
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalyst sites and active species in the early stages of MTO conversion over cobalt AlPO-18 followed by IR spectroscopy†
Alessia
Airi
 a,
Alessandro
Damin
a,
Jingxiu
Xie
b,
Unni
Olsbye
b and
Silvia
Bordiga
*a
a,
Alessandro
Damin
a,
Jingxiu
Xie
b,
Unni
Olsbye
b and
Silvia
Bordiga
*a
aDepartment of Chemistry, NIS, and INSTM Reference Centre, University of Turin, Via Quarello 15/A, 10135, Turin, Italy. E-mail: silvia.bordiga@unito.it
bCentre for Materials Science and Nanotechnology, Department of Chemistry, University of Oslo, Oslo N-0315, Norway
First published on 19th April 2022
Abstract
The isomorphic insertion of cobalt(II) confers strong acidity to the AlPO-18 zeotype. This makes it a suitable catalyst for the acid catalysed conversion of methanol to olefins (MTO). However, due to the tendency of Co(II) to be oxidised into Co(III) or to acquire distorted tetrahedral coordination, the concentration of strong Brønsted acidic sites (BAS) in CoAPO-18 is highly susceptible to catalyst activation conditions. The use of vibrational and electron spectroscopy coupled with the use of probe molecules sheds light on the conditions under which the concentration of active sites can be maximized, limiting the formation of species of different nature. The MTO reaction was followed by in situ FT-IR spectroscopy, revealing that the population of strong BAS profoundly affects the methanol conversion in the very early stages of the process. Indeed, spectroscopic evidence of the consumption of CO (produced in situ from the decomposition of methanol itself) was obtained, when the concentration and availability of BAS are optimized. This gives support to the occurrence of Koch carbonylation of surface methoxides, at the early stages of MTO.
Introduction
The process of methanol conversion into different valuable hydrocarbons (MTH, specifically: to olefins MTO, to gasoline MTG, to aromatics MTA) catalysed by zeolitic materials has been of high industrial relevance since the 1970s (ref. 1) and nowadays it is gaining even more importance as a sustainable alternative to fossil derivatives in the production of consumer chemicals. The reaction, in fact, can exploit methanol of non-extractive carbon sources (as generated from CO2 hydrogenation),2 and it is generally performed under relatively mild pressure (1.5–5 bar) and temperature (350–500 °C) conditions.3 The choice of catalyst in this process plays a key role,4 since the zeolite topology affects the selectivity to the products and the catalyst lifetime, while its acidity together with the operating conditions determines the catalytic activity and yield.5–7 Although the engineering of zeolite catalysts for MTO is in constant development, two frameworks are routinely used in industrial processes allowing the reaction to be pushed to the two opposite extremes of the so-called dual-cycle mechanism driving the methanol conversion.8 H-ZSM-5 (MFI framework, 3D 10MR zeolite) operates at temperature around 500 °C and promotes the formation of propylene from the alkene cycle, while SAPO-34 (CHA framework 3D 12-8MR zeotype) is selective for C2–C4 olefins at 350–400 °C passing through the aromatic cycle. Despite the long history of this industrial process, some aspects of the chemistry of MTH remain unresolved and continue to attract great interest in the academic world.7 The overall MTH process involves a complex interplay of different events within the zeolite cavities that include alkylation–dealkylation as well as H-transfer reactions.3 In the case of MTO, light olefins can be released after intermediate methylation or cracking following the dual-cycle process. The steady state of the reaction is widely accepted and defined as the “hydrocarbon pool” (HCP) mechanism, during which long hydrocarbons and coke like species are continuously forming.9 Although these species are not completely listed, there is a wide range of experimental evidence that has outlined their nature,10–12 recognising in methylated cyclic carbocations the intermediates that are constantly growing and being transformed by the zeolite catalyst (via protonations, alkylations and cracking) whose pore system determines their size and nature during the reaction steady state.13 The autocatalytic growth of the aromatic system representing the HCP species determines at the same time the deactivation of the catalyst by pore obstruction due to the polymerization of the intermediates to form coke.7 The instauration of the HCP involves an induction period, during which the first C–C bond must be formed to initiate the cascade reactions. The mechanism with which the first C–C bond is established is the source of an intense debate.14 The hypothesis of a direct coupling of C1 species induced by impurities in the precursors, although long supported, is now strongly refuted.15,16 So far more than 20 different mechanisms have been proposed, because of the lack of unambiguous experimental evidence due to the poor concentration of the intermediates and the rate of the transformations. Only very recently has direct C–C bond formation been outlined passing through a methoxide intermediate over the zeolite surface (Ze-OCH3), upon adsorption of CH3OH on the Brønsted acid sites.3,17 It is well established that during any hydrocarbon conversion process at high temperatures, the methoxide groups on the zeolite surface are able to establish new C–C bonds. What is still debated is the exact nature of the coupling process, the intermediates involved and what the actual mechanism is. Among the most recent mechanisms proposed,14 the hypothesis suggested by the group of Lercher18 in 2016 is particularly accredited, since it is supported by an extremely low calculated activation energy (80 kJ mol−1). They proposed the initiation of C–C bond formation via Koch carbonylation of methoxide species (Ze-OCH3) by the action of carbon monoxide (CO) generated in situ from the methanol degradation under process conditions, identifying methyl acetate as the first C–C containing molecule. The reaction involves the coupling of the electrophilic C atom of CH3OH (or its derivatives such as –OCH3, dimethyl ether (DME) and formaldehyde) with the nucleophilic C atom of the CO molecule, giving at the same time a reasonable explanation for the C–C formation and for the autocatalytic pathway for the MTH process. The group performed the reaction over H-ZSM-5 at 450 °C recording an increase in olefin production upon co-feeding the methanol source with CO, which was previously detected among the methanol degradation products under the same conditions. Moreover, the hypothesis is supported by the spectroscopic observation of carbonyl intermediates at the very early stages of the reaction, also by other groups.19–21 In parallel, Weckhuysen and co-workers provided evidence of Koch carbonylation occurring over SAPO-34, identifying methanol and formaldehyde as subsequent carbonylation agents for the acetate species produced by CO and surface Ze-OCH3.22,23In the present work, the FT-IR spectroscopic study operated in continuous mode provides additional observations that can be inserted in this scenario, giving support to the key role of the CO molecule in carrying out the methanol conversion. This aspect will be explored through the study of the reaction catalysed by CoAPO-18 compared with the corresponding SAPO-18, giving way to discuss at the same time the promotion of products by the two catalysts and to rationalize how the transition metal influenced the total process. Moreover, until now no evidence of Koch carbonylation has been provided for the reaction catalysed by metal aluminophosphate (MAPO) zeotypes. Experimental evidence in MAPOs increases the validity of the hypothesis, as it broadens the applicability of the Koch mechanism to other catalysts, thus making it general for the MTH process. Since the discovery of the AlPO-18 framework, its potential as a catalyst for the methanol to olefin reaction was rapidly investigated, being structurally related to SAPO-34.24 Metal AlPO-18 compounds have stability comparable to that of their silicon analogue and provide excellent performance as catalysts of the MTO reaction over a temperature range comparable to that of SAPO-34. The presence of the divalent metal confers to AlPO-18 increased acidity (due to the more ionic character of the anionic framework) that promotes the methanol conversion. It is attested that cobalt(II) containing AlPO-18 (CoAPO-18) reaches 100% conversion at 350 °C (the same for MgAPO and ZnAPO-18) while 70% selectivity for light olefins is observed.24
The insertion of cobalt in AlPO-18 represents a very interesting case study, since the transition metal can be oxidized from Co(II) to Co(III). In the second case, the charge neutrality is stabilised, and the material does not present any Brønsted acidic character. Compared to other AlPO frameworks, CoAPO-18 is particularly prone to change in oxidation state while maintaining a stable porous structure.25 The different oxidation states of cobalt can confer completely different chemical behaviour to the zeotype. Upon calcination (removal of the templating agent by combustion), the acidic form of the catalyst is generated through strongly reducing treatments (high temperature in presence of pure H2) that would induce the stabilization of the framework Co(II) state responsible for the presence of the Brønsted site. The catalyst in this form is generally applied in acid-catalysed reactions, such as MTO.26 In the absence of a subsequent reductive treatment, the oxidative conditions of the calcination can favour the presence of Co(III). In this case the zeotype finds application as an oxidation catalyst in reactions such as oxyfunctionalization of alkanes, epoxidation of alkenes or Baeyer–Villiger oxidations of ketones.27,28 The first part of this work will be devoted to outlining the spectroscopic features of cobalt in the AlPO-18 framework and to identifying which are closely related to the oxidation state of the heteroatom, on which the reactivity of the material depends. Since the early stages of this research, the process of template removal has turned out to be crucial in the formation of the active site and in influencing the oxidation state and the coordination sphere of the metal. Through the combination of different spectroscopic techniques and the investigation of samples that have undergone different activation treatments, the characterization of the CoAPO-18 active sites has been performed.
In the second part, the MTO reaction over CoAPO-18 and SAPO-18 will be followed by FT-IR spectroscopy in continuo, by means of a custom set-up constituted by a mass flow controller for the direct methanol dosage into a temperature-controlled IR cell operating in transmission mode (all the details are listed in the Experimental section). No outlet flow analyser was connected to the system, since the object of interest of this analysis was the identification of the species adsorbed on the catalyst surface and their evolution within the porous system during the reaction. The nature and the evolution of the hydrocarbons within the catalyst pore system, the species and the mechanism involved in the first C–C bond formation in relation to the acidic active sites will be the main focus of this study.
Experimental
The sample under investigation is a cobalt containing AlPO-18 (CoAPO-18), synthesized at the Oslo University and preliminary characterized as reported in ref. 29. Two SAPO-18 catalysts with different silicon contents were selected for comparison (synthesis and characterization of SAPO-18 are also reported in ref. 29). The elemental analysis performed by ICP-OES revealed for CoAPO-18 a Co/(Al + P) ratio equal to 0.65. The sample is microcrystalline, organized in cubic particles of about 0.5 μm extended; it presents the diffraction pattern of a pure AEI framework and a specific surface area of 480 m2 g−1 after calcination. In the present work the CoAPO-18 sample has been presented in two different forms:• CoAPO-18u (uncalcined). Calcined in a closed line system at 550 °C, connected to vacuum pumping until reaching 10−4 mbar internal pressure, in small aliquots under a controlled atmosphere only prior to each experiment to meticulously avoid contact with atmospheric oxygen and moisture.
• CoAPO-18c (calcined). Calcined in an oven at 550 °C for 4 hours and stocked in air for months, without any preservation from atmospheric agents.
We compared CoAPO-18u and CoAPO-18c in different spectroscopic experiments in order to rationalize their differences and try to relate them with the cobalt state.
With regard to SAPO-18:
• SAPO-18_0.4, previously calcined sample with Si/(Al + P) = 0.02, has been used as a reference for Raman spectra.
• SAPO-18, uncalcined sample with Si/(Al + P) = 0.6, has been compared with CoAPO-18 in MTO experiments.
Ex situ high temperature treatments
The samples have been treated before each experiment of FT-IR, UV-vis and Raman spectroscopy (except for in continuo FT-IR experiments, see the relative paragraph) directly in the customized sample holder (cell) of home-made design specific for each technique. The quartz cells were connected to a glass-line equipped with vacuum pumps, allowing the thermal treatments and gas dosage. The template removal of the uncalcined sample (referred also as high temperature O2 treatment in the next paragraphs) was conducted by controlled heating at 5 °C per minute until 550 °C, with intermediate steps of 50 mbar O2 dosages alternated to vacuum removal of gaseous species, following a literature reference.30 When the temperature of 550 °C was reached, the sample was kept in pure oxygen atmosphere for 2 hours, then cooled to room temperature in O2. Finally, the system was outgassed by vacuum pumps, until reaching 5 × 10−4 mbar internal pressure of the cell. High temperature H2 treatments have been performed over the calcined samples, by first outgassing the system while raising the temperature at 5 °C per minute until 400 °C. Once the internal pressure of 5 × 10−4 mbar was reached, 50 mbar of pure H2 was introduced in the system and kept in contact with the samples for 1 hour, followed by outgassing and cooling down to room temperature.Simple activation involved heating treatments of 5 °C per minute until 400 °C under dynamic vacuum outgassing conditions until reaching 5 × 10−4 mbar pressure.
DR-UV-visible spectroscopy
DR-UV-visible spectra have been collected with a Varian Cary 5000 UV-vis-NIR spectrophotometer working in diffuse reflectance mode in the 200–2500 nm range, with a resolution of 1 nm.Infra-red (IR) spectroscopy
Experiments of FT-IR spectroscopy “in continuo” (i.e. repeated, not delayed in situ measurements) have been performed using a Bruker Invenio R spectrophotometer with an MCT cryodetector working at 40 kHz, equipped with an AABSPEC 2000 multi-mode FT-IR cell operating in transmission mode, allowing high temperature and high pressure in situ measurements. The cell has been connected to a mass flow controller for direct gas dosage within the measurement chamber where the sample is located in the form of a self-supporting pellet of about 10 mg of pressed powder.
Raman spectroscopy
The Raman spectra of the activated sample in the form of a self-supporting pellet have been collected with a Renishaw InVia microRaman with 20× magnification and adopting a 442 nm He–Cd laser as the excitation source. 20 spectra of 20 scans for each have been accumulated at every measurement point. The spectra reported correspond to the average of three different points.Computational details
Periodic DFT32 based calculations were performed by exploiting the pure PBE33 functional and the empirical D3(BJ)34–36 scheme for the description of dispersive interactions and including the Axilrod–Teller–Muto type three-body term. In the whole set of calculations, the CRYSTAL software37,38 package was employed: this allows a complete treatment of periodic systems through the use of atom-centred linear combinations of Gaussian type functions for the description of atomic orbitals (basis set).a) Co atoms have been described by an all electron (8s12sp4d)/[1s4sp2d] basis set. The same scheme was already employed in ref. 39. See also https://www.crystal.unito.it/basis-sets.php.
b) Al atoms have been described by an all electron (8s7sp1d)/[1s3sp1d] basis set. The same scheme was already employed in ref. 40. See also https://www.crystal.unito.it/basis-sets.php.
c) P atoms were described by an all electron (8s8sp2d)/[1s3sp2d] basis set. The same scheme was already employed in ref. 41. The original d shell has been doubled through an even-tempered recipe. See also https://www.crystal.unito.it/basis-sets.php.
d) For O atoms, the adopted all electron basis set was a (8s5sp2d)/[1s3sp2d] one already adopted in ref. 42. The original d shell has been doubled through an even tempered recipe. See also https://www.crystal.unito.it/basis-sets.php.
Numerical accuracy in energy calculation was determined by setting the thresholds for mono- and bi-electronic integral to {888816} through the keyword (TOLINTEG). The shrinking factor parameter (keyword SHRINK), determining the k-point sampling in the reciprocal space, was set to 2. The default values for all the unreported computational parameters concerning the structure optimization, frequency and associated Raman intensities were used.
Finally, calculations have been performed by adopting AlPO-18 (ref. 43) as the starting structure for optimization, frequency measurement and Raman spectra calculation.
Results and discussion
Since the first synthesis of CoAPO-18,24 it has been deeply characterized by spectroscopic tools highlighting how the calcination process represents a more complex and peculiar case with respect to other zeotypes, because of the cobalt propensity to be oxidised in a combustion atmosphere (high temperature and O2).24,31,44 A preliminary indication of the redox process involving Co is given by the pronounced colour change affecting the material. Accordingly, as-synthesized CoAPO-18 appears as a blue powder, indicative of the tetrahedral Co(II). Upon calcination the material presents a green coloration which turns to blue after reduction.In contrast to the first XAS models,45–47 the UV-vis spectroscopy studies of these materials evidenced that the redox process does not involve the totality of the metal centres. A portion of the Co(II) present in as-synthesized CoAPO-18 does not undergo oxidation but more likely a distortion from the perfect tetrahedral coordination (Lewis site) and extra-framework Co(II).31,48 The conditions that favour the oxidation state changes and the modifications of the active sites will be discussed in the following sections through the analysis of spectroscopic results of samples after different activation treatments.
Cobalt states and active sites in CoAPO-18
The DR-UV-visible spectra collected in air atmosphere of CoAPO-18u and CoAPO-18c and their corresponding spectra upon ex situ high temperature O2 and H2 treatments (see the Experimental section for the details) are reported in Fig. 1 and discussed. The data collected here find good agreement with those reported in the literature.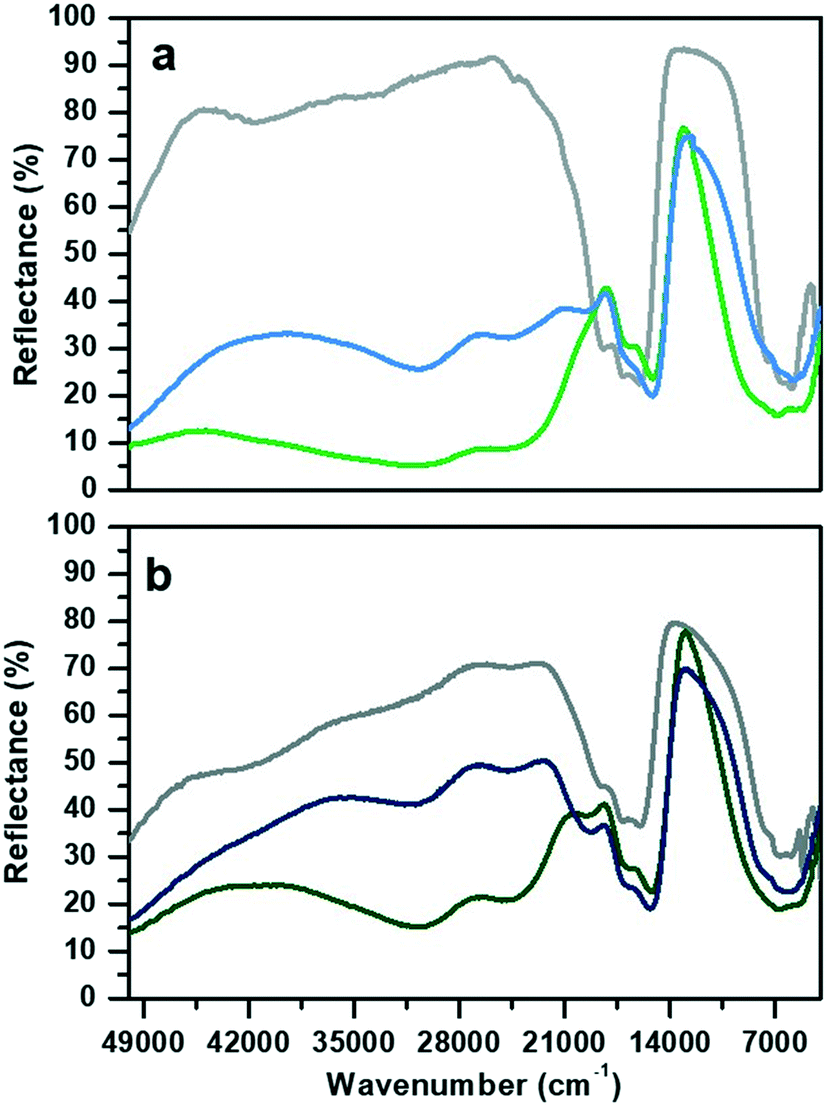 | ||
| Fig. 1 Diffuse reflectance UV-visible spectra of a) CoAPO-18u and b) CoAPO-18c. Grey lines refer to the spectra collected in air on the pristine samples, green after O2 treatment at 550 °C and blue to H2 treatment at 400 °C. | ||
The spectrum of CoAPO-18u before any treatment (Fig. 1a, grey line) presents the principal absorption bands of the tetrahedral Co(II) as previously reported for CoAPO-18 before template removal.49 The broad band in the UV region, around 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1, is compatible with the LMCT, while the visible region is dominated by the narrow triplet between 18500 and 16000 cm−1 typical of the Co(II) d–d transition in tetrahedral coordination, which is the result of the combination of ν2 ≡ 4A2 (F) → 4T1 (F) and ν3 ≡ 4A2 (F) → 4T1 (P). A broad signal is present in the NIR region, around 7000 cm−1 due to the ν1 ≡ 4A2 (F) → 4T2 (F) transition.31,50 The sample was then subjected to calcination at 550 °C in a pure oxygen atmosphere within a closed system (see the Experimental section for further details). The second spectrum (Fig. 1a, green line) was then recorded while maintaining the conditions of isolation from the external atmosphere. A deep variation of the absorption profile in the UV-visible region is observed with the establishment of two very broad and intense bands that have approximate maxima at 30600 and 24600 cm−1. These are compatible with the doublet corresponding to the LMCT transition of Co(III) in tetrahedral coordination.48,51–53 In the visible region, the signal associated with Co(II) has undergone some changes, being distorted in a band with two maxima at lower frequencies with respect to the triplet of the first spectrum (1700–15200 cm−1). In the NIR region, the absorption is more extended and more intense. The third spectrum (Fig. 1a, blue line) was collected after treatment in pure H2 at 400 °C under the same conditions of atmospheric isolation (see the Experimental section). A decrease in the intensity of the bands at 30600 and 24600 cm−1 can be observed, indicating a lower amount of Co(III) species; meanwhile in the visible zone a partial restoration of the triplet can be seen with the appearance of a signal at 19700 cm−1 which is added to the signals at 17000 and 15200 cm−1. These observations indicate an incomplete process of reduction, as Co(III) is still present, while Co(II) is being restored by passing through a distorted intermediate coordination, compared to the one of the starting spectrum.
000 cm−1, is compatible with the LMCT, while the visible region is dominated by the narrow triplet between 18500 and 16000 cm−1 typical of the Co(II) d–d transition in tetrahedral coordination, which is the result of the combination of ν2 ≡ 4A2 (F) → 4T1 (F) and ν3 ≡ 4A2 (F) → 4T1 (P). A broad signal is present in the NIR region, around 7000 cm−1 due to the ν1 ≡ 4A2 (F) → 4T2 (F) transition.31,50 The sample was then subjected to calcination at 550 °C in a pure oxygen atmosphere within a closed system (see the Experimental section for further details). The second spectrum (Fig. 1a, green line) was then recorded while maintaining the conditions of isolation from the external atmosphere. A deep variation of the absorption profile in the UV-visible region is observed with the establishment of two very broad and intense bands that have approximate maxima at 30600 and 24600 cm−1. These are compatible with the doublet corresponding to the LMCT transition of Co(III) in tetrahedral coordination.48,51–53 In the visible region, the signal associated with Co(II) has undergone some changes, being distorted in a band with two maxima at lower frequencies with respect to the triplet of the first spectrum (1700–15200 cm−1). In the NIR region, the absorption is more extended and more intense. The third spectrum (Fig. 1a, blue line) was collected after treatment in pure H2 at 400 °C under the same conditions of atmospheric isolation (see the Experimental section). A decrease in the intensity of the bands at 30600 and 24600 cm−1 can be observed, indicating a lower amount of Co(III) species; meanwhile in the visible zone a partial restoration of the triplet can be seen with the appearance of a signal at 19700 cm−1 which is added to the signals at 17000 and 15200 cm−1. These observations indicate an incomplete process of reduction, as Co(III) is still present, while Co(II) is being restored by passing through a distorted intermediate coordination, compared to the one of the starting spectrum.
The CoAPO-18c spectrum collected in air (Fig. 1b, grey line) shows at the same time features relatable to both Co(II) and Co(III). The transition at 50000 cm−1 and the frequencies of the three bands constituting the visible triplet indicate the presence of tetrahedral Co(II) as in the CoAPO-18u first spectrum (Fig. 1a, grey line). However, it is evident that these bands in the CoAPO-18c spectrum have a lower intensity and thus a lower concentration of species, with respect to the case of CoAPO-18u. This observation finds agreement with the contemporary presence of signals related to Co(III) (UV region, 30000–24000 cm−1). It follows that the total cobalt in CoAPO-18c is simultaneously present in the structure with both oxidation states. An extra band is present around 42000 cm−1 probably due to π → π* transitions of polymerised hydrocarbon structures trapped in the CoAPO-18 pores, deriving from the template combustion, as it disappears after the subsequent O2 treatment at 400 °C (Fig. 1b, green line). This process causes the growth of the Co(III) UV bands, as the concentration of the species is increased, even if the signal is less intense in CoAPO-18c than in CoAPO-18u. Indeed, a portion (greater than the case of oxidized CoAPO-18u) of the total Co is still present as Co(II), as evidenced by the triplet. This signal is broader with respect to the grey spectrum, suggesting a slight distortion of the tetrahedral coordination sphere; its occurrence is even further pronounced after the H2 treatment (Fig. 1b, blue line), indicating that the Co(II) fraction is rising. Also in this last case the persistence of the Co(III) bands indicates a not fully completed reduction process.
Fig. 2 reports the Raman spectra of CoAPO-18u and an as-synthesized low Si containing SAPO-18 (SAPO-18_0.4) (blue line (a) and yellow line (b), respectively) and after template removal by calcination (green line (a) and red line (b), respectively) collected with incident λ = 442 nm. Both samples are compared with the computed heteroatom free AlPO-18 Raman spectrum (black lines, Fig. 2a and b). The comparison with both SAPO-18_0.4 (experimental) and AlPO-18 (computed) spectra allowed discriminating between contributions belonging to the template molecule (N,N-diisopropylethylamine) interacting with the framework and the framework vibrations, aiming to highlight the Raman active modes of CoAPO-18u involving the Co atoms and recognise the changes due to the activation treatments.
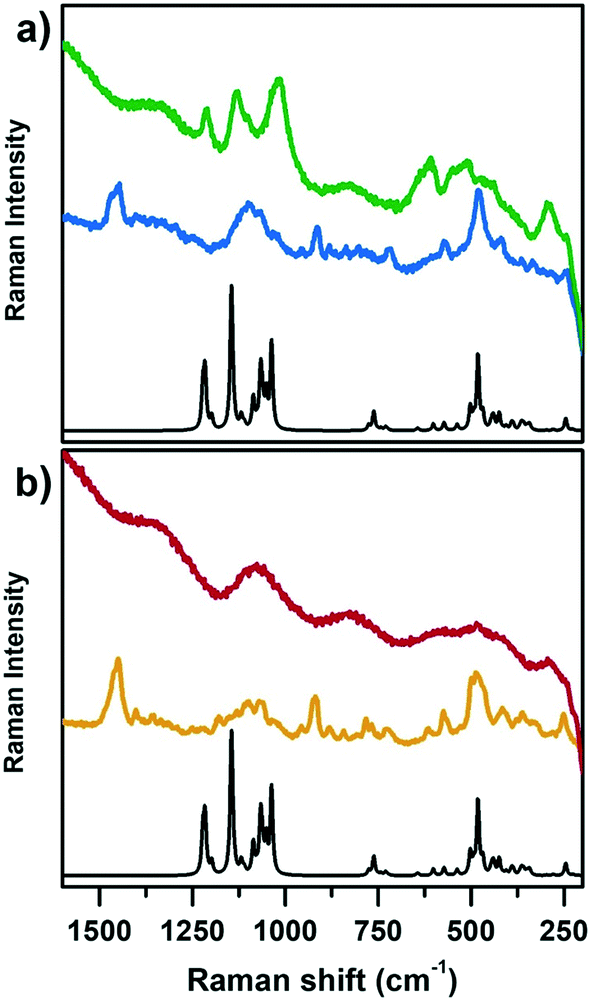 | ||
| Fig. 2 Computed Raman spectrum of AlPO-18 (black lines a and b) compared with experimental Raman spectra collected with incident λ = 442 nm of a) CoAPO-18u: template containing sample (blue spectrum) and after calcination in pure O2 (green spectrum); b) SAPO-18_0.4: template containing sample (yellow spectrum) and after calcination (red spectrum) in the Raman shift range of 1600–200 cm−1. | ||
On comparing first the spectra of the as-synthesized materials (blue (a) and yellow (b)) with the AlPO-18 computed one, different contributions can be identified. The families of signals positioned around 1460 cm−1 and 900 cm−1 must be traced to N,N-diisopropylethylamine since they are completely missing in the computed AlPO-18 spectrum. The latter is characterized by two main types of vibrational modes, the asymmetric and symmetric stretching of TO4 tetrahedra between 1250 cm−1 and 1000 cm−1 (νTO4) and lattice out-of-plane and in-plane bending below 700 cm−1 (δTO4),54,55 to which the signal at 250 cm−1 must be added. Reasonably it corresponds to the channel ring's breathing mode.56 All these signals are present in the spectra of CoAPO-18u and SAPO-18_0.4, however the intensity ratios do not correspond to those of AlPO-18 although they coincide in the two experimental spectra. It is possible that the relative intensities are affected by the presence of the templating amine that occupies the pores of the material, modifying the local symmetry. One general observation in Fig. 2, regarding both CoAPO-18u and SAPO-18_0.4, is the occurrence of an intense fluorescence emission that overcomes the Raman signals, when the samples are subjected to laser excitation after template removal (particularly visible in the red spectrum, Fig. 2b, calcined SAPO-18_0.4). This hampered the recording of the Raman spectrum of the reduced CoAPO-18u, for which the effect was even enhanced, therefore it is not reported in Fig. 2. On the contrary, in the spectrum of the calcined CoAPO-18u (green line (a)), some signals emerge from the fluorescence emission profile. These signals correspond to the more intense ones of the AlPO-18 computed spectrum. It is reasonably assumed that the modes involved are enhanced by the effect of the resonance established between the Raman and the electronic transitions of tetrahedral Co(III) since the wavelength of the incident radiation λ = 442 nm is in the same range of the Co(III) LMCT transition observed in Fig. 1 (doublet around 30600–24000 cm−1 = 326–406 nm). The presence of Co(III), responsible for the Raman resonant effect, enhances the vibrational modes (νTO4) and (δTO4) perturbed by the vicinal Co, in the regions 1250–1000 cm−1 and below 700 cm−1, respectively, approaching the intensity ratio computed for the APO-18 spectrum (black curve). The intense signal at 611 cm−1 can be reasonably attributed to a vibrational mode involving directly the Co(III)–O bonds, as observed from the analysis of the computed frequencies for the Co(III)APO-18 model (see Computational details). The resonant effect can be appreciated also considering that the intensity of the signals around 1000 cm−1 and the one at 611 cm−1 in Co(III)APO-18 significantly overcomes the one at 487 cm−1, which is supposed to be the more intense framework signal after calcination, since it is the only one emerging from the fluorescence profile in the SAPO-18_0.4 spectra (red curve, Fig. 2b).
In Fig. 3a and b, the IR spectra of CoAPO-18u and CoAPO-18c are shown upon ex situ high temperature O2 (green lines) and subsequent H2 (blue lines) treatments. The spectra of CoAPO-18 present different vibrational bands related to surface hydroxyl species visible after catalyst activation, as reported in Fig. 3.
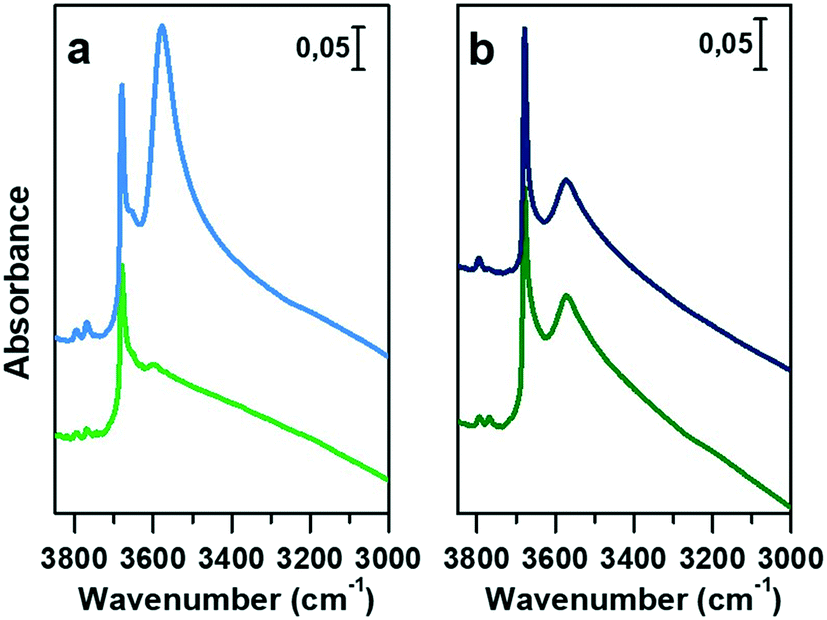 | ||
| Fig. 3 IR spectra in the hydroxyl stretching region (3850 to 3000 cm−1) of a) CoAPO-18u upon calcination (green line) and reduction (blue line) and b) CoAPO-18c after O2 (green line) and H2 (blue line) treatments. The spectra are normalized to the overtone's intensity for the sake of comparison. | ||
At the highest frequencies, two bands ascribable to Al–OH groups in different framework positions fall at 3793 and 3768 cm−1 in each spectrum.57 At 3678 cm−1 an intense and sharp band is assigned to the P–OH groups (free phosphanols) typical of all the aluminophosphate materials which represent framework local defects. The large signal at 3578 cm−1 is due to the stretching mode of bridged P–(OH)–Co hydroxyls which represent the Brønsted acidic sites (BAS).30 The intensity of the latter two bands significantly differs in the various spectra reported in Fig. 3 upon the different treatments the samples have been subjected to. Looking at the CoAPO-18u spectra (Fig. 3a) some differences are clearly visible. Upon O2 treatment (green line) the BAS signal in CoAPO-18u is almost absent, while the same band is extremely intense and overcomes the P–OH one upon H2 action (blue line). These results are in line with that reported in the literature.44 The case of CoAPO-18c is different (Fig. 3b) as the compared spectra show no notable changes.
The band at 3578 cm−1 is evidence of the tetrahedral Co(II) in the framework (as the charge compensating proton is needed to balance the charge of the phosphate groups [3−]), and its variations give information about the Co state, especially if the analysis is coupled with the UV-vis spectroscopy results. On the other hand, there is no direct evidence of the presence of Co(III), since there are no vibrational bands traceable to it, but only indirect considerations can be raised. From these statements, it is reasonable to assume that the cobalt in CoAPO-18u after calcination in pure O2 (Fig. 3a green spectrum) is present almost entirely in the oxidised Co(III) state, given the absence of the band associated with BAS. In contrast, the presence of a relatively intense band at 3578 cm−1 in the case of CoAPO-18c (Fig. 3b green line) evidences the persistence of a fraction of Co(II), in agreement with the UV-vis results. The remaining fraction may be Co(III). It should be noted, however, that the intensity of the band related to P–OH is higher than in the case of CoAPO-18u, indicating an increase in these defective species. This finding suggests that a fraction of Co may have been leached from the framework generating free P–OH. Consequently, the fraction of the heteroatom that does not contribute to the formation of BAS reasonably may consist partly of tetrahedral Co(III) and out-of-structure Co(II). Again, this description is in full agreement with the UV-vis data. After treatment in H2 of CoAPO-18u, it can be presumed that all Co(III) is reduced back to framework Co(II), as a huge increase in the BAS signal is observed, while leaching can be essentially excluded since the phosphanol band does not vary in intensity. If the intensity of the CoAPO-18u BAS band is considered an indication of the maximum concentration of in-framework Co(II) species, it is confirmed that this situation is never reached by CoAPO-18c. Indeed, the spectrum collected after treatment in H2 leaves the intensity of the BAS band essentially unchanged. As the maximum level of Co(II) is reached after treatment in H2, the samples at this stage will be considered “activated” hereafter.
The vibrational features of activated samples are summarised in Table S2 and highlighted in Fig. S1,† where CoAPO-18u and CoAPO-18c are also compared in the “transparent window region” (Fig. S1b†) i.e. the spectral portion enclosed between νasymmetric and νsymmetric of framework T-O4 units, where some diagnostic modes fall. The three bands visible in the CoAPO-18u spectrum (Fig. S1b, light line†) find correspondence with ones that have been assigned by Marchese et al.30,58 as CoAPO-18 fingerprints. Following their interpretation, the 955 cm−1 and 905 cm−1 signals correspond to δ(P–OH) and δ(P–OH–Co), respectively, while the 945 cm−1 band would be the perturbation of νT-O4 due to the presence of the heavier Co atom in the framework. The last attribution is based on the intensification of this band when the concentration of Co(II) is increased in CoAPO-18 and it is considered as the corresponding band of the 960 cm−1 band in TS-1 zeolites.59 In CoAPO-18c (Fig. S1b dark line†) these three components are not distinguishable, as they probably take part in the larger νasym(TO4) mode. This represents a further relevant difference in the CoAPO-18c nature with respect to the fresh sample.
The interaction of adsorbed probe molecules with the CoAPO-18 surface during in situ FT-IR spectroscopy experiments provides a wide range of information about the possible active sites. The activated materials are shown to interact with N2 at liquid nitrogen temperature (LNT) (Fig. S2†), CO at RT (Fig. 4) and CO at LNT (Fig. 5). The different proton affinity and the different degree of adsorption due to the temperature highlight the behaviour of active species allowing their better identification.60,61 In Fig. S2† the vibrational spectra of the activated samples in the presence of N2 adsorbed at LNT are shown. At low temperature, the interaction of N2 with the surface hydroxyl groups can be observed, as the establishment of a weak H-bond induces a downward shift of the ν(OH) involved and an upward shift of ν(N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), which is proportional to the acidic character of the group.62 Also Lewis acidity can be detected by observing the shift of ν(NN) (only the Raman active mode, falling at 2321 cm−1) due to electronic σ-donation or π-backdonation of transition metals to the diatomic molecule.63
N), which is proportional to the acidic character of the group.62 Also Lewis acidity can be detected by observing the shift of ν(NN) (only the Raman active mode, falling at 2321 cm−1) due to electronic σ-donation or π-backdonation of transition metals to the diatomic molecule.63
 | ||
| Fig. 4 IR spectra of a) CoAPO-18u and b) CoAPO-18c in the presence of adsorbed CO at RT in the OH stretching region (left panels), CO stretching region (right panels) and transparent window region (insets). Blue lines are the activated sample spectra, grey lines correspond to progressive CO dosage and red lines to the maximum of coverage (15 mbar). | ||
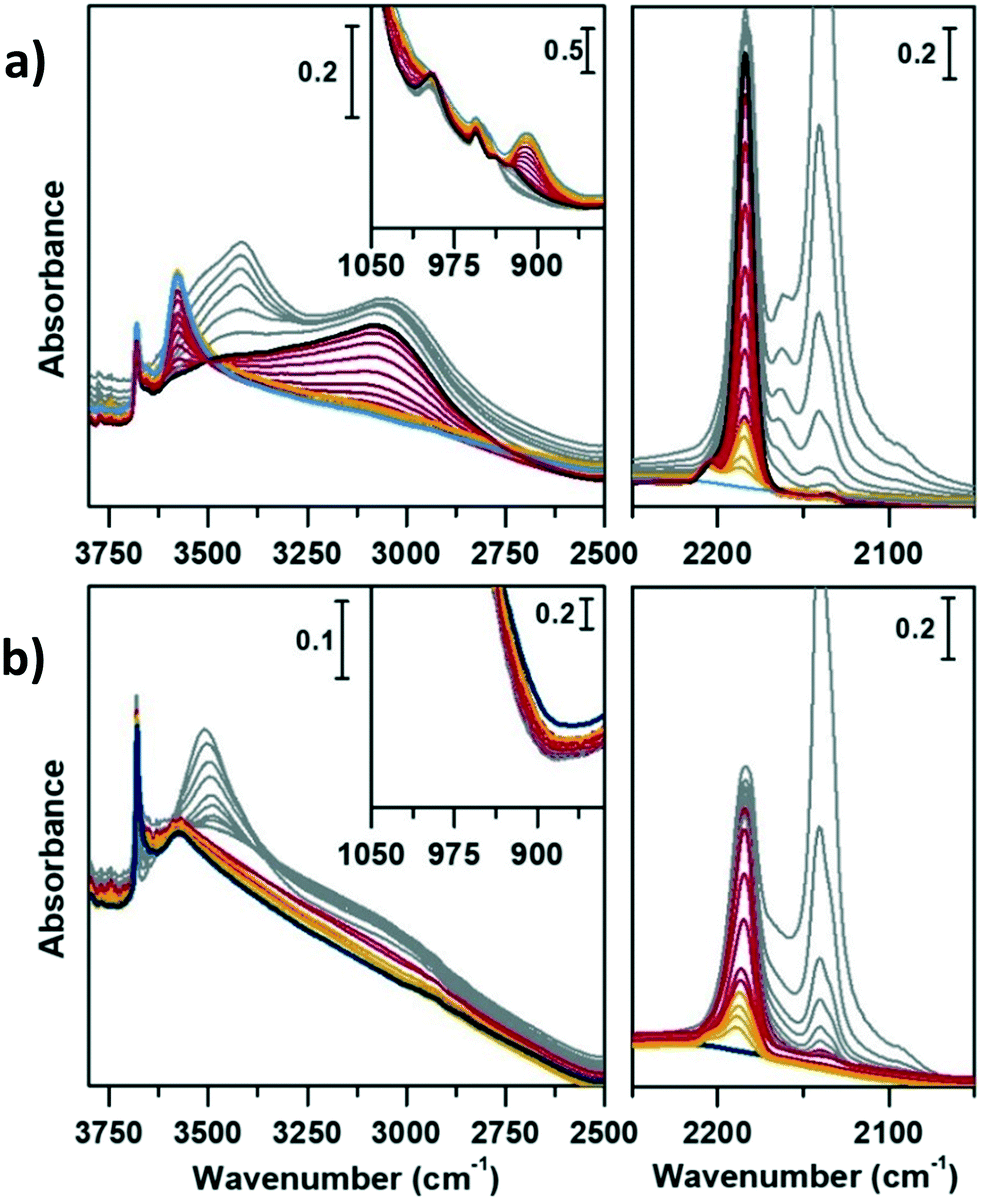 | ||
| Fig. 5 IR spectra of a) CoAPO-18u and b) CoAPO-18c in the presence of adsorbed CO at LNT in the OH stretching region (left panels), CO stretching region (right panels) and transparent window region (insets). Blue lines are the activated sample spectra, grey lines correspond to decreasing CO ep from 30 mbar to 0.3 mbar, black line to CO ep 0.3 mbar and red lines to decreasing CO ep from 0.2 mbar to 0.06 mbar by static vacuum evacuation; yellow lines correspond to decreasing CO below ep < 0.06 mbar by dynamic vacuum outgassing. | ||
The adsorption of N2 reveals in general that the BAS remains unaltered in the two forms of the sample and that it presents a significant acidic strength. Indeed, the 3578 cm−1 band (BAS) undergoes a Δν, which defines a spectroscopic acidity scale (lability of the O–H bond, excluding spatial constraints), equivalent to 110 cm−1, and its extent is comparable to that of ZSM-5, one of the most acidic solid catalysts.58 Moreover, N2 can directly interact with the electron density of Co(II) resulting in a persistent band at 2339 cm−1 which, consistent with the UV-vis spectroscopy results, is ascribed to distorted tetrahedral Co(II) centres.
The case of CoAPO-18 is again peculiar with respect to other cobalt free analogous materials (zeolites and zeotypes), as the transition metal easily coordinates basic molecules leading to the breakage of the (Co–OH)–P bond.31,44,64 In other words, it has been observed that different molecules presenting Lewis basicity can coordinate the tetrahedral cobalt and substitute the bridged –OH group. This implies the loss of the BAS, and the occurrence of a Lewis acid–base couple (Scheme 1) instead; the interaction is reversible after the desorption of the molecule.
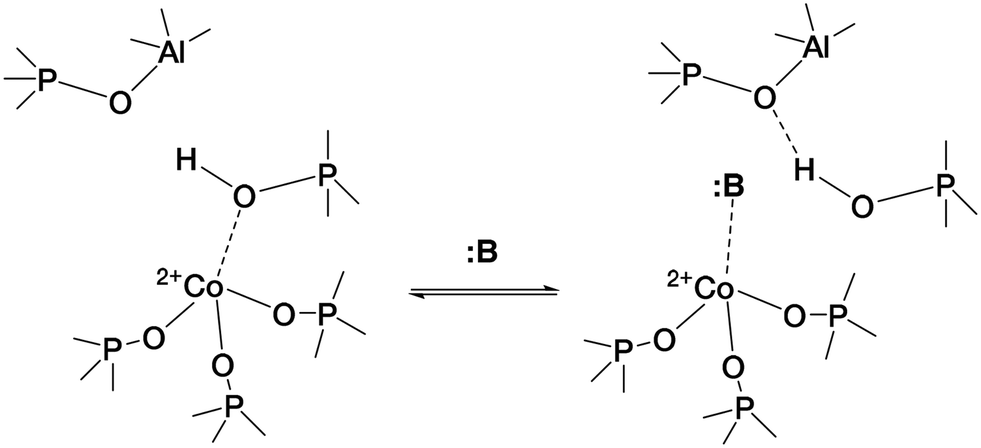 | ||
| Scheme 1 Establishment of the Lewis acid–base couple represented by framework Co(II) and a generic Lewis base :B, inducing the (Co–OH)–P bond displacement.31 | ||
This behaviour justifies some observations in the IR spectra of CoAPO-18 in the presence of relatively strong bases such as H2O, CH3CN and NH3, while what is observed in the case of a weak base such as CO is not totally explained. Due to the poor reactivity of CO, its adsorption at RT allows evidencing only the strongest interaction sites with the material surface, which are generally represented by coordinative unsaturated metals (LAS) or the localized density of free cations.
Fig. 4 displays the interaction of activated CoAPO-18u (a) and CoAPO-18c (b) with CO at RT.
In the left panel of Fig. 4 a little perturbation of the OH stretching is visible at 3578 cm−1 (BAS), whose band is slightly eroded and downward shifted to 3060 cm−1, while in inset a, the associated bending mode (905 cm−1) undergoes a comparable decrease in intensity (the supposed upward shifting is not distinguishable from the dominating νasym(TO4)). This effect is more pronounced for CoAPO-18u (a), since it presents a significantly higher concentration of BAS compared to CoAPO-18c (b) and an intense and visible bending mode. Although the perturbation of the spectroscopic modes appears to be weak, it reflects an extraordinary strong interaction, as generally no shift of the hydroxyls is observed upon CO at RT, but only at low temperature. In the ν(CO) region (Fig. 5a and b, right panels) two contributions for each sample clearly appear upon increasing the dose of adsorbed CO (from grey to red spectra; red spectra correspond to an equilibrium pressure of 15 mbar CO). These bands, falling at 2206 and 2183 cm−1, are generated by the upward shift of the free CO vibration interacting with a localized electron density. The 2206 cm−1 band finds correspondence with the direct interaction of the probe molecule with Co(II). Since the same signal is recorded also in Co(II) exchanged zeolites, it is reasonably identified as a Co(II) cationic site, in counter-ion position.65,66 This signal is more intense in the CoAPO-18c spectra, indicating again a higher concentration of the extra-framework species in that sample. The attribution of the 2183 cm−1 band is of more complex interpretation and a more complete description will be given by coupling these with the results of the experiments at LNT. This signal is commonly attributed to CO linearly interacting with a Co(II) LAS centre, stable in different systems.31,64,65 Marchese et al.31 explained the presence of such species with the mechanism reported in Scheme 1, for which a fraction of the framework Co(II) undergoes a ligand displacement of the bridged OH groups by CO action. Under this hypothesis, the formation of the Co(II)–CO adduct would justify at the same time the perturbation of the stretching and bending modes of bridged OH, which are forced to interact with framework oxygen atoms by H-bonding, stiffening the vibration. It must be noted also that the lattice vibration at 945 cm−1 is slightly shifted to 938 cm−1 by the probe adsorption (Fig. 3a, right panel). This description agrees with Marchese's results, which are valid for CoAPO-18u, as already occurred in the case of previous experiments (DR-UV-vis and ex situ FT-IR spectroscopy). According to this interpretation, the band at 2183 cm−1 would be generated by the action of CO adsorption and not by the pre-existing distorted Co(II). With all this, however, the behaviour of CoAPO-18c once again different. Indeed, as observed in the left panel of Fig. 4b, it exhibits a band at 2183 cm−1 which is more intense (or at least comparable) than that of CoAPO-18u. If this band was generated by ligand displacement of BAS, the behaviour should be exactly opposite, as it is well established that CoAPO-18c has a lower concentration of tetrahedral centres. In addition, the higher intensity of this band also clashes with the lower perturbation of the hydroxyls of CoAPO-18c. Therefore, in this case the LAS centre would not be generated by CO, but more reasonably would correspond to Co(II) in distorted coordination, of which there is UV-vis evidence. More consideration can be made from the analysis of the IR spectra recorded in the presence of CO adsorption at LNT. The low temperature enhances the adsorption rate allowing the determination of weaker interactions which can be discriminated following the progressive probe desorption from the zeotype surface. In Fig. 5, the activated samples CoAPO-18u (a) and CoAPO-18c (b) are reported as blue spectra, and they are then subjected to 30 mbar CO equilibrium pressure (ep) at LNT (first grey spectrum) and at progressive CO desorption (from grey to red spectra) by static vacuum evacuations until a CO ep < 0.06 mbar when a dynamic vacuum outgassing regime has been imposed (yellow spectra) at the same temperature.
The strongest interaction, with respect to RT adsorption, is visible in a more pronounced perturbation of both the O–H (left panels) and CO (right panels) spectral regions. At higher CO ep (30 mbar), when the cryogenic temperature is reached (first grey spectrum) it is possible to observe the erosion of both the phosphanol and BAS signals which are downward shifted. Simultaneously in the CO region, the doublet band centred at 2138 cm−1 corresponding to the liquid-like vibration of the molecule and two other bands at 2167 cm−1 and at 2183 cm−1 (very intense) can be noted. The attributions of these bands will be described in detail.
At LNT the weakly acidic P–OH of both samples (Fig. 5a and b) can establish a H-bonding with the probe; the 3678 cm−1 band is eroded and downward shifted to 3422 cm−1, while the νCO band is upward shifted at 2167 cm−1 by the same interaction (first grey spectrum CO ep 30 mbar), explaining the lower frequency component in the CO spectral region. This weak interaction is underlined by the grey lines of Fig. 5 and represents the first site of CO desorption, as the related bands are easily consumed with outgassing the probe by static vacuum. At CO ep 0.3 mbar (black spectrum) the pristine band of free phosphanols is completely restored while the 3422 and 2167 cm−1 components are consumed. At the same time, the disappearance of the liquid-like CO signal (2138 cm−1) is also attested, while the component at 2183 cm−1 persists as a very intense and narrow signal. As already noticed with the RT experiments, the interaction of CO with the BAS results in an outstanding spectral modification; the effect is more pronounced for CoAPO-18u where the concentration of the sites is much higher. At the maximum coverage (CO ep 30 mbar) the 3578 cm−1 band is consumed while the very intense and broad band at 3059 cm−1 is stabilized. Thus, the νP–(OH)–Co band undergoes a downward shift of 518 cm−1 (Table S3†) indicating an extremely acidic site, since the Δν value significantly overcomes that of common protonic zeolites. Therefore, it can be stated that the strongly ionic nature of the Co–O bond weakens the neighbouring O–H bond, while increasing the acidic strength of the site. During the probe outgassing, the shifted component of the νP–(OH)–Co mode persists up to low CO ep (0.3 mbar, black spectrum), indicating a relatively strong adsorption site. This band is gradually consumed only below 0.2 mbar CO ep while the 3578 cm−1 band is recovered with the same trend (red spectra). Looking at the right panels of Fig. 4, where the CO mode region is reported, at the same time it is possible to observe the start of the parallel consumption of the 2183 cm−1 band (red spectra), while the BAS signal is being restored. This component is much more intense in the CoAPO-18u spectra, but in both cases its decrease has the same rate as the BAS band regrowth. Once the pressure of 0.06 mbar is reached, the complete restoration of the 3578 cm−1 band (BAS signal) is attested for both samples but a fraction of the 2183 cm−1 band is still persistent (yellow spectra). Moreover, an additional band at 2206 cm−1 becomes visible. The frequency of these two bands and their shape (the band at 2206 cm−1 is lower and wider) coincide perfectly with the signals recorded in the RT experiment and therefore represent the interaction of CO with the sites of strongest adsorption. Indeed, these signals remain persistent even after prolonged dynamic vacuum degassing. Given the frequency and the strength of the site, it is reasonable to attribute them to Lewis sites, as already assumed in the case of CO at RT. The band at 2206 cm−1 probably corresponds to the interaction with Co(II) as a counter-ion, while the persistent fraction of the one at 2183 cm−1 corresponds to Co(II) in slightly distorted tetrahedral coordination toward the octahedral one, able to coordinate CO by back donation. This interpretation of the IR spectra would disprove the hypothesis that CO adsorption induces the mechanism in Scheme 1. The band at 2183 cm−1 is the result of the convolution of two contributions, one given by the adsorption of CO on the Brønsted site and one due to the Lewis site, which appear to overlap at high pressures and can only be distinguished with a slow progressive degassing of the probe. This interpretation (summarized in Scheme 2) is in perfect agreement with the trend of the signals in the OH region, which otherwise could hardly be explained by the mechanism in Scheme 1.
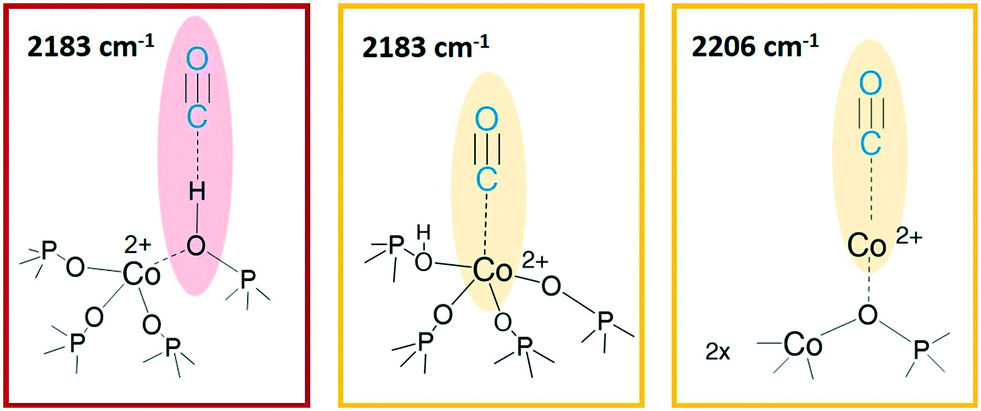 | ||
| Scheme 2 CO interaction sites at LNT. From left to right: Brønsted acid site, Co(II) as a generic Lewis acid site, Co(II) in counter-ion position. The images are only intended to indicate the vibrational modes highlighted, no suggestion of structure is provided here. | ||
The adsorption of CO over CoAPO-18u also induces the perturbation of the low frequency modes enclosed in the transparent window region (Fig. 5a, inset). The δ(P–OH–Co) band at 905 cm−1 is suddenly eroded by the first CO interaction and upward shifted to 995 cm−1, and the pristine band is recovered only at very low CO ep in parallel with the νP–(OH)–Co component. At the same time, the ν(TO3–Co) band is first slightly downward shifted to 938 cm−1 and then restored after the complete CO desorption (yellow spectra), indicating again the establishment of a CO– adduct with the Lewis site.
The perturbative effect of the different probe molecules on the activated CoAPO-18u is summarized in Table S3† and expressed as Δν of the pristine vibration frequency upon the surface interaction.
Methanol conversion over SAPO-18 and CoAPO-18
Before fluxing methanol and initiating the MTH reaction, the sample was specifically calcined within the same set-up, simulating a catalyst activation step within the reactor, common to many catalytic processes. The calcination process of CoAPO-18 has been monitored in continuo by variable temperature FT-IR spectroscopy from 30 °C to 550 °C (with a 5 °C per minute rate) under synthetic air flux (mixture of N2 60 mL min−1 and O2 20 mL min−1), see the Experimental section for further details. The evolution of the IR spectra during the template decomposition from the CoAPO-18 pore system is summarized in Fig. S3,† in which the effect of the rising temperature and duration of the treatment are highlighted.The reaction of methanol conversion has been performed over SAPO-18 (Fig. 6) and CoAPO-18 (Fig. 7) monitoring the process by means of transmission FT-IR spectroscopy under continuous MeOH vapour stream (vapour pressure 0.167 bar at 25 °C), simulating the operative conditions of the MTO reaction at 400 °C (see the Experimental section for further details). The development of the MeOH conversion is monitored with a time-resolution of 16 seconds, the time needed for each spectrum acquisition, which is collected continuously. The evolution of the surface adsorbed species during the progress of the reaction is here described as spectral changes. Catalytic data obtained when running the conversion at 350 °C can be found in ref. 29, for the sake of comparison. Herein, the description is focused on the transformation of active species before and during the mechanism of HCP, starting from the earliest interactions of methanol with active sites. The interpretation of the results obtained can be based on the most recent literature that has been able to spectroscopically give a profile to the complex set of species comprising the HCP within different systems, such as acidic zeolites (H-ZSM-5,67,68 H-beta,10,67 H-SSZ-13 (ref. 69)) or AlPOs (SAPO-34).70,71 Two spectral portions mainly involved can therefore be identified, namely that of ν(OH) and ν(CH) stretching (3850–2450 cm−1) and the region of ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) stretching and C–H bending modes (1900–1300 cm−1). Fig. 6 and 7 report in addition the ν(CO) IR spectral region (2250–2000 cm−1), which requires specific description.
C) stretching and C–H bending modes (1900–1300 cm−1). Fig. 6 and 7 report in addition the ν(CO) IR spectral region (2250–2000 cm−1), which requires specific description.
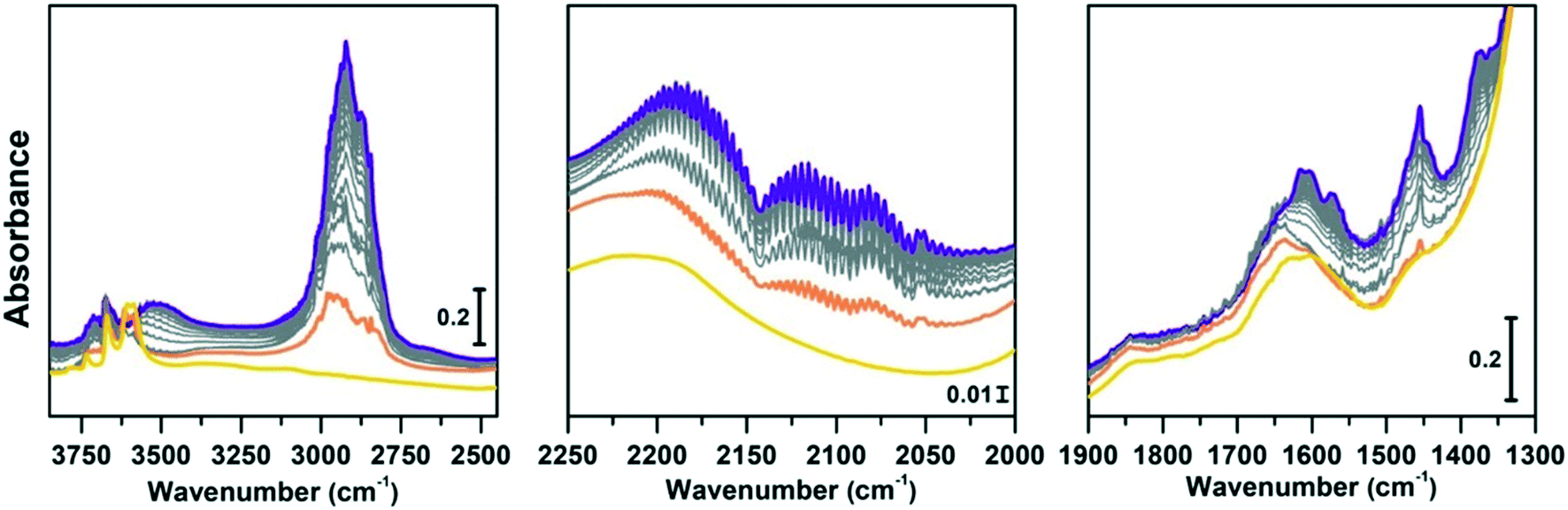 | ||
| Fig. 6 Conversion of methanol catalysed by SAPO-18 at 400 °C followed by IR spectra acquired in continuo. Left panel: O–H and C–H stretching regions (3850–2450 cm−1); central panel: CO stretching region (2250–2000 cm−1); right panel: CC region (1900–1300 cm−1). The yellow spectrum corresponds to the activated SAPO-18, orange is the first interaction with methanol, grey are subsequent acquisitions at increasing time of methanol stream, violet after 1 h conversion. | ||
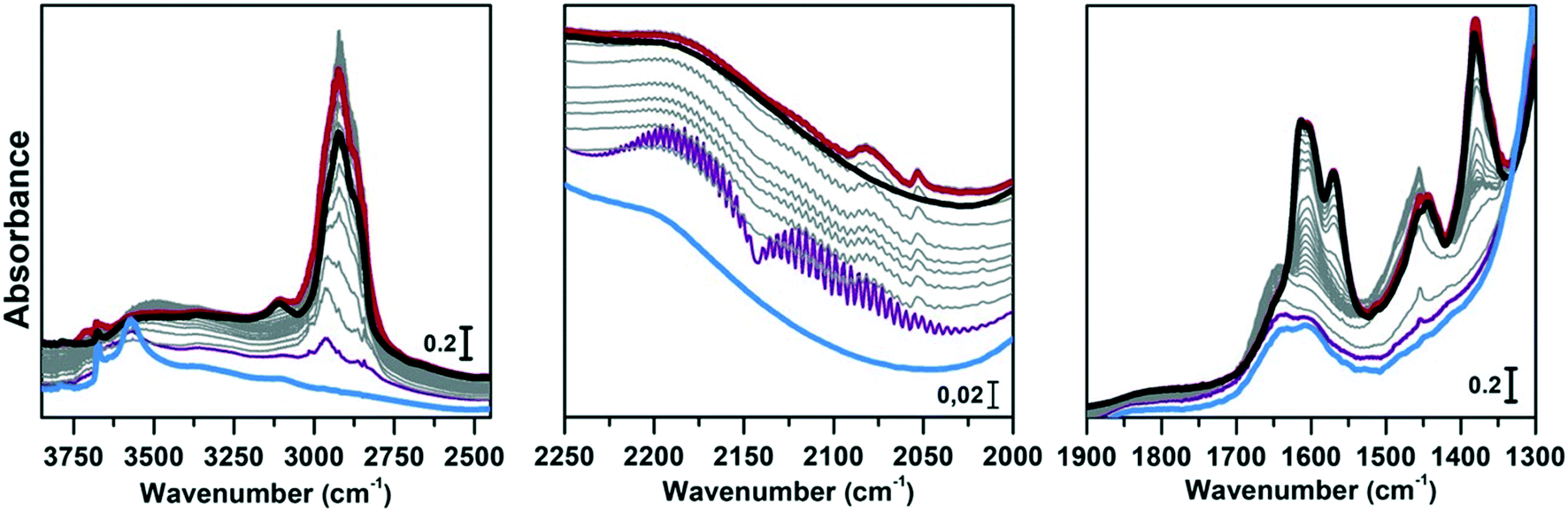 | ||
| Fig. 7 Conversion of methanol catalysed by CoAPO-18 at 400 °C followed by IR spectra acquired in continuo. Left panel: O–H and C–H stretching regions (3850–2450 cm−1); central panel: CO stretching region (2250–2000 cm−1); right panel: CC mode region (1900–1300 cm−1). The blue spectrum corresponds to CoAPO-18, purple is the first interaction with methanol, grey are subsequent acquisitions at increasing time of methanol stream, red after 4 h conversion, black after inert gas flux. | ||
Comparing Fig. 6 and 7 it is possible to qualitatively observe a similar behaviour of SAPO-18 (Fig. 6) and CoAPO-18 (Fig. 7) catalysts in terms of species produced, identifiable through the frequency of the vibrational modes involved. Meanwhile, a profound difference can be observed in the growth rate and intensity of signals corresponding to HCP species adsorbed on the catalyst surface. For rationalising the results, it is also important to consider that CoAPO-18 contains stronger Brønsted acid sites compared with SAPO-18, as demonstrated by FT-IR spectroscopy performed in the presence of basic probe molecules (the materials are compared in Tables S3, S4 and Fig. S4†).
The activated samples, before interaction with the substrate, are highlighted in Fig. 6 as the yellow line (SAPO-18) and in Fig. 7 as the blue line (CoAPO-18) and have been obtained as reported in the Experimental section. The CoAPO-18 spectrum corresponds to the blue spectrum b of Fig. S3.† The spectra have in common the presence of an intense signal due to ν(OH), corresponding to the BAS at 3620 cm−1 and 3580 cm−1 for SAPO-18 and CoAPO-18, respectively.29 The very first interaction with methanol vapours (orange spectrum in Fig. 6 and purple in Fig. 7) suddenly induces, in both cases, a partial consumption of the BAS band accompanied by the formation of two families of new signals. In the ν(CH)zone, bands (left panels) at 3012, 2981, 2950, 2923, 2867, and 2819 cm−1 are recorded, while in the low frequency region (right panels) a weak signal emerged at 1454 cm−1, and all signals appear intensified in the spectrum immediately afterward. These signals are associated with small methylated molecules, deriving from the instantaneous degradation of methanol and adsorption on the catalyst's active sites, which indeed decreases in intensity. The 3012–2819 cm−1 group of signals corresponds to the vibrations of CH3 of dimethyl ether complicated by Fermi resonance between stretching and overtones modes,10,21 among which the components at 2981 and 2867 cm−1 emerge better in the second spectrum. These are attributed to CH asymmetric and symmetric stretching of methoxy groups (OCH3) with their bending counterpart at 1454 cm−1 (symmetric deformation mode) reasonably corresponding to (Ze)-OCH3.70–75 This very first phase corresponds to the induction period, in which the building blocks of the HCP species are formed, whose evidence has allowed the direct coupling theory to be postulated.3 It is very interesting to note that in this phase, when the methanol interacts with the catalyst surface, the typical roto-vibrational profile of carbon monoxide is recorded, characterized by two relatively intense maxima at 2183 and 2114 cm−1 (central panels). The intensity of this signal is comparable only in the first spectrum recorded for the two samples (orange and purple, respectively) while it is significantly reduced in the case of CoAPO-18 remaining equally intense in the case of SAPO-18 (grey spectra). This signal is associated with two others that retain the same rotational profile at 2080 and 2060 cm−1, which still requires a proper attribution. In the following spectra the behaviour of the two catalysts is deeply different. In both cases, we observe the growth of a signal at 1645 cm−1 that can be attributed to CC stretching of linear olefins73 indicative of the initiation of chain reactions that drive the HCP. In the case of SAPO-18, the changes are very slow, while for CoAPO-18 they are instantaneous. During the continuation of the reaction, the signal at 1640 cm−1 is gradually substituted by the bands at 1614–1600 and 1597 cm−1, which become extremely intense in the case of CoAPO-18, indicating the condensation of olefins to form aromatic cycles. In parallel a component at 1382 cm−1 grows and that at 1454–1443 cm−1 is being partially consumed (violet spectrum in Fig. 6, red in Fig. 7). In the case of SAPO-18 there is a correspondence of frequencies, but not in the intensity of the signals and their growth rate. The first two bands around 1600 cm−1 are generated by the formation of aromatics (CC ring vibrations), which shift at 1597 cm−1 with evolution into polycondensed structures (coke precursor species), already observed within the HCP induced by SAPO-34. Meanwhile the lower frequency one corresponds to the bending modes of the alkyl substituents on the aromatic structures. This is also confirmed by the structuration of the signals in the stretching region, which acquire three defined maxima at 2955, 2925 and 2870 cm−1, corresponding to the ν(C–H) stretching of the same groups.69,70 In the case of CoAPO-18, a completely new signal is visible in the last stages of the conversion at 3109 cm−1, corresponding to νC–H aromatic, which will be described later. It is worth noting that the formation of aromatic structures in SAPO-18 is very low and the associated bands (skeletal modes at 1614 and 1597 cm−1 and bending of methyl substituents at 1382 cm−1) are poorly intense and their growth is slow (little changes in grey spectra), while the band at 1453 cm−1, due to the CH3 deformation of virtually isolated methoxy groups, continuously grows, overcoming the intensity of the other signals in the region. The same signal is instead reduced on CoAPO-18. This reasonably indicates that the SAPO-18 cavities keep trapping the unreacted methanol and its degradation products, which continuously occurs during all the time of stream (as attested by the intense roto-vibrational signal of CO), while the autocatalytic transformation of the HCP aromatic species is slower and inhibited. On the contrary CoAPO-18 quickly converts the small methyl intermediates which are involved in the HCP formation, which produces aromatic species dominating at the end of the conversion (red spectrum). The intensity of the CO signals identified in the central panels gives support to this observation. A correlation can be found between the different capacities of the two catalysts in producing hydrocarbons and the trend of the bands of the precursors of the first C–C bond, which according to the most recent theories correspond to CO and the surface methoxy groups –OCH3.18 The higher conversion showed by CoAPO-18 (which gives rise to significantly more intense signals in the aromatic zone, with a higher growth rate) coincides with a decrease in the signal intensity of free CO and a stop in the growth of the signal at 1454 cm−1 related to the CH3 bending of surface adsorbed methanol (Ze-OCH3), at the progress of the conversion. It is reasonable to think that the CO is produced in situ from the decomposition of the methanol and it is continuously consumed during the process by the reaction with the –OCH3 groups, explaining the modifications of the signals. This observation finds a logical answer in the Koch carbonylation mechanism, which involves the formation of a carbonyl by direct interaction of CO with a methoxy group, as proposed by Lercher et al.18 As a further confirmation, the black spectrum in Fig. 7 has been collected at the end of the conversion in the absence of methanol stream, under inert flow only. The total absence of the CO signal in this spectrum demonstrates that the gas is instantaneously produced in situ by methanol degradation, and it stops when the methanol stream is off. The CO is continuously generated by the methanol excess, and it is trapped in the catalyst cages throughout the process. Reasonably it takes part in the conversion not only in the first stage but also in the HCP growth as the carbon source.18 The minor conversion by SAPO-18 is accompanied by the constant presence of free CO, indicated by the roto-vibrational gas signal with unchanged intensity. The lower production of hydrocarbons by the action of this catalyst is accompanied by a larger presence of unreacted precursors CO and –OCH3, giving support to what is described above. Further considerations will be provided in the next paragraphs.
Regeneration of the CoAPO-18 catalyst after methanol conversion
The same spectroscopic approach has been applied during the regeneration of the spent CoAPO-18. The full process is reported in Fig. 8. The experiment described in Fig. 8 is composed of three phases: the methanol conversion on CoAPO-18 (Fig. 8a), the regeneration of the spent catalyst by thermal treatment at 550 °C for 4 hours (Fig. 8b) and the repetition of the reaction over the regenerated catalyst (Fig. 8c). Moreover, the full experiment provides an opportunity to gain insight into various aspects of methanol conversion and on CoAPO-18 as a catalyst. First, the reaction was carried out on the zeotype presenting the blue spectrum a of Fig. S3† (it has undergone a rapid calcination, see the Experimental section) which thus makes it possible to assess how the catalyst activation process affects the reaction. Secondly, by observing in real time how the species adsorbed in the porous system evolve during a thermal treatment, it is possible to obtain information on the deactivation/reactivation processes of the material. Finally, by subjecting the regenerated catalyst to a second cycle of catalysis, it is possible to observe how it influences the product formation. | ||
| Fig. 8 Regeneration of CoAPO-18. a) Conversion of methanol catalysed by CoAPO-18 at 400 °C followed by IR spectra acquired in continuo before regeneration. Left panel: O–H and C–H stretching regions (3850–2450 cm−1); central panel: CO stretching region (2250–2000 cm−1); right panel: CC mode region (1900–1300 cm−1). The blue spectrum corresponds to CoAPO-18, purple is the first interaction with methanol, red after 4 h conversion. b) Regeneration of CoAPO-18 followed by IR spectra acquired in continuo for 4 h at 550 °C; black spectra have been acquired with raising the temperature from RT to 550 °C; orange spectra represent the first hour at 550 °C; in purple the spectra acquired in the last 3 hours at 550 °C; in red is reported the spectrum of CoAPO-18 after 4 h of regeneration. Inset: CO stretching region (2250–2000 cm−1) c) Conversion of methanol catalysed by regenerated CoAPO-18 at 400 °C followed by IR spectra acquired in continuo. Right panel: O–H and C–H stretching regions (3850–2450 cm−1); central panel: CO stretching region (2250–2000 cm−1); right panel: CC mode region (1900–1300 cm−1). The red spectrum corresponds to the regenerated CoAPO-18; purple is after 1 h conversion. | ||
Fig. 8a must be compared with Fig. 7, as the conversion of methanol has been performed over CoAPO-18 under two different conditions. In the case of Fig. 7, the sample has been activated by operating a 4-hour calcination at 550 °C under synthetic air flow and subsequently it has been fluxed with pure N2 overnight at RT, while in Fig. 8a the experiment has been carried out directly after 4 hours of calcination. In Fig. S3† the result of the different calcinations is reported as blue spectrum a (the one in Fig. 8a) and blue spectrum b (Fig. 7). It is evident from Fig. S3† that the process of “fast calcination” does not lead to the complete desorption of the species originating from the template combustion, as the band at 3100 cm−1 due to the ν(N–H) is still present in blue spectrum a. The amine interacts with CoAPO-18 by H-bonding involving the Brønsted sites, which results in the ν(O–H) band being less intense with respect to blue spectrum b and generates a downward shifted component at 3356 cm−1. This implies a reduced availability of the active sites to take part in the methanol adsorption and the catalytic conversion when the stream is on. It results in a slower conversion of the substrate especially at the early stage, as the close-up spectra in Fig. 8a indicate less pronounced changes with respect to the ones in Fig. 7. The vibrational profile of the adsorbed species after 4 hours of reaction (red spectra Fig. 7 and 8a) appears the same, with the prevalence of methylated aromatic structures. However, the higher intensity of the ring modes at 1614–1600 and 1597 cm−1 together with the methyl bending at 1380 cm−1 of Fig. 7 suggests a more advanced level of development of aromatic species. Meanwhile in Fig. 8a the same signals present a comparable intensity with the band at 1454 cm−1 of isolated OCH3, as similarly observed for SAPO-18 (Fig. 6). This could suggest that the less availability of active sites (or weaker acidic strength in the case of SAPO-18) limits the conversion to aromatics and so the HCP development. The reaction relies at the stage of methanol decomposition which does not require strong acidity (it occurs also on pure silicalite-1 at comparable temperature).18 It must be underlined that during all the processes the signal of CO is recorded with unchanged intensity. Again, this trend agrees better with that of SAPO-18 with respect to the CoAPO-18 in Fig. 7. The precursors remain unreacted in the catalyst cages giving rise to the vibrational mode of isolated species, while the hydrocarbon species have a slower growth rate. From these observations, it is allowed to assume that the rate of Koch carbonylation strongly depends on the catalyst composition and thus on the acidic strength, as observed from the comparison between SAPO-18 and CoAPO-18, in a parallel trend with that reported for SAPO-34 and the more acidic SSZ-13.76 Moreover, comparing the two differently activated CoAPO-18s, an influence on the density of active sites can be highlighted. Their amount determines the surface saturation of available –OCH3 groups and therefore the carbonylation yield for the same acid strength, again in accordance with that reported for different Si/Al containing SSZ-13.76
During the regeneration of the sample, the adsorbed species have been followed with the IR spectra reported in Fig. 8b. The sample has been heated from RT to 550 °C acquiring the black spectra. The first phenomenon observed is the desorption of adsorbed atmospheric water (the sample has been exposed to air before its regeneration) through the rapid decrease of the very intense and broad band in the ν(OH) region, generated by the interaction of H2O with surface hydroxyls and the corresponding bending signal at 1600 cm−1. Only in the last moments of this phase does the signal related to the adsorbed methanol disappear (1454 cm−1) while a significant decrease in the C–(CH3) signals (2960–2860 and 1380 cm−1) and aromatic ring modes is observed. The latter evolves into a signal that is gradually structured in three defined maxima at 1624, 1596 and 1572 cm−1 indicating the increasing condensation of the aromatic systems. The orange spectra describe the first hour at 550 °C. An increase in the intensity of the ring modes of the condensed molecules (coke formation) is initially observed. At the same time there is an indication of the loss of ring substituents as the first step of degradation: the CH3 stretching modes are progressively decreasing, leaving space to the appearance of 3107 cm−1 and 1495 cm−1 bands that correspond to aromatic C–H stretching and bending, respectively. The 3107 cm−1 signal has already been recorded in the last stages of Fig. 7 corresponding to an advanced stage of thermal evolution within the pores, which can be identified as a sign of recognition for the deactivating species. Completely new signals appear at 1857 and 1781 cm−1 corresponding to the νasym and νsym CO stretching of carbonyl substituents of a five-membered aromatic ring.73 During the progress of the thermal treatment, underlined by violet spectra, the progressive consumption of the coke related bands is observed until reaching, after 4 hours, the situation represented by the red spectrum. In this, few signals can be identified in the region of aromatic ν(C–H) and carbonyl ν(CO) stretching, indicating the nature of the persistent species at the end of the calcination treatment. It is worth noting that the roto-vibrational signal of CO is never recorded during the catalyst regeneration, during which the sample is subjected only to N2 and O2 flow (Fig. 8b, inset). This attests that the combustion of the hydrocarbon species does not produce CO, which is, on the contrary, generated exclusively from methanol decomposition. The sample was then subjected to a new cycle of methanol conversion, under the same conditions used in the previous cases, obtaining the results shown in Fig. 8c. The reaction proceeds, as already observed, starting with the formation of CO and surface (Ze)-OCH3 groups and then giving rise to bands related to polycondensed aromatic systems, which probably grow from the pre-existing aromatic fragments, surviving the calcination process (witnessed by the bands at 3107 and 1495 cm−1) and not from the olefin fragments (the 1645 cm−1 signal is missing). The conversion is slower compared to that carried out by the fresh catalyst, but still gives evidence for the possibility of using the regenerated one, despite a lower product formation (in terms of IR signal intensity).
Conclusions
The overall analysis of the spectroscopic data demonstrates that the conditions under which the calcination treatment is performed induce relevant modification to the active site population of CoAPO-18. The amount of BAS, which strictly depends on the presence of Co(II) in the framework position (tetrahedral coordination), is subjected to variations derived from the calcination/activation treatments to which the material is exposed. The zeotype, synthesized in the presence of solely Co(II), can be subjected to redox cycles involving the metal centres in a different amount if the treatments are performed in a controlled atmosphere or open air. It is possible to selectively obtain the oxidized form Co(III)APO-18 by pure oxygen treatment or the acid catalyst H–Co(II)APO-18 by after-calcination reductive treatment with pure H2, as long as the cobalt sites retain the tetrahedral coordination, in accordance with the reactions in Scheme 3, valid for CoAPO-18u.48 | ||
| Scheme 3 a) Oxidation reaction involving Co(II) by the action of molecular oxygen. b) Reduction of Co(II) in H2 to form Co(II) and the zeolitic BAS. | ||
This is guaranteed by controlled processes that avoid the interaction of the material with atmospheric moisture or with the H2O molecules produced during the combustion of the template. The water molecules are believed to coordinate the Co(II) vicinal oxygen atoms, changing the coordination from tetrahedral to octahedral.52 For this reason, exposing the template-free material to air (CoAPO-18c) and then removing the adsorbed water by thermal treatment irreversibly increases the amount of coordinative distorted Co(II), decreasing the number of redox active centres. This corresponds to an irreversibly lower amount of BAS and a corresponding enhancement in concentration of LAS. These species can be reasonably represented by punctual formation of dehydroxylated Co(II) aluminates or phosphates,48 where the octahedral metal is able to coordinate linear diatomic molecules such as CO and N2, as probed by IR spectroscopy. It is worth noticing also that the reactions described in Scheme 3, in particular the oxidation reaction, require prolonged exposure to oxidative/reductive treatment for being quantitative, such as that carried out in the closed system adopted on the CoAPO-18u sample, through which temperature and atmosphere homogeneity are ensured and the combustion products are removed by pumping. In the case of natural air treatment of the sample (as in the case of CoAPO-18c) or under synthetic air flow, an intermediate and complex situation is reached. In addition to tetrahedral Co(II), several forms of cobalt are reasonably present at the same time, decreasing the nominal amount of BAS, i.e. distorted tetrahedral Co(II), which is responsible for LAS sites, Co(III) derived from partial oxidation and free Co(II) in the counter ion position derived directly from the synthetic precursor.
The role of carbon monoxide in the MTH conversion
The MTH conversion takes place when the methanol is adsorbed on the catalyst active sites producing surface methoxy species –OCH3 and releasing water, in accordance with Scheme 4.77,78 | ||
| Scheme 4 Methoxy group formation upon methanol adsorption on CoAPO-18 BAS. | ||
Reasonably, the excess of MeOH is decomposed, giving rise to small methylated molecules such as formaldehyde and dimethyl ether and CO, which are spectroscopically identified in this work, as mentioned earlier. It is not surprising to observe the formation of CO during the conversion of methanol at 400 °C over the surface of an acidic zeolite/zeotype. It is attested that under such conditions MeOH degrades, by the action of the acidic functionalities combined with the high temperature, forming formaldehyde, CH4, H2, CO and water.18 What we observed in the experiments of methanol conversion over the AlPO-18 catalysts is the presence of the free-vibrating gas CO entrapped within the pore structure at working temperature, throughout the reaction. Considering that at 400 °C the adsorption is thermodynamically inhibited, the appearance of the roto-vibrational profile of the carbon monoxide can be explained only with a continuous production of the gas. This signal is recorded in every experiment of methanol conversion reported in Fig. 6, 7, and 8a and c. The rate of decomposition of methanol is probably higher than the rate of acquisition of the IR signal, so the evaluations that will be made below must be considered to be entirely qualitative, as they are not supported by kinetic studies. In the central panel of the four figures above, the roto-vibrational CO signal presents comparable intensity in the first spectrum acquired for each, corresponding to the very first interaction between the catalyst and the substrate. At the same time, reasonably, the breaking of the methanol bonds occurs, then the coupling reaction cascade to form the HCP begins. This is coherent with that observed by Howe et al. as the same time resolution scale is adopted, with the acquisition time of each IR measurement (32 scans) being significantly below 1 minute.21
In this work, a correlation between the trend of the CO bands, generated by the methanol decomposition, and the band at 1454 cm−1, which must be considered diagnostic of the presence of surface methoxy species –OCH3,75 has been identified. Thus, the spectroscopical evidence reported in the present work can give support to the occurrence of a Koch carbonylation involving the –OCH3 and the CO generated in situ, in accordance with the newest theories in the field. The role of CO in the formation of the first C–C bond has never been ignored or underestimated, but often rejected because it was believed that its concentration in the reaction environment could not be high enough. Moreover, prior to the decisive work of Lercher et al.18 it was considered that the presence of CO could only result from impurities in the reactants. This was compounded by the lack of monitoring of CO production during catalytic experiments. The approach adopted in this work provides a new point of view. Showing what is formed within the system and not what it releases, it can be helpful in completing the overall picture. We believe that the experiments reported in this paper provide robust evidence of the generation of CO in situ from methanol degradation and its subsequent involvement in the hydrocarbon formation starting from the carbonylation of the –OCH3 groups. Summarising what is previously noticed, in the conversion catalysed by properly activated CoAPO-18 (Fig. 7), a significant reduction of the intensity of the roto-vibrational profile of the gaseous CO is observed at the progress of reaction while the diagnostic –OCH3 band appears constant in intensity. This finds a perfect accordance with the Koch carbonylation mechanism, for which the two species react in a 1:1 ratio. The –OCH3 species is the limiting reagent since its concentration matches with the number of available BAS, while CO is the excess reagent.
The behaviour of the spectroscopic signals suggests a continuous consumption and regeneration of the species involved which facilitates the progress of the conversion into aromatic methylated hydrocarbons. The fact that a constant growth of the same two signals (CO and –OCH3 diagnostic bands) is attested in the presence of SAPO-18 (Fig. 6) and CoAPO-18 rapidly activated or regenerated (Fig. 8a and c) indicates the presence of the unreacted precursors within the porous system, whose evolution into HCP species is more restricted. The reason can be traced to the higher acidity and availability of the active sites of CoAPO-18 (Fig. 7) that continuously allow the formation of –OCH3 and release of a carbonyl intermediate instantaneously converted into hydrocarbon molecules releasing water. The free BAS of CoAPO-18 in Fig. 8a and c are in lower concentration with respect to the sample in Fig. 7, and thus they are more easily saturated by methanol resulting in a lower concentration of the limiting reagent. It follows that the decomposition into CO prevails over the carbonylation.
It is not surprising that carbonyls are not detected by the set up adopted in this work, because molecules such as ketones, esters or carbonyl acids can easily leave the surface of the catalyst.
In conclusion, this work provides evidence of the involvement of carbon monoxide in the methanol conversion operated by different acidic APO-18, giving support to the occurrence of the Koch carbonylation process to form hydrocarbons, since the earliest stages of the reaction. The materials described in this work have characteristics known to promote the carbonylation, which could increase the potential of CoAPO-18 as a catalyst for MTO conversion. In fact, the carbonylation requires two distinct sites, one site to stabilize acidic protons, methyl and acetyl species, while the other site is responsible for binding CO. The BAS enclosed in the 8MR zeolites cages (as in MOR or CHA and reasonably in the AEI framework) are able to better stabilise the cationic intermediate by the action of the surrounding anionic O atoms.76,79,80 The rate-determining step is agreed to be the CO insertion to the surface methoxy group, so a common strategy is to introduce LAS in the catalyst to improve the CO binding.81 The Co(II) centres tending towards the octahedral geometry revealed in CoAPO-18 would therefore participate in further promotion of carbonylation, also considering that homogeneous Co-based catalysts were previously commercialized for the methanol carbonylation reaction in the presence of gaseous CO.82 It is possible that the cooperation of vicinal BAS and LAS in CoAPO-18 is able to increase the carbonylation rate and therefore further promote the overall MTO process. This hypothesis is not supported by direct evidence in this work, but deserves further investigation in the future. With a view of this finding, more information about the chemistry of the methanol conversion can be achieved with the same approach if combined with an outlet analyser for product identification.
Author contributions
Alessia Airi: FTIR and UV-vis experiments, data elaboration and interpretation, writing original draft; Dr. Alessandro Damin: Raman experiments and computational modelling, Dr. Jingxiu Xie: synthesis of the materials; Prof. Unni Olsbye conceptualization; and Prof. Silvia Bordiga: supervision of all the activities. All the authors contributed to the result discussion and review & editing of the manuscript.Conflicts of interest
The authors do not have any conflict of interests to be declared.Acknowledgements
This work is part of the project funded from the European Union‘s Horizon 2020 Research and Innovation Programme under grant agreement no. 837733 (COZMOS). The authors are grateful to Natale Gabriele Porcaro for the help in the realization of the MTO-IR experimental set-up.Notes and references
- C. Chang et al., Process for Manufacturing Olefins, US4025576 A, 1977, p. 682.
- R. P. Ye, J. Ding and W. Gong, et al. , Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- I. Yarulina, A. D. Chowdhury, F. Meirer, B. M. Weckhuysen and J. Gascon, Nat. Catal., 2018, 1, 398–411 CrossRef CAS.
- I. Yarulina, K. De Wispelaere, S. Bailleul, J. Goetze, M. Radersma, E. Abou-Hamad, I. Vollmer, M. Goesten, B. Mezari, E. J. M. Hensen, J. S. Martínez-Espín, M. Morten, S. Mitchell, J. Perez-Ramirez, U. Olsbye, B. M. Weckhuysen, V. Van Speybroeck, F. Kapteijn and J. Gascon, Nat. Chem., 2018, 10, 804–812 CrossRef CAS PubMed.
- M. Stöcker, Microporous Mesoporous Mater., 1999, 29, 3–48 CrossRef.
- D. Chen, K. Moljord and A. Holmen, Microporous Mesoporous Mater., 2012, 164, 239–250 CrossRef CAS.
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155–7176 RSC.
- U. Olsbye, S. Svelle, M. Bjrgen, P. Beato, T. V. W. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 CrossRef CAS PubMed.
- I. M. Dahl and S. Kolboe, Catal. Lett., 1993, 20, 329–336 CrossRef CAS.
- M. Bjørgen, F. Bonino, S. Kolboe, K.-P. Lillerud, A. Zecchina and S. Bordiga, J. Am. Chem. Soc., 2003, 125, 15863–15868 CrossRef PubMed.
- M. Bjørgen, F. Bonino, B. Arstad, S. Kolboe, K. P. Lillerud, A. Zecchina and S. Bordiga, ChemPhysChem, 2005, 6, 232–235 CrossRef PubMed.
- Saepurahman, M. Visur, U. Olsbye, M. Bjørgen and S. Svelle, Top. Catal., 2011, 54, 1293–1301 CrossRef CAS.
- I. M. Dahl and S. Kolboe, Catal. Lett., 1993, 20, 329–336 CrossRef CAS.
- T. Li, T. Shoinkhorova, J. Gascon and J. Ruiz-Martinez, ACS Catal., 2021, 11, 7780–7819 CrossRef CAS.
- W. Song, D. M. Marcus, H. Fu, J. O. Ehresmann and J. F. Haw, J. Am. Chem. Soc., 2002, 124, 3844–3845 CrossRef CAS PubMed.
- Y. Jiang, W. Wang, V. Reddymarthala, J. Huang, B. Sulikowski and M. Hunger, J. Catal., 2006, 238, 21–27 CrossRef CAS.
- A. J. O'Malley, S. F. Parker, A. Chutia, M. R. Farrow, I. P. Silverwood, V. García-Sakai and C. R. A. Catlow, Chem. Commun., 2016, 52, 2897–2900 RSC.
- Y. Liu, S. Müller, D. Berger, J. Jelic, K. Reuter, M. Tonigold, M. Sanchez-Sanchez and J. A. Lercher, Angew. Chem., Int. Ed., 2016, 55, 5723–5726 CrossRef CAS PubMed.
- L. Yang, T. Yan, C. Wang, W. Dai, G. Wu, M. Hunger, W. Fan, Z. Xie, N. Guan and L. Li, ACS Catal., 2019, 9, 6491–6501 CrossRef CAS.
- S. K. Matam, R. F. Howe, A. Thetford and C. R. A. Catlow, Chem. Commun., 2018, 54, 12875–12878 RSC.
- I. B. Minova, S. K. Matam, A. Greenaway, C. R. A. Catlow, M. D. Frogley, G. Cinque, P. A. Wright and R. F. Howe, ACS Catal., 2019, 9, 6564–6570 CrossRef CAS.
- A. D. Chowdhury, K. Houben, G. T. Whiting, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 15840–15845 CrossRef CAS PubMed.
- A. D. Chowdhury, A. L. Paioni, K. Houben, G. T. Whiting, M. Baldus and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2018, 57, 8095–8099 CrossRef CAS PubMed.
- J. Chen and J. M. Thomas, J. Chem. Soc., Chem. Commun., 1994, 603 RSC.
- G. Sankar and R. Raja, in Nanostructured Catalysts, Springer US, Boston, MA, 2008, pp. 195–212 Search PubMed.
- I. L. Franklin, A. M. Beale and G. Sankar, Catal. Today, 2003, 81, 623–629 CrossRef CAS.
- J. M. Thomas, Angew. Chem., Int. Ed., 1999, 38, 3588–3628 CrossRef PubMed.
- J. M. Thomas and R. Raja, Chem. Commun., 2001, 675–687 RSC.
- J. Xie, D. S. Firth, T. Cordero-lanzac, A. Airi, C. Negri, S. Øien-ødegaard, K. P. Lillerud, S. Bordiga and U. Olsbye, ACS Catal., 2022, 12(2), 1520–1531 CrossRef CAS PubMed.
- L. Marchese, J. Chen, J. M. Thomas, S. Coluccia and A. Zecchina, J. Phys. Chem., 1994, 98, 13350–13356 CrossRef CAS.
- L. Marchese, G. Martra, N. Damilano, S. Coluccia and J. M. Thomas, Stud. Surf. Sci. Catal., 1996, 101, 861–870 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Chem. Rev., 2016, 116, 5105–5154 CrossRef CAS PubMed.
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, 1–36 CAS.
- S. C. R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, Ph. D'Arco, M. Llunel, M. Causà, Y. Noel, L. Maschio, A. Erba and M. Rérat, CRYSTAL17 User's Manual, 2018 Search PubMed.
- R. Dovesi, F. Freyria Fava, C. Roetti and V. R. Saunders, Faraday Discuss., 1997, 106, 173–187 RSC.
- M. Catti, G. Valerio, R. Dovesi and M. Causà, Phys. Rev. B, 1994, 49, 14179–14187 CrossRef CAS PubMed.
- M. Corno, C. Busco, B. Civalleri and P. Ugliengo, Phys. Chem. Chem. Phys., 2006, 8, 2464 RSC.
- C. Gatti, V. R. Saunders and C. Roetti, J. Chem. Phys., 1994, 101, 10686–10696 CrossRef CAS.
- A. Simmen, L. B. McCusker, C. Baerlocher and W. M. Meier, Zeolites, 1991, 11, 654–661 CrossRef CAS.
- J. Chen, J. M. Thomas and G. Sankar, J. Chem. Soc., Faraday Trans., 1994, 90, 3455 RSC.
- G. Sankar, J. M. Thomas, J. Chen, P. A. Wright, P. A. Barrett, G. N. Greaves and C. R. A. Catlow, Nucl. Instrum. Methods Phys. Res., Sect. B, 1995, 97, 37–40 CrossRef CAS.
- P. A. Barrett, G. Sankar, C. R. A. Catlow and J. M. Thomas, J. Phys. Chem., 1996, 100, 8977–8985 CrossRef CAS.
- V. Martis, M. Martis, J. Lipp, D. Detollenaere, T. Rayment, G. Sankar and W. Bras, J. Synchrotron Radiat., 2014, 21, 744–750 CrossRef CAS PubMed.
- B. Modén, L. Oliviero, J. Dakka, J. G. Santiesteban and E. Iglesia, J. Phys. Chem. B, 2004, 108, 5552–5563 CrossRef.
- A. Frache, E. Gianotti and L. Marchese, Catal. Today, 2003, 77, 371–384 CrossRef CAS.
- A. A. Verberckmoes, B. M. Weckhuysen and R. A. Schoonheydt, Microporous Mesoporous Mater., 1998, 22, 165–178 CrossRef CAS.
- L. E. Iton, I. Choi, J. A. Desjardins and V. A. Maroni, Zeolites, 1989, 9, 535–538 CrossRef CAS.
- K. Nakashiro and Y. Ono, Bull. Chem. Soc. Jpn., 1993, 66, 9–17 CrossRef CAS.
- A. A. Verberckmoes, M. G. Uytterhoeven and R. A. Schoonheydt, Zeolites, 1997, 19, 180–189 CrossRef CAS.
- P. Prado-Herrero, J. Garcia-Guinea, E. Crespo-Feo and V. Correcher, Phase Transitions, 2010, 83, 440–449 CrossRef CAS.
- M. Zaarour, O. Perez, P. Boullay, J. Martens, B. Mihailova, K. Karaghiosoff, L. Palatinus and S. Mintova, CrystEngComm, 2017, 19, 5100–5105 RSC.
- F. Fan, Z. Feng, K. Sun, M. Guo, Q. Guo, Y. Song, W. Li and C. Li, Angew. Chem., Int. Ed., 2009, 48, 8743–8747 CrossRef CAS PubMed.
- C. Morterra and G. Magnacca, Catal. Today, 1996, 27, 497–532 CrossRef CAS.
- L. Marchese, E. Gianotti, N. Damilano, S. Coluccia and J. M. Thomas, Catal. Lett., 1996, 37, 107–111 CrossRef CAS.
- G. Ricchiardi, A. Damin, S. Bordiga, C. Lamberti, G. Spanò, F. Rivetti and A. Zecchina, J. Am. Chem. Soc., 2001, 123, 11409–11419 CrossRef CAS PubMed.
- S. Bordiga, G. Turnes Palomino, C. Pazè and A. Zecchina, Microporous Mesoporous Mater., 2000, 34, 67–80 CrossRef CAS.
- H. Knözinger and S. Huber, J. Chem. Soc., Faraday Trans., 1998, 94, 2047–2059 RSC.
- C. Pazé, S. Bordiga, C. Lamberti, M. Salvalaggio, A. Zecchina and G. Bellussi, J. Phys. Chem. B, 1997, 101, 4740–4751 CrossRef.
- F. Giordanino, P. N. R. Vennestrøm, L. F. Lundegaard, F. N. Stappen, S. Mossin, P. Beato, S. Bordiga and C. Lamberti, Dalton Trans., 2013, 42, 12741 RSC.
- E. Gianotti, M. Vishnuvarthan, G. Berlier, L. Marchese and S. Coluccia, Catal. Lett., 2009, 133, 27–32 CrossRef.
- F. Geobaldo, B. Onida, P. Rivolo, F. Di Renzo, F. Fajula and E. Garrone, Catal. Today, 2001, 70, 107–119 CrossRef CAS.
- K. Góra-Marek, B. Gil, M. Śliwa and J. Datka, Appl. Catal., A, 2007, 330, 33–42 CrossRef.
- M. J. Wulfers and F. C. Jentoft, ACS Catal., 2014, 4, 3521–3532 CrossRef CAS.
- E. D. Hernandez and F. C. Jentoft, ACS Catal., 2020, 10, 5764–5782 CrossRef CAS.
- Q. Qian, J. Ruiz-Martínez, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel and B. M. Weckhuysen, Catal. Today, 2014, 226, 14–24 CrossRef CAS.
- Q. Qian, J. Ruiz-Martínez, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel, H. E. van der Bij, J. Kornatowski and B. M. Weckhuysen, Chem. – Eur. J., 2013, 19, 11204–11215 CrossRef CAS PubMed.
- Q. Qian, J. Ruiz-Martínez, M. Mokhtar, A. M. Asiri, S. A. Al-Thabaiti, S. N. Basahel and B. M. Weckhuysen, ChemCatChem, 2014, 6, 772–783 CrossRef CAS.
- F. F. Madeira, N. S. Gnep, P. Magnoux, S. Maury and N. Cadran, Appl. Catal., A, 2009, 367, 39–46 CrossRef CAS.
- N. B. Colthup, L. H. Daly and S. E. Wiberley, Introduction to Infrared and Raman Spectroscopy, Elsevier, 2nd edn, 1975 Search PubMed.
- T. R. Forester, S. Wong and R. F. Howe, J. Chem. Soc., Chem. Commun., 1986, 1611 RSC.
- T. R. Forester and R. F. Howe, J. Am. Chem. Soc., 1987, 109, 5076–5082 CrossRef CAS.
- M. Lusardi, T. T. Chen, M. Kale, J. H. Kang, M. Neurock and M. E. Davis, ACS Catal., 2020, 10, 842–851 CrossRef CAS.
- Y. Ono and T. Mori, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 2209–2221 RSC.
- H. Yamazaki, H. Shima, H. Imai, T. Yokoi, T. Tatsumi and J. N. Kondo, Angew. Chem., Int. Ed., 2011, 50, 1853–1856 CrossRef CAS PubMed.
- M. Boronat, C. Martínez-Sánchez, D. Law and A. Corma, J. Am. Chem. Soc., 2008, 130, 16316–16323 CrossRef CAS PubMed.
- P. Cheung, A. Bhan, G. J. Sunley and E. Iglesia, Angew. Chem., Int. Ed., 2006, 45, 1617–1620 CrossRef CAS PubMed.
- T. Blasco, M. Boronat, P. Concepción, A. Corma, D. Law and J. A. Vidal-Moya, Angew. Chem., Int. Ed., 2007, 46, 3938–3941 CrossRef CAS PubMed.
- P. Kalck, C. Le Berre and P. Serp, Coord. Chem. Rev., 2020, 402, 213078 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00303a |
| This journal is © The Royal Society of Chemistry 2022 |