
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, electronic nature, and reactivity of selected silylene carbonyl complexes†
Juliane
Schoening
a,
Chelladurai
Ganesamoorthy
a,
Christoph
Wölper
a,
Ephrath
Solel
 b,
Peter R.
Schreiner
bc and
Stephan
Schulz
*ad
b,
Peter R.
Schreiner
bc and
Stephan
Schulz
*ad
aInorganic Chemistry, University of Duisburg-Essen, Universitätsstr. 7, 45141 Essen, Germany. E-mail: stephan.schulz@uni-due.de
bInstitute of Organic Chemistry, Justus Liebig University, Heinrich-Buff-Ring 17, D-35392 Giessen, Germany
cCenter for Materials Research (LaMa), Justus Liebig University, Heinrich-Buff-Ring 16, 35392 Giessen, Germany
dCenter for Nanointegration Duisburg-Essen (Cenide), University of Duisburg-Essen, Carl-Benz-Straße 199, 47057 Duisburg, Germany
First published on 11th May 2022
Abstract
Room-temperature stable main group element carbonyl complexes are rare. Here we report on the synthesis of two such complexes, namely gallium-substituted silylene-carbonyl complexes [L(X)Ga]2SiCO (X = I 2, Me 3; L = HC[C(Me)NDipp]2, Dipp = 2,6-iPr2C6H3) by reaction of three equivalents of LGa with IDippSiI4 (IDipp = 1,3-bis(2,6-iPr2C6H3)-imidazol-2-ylidene) or by salt elimination from [L(Br)Ga]2SiCO with MeLi. Both silylene carbonyl complexes were spectroscopically characterized as well as with single crystal X-ray diffraction (sc-XRD), while their electronic nature and the specific influence of the Ga-substituents X was evaluated by quantum chemical computations. In addition, we report the oxidative addition reaction of [L(Br)Ga]2SiCO with NH3, yielding [L(Br)Ga]2Si(H)NH24, demonstrating the promising potential of such complexes for small molecule activation.
Introduction
Low-valent main group metal compounds with an energetically high lying HOMO and a low lying LUMO are electronically similar to transition metal complexes and therefore provide alternative approaches to reactions previously only possible with use of the latter.1 In particular, monovalent group 13 diyls LM (M = Al, Ga) and divalent silylenes R2Si: have proven to be promising reagents in small molecule activation,2e.g., H2, CO2, ethylene, acetylene, and others, including catalytic processes.3 One of the most typical reactions of transition metals is their capability to form stable carbonyl complexes: σ-donation of the CO electron lone pair (HOMO) into an empty acceptor orbital of the transition metal and π-backbonding from a filled d-orbital into the empty π* orbital (LUMO) results in the formation of a very strong metal–CO bond. In marked contrast, only a handful of stable main group element carbonyl complexes have been isolated, since main group elements typically do not have suitable orbitals for π-backbonding.4 Matrix isolation experiments yielded a small number of main group element carbonyl complexes, including s-block metal complexes M(CO)8 (M = Ca, Sr, Ba) as well as carbonyl ions of barium Ba(CO)q (q = 1+/1−),5a,b whose bonding situation has been controversially discussed,5c,d and boron B(CO)2−.5e In marked contrast, room-temperature stable carbonyl complexes only formed with strongly Lewis acidic boranes such as (X2B)3BCO (X = F, Cl),6a B(CF3)3A,6b and B3H7B,6c respectively. In 2015, Braunschweig et al. reported the only dicarbonyl main group element complex known to date. Borylene dicarbonyl TpB(CO)2C (Tp = 2,6-Trip2-C6H3, Trip = 2,4,6-iPr3-C6H2), which releases CO upon irradiation as is known for transition metal carbonyl complexes, formed upon thermal treatment of a solution of TpBCr(CO)5 under CO atmosphere.7 Very recently, the same group isolated a carbene-stabilized boron-carbonyl complex LB(H)CO D that formed as an intermediate in the reaction of the carbene-coordinated diborene L2B2H2 with CO2 (Chart 1).8a However, due to the rather short B–C and long C–O bond, the electronic structure of D is better described as a borylketene, similar to aryl-substituted cAAC-bound arylborylenes LB(Ar)CO.8b,c The same is true for room-temperature stable sodium phosphaethynolate [Na(OCP)(dme)2]29 as well as group 13, 14, and 15 element phosphaketenes (R2PPCO),10 which serve as “masked” carbonyls since they react with release of CO in a variety of reactions.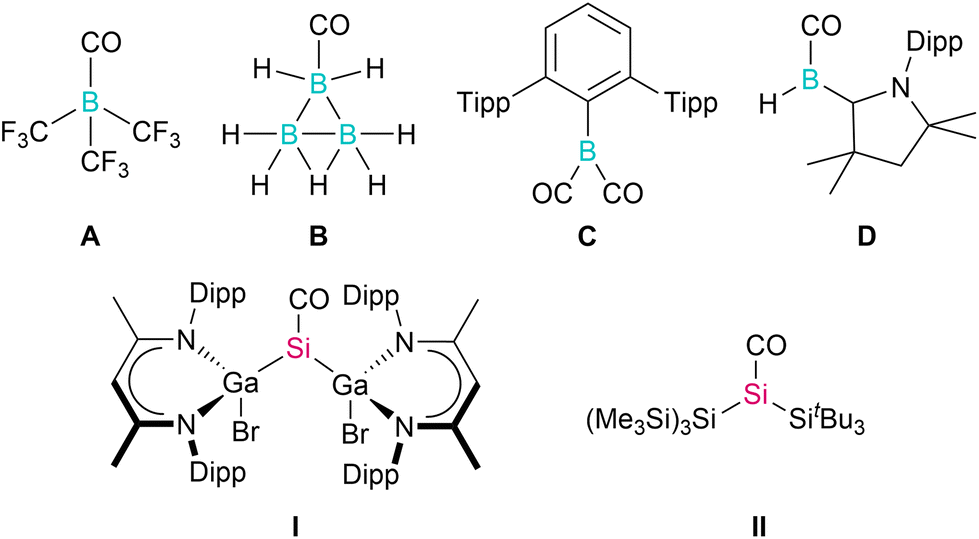 | ||
| Chart 1 Selected p-block element carbonyl complexes stable at room temperature. | ||
In group 14 element chemistry, the synthesis of carbonyl complexes has been frequently attempted using carbene-type compounds. However, the reaction of carbenes R2C: with CO yielded thermodynamically favoured ketenes rather than carbene-carbonyls adducts.11 In contrast, silylenes reacted with CO with formation of silylene-carbonyl complexes R2Si–CO, but these were so unstable that they could only be spectroscopically characterized in matrix isolation studies.12 In remarkable contrast, room-temperature stable carbonyl complexes remained unknown, until very recently the Schulz and Inoue groups reported on the synthesis and single crystal X-ray structures of [L(Br)Ga]2SiCO I13 and [(Me3Si)3Si](tBu3Si)SiCO II,14 respectively. The use of sterically demanding and electron-rich silylenes, which was achieved by introducing L(Br)Ga and tBu3Si ligands to the central Si atom, turned out to be crucial for the generation of these carbonyl complexes. Quantum chemical computations revealed that CO serves as an electron donor to the silylene, forming a σ-C–Si bond, while the presence of the L(Br)Ga (I) and silyl substituents (II) increase the electron density at the silicon atom, enabling the π-backbonding to the π*-orbital of the CO, resulting in a bonding motif similar to that of transition metal carbonyl complexes.13,14 Andrada et al. further pointed to an additional π back donation contribution from the HOMO−1 σGaSi orbital into the π*CO orbital in [L(Br)Ga]2SiCO I. With stronger σ-donating substituents, this contribution was found to increase to up to 20% of the total orbital interaction.15
We now report on the syntheses and solid-state structures of two additional silylene carbonyl complexes [L(X)Ga]2SiCO (X = I 2, Me 3) using alternative synthetic methods, and the influence of different substituents X (X = F, Cl, Br, I, Me, OMe, NMe2) on their electronic structures is shown. In addition, reactions of [L(Br)Ga]2SiCO I and ([L(Me)Ga]2SiCO 3 with H2, NH3, and other reagents (LiAlH4, SnCl4) is compared. [L(Br)Ga]2SiCO I was found to react via oxidative addition with all reagents including NH3, yielding [L(Br)Ga]2Si(H)NH24, whereas complex 3 failed to react, illustrating the distinct role of the substituent X on the chemical reactivity of such silylene carbonyl complexes.
Results and discussion
Silicon tetrahalides SiX4 (X = Cl, Br, I) differently react with LGa (Scheme 1) due to the different Si–X bond strengths. Oxidative addition reactions of LGa with SiX4 (X = Cl, Br) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 2:1 molar ratios yielded L(X)GaSiX3 (X = Cl,16 Br13) and [L(X)Ga]2SiX2 (X = Cl,17 Br13), but only [L(Br)Ga]2SiBr2 reacted with LGa to {[L(Br)Ga]Si[Ga(Br)][CHC(Me)NAr][C(Me)NAr]}.13 Analogous reactions of SiI4 failed to give [L(I)Ga]SiI3 and [L(I)Ga]2SiI2, but {[L(I)Ga]Si[Ga(I)][CHC(Me)NAr][C(Me)NAr]} (1) was isolated from the reaction in a 3:1 molar ratio. In addition, only [L(Br)Ga]2SiBr2 reacted with LGa under CO atmosphere to [L(Br)Ga]2SiCO I,13 whereas any attempts to reduce [L(Cl)Ga]2SiCl2 under CO atmosphere with a variety of reducing agents, e.g., Na, K, KC8, L2Mg2, and LGa, respectively, as well as to react SiI4 with three equivalents of LGa under CO atmosphere failed to give the desired silylene carbonyl complexes [L(Cl)Ga]2SiCO and [L(I)Ga]2SiCO (2). In marked contrast, the reaction of the carbene adduct IDippSiI4 with three equivalents of LGa under CO atmosphere gave [L(I)Ga]2SiCO (2) in reasonable yield (Scheme 1).
:1 and 2:1 molar ratios yielded L(X)GaSiX3 (X = Cl,16 Br13) and [L(X)Ga]2SiX2 (X = Cl,17 Br13), but only [L(Br)Ga]2SiBr2 reacted with LGa to {[L(Br)Ga]Si[Ga(Br)][CHC(Me)NAr][C(Me)NAr]}.13 Analogous reactions of SiI4 failed to give [L(I)Ga]SiI3 and [L(I)Ga]2SiI2, but {[L(I)Ga]Si[Ga(I)][CHC(Me)NAr][C(Me)NAr]} (1) was isolated from the reaction in a 3:1 molar ratio. In addition, only [L(Br)Ga]2SiBr2 reacted with LGa under CO atmosphere to [L(Br)Ga]2SiCO I,13 whereas any attempts to reduce [L(Cl)Ga]2SiCl2 under CO atmosphere with a variety of reducing agents, e.g., Na, K, KC8, L2Mg2, and LGa, respectively, as well as to react SiI4 with three equivalents of LGa under CO atmosphere failed to give the desired silylene carbonyl complexes [L(Cl)Ga]2SiCO and [L(I)Ga]2SiCO (2). In marked contrast, the reaction of the carbene adduct IDippSiI4 with three equivalents of LGa under CO atmosphere gave [L(I)Ga]2SiCO (2) in reasonable yield (Scheme 1).
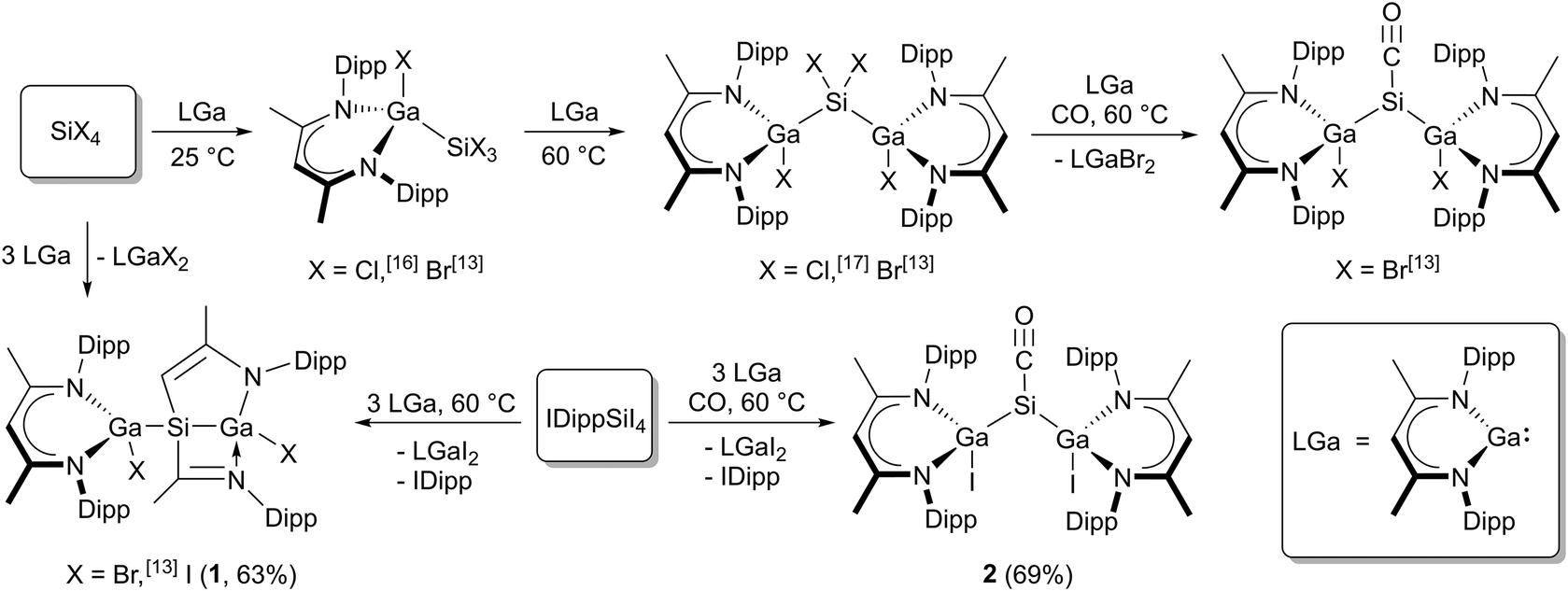 | ||
| Scheme 1 Overview of reactions of SiX4 (X = Cl, Br, I) or IDippSiI4 with LGa in different molar ratios (1:1 to 1:3) including the synthesis of new complexes (1) and (2). | ||
The 1H NMR and 13C NMR spectra of 1 and 2 are similar to those of analogous Br-substituted compounds I and III.13 The 1H NMR spectrum of 2 shows a singlet at 5.02 ppm of the γ-H proton, while the 13C{1H} NMR spectrum shows a signal at 207.0 ppm for the CO group. The resonance in the 29Si{1H} NMR at −249.9 ppm is slightly shifted to higher field compared to that of [L(Br)Ga]2SiCO I (−256.5 ppm).13 The IR spectrum of 2 exhibits a strong absorption band at 1934 cm−1, which is shifted to a lower wavenumber compared to [L(Br)Ga]2SiCO I (1945 cm−1).13 These experimental findings indicate a slightly stronger π-backbonding contribution in 2.
To study the influence of the ligand X in [L(X)Ga]2SiCO on their electronic nature, we became interested in replacing the electron-withdrawing (–I effect) halide substituents X (X = Br, I) by electron-donating (+I effect) alkyl groups, but unfortunately, LGa failed to oxidatively add Si–C bonds. Me/halide exchange reactions of [L(X)Ga]2SiCO (X = Br I, I 2) with AlMe3 and MeMgBr also failed, but salt elimination reaction of I with MeLi gave [L(Me)Ga]2SiCO 3 in 63% yield (Scheme 2). Any further attempts to introduce other substituents X (X = OMe, NMe2) by salt elimination using the corresponding alkaline metal salt MX (M = Na, Li) to electronically modify the carbonyl complexes [L(X)Ga]2SiCO remained unsuccessful.
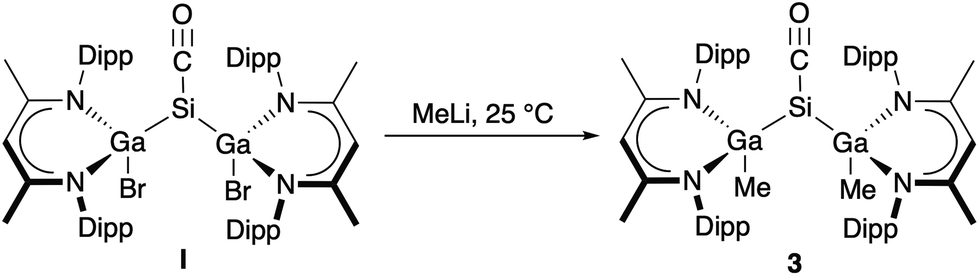 | ||
| Scheme 2 Synthesis of [L(Me)Ga]2SiCO (3). | ||
Carbonyl complexes 2, 3, and I form stable solutions in benzene and toluene at ambient temperature, whereas only complex 3 is stable in polar solvents such as CH2Cl2. [L(Me)Ga]2SiCO 3 is also thermally far more stable (Tdec. = 249 °C) than complexes 2 (Tdec. 168–169 °C) and I (Tdec. 176–177 °C), indicating a stronger Si–CO bond due to a larger π backbonding character of [L(Me)Ga]2Si compared to [L(X)Ga]2Si (X = Br, I), respectively. This is confirmed by IR spectroscopy, since 3 shows a strong stretching vibration of the CO group at 1906 cm−1. [(Me3Si)3Si](tBu3Si)SiCO II shows an almost identical adsorption frequency (1908 cm−1),14 whereas those of 2 (1934 cm−1) and I (1945 cm−1) are blueshifted.
The 1H NMR spectrum of 3 is similar to those of 2 and I and only shows an additional singlet at 0.32 ppm due to the Me group. The 13C{1H} NMR spectrum of 3 shows a resonance at 207.5 ppm (CO group), which is comparable to that observed for I (δ 206.5) and 2 (δ 207.0) but is shifted to higher field compared to [(Me3Si)3Si)(tBu3Si)SiCO II (226.1 ppm). In contrast, the singlet at −285.2 ppm in the 29Si{1H} NMR spectrum of 3 is shifted to higher field compared to 2 (−249.9 ppm), I (−256.5 ppm), and II (−228.5 ppm), respectively.
[L(Br)Ga]2SiCO I was shown to react as masked silylene.13 To reveal the influence of the substituent X, we compared reactions of [L(Br)Ga]2SiCO I and [L(Me)Ga]2SiCO 3, which contain the most electronegative (X = Br) and most electropositive (X = Me) substituent. Due to the larger π-backbonding contribution in 3 as was proven by the stronger redshift in the IR spectrum of 3, complex 3 was expected to be less reactive than complex I.
Complexes I and 3 both react with two equivalents of SnCl4 at ambient temperature with oxidation and almost quantitative formation of [L(Cl)Ga]2SiCl2 (Fig. S22, S23†). The formation of [L(Cl)Ga]2SiCl2 reflects the greater stability of Sn(II) vs. Si(II), and the Br/Cl and Me/Cl exchange at the Ga centers is also not unexpected due to the thermodynamically favored Ga–Cl bond. In contrast, only complex I reacted with H2, LiAlH4, and PBr3 to [L(Br)Ga]2SiH2 and [L(Br)Ga]2SiBr2,13 respectively, whereas complex 3 failed to react even at elevated temperatures (120 °C). Only LGa was formed in the reaction of 3 with H2 after several days and identified by in situ1H NMR spectroscopy (Fig. S21†). These findings prove the reduced reactivity of carbonyl complex 3 compared to complex I most likely resulting from the more strongly bound CO group in 3. We also reacted complexes I and 3 with NH3 since silylenes are known to activate the rather strong N–H bond,18i.e., Inoue's bis(silyl)silylene reacted already at −80 °C. I immediately reacted with NH3 at −20 °C (Scheme 3) to [L(Br)Ga]2Si(H)NH24, whereas 3 did not react even at elevated temperatures up to 70 °C. Removal of unreacted NH3 after several minutes is crucial to avoid the formation of a complex mixture of yet unidentified products, as was reported by Inoue and coworkers.19
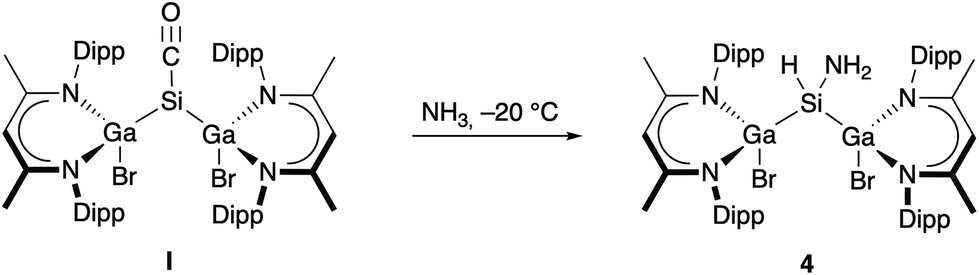 | ||
| Scheme 3 Synthesis of [L(Br)Ga]2Si(H)NH2 (4). | ||
The 1H NMR spectrum of compound 4 shows a triplet at 4.67 ppm (Si–H) and a doublet at −1.67 ppm (NH2), which are both in comparable regimes to previously reported complexes formed in NH3 activation reactions (Si–H: 4.72–5.22 ppm; NH2 –0.05 to 0.99 ppm).18 In addition, the 1H and 13C NMR spectra of 4 show typical resonances of the β-diketiminate ligand. The N–H stretching bands of 4 in the IR spectrum at 3428 and 3345 cm−1 are close to those of previously reported compounds, as is also true for the Si–H stretching band at 2060 cm−1.18
Single crystals of compounds 1–3 were obtained from benzene solutions upon storage at 4 °C for 72 h (1) and 24 h (2, 3) or from toluene solution upon storage at −18 °C (4). Complexes 1–3 crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one molecule in the asymmetric unit, and 4 in the monoclinic space group C2/c (Fig. 1) with the molecule placed on a special position (twofold rotational axis). The Ga–Si (2.3805(9) Å; 2.3599(9) Å) and Si–C (1.849(3) Å, 1.932(3) Å) bond lengths in 1 are almost identical to the Ga–Si (2.3919(7) Å; 2.3628(7) Å) and Si–C bond lengths (1.864(2) Å, 1.939(2) Å) reported for {[L(Br)Ga]Si[Ga(Br)][CHC(Me)NDipp][C(Me)NDipp]}.13 The Ga–Si bond lengths in [L(I)Ga]2SiCO 2 (2.4230(8) Å, 2.4348(9) Å) and [L(Me)Ga]2SiCO 3 (2.4314(4) Å, 2.4213(4) Å) are similar to those in [L(Br)Ga]2SiCO I (2.4204(5) Å), whereas the Si–C bond lengths in 2 (1.808(9) Å) and 3 (1.770(2) Å) are much shorter than those of complexes I (1.865(6) Å) and II (1.794(2) Å), respectively (Table 1).
with one molecule in the asymmetric unit, and 4 in the monoclinic space group C2/c (Fig. 1) with the molecule placed on a special position (twofold rotational axis). The Ga–Si (2.3805(9) Å; 2.3599(9) Å) and Si–C (1.849(3) Å, 1.932(3) Å) bond lengths in 1 are almost identical to the Ga–Si (2.3919(7) Å; 2.3628(7) Å) and Si–C bond lengths (1.864(2) Å, 1.939(2) Å) reported for {[L(Br)Ga]Si[Ga(Br)][CHC(Me)NDipp][C(Me)NDipp]}.13 The Ga–Si bond lengths in [L(I)Ga]2SiCO 2 (2.4230(8) Å, 2.4348(9) Å) and [L(Me)Ga]2SiCO 3 (2.4314(4) Å, 2.4213(4) Å) are similar to those in [L(Br)Ga]2SiCO I (2.4204(5) Å), whereas the Si–C bond lengths in 2 (1.808(9) Å) and 3 (1.770(2) Å) are much shorter than those of complexes I (1.865(6) Å) and II (1.794(2) Å), respectively (Table 1).
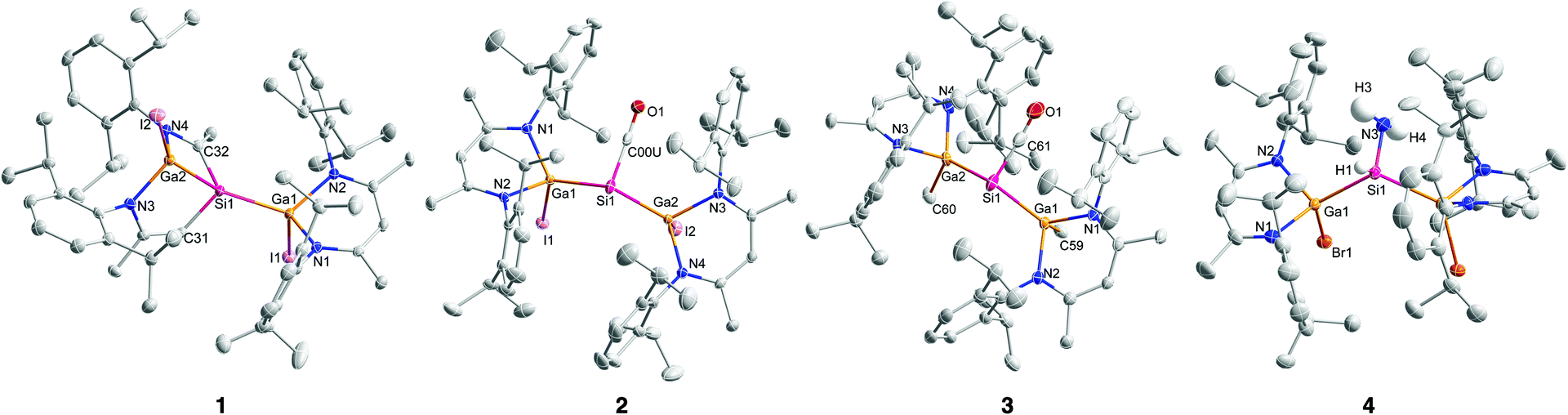 | ||
| Fig. 1 Molecular structures of {[L(I)Ga]Si[Ga(I)][CHC(Me)NDipp][C(Me)NDipp]} (1), [L(I)Ga]2SiCO (2), [L(Me)Ga]2SiCO (3), and [L(Br)Ga]2Si(H)NH2 (4); ellipsoids set at 50% probability level; hydrogen atoms (except SiH and NH2 in 4) and solvent molecules (benzene) are omitted for clarity. | ||
| [L(I)Ga]2SiCO 2 | [L(Me)Ga]2SiCO 3 | [L(Br)Ga]2SiCO I | [(Me3Si)3Si](tBu3Si)SiCO II | |
|---|---|---|---|---|
| Ga–Si [Å] | 2.4230(8), 2.4348(9) | 2.4314(4), 2.4213(4) | 2.4204(5) | — |
| Si–C [Å] | 1.808(2) | 1.770(2) | 1.865(6) | 1.794(2) |
| C–O [Å] | 1.149(3) | 1.144(2) | 1.136(7) | 1.153(2) |
| Ga–X [Å] | 2.6012(5), 2.5840(5) | 1.990(2), 1.992(2) | 2.3777(3) | — |
| Ga–Si–Ga [°] | 119.22(3) | 116.10(2) | 122.73(4) | — |
| Si–C–O [°] | 172.2(2) | 170.8(2) | 172.7(6) | 171.3(1) |
The Si–C bond length in 3 is also substantially shorter than Si–C single bonds (1.87–1.93 Å)20 and close to Si–C double bond distances, which range from 1.70 to 1.76 Å.20 These structural parameters indicate a higher π-backbonding contribution from the filled silicon sp2 orbital to the π*CO antibonding orbital, probably resulting from the more electropositive character of the iodine and methyl groups compared to the bromine substituent in complex I, resulting in a slightly higher electron density at the silicon center. The elongated C–O bond lengths in 2 (1.149(3) Å) and 3 (1.144(2) Å) compared to that in I (1.136(7) Å) agrees with this description. Complex II showed an even longer C–O bond of 1.153(2) Å.
The Ga and Si atoms in silane complex [L(Br)Ga]2Si(H)NH24 feature distorted tetrahedral geometries and the GaN2C3 rings adopt the typical boat-type conformation. The Ga–Si bond length (2.4471(19) Å) is comparable to those of 1–3 and other compounds with Ga–Si single bonds (2.33–2.53 Å).21 The Ga–Si–Ga bond angle (126.16(8)°) is larger than the ideal tetrahedral angle and clearly reflects the repulsive interactions between the sterically demanding L(Br)Ga substituents. The Si–NH2 (1.716(6) Å) bond lengths agree with those previously reported for these type of compounds (Si–N 1.729(1) Å,18a 1.711(3) Å, 1.703(3) Å18b 1.653(3) Å,18c 1.691(4) Å18d).
To further investigate the role of the X substituents on the bonding nature of these types of silylene carbonyl complexes, we performed quantum chemical computations. We optimized the silylene carbonyl complexes with different atoms or groups attached to Ga: X = F, Cl, Br, I, Me, OMe, NMe2. All DFT computations were carried out using B3LYP22 with Grimme's D3BJ dispersion correction,23 as implemented in Gaussian16 revision C.01.24 The 6-311G(d,p) basis set was used for all atoms except Ga, I and Br, for which the def2-TZVP25 basis set was employed. All minima were characterized with analytical Hessian calculations to ensure minima (i.e., no imaginary frequencies). NBO analyses were performed with NBO 7.026 as implemented in Gaussian16.
Our computations suggest that the electronic effect of the substituent X on the C–O bond length is rather small. However, while the differences between the different halogen atoms are negligible, electron donating groups such as Me, OMe, NMe2 slightly shorten the Si–C bond and increase the C–O bond length (Table 2). This is reflected by the bond lengths, Wiberg bond orders and by the computed IR shifts for the C–O bond. Our computed C–O bond lengths somewhat differ from experiment since the computed C–O bond in 3 is longer than in I (matches experiment) and 2 (unlike experiment). However, the computed trends in the IR νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O match the experimental trends.
O match the experimental trends.
| X= | F | Cl | Br | Br I (exp.) | I | I 2 (exp.) | Me | Me 3 (exp.) | OMe | NMe2 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Average value of the two bonds. b Pyramidalization angle, as measured from the angle between the Si–C bond and the centroid of the two Ga atoms. c WBI, Wiberg bond index. d Scaled with a factor of 0.967. e Orbital energies in eV. | ||||||||||
| RMSD | 0.533 | 0.414 | 0.364 | |||||||
| r Ga–X | 1.815 | 2.228 | 2.382 | 2.3777(3) | 2.595 | 2.6012(5) 2.5840(5) | 2.001 | 1.9898(13) 1.9923(14) | 1.836 | 1.873 |
| r Ga–Si | 2.437 | 2.437 | 2.440 | 2.4203(5) | 2.441 | 2.4230(7) 2.4348(7) | 2.439 | 2.4213(4) 2.4314(4) | 2.419 | 2.436 |
| r Si–C | 1.815 | 1.811 | 1.811 | 1.865(6) | 1.811 | 1.808(3) | 1.791 | 1.7703(18) | 1.804 | 1.790 |
| r C–O | 1.150 | 1.151 | 1.151 | 1.136(7) | 1.151 | 1.149(3) | 1.157 | 1.144(2) | 1.153 | 1.156 |
| ∠Ga–Si–Ga | 111.9 | 113.0 | 113.9 | 122.73(4) | 115.0 | 119.22(3) | 116.0 | 116.105(16) | 108.4 | 122.9 |
| ∠Ga–Si–Ca | 88.9 | 90.2 | 90.6 | 92.97(15) | 91.0 | 90.81(7) 91.02(8) | 92.4 | 95.16(5) 94.13(5) | 90.6 | 91.6 |
| ∠Si–C–O | 171.7 | 171.2 | 171.0 | 172.7(6) | 170.8 | 172.2(2) | 171.3 | 170.82(19) | 169.8 | 171.9 |
| ∠pb | 88.0 | 90.3 | 91.0 | 91.9 | 94.61 | 91.0 | 93.4 | |||
| WBIGa–Rc | 0.43 | 0.67 | 0.75 | 0.84 | 0.60 | 0.43 | 0.48 | |||
| WBIGa–Sic | 0.79 | 0.81 | 0.81 | 0.82 | 0.79 | 0.80 | 0.80 | |||
| WBISi–Cc | 1.30 | 1.29 | 1.29 | 1.28 | 1.36 | 1.33 | 1.36 | |||
| WBICOc |
2.02 | 2.01 | 2.02 | 2.02 | 1.95 | 1.98 | 1.95 | |||
|
ν
CO
|
1966 | 1959 | 1958 | 1945 | 1956 | 1934 | 1925 | 1906 | 1946 | 1934 |
| HOMOe | −5.50 | −5.53 | −5.48 | −5.42 | −4.91 | −5.34 | −4.88 | |||
| LUMOe | −1.37 | −1.41 | −1.44 | −1.51 | −1.22 | −1.27 | −1.18 | |||
Wiberg bond indices suggest that the Ga–X bond is weaker than a single bond, and the bond order increases with decreasing electronegativity along F, O < N < C, and increases when going down the halogen series F < Cl < Br < I. The bonds further away from the X group are less affected by electronegativity, as for Me, OMe, and NMe2 the Si–C bond order is higher and the C–O bond lower relative to the other groups. From these results the most significant change in νCO, in bond lengths and in bond orders is a reduction observed for electron donating groups (either inductively or resonatively) such as X = Me and NMe2. Interestingly, OMe has a very similar influence. The νCO vibration correlates well with σF27 (Fig. 2), and thus operates as a measure of field/inductive effects of the groups. σF matches the F parameter presented in Table I of ref. 27b.
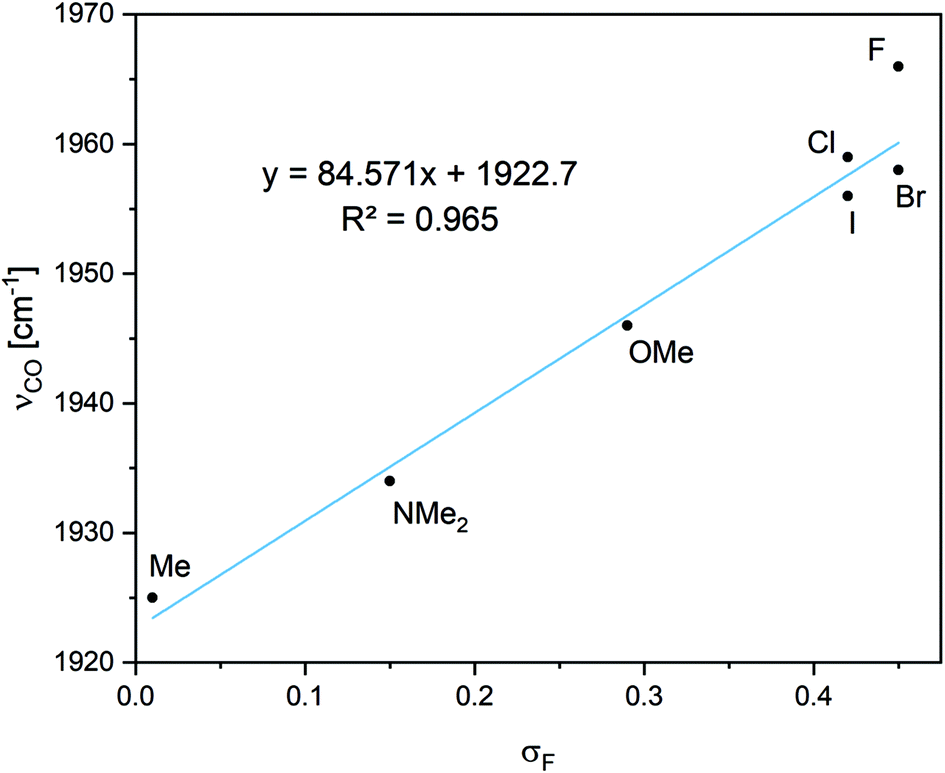 | ||
| Fig. 2 The correlation between νCO and the field inductive effects of the various groups (σF).27b. | ||
Field/inductive effects are long range through-space or through-bond dipolar interactions, different from the short-range effect of electronegativity. As this effect arises from dipole interactions and can roughly be related to the size of the dipole moment,27a we can rationalize this as the interaction between the Ga–X and the Si–C–O dipole moments. A larger Gaδ+–Xδ− dipole moment would interact more favorably with a smaller Cδ+–Oδ− dipole moment. Free CO has a small dipole moment of 0.107 D,28 pointing towards the carbon atom (i.e., δ−COδ+) while carbonyls with C–O double bonds have larger dipole moments (with the partial negative charge on oxygen).
This would suggest that larger Gaδ+–Xδ− dipole moments would destabilize the longer, more polar C–O bonds, resulting in shorter bonds. Alternatively, this can be explained by considering the π-back-donation of electrons from silicon, which is expected to result in larger dipole moments pointing towards the oxygen, being destabilized as the Gaδ+–Xδ− dipole moment becomes larger. This concept has also been reported from other groups.29 However, the orbital interaction energies from second order perturbation theory analysis do not show a good correlation with σF, suggesting a more complex orbital picture (Table S2†). Also, in contrast to the field/inductive effect, the νCO vibration does not show good correlations with measurements of resonance interactions (see ESI for details†).27b
The HOMO of all complexes is a bonding orbital between the Si atom's lone pair and the π*CO (Fig. S25†). Table 2 shows that for X = Me and NMe2 both HOMO and LUMO are slightly higher in energy than for the other complexes, indicating that the Si–C–O unit is more electron rich. This can be also observed from the NBO charges (Table S2†), showing a more negative charge on the CO for X = Me, NMe2, OMe. Hirshfeld charges (Table S3†) show a similar picture. Note that for both charge partitioning methods the charge on the silicon does not follow the electronegativity order (NBO charge is the most negative for F > Cl > Br > I for example) or the electron donation/withdrawal ability (very similar NBO charges for Me, OMe, NMe2), or the field effect.
Table S2† also shows the results of second order perturbation theory for the interaction energies between the orbitals. Both the LPSi → π*CO and σSi–Ga → π*CO interactions give generally higher values for X = Me, NMe2, and OMe than for the halogens, in accordance with the longer C–O bond length for the electron donating groups. However, no such correlation between the electron donating groups, or between the different halogen substitutents was observed. To conclude, our computations demonstrate that groups attached to the Ga atoms have only relatively small effects on the Si–C and C–O bond lengths. These groups do not interact via resonance with the SiCO unit, but instead their main influence is through field/inductive interactions.
Conclusions
Two new room-temperature stable silylene carbonyl complexes [L(X)Ga]2SiCO (X = I 2, Me 3) were synthesized and structurally characterized by single crystal X-ray diffraction. Quantum chemical computations were employed to analyze the influence of different substituents X on the electronic nature of the complexes and the strength of the π backbonding interaction from the silylene to the carbonyl ligand, hence resulting in different Si–CO bond strengths and, consequently, different activities of [L(X)Ga]2SiCO with respect to small molecule activations. This was revealed in several reactions including those with ammonia, in which only [L(Br)Ga]2SiCO I was found to undergo oxidative addition of the N–H bond with subsequent formation of [L(X)Ga]2Si(H)NH24, whereas [L(Me)Ga]2SiCO 3 failed to react.The halide-substituted carbonyl complex I turned out to be more reactive than the Me-substituted complex 3. To increase the reactivity of such silylene carbonyl complexes, the Si–CO bond needs to be weakened, which can be achieved by hampering the π-backbonding contribution of the silylene. The synthesis of heteroleptic silylenes containing only one electropositive L(X)Ga and one electronegative ligand, i.e., β-diketiminate or amidinate substituent, might therefore be a promising strategy.
Experimental
General procedures
All manipulations were performed in an atmosphere of purified argon using standard Schlenk and glove-box techniques. NMR-scale reactions were carried out in J-Young NMR tubes. n-Hexane was dried using mBraun Solvent Purification System (SPS). Benzene was dried over potassium. Deuterated solvents were dried over activated molecular sieves (4 Å) and degassed prior to use. The anhydrous nature of the solvents was verified by Karl Fischer titration. LGa {L = HC[C(Me)N(2,6-iPr2C6H3)]2},30 [L(Br)Ga]2SiCO13 and DippNHC-SiI431 were prepared according to literature methods and other chemicals were obtained from commercial sources and purified prior to use in necessary cases. Microanalyses were performed at the elemental analysis laboratory of University of Duisburg-Essen. Melting points were measured using a Thermo Scientific 9300 apparatus.1H (300.1 MHz; 400 MHz),13C{1H} (75.5 MHz; 101 MHz) and 29Si{1H} (59.1 MHz; 79 MHz) NMR spectra were recorded using a Bruker Avance DPX-300 spectrometer or AscendTM 400 spectrometer. 1H and 13C{1H} spectra were referenced to internal C6D5H (1H: δ = 7.154; 13C: δ = 128.39), while 29Si{1H} NMR and 29Si spectra were referenced using IUPAC recommendation of NMR nomenclature.32 IR spectra were recorded with an ALPHA-T FT-IR spectrometer equipped with a single reflection ATR sampling module, which was placed in a glovebox to guarantee measurements under inert gas conditions.
Synthesis procedures
Yield: 0.162 mg (0.129 mmol, 63%). M.p. 188–189 °C (dec.). Anal. Calcd for C58H82N4I2Ga2Si: C, 55.44; H, 6.58; N, 4.46. Found: C, 55.49; H, 6.63; N, 4.41%. IR (neat): ν 2961, 2922, 2866, 1517, 1461, 1437, 1386, 1362, 1314, 1251, 1200, 1176, 1128, 1100, 1052, 1021, 933, 866, 838, 802, 766, 715, 632, 524, 501, 425 cm−1. 1H NMR (C6D6, 300.1 MHz): δ 7.14–6.94 (m, 12 H, C6H3(iPr)2), 5.03 (s, 1 H, γ-CH), 4.57 (s, 1 H, Si–CH), 4.11 (sept, 3JHH = 6.6 Hz, 1 H, CH(CH3)2), 3.82 (sept, 3JHH = 6.9 Hz, 1 H, CH(CH3)2), 3.56 (sept, 3JHH = 6.9 Hz, 1 H, CH(CH3)2), 3.47 (sept, 3JHH = 6.6 Hz, 1 H, CH(CH3)2), 3.30 (sept, 3JHH = 6.9 Hz, 1 H, CH(CH3)2), 3.14 (sept, 3JHH = 6.9 Hz, 1 H, CH(CH3)2), 2.91 (sept, 3JHH = 6.9 Hz, 1 H, CH(CH3)2), 2.53 (sept, 3JHH = 6.9 Hz, 1 H, CH(CH3)2), 1.82 (s, 3 H, SiCHC(CH3)NAr), 1.59 (d, 3JHH = 6.6 Hz, 3 H, CH(CH3)2), 1.54 (m, 15 H, CH(CH3)2, ArNCCH3), 1.38 (d, 3JHH = 6.6 Hz, 3 H, CH(CH3)2), 1.28 (s, 3 H, SiC(CH3)NAr), 1.24 (d, 3JHH = 6.6 Hz, 3 H, CH(CH3)2), 1.23 (d, 3JHH = 6.6 Hz, 3 H, CH(CH3)2), 1.19 (d, 3JHH = 6.9 Hz, 6 H, CH(CH3)2), 1.14 (d, 3JHH = 6.6 Hz, 3 H, CH(CH3)2), 1.08 (d, 3JHH = 6.9 Hz, 3 H, CH(CH3)2), 1.04 (d, 3JHH = 6.9 Hz, 3 H, CH(CH3)2), 1.01 (d, 3JHH = 6.9 Hz, 3 H, CH(CH3)2), 0.95 (d, 3JHH = 6.6 Hz, 3 H, CH(CH3)2), 0.94 (d, 3JHH = 6.6 Hz, 3 H, CH(CH3)2), 0.85 (d, 3JHH = 6.9 Hz, 3 H, CH(CH3)2). 13C NMR (C6D6, 75.5 MHz): δ 170.1 (ArNCCH3), 169.6 (SiCHC(CH3)NAr), 168.1 (SiC(CH3)NAr), 147.1, 146.6, 146.5, 146.4, 143.6, 143.0, 142.5, 142.4, 141.9, 141.8, 141.5, 141.4, 140.9, 128.3, 128.1, 127.3, 126.2, 126.0, 125.6, 124.7, 124.6, 123.9, 123.8, 123.7 (C6H3), 99.2 (γ-CH), 85.7 (–Si–CH), 30.2, 29.5, 29.2, 28.7, 28.2, 27.9, 27.8, 27.6 (CH(CH3)2), 27.4, 27.2, 26.7, 26.6, 25.8, 25.7, 25.5, 25.1, 25.0, 24.9, 24.9, 24.8, 24.7, 24.5, 24.4, 24.2 (CH(CH3)2), 24.1 (ArNCCH3), 23.9 (SiCHC(CH3)NAr), 23.6 (SiC(CH3)NAr). 29Si NMR (C6D6, 59.6 MHz): δ −31.9.
Yield: 91 mg (0.071 mmol, 69%). M.p. 168–169 °C (dec.). Anal. Calcd for C59H82N4I2Ga2OSi: C, 55.16; H, 6.43; N, 4.36. Found: C, 55.90; H, 6.49; N, 4.21%. IR (neat): ν 2961, 2922, 2862, 1934, 1528, 1461, 1434, 1382, 1319, 1255, 1176, 1100, 1021, 937, 858, 794, 759, 640, 576, 532, 505, 438 cm−1. 1H NMR (C6D6, 300.1 MHz): δ 7.14−6.96 (m, 12 H, C6H3(iPr)2), 5.02 (s, 2 H, γ-CH), 3.85 (sept, 3JHH = 6.9 Hz, 4 H, CH(CH3)2), 3.20 (sept, 3JHH = 6.9 Hz, 4 H, CH(CH3)2), 1.48 (s, 12 H, ArNCCH3), 1.35 (d, 3JHH = 6.6 Hz, 12 H, CH(CH3)2), 1.25 (d, 3JHH = 6.9 Hz, 12 H, CH(CH3)2), 1.21 (d, 3JHH = 6.9 Hz, 12 H, CH(CH3)2), 0.96 (d, 3JHH = 6.9 Hz, 12 H, CH(CH3)2). 13C NMR (C6D6, 75.5 MHz): δ 207.0 (SiCO), 169.4 (ArNCCH3), 146.6, 142.9, 141.7, 127.7, 125.9, 123.8 (C6H3), 99.0 (γ-CH), 30.4, 29.7 (CH(CH3)2), 28.6, 25.2, 24.4, 24.3 (CH(CH3)2), 24.1 (ArNCCH3). 29Si NMR (C6D6, 59.6 MHz): δ −249.9.
Yield: 56.2 mg (0.053 mmol, 63%). M.p. 249 °C (dec.). Anal. Calcd for C59H82N4I2Ga2OSi: C, 69.06; H, 8.36; N, 5.28. Found: C, 68.7; H, 8.48; N, 5.33%. IR (neat): ν 3060, 2958, 2925, 2868, 1942, 1906, 1549, 1517, 1435, 1383, 1314, 1254, 1177, 1100, 1017, 935, 855, 794, 757, 697, 636, 536, 444 cm−1. 1H NMR (C6D6, 400 MHz): δ 7.15−6.97 (m, 12 H, C6H3(iPr)2), 4.76 (s, 2 H, γ-CH), 3.44 (sept, 3JHH = 6.8 Hz, 4 H, CH(CH3)2), 3.17 (sept, 3JHH = 6.8 Hz, 4 H, CH(CH3)2), 1.47 (s, 12 H, ArNCCH3), 1.34 (d, 3JHH = 6.8 Hz, 12 H, CH(CH3)2), 1.18 (d, 3JHH = 6.8 Hz, 12 H, CH(CH3)2), 1.10 (d, 3JHH = 6.8 Hz, 12 H, CH(CH3)2), 1.07 (d, 3JHH = 6.8 Hz, 12 H, CH(CH3)2), 0.32 (s, 6 H, GaCH3). 13C NMR (C6D6, 101 MHz): δ 207.5 (SiCO), 168.0 (ArNCCH3), 144.6 (NCC(CH(CH3)2)), 143.2, 143.0 (NCC(CH(CH3)2)), 126.8, 124.4, 124.1 (C6H3), 96.7 (γ-CH-), 29.4 (CH(CH3)2), 27.7 (CH(CH3)2), 27.6 (CH(CH3)2), 25.2, 24.3, 24.2, (CH(CH3)2), 24.2 (ArNCCH3), −0.26 (GaCH3). 29Si NMR (C6D6, 79 MHz): δ −285.2.
Yield: 42.4 mg (0.036 mmol, 43%). M.p. 189 °C (dec.). Anal. Calcd for C58H85Br2Ga2N5Si: C, 59.05; H, 7.24; N, 5.94. Found: C, 59.58; H, 6.92; N, 4.73%. IR (neat): ν 3428, 3345, 3127, 3020, 2953, 2859, 2060, 1520, 1453, 1430, 1377, 1311, 1254, 1172, 1100, 1016, 937, 856, 794, 756, 668, 625, 534, 442 cm−1. 1H NMR (C6D6, 400 MHz): δ 7.15−6.97 (m, 12 H, C6H3(iPr)2), 4.96 (s, 2 H, γ-CH), 4.67 (t, 3JHH = 4.6 Hz, 1H, SiH), 3.94 (sept, 3JHH = 6.7 Hz, 2 H, CH(CH3)2), 3.67 (sept, 3JHH = 6.7 Hz, 2 H, CH(CH3)2), 3.23 (sept, 3JHH = 6.7 Hz, 4 H, CH(CH3)2), 1.54 (s, 6 H, ArNCCH3), 1.51 (s, 6 H, ArNCCH3), 1.42 (d, 3JHH = 6.6 Hz, 6 H, CH(CH3)2), 1.30 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 1.23 (d, 3JHH = 6.6 Hz, 6 H, CH(CH3)2), 1.21 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 1.20 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 1.13 (d, 3JHH = 6.8 Hz, 6 H, CH(CH3)2), 0.98 (d, 3JHH = 6.8 Hz, 12 H, CH(CH3)2), −1.67 (d, 3JHH = 4.4 Hz, 2 H, SiNH2). 13C NMR (C6D6, 101 MHz): δ 168.8 (ArNCCH3), 147.0, 146.2 (NCC(CH(CH3)2)), 143.2, 143.0, 142.3, 141.5 (NCC(CH(CH3)2)), 127.5, 127.3, 125.6, 125.4, 123.4, 123.4 (C6H3), 98.6 (γ-CH), 29.7 (CH(CH3)2, 29.4, 29.2, 28.7 (CH(CH3)2), 28.4, 28.0 (CH(CH3)2, 25.2, 24.9, 24.6, 24.2, 24.0 (CH(CH3)2), 23.8, 23.7(ArNCCH3), 23.3 (CH(CH3)2). 29Si NMR (C6D6, 119 MHz, DEPT90): −39.6 (1JHSi = 175.6 Hz).
Data availability
All additional data part of the ESI.†Author contributions
J. S. and C. G. performed the experiments, E. S. the quantum chemical calculations, C. W. the single crystal X-ray diffraction. The work was supervised by St. S. and P. R. S. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Deutsche Forschungsgemeinschaft DFG (St. S., SCHU 1069/26-1), the University of Duisburg-Essen (St. S.), the Justus Liebig University (P. R. S.), and the Alexander-von-Humboldt-Foundation (Fellowship to E. S.).Notes and references
- (a) P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed; (b) C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS; (c) R. L. Melen, Science, 2019, 363, 479–484 CrossRef CAS PubMed.
- (a) M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed; (b) C. Shan, S. Yao and M. Driess, Chem. Soc. Rev., 2020, 49, 6733–6754 RSC.
- (a) S. Yadav, S. Saha and S. S. Sen, ChemCatChem, 2016, 8, 468–501 Search PubMed; (b) T. J. Hadlington, M. Driess and C. Jones, Chem. Soc. Rev., 2018, 47, 4176–4197 RSC.
- (a) N. Wiberg, Lehrbuch der Anorganischen Chemie, De Gruyter, Berlin, Boston, 2008 Search PubMed; (b) S. Fuhimori and S. Inoue, J. Am. Chem. Soc., 2022, 144, 2034–2050 CrossRef PubMed.
- (a) X. Wu, L. Zhao, D. Jiang, I. Fernández, R. Berger, M. Zhou and G. Frenking, Angew. Chem., Int. Ed., 2018, 57, 3974–3980 CrossRef CAS PubMed; (b) X. Wu, L. Zhao, J. Jin, S. Pan, W. Li, X. Jin, G. Wang, M. Zhou and G. Frenking, Science, 2018, 361, 912–916 CrossRef CAS PubMed; (c) C. R. Landis, R. P. Hughes and F. Weinhold, Science, 2019, 365, 2355 CrossRef PubMed; (d) L. Zhao, S. Pan, M. Zhou and G. Frenking, Science, 2019, 365, 5021 CrossRef PubMed; (e) Q. Zhang, W.-L. Li, C.-Q. Xu, M. Chen, M. Zhou, J. Li, D. M. Andrada and G. Frenking, Angew. Chem., Int. Ed., 2015, 54, 11078–11083 CrossRef CAS PubMed.
- (a) J. C. Jeffery, N. C. Norman, J. A. J. Pardoe and P. L. Timms, Chem. Commun., 2000, 2367–2368 RSC; (b) M. Finze, E. Bernhardt, A. Terheiden, M. Berkei, H. Willner, D. Christen, H. Oberhammer and F. Aubke, J. Am. Chem. Soc., 2002, 124, 15385–15398 CrossRef CAS PubMed; (c) J. D. Glore, J. W. Rathke and R. Schaeffer, Inorg. Chem., 1973, 12, 2175–2178 CrossRef CAS.
- (a) H. Braunschweig, R. D. Dewhurst, F. Hupp, M. Nutz, L. Radacki, C. W. Tate, A. Vargas and Q. Ye, Nature, 2015, 522, 327–330 CrossRef CAS PubMed; (b) M. Rang, F. Fantuzzi, M. Arrowsmith, E. Beck, R. Witte, A. Matler, A. Rempel, T. Bischof, K. Radacki, B. Engels and H. Braunschweig, Angew. Chem., Int. Ed., 2021, 60, 2963–2968 CrossRef CAS PubMed.
- (a) A. Stoy, M. Härterich, R. D. Dewhurst, J. O. C. Jiménez-Halla, P. Endres, M. Eyßelein, T. Kupfer, A. Deissenberger, T. Thiess and H. Braunschweig, J. Am. Chem. Soc., 2022 Search PubMed; (b) A. Hofmann, M.-A. Légaré, L. Wüst and H. Braunschweig, Angew. Chem., Int. Ed., 2019, 58, 9776–9781 CrossRef CAS PubMed; (c) H. Braunschweig, I. Krummenacher, M.-A. Légaré, A. Matler, K. Radacki and Q. Ye, J. Am. Chem. Soc., 2017, 139, 1802–1805 CrossRef CAS PubMed.
- F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS PubMed.
- Phosphaketenes of p-block elements: Group 13: (a) M. K. Sharma, C. Wölper, G. Haberhauer and S. Schulz, Angew. Chem., Int. Ed., 2021, 60, 6784 CrossRef CAS; (b) D. Wilson, W. Myers and J. M. Goicoechea, Dalton Trans., 2020, 49, 15249–15255 RSC; (c) D. W. N. Wilson, M. P. Franco, W. K. Myers, J. E. McGrady and J. M. Goicoechea, Chem. Sci., 2020, 11, 862–869 RSC; (d) W. Yang, K. E. Krantz, D. A. Dickie, A. Molino, D. J. D. Wilson and R. J. Gilliard, Angew. Chem., Int. Ed., 2020, 59, 3971–3975 CrossRef CAS; (e) Y. Mei, J. E. Borger, D. J. Wu and H. Grützmacher, Dalton Trans., 2019, 48, 4370–4374 RSC. Group 14: (f) S. Yao, Y. Xiong, T. Szilvási, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 4781–4785 CrossRef CAS PubMed; (g) Y. Xiong, S. Yao, T. Szilvási, E. Ballestero-Martínez, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 4333–4336 CrossRef CAS PubMed; (h) S. Bestgen, M. Mehta, T. C. Johnstone, P. W. Roesky and J. M. Goicoechea, Chem. Eur. J., 2020, 26, 9024–9031 CrossRef CAS PubMed. Group 15: (i) Z. Li, X. Chen, M. Bergeler, M. Reiher, C. Y. Su and H. Grützmacher, Dalton Trans., 2015, 44, 6431–6438 RSC; (j) L. Liu, D. A. Ruiz, D. Munz and G. A. Bertrand, Chem., 2016, 1, 147–153 CrossRef CAS; (k) Z. Li, X. Chen, L. Liu, D. A. Ruiz, J. L. Peltier, G. Bertrand, C.-Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 55, 6018–6022 CrossRef CAS PubMed; (l) M. M. Hansmann, D. A. Ruiz, L. Liu, R. Jazzar and G. Bertrand, Chem. Sci., 2017, 8, 3720–3725 RSC.
- (a) V. Lavallo, Y. Canac, B. Donnadieu, W. W. Schoeller and G. Bertrand, Angew. Chem., Int. Ed., 2006, 45, 3488–3491 CrossRef CAS PubMed; (b) C. Goedecke, M. Leibold, U. Siemeling and G. Frenking, J. Am. Chem. Soc., 2011, 133, 3557–3569 CrossRef CAS PubMed.
- (a) R. R. Lembke, R. F. Ferrante and W. Weltner, Jr., J. Am. Chem. Soc., 1977, 99, 416–423 CrossRef CAS; (b) R. S. Grev and H. F. Schaefer III, J. Am. Chem. Soc., 1989, 111, 5687–5691 CrossRef CAS; (c) M. Zhou, L. Jiang and Q. Xu, J. Chem. Phys., 2004, 121, 10474–10482 CrossRef CAS PubMed; (d) G. Maier, H. P. Reisenauer and H. Egenolf, Organometallics, 1999, 18, 2155–2161 CrossRef CAS; (e) C. A. Arrington, J. T. Petty, S. E. Payne and W. C. K. Haskins, J. Am. Chem. Soc., 1988, 110, 6240–6241 CrossRef CAS PubMed; (f) M. A. Pearsall and R. West, J. Am. Chem. Soc., 1988, 110, 7228–7229 CrossRef CAS; (g) M. Tacke, C. Klein, A. J. Stufkens, A. Oskam, P. Jutzi and E. A. Bunte, Z. Anorg. Allg. Chem., 1993, 619, 865–868 CrossRef CAS; (h) H. Bornemann and W. Sander, J. Organomet. Chem., 2002, 641, 156–164 CrossRef CAS.
- C. Ganesamoorthy, J. Schoening, C. Wölper, L. Song, P. R. Schreiner and S. Schulz, Nat. Chem., 2020, 12, 608–614 CrossRef CAS PubMed.
- D. Reiter, R. Holzner, A. Porzelt, P. Frisch and S. Inoue, Nat. Chem., 2020, 12, 1131–1135 CrossRef CAS PubMed.
- T. Sergeieva, D. Mandal and D. M. Andrada, Chem. – Eur. J., 2021, 27, 10601–10609 CrossRef CAS PubMed.
- A. Kempter, C. Gemel and R. A. Fischer, Inorg. Chem., 2008, 47, 7279–7285 CrossRef CAS PubMed.
- C. Helling, C. Ganesamoorthy, C. Wölper and S. Schulz, Dalton Trans., 2022, 51, 2050–2058 RSC.
- (a) A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS PubMed; (b) T. J. Hadlington, J. A. B. Abdalla, R. Tirfoin, S. Aldridge and C. Jones, Chem. Commun., 2016, 52, 1717–1720 RSC; (c) A. Jana, C. Schulzke and H. W. Roesky, J. Am. Chem. Soc., 2009, 131, 4600–4601 CrossRef CAS PubMed; (d) N. Weyer, M. Heinz, J. I. Schweizer, C. Bruhn, M. C. Holthausen and U. A. Siemeling, Angew. Chem., Int. Ed., 2021, 60, 2624–2628 CrossRef CAS PubMed; (e) D. Reiter, R. Holzner, A. Porzelt, P. J. Altmann, P. Frisch and S. Inoue, J. Am. Chem. Soc., 2019, 141, 13536–12546 CrossRef CAS PubMed.
- D. Reiter, P. Frisch, D. Wendel, F. M. Hörmann and S. Inoue, Dalton Trans., 2020, 49, 7060–7068 RSC.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- Search for Ga–Si single bonds in the CSD gave 86 hits ranging from 2.3290 Å to 2.5360 Å (mean 2.442 Å). C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Cryst., 2016, B72, 171–179.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Revision C.01), Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- (a) E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO Version 7.0; TCI, University of Wisconsin, Madison, WI, 2018 Search PubMed; (b) E. D. Glendening, C. R. Landis and F. Weinhold, Comput. Mol. Sci., 2012, 2, 1–42 CrossRef CAS.
- (a) R. W. Taft and R. D. Topsom, The Nature and Analysis of Substitutent Electronic Effects, in Progress in Physical Organic Chemistry, John Wiley & Sons, Ltd, 1987, pp. 1–83 CrossRef; (b) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- G. Tsankova, M. Richter, P. L. Stanwix, E. F. May and R. Span, ChemPhysChem, 2018, 19, 784–792 CrossRef CAS PubMed . Other calculated and experimental values can be found in this reference.
- (a) N. S. Hush and M. L. Williams, J. Mol. Spectrosc., 1974, 50, 349–368 CrossRef CAS; (b) A. S. Goldman and K. Krogh-Jespersen, J. Am. Chem. Soc., 1996, 118, 12159–12166 CrossRef CAS; (c) G. Bistoni, S. Rampino, N. Scafuri, G. Ciancaleoni, D. Zuccaccia, L. Belpassi and F. Tarantelli, Chem. Sci., 2013, 7, 1174–1184 RSC.
- N. J. Hardman, B. E. Eichler and P. P. Power, Chem. Commun., 2000, 1991–1992 RSC.
- A. C. Filippou, Y. N. Lebedev, O. Chernov, M. Straßmann and G. Schnakenburg, Angew. Chem., Int. Ed., 2013, 52, 6974–6978 CrossRef CAS PubMed.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Magn. Reson. Chem., 2002, 40, 489–505 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures, analytical data, NMR, IR, computational details and cif files. CCDC 2144415 (1), 2144416 (2), 2144417 (3) and 2144418 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt01335e |
| This journal is © The Royal Society of Chemistry 2022 |