
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Magnetic interactions controlled by light in the family of Fe(II)–M(IV) (M = Mo, W, Nb) hybrid organic–inorganic frameworks†
Michał
Magott
 *a,
Magdalena
Ceglarska
b,
Michał
Rams
b,
Barbara
Sieklucka
a and
Dawid
Pinkowicz
a
*a,
Magdalena
Ceglarska
b,
Michał
Rams
b,
Barbara
Sieklucka
a and
Dawid
Pinkowicz
a
aFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Kraków, Poland. E-mail: michal.magott@uj.edu.pl
bJagiellonian University, Institute of Physics, Łojasiewicza 11, 30-348 Kraków, Poland
First published on 24th May 2022
Abstract
Three new hybrid organic–inorganic frameworks employing octacyanidometallates and 4,4′-bypiridine dioxide (4,4′-bpdo) as bridging molecules were prepared and characterized. The three-dimensional coordination frameworks {[FeII(μ-4,4′-bpdo)(H2O)2]2[MIV(CN)8]·9H2O}n (Fe2Mo, Fe2W and Fe2Nb; M = Mo, W and Nb) are composed of cyanido-bridged chains, which are interconnected by the organic linkers. Magnetic measurements for Fe2Nb show a two-step transition to the antiferromagnetic state, which results from the cooperation of antiferromagnetic intra- and inter-chain interactions. Fe2Mo and Fe2W, on the other hand, behave as paramagnets at 2 K because of the diamagnetic character of the corresponding octacyanidometallate(IV) building units. However, after 450 nm light irradiation they show transition to the metastable high spin MoIV or WIV states, respectively, with distinct ferromagnetic intrachain spin interactions, as opposed to the antiferromagnetic ones observed in the Fe2Nb framework.
Introduction
One-dimensional coordination polymers play a central role in the field of molecular magnetism, which emerged with studies of chain assemblies based on radical molecules1,2 and later expanded with the discovery of Single Chain Magnets (SCMs),3–8 one-dimensional analogues of Single Molecule Magnets (SMMs).9 These materials are expected to enable high-density information storage in future devices, due to their molecular character.10–12 However, in order to achieve functional control over these units, the appropriate chemical tools need to be developed. In the case of SMMs, ordered molecular architectures were prepared by embedding such molecules in metal–organic frameworks.13–16 Among SCMs, a similar strategy was employed for cobalt(II) compounds17 and only a few examples of iron(II) chains ‘trapped’ inside higher dimensional frameworks were described so far.18–20 The alternative approach to future magnetic memory devices is based on photo-switchable assemblies, where multiple magnetic states may be addressed with light.21–24 Introduction of suitable building units into 1-D coordination polymers led to the discovery of photo-switchable chain compounds showing light-induced changes in the paramagnetic properties25–27 and in some cases slow magnetic relaxation triggered by UV-vis irradiation.28–32Herein we demonstrate three analogous cyanido-bridged chains based on iron(II) and three different octacyanidometallates(IV), which are organized into a three-dimensional (3D) architecture by an organic linker molecule – 4,4′-bipyridine dioxide (4,4′-bpdo): {[FeII(μ-4,4′-bpdo)(H2O)2]2[MIV(CN)8]·9H2O}n (M = Mo, W and Nb; Fe2Mo, Fe2W and Fe2Nb). In the case of Fe2Nb, the intrachain antiferromagnetic (AF) exchange interactions mediated by the cyanido ligands between the NbIV and FeII centers, and interactions transmitted through the long 4,4′-bpdo molecules linking FeII centres and through space, cause a transition to the AF phase below 5.6 K. On the other hand, for compounds comprising diamagnetic octacyanidomolybdate(IV) (Fe2Mo) or octacyanidotungstate(IV) (Fe2W), the irradiation by 450 nm light activates ferromagnetic intrachain interactions. To our knowledge, this is the first demonstration of the octacyanidometallate-centred photomagnetism in FeII–M(CN)8 coordination polymers.
Experimental section
K4[NbIV(CN)8]·2H2O,33 K4[MoIV(CN)8]·2H2O,33 K4[WIV(CN)8]·2H2O34 and 4,4′-bypiridine dioxide35 (4,4′-bpdo) were obtained according to published procedures. All other reagents were purchased from Sigma-Aldrich and used as supplied.{[FeII(μ-4,4′-bpdo)(H2O)2]2[MIV(CN)8]·9H2O}n (Fe2Mo for M = Mo, Fe2W for M = W, and Fe2Nb for M = Nb)
A solution of Fe(NH4)2(SO4)2·6H2O (0.084 mmol, 33 mg) and 4,4′-bypiridine dioxide hemihydrate (0.4 mmol, 80 mg) in 12 mL of water was dropwise added to a solution of the respective potassium octacyanidometallate(IV) (0.05 mmol, 25 mg for octacyanidomolybdate(IV) and octacyanidoniobate(IV), 28 mg for octacyanidotungstate(IV)) dissolved in 10 mL of water. After 24 hours needle-shaped crystals were collected by decantation. Typical yield in 10 repetitions: 15–20 mg (30–40% based on Fe). Sample purity was confirmed by powder X-ray diffraction (Fig. S1–3†) and elemental analysis. EA Found for Fe2Mo: C 32.69, H 3.93, N 16.31; calculated for Fe2MoC28N12O17H42: C 32.77, H 4.12, N 16.38. Found for Fe2W: C 29.74, H 4.08, N 15.05; calculated for Fe2WC28N12O17H42: C 30.18, H 3.80, N 15.08. Found for Fe2Nb: C 32.95, H 3.88, N 16.52; calculated for Fe2NbC28N12O17H42: C 32.83, H 4.14, N 16.43. The infrared spectra of the three compounds are demonstrated in the ESI (Fig. S4–6†).Results and discussion
Crystal structures
The three coordination polymers Fe2Mo, Fe2W and Fe2Nb are isomorphous and crystallize in a self-assembly process as needle-shaped crystals in C2/c space group (Table S1† in the ESI†). Iron(II) cations are linked by octacyanidometallate(IV) anions to form vertex-sharing coordination chains (Fig. 1a). Within each chain, the [MIV(CN)8]4− moieties form four coordination bridges to four iron(II) centres, while the four remaining cyanido ligands are engaged in hydrogen bonds with crystallization water molecules. The coordination sphere of Fe(II) consists of two nitrogen atoms of the bridging CN− ligands, two oxygen atoms of 4,4′-bipyridine dioxide molecules in the trans geometry and is completed by two water molecules in the cis configuration (Fig. S7†). Organic linkers bridge iron(II) cations in the anti-configuration,36 connecting inorganic {FeII–NC–MIV} chains into a three-dimensional I1O2 organic–inorganic framework37 depicted in Fig. 1b. In all three reported compounds, the octacyanidometallate moiety attains a dodecahedral geometry and the iron(II) centres exhibit a slightly distorted octahedral environment, as verified by the continuous symmetry measures using SHAPE software38 (Table S2†). The coordination frameworks are stabilized by numerous hydrogen bonds between the water molecules, nitrogen atoms of terminal cyanides and oxygen atoms of 4,4′-bipyridine dioxide. Interestingly, there are also relatively short contacts between terminal cyanides and centroids of bpdo aromatic rings (Fig. S8†). The corresponding N3–centroid distances are 3.290(3) Å for Fe2Mo and Fe2W, and 3.230(2) Å for Fe2Nb. The difference of 0.06 Å may be explained by the elongation of the appropriate MIV–CN bond from 2.160(3) Å to 2.255(3) Å upon substitution of Mo/W with Nb or by a larger electron density at the N3 atom of the [NbIV(CN)8] moiety in Fe2Nb. The presence of these structural motifs may indicate a significant role of anion–pi interactions in the self-assembly process, which was previously studied for other cyanidometallate-based assemblies with π-deficient molecules/ligands.39,40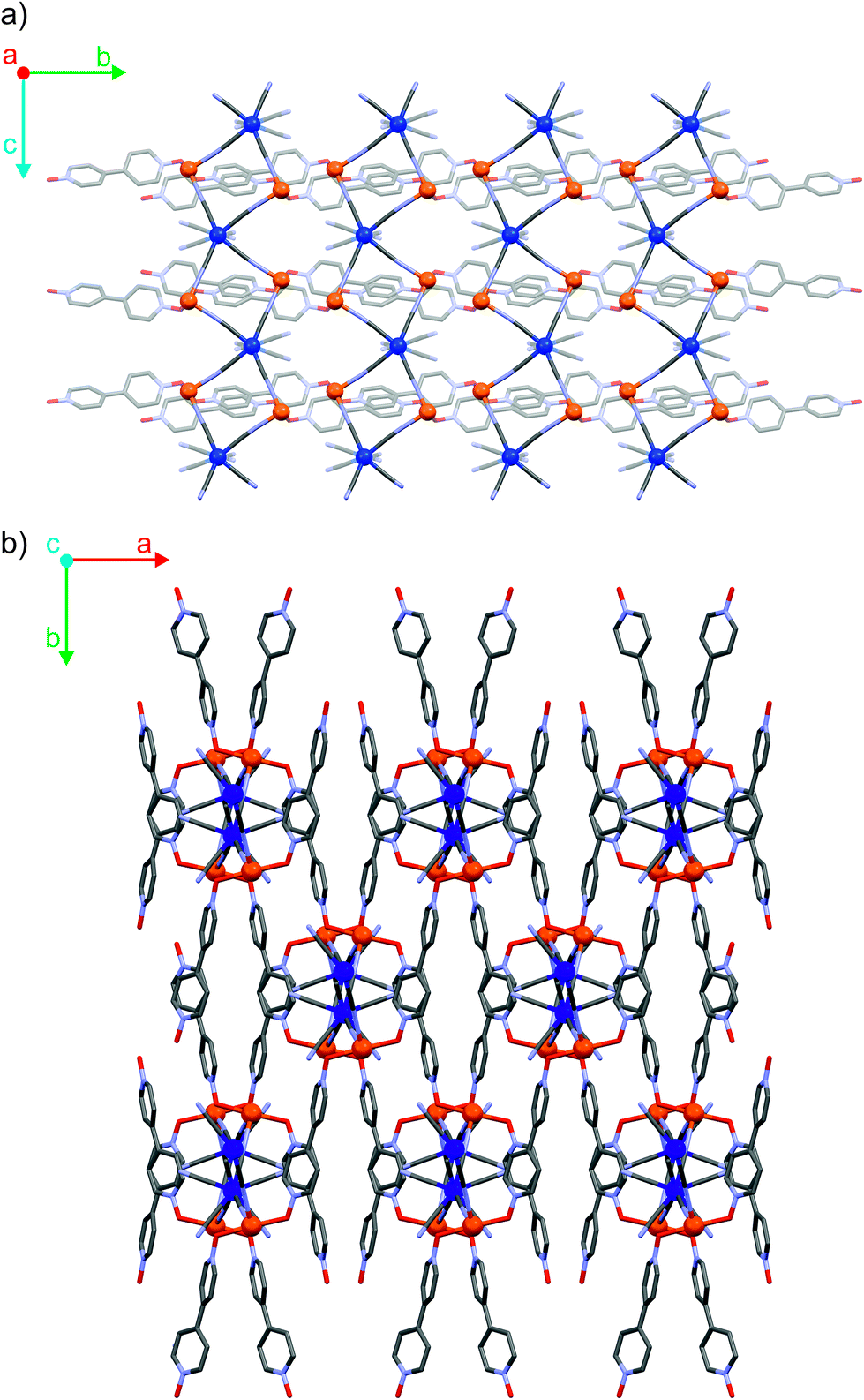 | ||
| Fig. 1 Schematic representation of cyanido-bridged coordination chains along crystallographic a axis (a) and coordination layers formed by 4,4′-bpdo linker molecules along crystallographic c direction (b) in the structure of Fe2Mo. | ||
Magnetic properties
Fe2Mo and Fe2W comprise paramagnetic iron(II) centres linked by diamagnetic octacyanidometallates(IV). At 250 K they show the χT product of 7.4 cm3 K mol−1 (Fig. 2), indicating that both iron(II) cations are in the high spin state, as can be anticipated for the N2O4 coordination environment of FeII based on the structural data. Below 50 K, these compounds show a distinct decrease of χT, down to 2.0 cm3 K mol−1 at 1.8 K for Fe2Mo and 2.3 cm3 K mol−1 at 1.8 K for Fe2W due to the zero field splitting (zfs) effect and antiferromagnetic interactions between the FeII centres. The magnetization versus field curves recorded at 1.8 K reach at 7 T values 5.4 Nβ and 5.7 Nβ for Fe2Mo and Fe2W, respectively (Fig. 2, insets). These values are far from the saturation value of 8.8 Nβ expected for two iron(II) ions with g = 2.2, and such behaviour is in line with zfs effect. Measured dependences were quantitatively analysed with the following Hamiltonian:assuming that the iron(II) cations with S = 2, as linked by bpdo ligands, are AF coupled into chains. To facilitate calculations, a finite model of a ring comprising 4 such spins was used to fit simultaneously χT(T) and M(H) dependences using MAGPACK41 based program. The best fit parameters JFeFe (superexchange coupling constant) and D (zfs parameter) summarized in Table 1 are similar for both compounds. The reported JFeFe values are in line with previous studies of systems bridged by the 4,4′-bpdo linker,42,43 as well as D parameters falling in the expected range for iron(II) systems.44–47 Nonetheless, the obtained J and D should be treated rather as approximate values, as our model does not account for superexchange interactions transferred by long [–NC–MIV(CN)6–CN–] linkers and dipole–dipole interactions between neighbouring iron(II) ions.
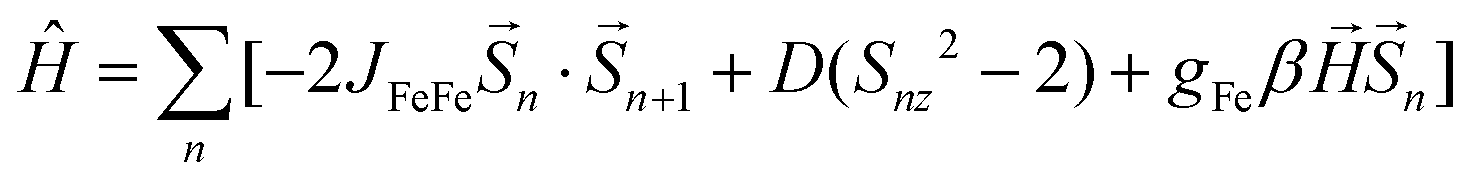
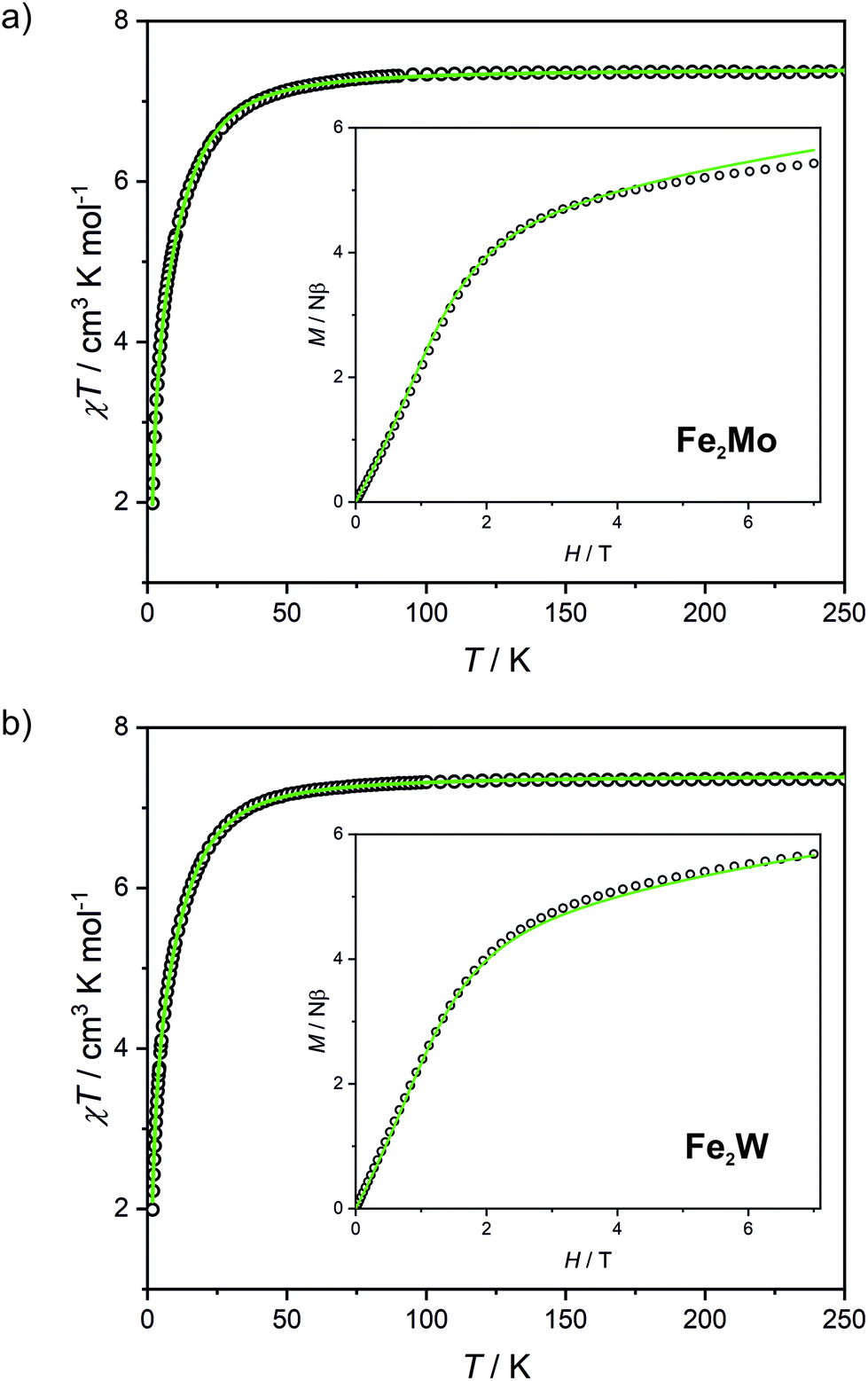 | ||
| Fig. 2 Temperature dependence of χT at 0.1 T and magnetic field dependence of magnetization at 1.8 K (inset) recorded for Fe2Mo (a) and Fe2W (b). Green lines demonstrate the result of χT(T) and M(H) fitting. | ||
| Compound | g | D/cm−1 | J FeFe/cm−1 |
|---|---|---|---|
| Fe2Mo | 2.22(3) | −10.1(1) | −0.073(5) |
| Fe2W | 2.22(2) | −10.0(1) | −0.069(3) |
The peaks of specific heat (Fig. 3), observed at Tc = 1.20 K and 1.30 K for Fe2Mo and Fe2W, respectively, demonstrate the magnetic ordering of these compounds. The entropy change for Fe2Mo is 5.71 J mol−1 K−1 from 0.5 to 5 K, which is very close to Rln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 corresponding to ordering of a spin doublet system, such as the ground state Sz = ±2 doublet of FeII in Fe2Mo. In both cases, over 60% of the entropy change is observed above Tc, in line with the dominant exchange within Fe-bpdo-Fe chains, and much weaker inter-chain interactions.48 As aforementioned, the iron(II) cations in Fe2Mo and Fe2W are connected via diamagnetic octacyanidomolybdate(IV) and octacyanidotungstate(IV) linkers. For the Fe2Nb analogue, the presence of the spin s = 1/2 of the octacyanidoniobate(IV), results in higher χT = 7.6 cm3 K mol−1 at 250 K and most importantly, leads to the decrease of χT that starts already below 100 K, indicating antiferromagnetic exchange, JFeNb, via FeII–NC–NbIV bridges (Fig. S9†). Upon further temperature decrease, the χ(T) measured in fields below 0.8 T reaches a maximum at Tc2 = 5.6 K (Fig. 4a), and another inflection at Tc1 = 3.7 K is observed, resulting in the even faster susceptibility decrease. The specific heat of Fe2Nb (Fig. 3) shows a λ-shaped peak typical for 3D ordering at 5.50 K and also a small feature at 3.55 K. Zero-field cooled (zfc) and field cooled (fc) magnetization curves for Fe2Nb show no divergence (Fig. S10†), there is no frequency dependence of AC magnetic signal (Fig. S11†) and magnetic hysteresis loop is not observed at 1.8 K (Fig. S12†). A two-step transition is also present in the magnetization field dependence (Fig. 4b). The first step is visible around μ0Hc1 = 0.95 T at 1.8 K, while a more subtle step starts around μ0Hc2 = 3.8 T (Fig. S13†). All these measurements consistently show that Fe2Nb has an AF ground state magnetic structure, but there is also another ordered structure between Tc1 and Tc2, that is also fully compensated AF at H = 0. An identification of involved metamagnetic processes can be inferred by comparison with Fe2Mo, where at a field 0.5 T a similar step in M(H) curve starts to form (Fig. 2a, inset). This suggests that the transition at Hc1 in Fe2Nb is caused by overcoming of JFeFe interaction by external field, i.e. flipping spins of whole Fe2Nb chains. The condition for such spin–flip: −4zJFeFeS2 = 2βHc1(2gFeS − gNbs) takes into account z = 4 spin interaction paths JFeFe per one Fe2Nb unit, and effectively Ising-like exchange in the zfs ground state. This, for S = 2, gFe = 2.2 and gNb = 2.0, leads to JFeFe = −0.10 cm−1, slightly stronger than for Fe2Mo.
2 corresponding to ordering of a spin doublet system, such as the ground state Sz = ±2 doublet of FeII in Fe2Mo. In both cases, over 60% of the entropy change is observed above Tc, in line with the dominant exchange within Fe-bpdo-Fe chains, and much weaker inter-chain interactions.48 As aforementioned, the iron(II) cations in Fe2Mo and Fe2W are connected via diamagnetic octacyanidomolybdate(IV) and octacyanidotungstate(IV) linkers. For the Fe2Nb analogue, the presence of the spin s = 1/2 of the octacyanidoniobate(IV), results in higher χT = 7.6 cm3 K mol−1 at 250 K and most importantly, leads to the decrease of χT that starts already below 100 K, indicating antiferromagnetic exchange, JFeNb, via FeII–NC–NbIV bridges (Fig. S9†). Upon further temperature decrease, the χ(T) measured in fields below 0.8 T reaches a maximum at Tc2 = 5.6 K (Fig. 4a), and another inflection at Tc1 = 3.7 K is observed, resulting in the even faster susceptibility decrease. The specific heat of Fe2Nb (Fig. 3) shows a λ-shaped peak typical for 3D ordering at 5.50 K and also a small feature at 3.55 K. Zero-field cooled (zfc) and field cooled (fc) magnetization curves for Fe2Nb show no divergence (Fig. S10†), there is no frequency dependence of AC magnetic signal (Fig. S11†) and magnetic hysteresis loop is not observed at 1.8 K (Fig. S12†). A two-step transition is also present in the magnetization field dependence (Fig. 4b). The first step is visible around μ0Hc1 = 0.95 T at 1.8 K, while a more subtle step starts around μ0Hc2 = 3.8 T (Fig. S13†). All these measurements consistently show that Fe2Nb has an AF ground state magnetic structure, but there is also another ordered structure between Tc1 and Tc2, that is also fully compensated AF at H = 0. An identification of involved metamagnetic processes can be inferred by comparison with Fe2Mo, where at a field 0.5 T a similar step in M(H) curve starts to form (Fig. 2a, inset). This suggests that the transition at Hc1 in Fe2Nb is caused by overcoming of JFeFe interaction by external field, i.e. flipping spins of whole Fe2Nb chains. The condition for such spin–flip: −4zJFeFeS2 = 2βHc1(2gFeS − gNbs) takes into account z = 4 spin interaction paths JFeFe per one Fe2Nb unit, and effectively Ising-like exchange in the zfs ground state. This, for S = 2, gFe = 2.2 and gNb = 2.0, leads to JFeFe = −0.10 cm−1, slightly stronger than for Fe2Mo.
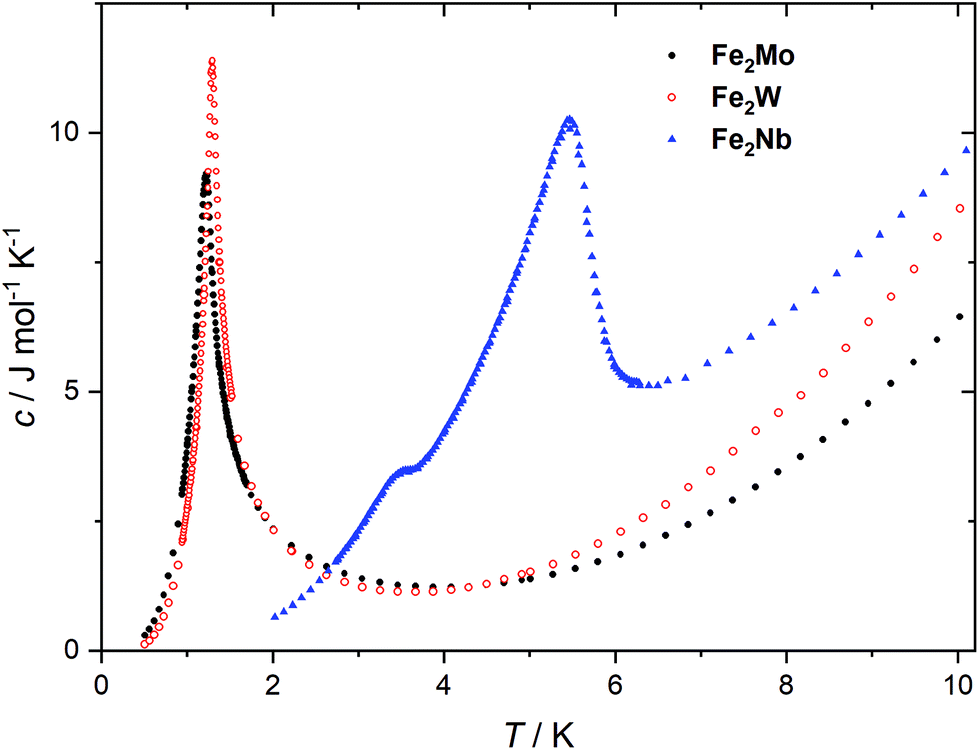 | ||
| Fig. 3 Specific heat of Fe2Mo, Fe2W and Fe2Nb, measured at zero magnetic field. | ||
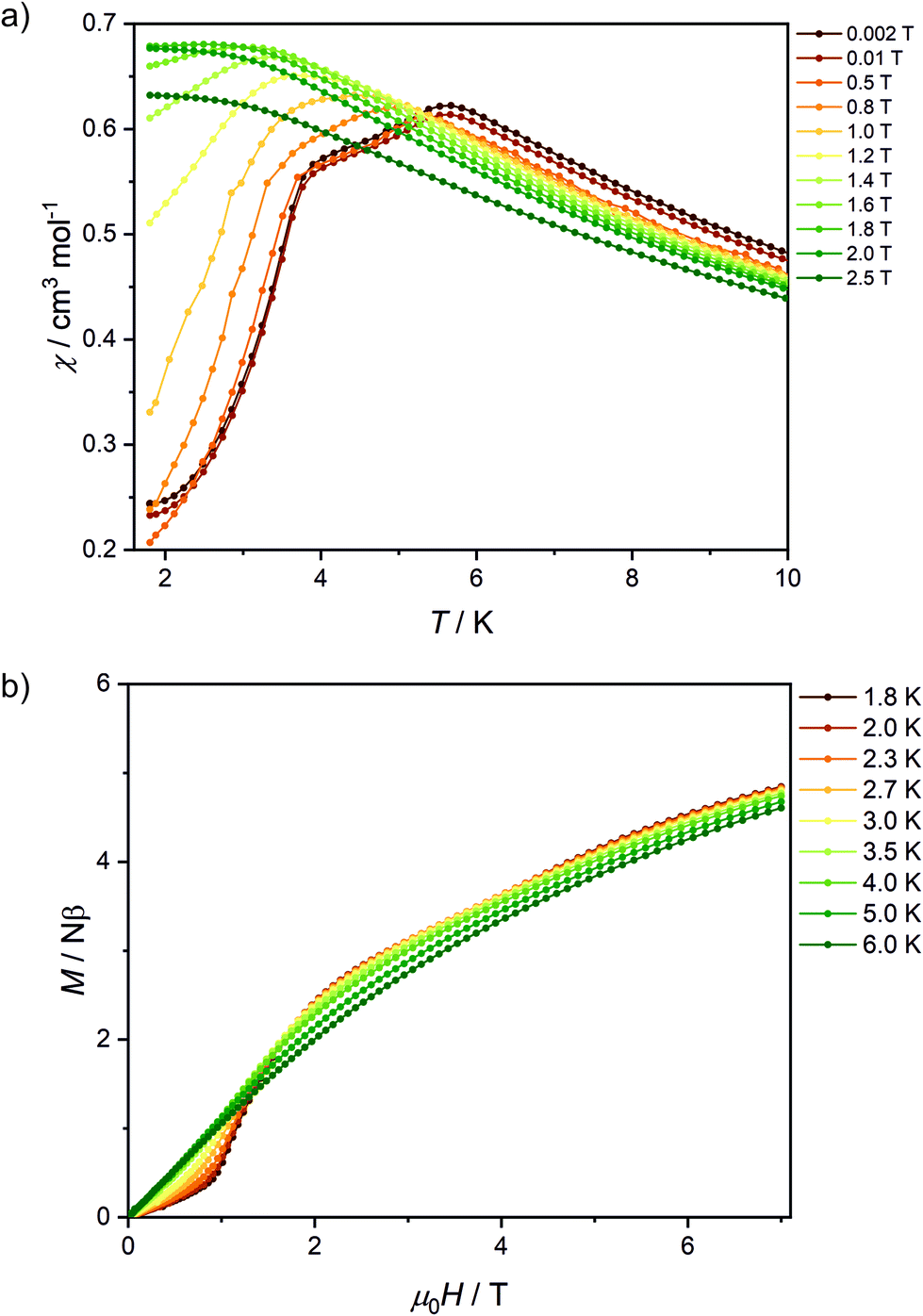 | ||
| Fig. 4 Temperature dependence of χ for various magnetic fields (a) and magnetic field dependence of magnetization at various temperatures (b) recorded for Fe2Nb. Solid lines are guides for an eye. | ||
A smaller M(H) step at Hc2 remains to be related to flipping of Nb spins when the field overcomes the JFeNb interaction. A similar estimation as before leads to JFeNb = −0.24 cm−1 using −4zJFeNbSs = 2βgNbHc2s for s = 1/2 and z = 4.
Assuming similar zfs in Fe2Nb as in Fe2Mo, the excited zfs state of Fe(II) is above 30 cm−1, which excludes its direct interfering with the ground magnetic state in the measured field range. However, still more complicated scenarios of magnetic structures sequence are possible, such as spin rearrangement, because the AF structure is expected to be non-collinear, due to presence of two different orientations of Fe(II) in the crystal lattice, and anisotropic zfs ground state of Fe(II).
Photomagnetic properties
Octacyanidomolybdate(IV) and octacyanidotungstate(IV) were demonstrated to behave as photomagnetic chromophores (intrinsic photomagnetic units).27,49–52 As aforementioned, both [MoIV(CN)8]4− and [WIV(CN)8]4− are characterized by diamagnetic S = 0 ground state, where d2 electrons are paired on a single lowest lying orbital. However, after blue light irradiation MoIV and WIV may be excited to the metastable S = 1 state, which in the case of K4[MoIV(CN)8]·2H2O was demonstrated to arise from the photoinduced cyanide dissociation, yielding heptacyanidomolybdate(IV).53 The initial diamagnetic state may be recovered by heating the sample above a certain temperature at which the thermal relaxation is very fast.In order to test the possibility of a photomagnetic switching in Fe2Mo and Fe2W, the samples of both compounds were cooled to 10 K and subjected to 450 nm light irradiation. This irradiation wavelength was selected based on UV-vis spectra of Fe2Mo and Fe2W depicted in Fig. S14.† Both compounds show broad transition in 450–650 nm range. This band is associated with iron(II)–centred d–d transitions and the d–d transition of the [MoIV(CN)8] and [WIV(CN)8] moieties, respectively.27,53,54 After turning the light on, the sudden drop of the χT is observed (Fig. S15†), which is related to the heating of the samples by ca. 2 K due to the light absorption. Upon continued irradiation, the χT slowly increases as the octacyanidometallate(IV) moieties are gradually converted from the S = 0 to the photoexcited S = 1 state. However, even after prolonged irradiation (20 h and nearly 40 h for Fe2Mo and Fe2W, respectively) no saturation has been observed, indicating that the photo-conversion to the high-spin state could not be fully completed. Finally, turning the light off leads to the sudden increase of the χT, resulting from the sample temperature equilibration. For Fe2Mo, a total increase of χT from 5.4 to 11.4 cm3 K mol−1 is observed after 23 hours of 450 nm irradiation at 10 K, while in the case of Fe2W, the χT product changes from 5.4 to 9.4 cm3 K mol−1 as a result of 39 hours of light exposure. After turning the light off both compounds show no evolution of the magnetic signal (within 30 minutes). Therefore, both Fe2Mo and Fe2W clearly show light-induced transition to the metastable state with negligible relaxation at 10 K.
After light irradiation at 10 K both Fe2Mo and Fe2W were cooled to 2 K and magnetic field dependence of magnetization was recorded (Fig. 5). In the case of Fe2W, the magnetization after irradiation is higher in the whole 0–7 T magnetic field range, reaching 6.15 Nβ as compared to 5.65 Nβ at 7 T before irradiation. The M(H) dependence with M values higher in the whole 0–7 T magnetic field range as compared with the “dark” initial state, indicates ferromagnetic superexchange interactions between the FeII and photo-induced S = 1 WIV centres, as opposed to antiferromagnetic superexchange observed in Fe2Nb. The M(H) curve for Fe2Mo also shows higher magnetization values after irradiation, with the value at saturation of 6.1 Nβ at 7 T as compared to 5.5 Nβ for the initial “dark” state. However, in the 2–3.5 T magnetic field range there is a distinct overlap of the experimental points recorded before and after light irradiation. This might result from the unaccounted anisotropy of the photo-induced S = 1 MoIV centres53 and/or antiferromagnetic interactions between the photo-induced chain fragments in Fe2Mo, which seem to be responsible for the long-range antiferromagnetic ordering in Fe2Nb.
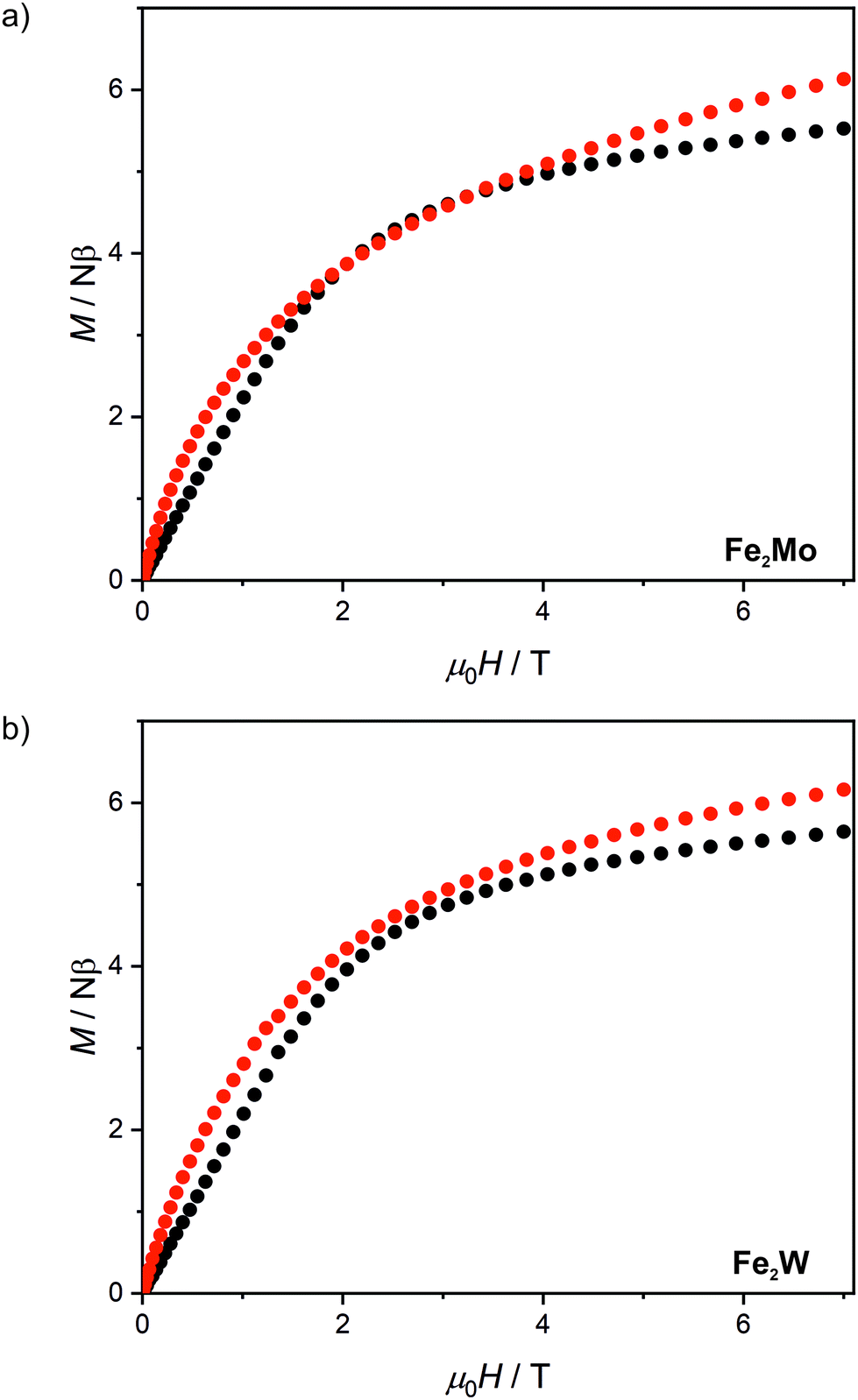 | ||
| Fig. 5 Magnetic field dependence of magnetization at 2.0 K recorded for Fe2Mo (a) and Fe2W (b) before light irradiation (black points) and after 450 nm light irradiation (red points). | ||
Measurements of magnetization field dependence after blue light irradiation were followed by χT(T) measurements at 0.1 T in 2–150 K range, which are depicted in Fig. 6 (above 150 K the signal of the sample is almost completely obscured by the diamagnetism of the sample holder, hence, the measurements were stopped at this temperature). The χT for the irradiated states of both compounds shows a significant increase upon heating starting from 2 K and reaching the maximum value of 11.1 cm3 K mol−1 at 11 K for Fe2Mo and 9.5 cm3 K mol−1 at 15 K for Fe2W. The appearance of the maximum indicates ferromagnetic intra-chain interactions via cyanido bridges that compete with the antiferromagnetic inter-chain ones via 4,4′-bpdo linkers. Further heating leads to the gradual decrease of χT for both compounds, at 150 K reaching 8.7 and 7.6 cm3 K mol−1 for Fe2Mo and Fe2W, respectively. Both values are noticeably higher than 7.3 cm3 K mol−1 observed before irradiation, indicating that at 150 K the thermal relaxation is still slow and the initial state is not fully restored. The analysis of the slope of the χT curves after irradiation presented in Fig. 6 suggests that Fe2W undergoes thermal relaxation more readily than Fe2Mo. In order to enforce complete restoration of the initial state, both compounds were heated to 240 K and kept at this temperature for 1 hour (higher temperatures were avoided to prevent dehydration of the compounds). The χT(T) curve obtained for Fe2Mo after thermal relaxation still does not overlap perfectly with the initial one below 80 K, indicating the presence of the “remnant” paramagnetic molybdenum centres still present after the irradiation. The χT(T) curve recorded after the thermal relaxation for Fe2W, on the other hand, shows perfect agreement with that recorded before the irradiation experiment. This indicates that octacyanidotungstate(IV) either shows better reversibility of the photo-induced transition or undergoes thermal relaxation in lower temperature range than the octacyanidomolybdate(IV), which is in line with similar previous observations for {[MnII(imH)]2[MIV(CN)8]}n (M = Mo, W) compounds.55,56
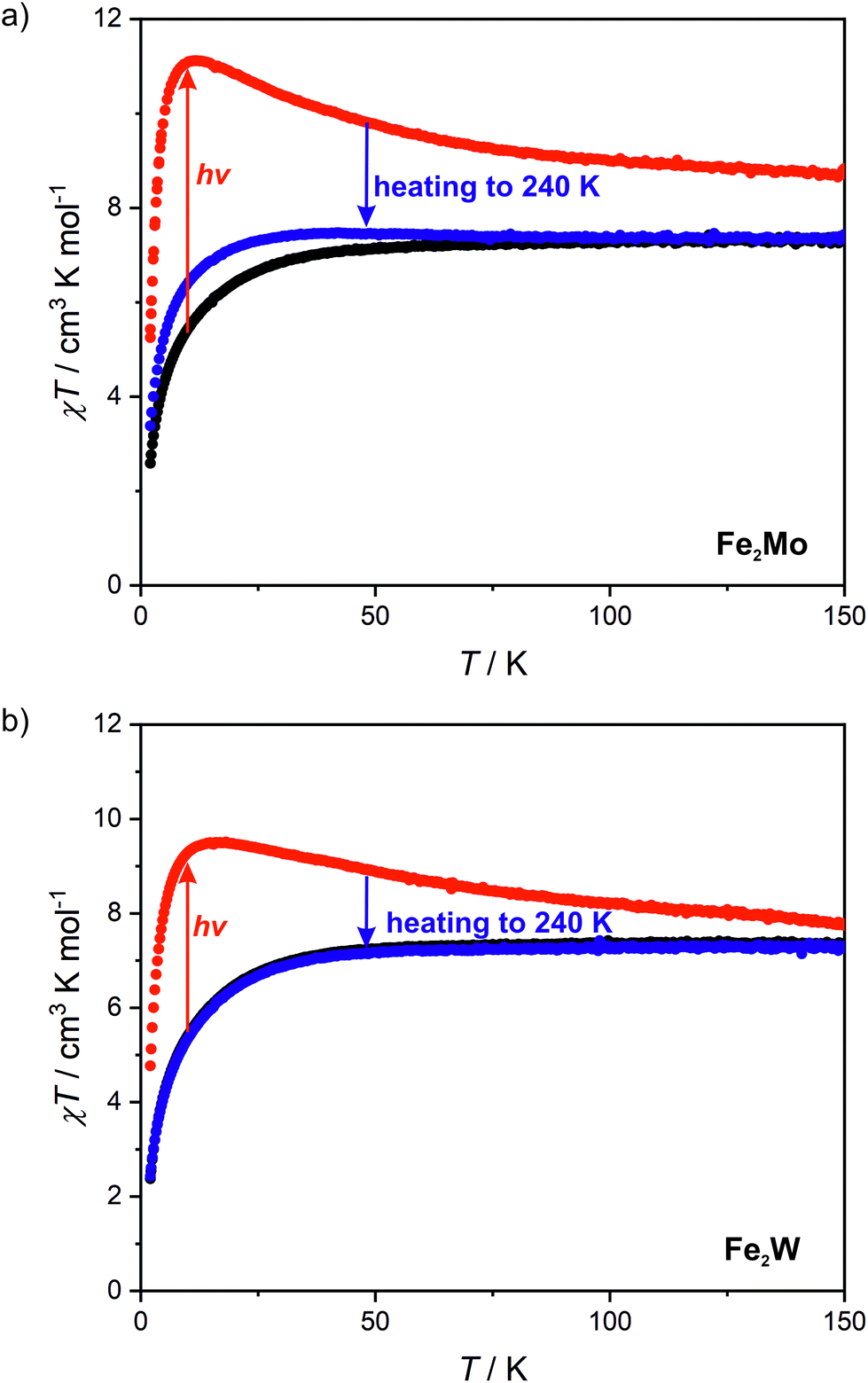 | ||
| Fig. 6 Temperature dependence of χT at 0.1 T recorded for Fe2Mo (a) and Fe2W (b) before light irradiation (black points), after 450 nm light irradiation (red points) and after thermal relaxation at 240 K (blue points). | ||
Conclusions
We have designed, obtained and characterized three new hybrid organic–inorganic I1O2 coordination frameworks composed of inorganic cyanido-bridged chains interconnected by organic linker molecules to form a 3D coordination architecture. In the case of octacyanidomolybdate(IV)- and octacyanidotungstate(IV)-based Fe2Mo and Fe2W compounds, the iron(II) centres are only weakly interacting through the 4,4′-bpdo ligands and their magnetic behaviour is governed mainly by significant zero-field splitting effects associated with FeII. In the case of Fe2Nb, the magnetic interactions between spins of iron(II) and niobium(IV) transmitted through the short CN-bridges lead to the emergence of an antiferromagnetic ordering below 5.6 K with an additional magnetic transition. The complex magnetic behaviour of Fe2Nb results most probably from the hybrid organic–inorganic architecture of this compound with the antiferromagnetic FeII–NC–NbIV interactions giving rise to the long-range ferrimagnetic correlation along the inorganic chains, which are further antiferromagnetically coupled to yield a complex 3D magnetic structure. Fe2Mo and Fe2W, on the other hand, show photomagnetic behaviour. The light-induced activation of high spin MoIV or WIV centres, respectively, leads to the appearance of ferromagnetic FeII–NC–MoIV/WIV superexchange interactions. Therefore, by the appropriate combination of the chemical design and physical stimuli (light and temperature) one can achieve paramagnetic (Fe2Mo and Fe2W before irradiation), antiferromagnetic (Fe2Nb) and ferromagnetically-coupled states (Fe2Mo and Fe2W after irradiation) within the {[FeII(μ-4,4′-bpdo)(H2O)2]2[MIV(CN)8]·9H2O}n (M = Nb, Mo, W) family of compounds. Moreover, when the photo-irradiated Fe2Mo and Fe2W are heated to 240 K in order to reverse the photo-induced changes, the ground diamagnetic state of the octacyanidometallate(IV) chromophores can be fully recovered only for Fe2W. This is in line with our previous studies of MoIV/WIV photomagnetic assemblies55,56 and may indicate better reversibility of the photomagnetic effects in octacyanidotungstate(IV)-based compounds.Author contributions
M. Magott: conceptualization, investigation, writing – original draft, writing – review & editing; M. Ceglarska: investigation, funding acquisition; M. Rams: investigation, writing – review & editing; B. Sieklucka: funding acquisition, supervision, writing – review & editing; D. Pinkowicz: conceptualization, funding acquisition, project administration, supervision, writing – review & editing. All authors reviewed and agreed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the National Science Centre (Poland) within the OPUS-20 project (Grant No. 2020/39/B/ST5/02815). MC thanks Priority Research Area SciMat under the program Excellence Initiative – Research University at the Jagiellonian University in Kraków. The single crystal X-ray diffraction experiments for Fe2Nb were performed on beamline BM01 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France (experiment no. SC-4651). We are grateful to Dr Vadim Dyadkin at the ESRF for providing assistance in using the beamline BM01.Notes and references
- G. A. Candela, L. J. Swartzendruber, J. S. Miller and M. J. Rice, J. Am. Chem. Soc., 1979, 101, 2755–2756 CrossRef CAS.
- A. H. Reis, L. D. Preston, J. M. Williams, S. W. Peterson, G. A. Candela, L. J. Swartzendruber and J. S. Miller, J. Am. Chem. Soc., 1979, 101, 2756–2758 CrossRef CAS.
- A. Caneschi, D. Gatteschi, N. Lalioti, C. Sangregorio, R. Sessoli, G. Venturi, A. Vindigni, A. Rettori, M. G. Pini and M. A. Novak, Angew. Chem., Int. Ed., 2001, 40, 1760–1763 CrossRef CAS PubMed.
- R. Clérac, H. Miyasaka, M. Yamashita and C. Coulon, J. Am. Chem. Soc., 2002, 124, 12837–12844 CrossRef PubMed.
- R. Lescouëzec, J. Vaissermann, C. Ruiz-Pérez, F. Lloret, R. Carrasco, M. Julve, M. Verdaguer, Y. Dromzee, D. Gatteschi and W. Wernsdorfer, Angew. Chem., Int. Ed., 2003, 42, 1483–1486 CrossRef PubMed.
- M. Ferbinteanu, H. Miyasaka, W. Wernsdorfer, K. Nakata, K.-i. Sugiura, M. Yamashita, C. Coulon and R. Clérac, J. Am. Chem. Soc., 2005, 127, 3090–3099 CrossRef CAS PubMed.
- H. Miyasaka, M. Julve, M. Yamashita and R. Clérac, Inorg. Chem., 2009, 48, 3420–3437 CrossRef CAS PubMed.
- R.-M. Wei, F. Cao, J. Li, L. Yang, Y. Han, X.-L. Zhang, Z. Zhang, X.-Y. Wang and Y. Song, Sci. Rep., 2016, 6, 24372 CrossRef CAS PubMed.
- S. M. J. Aubin, M. W. Wemple, D. M. Adams, H.-L. Tsai, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1996, 118, 7746–7754 CrossRef CAS.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- L. Bogani, A. Vindigni, R. Sessoli and D. Gatteschi, J. Mater. Chem., 2008, 18, 4750–4758 RSC.
- J. M. Frost, K. L. M. Harriman and M. Murugesu, Chem. Sci., 2016, 7, 2470–2491 RSC.
- G. Huang, G. Fernandez-Garcia, I. Badiane, M. Camarra, S. Freslon, O. Guillou, C. Daiguebonne, F. Totti, O. Cador, T. Guizouarn, B. Le Guennic and K. Bernot, Chem. – Eur. J., 2018, 24, 6983–6991 CrossRef CAS PubMed.
- L. H. G. Kalinke, D. Cangussu, M. Mon, R. Bruno, E. Tiburcio, F. Lloret, D. Armentano, E. Pardo and J. Ferrando-Soria, Inorg. Chem., 2019, 58, 14498–14506 CrossRef CAS PubMed.
- D. Aulakh, L. Liu, J. R. Varghese, H. Xie, T. Islamoglu, K. Duell, C.-W. Kung, C.-E. Hsiung, Y. Zhang, R. J. Drout, O. K. Farha, K. R. Dunbar, Y. Han and M. Wriedt, J. Am. Chem. Soc., 2019, 141, 2997–3005 CrossRef CAS PubMed.
- S. Chorazy, R. Podgajny, W. Nitek, M. Rams, S.-i. Ohkoshi and B. Sieklucka, Cryst. Growth Des., 2013, 13, 3036–3045 CrossRef CAS.
- M. Wang, X. Gou, W. Shi and P. Cheng, Chem. Commun., 2019, 55, 11000–11012 RSC.
- Y.-Z. Zheng, W. Xue, M.-L. Tong, X.-M. Chen, F. Grandjean and G. J. Long, Inorg. Chem., 2008, 47, 4077–4087 CrossRef CAS PubMed.
- Y.-Q. Wang, Q. Yue, Y. Qi, K. Wang, Q. Sun and E.-Q. Gao, Inorg. Chem., 2013, 52, 4259–4268 CrossRef CAS PubMed.
- J. Chen, Y. Sekine, Y. Komatsumaru, S. Hayami and H. Miyasaka, Angew. Chem., Int. Ed., 2018, 57, 12043–12047 CrossRef CAS PubMed.
- N. O. Moussa, G. Molnár, S. Bonhommeau, A. Zwick, S. Mouri, K. Tanaka, J. A. Real and A. Bousseksou, Phys. Rev. Lett., 2005, 94, 107205 CrossRef PubMed.
- T. Matsumoto, G. N. Newton, T. Shiga, S. Hayami, Y. Matsui, H. Okamoto, R. Kumai, Y. Murakami and H. Oshio, Nat. Commun., 2014, 5, 3865 CrossRef CAS PubMed.
- M. Arczyński, J. Stanek, B. Sieklucka, K. R. Dunbar and D. Pinkowicz, J. Am. Chem. Soc., 2019, 141, 19067–19077 CrossRef PubMed.
- J.-P. Launay and M. Verdaguer, Electrons in Molecules: From Basic Principles to Molecular Electronics, Oxford University Press, 2014 Search PubMed.
- J. Long, L.-M. Chamoreau, C. Mathonière and V. Marvaud, Inorg. Chem., 2009, 48, 22–24 CrossRef CAS PubMed.
- T. Korzeniak, S. Sasmal, D. Pinkowicz, W. Nitek, R. Pełka, D. Czernia, O. Stefańczyk and B. Sieklucka, Inorg. Chem., 2020, 59, 5872–5882 CrossRef CAS PubMed.
- M. Magott, O. Stefańczyk, B. Sieklucka and D. Pinkowicz, Angew. Chem., Int. Ed., 2017, 56, 13283–13287 CrossRef CAS PubMed.
- T. Liu, Y.-J. Zhang, S. Kanegawa and O. Sato, J. Am. Chem. Soc., 2010, 132, 8250–8251 CrossRef CAS PubMed.
- R. Ababei, C. Pichon, O. Roubeau, Y.-G. Li, N. Bréfuel, L. Buisson, P. Guionneau, C. Mathonière and R. Clérac, J. Am. Chem. Soc., 2013, 135, 14840–14853 CrossRef CAS PubMed.
- T. Liu, H. Zheng, S. Kang, Y. Shiota, S. Hayami, M. Mito, O. Sato, K. Yoshizawa, S. Kanegawa and C. Duan, Nat. Commun., 2013, 4, 2826 CrossRef.
- M. Hojorat, H. Al Sabea, L. Norel, K. Bernot, T. Roisnel, F. Gendron, B. L. Guennic, E. Trzop, E. Collet, J. R. Long and S. Rigaut, J. Am. Chem. Soc., 2020, 142, 931–936 CrossRef CAS PubMed.
- Y.-J. Ma, J.-X. Hu, S.-D. Han, J. Pan, J.-H. Li and G.-M. Wang, J. Am. Chem. Soc., 2020, 142, 2682–2689 CrossRef CAS PubMed.
- G. Handzlik, M. Magott, B. Sieklucka and D. Pinkowicz, Eur. J. Inorg. Chem., 2016, 2016, 4872–4877 CrossRef CAS.
- D. Matoga, J. Szklarzewicz and M. Mikuriya, Inorg. Chem., 2006, 45, 7100–7104 CrossRef CAS PubMed.
- D. Wang, W. E. Crowe, R. M. Strongin and M. Sibrian-Vazquez, Chem. Commun., 2009, 1876–1878 RSC.
- J. Jia, A. J. Blake, N. R. Champness, P. Hubberstey, C. Wilson and M. Schröder, Inorg. Chem., 2008, 47, 8652–8664 CrossRef CAS PubMed.
- A. K. Cheetham, C. N. R. Rao and R. K. Feller, Chem. Commun., 2006, 4780–4795 RSC.
- P. Alemany, D. Casanova, S. Alvarez, C. Dryzun and D. Avnir, in Reviews in Computational Chemistry, 2017, pp. 289–352 Search PubMed.
- R. Podgajny, D. Pinkowicz, B. Czarnecki, M. Kozieł, S. Chorąży, M. Wis, W. Nitek, M. Rams and B. Sieklucka, Cryst. Growth Des., 2014, 14, 4030–4040 CrossRef CAS.
- E. Kuzniak-Glanowska, J. Kobylarczyk, K. Jedrzejowska, D. Glosz and R. Podgajny, Dalton Trans., 2021, 50, 10999–11015 RSC.
- J. J. Borrás-Almenar, J. M. Clemente-Juan, E. Coronado and B. S. Tsukerblat, J. Comput. Chem., 2001, 22, 985–991 CrossRef.
- M. Yuan, S. Gao, F. Zhao, W. Zhang and Z. Wang, Sci. China, Ser. B: Chem., 2009, 52, 266–275 CrossRef CAS.
- D. Pinkowicz, M. Rams, W. Nitek, B. Czarnecki and B. Sieklucka, Chem. Commun., 2012, 48, 8323–8325 RSC.
- R. Boča, Coord. Chem. Rev., 2004, 248, 757–815 CrossRef.
- A. Ozarowski, S. A. Zvyagin, W. M. Reiff, J. Telser, L.-C. Brunel and J. Krzystek, J. Am. Chem. Soc., 2004, 126, 6574–6575 CrossRef CAS PubMed.
- G. Novitchi, S. Jiang, S. Shova, F. Rida, I. Hlavička, M. Orlita, W. Wernsdorfer, R. Hamze, C. Martins, N. Suaud, N. Guihéry, A.-L. Barra and C. Train, Inorg. Chem., 2017, 56, 14809–14822 CrossRef CAS PubMed.
- C. Rojas-Dotti, N. Moliner, R. González and J. Martínez-Lillo, J. Coord. Chem., 2018, 71, 737–747 CrossRef CAS.
- R. L. Carlin, Magnetochemistry, Springer, Berlin, Heidelberg, 1986 Search PubMed.
- N. Bridonneau, J. Long, J. L. Cantin, J. von Bardeleben, S. Pillet, E. E. Bendeif, D. Aravena, E. Ruiz and V. Marvaud, Chem. Commun., 2015, 51, 8229–8232 RSC.
- T. Korzeniak, R. Jankowski, M. Kozieł, D. Pinkowicz and B. Sieklucka, Inorg. Chem., 2017, 56, 12914–12919 CrossRef CAS PubMed.
- M. Magott, M. Sarewicz, S. Buda and D. Pinkowicz, Inorg. Chem., 2020, 59, 8925–8934 CrossRef CAS PubMed.
- X. Qi, P. Guionneau, E. Lafon, S. Perot, B. Kauffmann and C. Mathonière, Magnetochemistry, 2021, 7, 97 CrossRef CAS.
- X. Qi, S. Pillet, C. de Graaf, M. Magott, E.-E. Bendeif, P. Guionneau, M. Rouzières, V. Marvaud, O. Stefańczyk, D. Pinkowicz and C. Mathonière, Angew. Chem., Int. Ed., 2020, 59, 3117–3121 CrossRef CAS PubMed.
- M. F. A. Hendrickx, V. S. Mironov, L. F. Chibotaru and A. Ceulemans, Inorg. Chem., 2004, 43, 3142–3150 CrossRef CAS PubMed.
- M. Magott, M. Reczyński, B. Gaweł, B. Sieklucka and D. Pinkowicz, J. Am. Chem. Soc., 2018, 140, 15876–15882 CrossRef CAS PubMed.
- M. Magott, B. Gaweł, M. Sarewicz, M. Reczyński, K. Ogorzały, W. Makowski and D. Pinkowicz, Chem. Sci., 2021, 12, 9176–9188 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, powder X-ray diffraction patterns, magnetic measurements, UV-vis and IR spectra, and CIF files. CCDC 2155441–2155443. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00777k |
| This journal is © The Royal Society of Chemistry 2022 |