
Redox-neutral ketyl radical coupling/cyclization of carbonyls with N-aryl acrylamides through consecutive photoinduced electron transfer†
Zhonghua
Qu
 a,
Tong
Tian
a,
Yongbo
Tan
a,
Xiaochen
Ji
a,
Guo-Jun
Deng
ab and
Huawen
Huang
*ab
a,
Tong
Tian
a,
Yongbo
Tan
a,
Xiaochen
Ji
a,
Guo-Jun
Deng
ab and
Huawen
Huang
*ab
aKey Laboratory for Green Organic Synthesis and Application of Hunan Province, Key Laboratory of Environmentally Friendly Chemistry and Application of Ministry of Education, College of Chemistry, Xiangtan University, Xiangtan 411105, China. E-mail: hwhuang@xtu.edu.cn; Fax: (+86)0731-5829-2251; Tel: (+86)0731-5829-8601
bSchool of Chemistry and Chemical Engineering, Henan Normal University. Xinxiang, 453007, P. R. China
First published on 13th July 2022
Abstract
While the reductive ketyl couplings of carbonyls have been widely explored, we report in this work on a redox-neutral umpolung carbonyl coupling reaction through ketyl radical formation by consecutive photoinduced electron transfer (ConPET) under metal- and additive-free conditions. The donor–acceptor cyanoarene-based fluorophore, 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN), was used as an efficient photocatalyst. It undergoes ConPET to form the excited radical anion (4CzIPN˙−*) possessing high reductive potential without an external electron donor. This mild and simple photocatalytic system allows highly efficient coupling/cyclization of N-aryl acrylamides and carbonyls and provides straightforward access to structurally useful hydroxyalkyl oxindoles with formal 100% atom economy.
Introduction
Reactivity discovery with polarity reversal of carbonyl compounds is a fascinating topic among methodology development in molecular synthesis. Ketyl radicals are substantial active intermediates in umpolung carbonyl coupling reactions. Traditionally, the formation of ketyl radicals from carbonyls relies on single-electron reduction by stoichiometric low-valent metal reagents such as samarium and titanium.1 Recent advances in powerful photoredox catalysis2 reveal that certain excited metal complexes and organic photocatalysts with high reductive potential could initiate electron transfer to protonated carbonyls (proton-coupled electron transfer, PCET). This strategy was pioneered by the Knowles group3 and has been widely exploited in recent reductive carbonyl umpolung addition reactions to unsaturated bonds such as carbonyls,4 alkenes5 and nitrones6 among others7 (Scheme 1a). With the aid of chiral Brønsted acids, Knowles,3a Jiang,5c and Huang6et al. have developed highly enantioselective catalytic protocols. In spite of these utilities, the redox-neutral couplings of carbonyls with unsaturated compounds through ketyl radicals are rarely explored.8 This is probably because electron-sacrificial agents such as Hantzsch ester and amines are generally required to enable the reductive subprocess but they also quench the oxidative species. In addition, while protonation by acid additives could increase the reductive potential of carbonyls in a concerted PCET process, it also complicates reaction systems and thereby limits the substrate scope and broader applications. Hence, it is highly desirable to develop new strategies to expand the applications of carbonyl-based ketyl couplings under facile redox-neutral conditions.9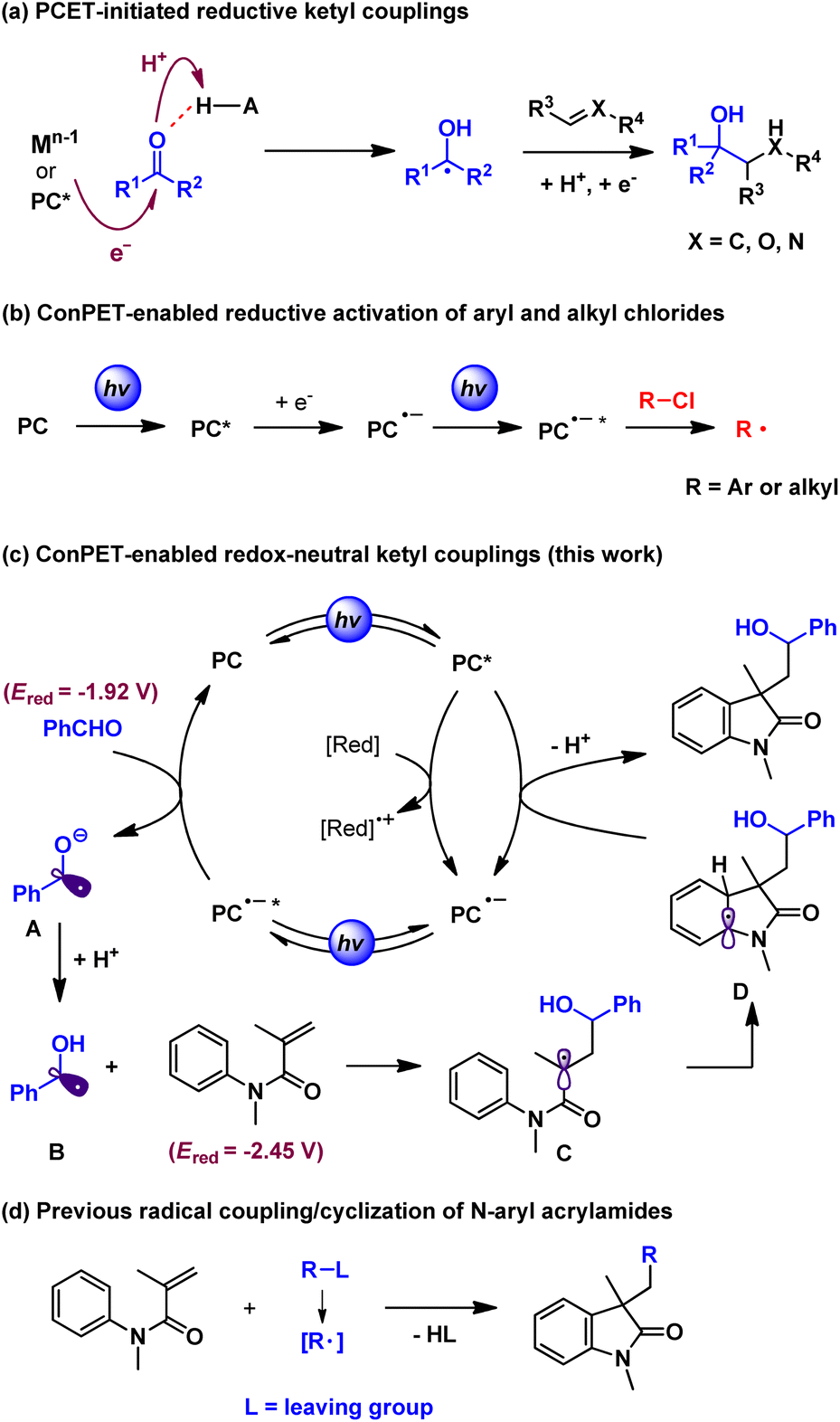 | ||
| Scheme 1 Reaction profile of the present work. | ||
Consecutive photoinduced electron transfer (ConPET) has recently emerged as a robust technique in visible-light-driven photocatalysis, especially to overcome thermodynamic limits set by the accessible redox potentials of common photocatalysts.10 Specifically, the two-photon excitation of photocatalysts with an electron donor could form excited radical anion species (*PC˙−) with extreme reduction potentials. In this context, a range of organic photocatalysts including perylene diimide (PDI),11 rhodamine 6G (Rh-6G),12 and 9,10-dicyanoanthracene (DCA),13 as well as Ir and Ru complexes,14 were employed to activate reductively recalcitrant aryl and alkyl chlorides (Ered ≈ −1.9 to −2.9 V vs. SCE) (Scheme 1b). We suspect that this multi-photon excitation strategy could provide complementary access to ketyl radicals from carbonyls (e.g. Ered = −1.92 V vs. SCE for benzaldehyde)15 beyond the PCET process. However, the development of redox-neutral ketyl couplings is still challenging because of the inevitable use of common electron donors that thermodynamically favor reductive reactions. To this end, N-aryl acrylamides can be selected as the ketyl radical coupling partner because the resultant amide α carbon radical would undergo rapid intramolecular cyclization with the ortho position of the aniline moiety. The dearomative intermediate, following single-electron oxidation by the excited photocatalyst and deprotonation, then readily affords the structurally valuable hydroxyalkyl oxindoles16 (Scheme 1c). Notably, while the radical addition/cyclization reactions of N-aryl acrylamides have been widely explored (Scheme 1d),17 our method represents the first facile protocol with formal 100% atom economy of this kind of transformation.
Results and discussion
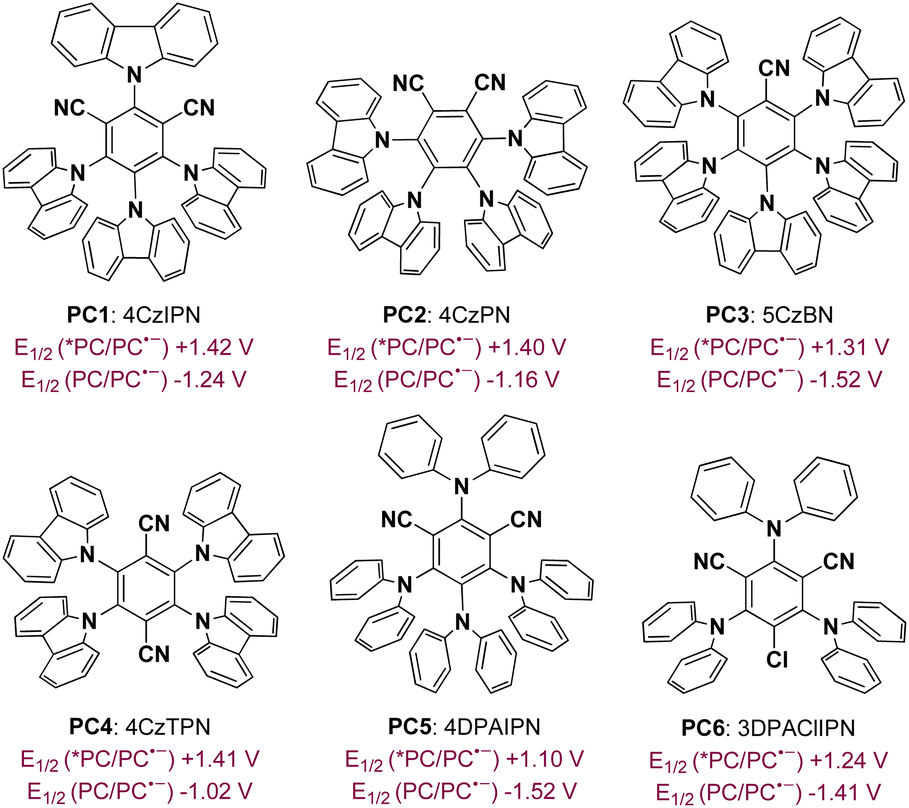
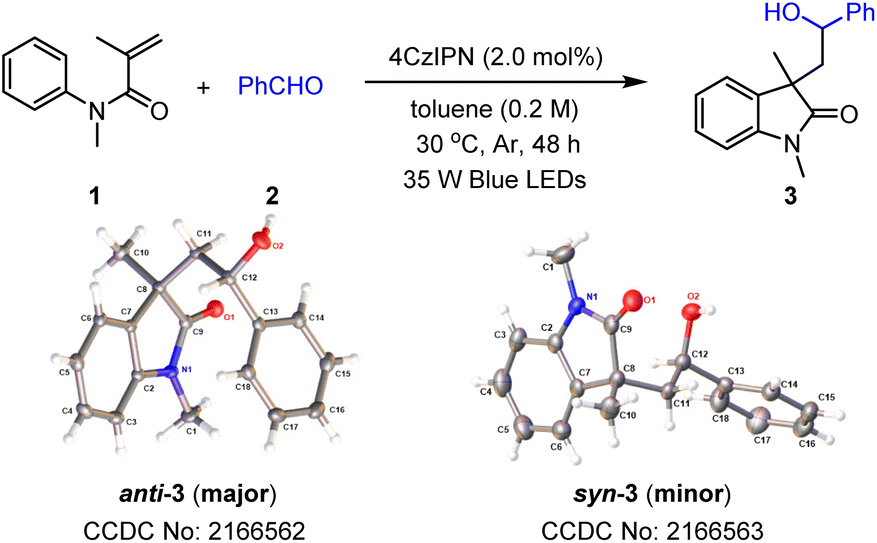
To achieve a photoredox neutral ketyl coupling of benzaldehyde (2) with N-phenyl acrylamide (1), the use of a suitable photocatalyst with both high oxidative potential in its excited state (PC*) and high reductive potential in its reductive state (PC˙−) should be the key factor. The ConPET process described by Wu18 as well as our previous studies on cyclization reactions of N-phenyl acrylamides19 revealed that donor–acceptor cyanoarenes20 would probably be a viable impetus. Our initial efforts to optimize reaction conditions thus focused on screening this type of photosensitizer with visible-light stimulation. In line with our deduction, 4CzIPN (PC1), which exhibits both high oxidative potential in its excited state [E1/2(PC*/PC˙−) = +1.42 V vs. SCE] and high reductive potential in its reductive state [E1/2(PC˙−/PC) = −1.24 V vs. SCE],20a was found to be the best photocatalyst when toluene was combined as the reaction media under blue light irradiation (Table 1, entry 1). In comparison, cyanoarenes with slightly lower reductive potential such as 4CzPN [PC2, E1/2(PC˙−/PC) = −1.16 V vs. SCE] or oxidative potential such as 5CzBN [PC3, E1/2(PC*/PC˙−) = +1.31 V vs. SCE]21 decreased the yield to 26% and 68%, respectively. Moreover, no product or a trace amount of the product was detected using other photosensitisers (PC4–PC6) with a bigger disparity of the redox potential to 4CzIPN. Screening solvents revealed that methyl benzenes such as toluene, mesitylene, and p-xylene exhibited excellent productivities, while others including PhCl, CH2Cl2, and CH3CN gave very low yields of the product. The addition of electron donors such as diisopropyl ethylamine (DIPEA) did not benefit the present reaction, suggesting that the solvent or one of the reactants provided electrons in the initial step of the catalytic cycle. The protonation activation of benzaldehydes was also not required, which rules out a PCET process of the current transformation. Control experiments demonstrated the critical role of visible light and a photocatalyst. Finally, air atmosphere dramatically decreased the efficiency of the ketyl coupling/cyclization reaction cascade probably because of the radical trapping capacity of dioxygen. Interestingly, the diastereomeric ratio (dr) was found to be around 4.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 under these conditions. We suspect that this diastereoselectivity is derived from the steric hindrance effect in the radical cyclization process (see the ESI† for detailed analysis).
:1 under these conditions. We suspect that this diastereoselectivity is derived from the steric hindrance effect in the radical cyclization process (see the ESI† for detailed analysis).
|
|
||
|---|---|---|
| Entry | Variation from the standard conditions | Yieldb (%) |
|
a Reaction conditions: 1 (0.24 mmol, 1.2 equiv.), 2 (0.2 mmol), and photocatalyst (PC, 0.004 mmol, 2 mol%) in solvent (1.0 mL, 0.2 M) at 30 °C under an Ar atmosphere and 35 W blue LED irradiation for 48 h. ND refers to not detected.
b Isolated yield. The diastereomeric ratio (dr) is around 4.0:1 under the conditions given in this table.
|
||
| 1 | None | 94 |
| 2 | PC2, PC3 instead of PC1 | 26, 68 |
| 3 | PC4–PC6 instead of PC1 | ND or trace |
| 4 | Mesitylene, p-xylene as the solvent | 84, 87 |
| 5 | PhCl, CH2Cl2, CH3CN as the solvent | <25 |
| 6 | With additional DIPEA (5 mol%) | 26 |
| 7 | With additional AcOH (5 mol%) | 65 |
| 8 | No light or no PC1 | ND |
| 9 | Under an air atmosphere | 44 |
|
||
With the optimized reaction conditions in hand, we next probed the substrate tolerance of the current photoredox neutral metal-free ketyl couplings. Our ketyl coupling system was found to accommodate a broad range of functional groups attached with the N atom of acrylamides, including bulky alkyl, benzyl, nitrile, alkene, alkyne, ether, and even silane (Table 2, products 4–12). The corresponding cyclized products were obtained with yields ranging from 51% to 91% and generally moderate levels of stereocontrol (2.7:1 to 5:1). Among substituted N-phenyl acrylamides at the benzene ring (13–24), a distinct reactivity was observed when using anisidine (16). Therein, dihydroquinolinone was generated as the major product through, as previously proposed, energy transfer-enabled self-cyclization (see the ESI for details†).19b Gratifyingly, aryl halides, even with iodine, were compatible without reductive protodehalogenation, which allows further convenient manipulations in organometallic cross couplings. The generality of other α-substituted acrylamides (25–28) was clarified by those with electron-withdrawing trifluoromethyl (27) and electron-donating hydroxymethyl (28), both of which featured the same excellent efficiency (94% yield). Expectedly, the spiro oxindole product 29′ was delivered as the major product when the acrylamide substrate was attached with an ester group (29).
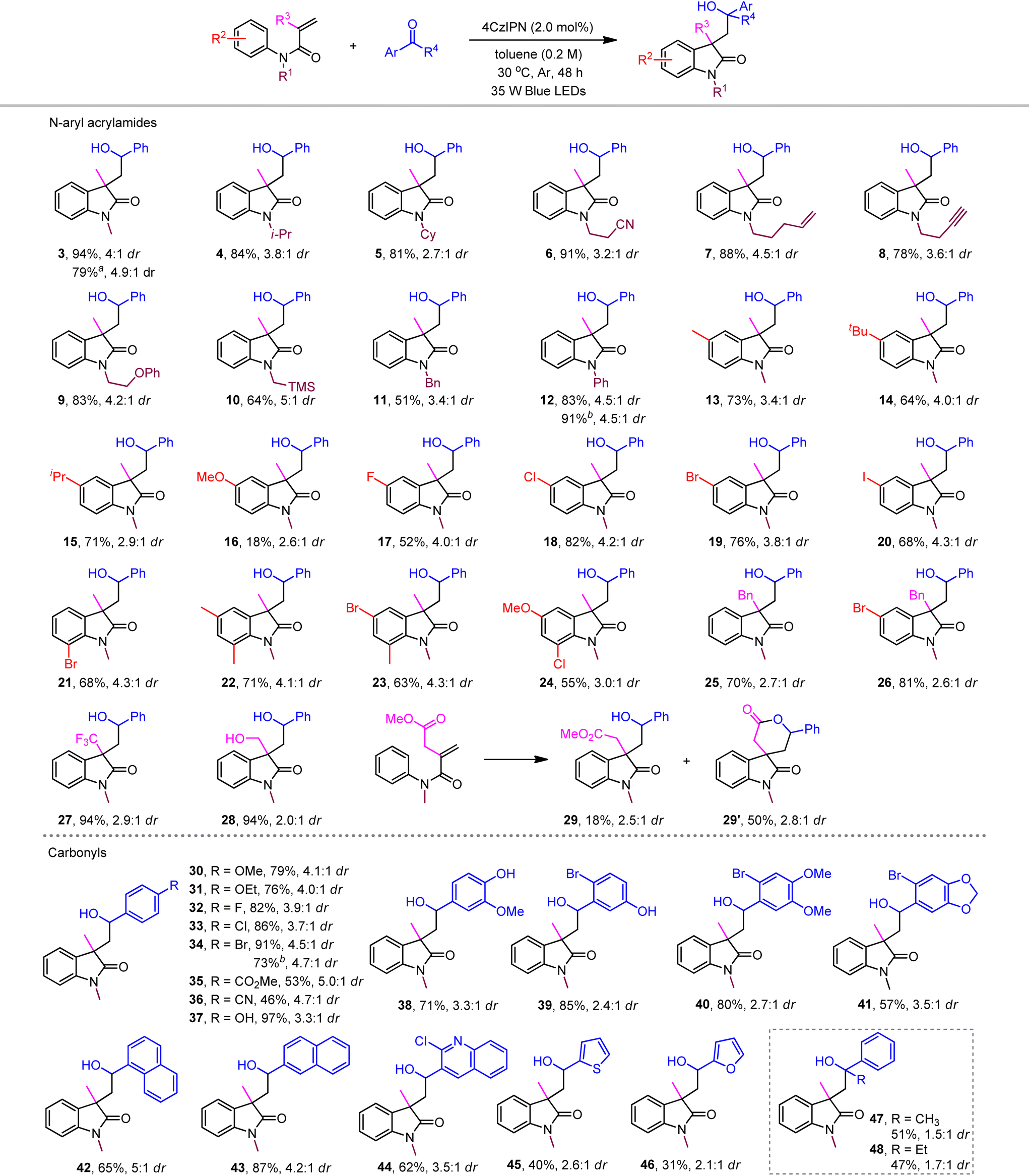
With respect to aldehyde reactants, benzaldehydes bearing various functional groups coupled smoothly to produce the desired oxindoles in moderate to excellent yields (30–41). However, their electronic nature responds to the nucleophilicity of the in situ generated ketyl radicals, which led to relatively lower productivity of electron-deficient benzaldehydes (35 and 36). The free hydroxyl benzaldehydes exhibited excellent productivities (37–39). In this case, the acidic substrates may also undergo a PCET process to afford ketyl radical intermediates.22 While sterically bulky 1-naphthaldehyde afforded the corresponding product in 65% yield (42), 2-naphthaldehyde enhanced the yield to 87% (43). Heteroaryl aldehydes furnished the target product in generally moderate yields (44–46). Finally, aryl ketones such as acetophenone and propiophenone featured modest productivity with poor stereoselectivity (47 and 48). Unexpectedly, neither aliphatic aldehydes nor ketones coupled in the present systems, indicating that the stability of a benzylic ketyl radical plays a crucial role in successful coupling.
The synthetic practicality of our method was demonstrated by the effective gram-scale synthesis of hydroxyl indoles under more intensive light irradiation (79% yield on a 15 mmol scale for 3, 91% yield on a 5 mmol scale for 12, and 73% yield on a 5 mmol scale for 34).
The robust nature of the redox neutral systems was further reflected by the effective couplings with a range of substrates bearing biologically active molecules (Table 3). While loxoprofen- and naproxen-derived α-substituted acrylamides produced products 49 and 50, respectively, the reactants with ibuprofen and ciprofibrate showed slightly higher reactivities and moderate stereoselectivity (51 and 52). Benzaldehydes attached with some drug molecules, including indometacin, ciprofibrate, loxoprofen, proline, and estrone, were also tolerated to generate the desired products with yields ranging from 61% to 80% (53–57).
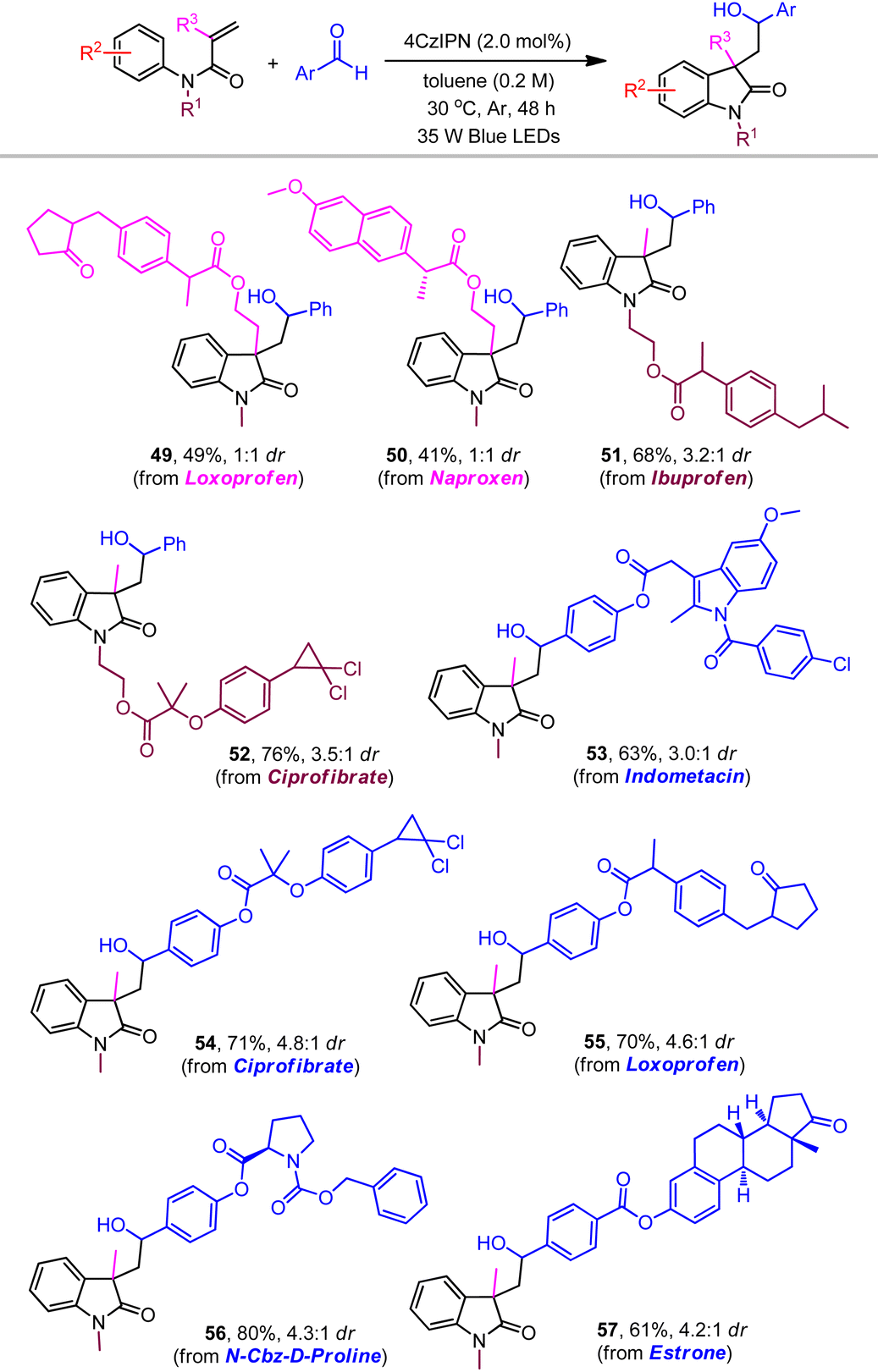
|
The resultant hydroxyl oxindoles allow diverse late-stage derivations (Scheme 2). For example, the oxidative dehydrogenation of anti-3 delivered acyl oxindole 58 in a quantitative yield (Scheme 2a). Esterification with the treatment of Ac2O afforded acetate 59 (Scheme 2b). Cyclizations by reductive LiAlH4 and nucleophilic n-BuLi readily occurred to efficiently generate tetrahydrofuroindoles 60 (Scheme 2c) and 61 (Scheme 2d), respectively. Besides, the Brønsted acid-promoted dehydration reactions of hydroxyl oxindoles afforded structurally significant vinyl oxindoles (62–64) (Scheme 2e). Furthermore, the olefination products could readily transform into broader oxindole products through cyclopropanation (65) (Scheme 2f), epoxidation (66) (Scheme 2g), hydroxybromination (67) (Scheme 2h), and arylation (68) (Scheme 2i). Ultimately, the dehydrative cyclization of oxindole 28 generated the spiro oxindole product 69 in an excellent yield (Scheme 2j).
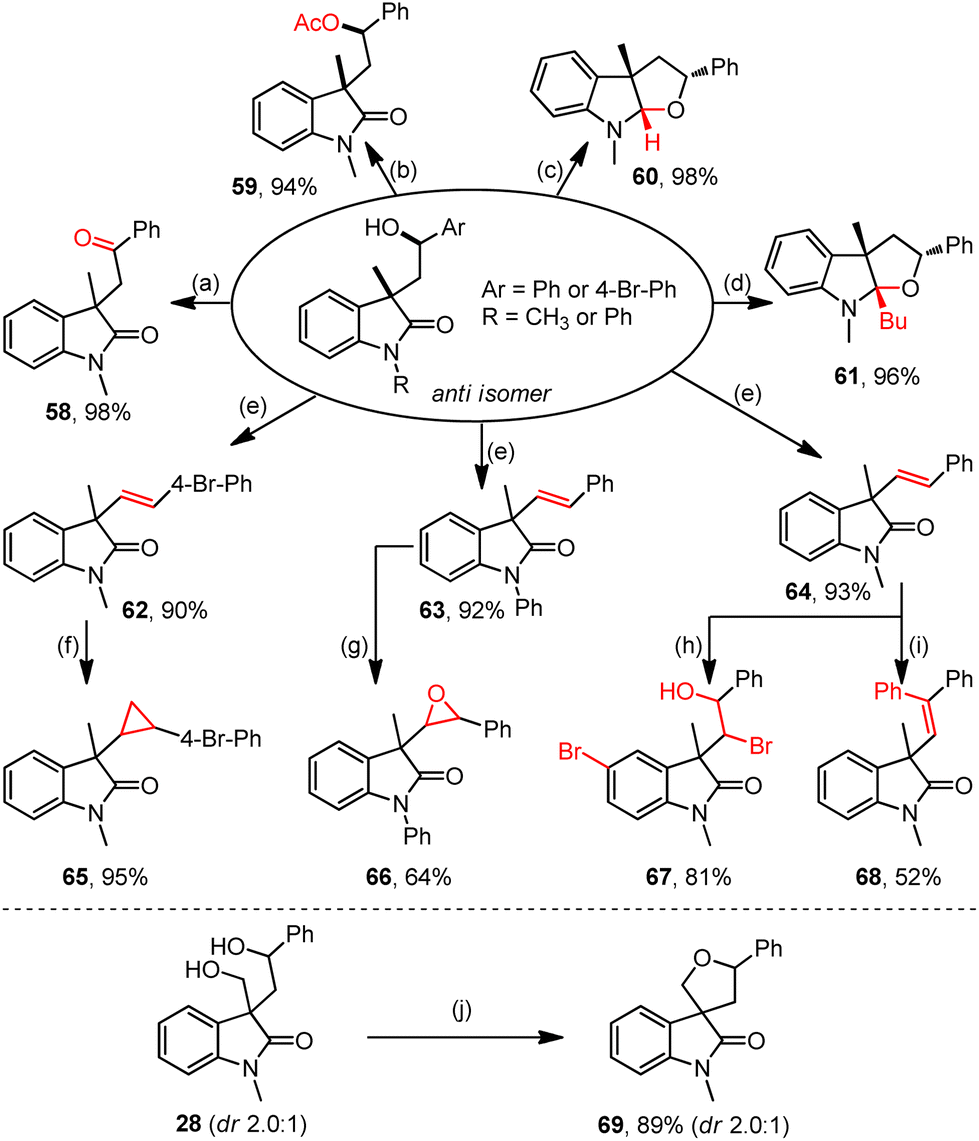 | ||
| Scheme 2 Late-stage applications of the resultant oxindoles. Reaction conditions: (a) DMP, DCM, rt, 1 h; (b) Ac2O, pyridine, rt, 24 h; (c) LiAlH4, THF, 0 °C, 1 h; (d) n-BuLi, THF, Ar, 0 °C–rt, 6 h; (e) H2SO4, 1,4-dioxane, 120 °C, 6 h; (f) Et2Zn, CF3COOH, CH2I2, DCM, 0 °C–rt, 20 h; (g) m-CPBA, NaHCO3, DCM, rt, 24 h; (h) NaNO2, HBr, CH3CN, O2, rt, 12 h; (i) Pd(OAc)2, AgOAc, PhI, HOAc, 120 °C, 12 h; (j) BF3·OEt2, DCM, rt, 4 h. | ||
To obtain mechanistic insight into the proposed ConPET process in the present ketyl coupling reactions, some control experiments and spectroscopic studies were carried out. First, in the absence of the acrylamide substrate, the reductive homo-coupling product 1,2-diphenylethane-1,2-diol was generated (Scheme 3a).4a The Stern–Volmer quenching experiments (Fig. S18–S25, ESI†) reveal that only benzaldehyde had a quenching effect on the emission of excited 4CzIPN and its combination with toluene slightly enhanced such an effect (Fig. S25†). When the radical quencher TEMPO was added under the standard conditions, the product 3 and TEMPO-trapped product were not detected. The benzyl radical was trapped by 1,1-diphenylethylene (DPE), which significantly decreased the yield of the cyclization product (Scheme 3b). Furthermore, the addition of butylated hydroxytoluene (BHT) dramatically decreased the productivity of the desired transformation, along with the detection of the ketyl radical-trapped product by BHT (Scheme 3c). Additionally, we found that when substrate 1 reacted with toluene or mesitylene in the absence of benzaldehyde, the product of benzyl radical addition to substrate 1 could be detected by GCMS (Fig. S8, ESI†). These results reveal the formation of a ketyl radical and the capability of toluene as an electron donor in the current system. While the oxidative potential of toluene is very high [Eox = +2.28 V vs. SCE],23 its direct electron transfer to the excited 4CzIPN [E1/2(PC*/PC˙−) = +1.42 V vs. SCE] is impossible. We speculate that an in situ formed radical species in the system abstracts the H atom from toluene through HAT to form a benzyl radical (toluene C(sp3)–H bond BDE = 89.7 kcal mol−1).
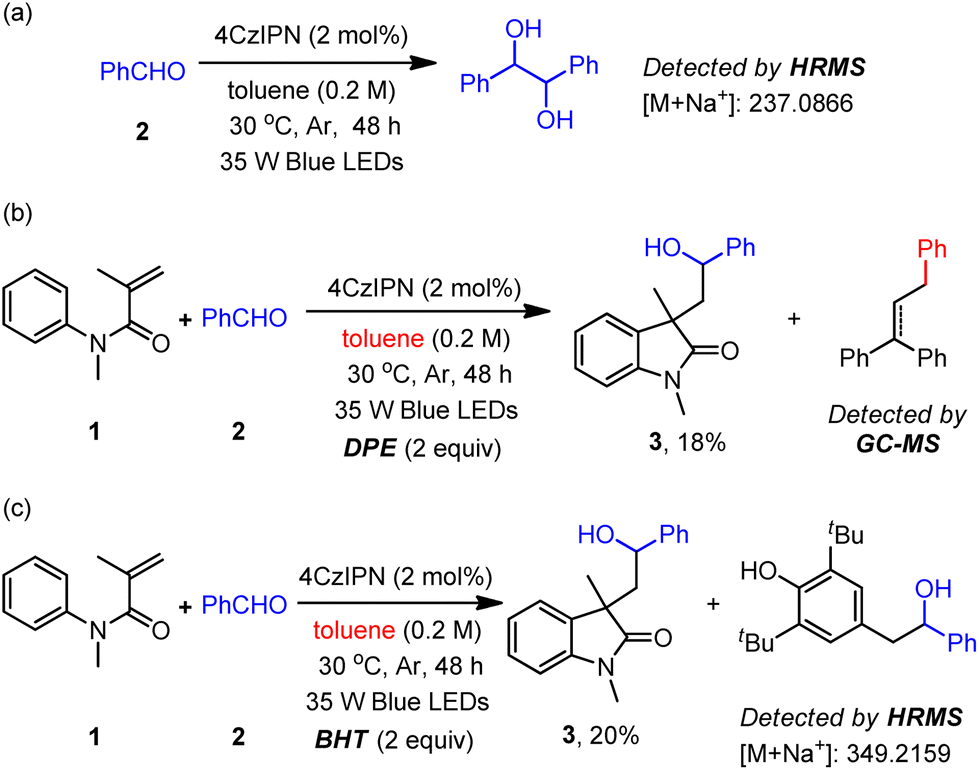 | ||
| Scheme 3 Control experiments. | ||
Moreover, the highest emission with a slight blue shift was observed with the toluene solution of 4CzIPN compared with other solvents (Fig. 1a). Additionally, when the solution of 4CzIPN and toluene was irradiated with blue LED light, a blue-shift of the maximum absorption peak was observed in its UV–vis spectra and the emission of this solution obviously appeared with a hypsochromic shift (λmax from 537 to 488 nm, Fig. 1b). These results indicate the formation of the radical anion species of the photocatalyst (4CzIPN˙−), in line with Wu's observations with DIPEA as the electron donor.18
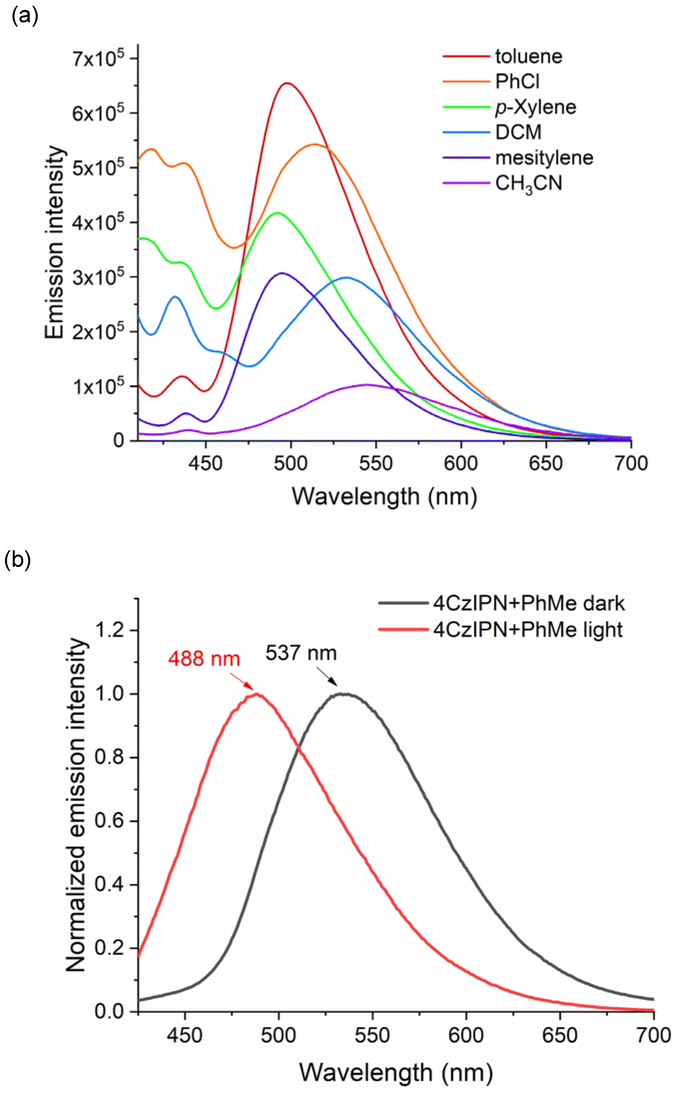 | ||
| Fig. 1 Spectroscopic experiments. | ||
Then, the experiment of the dependence of the yield on the excitation power was carried out; the initial reaction rate was found to be nearly quadratically dependent on the irradiation density (Fig. 2), further demonstrating the consecutive two-photon irradiation of the transformation.10a
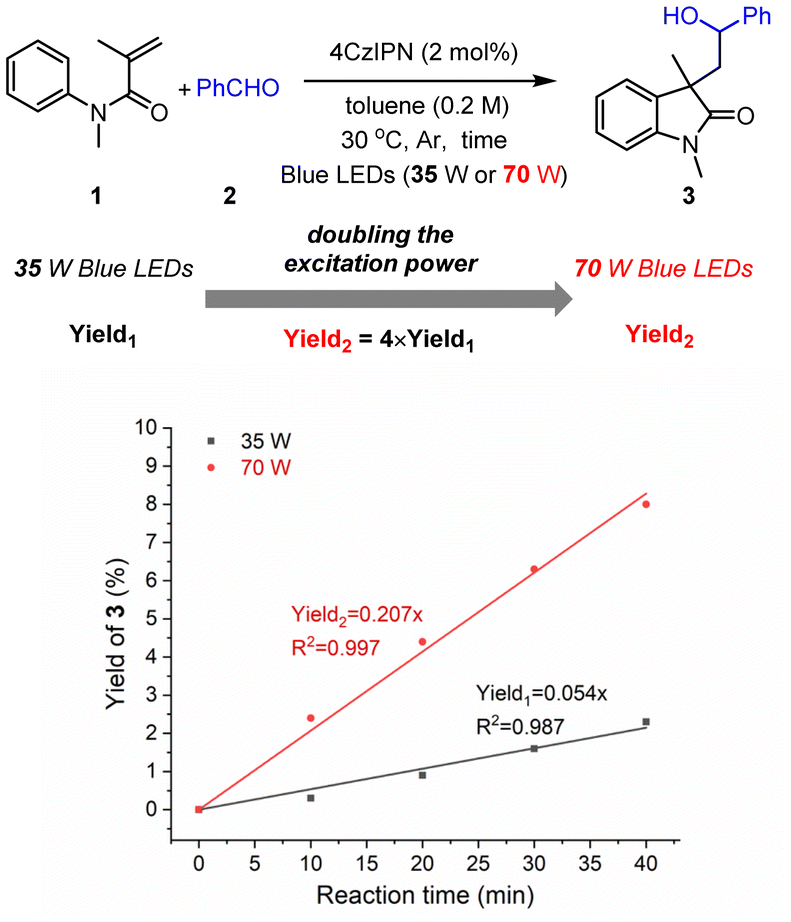 | ||
| Fig. 2 Dependency of the yield on the irradiation density. | ||
Conclusions
In summary, we have developed a mild and simple photocatalytic system that enables umpolung carbonyl coupling reactions through ConPET-initiated ketyl radical formation. Hence, the redox-neutral coupling/cyclization of N-aryl acrylamides with carbonyls has been achieved to provide new access to structurally valuable hydroxyalkyl oxindoles with generally good to excellent yields. This metal-, base-, acid-, and oxidant-free reaction system was found to be scalable and tolerate an extremely broad range of functionalities and bio-active molecules attached with the substrates. The application of this method has also been demonstrated by diverse late-stage derivations of the resultant hydroxyl oxindoles which successfully transformed into a range of structurally diverse oxindoles.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The support by the National Natural Science Foundation of China (22071211), the Science and Technology Planning Project of Hunan Province (2019RS2039), the Hunan Provincial Natural Science Foundation of China (2020JJ3032), the Scientific Research Fund of Education Department of Hunan Province (21A0079), the Hunan Provincial Innovation Foundation for Postgraduate (XDCX2021B155), and the Open Research Fund of School of Chemistry and Chemical Engineering of Henan Normal University (2022C02) is gratefully acknowledged.Notes and references
- (a) B. E. Kahn and R. D. Rieke, Chem. Rev., 1988, 88, 733–745 CrossRef CAS; (b) J. E. McMurry, Chem. Rev., 1989, 89, 1513–1524 CrossRef CAS; (c) G. A. Molander and C. R. Harris, Chem. Rev., 1996, 96, 307–338 CrossRef CAS PubMed; (d) M. Szostak, N. J. Fazakerley, D. Parmar and D. J. Procter, Chem. Rev., 2014, 114, 5959–6039 CrossRef CAS PubMed.
- (a) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed; (b) J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (c) J. Xuan and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed; (d) C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (e) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed; (f) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (g) M. Silvi and P. Melchiorre, Nature, 2018, 554, 41–49 CrossRef CAS PubMed; (h) Y. Chen, L.-Q. Lu, D.-G. Yu, C.-J. Zhu and W.-J. Xiao, Sci. China: Chem., 2018, 62, 24–57 CrossRef; (i) K. Xie, M. Jiang, X. Chen, Q. Lü and B. Yu, Chin. J. Org. Chem., 2021, 41, 4575–4587 CrossRef; (j) B. Cai and J. Xuan, Chin. J. Org. Chem., 2021, 41, 4565–4574 CrossRef.
- (a) L. J. Rono, H. G. Yayla, D. Y. Wang, M. F. Armstrong and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 17735–17738 CrossRef CAS PubMed; (b) K. T. Tarantino, P. Liu and R. R. Knowles, J. Am. Chem. Soc., 2013, 135, 10022–10025 CrossRef CAS PubMed.
- (a) M. Nakajima, E. Fava, S. Loescher, Z. Jiang and M. Rueping, Angew. Chem., Int. Ed., 2015, 54, 8828–8832 CrossRef CAS PubMed; (b) C. M. Wang, P. J. Xia, J. A. Xiao, J. Li, H. Y. Xiang, X. Q. Chen and H. Yang, J. Org. Chem., 2017, 82, 3895–3900 CrossRef CAS PubMed; (c) J. Zhu, Y. Yuan, S. Wang and Z. J. Yao, ACS Omega, 2017, 2, 4665–4677 CrossRef CAS PubMed.
- (a) K. N. Lee, Z. Lei and M. Y. Ngai, J. Am. Chem. Soc., 2017, 139, 5003–5006 CrossRef CAS PubMed; (b) L. Pitzer, F. Sandfort, F. Strieth-Kalthoff and F. Glorius, J. Am. Chem. Soc., 2017, 139, 13652–13655 CrossRef CAS PubMed; (c) K. Cao, S. M. Tan, R. Lee, S. Yang, H. Jia, X. Zhao, B. Qiao and Z. Jiang, J. Am. Chem. Soc., 2019, 141, 5437–5443 CrossRef CAS PubMed; (d) T. Rossolini, J. A. Leitch, R. Grainger and D. J. Dixon, Org. Lett., 2018, 20, 6794–6798 CrossRef CAS PubMed; (e) E. Fava, M. Nakajima, A. L. Nguyen and M. Rueping, J. Org. Chem., 2016, 81, 6959–6964 CrossRef CAS PubMed; (f) L. Qi and Y. Chen, Angew. Chem., Int. Ed., 2016, 55, 13312–13315 CrossRef CAS PubMed.
- C. X. Ye, Y. Y. Melcamu, H. H. Li, J. T. Cheng, T. T. Zhang, Y. P. Ruan, X. Zheng, X. Lu and P. Q. Huang, Nat. Commun., 2018, 9, 410 CrossRef PubMed.
- (a) E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed; (b) N. Hoffmann, Eur. J. Org. Chem., 2017, 1982–1992 CrossRef CAS; (c) J. D. Yang, P. Ji, X. S. Xue and J. P. Cheng, J. Am. Chem. Soc., 2018, 140, 8611–8623 CrossRef CAS PubMed; (d) Q. Xia, J. Dong, H. Song and Q. Wang, Chem. – Eur. J., 2019, 25, 2949–2961 CAS; (e) M. Chen, X. Zhao, C. Yang and W. Xia, Org. Lett., 2017, 19, 3807–3810 CrossRef CAS PubMed; (f) T. Ju, Q. Fu, J. H. Ye, Z. Zhang, L. L. Liao, S. S. Yan, X. Y. Tian, S. P. Luo, J. Li and D. G. Yu, Angew. Chem., Int. Ed., 2018, 57, 13897–13901 CrossRef CAS PubMed; (g) Z. Wang, Q. Liu, X. Ji, G.-J. Deng and H. Huang, ACS Catal., 2020, 10, 154–159 CrossRef CAS.
- L. Wang, J. Lear, S. M. Rafferty, S. C. Fosu and D. A. Nagib, Science, 2018, 362, 225–229 CrossRef CAS PubMed.
- K. D. Nguyen, B. Y. Park, T. Luong, H. Sato, V. J. Garza and M. J. Krische, Science, 2016, 354, aah5133 CrossRef PubMed.
- (a) F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed; (b) Y. Kobayashi, K. Mutoh and J. Abe, J. Photochem. Photobiol., C, 2018, 34, 2–28 CrossRef CAS.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016, 55, 7676–7679 CrossRef CAS PubMed.
- M. Neumeier, D. Sampedro, M. Májek, V. A. de la Peña O’Shea, A. Jacobi von Wangelin and R. Pérez-Ruiz, Chem. – Eur. J., 2018, 24, 105–108 CrossRef CAS PubMed.
- (a) R. Naumann, C. Kerzig and M. Goez, Chem. Sci., 2017, 8, 7510–7520 RSC; (b) J. I. Bardagi, I. Ghosh, M. Schmalzbauer, T. Ghosh and B. König, Eur. J. Org. Chem., 2018, 34–40 CrossRef CAS; (c) M. Giedyk, R. Narobe, S. Weiß, D. Touraud, W. Kunz and B. König, Nat. Catal., 2020, 3, 40–47 CrossRef CAS.
- H. G. Roth, N. A. Romero and D. A. Nicewicz, Synlett, 2016, 714–723 CAS.
- (a) W. Kong, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2015, 137, 16028–16031 CrossRef CAS PubMed; (b) G. S. Singh and Z. Y. Desta, Chem. Rev., 2012, 112, 6104–6155 CrossRef CAS PubMed; (c) R. Dalpozzo, G. Bartoli and G. Bencivenni, Chem. Soc. Rev., 2012, 41, 7247–7290 RSC.
- (a) R.-J. Song, Y. Liu, Y.-X. Xie and J.-H. Li, Synthesis, 2015, 1195–1209 CAS; (b) Y. Meng, L.-N. Guo, H. Wang and X.-H. Duan, Chem. Commun., 2013, 49, 7540–7542 RSC; (c) M.-B. Zhou, R.-J. Song, X.-H. Ouyang, Y. Liu, W.-T. Wei, G.-B. Deng and J.-H. Li, Chem. Sci., 2013, 4, 2690–2694 RSC; (d) Z.-L. Wu, J.-Y. Chen, X.-Z. Tian, W.-T. Ouyang, Z.-T. Zhang and W.-M. He, Chin. Chem. Lett., 2022, 33, 1501–1504 CrossRef CAS; (e) L. Yuan, S.-M. Jiang, Z.-Z. Li, Y. Zhu, J. Yu, L. Li, M.-Z. Li, S. Tang and R.-R. Sheng, Org. Biomol. Chem., 2018, 16, 2406–2410 RSC; (f) S. Tang, L. Yuan, Z.-Z. Li, Z.-Y. Peng, Y.-L. Deng, L.-N. Wang, G.-X. Huang and R.-L. Sheng, Tetrahedron Lett., 2017, 58, 2127–2130 CrossRef CAS; (g) D. Zhou, Z.-H. Li, J. Li, S.-H. Li, M.-W. Wang, X.-L. Luo, G.-L. Ding, R.-L. Sheng, M.-J. Fu and S. Tang, Eur. J. Org. Chem., 2015, 1606–1612 CrossRef CAS; (h) S. Tang, Z.-H. Li, M.-W. Wang, Z.-P. Li and R.-L. Sheng, Org. Biomol. Chem., 2015, 13, 5285–5288 RSC; (i) S. Tang, D. Zhou and Y.-C. Wang, Eur. J. Org. Chem., 2014, 3656–3661 CrossRef CAS.
- J. Xu, J. Cao, X. Wu, H. Wang, X. Yang, X. Tang, R. W. Toh, R. Zhou, E. K. L. Yeow and J. Wu, J. Am. Chem. Soc., 2021, 143, 13266–13273 CrossRef CAS PubMed.
- (a) Z. Liu, S. Zhong, X. Ji, G.-J. Deng and H. Huang, ACS Catal., 2021, 11, 4422–4429 CrossRef CAS; (b) Z. Liu, S. Zhong, X. Ji, G. J. Deng and H. Huang, Org. Lett., 2022, 24, 349–353 CrossRef CAS PubMed.
- (a) J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS; (b) T. Y. Shang, L. H. Lu, Z. Cao, Y. Liu, W. M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC; (c) Y. Liu, X. L. Chen, X. Y. Li, S. S. Zhu, S. J. Li, Y. Song, L. B. Qu and B. Yu, J. Am. Chem. Soc., 2021, 143, 964–972 CrossRef CAS PubMed.
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
- G. Qiu and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 2721–2730 CrossRef CAS PubMed.
- F. Li, D. Tian, Y. Fan, R. Lee, G. Lu, Y. Yin, B. Qiao, X. Zhao, Z. Xiao and Z. Jiang, Nat. Commun., 2019, 10, 1774 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2166562 and 2166563. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2gc01966c |
| This journal is © The Royal Society of Chemistry 2022 |