
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in solid-phase peptide synthesis in aqueous media (ASPPS)
Da'san M. M.
Jaradat
 *a,
Othman
Al Musaimi
*b and
Fernando
Albericio
*cde
*a,
Othman
Al Musaimi
*b and
Fernando
Albericio
*cde
aDepartment of Chemistry, Faculty of Science, Al-Balqa Applied University, P.O. Box 19117, Al-Salt, Jordan. E-mail: dasan.jaradat@bau.edu.jo; jaradatdasan@gmail.com; Tel: +962 775477262
bDepartment of Chemical Engineering, Imperial College London, London SW7 2AZ, UK. E-mail: o.al-musaimi@imperial.ac.uk; musamiau@gmail.com
cSchool of Chemistry and Physics, University of KwaZulu-Natal, Durban 4001, South Africa. E-mail: albericio@ukzn.ac.za
dCIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine and Department of Organic Chemistry, University of Barcelona, 08028 Barcelona, Spain
eInstitute for Advanced Chemistry of Catalonia (IQAC-CSIC), 08034 Barcelona, Spain
First published on 6th August 2022
Abstract
Peptides have been gaining ground in the pharmaceutical arena, with a total of 22 approvals over the last six years. These molecules are also present in antibody–drug conjugate constructs as linkers or payloads, or both. Solid-phase peptide synthesis (SPPS) is the method of choice for peptide synthesis. The introduction of an automatic synthesizer facilitated this methodology and helped in reducing the amounts of the required solvents. However, there are still concerns regarding the amounts, as well as the safety profiles, of the solvents involved in SPPS. Here, we discuss the work addressing the use of water as the greenest alternative to the common non-green solvents employed in various steps of the SPPS methodology. Various technologies were introduced which enabled the synthesis of di- and up to decapeptides in aqueous media with satisfactory yields and purities.
 Da'san M. M. Jaradat | Da'san Jaradat is an Associate Professor of Bioorganic Chemistry at Al-Balqa Applied University, Jordan. He received his Ph.D. in Bioorganic Chemistry and M.Sc. in Crystallography from Freie Universität Berlin, Germany. He was a visiting research scholar at the University of California, Berkeley and Lawrence Berkeley National Laboratory, USA. His major scientific efforts have been concentrated on the development of new synthetic routes to access bioactive peptides and small molecules. Currently, he is working on developing new therapeutic peptides and small molecules acting as inhibitors of efflux pumps of several multidrug-resistant bacteria. |
 Othman Al Musaimi | Othman Al Musaimi obtained his Ph.D. in Pharmaceutical Chemistry from the University of Kwazulu-Natal – South Africa under the supervision of Prof. Fernando Albericio and Prof. Beatriz G. de l a Torre. Othman is a Research Associate at the Department of Chemical Engineering – Imperial College London. His research interests are focused on developing synthetic, separation, and purification methodologies for therapeutic peptide entities. Currently, he is working on expanding the hydrophobicity index of amino acids and delivering blood–brain barrier shuttle peptides to target brain-related diseases. |
 Fernando Albericio | Fernando Albericio has developed his academic and professional career in Europe, USA, Latin America, Asia, and presently in Africa (Research Professor at the University of KwaZulu-Natal (Durban, 803 South Africa)). His primary research interests cover practically all aspects of peptide synthesis, as well as the synthesis of peptides and small molecules with therapeutic activities, especially tuberculosis, infectious diseases, and cancer. His group is also involved in the development of new strategies for drug delivery. He is deeply involved in the development of the third mission of the university: the transference of knowledge and technology to society. He was the co-founder and general director of the Barcelona Science Park and he is the co-founder of BioDurban. |
1. Introduction
Peptides and proteins play key molecular roles in sustaining life.1,2 They are the final products of the central dogma of molecular genetics, which involves DNA transcription and mRNA translation to afford proteins3 that can be modified during or after the translation process, namely co- or post-translational modifications, respectively.4 Proteins and peptides are present in all living organisms and play major roles in many biological processes, including protection by acting as antibodies within the immune system, communication between cells, and the transport of a wide spectrum of substances through membranes. Therefore, countless studies have been carried out to gain insights into the mechanisms and principles underlying the structural and functional characteristics of biologically active proteins and peptides, which can ultimately be utilized for applications in the medicinal and pharmaceutical industries.5,6 Such scientific studies require the availability of pure peptides and proteins in reasonable amounts with purity. In this regard, significant research efforts have pursued such access. Generally, these molecules can be achieved by isolating them from natural sources and by using the recombinant techniques employed in biochemical protein expression and chemical synthesis, including chemical peptide synthesis and ligation, or a combination of all or some of these approaches.7 Beyond human peptides, medicinal chemistry has also played a key role in delivering peptides with diverse structures and applications.8 Interestingly, therapeutic peptides are considered a complement and, in several circumstances, more favorable alternatives to small molecules and biologics, where they provide a tolerable safety profile and can be used to address unmet medical challenges.9 More than 120 peptides have reached the market for the treatment of a wide spectrum of diseases, including diabetes, cancer, and HIV, among others.10 Of note, 22 peptides have been approved by the FDA over the last six years, either as peptide-based drugs or as payloads and/or linkers in antibody–drug conjugates (ADCs).11Given the above developments, there is now a strong demand for peptides for both research and production purposes. Thus, in 2018, peptides accounted for USD 25 billion in the global therapeutic market and they are expected to reach an estimated value of USD 27 billion by 2027.12,13 The main focus of the industrial era was to cover the needs of the population. In contrast, the focus of current times is moving towards achieving sustainability, taking into account the 12 principles of Green Chemistry.14 The main goals of Green Chemistry are to protect humans and the environment and to comply with the legislation set out by regulatory agencies.15 Furthermore, if the circular economy (CE) is followed, Green Chemistry will be considered a truly significant improvement with respect to the cost of the overall manufacturing process (Fig. 1).16,17
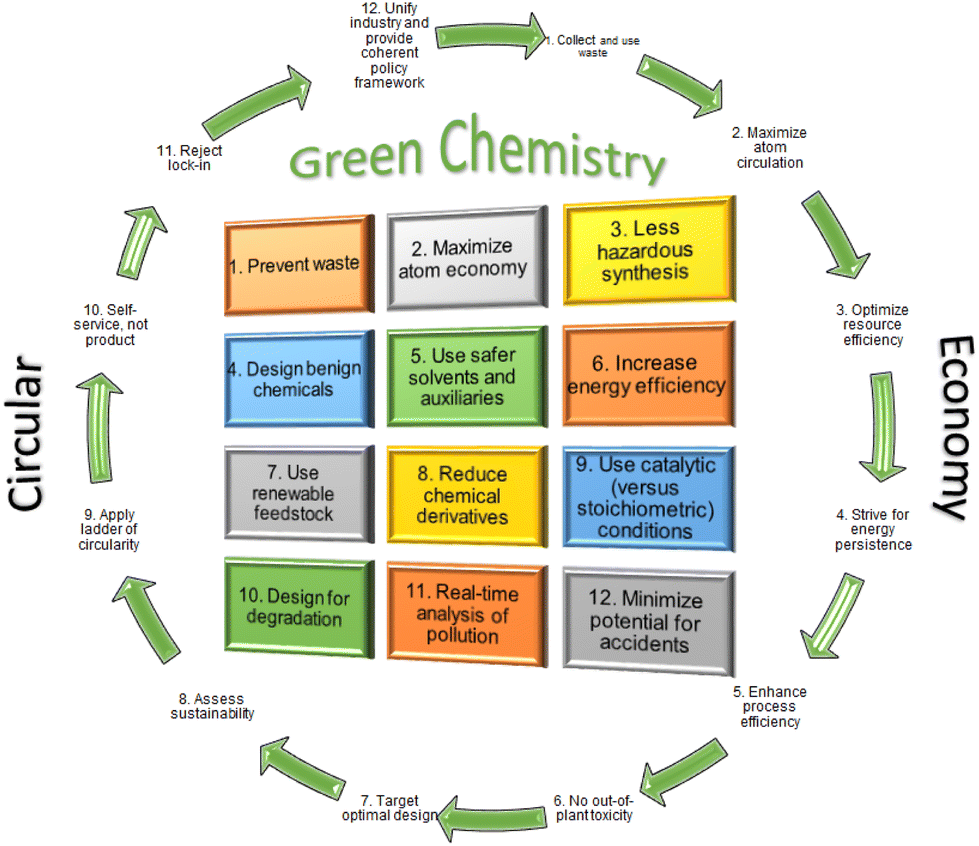 | ||
| Fig. 1 The “24 principles” of green chemistry and the circular economy. Reproduced from ref. 17 with permission from the TKS publisher, copyright 2021. | ||
Solid-phase peptide synthesis (SPPS)18 is the most attractive method for preparing peptides. The 9-fluorenylmethoxycarbonyl solid-phase peptide synthesis (Fmoc-SPPS) approach19 is currently the most commonly used method for this purpose. Fmoc-SPPS is of friendlier nature in comparison with tert-butoxycarbonyl (Boc)-SPPS, which requires much harsher acidic treatments for the final global deprotection. For the recent advances in the SPPS field, consult the excellent review of Behrendt et al.20
Solvents are considered the major components across all synthetic fields, but are more important in SPPS.21 In SPPS, the intermediates are not isolated and the only purification carried out through the elongation of the peptide chain is done by extensive washing. In general, the solvents widely used in SPPS are environmentally unfriendly.22–27 It involves the use of N,N-dimethyl formamide (DMF) and/or N-methyl pyrrolidone (NMP) as the main solvents for coupling and deprotection steps. A trending study performed by Ashcroft et al. concluded that the use of NMP increased three-fold between 1997 and 2002, while the use of DMF and DCM remained the same.28
This excessive use of solvents in SPPS makes a significant contribution to industrial waste. In this context, sustainable green alternatives are highly desirable.13,29 Therefore, many research groups have directed their attention to switching to eco-friendly alternatives.30–39 Fully switching to water – the biochemical solvent for the biosynthesis of biomolecules, including peptides and proteins – is one of the most fascinating and promising unsolved challenges in SPPS.40,41 Thus, the ultimate goal in Green SPPS is the so-called Aqueous SPPS (ASPPS) to achieve sustainable peptide synthesis.42–48
This review addresses several encouraging and important aspects of ASPPS, starting with the early efforts made by Kawasaki and co-workers49 and ending with recent advances in this field. In the following sections, the key features of ASPPS will be described, including resins, coupling reagents, building blocks, and protecting groups, as well as other aspects.
2. Resins used in ASPPS
Solid-phase synthesis, including SPPS and solid-phase organic synthesis (SPOS), requires solid support, which is bound permanently to a linker, which in turn facilitates permanent anchoring of a target peptide to it. 1–2% divinylbenzene – cross-linked polystyrene is one of the most common resins used in SPPS.50,51 Others include the following: polyethylene glycol (PEG), such as ChemMatrix;52 polyethylene glycol-polystyrene graft solid supports, such as PEG-PS, Champion support and TentaGel;53–55 polyamide solid supports;56 beaded polyethylene glycol-acrylamide (PEGA) copolymers;57 and polyethylene-polystyrene (PE-PS) films.58 For alternative polymeric solid supports, the reader is encouraged to refer to the following review.59ASPPS requires solid support that is compatible with water and efficiently swollen in it. The common divinylbenzene – cross-linked polystyrene polymeric solid support is not suitable for this synthetic approach because it has poor swelling capacity in water. Fortunately, several water-compatible solid supports are available.
ChemMatrix52 belongs to the family of polyethene cross-linked with PEG supports. It contains only PEG units with no polyacrylamide or polystyrene backbones. The amphiphilic nature of this solid support makes it an excellent resin for ASPPS since it swells extensively in a wide range of solvents, including water, tetrahydrofuran (THF), methanol, dichloromethane (DCM), and DMF.
Another family of excellent water-compatible supports is PEGA.57 Unlike ChemMatrix, PEGA has a polyacrylamide backbone copolymerized with PEG units. PEGA solid support beads have similar swelling properties to ChemMatrix beads.
Alternative polymeric solid supports for ASPPS are PEG grafted on cross-linked polystyrene (PEG-PS)53,60 and TentaGel.55,61 PEG-PS is prepared by adding PEG to aminomethyl-PS resins, while TentalGel is made up of a composite of polyethylene glycol and low cross-linked hydroxyethylpolystyrene, which can be terminally functionalized.
Different types of linkers can be attached to these solid supports. Linkers can be categorized into different types based on peptide cleavage conditions (nucleophilic displacement, basic, acidic, photolytic, etc.). Examples in the literature include the use of ChemMatrix, TentaGel of the PEG-PS graft with Rink amide, hydroxymethyl-3-methoxyphenoxy-butyric acid (HMPB) and hydroxymethyl-benzoic acid (HMBA) linkers.62–65 In general, there is no difference between the linkers used for ASPPS and those used for conventional SPPS involving DMF or NMP as solvents. Therefore, upon the completion of chain elongation, the peptides are cleaved from the resin using common cleavage cocktails used in conventional Fmoc- and Boc-SPPS.
3. Coupling reagents used in ASPPS for stepwise peptide chain elongation
A wide range of coupling reagents are available for their use in peptide synthesis. The conditions employed during a coupling reaction (peptide bond formation) determine the rate of acylation and the extent of side reactions such as racemization.1Conventional coupling reagents include carbodiimides66–68 in conjunction with substituted phenols or substituted hydroxylamines, forming active esters,69–73 phosphonium salts,74–79 uronium salts,80,81 and aminium/guanidinium salts,82–88 all of which also render active esters.
To carry out ASPPS, the coupling reagent should be soluble or at least compatible with water. To date, a few efficient coupling reagents have been available for ASPPS, including N-ethyl-N’-(3-dimethylaminopropyl)carbodiimidehydrochloride66 (EDC·HCl), also known as water-soluble carbodiimide (WSCD) (Fig. 2A). For efficient coupling rates, the EDC·HCl coupling reaction should be carried out under basic conditions in the presence of additives, including N-hydroxy-5-norbornene-2,3-dicarboximide89 (HONB), ethyl-2-cyano-2-(hydroxyimino)acetate79 (OxymaPure), 3-sulfo-N-hydroxysuccinimide64 (sulfo-HOSu), (Fig. 2B) and among others. Moreover, the following stand-alone coupling reagents have also been used: 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride90 (DMT-MM); the aminium salt 2-(5-norbornene-2,3-dicarboximido)-1,1,3,3-tetramethyluronium tetrafluoroborate91 (TNTU); and (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate46 (COMU) (Fig. 2A).
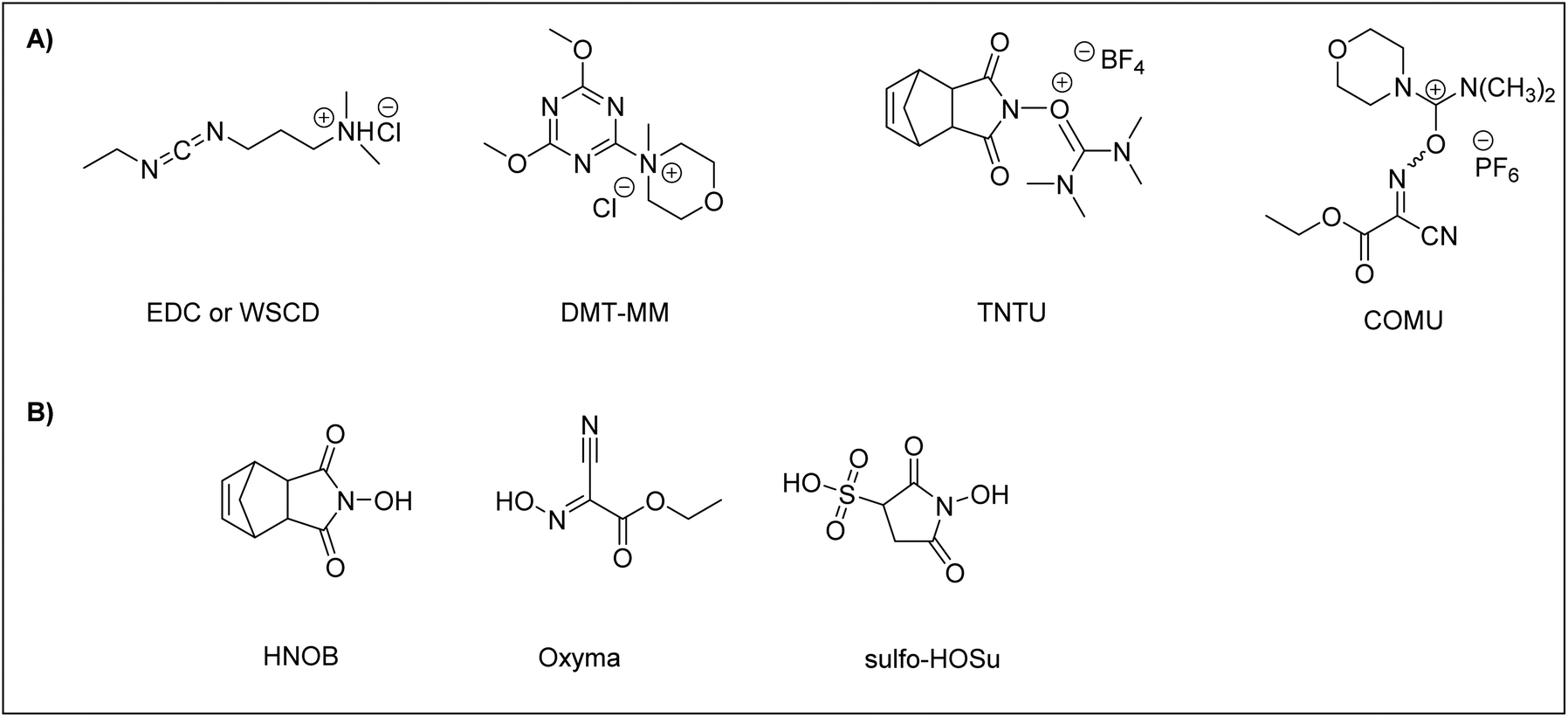 | ||
| Fig. 2 (A) Efficient coupling reagents for aqueous solid-phase peptide synthesis. (B) Common additives used in coupling reactions of aqueous solid-phase peptide synthesis. ECD = N-ethyl-N’-(3-dimethylaminopropyl)carbodiimidehydrochloride; WSCD = water-soluble carbodiimide; COMU = (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate. | ||
To date, WSCD (EDC·HCl) has been the coupling reagent of choice for the synthesis of numerous peptides reported in the literature using ASPPS.46,62,63,91–99 However, as mentioned earlier, additives should be employed in order to improve the coupling efficiency of WSCD.
Cortes-Clerget et al. showed that some well-established coupling reagents used in peptide synthesis such as 1-((dimethylamino)(dimethyliminio)methyl)-1H-[1,2,3]triazolo[4,5-b]pyridine 3-oxide hexafluorophosphate (HATU) and benzotriazol-1-yloxytri(pyrrolidino) phosphonium hexafluorophosphate (PyBOP) proved to be less efficient than COMU. However, COMU paired with 2,6-lutidine led to highly efficient couplings furnishing amides in high yields. 2,6-Lutidine, as a base, eliminates concerns about epimerization and decreases side reactions since it allows the use of mild conditions (pH 6–7). This coupling reagent enabled them to produce nonpolar peptides in excellent yields in the course of ASPPS using a nano-micelles strategy.40,100 Recently, N,N,N′,N′-tetramethylchloroformamidinium hexafluorophosphate (TCFH) paired with collidine has been proven to be a more efficient coupling agent than COMU for SPPS when aqueous 20% PolarClean was used as a solvent and TentaGel S as a resin.41
DMT-MM and TNTU were examined for their ability as coupling reagents for peptide synthesis in aqueous media. In a comparison study with WSCD, TNTU was ineffective as a coupling reagent when the reaction was attempted in water even when HONB was employed as an additive, resulting in poor coupling due to its degradation in water. On the other hand, DMT-MM proved to be a satisfactory coupling reagent in aqueous 50% EtOH. However, like TNTU, DMT-MM did not work well when the reaction was attempted in water alone.91
4. Protecting groups used in ASPPS
4.1 New Nα-amino protecting groups to be used specifically in ASPPS
Many research groups have synthesized short peptides using solvents that are greener alternatives to those commonly used in Fmoc-SPPS.27,31,33,36,39,41,48,101–103 However, current Fmoc-protected amino acids show low to poor solubility in these greener solvents, thus limiting their use of these solvents.104 Therefore, there is a need for greener solvents for current Fmoc-protected amino acids. In this regard, water heads the list of green solvents. Here, we discuss just the currently available Nα-amino protected amino acids that are described to be compatible with water.In a pioneering study in this field, Kawasaki and co-workers introduced several Nα-amino protecting groups that could facilitate and improve the solubility of amino acids in water. They prepared Nα-protected amino acids with 2-[phenyl(methyl)sulfonio]ethyloxycarbonyl tetrafluoroborate (Pms),92,93,95N-ethanesulfonylethoxycarbonyl (Esc) amino acids,94 and 2-(4-sulfophenylsulfonyl)ethoxycarbonyl (Sps) amino acids.105Fig. 3 shows the chemical structures of these Nα-amino protecting groups (Fig. 3A) and the approach used for their removal under basic conditions through a β-elimination is similar to that for Fmoc removal (Fig. 3B).
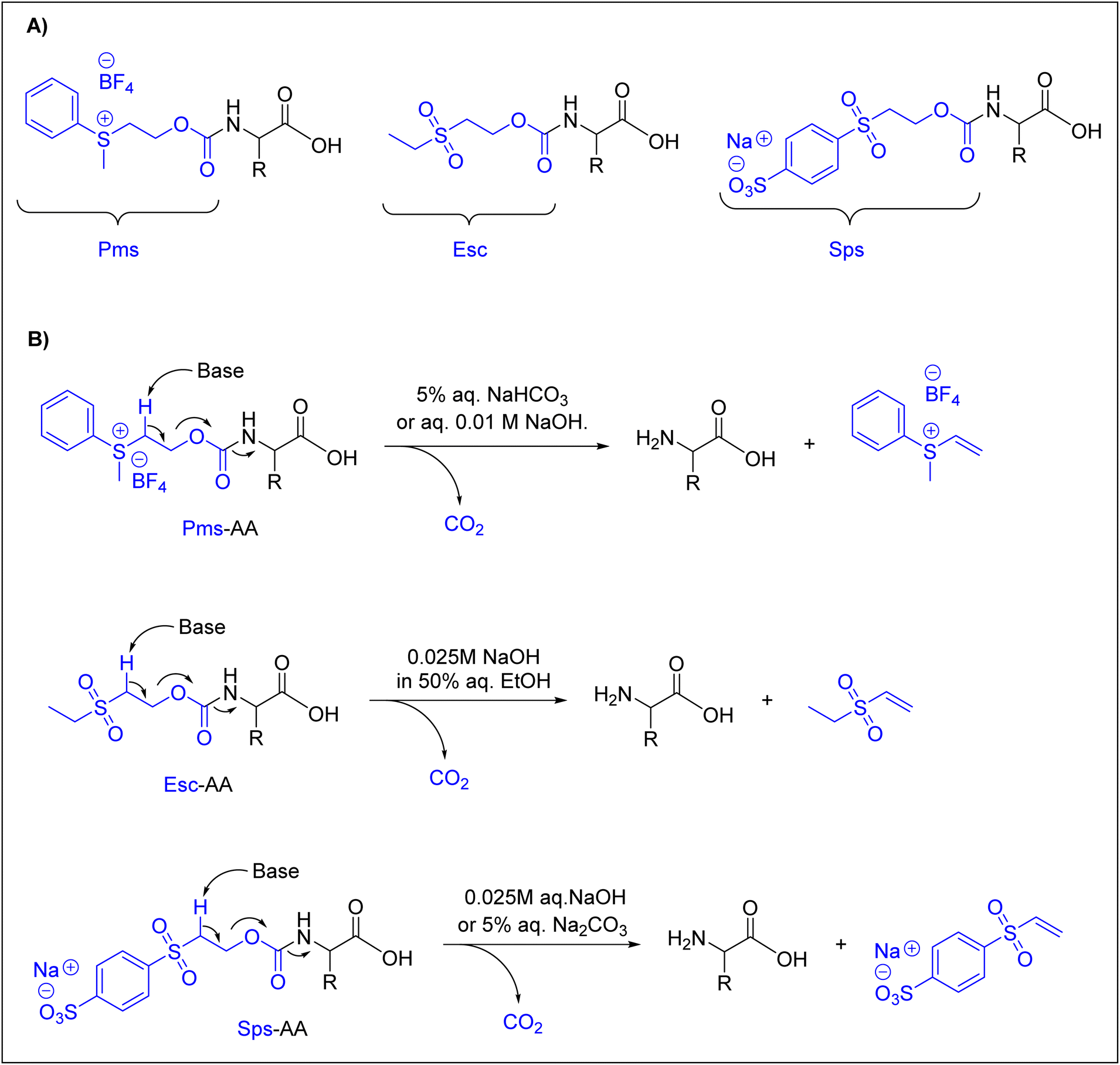 | ||
| Fig. 3 (A) Nα-Amino protecting groups (Pms, Esc, and Sps) introduced by Kawasaki and co-workers. (B) Base-mediated removal of Nα-amino protecting groups. Pms = 2-[phenyl(methyl)sulfonio]ethyloxycarbonyl tetrafluoroborate; Esc = N-ethanesulfonylethoxycarbonyl; Sps = 2-(4-sulfophenylsulfonyl)ethoxycarbonyl; aq. = aqueous. | ||
With these Nα-amino protecting groups, Kawasaki and co-workers were able to prepare only short peptides, such as Met-enkephalin and Leu-enkephalin. However, Tyr was incorporated without side-chain protection.106 These authors prepared short peptides using a PEG-PS resin. The coupling reaction was performed using WSCD, DMT-MM, or TNTU as a coupling agent, 4-methylmorpholine (NMM) or N,N-diisopropylethylamine (DIEA) as a base, and HONB or sulfo-HOSu as an additive.63,64,91,96,107
For amino acids protected with Pms, coupling was achieved using WSCD in combination with HNOB. The addition of DIEA was considered in the case of amino acids protected with Esc and Sps.94,105,106 In all cases, water was considered as the washing solvent, in addition to an aqueous 0.2% Triton X solution, which was used to enhance the swelling of the resin, as well as the solubility of amino acids protected by the Sps and Esc groups. The peptides were cleaved from the resin using TFA, achieving overall yields of 61%, 32%, 71%, and 61% in the case of Pms, Pms-ONp, Esc, and Sps, respectively.92–95,105
Although these groups were very well designed, they had several drawbacks, mainly the lack of stability, as shown by Pms, and insufficient removal with an aqueous solution, as in the case of Esc.38
Recently, Kolmar and co-workers reported an interesting and promising Nα-amino protecting group that can be used efficiently in ASPPS.62 They introduced a water-soluble 2,7-disulfo-9-fluorenylmethoxycarbonyl (Smoc) Nα-amino protecting group, which is a derivative of the water-insoluble Fmoc group. As shown in (Fig. 4A), 2,7-disulfo-9-fluorenylmethoxycarbonyl chloride (Smoc-Cl) was synthesized by reacting 9-fluorenylmethoxycarbonyl chloride (Fmoc-Cl) with oleum (SO3/H2SO4), with an overall yield of about 75%. Next, Nα-Smoc-protected amino acids were achieved by the reaction between Nα-unprotected amino acids and Smoc-Cl (Fig. 4B). This synthetic approach was successfully applied to all common amino acids and also to a number of important uncommon ones to obtain the desired Nα-Smoc protected amino acids in yields exceeding 85% in most cases.
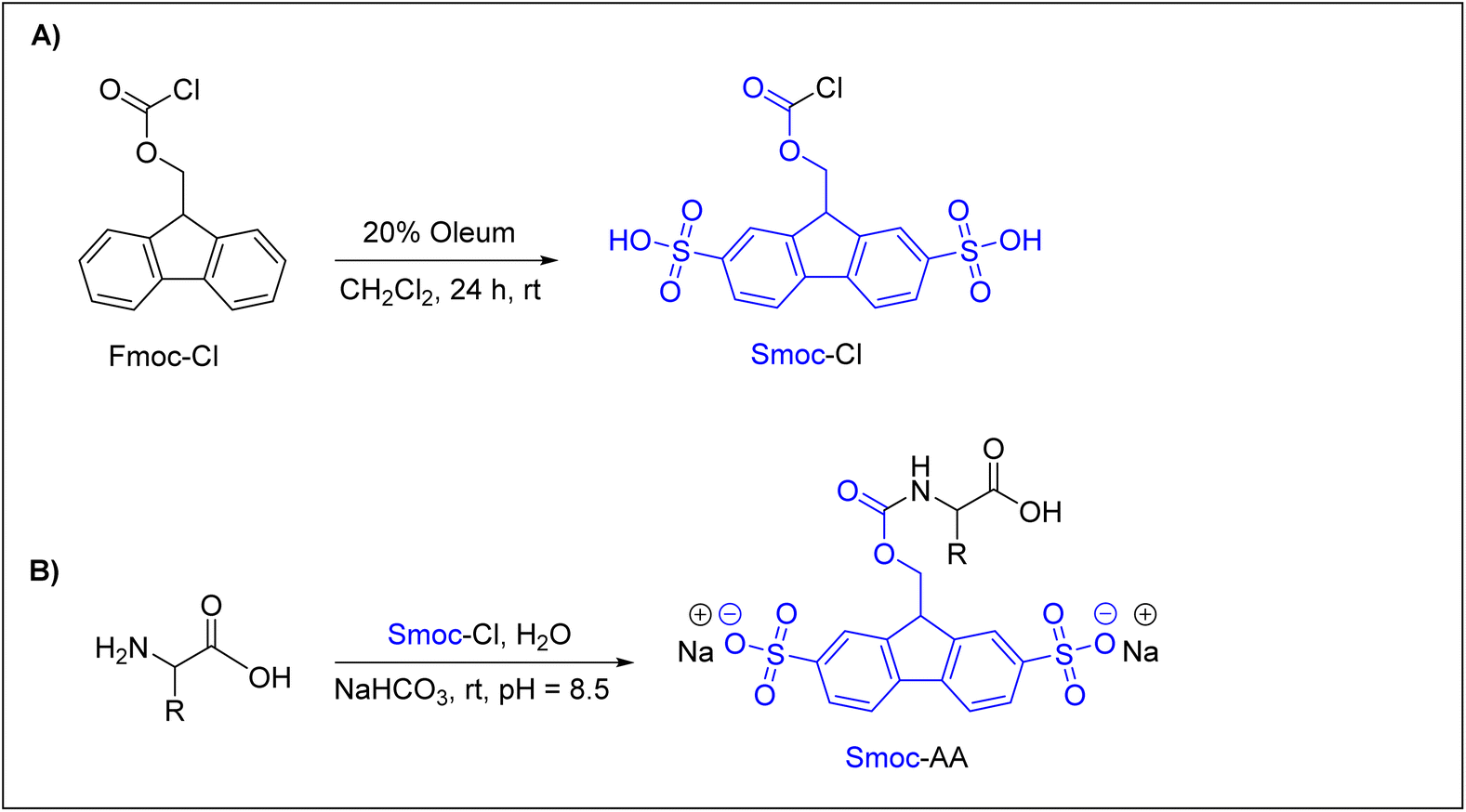 | ||
| Fig. 4 (A) Preparation of Smoc-Cl, 28. (B) Preparation of Smoc-protected amino acids 30, Smoc-AA. | ||
As Smoc is a derivative of the Fmoc group, it follows the same base-mediated removal mechanism described for Fmoc. The base-mediated removal of Smoc leads to the formation of the desired free amino component and the formation of a 2,7-disulfo-fulvene–base adduct as a result of the reaction between 2,7-disulfo-fulvene and the base used. Using these building blocks, Kolmar and co-workers applied ASPPS to prepare several short linear and cyclic peptides, such as acyl carrier protein (ACP) 65–74 decapeptide (10 residues), oxytocin, and vasopressin, in promising overall yields of up to ∼72%. The authors91 used WSCD as a coupling reagent with different additives but found that OxymaPure was the most efficient additive, followed by HOPO. They used either the HMPB-ChemMatrix resin or H-Rink amide-ChemMatrix resin. All washing steps were performed with water and Smoc was removed with aqueous 1 M NaOH when a Rink amide-ChemMatrix resin was used and with 25% aqueous ethanolamine when the HMPB-ChemMatrix resin was used. 5–10% aqueous piperazine was also used to remove the Smoc group from both resins. Of note, NaOH should be avoided in ASPPS if esters are applied as side-chain protecting groups or linkers.62 It is interesting to highlight that these authors have created a biotech company, Sulfotools GmbH, to develop its chemistry for manufacturing purposes.
4.2 Use of conventional Nα-amino protecting groups (Fmoc, Boc, Z, and N3) in ASPPS
A sustainable alternative to the conventional SPPS approach requires the availability of water-compatible amino acid building blocks with Nα-amino protecting groups and side-chain protecting groups that are relatively polar to facilitate solubility in water.Although commonly used Fmoc- and Boc-amino acids are sparingly water-soluble, Hojo et al. described ASPPS using water-dispersible Fmoc- and Boc-amino acid nanoparticles obtained via their innovative technology.63–65,107 They prepared these nanoparticles by pulverization using a ball mill equipped with zirconium oxide beads in the presence of PEG as a dispersion agent. Fmoc-amino acid nanoparticles ranged from 250 to 500 nm while Boc-amino acid nanoparticles were between 500 to 750 nm (Fig. 5).
 | ||
| Fig. 5 Water-based SPPS using water-dispersible Fmoc- and Boc-amino acid nanoparticles. | ||
Using this technology, Hojo et al. prepared short peptides on the TentaGel resin, and the coupling was carried out using WSCD, HONB, and DIEA. The washing step was performed with water, and Fmoc was removed by treatment with 0.1 M NaOH in 90% ethanol. Parallelly, two syntheses were performed, one with nanodispersed Fmoc-amino acids and a second one with the unprocessed ones. In this regard, a 67% yield of the pentapeptide was achieved with the former, while no peak was observed by HPLC while using the latter. Furthermore, the technology was also investigated in Boc-SPPS and an 89% yield of Leu-Enkephalin was achieved.107 In the Boc mode, the coupling reaction was performed using DMT-MM along with NMM. The Boc group was removed with the aid of TFA and 4 M HCl in dioxane. As expected, these results can be attributed to the less hydrophobicity of Boc vs. Fmoc, hence better solubility and efficiency in aqueous settings. These authors were also able to extend this technology and synthesize more difficult longer peptides using a microwave-assisted mode.98 However, in this case, and after testing several coupling reagents, they found that a combination of DMT-MM and NMM was the best choice.63,98 Furthermore, the aqueous microwave-assisted paradigm renders lower epimerization in comparison to the common paradigm with organic solvents. Provided that, in case of Cys(Acm), the epimerization was as low as 0.50% vs. 10.5%108 and in the case of His(Trt); 3.3% vs. 13.8%.99 Although this technology is promising, these authors reported the synthesis of only short peptides such as Leu-enkephalin amide, and upscaling was also limited.
Cortes-Clerget et al. used the carbobenzoxy group (Z) as an Nα-amino protecting group for the amino acids employed.40,100 They successfully prepared di- and tri-peptides in an aqueous micellar medium. They used 2% (w/w) TPGS-750-M–H2O nano-micelles, whose core is lipophilic and can therefore accommodate the peptide synthesis reaction, as shown in Fig. 6.
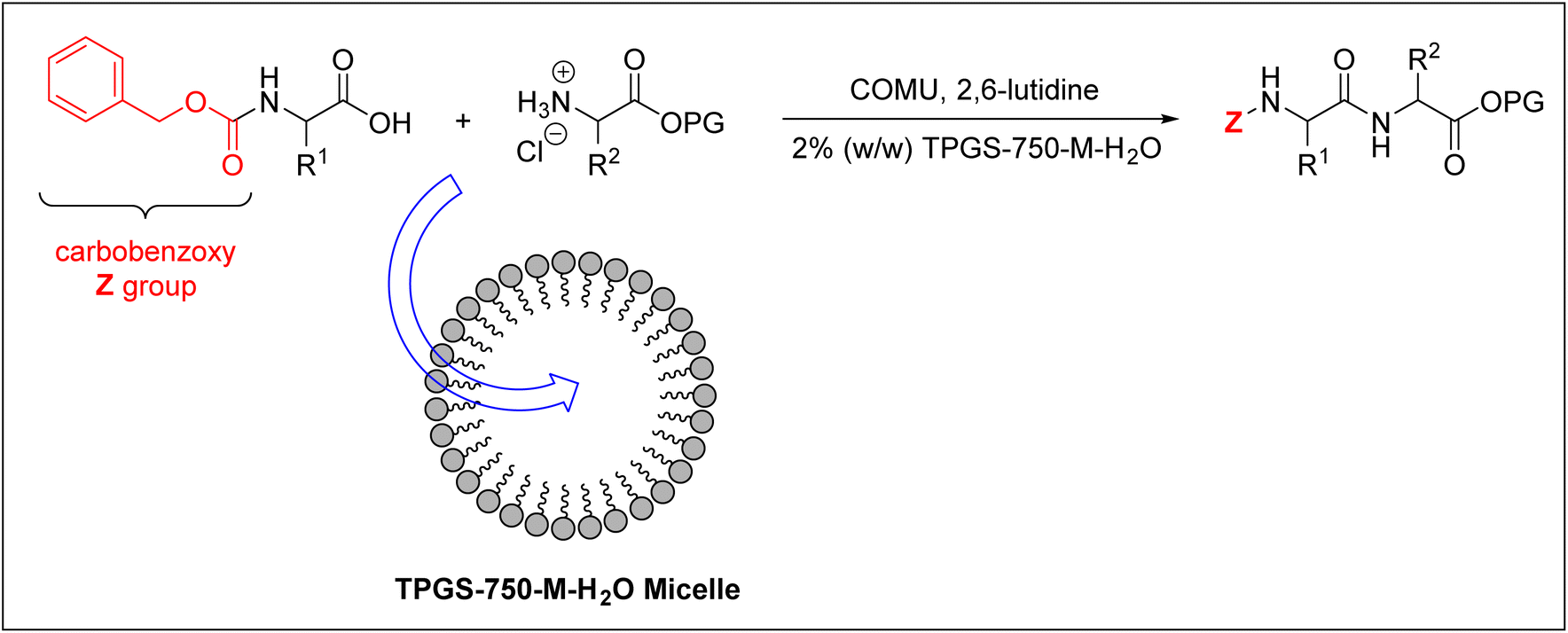 | ||
| Fig. 6 Peptide synthesis inside TPGS-750-M–H2O micelles. COMU = (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate; PG = protecting group. | ||
On the other hand, the surface of this micelle is polar, thus enabling it to be solvated in water. This approach effectively overcomes the insolubility challenge of protected amino acids in water, hence it has been employed for synthesising decapeptide as well.40
Grøtli and co-workers reported the synthesis of Leu-enkephalin using ASPPS on Tentagel and CM resins using Boc, Fmoc and azido amino acids. After studying several coupling reagents, WSCD in combination with HNOB was found to be the best choice. Washing was performed with water and Boc was removed with 1 M HCl. Subsequent neutralization was achieved with 10% DIEA in water. Fmoc- or azido-rendered lower yields than the Boc-amino acid paradigm. This observation can be attributed to the higher hydrophilic nature of the Boc group in comparison with Fmoc or azido. Taking advantage of the microwave, the authors attempted this ASPPS with PS resins, obtaining low coupling efficiency, which was attributed to the lack of the PS resin swelling in water.46
The superiority of microwave power was demonstrated by the relatively poor purity obtained for the same peptide using a normal heating protocol (hot plate) and the same temperature that was selected in the microwave synthesis (75 °C) was considered, in which, in case of normal heating, a higher temperature was needed to achieve comparable purity (83 °C).
The authors noticed the rapid formation of the active ester in the presence of HNOB. Thus, a short reaction time and short activation time (1 min) are recommended. HNOB undergoes a Loosen rearrangement after the protected-AA-ONB active ester is attacked by a nucleophile. A by-product with a mass of the desired peptide + 135, which corresponded to a β-alanine derivative, was detected during Leu-enkephalin synthesis in water using WSCD along with HNOB as an additive.
Recently, by applying SPPS, Pawlas and Rasmussen used a binary mixture consisting of water–PolarClean (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to prepare Leu-enkephalin amides, achieving up to 86% purity and 85% conversion.109
:1) to prepare Leu-enkephalin amides, achieving up to 86% purity and 85% conversion.109
Interestingly, these values were obtained by considering as low as 1.3 eq. of amino acids. They considered TentaGel S resin, TCFH and 2,4,6-collidine as the model resin, coupling reagent, and base, respectively. Moreover, they recycled the waste with the aid of a mixed ion exchange resin. They were able to reuse the recycled solvent in further syntheses, achieving purity and yields compared to those obtained from the virgin solvent.
4.3 Side-chain protecting groups used in ASPPS
Among the common 20 α-amino acids, only 12 should be considered in terms of side-chain protection; His, Arg, Lys, Asp, Glu, Trp, Cys, Ser, Thr, Tyr, Asn, and Gln. However, not all of these 12 amino acids require a mandatory protecting group. For instance, Trp, Asn, and Gln can be incorporated without side-chain protection, and even Ser, Thr, and Tyr don't require protection in the so-called minimal protection scheme.110–112 To date, ASPPS approaches have used the conventional side-chain protecting groups employed in Fmoc- and Boc-SPPS strategies. Knauer et al.62 found that only the side chains of four amino acids should be protected in the course of a water-based Smoc-SPPS approach. Provided that Hojo and co-worker have considered Pms-Tyr-OH and Esc-Tyr-OH with a free side chain and successfully synthesised Met-Enkephalin and Leu-Enkephalin, respectively.94,95Indeed, Lys, Glu, Asp and Cys need side-chain protection under aqueous conditions. However, such protection is recommended for other amino acids (His, Arg, Trp, Ser, Thr, Tyr, Asn, and Gln) to overcome potential challenges associated with unprotected side chains. Table 1 lists the recommended side-chain protecting groups for Fmoc-SPPS, and Smoc-SPPS and highlights the mandatory and optional protection possibilities. As appreciated, Smoc employs side-chain protecting groups similar to Fmoc-SPPS.
| Amino acid | Fmoc strategya | Smoc strategya | Protectionb |
|---|---|---|---|
| a t-Bu = tert-butyl; Boc = tert-butyloxycarbonyl; Trt = Trityl; Acm = acetamidomethyl; Mtr = 4-methoxy-2,3,6-trimethyl-benzenesulfonyl; Pmc = 2,2,5,7,8-pentamethylchroman-6-sulfonyl; Pbf = 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl. b Side-chain protection is mandatory for these amino acids. | |||
| Asp (D) | t-Bu | t-Bu | Mandatory |
| Glu (E) | t-Bu | t-Bu | Mandatory |
| Trp (W) | Boc | Boc | Optional |
| Ser (S) | t-Bu | t-Bu | Optional |
| Thr (T) | t-Bu | t-Bu | Optional |
| Tyr (Y) | t-Bu | t-Bu | Optional |
| Cys (C) | Trt, Acm | Trt | Mandatory |
| His (H) | Trt | Trt | Optional |
| Lys (K) | Boc | Boc | Mandatory |
| Arg (R) | Mtr, Pmc, Pbf | — | Optional |
| Asn (N) | Trt | — | Optional |
| Gln (Q) | Trt | — | Optional |
5. Examples of peptides synthesized by following the ASPPS strategy
Table 2 summarizes various peptides synthesised by following the ASPPS along with the resin used, washing solvent and the cleavage protocol.| Entry | Peptide sequence | Peptide size | Synthesis strategy | Resin | Solvent | Washing | Cleavage | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Cleavage cocktail is composed of 0.5 eq. of Cu(OAc)2, 10 eq. of pyridine and 5 eq. of H2O. CLEAR = Crosslinked ethoxylate acrylate resin; MeCN = Acetonitrile; i-PrOH = Isopropylalcohol, D-A = D-Alanine; Smoc = 2,7-disulfo-9-fluorenylmethoxycarbonyl; Esc = N-ethanesulfonylethoxycarbonyl; HZB-NovaGel = Hydrazinobenzoyl NovaGel; PEG = Poly(ethylene glycol); Pms = 2-[phenyl(methyl)sulfonio]ethyloxycarbonyl tetrafluoroborate; Sps = 2-(4-sulfophenylsulfonyl)ethoxycarbonyl; Orn(Boc) = Boc-protected Ornithine amino acid; HFIP = Hexafluoroisopropanol, TFA = Trifluoroacetic acid. | ||||||||
| 1 | Z-PL-OEt | 2 | Nano-micelles | TPGS-750-M (Surfactant) | THF:Water (10%:90%) |
-1 M HCl | — | 40 and 100 |
| -aq.Na2CO3 | ||||||||
| 2 | Z-AFL-OEt | 3 | Nano-micelles | TPGS-750-M (Surfactant) | THF:Water (10%:90%) |
-1 M HCl | — | 100 |
| -aq.Na2CO3 | ||||||||
| 3 | Smoc-VVIA-NH2 | 4 | Smoc-SPPS | Rink amide-ChemMatrix | Water | Water | 95% TFA | 62 |
| 4 | H-YGGFL-NH2 (Leu-Enkephalin amide) | 5 | Smoc-SPPS | Rink amide-ChemMatrix | Water | Water | 96% TFA | 62 |
| 5 | H-YGGFM-OH (Met-Enkephalin) | 5 | Smoc-SPPS | HMPB-ChemMatrix resin | MeCN:H2O (30%:70%) |
Water | 20% HFIP (in DCM) | 62 |
| 6 | H-YGGFL-NH2 (Leu-Enkephalin amide) | 5 | Fmoc-SPPS | TentaGel S | PolarClean:Water (20%:80) |
i-PrOH | 90% TFA | 41 |
| 7 | H-YGGFL-NH2 (Leu-Enkephalin amide) | 5 | Esc-SPPS | Rink amide-PEG resin | Water | Water | 94% TFA | 94 and 96 |
| 8 | H-YGGFL-NH2 (Leu-Enkephalin amide) | 5 | Pms-SPPS | PEG-grafted Rink amide resin | Water | Water | 94% TFA | 92 |
| 9 | H-YGGFL-NH2 (Leu-Enkephalin amide) | 5 | Sps-SPPS | Rink amide-TentaGel resin | EtOH:H2O (50%:50%) |
Water | 94% TFA | 91 |
| 10 | H-YGGFM-NH2 (Met-enkephalin amide) | 5 | Pms-SPPS | CLEAR resin | 0.5% Triton X in water | Water | 94% TFA | 93 |
| 11 | H-Y(D-A)GFL-OH (Leuphasyl) | 5 | Smoc-SPPS | HMPB-ChemMatrix | MeCN:H2O (20%:80%) |
Water | 1% TFA | 62 |
| 12 | H-YGGFL-NH2 (Leu-Enkephalin amide) | 5 | Fmoc-SPPS (using water-dispersible nanoparticulate Fmoc-amino acids) | Rink amide TentaGel resin | Water | -Water | 92% TFA | 97 and 98 |
| -EtOH:H2O (50%:50%) |
||||||||
| 13 | H-VAVAG-OH | 5 | Boc-SPPS (using water-dispersible nanoparticulate Boc-amino acids) | HMBA-PEG grafted resin | Water | 0.05 M NaHCO3 solution (Neutralization), followed by washing with H2O | 0.5 M NaOH | 63 and 64 |
| 14 | H-YGGFL-OH (Leu-Enkephalin) | 5 | Boc-SPPS (using water-dispersible nanoparticulate Boc-amino acids) | HMBA-PEG grafted resin | Water | 0.05 M NaHCO3 solution (Neutralization), followed by washing with H2O | 0.5 M NaOH | 64 |
| 15 | H-YGGFL-OH (Leu-Enkephalin) | 5 | Boc-SPPS (Using microwave-assisted heating) | HZB-NovaGel | Water | -Water | Cu(OAc)2a pyridine H2O |
46 |
| -0.5% Triton-X100 (aq) | ||||||||
| 16 | H-YHTVGR-NH2 (Neuropeptide W-30 (10–15)) | 6 | Fmoc-SPPS (using water-dispersible nanoparticulate Fmoc-amino acids and microwave-assisted heating) | Rink amide-PEG grafted resin | Water | Water then DMF | 94% TFA | 99 |
| 17 | H-Y(D-A)FGYPS-NH2 (Dermorphinamide) | 7 | Fmoc-SPPS (using water-dispersible nanoparticulate Fmoc-amino acids) | Rink amide TentaGel resin | Water | -Water | 92% TFA | 98 |
| -EtOH:H2O (50%:50%) |
||||||||
| 18 | H-RRRRRRR-NH2 (heptaarginine cell-penetrating peptide) | 7 | Smoc-SPPS | Rink amide-ChemMatrix | MeCN:H2O (30%:70%) |
Water | 95% TFA | 62 |
| 19 | Z-(D-F)PV-Orn(Boc)L(D-F)PV-OMe | 8 | Nano-micelles | TPGS-750-M (Surfactant) | THF:Water (10%:90%) |
-1 M HCl | — | 40 and 100 |
| -aq.Na2CO3 | ||||||||
| 20 | Vasopressin | 9 | Smoc-SPPS | Rink amide-ChemMatrix | MeCN:H2O (30%:70%) |
Water | 90% TFA | 62 |
| 21 | Oxytocin | 9 | Smoc-SPPS | Rink amide-ChemMatrix | MeCN:H2O (30%:70%) |
Water | 90% TFA | 62 |
| 22 | H-VQAAIDYING-NH2 ((ACP) 65–74) | 10 | Smoc-SPPS | Rink amide-ChemMatrix | MeCN:H2O (50%:50%) |
Water | 95% TFA | 62 |
| 23 | Z-(D-F)PV-Orn(Boc)L(D-F)PVOrn(Boc)L-OMe | 10 | Nano-micelles | TPGS-750-M (Surfactant) | THF:Water (10%:90%) |
-1 M HCl | — | 40 and 100 |
| -aq.Na2CO3 | ||||||||
| 24 | H-VQAAIDYING-NH2 ((ACP) 65–74) | 10 | Fmoc-SPPS (using water-dispersible nanoparticulate Fmoc-amino acids) | Rink amide TentaGel resin | Water | -Water | 92% TFA | 98 |
| -EtOH:H2O (50%:50%) |
||||||||
As it can be noticed from Table 2, ASPPS is able to deliver various peptides from 2-mer to 10-mer in length. Provided that some syntheses showed lower racemization than those carried out in common organic solvents.99,108 Moreover, with the minimal protection strategy, the amount of TFA could be lowered as well leading to higher productivity and reducing the unnecessary consumption of solvents and the generated waste afterwards.94,95,112
6. Conclusion and outlook
In the context of making SPPS greener, various groups have investigated the use of water as the principal solvent in the so-called ASPPS. As a starting point, reagents and strategies already available in the conventional SPPS toolbox have been used with some adaption to the presence of water. Thus, from the resin perspective, PEG-based resins such as ChemMatrix, PEG-PS, TentaGel, and PEGA emerge as the most promising options. However, the recently described Li-resin,113 which is a fiber-based polyacrylamide resin with excellent swelling capacity in water, could provide an optimal alternative to PEG-based resins. All the above-mentioned resins are microporous and show good swelling in solvents used in SPPS. However, it would be interesting to address the use of solid supports that do not swell in any solvent. Regarding the coupling cocktail for ASPPS, while EDC·HCl emerges as the reagent of choice, the search for a more water-friendly additive is recommendable. While Sulfo-HOSu is water-compatible, HOSu derivatives are known to show poor reactivity compared with other additives.114 Thus, the use of glyceroacetonide-Oxyma, which is the water-compatible version of OxymaPure, deserves further attention. To date, up to decapeptides have been synthesized using this reagent and Z-, Boc-, Fmoc-, and Ac-amino acids.115 Furthermore, the development of some stand-alone water-compatible coupling reagents should be pursued. In this regard, a major transformation of the full protection scheme should be attempted. The pioneering work of Kawasaki and co-workers, followed by Hojo, and finally by Kolmar should materialize in the development of water-compatible side-chain protecting groups. Of course, the protection of the α-amino protecting group should be further evaluated and an acid-labile group may be relevant. This strategy would imply the development of side-chain protecting groups and linkers that are labile through another chemical mechanism. It is important to have in consideration that some research groups have proved the applicability of the so-called “minimal protection approach” in ASPPS as well.94,95 It is also important to highlight that the implementation of techniques and strategies from nanotechnology should continue.In conclusion, although progress has been made in ASPPS, there is still considerable scope for both the optimization of current methodologies and the development of new approaches.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work in the author's laboratory is partially funded by the National Research Foundation (Blue Skies) (South Africa) and Al-Balqa Applied University, Jordan.References
- D. M. M. Jaradat, Amino Acids, 2018, 50, 39–68, DOI:10.1007/s00726-017-2516-0.
- D. M. M. Jaradat, N. Al-Karablieh, B. H. M. Zaarer, W. Li, K. K. Y. Saleh, A. J. Rasras, S. Abu-Romman, N. M. O'Brien-Simpson and J. D. Wade, Biol. Chem., 2021, 402, 513–524, DOI:10.1515/hsz-2020-0351.
- M. Cobb, PLoS Biol., 2017, 15, 1–8, DOI:10.1371/journal.pbio.2003243.
- T. Shandala, E. J. Parkinson-Lawrence and D. A. Brooks, Encycl. Life Sci., 2011, 1–9, DOI:10.1002/9780470015902.a0005716.pub2.
- D. M. M. Jaradat, Methods Mol. Biol., 2022, 2371, 19–29, DOI:10.1007/978-1-0716-1689-5_2.
- D. M. M. Jaradat, K. K. Y. Saleh, B. H. M. Za'arir, T. Arafat, K. H. Alzoubi, S. A. Al-Taweel, E. Mallah, M. A. Haddad and B. A. K. Haimur, Int. J. Pept. Res. Ther., 2019, 25, 1095–1102, DOI:10.1007/s10989-018-9757-y.
- T. Kimmerlin and D. Seebach, J. Pept. Res., 2005, 65, 229–260, DOI:10.1111/j.1399-3011.2005.00214.x.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2018, 26, 2700–2707, DOI:10.1016/j.bmc.2017.06.052.
- A. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382–1414, DOI:10.1021/acs.jmedchem.7b00318.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325, DOI:10.1038/s41573-020-00135-8.
- D. Al Shaer, O. Al Musaimi, F. Albericio and B. G. de la Torre, Pharmaceuticals, 2022, 15, 222, DOI:10.3390/ph15020222.
- Peptide Therapeutics Market, Global Industry Analysis, Size, Share, Growth, Trends and Forecast 2019–2027, Transparency Market Research, 2019 Search PubMed.
- L. Ferrazzano, M. Catani, A. Cavazzini, G. Martelli, D. Corbisiero, P. Cantelmi, T. Fantoni, A. Mattellone, C. De Luca, S. Felletti, W. Cabri and A. Tolomelli, Green Chem., 2022, 24, 975–1020, 10.1039/D1GC04387K.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312, 10.1039/b918763b.
- J. Sherwood, H. L. Parker, K. Moonen, T. J. Farmera and A. J. Hunt, Green Chem., 2016, 18, 3990–3996, 10.1039/c6gc00932h.
- J. Garcia-Martinez, Angew. Chem., Int. Ed., 2021, 60, 4956–4960, DOI:10.1002/anie.202014779.
- O. Al Musaimi, D. Al Shaer, B. G. De la Torre and F. Albericio, Chim. Oggi, 2021, 39, 42–45 Search PubMed https://www.teknoscienze.com/tks_article/green-circular-economy-applied-to-peptide-synthesis/ .
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149–2154, DOI:10.1021/ja00897a025.
- L. A. Carpino and G. Y. Han, J. Am. Chem. Soc., 1970, 92, 5748–5749, DOI:10.1021/ja00722a043.
- R. Behrendt, P. White and J. Offer, J. Pept. Sci., 2016, 22, 4–27, DOI:10.1002/psc.2836.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137, DOI:10.1021/op060170h.
- J. Hachmann and M. Lebl, J. Comb. Chem., 2006, 8, 149, DOI:10.1021/cc050123l.
- R. Wohlgemuth, Curr. Opin. Biotechnol., 2010, 21, 713–724, DOI:10.1016/j.copbio.2010.09.016.
- A. Přibylka, V. Krchňák and E. Schütznerová, Green Chem., 2019, 21, 775–779, 10.1039/c8gc03778g.
- M. Alhassan, O. Al Musaimi, J. M. Collins, F. Albericio and B. G. de la Torre, Green Chem., 2020, 22, 2840–2845, 10.1039/D0GC00834F.
- A. Kumar, Y. E. Jad, J. M. Collins, F. Albericio and B. G. de la Torre, ACS Sustainable Chem. Eng., 2018, 6, 8034–8039, DOI:10.1021/acssuschemeng.8b01531.
- S. B. Lawrenson, R. Arav and M. North, Green Chem., 2017, 19, 1685–1691, 10.1039/c7gc00247e.
- C. P. Ashcroft, P. J. Dunn, J. D. Hayler and A. S. Wells, Org. Process Res. Dev., 2015, 19, 740–747, DOI:10.1021/op500276u.
- O. Al Musaimi, B. G. de la Torre and F. Albericio, Green Chem., 2020, 22, 996–1018, 10.1039/c9gc03982a.
- Y. E. Jad, T. Govender, H. G. Kruger, A. El-Faham, B. G. de la Torre and F. Albericio, Org. Process Res. Dev., 2017, 21, 365–369, DOI:10.1021/acs.oprd.6b00439.
- J. Pawlas and J. H. Rasmussen, Green Chem., 2019, 21, 5990–5998, 10.1039/c9gc02775k.
- A. Přibylka, M. Pastorek, M. Grepl and E. P. Schütznerová, Tetrahedron, 2021, 99, 132452, DOI:10.1016/j.tet.2021.132452.
- J. Pawlas, B. Antonic, M. Lundqvist, T. Svensson, J. Finnman and J. H. Rasmussen, Green Chem., 2019, 21, 2594–2600, 10.1039/c9gc00898e.
- L. Ferrazzano, D. Corbisiero, G. Martelli, A. Tolomelli, A. Viola, A. Ricci and W. Cabri, ACS Sustainable Chem. Eng., 2019, 7, 12867–12877, DOI:10.1021/acssuschemeng.9b01766.
- Y. Ran, F. Byrne, I. D. V. Ingram and M. North, Chemistry, 2019, 25, 4951–4964, DOI:10.1002/chem.201900228.
- S. Lawrenson, M. North, F. Peigneguy and A. Routledge, Green Chem., 2017, 19, 952–962, 10.1039/c6gc03147a.
- O. Al Musaimi, Y. E. Jad, A. Kumar, J. M. Collins, A. Basso, B. G. de la Torre and F. Albericio, Curr. Opin. Green Sustainable Chem., 2018, 11, 99–103, DOI:10.1016/j.cogsc.2018.06.017.
- Y. E. Jad, A. Kumar, A. El-Faham, B. G. de la Torre and F. Albericio, ACS Sustainable Chem. Eng., 2019, 7, 3671–3683, DOI:10.1021/acssuschemeng.8b06520.
- Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2015, 13, 2393–2398, 10.1039/c4ob02046d.
- M. Cortes-Clerget, J. Y. Berthon, I. Krolikiewicz-Renimel, L. Chaisemartin and B. H. Lipshutz, Green Chem., 2017, 19, 4263–4267, 10.1039/c7gc01575e.
- J. Pawlas and J. H. Rasmussen, ChemSusChem, 2021, 14, 3231–3236, DOI:10.1002/cssc.202101028.
- Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Amino Acids, 2016, 48, 419–426, DOI:10.1007/s00726-015-2095-x.
- G. A. Acosta, M. del Fresno, M. Paradis-Bas, M. Rigau-DeLlobet, S. Côte, M. Royo and F. Albericio, J. Pept. Sci., 2009, 15, 629–633, DOI:10.1002/psc.1158.
- A. Kumar, A. Niyi, K. P. Nandhini, J. M. Collins, F. Albericio and B. G. de la Torre, Org. Process Res. Dev., 2019, 1096–1100, DOI:10.1021/acs.oprd.9b00073.
- Y. E. Jad, G. A. Acosta, T. Govender, H. G. Kruger, A. El-Faham, B. G. de la Torre and F. Albericio, ACS Sustainable Chem. Eng., 2016, 4, 6809–6814, DOI:10.1021/acssuschemeng.6b01765.
- A. S. Galanis, F. Albericio and M. Grøtli, Org. Lett., 2009, 11, 4488–4491, DOI:10.1021/ol901893p.
- A. Kumar, Y. E. Jad, A. El-Faham, B. G. de la Torre and F. Albericio, Tetrahedron Lett., 2017, 58, 2986–2988, DOI:10.1016/j.tetlet.2017.06.058.
- J. Lopez, S. Pletscher, A. Aemissegger, C. Bucher and F. Gallou, Org. Process Res. Dev., 2018, 22, 494–503, DOI:10.1021/acs.oprd.7b00389.
- K. Kawasaki, T. Tsuji, M. Maeda, T. Matsumoto and K. Hirase, Chem. Pharm. Bull., 1987, 35, 1044–1048 CrossRef CAS https://www.jstage.jst.go.jp/article/cpb1958/35/3/35_3_1044/_pdf .
- A. R. Vaino and K. D. Janda, J. Comb. Chem., 2000, 2, 579–596, DOI:10.1021/cc000046o.
- C. Blackburn, Pept. Sci., 1998, 47, 311–351 CrossRef CAS.
- F. García-Martín, M. Quintanar-Audelo, Y. García-Ramos, L. J. Cruz, C. Gravel, R. Furic, S. Côté, J. Tulla-Puche and F. Albericio, J. Comb. Chem., 2006, 8, 213–220, DOI:10.1021/cc0600019.
- S. Zalipsky, J. L. Chang, F. Albericio and G. Barany, React. Polym., 1994, 22, 243–258, DOI:10.1016/0923-1137(94)90122-8.
- J. H. Adams, R. M. Cook, D. Hudson, V. Jammalamadaka, M. H. Lyttle and M. F. Songster, J. Org. Chem., 1998, 63, 3706–3716, DOI:10.1021/jo9802269.
- E. Bayer, Angew. Chem., Int. Ed. Engl., 1991, 30, 113–216, DOI:10.1002/anie.199101133.
- E. Atherton, D. L. J. Clive and R. C. Sheppard, J. Am. Chem. Soc., 1975, 97, 6584–6585 CrossRef CAS PubMed.
- M. Meldal, Tetrahedron Lett., 1992, 33, 3077–3080, DOI:10.1016/S0040-4039(00)79604-3.
- R. H. Berg, K. Almdal, W. B. Pedersen, A. Holm, J. P. Tam and R. B. Merrifield, J. Am. Chem. Soc., 1989, 111, 8024–8026, DOI:10.1021/ja00202a059.
- F. García-Martin and F. Albericio, Chim. Oggi, 2008, 26, 29–34 Search PubMed.
- S. A. Kates, B. F. McGuinness, C. Blackburn, G. W. Griffin, N. A. Solé, G. Barany and F. Albericio, Biopolymers, 1998, 47, 365–380, DOI:10.1002/(sici)1097-0282(1998)47:5<365::Aid-bip4>3.0.Co;2-8.
- R. Quarrell, T. D. W. Claridge, G. W. Weaver and G. Lowe, Mol. Diversity, 1996, 1, 223–232, DOI:10.1007/BF01715526.
- S. Knauer, N. Koch, C. Uth, R. Meusinger, O. Avrutina and H. Kolmar, Angew. Chem., Int. Ed., 2020, 59, 12984–12990, DOI:10.1002/anie.202003676.
- K. Hojo, N. Shinozaki, Y. Nozawa, Y. Fukumori and H. Ichikawa, Appl. Sci., 2013, 3, 614–623, DOI:10.3390/app3030614.
- K. Hojo, S. Fujiwara, H. Inai, Y. Manabe and Y. Tsuda, Int. J. Pept. Res. Ther., 2022, 28, 1–10, DOI:10.1007/s10989-021-10354-1.
- K. Hojo, H. Ichikawa, M. Maeda, S. Kida, Y. Fukumori and K. Kawasaki, J. Pept. Sci., 2007, 13, 493–497, DOI:10.1002/psc.874.
- J. Sheehan, P. Cruickshank and G. Boshart, J. Org. Chem., 1961, 26, 2525–2528, DOI:10.1021/jo01351a600.
- J. C. Sheehan and G. P. Hess, J. Am. Chem. Soc., 1955, 77, 1067–1068, DOI:10.1021/ja01609a099.
- N. L. Benoiton and F. M. F. Chen, J. Chem. Soc., Chem. Commun., 1981, 543–545, 10.1039/C39810000543.
- M. Bodanszky and V. Du Vigneaud, Nature, 1959, 183, 1324–1325, DOI:10.1038/1831324b0.
- W. Konig and R. Geiger, Chem. Ber., 1970, 103, 788–798 CrossRef CAS PubMed https://www.ncbi.nlm.nih.gov/pubmed/5436656 .
- J. Kovacs, L. Kisfaludy and M. Q. Ceprini, J. Am. Chem. Soc., 1967, 89, 183–184, DOI:10.1021/ja00977a059.
- J. Kovacs, M. Q. Ceprini, C. A. Dupraz and G. N. Schmit, J. Org. Chem., 1967, 32, 3696–3698, DOI:10.1021/jo01286a098.
- G. W. Anderson, J. E. Zimmerman and F. M. Callahan, J. Am. Chem. Soc., 1963, 85, 3039–3039, DOI:10.1021/ja00902a047.
- F. Albericio, M. Cases, J. Alsina, S. A. Triolo, L. A. Carpino and S. A. Kates, Tetrahedron Lett., 1997, 38, 4853–4856, DOI:10.1016/S0040-4039(97)01011-3.
- G. Gawne, G. W. Kenner and R. C. Sheppard, J. Am. Chem. Soc., 1969, 91, 5669–5671, DOI:10.1021/ja01048a057.
- B. Castro, J. R. Dormoy, G. Evin and C. Selve, Tetrahedron Lett., 1975, 16, 1219–1222, DOI:10.1016/S0040-4039(00)72100-9.
- J. Coste, E. Frerot and P. Jouin, J. Org. Chem., 1994, 59, 2437–2446 CrossRef CAS.
- J. Coste, D. Le-Nguyen and B. Castro, Tetrahedron Lett., 1990, 31, 205–208, DOI:10.1016/S0040-4039(00)94371-5.
- R. Subirós-Funosas, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2010, 8, 3665–3673, 10.1039/c003719b.
- R. Knorr, A. Trzeciak, W. Bannwarth and D. Gillessen, Tetrahedron Lett., 1989, 30, 1927–1930, DOI:10.1016/S0040-4039(00)99616-3.
- A. El-Faham, R. Subirós Funosas, R. Prohens and F. Albericio, Chemistry, 2009, 15, 9404–9416, DOI:10.1002/chem.200900615.
- V. Dourtoglou, B. Gross, V. Lambropoulou and C. Zioudrou, Synthesis, 1984, 572–574, DOI:10.1055/s-1984-30895.
- V. Dourtoglou, J.-C. Ziegler and B. Gross, Tetrahedron Lett., 1978, 15, 1269–1272, DOI:10.1016/0040-4039(78)80103-8.
- I. Abdelmoty, F. Albericio, L. A. Carpino, B. M. Foxman and S. A. Kates, Lett. Pept. Sci., 1994, 1, 57–67, DOI:10.1007/BF00126274.
- L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397–4398, DOI:10.1021/ja00063a082.
- R. F. Poulain, A. L. Tartar and B. T. P. Déprez, Tetrahedron Lett., 2001, 42, 1495–1498, DOI:10.1016/S0040-4039(00)02293-0.
- O. Marder, Y. Shvo and F. Albericio, Chim. Oggi, 2002, 20, 37–41 CAS.
- F. Albericio, J. M. Bofill, A. El-Faham and S. A. Kates, J. Org. Chem., 1998, 63, 9678–9683, DOI:10.1021/jo980807y.
- M. Fujino, S. Kobayashi, M. Obayashi, T. Fukuda, S. Shinagawa and O. Nishimura, Chem. Pharm. Bull., 1974, 22, 1857–1863, DOI:10.1248/cpb.22.1857.
- M. Kunishima, C. Kawachi, J. Morita, K. Terao, F. Iwasaki and S. Tani, Tetrahedron, 1999, 55, 13159–13170, DOI:10.1016/S0040-4020(99)00809-1.
- K. Hojo, M. Maeda, N. Tanakamaru, K. Mochida and K. Kawasaki, Protein Pept. Lett., 2006, 13, 189–192, DOI:10.2174/092986606775101607.
- K. Hojo, M. Maeda and K. Kawasaki, J. Pept. Sci., 2001, 7, 615–618, DOI:10.1002/psc.361.
- K. Hojo, M. Maeda, Y. Takahara, S. Yamamoto and K. Kawasaki, Tetrahedron Lett., 2003, 44, 2849–2851, DOI:10.1016/s0040-4039(03)00470-2.
- K. Hojo, M. Maeda, T. Smith, E. Kita, F. Yamaguchi, S. Yamamoto and K. Kawasaki, Chem. Pharm. Bull., 2004, 52, 422–427, DOI:10.1248/cpb.52.422.
- K. Hojo, M. Maeda and K. Kawasaki, Tetrahedron, 2004, 60, 1875–1886, DOI:10.1016/j.tet.2003.12.036.
- K. Hojo, H. Ichikawa, M. Maeda, S. Kida, Y. Fukumori and K. Kawasaki, J. Pept. Sci., 2007, 13, 493–497, DOI:10.1002/psc.874.
- K. Hojo, A. Hara, H. Kitai, M. Onishi, H. Ichikawa, Y. Fukumori and K. Kawasaki, Chem. Cent. J., 2011, 5, 49, DOI:10.1186/1752-153X-5-49.
- K. Hojo, H. Ichikawa, A. Hara, M. Onishi, K. Kawasaki and Y. Fukumori, Protein Pept. Lett., 2012, 19, 1231–1236, DOI:10.2174/092986612803217114.
- K. Hojo, N. Shinozaki, K. Hidaka, Y. Tsuda, Y. Fukumori, H. Ichikawa and J. D. Wade, Amino Acids, 2014, 46, 2347–2354, DOI:10.1007/s00726-014-1779-y.
- M. Cortes-Clerget, N. R. Lee and B. H. Lipshutz, Nat. Protoc., 2019, 14, 1108–1129, DOI:10.1038/s41596-019-0130-1.
- J. Pawlas, T. Nuijens, J. Persson, T. Svensson, M. Schmidt, A. Toplak, M. Nilsson and J. H. Rasmussen, Green Chem., 2019, 21, 6451–6467, 10.1039/c9gc03600h.
- G. Martelli, P. Cantelmi, A. Tolomelli, D. Corbisiero, A. Mattellone, A. Ricci, T. Fantoni, W. Cabri, F. Vacondio, F. Ferlenghi, M. Mor and L. Ferrazzano, Green Chem., 2021, 23, 4095–4106, 10.1039/d1gc00910a.
- O. Al Musaimi, Y. E. Jad, A. Kumar, A. El-Faham, J. M. Collins, A. Basso, B. G. de la Torre and F. Albericio, Org. Process Res. Dev., 2018, 22, 1809–1816, DOI:10.1021/acs.oprd.8b00335.
- A. Přibylka, V. Krchňák and E. Schütznerová, J. Org. Chem., 2020, 85, 8798–8811 CrossRef PubMed.
- K. Hojo, M. Maeda and K. Kawasaki, Tetrahedron Lett., 2004, 45, 9293–9295, DOI:10.1016/j.tetlet.2004.10.095.
- K. Hojo, M. Maeda and K. Kawasaki, Tetrahedron, 2004, 60, 1875–1886, DOI:10.1016/j.tet.2003.12.036.
- K. Hojo, A. Hara, H. Kitai, M. Onishi, H. Ichikawa, Y. Fukumori and K. Kawasaki, Chem. Cent. J., 2011, 5, 1–10, DOI:10.1186/1752-153X-5-49.
- K. Hojo, N. Shinozaki, A. Hara, M. Onishi, Y. Fukumori and H. Ichikawa, Protein Pept. Lett., 2013, 20, 1122–1128, DOI:10.2174/0929866511320100006.
- J. Pawlas and J. H. Rasmussen, ChemSusChem, 2021, 14, 3231–3236, DOI:10.1002/cssc.202101028.
- D. H. Coy and N. Branyas, Int. J. Pept. Protein Res., 1979, 14, 339–343, DOI:10.1111/j.1399-3011.1979.tb01941.x.
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504, DOI:10.1021/cr800323s.
- Y. Yang, L. Hansen and P. Ryberg, Org. Process Res. Dev., 2022, 26, 1520–1530, DOI:10.1021/acs.oprd.2c00083.
- D. C. Akintayo, B. G. de la Torre, Y. Li and F. Albericio, Polymers, 2022, 14, 928, DOI:10.3390/polym14050928.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602, DOI:10.1021/cr100048w.
- Y. Wang, B. A. Aleiwi, Q. Wang and M. Kurosu, Org. Lett., 2012, 14, 4910–4913, DOI:10.1021/ol3022337.
| This journal is © The Royal Society of Chemistry 2022 |