
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Factors influencing stoichiometry and stability of polyoxometalate – peptide complexes†
Björn H.
Greijer
,
Gustav
Nestor
 ,
Jan E.
Eriksson
,
Gulaim A.
Seisenbaeva
and
Vadim G.
Kessler
*
,
Jan E.
Eriksson
,
Gulaim A.
Seisenbaeva
and
Vadim G.
Kessler
*
Department of Molecular Sciences, Swedish University of Agricultural Sciences, Box 7015, 75007 Uppsala, Sweden. E-mail: vadim.kessler@slu.se
First published on 7th June 2022
Abstract
In the pursuit of understanding the factors guiding interactions between polyoxometalates (POMs) and biomolecules, several complexes between Keggin phosphomolybdate and diglycine have been produced at different acidity and salinity conditions, leading to difference in stoichiometry and in crystal structure. Principal factors determining how the POM and dipeptide interact appear to be pH, ionic strength of the medium, and the molar ratio of POM to peptide. An important effect turned out to be even the structure-directing role of the sodium cations coordinating carbonyl functions of the peptide bond. Given the interest in applying POMs in biological systems, these factors are highly relevant to consider. In the view of recent interest in using POMs as nano catalysts in peptide hydrolysis also the potential Keggin POM transformation in phosphate buffered saline medium was investigated leading to insight that nanoparticles of zirconium phosphate (ZrP) can be actual catalysts for breakdown of the peptide bond.
Introduction
Polyoxometallates (POMs) are molecular metal oxide oligomers, generally consisting of tungsten, molybdenum or vanadium oxides, commonly, in their highest oxidation state. Quite a number of different families of POMs have been reported, but in this study we have focused on the Keggin type hetero poly acid Hn[EM12O40] (E = P, Si; M = Mo, W) involving a central phosphorus atom surrounded by molybdenum oxide triades of edge-sharing octahedra, forming a tetrahedron-shaped construction, a phosphometallate (PM12). These species and their heterometallic derivatives possess spheroidal shape with a diameter of about 1.04 nm, making them attractive as potentially well structurally resolved models of metal oxide nanoparticles (NPs).Biological activity of small NPs and POMs has recently attracted a lot of attention.1–5 There are numerous PDB entries which contain POM ligands,6 and modified phosphotungstates have shown great catalytic abilities towards proteins7,8 and DNA analogues,9 acting as catalysts in hydrolysis of peptide and phosphoester bonds, respectively. It is, therefore, of interest to structurally characterize the interactions between POMs and various biomolecules in detail, in order to identify factors guiding formation of new structures and determining their activity. This can give insights into the factors guiding biological activity of metal oxide nanoparticles. In particular, the insights into the POMs’ interaction with biomolecules has potential to contribute to understanding the phenomenon of formation of the so-called “molecular corona”.
Nanoparticles typically form a protein corona,10,11 where protein molecules adhere to the surface of the particle. With regard to Keggin POMs, however, their small size (∼1 nm diameter) permits treating them as small molecule ligands, which can be directly visualized and provide quantitative information about the chemical bonding. The high negative charge of Keggin POMs allows formation of both electrostatic and hydrogen bonds with proteins targeting positively charged domains on the protein surface. Furthermore, the small size of Keggin POMs allows for interaction limited to small patches as well as fitting into cavities within protein structures.
Because of the POM's strong negative charge, the target in their interactions are thus primarily the positively charged regions. When no such regions exist on a protein, they may be created via protonation by lowering the pH and thus opening for new unexpected interactions. Keggin POM PMo12O403− anions have shown a tendency towards electrostatic interactions with oligoglycines, aside from the H-bonds one would expect.12,13 These electrostatic interactions may allow for contacts with uncharged or hydrophobic residues, increasing the versatility in comparison to the more studied PW12 species.
The Keggin POM is more stable under low pH,14,15 though we have previously demonstrated that oligopeptides have a stabilizing effect on the POM structure.12 The extent of stabilization is yet undetermined. Vandebroek and colleagues16 reported a structure of a zirconium-modified Keggin phosphotungstate [Zr(PW11O39)]−4 in complex with Hen Egg-White (HEW) Lysozyme (HEWL) protein, which was crystallized at neutral pH. Normally, the Keggin phosphomolybdate POM would break down above pH 4–5, depending on the composition,14,15 so preserving the intact POM at physiological pH would be highly beneficial in regards to in vivo applications. The new species formed reportedly in the course of the synthesis from (Et2NH2)8[{α-PW11O39Zr-(μ-OH)(H2O)}2]·7H2O used as reactant, and was considered as the potentially active species in protein hydrolysis.16
In our previous study, we have investigated the interaction of Keggin POMs, phosphomolybdic and phostungstic acid with diglycine peptide in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio and demonstrated that at pH between 3 and 4 the structures differed considerably, indicating stronger electrostatic interactions with Mo-POM and relatively stronger manifested H-bonding with W-POM.12 We noticed that heating of the reaction mixture led generally to products with lower crystal water contents. Increase in the length of the peptide to tri and tetra-glycine led to products with lower POM-peptide ratios and require lowering of pH for isolation of the products.13 We have demonstrated that the pH affected considerably the ability of a complex between tetraglycine and PMo12 to form crystals.13 This was hypothesized to become possible due to the protonation of the oligopeptide, which would make it stronger positively charged for better electrostatic interaction with POM. This peptide possessed also quite poor solubility under near-neutral pH conditions, which was strongly improved on lowering the pH. In order to ensure that it was fully protonated, the acid concentration was increased considerably, which yielded crystals fit for X-ray diffraction. In this study, we aimed to investigate this phenomenon further, by analyzing crystals derived from solutions with different ratios of diglycine-to-POM, acidity and also salinity.
:3 ratio and demonstrated that at pH between 3 and 4 the structures differed considerably, indicating stronger electrostatic interactions with Mo-POM and relatively stronger manifested H-bonding with W-POM.12 We noticed that heating of the reaction mixture led generally to products with lower crystal water contents. Increase in the length of the peptide to tri and tetra-glycine led to products with lower POM-peptide ratios and require lowering of pH for isolation of the products.13 We have demonstrated that the pH affected considerably the ability of a complex between tetraglycine and PMo12 to form crystals.13 This was hypothesized to become possible due to the protonation of the oligopeptide, which would make it stronger positively charged for better electrostatic interaction with POM. This peptide possessed also quite poor solubility under near-neutral pH conditions, which was strongly improved on lowering the pH. In order to ensure that it was fully protonated, the acid concentration was increased considerably, which yielded crystals fit for X-ray diffraction. In this study, we aimed to investigate this phenomenon further, by analyzing crystals derived from solutions with different ratios of diglycine-to-POM, acidity and also salinity.
Results and discussion
Synthesis
In our previous work,12 where we aimed to compare Mo and W Keggin POM bonding to diglycine, the compounds were prepared by mixing the POM and peptides in a 1:3 ratio without addition of acids for pH regulation. This resulted in isolation of compound 1, (HGlyGly)3[PMo12O40]·4H2O, when the reaction was carried out at room temperature for Mo-POM. Increase in hydrophobicity of a peptide led to decrease in POM:peptide ratio in the isolated products with 1:2 for Mo- and just 1:1 for W-POM, in spite of 1:3 composition in the reaction mixtures.13 Here, we aimed to reveal the action of such factors as POM-peptide ratio, salinity and pH on the composition and structure of emerging complexes. Interestingly, changing the ratio of Mo-POM and diglycine did not affect the composition of the complex. When subjecting the solutions with GlyGly:PM12 in 1:1 and 2:1 ratios to crystallization on cooling or solvent evaporation, the primary product that could be isolated at room temperature, was the earlier described (HGlyGly)3[PMo12O40]·4H2O (1).12 When the crystals of 1 were removed from solution and the ratio of GlyGly:PM12 decreased to less than 0.5:1, a new phase appeared denoted as compound 2, featuring the same composition, but with the structure analogous to that of earlier reported (HGlyGly)3[PW12O40]·4H2O.12 Heat-treatment of the reaction mixture led for 1:1 composition to the complex with the same peptide to POM ratio, but with a doubled unit cell and exact composition (HGlyGly)3[PMo12O40]·3H2O (3), i.e. containing less water per asymmetric unit. This change was in line with our earlier observations.12
Na(HGlyGly)2[PMo12O40]·8H2O (4) was obtained in attempt to crystallize iron-substituted POM and titanium substituted POM and can be reproduced, applying simply increased salinity. Na(HGlyGly)2[PMo12O40]·8H2O (5) was obtained from the titanium containing solution at lower pH. A different form of 1:1 complex, not containing any interstitial water molecules, Na(HGlyGly)(H3O)[PMo12O40]·3H2O (6), was obtained from the attempt to produce a zirconium-substituted POM (Fig. 1).
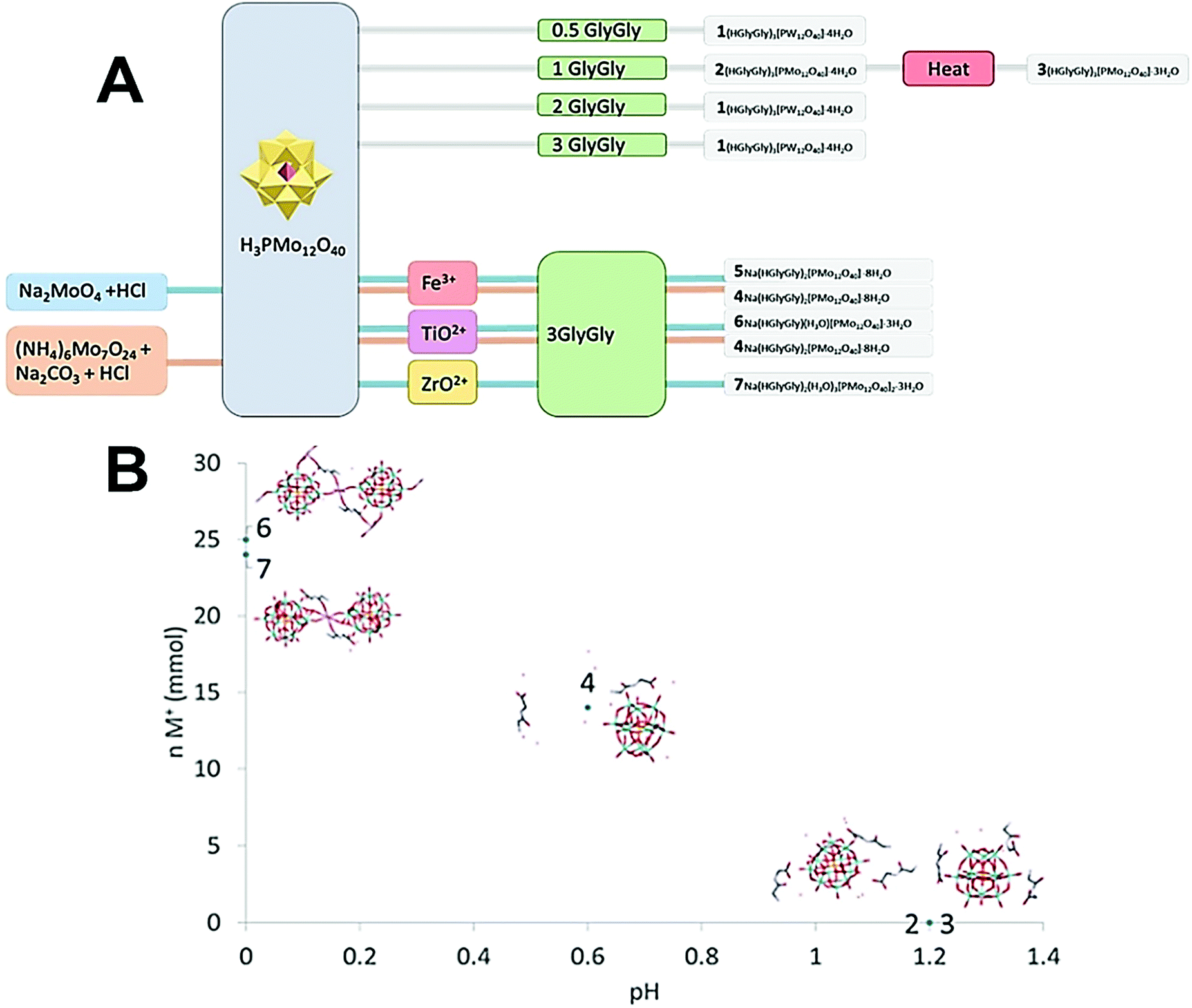 | ||
| Fig. 1 (A) Scheme of how synthesis conditions yields different crystal structures. (B) Plot of ionic strength of the medium vs. pH for compounds 2–7. | ||
The pH of 2 and 3 were approximately 1.2, for 4 pH was 0.6, and for 5 and 6 pH was approximately 0. It appears that pH is a major factor in deciding which structure forms, and how many peptide molecules are found in it. 2 and 3 are also distinct from the previously described structure of diglycine and phosphomolybdate.12 Though the older structure does contain three peptides, their conformation differ to that of those reported here. The structure of 2, obtained at lower pH is isomorphous to that of W-POM with more pronounced trend to H-bonding. The effect of pH may be due to the peptides being more, or less, protonated,17 or the POM being partially lacunary at certain pH.15 These equilibria may be one reason for the great diversity observed. 3, 6 and 7 also are obtained in syntheses involving multiply charged metal ions which, while not entering the crystal structures, might contribute to their formation.
The single crystal to bulk identity of products was confirmed by unit cell determination of several randomly chosen single crystals of each compound and also by matching X-ray powder diffractogram of each material to its theoretically calculated powder pattern generated from single crystal structure determination results. The theoretical and experimental powder patterns were in good agreement for all samples.
Crystal and molecular structures
(HGlyGly) 3 [PMo 12 O 40 ]·4H 2 O (2) (Fig. 2) is triclinic centrosymmetric, with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . It contains three protonated diglycine molecules, one phosphomolybdate Keggin anion, and five water molecules in an asymmetric unit, Z = 2.
. It contains three protonated diglycine molecules, one phosphomolybdate Keggin anion, and five water molecules in an asymmetric unit, Z = 2.
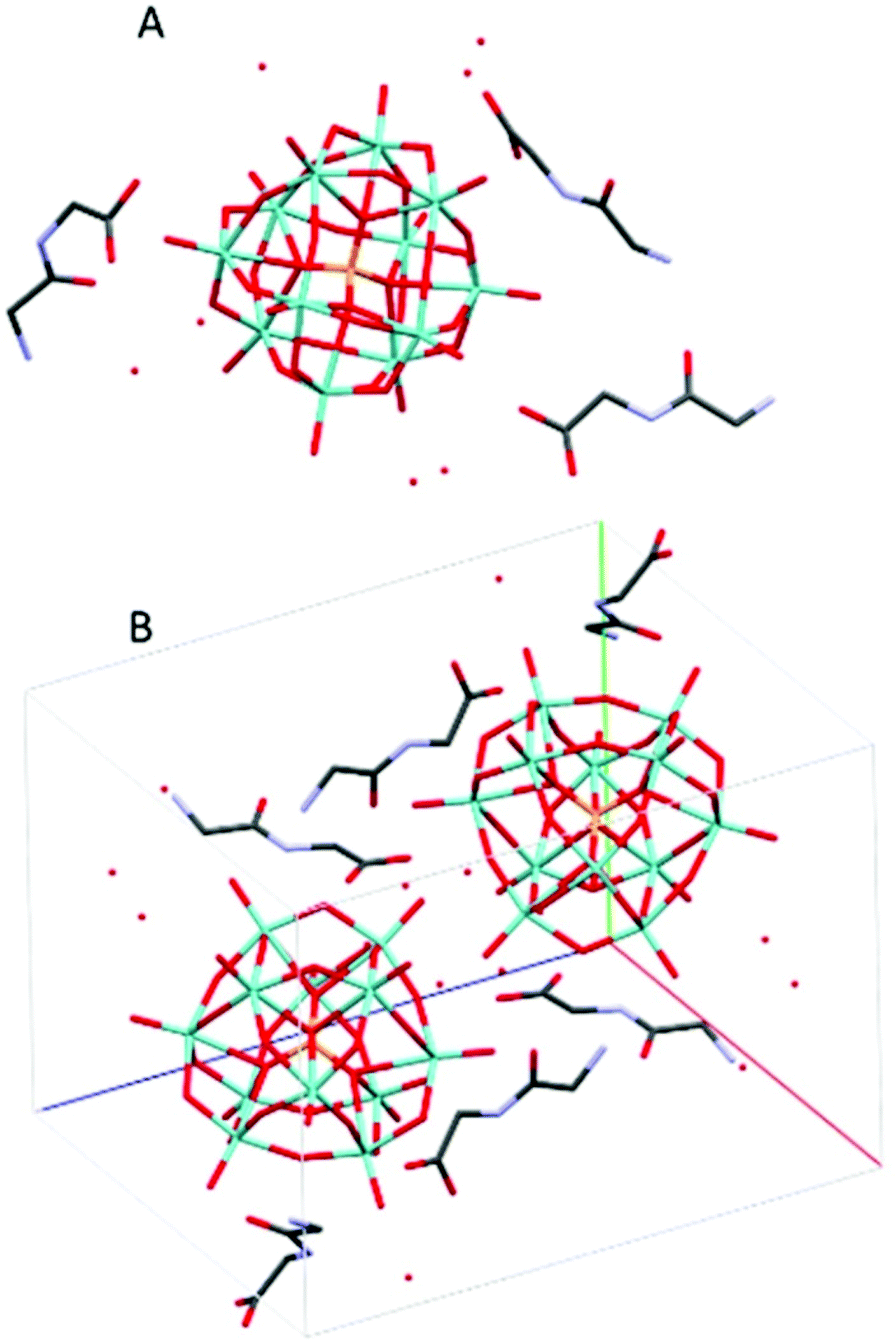 | ||
| Fig. 2 The molecular (A) and crystal (B) structure of compound (HGlyGly)3[PMo12O40]·4H2O (2). | ||
The strongest H-bonds are between water and carboxyl group (2B)–O(4D) 2.717(6) Å and between waters O(1D)–O(7D) 2705(7) Å. The next strongest bonds are between ammonium units and carbonyl groups N(2A)–O(3B) 2.776(4) Å and N(2B)–O(3C) 2.786(6) Å. Slightly weaker are the bonds between carboxyl groups and water O(1A)–O(5D) 2.795(6) Å and amino groups and water N(2B)–O(1D) 2.810(5) Å and N(2C)–O(2D) 2.861(8) Å as well as bonds between water molecules O(1D)–O(2D) 2.841(7) Å and O(2D)–O(3D) 2.832(6) Å and bridging oxygen atoms of the POM with water O(5D)–O(12) 2.836(6) Å, carboxyl oxygen O(1C)–O10 2.840(6) Å and an amino group N(2B)–O(38) 2.866(5) Å, and between water and carboxyl oxygen O(2D)–O(2A) 2.836(5) Å and between amino group and a carbonyl oxygen N(2A)–O(3A) 2.867(5) Å. Next comes bonds between an amide group and water N(1C)–O(5D) 2.941(7) Å, water and a carbonyl group O(7D)–O(3A) 2.929(8), bridging oxygen on the POM and a water O(7D)–O(40) 2.937(6) Å and an amino group N(2A)–O(32) 2.947(5), terminal oxygen on the POM and a carboxyl group O(1C)–O(22) 2.948(6) Å, an amino group and water molecules N(2C)–O(6D) 2.939(9) Å, and an amide group and a terminal oxygen on the POM N(1A)–O(34) 2.962(4) Å.
The weakest bonds are between water molecules O(7D)–O(4D) 3.028(9) Å, O(6D)–O(5D) 3.038(7) Å, an amino group and a bridging POM oxygen N(2C)–O(33) 3.207(5) Å and an amide group and a carboxyl group N(1B)–O(1C) 3.264(4) Å.
(HGlyGly) 3 [PMo 12 O 40 ]·3H 2 O (3) (Fig. 3) is monoclinic, space group P21/n, with one POM, three diglycine, and three water molecules in an asymmetric unit, Z = 4. The strongest H-bond is between carboxyl and carbonyl oxygen O(2D)–O(3B) 2.571(20) Å, a carboxyl group and water O(2B)–O(1A) 2.643(10), and corresponding amide groups N(1D)–N(1D) 2.747(30) Å. Next is amino groups with a carboxyl group N(2D)–O(1C) 2.802(30) Å or water N(2C)–O(3A) 2.849(30) Å, N(2C)–O(2A) 2.880(30) Å and between an amino group and a bridging POM oxygen N(2B)–O(16) 2.904(10) Å. Slightly weaker are a number of bonds between amino groups and terminal POM oxygen N(2D)–O(40) 2.948(30) Å, N(2C)–O(22) 3.043(20) Å, N(2B)–O(3A) 3.063(20) Å, N(2B)–O(14) 3.071(10) Å, or bridging POM oxygens N(2B)–O(1) 3.062(10) Å.
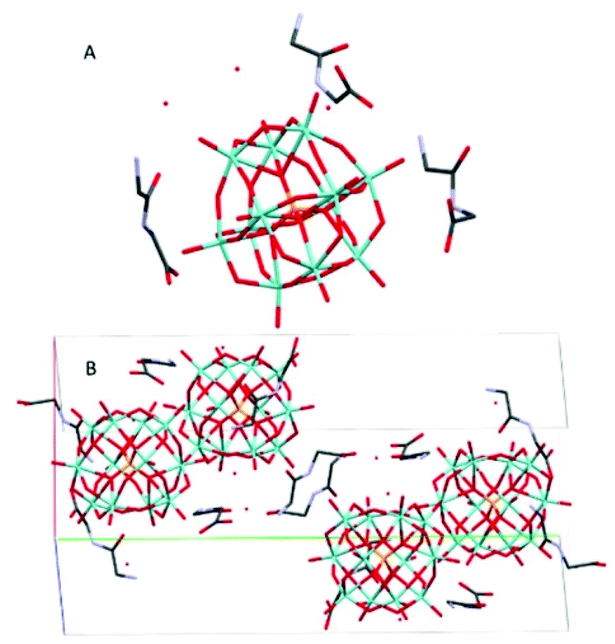 | ||
| Fig. 3 The molecular (A) and crystal (B) structure of compound (HGlyGly)3[PMo12O40]·3H2O (3). | ||
Na(HGlyGly) 2 [PMo 12 O 40 ]·8H 2 O (4) (Fig. 4) has an orthorhombic space group, Pbca, and contains one Keggin POM unit, two diglycine molecules, eight water molecules and a sodium ion in an asymmetric unit, Z = 8.
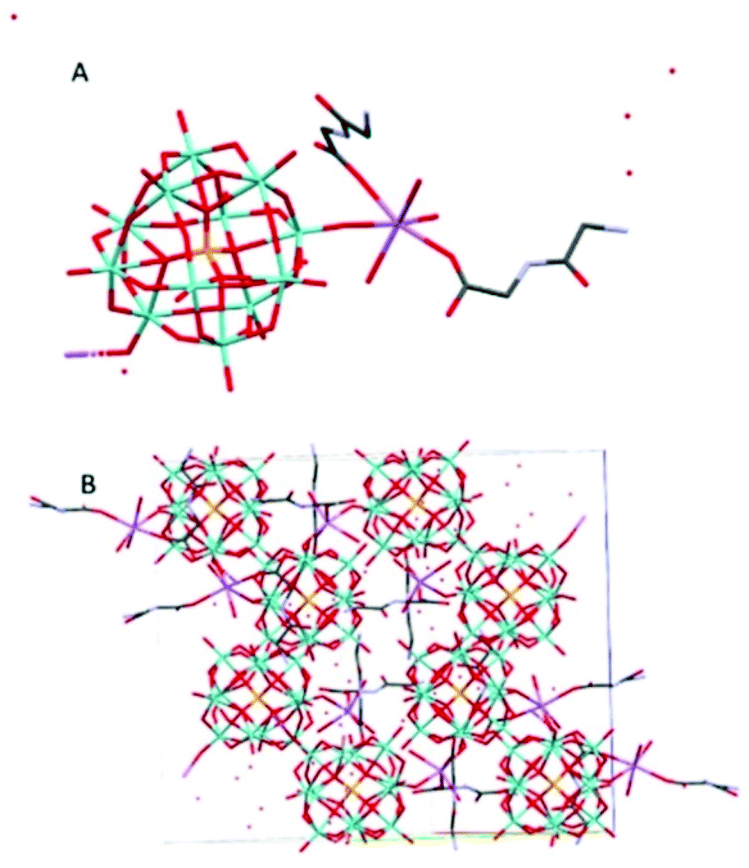 | ||
| Fig. 4 The molecular (A) and crystal (B) structure of compound Na(HGlyGly)2[PMo12O40]·8H2O (4). | ||
The shortest bonds are between oxygens and the hydrated sodium ion Na(1)–O(1C) 2.341(5) Å, Na(1)–O(1A) 2.346(5), Na(1)–O(40) 2.374(5) Å, Na(1)–O(1B) 2.395(5), Na(1)–O(35) 2.415(5) Å, Na(1)–O(2A) 2.453(5) Å. The strongest H-bonds are between a carboxyl group and water O(2C)–O(5A) 2.634(8) Å, and between amino groups and carbonyl groups N(2C)–O(3B) 2.729(8) Å, N(2B)–O(3C) 2.790(7) Å, which form a ring involving four adjacent diglycine molecules (Fig. S1†). Next, come bonds between water molecules O(5A)–O(7A) 2.796(9) Å, O(3A)–O(5A) 2.804(8) Å, O(4A)–O(7A) 2.833(8) Å O(2A)–O(3A) 2.870(8) Å, water and an amino group O(7A)–N(2C) 2.856(8) Å, and between water and bridging POM oxygen O(1A)–O(29) 2.863(6) Å, O(3A)–O(12) 2.892(6) Å.
The weakest H-bonds are between amino groups and water N(2B)–O(3A) 2.910(10) Å or terminal POM oxygens N(2B)–O(31) 2.919(8) Å, O(13)–N(2C) 2.966(8) Å.
Na(HGlyGly) 2 [PMo 12 O 40 ]·8H 2 O (5) Is monoclinic, space group P21/c. It contains one α-Keggin POM, two diglycine molecules, one sodium ion and eight water molecules, Z = 4 (Fig. 5).
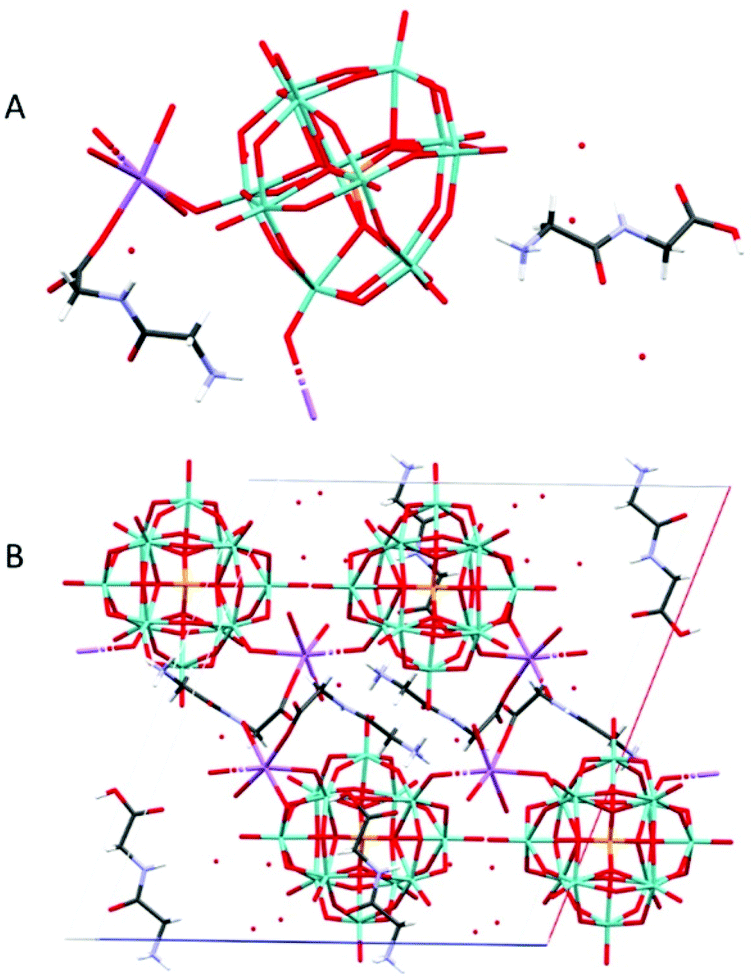 | ||
| Fig. 5 The molecular (A) and crystal (B) structure of Na(HGlyGly)2[PMo12O40]·8H2O (5). | ||
The shortest contacts are between the sodium ion and a carboxyl oxygen Na(1)–O(1C) 2.267(6) Å, water Na(1)–O(1A) 2.337(4) Å, Na(1)–O(4A) 2.344(5) Å, Na(1)–O(2A) 2.418(4) Å, or terminal POM oxygen Na(1)–O(27) 2.535(4) Å, Na(1)–O(14) 2.612(4) Å, and between carboxyl oxygen and water O(2C)–O(8A) 2.636(8), O(2C)–O(2A) 2.689(6) Å. Next are H-bonds between waters O(2A)–O(5A) 2.712(6) Å, O(3A)–O(5A) 2.820(6) Å, carboxyl and amino group O(1B)–N(2C) 2.731(8), bridging POM oxygen and water O(23)–O(1A) 2.797(5) Å, carbonyl oxygen and water O(3B)–O(3A) 2.823(6) Å, and amino and water N(2B)–O(7A) 2.827(6) Å. Slightly weaker H-bonds are between a bridging POM oxygen and water O(11)–O(4A) 2.906(5) Å, amino and water N(2B)–O(3A) 2.918(6) Å, N(2C)–O(1A) 2.938(7) Å, two water molecules O(6A)–O(4A) 2.918(6) Å, bridging POM oxygen and water O(36)–O(2A) 2.935(5) Å, bridging and terminal POM oxygen O(19)–O(10) 2.940(5) Å, terminal POM oxygen and water O(40)–O(5A) 2.988(5), and between carbonyl oxygen and water O(3C)–O(1A) 2.990(5) Å. The weakest H-bonds are between an amino group and a bridging POM oxygen N(2B)–O(12) 3.014(5) Å, amino and carbonyl groups N(2C)–O(3C), Carboxyl group and water O(2C)–O(1A) 3.015(6), bridging and terminal POM oxygen O(30)–O(33) 3.017(5) Å, two terminal POM oxygen O(19)–O(34) 3.021(4) Å, O(40)–O(28) 3.026(6) Å, terminal POM oxygen and water O(14)–O(4A) 3.031(5) Å, and between an amide group and water N(1C)–O(8A) 3.056(6) Å.
Na(HGlyGly)(H 3 O)[PMo 12 O 40 ]·3H 2 O (6) (Fig. 6) is orthorhombic, space group Pbcn, and contains one α-Keggin POM, one diglycine, four water molecules (two of which should actually oxonium ions) and one half sodium ion per asymmetric unit, Z = 8.
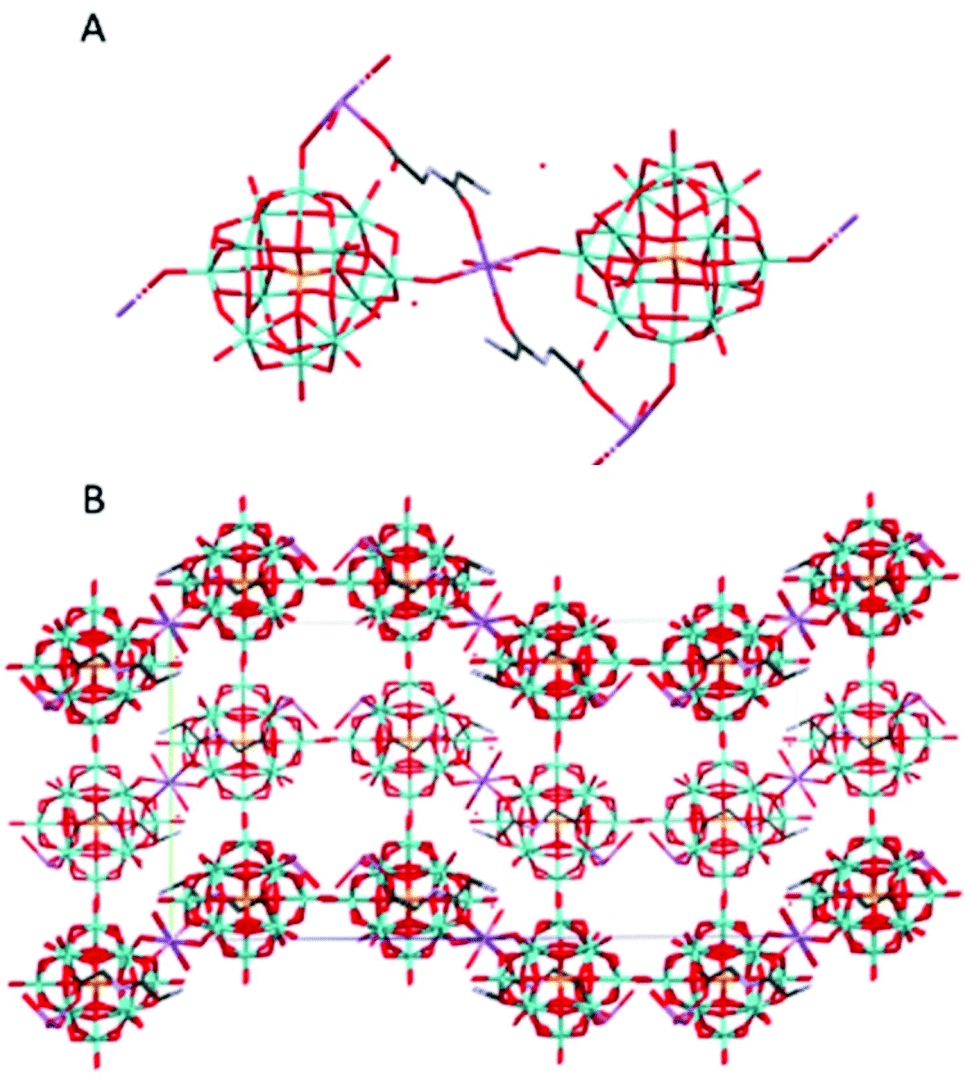 | ||
| Fig. 6 The molecular (A) and crystal (B) structure of compound Na(HGlyGly)(H3O)[PMo12O40]·3H2O (6). | ||
The shortest bonds are between a sodium ion and a carboxyl group Na(2)–O(2A) 2.164(20) Å, a carbonyl group Na(1)–O(3A) 2.35(20) Å, water Na(2)–O(4C) 2.41(30) Å, Na(1)–O(2C) 2.33(20) Å, and terminal POM oxygen Na(1)–O(26) 2.319() Å, Na(2)–O(38) 2.69(20) Å, Na(2)–O(35) 2.74(20) Å. Next come contacts between an amino group and water N(2A)–O(1C) 2.780(20) Å, N(2A)–O(2C) 2.920(20) Å, two equivalent water molecules O(4C)–O(4C) 2.870(20) Å, water and a bridging POM oxygen O(36)–O(4C) 2.980(20) Å, and water and an amide group O(3C)–N(1A) 2.99(20) Å. The weakest H-bonds are between terminal and bridging POM oxygen O(18)–O(27) 3.01(19 Å, water and a terminal POM oxygen O(1C)–O(30) 3.02(10), water and carboxyl group O(4C)–O(1A) 3.02(20), two terminal POM oxygen O(27)–O(35) 3.03(10) Å, O(31)–O(39) 3.04(10), and a terminal POM oxygen and water O(40)–O(3C) 3.03(20). The dipeptide lies flat against the POM, making contact via the sodium ions. The shortest distance between the two is greater than 3 Å, so there does not seem to be any direct interaction in this complex.
Na(HGlyGly) 2 (H 3 O) 3 [PMo 12 O 40 ] 2 ·3H 2 O (7) (Fig. 7) is monoclinic, space group P21/c. It contains one α-Keggin POM, one diglycine molecule, one half of sodium ion and three water molecules half of which statistically should be oxonium ions, Z = 4.
 | ||
| Fig. 7 The molecular (A) and crystal (B) structure of compound Na(HGlyGly)2(H3O)3[PMo12O40]2·3H2O (7). | ||
The strongest H-bonds are between the amino group and water molecules N(2B)–O(2A) 2.795(30) Å and N(2B)–O(1A) 2.899(30) Å. The weaker bonds are around the sodium ion, and connect to a terminal POM oxygen Na(1)–O(20) 2.27 Å, water Na(1)–O(1C) 2.37 Å, and a carbonyl group Na(1)–O(3B) 2.38 Å. Next come H-bonds between the amino group and water N(2B)–O(5C) 2.79(30) Å, N(2B)–O(1C) 2.82(30) Å and between the carboxyl group and a terminal POM oxygen O(4)–O(2B) 2.82(30) Å. Then come bonds between the amide group and water N(1B)–O(3C) 2.96(6) Å, amino group and terminal POM oxygen O(20)–N(2B) 2.98(3) Å, a terminal and a bridging POM oxygen O(11)–O(14) 2.99(2) Å, and a terminal POM oxygen and water O(5C)–O(19) 2.99(3) Å. The weakest H-bonds are between the amino group and a terminal POM oxygen N(2B)–O(25) 3.01(30) Å, two terminal POM oxygen O(11)–O(5) 3.01(20) Å and between terminal and bridging PM oxygen O(33)–O(18) 3.02(20) Å, O(35)–O(29) 3.02(29) Å and O(33)–O(36) 3.03(20) Å. The extensive POM–POM contact may imply protonation of the anion. It is worth noting that despite the similarity to 4, it is distinct on a few points. Here we see direct contact between the peptide and POM, as well as less sodium in the structure.
Compound 3–7 were synthesized using sodium salts of molybdenum oxides, and this shows up in the structures. 3 and 4 had relatively less sodium present, and are nearly identical to one another. 6 and 7 contained more sodium, and, while not identical, do appear very similar in the asymmetric unit. 5 appears to be an outlier, as it has a high amount of sodium, but a low amount of peptide in solution, yet resembles 4 more than 6 or 7. As both 4 and 5 involved iron cations in the synthesis, it might also have been a factor that together with the presence of nitrate influences polarity of the media. Nevertheless, the overall trend appear to be that more sodium in the solution means less peptide in the structure. While only diglycine is treated here, and the effect of the various side chains of other amino acids remain to be investigated, it is clear that the presence of sodium ions will interfere with POM-peptide interactions. This is significant, as the presence of sodium is nearly universal in biological systems. Compared to the other structures, the peptides of 3 and 7 appear more disordered (Fig. S4–S9†), though no obvious pattern can be gleaned from this.
Solution behavior
In order to determine whether diglycine has a stabilizing effect on the POM at higher pH, a mixture of PMo12 and diglycine was prepared and brought to pH 6, at which point the solution had become blue. The main signal in the 31P NMR spectra at 2.50–2.52 ppm (Fig. 10G and H) is probably from a lacunary Keggin species on the formula HxP2Mo5O23(6−x)−, which is formed at higher pH (Pettersson et al. 1986).24 The blue color may be due to reduced forms of Keggin type POM, which are present in very low concentrations and not observed in the NMR spectrum.Compound 7 was formed during an attempt to produce Zr-substituted PMo, as has been done with Keggin type phosphotungstate.18 The zirconium tended to form a precipitate with aggregates of particles 1–5 μm in size (Fig. S15†), which was identified as zirconium phosphate via EDS (Fig. S19 and Table S5†). Vanderbroek et al.16 reported a structure of Zr-substituted PW in complex with hen egg white lyzosyme (HEWL), however our results imply that under these conditions zirconium may be removed from complexes forming a separate phosphate phase, which in diluted solution was going to be present in nano form. Close analysis of the rather disordered structure published by Vanderbroek et al.16 reveals coordination of the site, interpreted as that of Zr(IV), very unusual for this cation with coordination number 5 and involving the carbonyl oxygen of the protein. The arrangement of Zr(IV) in its complexes in aqueous medium is usually following the Archimedes anti-prism with coordination number 819 and in complexes with chelating organic ligands can in some cases feature mono-20 or tricapped21 trigonal prism with coordination numbers 7 and 9 respectively. The coordination number 5 is unprecedented in unequivocally determined molecular structures of Zr(IV) in contact with O- and N-donor ligands. In the view that, on one hand, the complexation with peptides appears to enhance stability of Keggin POMs, and, on the other hand, Zr(IV) seems to be removed in the presence of phosphate, it would be plausible to consider a different interpretation of the structure reported by Vanderbroek et al.16 The POM site could rather be occupied by statistically disordered PW12O403− ions and the lacunary PW11O397−14 ion with predominance of the latter, resulting in a decreased “shady” population of the elusive 12th W-site approximately matching the electron density of Zr, atomic number 40, instead of W, atomic number 74.
The same group has reported the same ZrPW12 nanoparticles as catalyzing the peptide bond in horse heart myoglobin,22 transferin,8 and dipeptides.23 Should these catalytic properties be attributed to the Zr–P portion of the ZrPW particle, a pure zirconium phosphate (ZrP) material may display similar abilities. Therefore, we have investigated the catalytic properties of these ZrP particles. Formation of Gly was observed when ZrP was incubated with GlyGly at 37 °C for one week (Fig. S20†), though this was not observed for a similar experiment with tetraglycine. A second experiment was performed where diglycine was incubated with ZrP for 30 days at 37 °C with regular NMR spectra taken (Fig. 8A). Detectable quantities of glycine formed after seven days, and after 14 days, a linear increase in glycine formation was observed (Fig. 8B), implying a zero order reaction with a significant lag phase at the beginning. A control with identical conditions, excluding presence of ZrP, showed no detectable glycine formation.
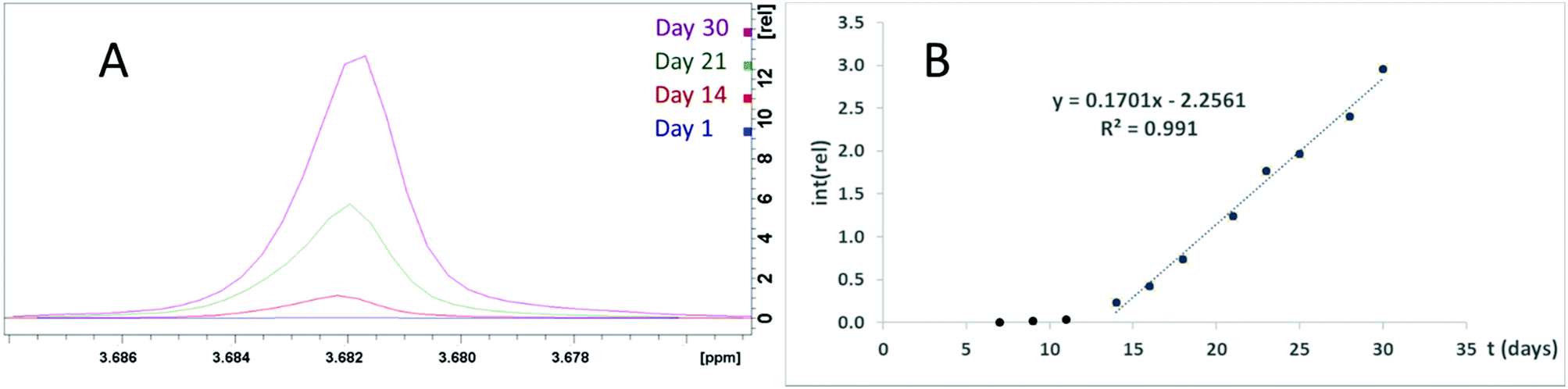 | ||
| Fig. 8 (A) The glycine signal in NMR spectra of ZrP and GlyGly incubated at 37 °C for 0, 14, 21, and 30 days. (B) Plot of the glycine signal integer normalized to the DSS signal. | ||
Thermal analysis
The TGA curves for compounds 1–4, 6, 7 (ESI Fig. S21–S26†) are resembling each other and contain rather distinct steps, corresponding to the loss of interstitial water (below 100 °C), then of water, resulting from condensation of bis-glycine and coordinated to Na+-cations (100–200 °C). Then the decomposition of organics occurs in several steps, finishing at about 500 °C. The residue, consisting of MoO3 and combination of MoO3 and Na2MoO4 volatizes essentially completely at temperatures over 700 °C.FTIR
FTIR showed that the characteristic bonds from Keggin phosphomolybdate and diglycine were present in the spectra of the samples (Table 1 and Fig. 9). In the spectra of 6 and 7 only one broad signal was observed for oxygen/nitrogen-proton bonds, which may result from facile proton exchange caused by formation of extensive network of hydrogen bonding as seen in the structures.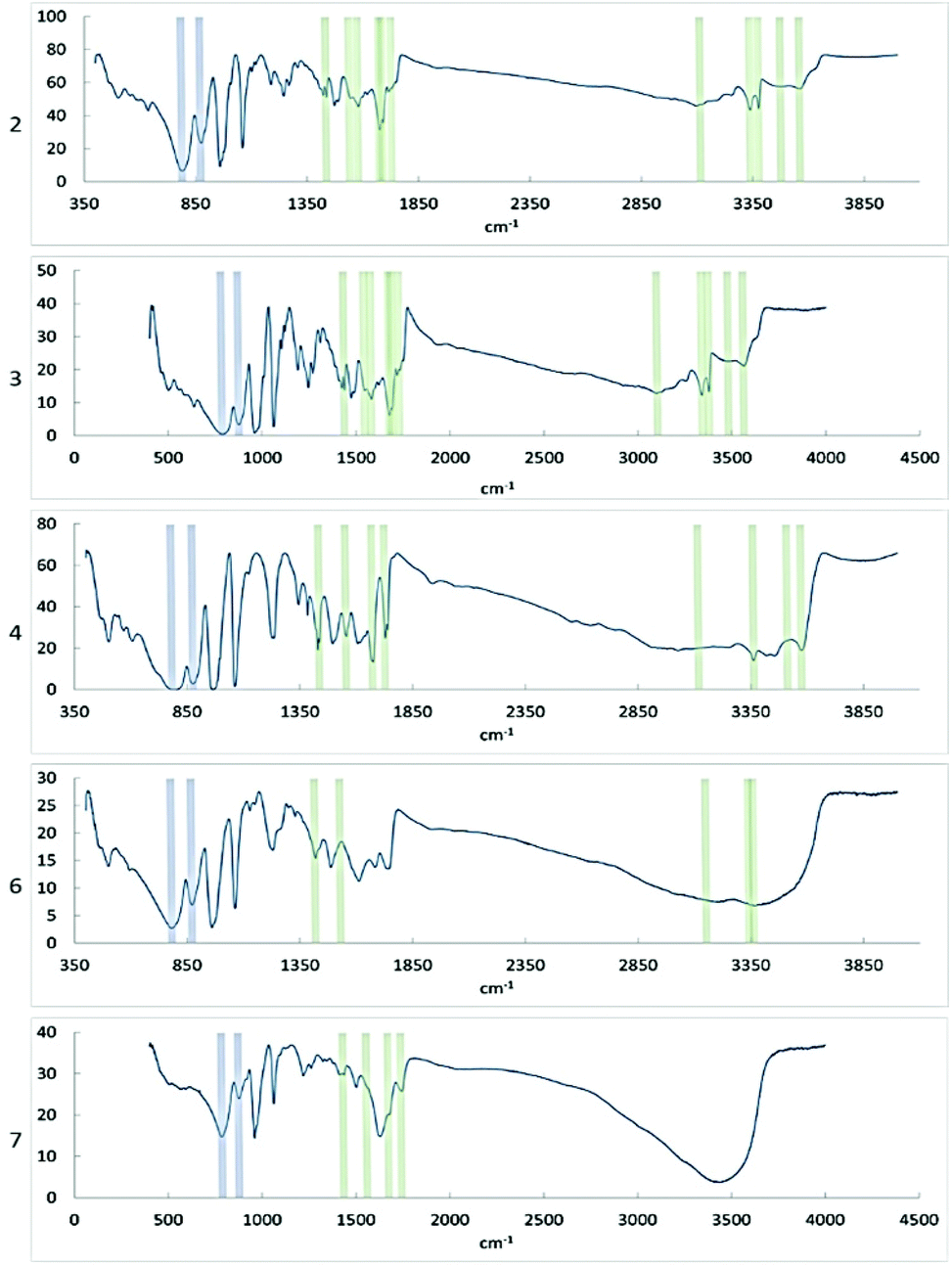 | ||
| Fig. 9 FT-IR spectra of compounds 2, 3, 4, 6, and 7. Signals corresponding to bonds in the PMo particle are indicated in blue, signals corresponding to diglycine bonds are indicated in green. | ||
| Bond | 2 | 3 | 4 | 6 | 7 |
|---|---|---|---|---|---|
| (−) indicates absent or poorly resolved peak (s). | |||||
| ν(P–O–M) | 1060 | 1060 | 1061 | 1063 | 1062 |
ν(M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) O) |
968 | 959 | 968 | 960 | 960 |
| ν(M–Od–M) | 875 | 875 | 876 | 872 | 876 |
| ν(M–Oc–M) | 790 | 787 | 797 | 782 | 786 |
|
ν(CO) |
1676, 1691, 1731 | 1731, 1691, 1676 | 1675, 1729 | 1738, 1684 | 1744, 1676 |
| ν(CO2)s | 1544, 1580 | 1543, 1580 | 1555 | 1532 | 1560 |
| ν(CO2)as | 1437 | 1437 | 1437 | 1419 | 1437 |
| ν(O–H) | 3560 | 3561 | 3574 | − | − |
| ν(N–H) | 3477 | 3482 | 3511 | − | − |
| ν(NH3+) | 3116, 3340, 3377 | 3370, 3340, 3102 | 3118, 3362 | 3361, 3342, 3152 | — |
Mass spectrometry
2–3 mg of 2 was dissolved in 1 mL water, 0.2 M HCl, or 5 mM phosphate buffer, pH 6.5 and mass spectra were obtained (Fig. 10A–C). In water, the signals corresponding to heavier compounds are roughly even in intensity, implying a variety of breakdown products in solution. In the sample dissolved in HCl, the signal at 1822 m/z, corresponding to the Keggin phosphomolybdate, is dominant, and there appears to be some amount of free molybdate (MoO4−2, 160 m/z). In the phosphate buffered sample, the extremes appear subdued, with the most prominent signals appearing between 500 and 1000 m/z. This may be due to the high pH destabilizing the heavier forms, while the lighter fragments aggregate onto the phosphate in the buffer, which results in mid-weight fragments being more prevalent.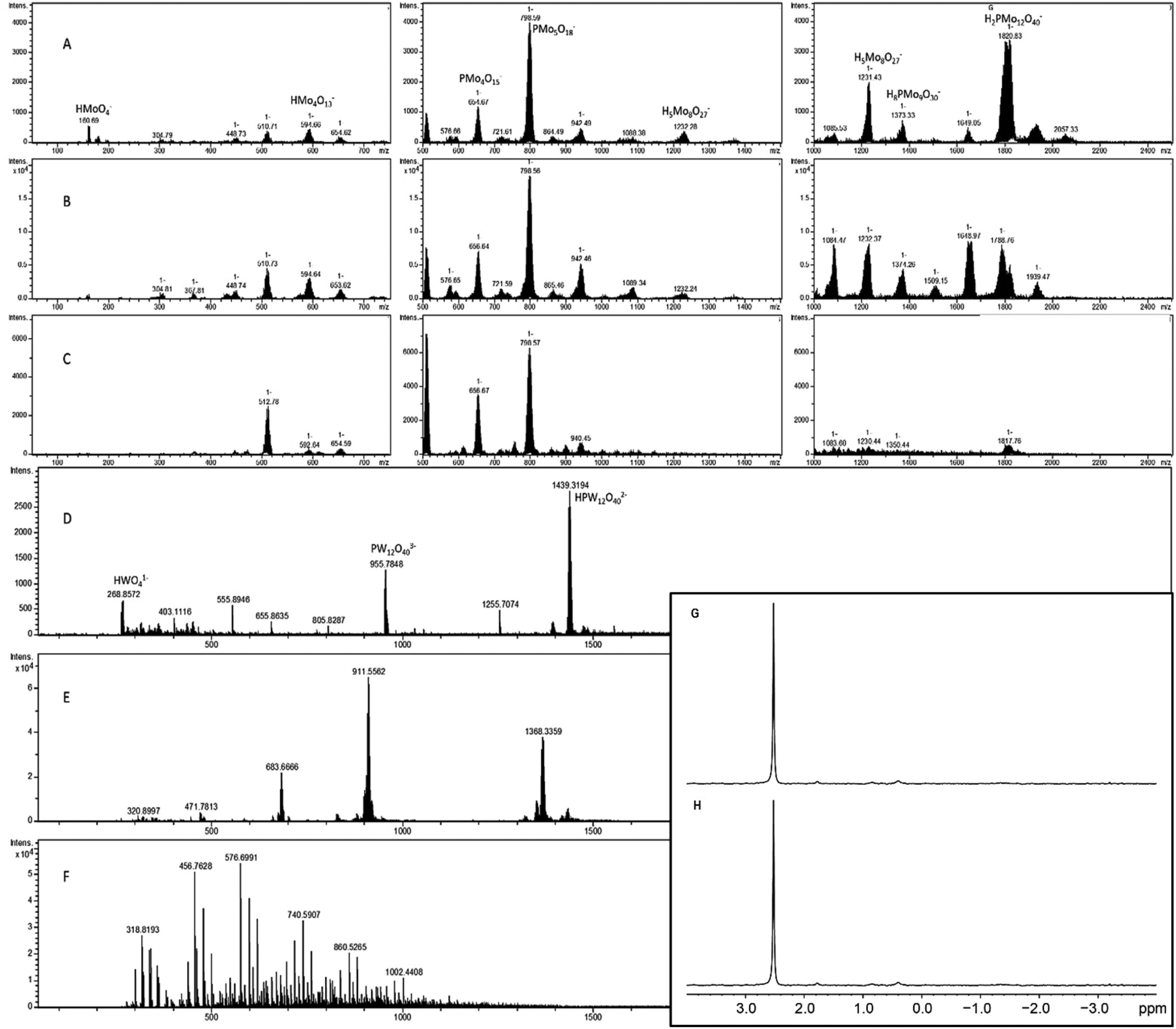 | ||
| Fig. 10 Mass and NMR spectra of POM-diglycine complexes dissolved in different media. Diglycine-PMo complex 2 (3:1) (1.5 mM) 2 was dissolved in (A) 0.2 M HCl, (B) water, and (C) phosphate buffer (5 mM, pH 6.5). Diglycine-PW complex (3:1) (14 uM) dissolved in (D) 0.2 M HCl, (E) water, and (F) phosphate buffer (5 mM, pH 6.5). (G) and (H) 31P NMR spectra of phoshomolybdate at pH 6 with diglycine (compound 2) and without diglycine respectively. | ||
3–5 mg of phosphotungstate – diglycine complex was dissolved in 1 mL water, 0.2 M HCl, or 5 mM phosphate buffer, then diluted one hundred times before collecting mass spectra (Fig. 10D–F). The sample in water shows some amount of double- and triple charged PW anions (1433 and 911 m/z, respectively), as well as the lacunary species [PW11O39] (1368 m/z). The sample in acid showed clear signals at 2879 m/z and 1439 m/z corresponding to the complete POM, as well as signals at 955 m/z (W4O14−2) and 268 m/z (WO4−2). The sample in phosphate buffer displayed no isotope patterns identifiable as WxOy species, implying complete hydrolysis of the POM at this high pH and very low concentration.
SEM-EDS
Crystals of complexes were placed on carbon tape fixed on aluminium holders and investigated with Hitachi Flex-SEM 1000 II. Elemental distribution maps were obtained to confirm the chemical homogeneity. The ratio of elements was in all cases matching well the chemical composition obtained from single crystal structure models.Experimental
Synthesis
All chemicals were analytical grade and used as received from Sigma Aldrich.2 was prepared by dissolving 0.03 mmol phosphomolybdic acid hydrate and 0.03 mmol diglycine in 0.5 mL 0.1 M HCl each. The POM solution was added dropwise to the peptide solution, after which it was mixed by inversion. The test tube was left open to let crystals form via solvent evaporation. 3 was prepared in a similar manner, but was heated in a water bath after mixing.
4 was prepared by dissolving 1.57 mmol ammonium heptamolybdate and 11 mmol sodium carbonate, which was boiled to remove the ammonia. 1 mmol iron(III) nitrate was dissolved in 2 mL 0.1 M HCl, 1 mmol sodium phosphate dibasic dehydrate was dissolved in 2 mL water, and 3 mmol Gly–Gly was dissolved in 2.5 mL water. The phosphate solution was added to the molybdate and concentrated HCl was added until it turned yellow. The resulting phosphomolybdate solution was added to the hot Fe(III) solution dropwise. The Gly–Gly was added, and the solution was allowed to cool slowly. Crystals appeared within hours.
5 was prepared by dissolving 11 mmol sodium molybdate and 1 mmol sodium dihydrogen phosphate in 10 mL water, 1 mmol iron nitrate nonahydrate in 5 mL 1 M HCl, and 1 mmol diglycine in 1 mL 0.1 M HCl. Acid was added to the phosphate and molybdate solution until it became yellow, at which point it was added dropwise to the Fe solution, which turned reddish brown. The diglycine solutionwas then added and the solution was evaporated by heating in a water bath. Crystals formed upon slow cooling.
6 was prepared by dissolving 11 mmol sodium molybdate and 1 mmol sodium dihydrogen phosphate in 10 mL water, and 1 mmol titanium(IV) oxysulfate in 5 mL concentrated HCl, by vigorous stirring at above 80 °C. HCl was added to the phosphate and molybdate solution until it became yellow (pH 4.8), at which point it was added dropwise to the titanium solution. A white precipitate was formed. Diglycine was added after the PMo, and the solution was slowly cooled to room temperature. Yellow crystals formed after several days, while the solution had become green.
7 was prepared by dissolving 1 mmol sodium dihydrogen phosphate and 11 mmol sodium molybdate in 5 mL water, and adding concentrated HCl until the solution turned yellow. 1 mmol zirconyl chloride octahydrate dissolved in 5 mL water was added to the phosphomolybdate solution, dropwise. A white precipitate was formed. 3 mmol diglycine dissolved in 2 mL 0.1 M HCl was added, and the solution was kept at 70–75 °C for several hours, until it was greenish blue. Blue crystals had formed after several weeks at room temperature.
Analysis
FTIR spectrometer Perkin–Elmer Spectrum-100 was used for infrared spectroscopy studies. Crystals were milled in paraffin oil and their spectra were recorded by a total of 8–16 scans in 400–4000 cm−1 range. SEM-EDS studies were performed with Hitach FlexSEM 1000 II instrument supplied by Oxford Instruments EDS analysis system operated by Aztec software.NMR spectroscopy
NMR spectra were acquired on a Bruker Avance III 600 MHz spectrometer using a 5 mm broadband observe detection SmartProbe. Samples were dissolved in D2O with DSS as 1H chemical shift reference and spectra were recorded at 37 °C.X-ray crystallography
Single-crystal X-ray diffraction data were recorded at room temperature with a Bruker D8 SMART APEX II CCD diffractometer (operating with graphite monochromated Mo-Kα radiation, λ = 0.71073 Å). The structures were solved by direct methods. Metal atom coordinates were obtained from the initial solutions and the other non-hydrogen atoms by Fourier synthesis. All non-hydrogen atoms were refined first isotropically and then anisotropically in full-matrix approximation. The hydrogen atom positions were calculated geometrically and they were added into the final refinement in isotropic approximation. For details, please, see Table 2. Powder X-ray diffraction patterns were registered on Bruker Discover diffractometer with theta-theta Bragg–Brentano geometry using CuKα radiation (λ = 1.54056 Å) and imaged using CCDC DASH software. Theoretical powder patterns were generated using CCDC Mercury software from the produced cif-files. Good matching between the experimental and theoretical X-ray powder patterns confirmed the identity of the bulk materials with the identified single crystal structures (see Figs. S27†).| Compound | 2 | 3 | 4 | 5 | 6 | 7 |
| Chemical composition | C12H41Mo12N6O56P | C12H43Mo12N6O57P | C8H36Mo12N4NaO54P | C8H34Mo12N4NaO54P | C4H23Mo12N2NaO47P | C4H25Mo12N2Na0.5O46P |
| Formula weight | 2347.76 | 2365.77 | 2250.66 | 2255.63 | 2065.49 | 2083.51 |
| Crystal system | Triclinic | Monoclinic | Orthorombic | Monoclinic | Orthorombic | Monoclinic |
| Space group |
P (2) |
P21/n (14) | Pbca (61) | P21/c (14) | Pbcn (60) | P21/c (14) |
| R1 | 0.0263 | 0.0494 | 0.0474 | 0.0304 | 0.0544 | 0.1018 |
| wR2 | 0.0656 | 0.1406 | 0.1377 | 0.1062 | 0.1733 | 0.2341 |
| a [Å] | 12.7308(12) | 12.788(2) | 17.3441(13) | 21.135(3) | 12.765(3) | 19.443(8) |
| b [Å] | 13.0510(12) | 31.897(5) | 23.078(2) | 12.9339(16) | 19.680(5) | 12.745(5) |
| c [Å] | 18.0332(17) | 13.843(2) | 25.295(2) | 19.455(2) | 37.663(10) | 19.657(8) |
| α [°] | 97.8970(10) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| β [°] | 105.2180(10) | 112.173(2) | 90.00 | 111.185(2) | 90.00 | 105.275(5) |
| γ [°] | 105.9070(10) | 90.00 | 90.00 | 90.00 | 90.00 | 90.00 |
| V [Å3] | 2708.8(4) | 5228.8(15) | 10124.8(14) |
4958.7(11) | 9462(4) | 4699(3) |
| T [K] | 296(2) | 296(2) | 296(2) | 296(2) | 296(2) | 296(2) |
| Z | 2 | 4 | 8 | 4 | 8 | 4 |
| Nr. of obs. independent refl., I > 2σ(I) | 9555 | 9193 | 13401 |
8719 | 8333 | 7421 |
| Residual electron density max | 1.086 | 2.190 | 1.115 | 0.754 | 2.700 | 0.686 |
Mass spectrometry
Negative electrospray ionization mass spectra were recorded with a Bruker Esquire 3000 ion trap mass spectrometer using direct infusion at 3 μL min−1. The spectrum acquisition was optimized using the built in target m/z feature together with an automated charge control value of 30000 for 1 and 3, or 15000 for 2 and 4. Negative electrospray ionization mass spectra of PW were recorded with a Bruker maXis impact TOF mass spectrometer under the same electrospray conditions as was the case for 2. The spectra were analyzed with the Bruker Compass Data Analysis 4.1 software.
Conclusion
In the absence of competing cations the PMo12 POM is forming a 1:3 complexes with the GlyGly peptide independently of the solution composition. The crystal structure of the products is, however, dependent on the acidity of the applied media, offering a form with enhanced hydrogen bonding isostructural to the earlier described PW12 analog in highly acidic medium. As in the case of earlier described complexes isolated from less acidic media, the intermediate heating is leading to a phase with lower water content. Increased salinity results in incorporation of sodium cations that have a structure-directing role via unexpectedly strong coordination of carbonyl oxygen atoms of protonated peptide molecules. This observation is important in the view of abundance of Na+ in biological media. Increase in pH to near-neutral values leads to dissociation of POMs partly stabilized by complexation with peptide. Zirconium cations form under these conditions, in a phosphate buffer, the nanosized zirconium hydrogen phosphate. The latter acts as (micro) heterogeneous catalyst of peptide hydrolysis.
Author contributions
BG performed synthesis of all compounds, contributed actively to structure solution and refinement and produced draft of the manuscript. GN helped with NMR investigation and JE – with MS studies. GS helped with SEM-EDS and FTIR investigation. VK proposed the project, finalized the X-ray structure determinations and supervised the whole study. All authors contributed actively to discussion of the results and editing the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Swedish Research Council Vetenskapsrådet for support to the project 2018-03811 Molecular mechanisms in nanoparticle-protein interactions. We express our sincerest gratitude to Dr Fredric Svensson for the aid in registering powder diffraction data.Notes and references
- W. Youn, E. H. Ko, M.-H. Kim, M. Park, D. Hong, G. A. Seisenbaeva, V. G. Kessler and I. S. Choi, Angew. Chem., Int. Ed., 2017, 56, 10702–10706 CrossRef CAS PubMed.
- B. Ekstrand-Hammarström, J. Hong, P. Davoodpour, K. Sandholm, K. N. Ekdahl, A. Bucht and B. Nilsson, Biomaterials, 2015, 51, 58–68 CrossRef PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- N. I. Gumerova and A. Rompel, Inorg. Chem., 2021, 60(9), 6109–6114, DOI:10.1021/acs.inorgchem.1c00125.
- L. S. V. Rompuy and T. N. Parac-Vogt, Chem. Commun., 2017, 53, 10600–10603 RSC.
- A. Bijelic and A. Rompel, Coord. Chem. Rev., 2015, 299, 22–38 CrossRef CAS PubMed.
- G. Absillis and T. N. Parac-Vogt, Inorg. Chem., 2012, 51, 9902–9910 CrossRef CAS PubMed.
- L. S. Van Rompuy, N. D. Savić, A. Rodriguez and T. N. Parac-Vogt, Molecules, 2020, 25, 3472 CrossRef CAS PubMed.
- S. Vanhaecht, G. Absillis and T. N. Parac-Vogt, Dalton Trans., 2012, 41, 10028–10034 RSC.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- K. M. Rominger, G. Nestor, J. E. Eriksson, G. A. Seisenbaeva and V. G. Kessler, Eur. J. Inorg. Chem., 2019, 2019, 4297–4305 CrossRef CAS.
- B. Greijer, T. D. Donder, G. Nestor, J. E. Eriksson, G. A. Seisenbaeva and V. G. Kessler, Eur. J. Inorg. Chem., 2021, 54–61, DOI:10.1002/ejic.202000855.
- Z. Zhu, R. Tain and C. Rhodes, Can. J. Chem., 2003, 81, 1044–1050 CrossRef CAS.
- D. Bajuk-Bogdanović, S. Uskoković-Marković, R. Hercigonja, A. Popa and I. Holclajtner-Antunović, Spectrochim. Acta, Part A, 2016, 153, 152–159 CrossRef PubMed.
- L. Vandebroek, L. Van Meervelt and T. N. Parac-Vogt, Acta Crystallogr., Sect. C: Struct. Chem., 2018, 74, 1348–1354 CrossRef CAS PubMed.
- K. Niwa, S. Toda, K. Fuwa and H. Haraguchi, Agric. Biol. Chem., 1977, 41, 1287–1294 CAS.
- M. N. Sokolov, E. V. Chubarova, E. V. Peresypkina, A. V. Virovets and V. P. Fedin, Russ. Chem. Bull., 2007, 56, 220–224 CrossRef CAS.
- C. Hagfeldt, V. Kessler and I. Persson, Dalton Trans., 2004, 2142–2151 RSC.
- V. G. Kessler, G. I. Spijksma, G. A. Seisenbaeva, S. Håkansson, D. H. A. Blank and H. J. M. Bouwmeester, J. Sol-Gel Sci. Technol., 2006, 40, 163–179 CrossRef CAS.
- G. I. Spijksma, L. Kloo, H. J. M. Bouwmeester, D. H. A. Blank and V. G. Kessler, Inorg. Chim. Acta, 2007, 360, 2045–2055 CrossRef CAS.
- H. G. T. Ly and T. N. Parac-Vogt, ChemPhysChem, 2017, 18, 2451–2458 CrossRef CAS PubMed.
- H. G. T. Ly, G. Absillis and T. N. Parac-Vogt, Dalton Trans., 2013, 42, 10929–10938 RSC.
- L Pettersson, I Andersson and L O Öhman, Inorganic Chemistry, 1986, 25, 4726–4733 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2151360-2151364 for compounds 2–4 and 6–7 respectively and 2156667 for compound 5. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00717g |
| This journal is © The Royal Society of Chemistry 2022 |