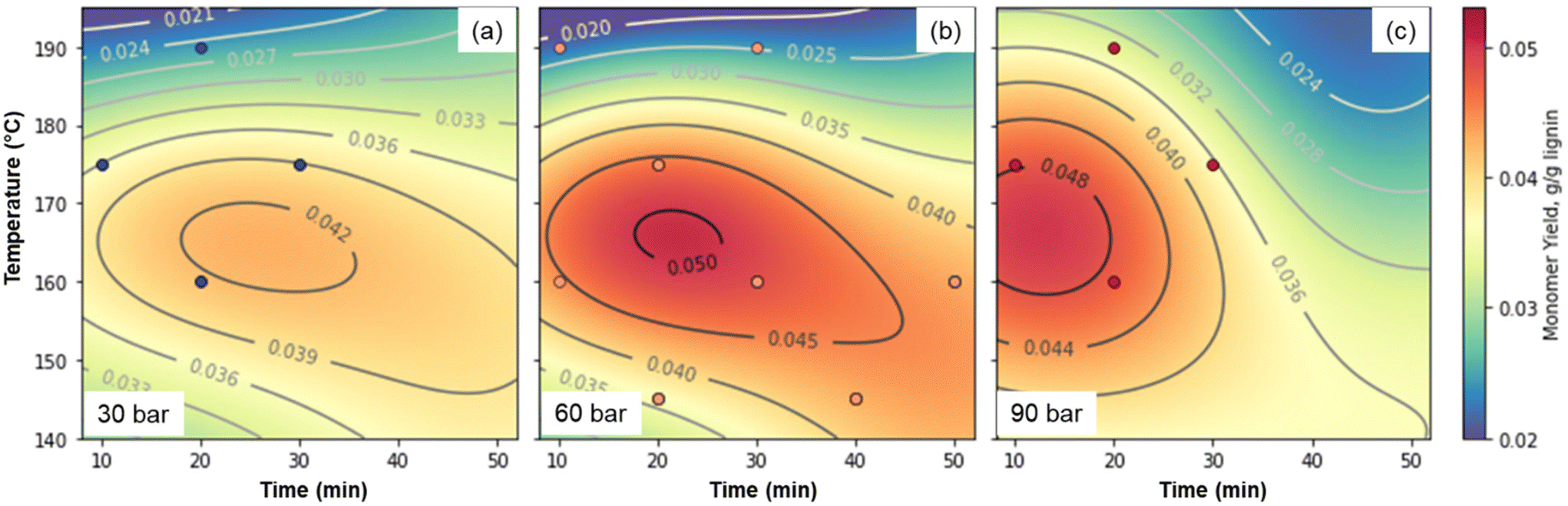 | ||
| Fig. 8 Optimization of Sr(OH)2-promoted alkaline oxidation of DMR-EH lignin. (a). 30 bar total pressure. (b). 60 bar total pressure. (c). 90 bar total pressure. The machine learning fit used to generate the contour plots is shown in Fig. S10† and the numerical data corresponding to the monomer yields are provided in Table S11.† | ||
Because the highest yielding point was near one of the extremes of the model, we added three additional points, which showed that the highest-yielding point in the initial design was indeed close to the optimum. For comparison, the monomer yield using NaOH as the alkali source under the same conditions that yielded 7.9% monomers. The difference is largely due to higher yields of hydroxycinnamates and S-type monomers with NaOH, as observed in Fig. 2 above. These values are considerably lower than the monomer yields reported above for the lower concentration of lignin, but as discussed below, the higher substrate concentration has significant economic and environmental advantages, despite the lower yields. As a comparison, industrial production of vanillin from softwood lignin using NaOH and Cu-based catalysts has been reported as high 15–20 wt% from sulfite pulping liquor,76,77 but more recently up to 8.0 wt% from lignosulfonates.35 However, we note that yields are highly dependent on the lignin source (biomass and isolation procedure),45 and while some authors have noted that the use of Cu catalysts increases monomer yields by up to a factor of two,45 other researchers have noted that the addition of Cu catalysts does not increase the maximum monomer yield achievable through alkaline oxidation.47
To facilitate economic and sustainability comparisons, we adapted process models from Davis et al.5 using the system boundaries shown in Fig. S11† and parameters described in Tables S12–S14.† We note that the process models do not include downstream monomer separations or valorization, which could provide additional benefits for Sr(OH)2 over NaOH. Combining the monomer yields and Sr recovery from the optimized conditions into a process model for techno-economic analysis (TEA) and life cycle assessment (LCA) suggests that at the monomer yields and Sr recovery level reported above for the 10 g L−1 lignin loading, the use of Sr(OH)2 will be economically advantaged over NaOH by at least 60%, from $34 per kg monomers to $13 per kg monomers, despite the higher cost of Sr(OH)2 and the additional capital and operating costs of the recycle operations, as shown for scenarios I, II, and III in Fig. 9 and Table S15.† For context, the selling price of synthetic vanillin is typically $10–20 per kg.78 The processing cost of the lignin monomers is decreased substantially further when moving from 10 g L−1 to 100 g L−1 of the DMR-EH substrate, though the higher relative monomer yield for NaOH (7.9 wt%) compared to Sr(OH)2 (4.9 wt%) at this lignin loading offsets some of the advantage of Sr(OH)2 (scenarios IV, V, and VI in Fig. 9). The importance of monomer yield on the monomer selling price is further highlighted in Table S16,† which shows that suboptimal monomer yield at lower pressure and reaction time, which generally correlate with lower reactor capital costs, results in an increased monomer price. It is worth noting that for other types of biomass with a lower relative abundance of hydroxycinnamate monomers (e.g., softwoods), the yields would likely be more similar. Conversely, the recycle of CO2 released during regeneration of SrCO3 to SrO has a relatively minor economic impact (scenarios II and III, V and VI in Fig. 9).
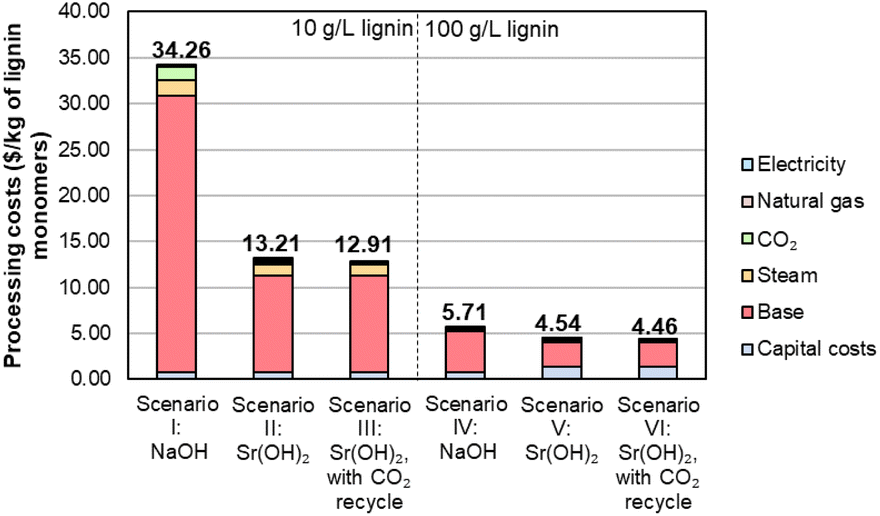 | ||
| Fig. 9 Economic analysis of lignin monomer production by alkaline oxidation promoted by Sr(OH)2 compared to NaOH. Numerical data for this figure are included in Table S15.† | ||
Similar improvements in moving to the higher concentration are observable in the LCA results, as shown in Fig. 10 and Table S17,† though the sustainability benefits are relatively minor for Sr(OH)2 compared to NaOH. Replacing NaOH with Sr(OH)2 and implementing a CO2 recycle stream decreases global warming potential (GWP, Fig. 10a) by almost 45% and cumulative energy demand (CED, Fig. 10b) by 36% at the 10 g L−1 lignin loading, but ecotoxicity (Fig. 10c) is increased by 67% (scenarios I and III) At the 100 g L−1 lignin loading, GWP is decreased by only 3.7% while both CED and ecotoxicity metrics favor NaOH even with CO2 recycle because of the higher relative monomer yield when using NaOH (scenarios IV and VI). These results suggest that the Sr regeneration step is a significant energy driver and that the sustainability of replacing NaOH with Sr(OH)2 could improve with optimization of this operation. It is worth noting that the ecotoxicity impacts of Sr are considerably larger than Na, mainly due to the extremely low toxicity of Na (Fig. 10c).
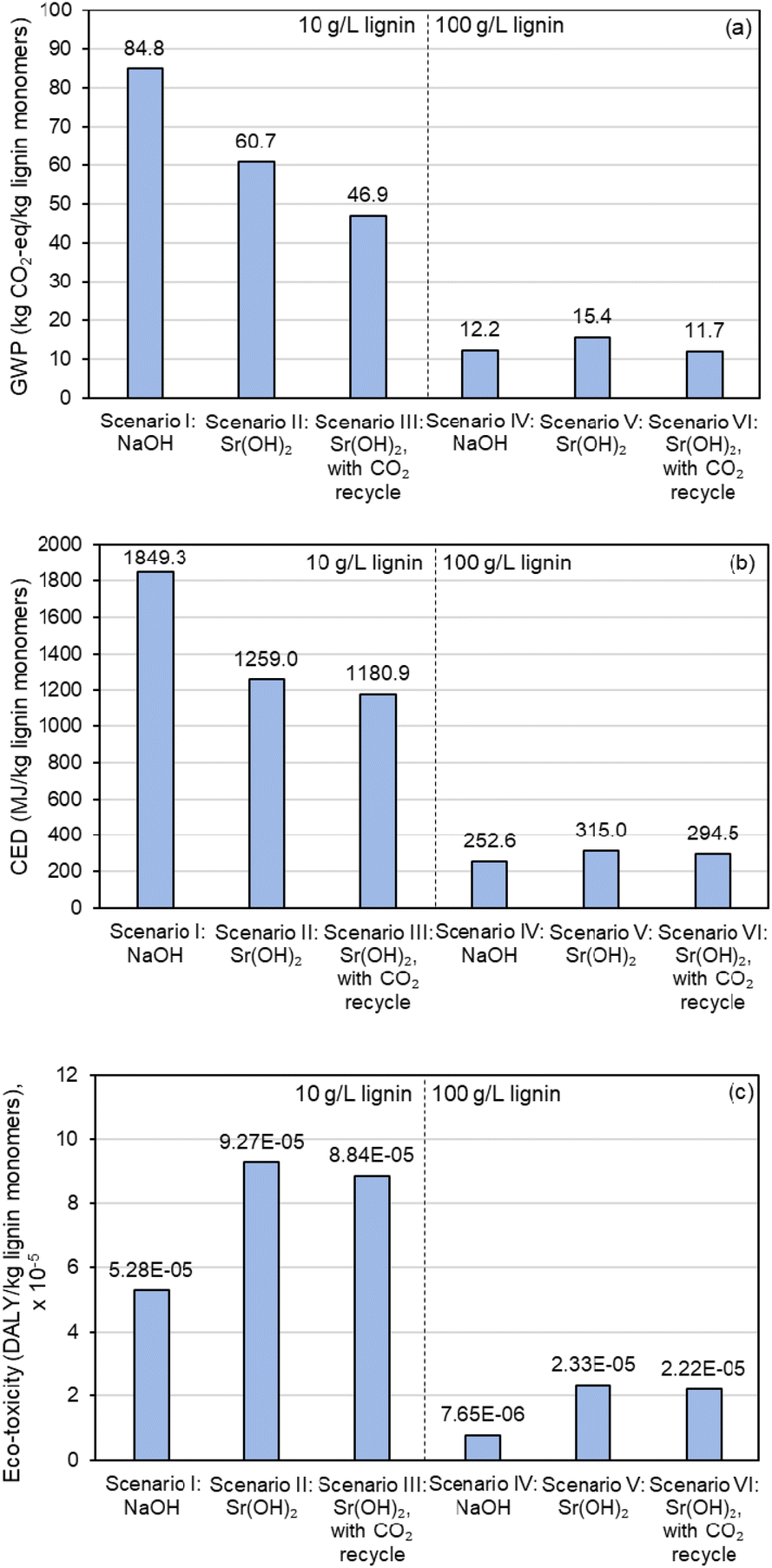 | ||
| Fig. 10 LCA comparison of lignin alkaline oxidation promoted by Sr(OH)2 in comparison to NaOH. (a) Global Warming Potential (GWP), as total greenhouse gas emissions normalized to CO2 equivalent impact. (b) Cumulative energy demand (CED), as MJ energy consumed. (c) Ecotoxicity, as disability-adjusted life years (DALY). Numerical data for this figure are included in Table S17.† | ||
Conclusions
Ba(OH)2 and Sr(OH)2 enable similar aromatic monomer yields as NaOH in the alkaline oxidation of lignin. Unlike NaOH, however, Ba(OH)2 and Sr(OH)2 are only soluble at high temperature and are mostly insoluble at room temperature, allowing for the high hydroxide ion concentrations necessary for high aromatic monomer yields at reaction temperature, and facile recovery of excess hydroxide via filtration and neutralization with CO2 at room temperature. After neutralization, the monomer solution is at pH 6–7, allowing for direct upgrading of monomers and/or extraction of non-carboxylic monomers. Preliminary optimization of the time, temperature, and air pressure suggests that a lignin concentration of 100 g L−1 and Sr(OH)2 concentration of 1 M (2 M hydroxide), optimal conditions are 160 °C, 22 bar air, and 30 min reaction time. TEA and LCA suggest that replacing NaOH with Sr(OH)2 can be beneficial in terms of process economics, despite the higher initial cost and more complex recycling required for implementation of Sr(OH)2. The process is predicted to be more sustainable at higher lignin loadings despite lower total monomer yields, but further process development and judicious selection of biomass type may be required to realize improvements in sustainability for Sr- over Na- promoted alkaline oxidation systems.Author contributions
Conceptualization: J. S. K., D. G. B., G. T. B. Investigation: J. S. K., R. J. D., D. G. W., A. J. R., K. L. N., and C. K. A. Formal analysis: D. G. B., K. J. R., S. J. H., R. M. H., N. S. C., E. D. C., and J. M. (experimental), B. C. K., R. D., and R. J. H. (T. E. A. and L. C. A.). Visualization and writing – original draft: J. S. K. Funding acquisition: G. T. B. writing – review & editing: all.Conflicts of interest
J. S. K., D. G. B., and G. T. B. have a patent pending on the use of reversible bases for biomass valorization. No other authors have conflicts of interest to declare.Acknowledgements
This work was authored by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under Contract No. DE-AC36-08GO28308. Funding provided by the U.S. Department of Energy Office of Energy Efficiency and Renewable Energy Bioenergy Technologies Office. The views expressed in the article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- S. P. S. Chundawat, G. T. Beckham, M. E. Himmel and B. E. Dale, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 121–145 CrossRef CAS PubMed.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 CrossRef PubMed.
- R. Davis, L. Tao, E. Tan, M. Biddy, G. Beckham, C. Scarlata, J. Jacobson, K. Cafferty, J. Ross and J. Lukas, Process design and economics for the conversion of lignocellulosic biomass to hydrocarbons: Dilute-acid and enzymatic Deconstruction of biomass to sugars and biological conversion of Sugars to Hydrocarbons, Report NREL/TP-5100-60223,National Renewable Energy Laboratory (NREL), Golden, CO, 2013.
- A. Corona, M. J. Biddy, D. R. Vardon, M. Birkved, M. Z. Hauschild and G. T. Beckham, Green Chem., 2018, 20, 3857–3866 RSC.
- R. E. Davis, N. J. Grundl, L. Tao, M. J. Biddy, E. C. Tan, G. T. Beckham, D. Humbird, D. N. Thompson and M. S. Roni, Process Design and economics for the conversion of lignocellulosic biomass to hydrocarbon fuels and coproducts: 2018 biochemical design case update; biochemical deconstruction and conversion of biomass to fuels and products via integrated biorefinery pathways. NREL/TP-5100-71949,National Renewable Energy Lab (NREL), 2018.
- R. Davis, A. Bartling and L. Tao, Biochemical Conversion of Lignocellulosic Biomass to Hydrocarbon Fuels and Products: 2019 State of Technology and Future Research. No. NREL/TP-5100-76567,National Renewable Energy Lab (NREL), Golden, CO (United States), 2020.
- K. Huang, P. Fasahati and C. T. Maravelias, iScience, 2020, 23, 100751 CrossRef CAS PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- H. Lange, S. Decina and C. Crestini, Eur. Polym. J., 2013, 49, 1151–1173 CrossRef CAS.
- R. Behling, S. Valange and G. Chatel, Green Chem., 2016, 18, 1839–1854 RSC.
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef CAS PubMed.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed.
- S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- T. Vangeel, W. Schutyser, T. Renders and B. F. Sels, Lignin Chem., 2020, 53–68 Search PubMed.
- M. I. F. Mota, P. C. Rodrigues Pinto, J. M. Loureiro and A. E. Rodrigues, Sep. Purif. Rev., 2016, 45, 227–259 CrossRef CAS.
- P. C. Rodrigues Pinto, E. A. B. da Silva and A. E. Rodrigues, in Biomass Conversion: The Interface of Biotechnology, Chemistry and Materials Science, ed. C. Baskar, S. Baskar and R. S. Dhillon, Springer Berlin Heidelberg, Berlin, Heidelberg, 2012, pp. 381–420. DOI:10.1007/978-3-642-28418-2_12.
- A. M. Shah, A. Abdul-Rahim, A. Mohamad and M. Hazwan, Int. J. Electrochem. Sci., 2017, 12, 9017–9039 CrossRef CAS.
- J. F. Stanzione III, J. M. Sadler, J. J. La Scala, K. H. Reno and R. P. Wool, Green Chem., 2012, 14, 2346–2352 RSC.
- A. L. Holmberg, J. F. Stanzione, R. P. Wool and T. H. Epps, ACS Sustainable Chem. Eng., 2014, 2, 569–573 CrossRef CAS.
- M. A. Lucherelli, A. Duval and L. Avérous, Prog. Polym. Sci., 2022, 101515 CrossRef CAS.
- O. Y. Abdelaziz, D. P. Brink, J. Prothmann, K. Ravi, M. Sun, J. García-Hidalgo, M. Sandahl, C. P. Hulteberg, C. Turner, G. Lidén and M. F. Gorwa-Grauslund, Biotechnol. Adv., 2016, 34, 1318–1346 CrossRef CAS PubMed.
- G. T. Beckham, C. W. Johnson, E. M. Karp, D. Salvachúa and D. R. Vardon, Curr. Opin. Biotechnol., 2016, 42, 40–53 CrossRef CAS PubMed.
- J. Becker and C. Wittmann, Biotechnol. Adv., 2019, 37, 107360 CrossRef CAS PubMed.
- C. W. Johnson, D. Salvachúa, N. A. Rorrer, B. A. Black, D. R. Vardon, P. C. S. John, N. S. Cleveland, G. Dominick, J. R. Elmore and N. Grundl, Joule, 2019, 3, 1523–1537 CrossRef CAS.
- A. Rahimi, A. Azarpira, H. Kim, J. Ralph and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 6415–6418 CrossRef CAS PubMed.
- M. Alherech, S. Omolabake, C. M. Holland, G. E. Klinger, E. L. Hegg and S. S. Stahl, ACS Cent. Sci., 2021, 7, 1831–1837 CrossRef CAS PubMed.
- X. Du, A. W. Tricker, W. Yang, R. Katahira, W. Liu, T. T. Kwok, P. Gogoi and Y. Deng, ACS Sustainable Chem. Eng., 2021, 9(23), 7719–7727 CrossRef CAS.
- H. Luo, E. P. Weeda, M. Alherech, C. W. Anson, S. D. Karlen, Y. Cui, C. E. Foster and S. S. Stahl, J. Am. Chem. Soc., 2021, 143, 15462–15470 CrossRef CAS PubMed.
- G. Wu, M. Heitz and E. Chornet, Ind. Eng. Chem. Res., 1994, 33, 718–723 CrossRef CAS.
- A. L. Mathias and A. E. Rodrigues, Holzforschung, 1995, 49, 273 CrossRef CAS.
- V. E. Tarabanko, N. A. Fomova, B. N. Kuznetsov, N. M. Ivanchenko and A. V. Kudryashev, React. Kinet. Catal. Lett., 1995, 55, 161–170 CrossRef CAS.
- G. Wu and M. Heitz, J. Wood Chem. Technol., 1995, 15, 189–202 CrossRef CAS.
- C. Fargues, Á. Mathias, J. Silva and A. Rodrigues, Chem. Eng. Technol., 1996, 19, 127–136 CrossRef CAS.
- H.-R. Bjørsvik and F. Minisci, Org. Process Res. Dev., 1999, 3, 330–340 CrossRef.
- V. E. Tarabanko, Y. V. Hendogina, D. V. Petuhov and E. P. Pervishina, React. Kinet. Catal. Lett., 2000, 69, 361–368 CrossRef CAS.
- Q. Xiang and Y. Y. Lee, Appl. Biochem. Biotechnol., 2001, 91, 71–80 CrossRef PubMed.
- V. E. Tarabanko and D. V. Petukhov, Chem. Sustainable Dev., 2003, 11, 655–667 Search PubMed.
- V. E. Tarabanko, D. V. Petukhov and G. E. Selyutin, Kinet. Catal., 2004, 45, 569–577 CrossRef CAS.
- J. Zhang, H. Deng and L. Lin, Molecules, 2009, 14, 2747–2757 CrossRef CAS PubMed.
- J. D. P. Araújo, C. A. Grande and A. E. Rodrigues, Chem. Eng. Res. Des., 2010, 88, 1024–1032 CrossRef.
- S. G. Santos, A. P. Marques, D. L. D. Lima, D. V. Evtuguin and V. I. Esteves, Ind. Eng. Chem. Res., 2011, 50, 291–298 CrossRef CAS.
- A. W. Pacek, P. Ding, M. Garrett, G. Sheldrake and A. W. Nienow, Ind. Eng. Chem. Res., 2013, 52, 8361–8372 CrossRef CAS.
- P. C. Rodrigues Pinto, C. E. Costa and A. E. Rodrigues, Ind. Eng. Chem. Res., 2013, 52, 4421–4428 CrossRef.
- V. E. Tarabanko and N. Tarabanko, Int. J. Mol. Sci., 2017, 18, 2421 CrossRef PubMed.
- G. Lyu, C. G. Yoo and X. Pan, Biomass Bioenergy, 2018, 108, 7–14 CrossRef CAS.
- W. Schutyser, J. S. Kruger, A. M. Robinson, R. Katahira, D. G. Brandner, N. S. Cleveland, A. Mittal, D. J. Peterson, R. Meilan and Y. Román-Leshkov, Green Chem., 2018, 20, 3828–3844 RSC.
- S. K. Singh, A. W. Savoy, Z. Yuan, H. Luo, S. S. Stahl, E. L. Hegg and D. B. Hodge, Ind. Eng. Chem. Res., 2019, 58, 15989–15999 CrossRef CAS.
- J. H. Fisher and H. B. Marshall, US Pat., 2576752, 1951 Search PubMed.
- J. H. Fisher and H. B. Marshall, US Pat., 2576753, 1951 Search PubMed.
- F. M. Casimiro, C. A. E. Costa, C. M. Botelho, M. F. Barreiro and A. E. Rodrigues, Ind. Eng. Chem. Res., 2019, 58, 16442–16449 CrossRef CAS.
- M. B. Hocking, J. Chem. Educ., 1997, 74, 1055 CrossRef CAS.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- Y. Jiang, X. Zeng, R. Luque, X. Tang, Y. Sun, T. Lei, S. Liu and L. Lin, ChemSusChem, 2017, 10, 3982–3993 CrossRef CAS PubMed.
- N. Ding, X. Song, Y. Jiang, B. Luo, X. Zeng, Y. Sun, X. Tang, T. Lei and L. Lin, Sustainable Energy Fuels, 2018, 2, 2206–2214 RSC.
- Z. Strassberger, P. Prinsen, F. V. D. Klis, D. S. V. Es, S. Tanase and G. Rothenberg, Green Chem., 2015, 17, 325–334 RSC.
- L. da Costa Sousa, M. Foston, V. Bokade, A. Azarpira, F. Lu, A. J. Ragauskas, J. Ralph, B. Dale and V. Balan, Green Chem., 2016, 18, 4205–4215 RSC.
- J. S. Kim, Y. Y. Lee and T. H. Kim, Bioresour. Technol., 2016, 199, 42–48 CrossRef CAS PubMed.
- A. Mittal, R. Katahira, B. S. Donohoe, S. Pattathil, S. Kandemkavil, M. L. Reed, M. J. Biddy and G. T. Beckham, ACS Sustainable Chem. Eng., 2017, 5, 2544–2561 CrossRef CAS.
- P. Prinsen, A. Narani and G. Rothenberg, ChemSusChem, 2017, 10, 1022–1032 CrossRef CAS PubMed.
- I. Lambert and H. L. Clever, Alkaline earth hydroxides in water and aqueous solutions, Elsevier, 1992 Search PubMed.
- E. D. Gomes and A. E. Rodrigues, Sep. Purif. Technol., 2019, 216, 92–101 CrossRef CAS.
- I. F. Mota, P. Rodrigues Pinto, J. M. Loureiro and A. E. Rodrigues, Sep. Purif. Technol., 2020, 234, 116083 CrossRef.
- J. Ralph, Phytochem. Rev., 2010, 9, 65–83 CrossRef CAS.
- X. Chen, E. Kuhn, E. W. Jennings, R. Nelson, L. Tao, M. Zhang and M. P. Tucker, Energy Environ. Sci., 2016, 9, 1237–1245 RSC.
- M. M. Abu-Omar, K. Barta, G. T. Beckham, J. S. Luterbacher, J. Ralph, R. Rinaldi, Y. Román-Leshkov, J. S. Samec, B. F. Sels and F. Wang, Energy Environ. Sci., 2021, 14, 262–292 RSC.
- S. D. Karlen, P. Fasahati, M. Mazaheri, J. Serate, R. A. Smith, S. Sirobhushanam, M. Chen, V. I. Tymokhin, C. L. Cass, S. Liu, D. Padmakshan, D. Xie, Y. Zhang, M. A. McGee, J. D. Russell, J. J. Coon, H. F. Kaeppler, N. de Leon, C. T. Maravelias, T. M. Runge, S. M. Kaeppler, J. C. Sedbrook and J. Ralph, ChemSusChem, 2020, 13, 2012–2024 CrossRef CAS PubMed.
- P. O. Saboe, E. G. Tomashek, H. R. Monroe, S. J. Haugen, R. L. Prestangen, N. S. Cleveland, R. M. Happs, J. Miscall, K. J. Ramirez, R. Katahira, E. C. D. Tan, J. Yan, N. Sun, G. T. Beckham and E. M. Karp, Green Chem., 2022, 24, 3152–3166 RSC.
- H. Luo, I. M. Klein, Y. Jiang, H. Zhu, B. Liu, H. I. Kenttämaa and M. M. Abu-Omar, ACS Sustainable Chem. Eng., 2016, 4, 2316–2322 CrossRef CAS.
- M. S. Holm, S. Saravanamurugan and E. Taarning, Science, 2010, 328, 602–605 CrossRef CAS PubMed.
- D. Salvachúa, E. M. Karp, C. T. Nimlos, D. R. Vardon and G. T. Beckham, Green Chem., 2015, 17, 4951–4967 RSC.
- A. Rodriguez, D. Salvachúa, R. Katahira, B. A. Black, N. S. Cleveland, M. Reed, H. Smith, E. E. Baidoo, J. D. Keasling and B. A. Simmons, ACS Sustainable Chem. Eng., 2017, 5, 8171–8180 CrossRef CAS.
- T. Aro and P. Fatehi, ChemSusChem, 2017, 10, 1861–1877 CrossRef CAS PubMed.
- A. Kalliola, T. Vehmas, T. Liitiä and T. Tamminen, Ind. Crops Prod., 2015, 74, 150–157 CrossRef CAS.
- B. Cao, L. A. Adutwum, A. O. Oliynyk, E. J. Luber, B. C. Olsen, A. Mar and J. M. Buriak, ACS Nano, 2018, 12, 7434–7444 CrossRef CAS PubMed.
- H. B. Marshall and C. A. Sankey, US Pat, 2544999, p. 1951.
- C. C. Bryan, US Pat., 2692291, 1954 Search PubMed.
- D. Havkin-Frenkel, in Kirk–Othmer Encyclopedia of Chemical Technology, 2018, pp. 1–12. DOI:10.1002/0471238961.2201140905191615.a01.pub3.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03333j |
| This journal is © The Royal Society of Chemistry 2022 |