
Zirconium-catalysed hydrosilylation of esters and depolymerisation of polyester plastic waste†
Marie
Kobylarski‡
,
Liam J.
Donnelly‡
 *,
Jean-Claude
Berthet
and
Thibault
Cantat
*
*,
Jean-Claude
Berthet
and
Thibault
Cantat
*
Université Paris-Saclay, CEA, CNRS, NIMBE, 91191 Gif-sur-Yvette, France. E-mail: liam.donnelly@strath.ac.uk; thibault.cantat@cea.fr
First published on 27th August 2022
Abstract
Schwartz's reagent, Cp2Zr(H)Cl, has traditionally been used as a stoichiometric reagent for the reduction of unsaturated organic molecules. Recently, methods to use Cp2Zr(H)Cl as a catalyst have been developed through turnover of the [Zr]–X intermediates, formed upon reaction with an organic substrate, by hydride reagents. Herein, we report the development of a new catalytic pathway for the reduction of esters that uses the bench-stable silane Me(OMe)2SiH (DMMS) as a mild stoichiometric reductant and Cp2Zr(H)Cl as the catalyst. This system exploits the regeneration of [Zr]–H through the σ-bond metathesis of [Zr]–OR with [Si]–H to achieve catalyst turnover. These reaction conditions tolerate a range of reducible functional groups (e.g. alkyne, alkene, and nitro) and give high yields of the corresponding alcohols (up to 91% isolated yield). This methodology also proved fruitful for the reductive depolymerisation of a variety of polyesters found in household plastic waste.
The monohydride Zr(VI) complex Schwartz's reagent (Cp2Zr(H)Cl) (1) was first reported by Wailes and Weigold in the 1970s.1–3 Complex 1 has been shown to be a versatile stoichiometric reducing agent for unsaturated organic compounds.4 This reactivity is enabled by the availability of an empty valence orbital on the zirconium centre that allows the coordination of the substrate, followed by reduction via a hydrozirconation pathway. Stoichiometric hydrozirconation using complex 1 was demonstrated for alkenes,5 alkynes,3 amides,6,7 nitriles,8 imines,9 isocyanates,10 phosphine oxides,11 and ketones.12 In 1996, Majoral and Skowronska developed the first stoichiometric application of complex 1 for the reductive cleavage of ester groups in lactones and cyclic anhydrides.13 They obtained the corresponding diol derivatives in excellent yields at room temperature after 1 h (Scheme 1). Although these results were limited in terms of scope and mechanistic investigation, they highlighted the efficiency of Schwartz's reagent (1) as a reducing agent for ester functionalities.
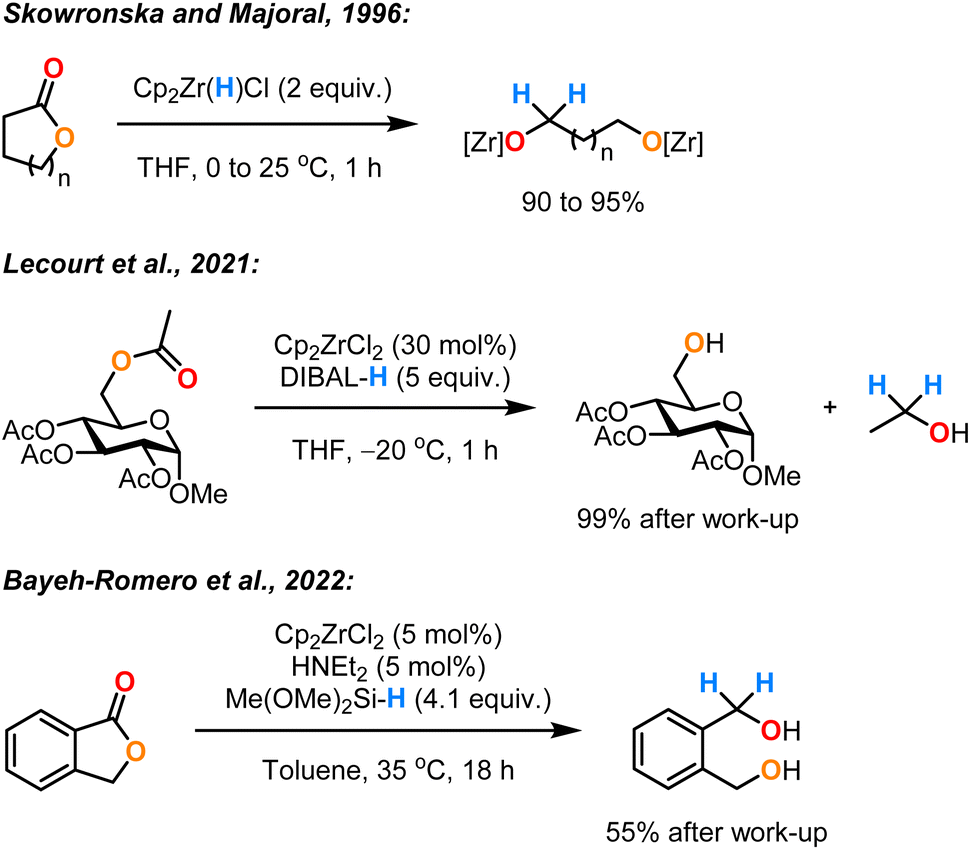 | ||
| Scheme 1 Limited previous examples of ester reduction mediated/catalysed by [Zr]–H. | ||
Only quite recently, there has been a growing interest in using zirconocene complexes as catalysts combined with hydride donors for the reduction of carbonyl groups. In 2019 and 2021, Lecourt et al. described the reduction of primary acetates using Cp2ZrCl2 as the catalyst and diisobutylaluminium hydride (DIBAL-H) as the reductant.14,15 However, this required an excess of pyrophoric reagents, a high catalyst loading (30 mol%) and cryogenic temperatures (−20 to −30 °C). In 2022, Bayeh-Romero et al. reported a single example of the reduction of an ester (a lactone) by the in situ preparation of the catalyst 1 from Cp2ZrCl2 and Me(OMe)2SiH (DMMS) in the presence of Et2NH.16 The desired diol was obtained in a moderate yield (55%) even after an extended reaction time (24 h).
Recently, we proved that Cp2Zr(H)Cl (1) could also catalyse the partial reduction of secondary amides to imines.17 This reaction proceeded with high chemoselectivity, avoiding over reduction to amine products. The reactivity was enabled by the exquisite inherent selectivity of the catalyst and the use of hydrosilanes as mild reductants. Given that amides with their poor carbonyl electrophilicity are more difficult to reduce than esters and inspired by the previous examples of Zr-catalysed/mediated ester reduction, we herein describe the first general and practical method for the [Zr]–H catalysed hydrosilylation of linear monomeric esters into their alcohol derivatives (Scheme 2). To demonstrate the robustness of our methodology, we expanded the substrate scope to include a range of polyester plastic materials. By overcoming the inherent low reactivity of these polymer materials due to their lack of solubility, inhomogeneity and relatively low purity, we provide an example of chemical upcycling by transforming waste materials into value-added monomer products.18–20
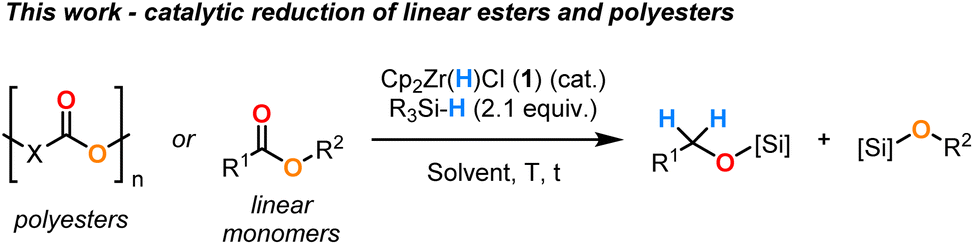 | ||
| Scheme 2 Development of a general procedure for Zr-catalysed ester hydrosilylation. | ||
Based on the work of Bayeh-Romero et al. and our previous work on the reduction of secondary amides,16,17 initial conditions were applied to the reduction of ethyl 4-methylbenzoate 2a, using 10 mol% of Cp2Zr(H)Cl (1) with 2.1 equiv. of DMMS in THF-d8 at 80 °C (Table 1, entry 1). After only 1 h, the corresponding 4-methylbenzyl silylated ether 3aSi was obtained in a yield of >95%. Reducing the catalyst loading to 5 mol% required an extended reaction time of 2 h to reach a similar yield in 3a. The conditions using 1 mol% of catalyst 1 led to the reaction stalling at 81% yield after 18 h (Table 1, entries 2 and 3). To study the effect of varying the reductant, we tested using 5 mol% of catalyst 1 with different hydrosilanes as reductants (Table 1, entries 4 to 7). Polymethylhydrosiloxane (PMHS), Et3SiH and Ph3SiH only gave trace yields of 3a, whereas (EtO)3SiH gave >95% yield after 5 h. This highlights the importance of having a more Lewis acidic silicon centre on the hydrosilane, as noted previously for the reduction of secondary amides.17 A solvent screen showed a rate increase in C6D6, with >95% yield of 3a already after 1 h (Table 1, entry 8). Similar reactivity to that in THF-d8 was observed in CD2Cl2 and in anisole (Table 1, entries 2, 9 and 10), which is regarded as a non-toxic and biodegradable solvent according to the CHEM21 guide.21 The influence of the temperature was investigated and full conversion was achieved at 25 °C after 12 h and at 60 °C after 2 h (Table 1, entries 11 and 12). Exchanging DMMS for pinacolborane (HBpin) resulted in the efficient reduction of ester 2a after 2 h at 60 °C, demonstrating that reductants based on boron are also applicable to this system, although only sluggish reactivity was observed using catecholborane (HBCat) (Table 1, entries 13 and 14). The reaction did not proceed in the absence of the catalyst while in the absence of hydrosilane, the 5 mol% of catalyst 1 reacted quantitatively with 2a to give the alkoxyzirconocene products in trace yield (see ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | X (mol%) | Solvent | E–H | Time (h) | Yielda (%) 3a |
| a Yields measured by 1H NMR against mesitylene as an internal standard. n.d. = not detected. b Reaction conducted at 25 °C. c Reaction conducted at 60 °C. | |||||
| 1 | 10 | THF-d8 | DMMS | 1 | >95 |
| 2 | 5 | THF-d8 | DMMS | 1 | 86 |
| 2 | >95 | ||||
| 3 | 1 | THF-d8 | DMMS | 1 | 48 |
| 18 | 81 | ||||
| 4 | 5 | THF-d8 | PMHS | 7 | <5 |
| 5 | 5 | THF-d8 | Et3SiH | 7 | n.d. |
| 6 | 5 | THF-d8 | Ph3SiH | 7 | n.d. |
| 7 | 5 | THF-d8 | (EtO)3SiH | 1 | 31 |
| 2 | 65 | ||||
| 5 | >95 | ||||
| 8 | 5 | C6D6 | DMMS | 1 | >95 |
| 9 | 5 | CD2Cl2 | DMMS | 1 | 72 |
| 2 | >95 | ||||
| 10 | 5 | Anisole | DMMS | 1 | 85 |
| 2 | >95 | ||||
| 11b | 5 | C6D6 | DMMS | 1 | 30 |
| 12 | >95 | ||||
| 12c | 5 | C6D6 | DMMS | 1 | 70 |
| 2 | >95 | ||||
| 13c | 5 | C6D6 | HBpin | 2 | >95 |
| 14 | 5 | C6D6 | HBCat | 2 | 8 |

To investigate the mechanism of this catalytic system, we first studied the elementary steps by a series of stoichiometric reactions (Fig. 1). The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 reaction of complex 1 with ester 2a in C6D6 resulted in almost 50% conversion (47%) of the ester 2a to the alkoxyzirconocene products 3aZr and 4aZr after 2 h at 80 °C. This ratio in 3aZr and 4aZr did not change upon heating for a further 16 h and at this stage, complex 1 was fully consumed. The addition of 1.1 equiv. of DMMS to this mixture gave full conversion of ester 2a with the formation of the two alkoxyzirconocene products 3aZr (36%) and 4aZr (21%) and the two silylethers 3aSi (61%) and 4aSi (78%) after 1 h at 80 °C. The ratio between these products did not change significantly upon heating for a further 25 h. The potential over reduced products (p-xylene and ethane) resulting from exhaustive deoxygenation were not detected during this experiment.
:1 reaction of complex 1 with ester 2a in C6D6 resulted in almost 50% conversion (47%) of the ester 2a to the alkoxyzirconocene products 3aZr and 4aZr after 2 h at 80 °C. This ratio in 3aZr and 4aZr did not change upon heating for a further 16 h and at this stage, complex 1 was fully consumed. The addition of 1.1 equiv. of DMMS to this mixture gave full conversion of ester 2a with the formation of the two alkoxyzirconocene products 3aZr (36%) and 4aZr (21%) and the two silylethers 3aSi (61%) and 4aSi (78%) after 1 h at 80 °C. The ratio between these products did not change significantly upon heating for a further 25 h. The potential over reduced products (p-xylene and ethane) resulting from exhaustive deoxygenation were not detected during this experiment.
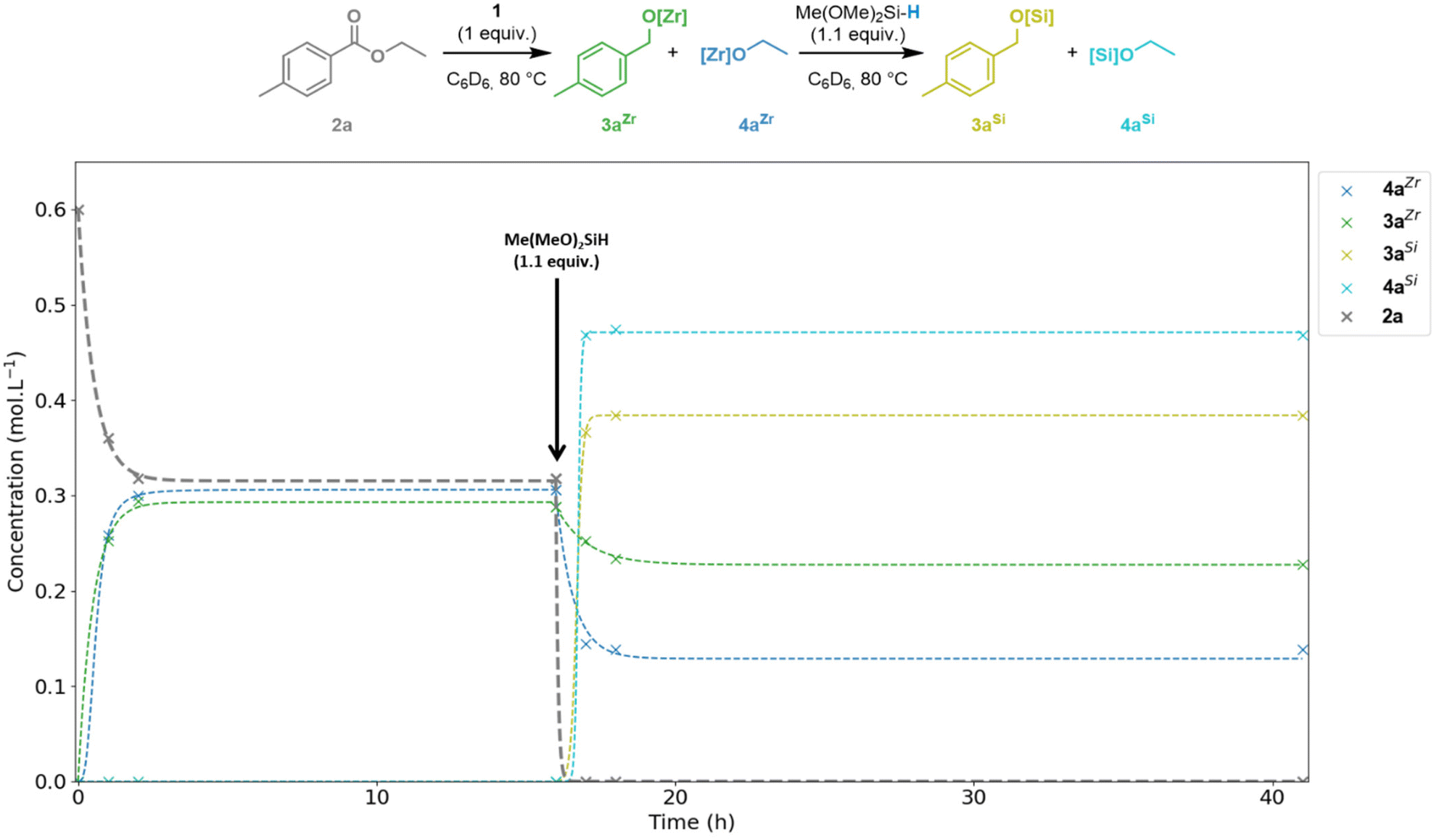 | ||
| Fig. 1 Time-step analysis by 1H NMR spectroscopy of the stepwise stoichiometric reduction of 2a by complex 1 and DMMS. | ||
As only 1.1 equiv. instead of 2 equiv. of DMMS were used, the yields in 3aSi and 4aSi cannot reach 100% and thus the presence of both zirconium- and silicon-alkoxides is expected. In line with the relative oxophilicities of Si and Zr,22 the complexes 3aZr and 4aZr are converted to the silyl ethers 3aSi and 4aSi by σ-bond metathesis of [Zr]–OR with [Si]–H with concomitant formation of Cp2Zr(H)Cl (1). The newly formed complex 1 then reacts with the remaining ∼50% of 2a. The distinct percentages in 3aZr/Si and 4aZr/Si mentioned above evidence that the metathesis reactions of the two Zr-alkoxide complexes 3aZr and 4aZr with the hydrosilane do not proceed at the same rate. The conversion from 4aZr to 4aSi is faster than 3aZr to 3aSi due to the greater affinity for the silicon atom of the more electron rich alkoxide EtO−. Buchwald and co-workers reported the use of Cp2TiCl2 in combination with hydrosilanes and n-BuLi as an activator for the selective reduction of esters.23,24 A ‘self-activating’ system was also developed using Ti(OiPr)4 as the catalyst which was proposed to proceed by σ-bond metathesis of the Ti–OiPr bond with the Si–H bond of the silane to generate a reactive Ti–H species.25 However, these methods can require high catalyst loadings (stoichiometric in some cases) and suffer from the potential generation of the flammable and toxic gas SiH4 depending on the hydrosilane used.25,26 We have now shown that Schwartz's reagent exhibits similar reactivity to these TiIV catalysts and related d0 catalysts in the hydrosilylation of esters.
Based on these results and the previous work in this area, we can propose a catalytic cycle (Scheme 3) which is similar to that proposed by Buchwald and co-workers for the catalytic hydrosilylation of esters with Ti(OiPr)4.25 After the addition of the Zr–H bond of the catalyst 1 to the carbonyl C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of the ester 2, the hemiacetal intermediate (Int-1) (not detected) degrades rapidly into the zirconium alkoxide 4Zr and aldehyde via a β-alkoxide elimination in a similar fashion to classical stoichiometric ester reduction reagents (e.g. LiAlH4). Under the catalytic conditions, the aldehyde (not detected) is immediately reduced by complex 1 to the Zr alkoxide complex 3Zr.16 The two alkoxyzirconocene complexes 3Zr and 4Zr perform a σ-bond metathesis with the hydrosilane to form the desired silyl ethers 3Si and 4Si, respectively, with concomitant regeneration of the catalyst 1.16,17
O bond of the ester 2, the hemiacetal intermediate (Int-1) (not detected) degrades rapidly into the zirconium alkoxide 4Zr and aldehyde via a β-alkoxide elimination in a similar fashion to classical stoichiometric ester reduction reagents (e.g. LiAlH4). Under the catalytic conditions, the aldehyde (not detected) is immediately reduced by complex 1 to the Zr alkoxide complex 3Zr.16 The two alkoxyzirconocene complexes 3Zr and 4Zr perform a σ-bond metathesis with the hydrosilane to form the desired silyl ethers 3Si and 4Si, respectively, with concomitant regeneration of the catalyst 1.16,17
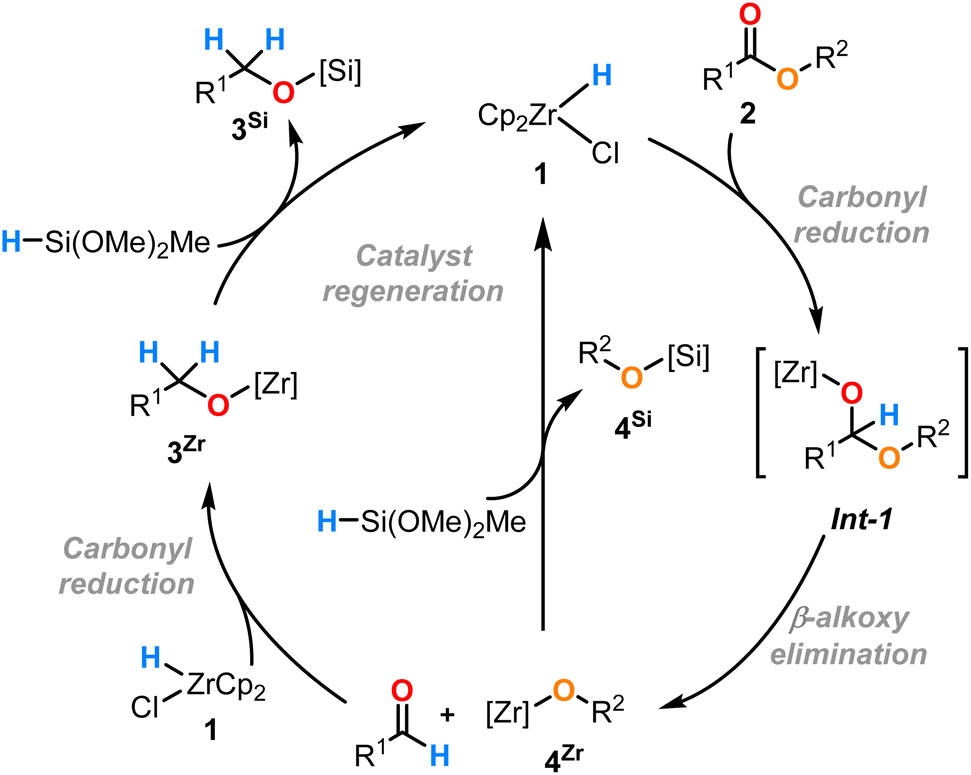 | ||
| Scheme 3 Proposed catalytic cycle for the reduction of esters 2 by complex 1. | ||
Using the optimised conditions defined above for ethyl 4-methylbenzoate 2a (Table 1, entries 2 and 8), the generality of this system was explored through a substrate scope (Scheme 4). The benzoate esters 2a and 2b and methyl phenylacetate 2c were cleanly reduced to the corresponding alcohols. Iodo- and amino-substituted esters 2d and 2e were well tolerated in this reaction with no observed hydrodehalogenation nor N–H silylation, respectively. The keto-substituted ester 2f was reduced to the diol by addition of 3.1 equiv. of DMMS. An ester containing a nitro group 2g required heating at lower temperature (40 °C) for an extended reaction time (16 h) to control the chemoselectivity. Alkynes and alkenes 2h and 2i were not reduced under these conditions with only selective ester reduction observed for these examples, although it was necessary to perform the reduction of 2h at a lower temperature of 40 °C for 16 h to ensure good chemoselectivity. ε-Caprolactone 2j was efficiently reduced to the corresponding diol. The electron-poor ester 2k was converted to the desired products but required an extended reaction time of 16 h to reach full conversion. The sterically congested ester 2l was also efficiently transformed to the alcohol products.
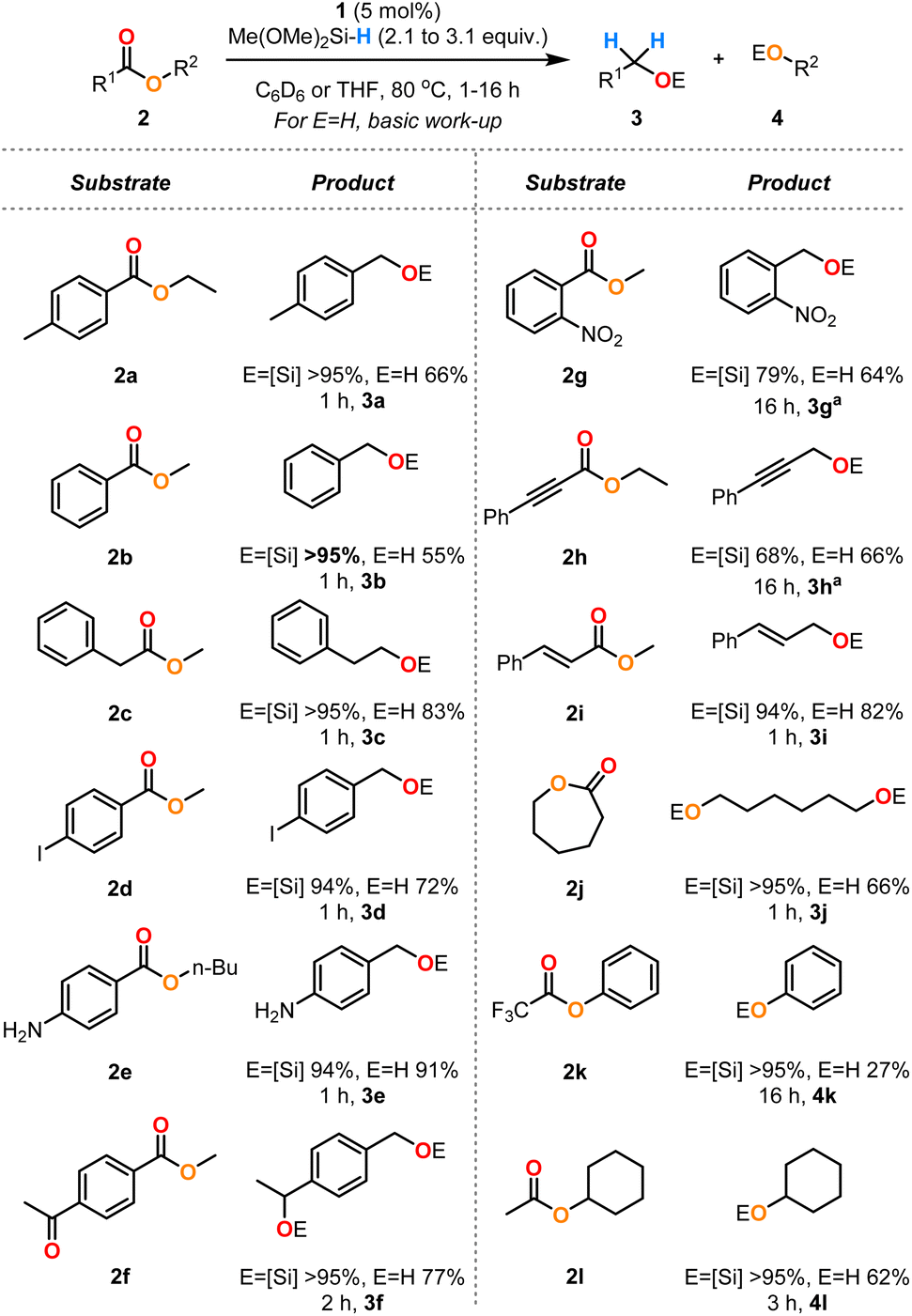 | ||
| Scheme 4 Substrate scope for the hydrosilylation of monomeric esters 2 catalysed by complex 1. The yields of the silyl ethers were measured by 1H NMR spectroscopy by integration against mesitylene as an internal standard (reactions conducted in C6D6). The yields of the alcohols are isolated yields from reactions in THF after basic workup (see ESI†). aReactions conducted at 40 °C. | ||
Given that this catalytic hydrosilylation system could reduce monomeric esters to alcohols in high efficiency, we were encouraged to apply it to the reductive depolymerisation of polyesters found in household plastic waste.27,28 Within this growing research field, our group previously reported the catalytic reductive depolymerisation of a variety of polyesters with the strongly Lewis acidic catalysts B(C6F5)3 and [Ir(POCOP)H(THF)][B(C6F5)4] (POCOP = 1,3-(tBu2PO)2C6H3) using hydrosilanes as hydride donors.29,30 Recently, Fernandes et al. performed the hydrosilylation of polyesters from commercial and waste samples using MoO2Cl2(H2O)2 and Zn(OAc)2 as catalysts.31,32 Related to this hydrosilylation reactivity, we have demonstrated that an f-element complex (La(N(SiMe3)2)3) could act as a catalyst to depolymerise polyesters and polycarbonates in the presence of hydroboranes at low catalyst loadings (1 mol%).33 However, this catalyst was inactive for the catalytic hydrosilylation of polyesters.
Therefore, we tested the above optimized conditions on three commonly used polyesters, the biodegradable synthetic polycaprolactone (PCL), two samples of polylactic acid (PLA) from blue and white 3D printer filaments and polyethylene terephthalate (PET) from an Evian© bottle (Scheme 5). Excellent yields of the desired disilyl ethers from PCL and PET were obtained with 94% of 3jSi and >95% of 3nSi and 4nSi after 4 h and 22 h, respectively, at 80 °C. Interestingly, this system evidenced a high tolerance toward additives contained in the white and blue PLA filaments, giving 3mSi as the sole product with distinct yields due to the differing additive content (in 75% and >95% respectively, after 4 h at 80 °C). In all cases, the pure diol products 3j, 3m and 3n could be readily isolated by hydrolysis of the O–Si bond under basic conditions. Hydroboranes (HBpin and HBCat) were tested in this system as boron-based reductants but were poorly reactive in comparison with DMMS (see ESI†). In the interest of improving the sustainability of this process, anisole was tested as a solvent for the depolymerisation of the three plastics. Similar yields of the corresponding disilyl ether products were reached (81–84%) but required extended reaction times (38–54 h) (see ESI†). Contrary to the hydrosilylation conditions with B(C6F5)3,30 Ir(III),29 Zn(II)31 and Mo(VI)32 catalysts which displayed poor chemoselectivity and were sensitive to the reaction conditions, the Cp2Zr(H)Cl (1) catalyst evidenced high selectivity toward the disilyl ether derivatives without formation of over-reduced products (alkanes or monosilyl ethers), which is reminiscent of the hydroboration conditions using a La(III) catalyst.33 Surprisingly, compared to the La(III)/HBpin system, the PCL depolymerisation required harsher conditions (4 h at 80 °C vs. 1 h at r.t.) and under similar experimental conditions, higher yields were obtained with PET (>95% after 22 h at 80 °C vs. 73% after 24 h at 100 °C).
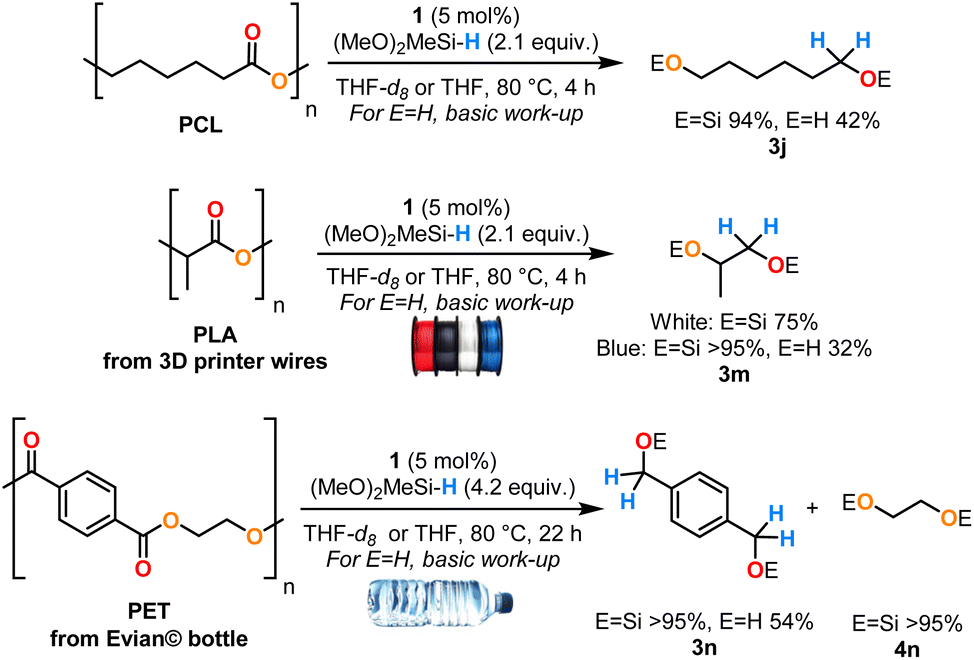 | ||
| Scheme 5 Reductive depolymerisation of polyesters catalysed by complex 1. The yields of silyl ethers were measured by 1H NMR spectroscopy by integration against mesitylene as an internal standard (reactions conducted in THF-d8). The yields of alcohols are isolated yields from reactions in THF after basic workup (see ESI†). | ||
Conclusions
In conclusion, we have developed a robust and efficient catalytic system based on the Earth-abundant metal complex Cp2Zr(H)Cl (1) with a mild and stable reductant (DMMS) for the hydrosilylation of ester compounds and materials into their alcohol derivatives. In the case of monomeric substrates, we have demonstrated excellent functional group tolerance (alkene, alkyne, amine, etc.) coupled with excellent chemoselectivity to form alcohol derivatives exclusively. With this catalytic system, a range of polyesters obtained from commercial and household waste samples (PCL, PLA and PET) were also successfully depolymerised into their monomeric diols in high yields. Mechanistic studies suggest that catalyst turnover is achieved via the metathesis of Zr–O and Si–H σ-bonds. This work highlights the use of the classical organometallic reagent Cp2Zr(H)Cl as a powerful catalyst for selective reduction reactions. New catalytic transformations using this complex are currently under investigation in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
For financial support we acknowledge CEA, CNRS, University of Paris-Saclay, European Research Council (ERC consolidator grant agreement no. 818260) and H2020 Sun-to-X project (grant agreement no. 883264). T. C. thanks the Fondation Louis D.–Institut de France for its support.References
- P. C. Wailes and H. Weigold, J. Organomet. Chem., 1970, 24, 405–411 CrossRef CAS.
- P. C. Wailes and H. Weigold, J. Organomet. Chem., 1970, 24, 413–417 CrossRef CAS.
- P. C. Wailes, H. Weigold and A. P. Bell, J. Organomet. Chem., 1971, 27, 373–378 CrossRef CAS.
- M. M. Więcław and S. Stecko, Eur. J. Org. Chem., 2018, 6601–6623 CrossRef.
- D. W. Hart and J. Schwartz, J. Am. Chem. Soc., 1974, 96, 8115–8116 CrossRef CAS.
- D. J. A. Schedler, A. G. Godfrey and B. Ganem, Tetrahedron Lett., 1993, 34, 5035–5038 CrossRef CAS.
- D. J. A. Schedler, J. Li and B. Ganem, J. Org. Chem., 1996, 61, 4115–4119 CrossRef CAS PubMed.
- G. Erker, W. Frömberg, J. L. Atwood and W. E. Hunter, Angew. Chem., Int. Ed. Engl., 1984, 23, 68–69 CrossRef.
- D. Vargová, B. Mudráková, I. Némethová and R. Šebesta, Eur. J. Org. Chem., 2019, 7606–7612 CrossRef.
- V. Pace, K. de la Vega-Hernández, E. Urban and T. Langer, Org. Lett., 2016, 18, 2750–2753 CrossRef CAS PubMed.
- M. Zablocka, B. Delest, A. Igau, A. Skowronska and J.-P. Majoral, Tetrahedron Lett., 1997, 38, 5997–6000 CrossRef CAS.
- E. Cesarotti, A. Chiesa, S. Maffi and R. Ugo, Inorg. Chim. Acta, 1982, 64, L207–L208 CrossRef CAS.
- N. Cénac, M. Zablocka, A. Igau, J.-P. Majoral and A. Skowronska, J. Org. Chem., 1996, 61, 796–798 CrossRef PubMed.
- M. Gavel, T. Courant, A. Y. P. Joosten and T. Lecourt, Org. Lett., 2019, 21, 1948–1952 CrossRef CAS PubMed.
- T. Courant, M. Gavel, R. M. Q. Renard, V. Gandon, A. Y. P. Joosten and T. Lecourt, J. Org. Chem., 2021, 86, 9280–9288 CrossRef CAS PubMed.
- R. A. Kehner, M. C. Hewitt and L. Bayeh-Romero, ACS Catal., 2022, 1758–1763 CrossRef CAS.
- L. J. Donnelly, J.-C. Berthet and T. Cantat, Angew. Chem., Int. Ed., 2022, 61, e202206170 CrossRef CAS PubMed.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- Q. Hou, M. Zhen, H. Qian, Y. Nie, X. Bai, T. Xia, M. Laiq Ur Rehman, Q. Li and M. Ju, Cell Rep. Phys. Sci., 2021, 2, 100514 CrossRef CAS.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- S. C. Berk, K. A. Kreutzer and S. L. Buchwald, J. Am. Chem. Soc., 1991, 13, 5093–5095 CrossRef.
- K. J. Barr, S. C. Berk and S. L. Buchwald, J. Org. Chem., 1994, 59, 4323–4326 CrossRef CAS.
- M. T. Reding and S. L. Buchwald, J. Org. Chem., 1995, 60, 7884–7890 CrossRef CAS.
- S. C. Berk and S. L. Buchwald, J. Org. Chem., 1992, 57, 3753–3755 CrossRef.
- C. Wang and O. El-Sepelgy, Curr. Opin. Green, 2021, 100547 CAS.
- A. C. Fernandes, Green Chem., 2021, 23, 7330–7360 RSC.
- L. Monsigny, J.-C. Berthet and T. Cantat, ACS Sustainable Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS.
- E. Feghali and T. Cantat, ChemSusChem, 2015, 8, 980–984 CrossRef CAS PubMed.
- A. C. Fernandes, ChemSusChem, 2021, 14, 4228–4233 CrossRef CAS PubMed.
- B. F. S. Nunes, M. C. Oliveira and A. C. Fernandes, Green Chem., 2020, 22, 2419–2425 RSC.
- M. Kobylarski, J.-C. Berthet and T. Cantat, Chem. Commun., 2022, 58, 2830–2833 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc02186b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |