
Enhanced electronic communication through a conjugated bridge in a porphyrin–fullerene donor–acceptor couple†‡
Rubén
Caballero
 a,
Joaquín
Calbo
b,
Juan
Aragó
b,
Pilar
de la Cruz
a,
Enrique
Ortí
*b,
Nikolai V.
Tkachenko
*c and
Fernando
Langa
*a
a,
Joaquín
Calbo
b,
Juan
Aragó
b,
Pilar
de la Cruz
a,
Enrique
Ortí
*b,
Nikolai V.
Tkachenko
*c and
Fernando
Langa
*a
aInstituto de Nanociencia, Nanotecnología y Materiales Moleculares (INAMOL), Universidad de Castilla-La Mancha, Campus de la Fábrica de Armas, 45071 Toledo, Spain. E-mail: fernando.langa@uclm.es
bInstituto de Ciencia Molecular, Universidad de Valencia, 46950 Paterna, Spain. E-mail: enrique.orti@uv.es
cFaculty of Engineering and Natural Science, Tampere University, P. O. Box 541, Tampere 33101, Finland. E-mail: nikolai.tkachenko@tuni.fi
First published on 25th June 2021
Abstract
A ZnP–2EDOTV–C60 triad, with enhanced electronic communication between terminus donor and acceptor moieties, was synthesized and studied both experimentally and theoretically. Electrochemical measurements and density functional theory calculations support that the first oxidation takes place on the 3,4-ethylenedioxythiophenevinylene (2EDOTV) bridge followed by the oxidation of the ZnP moiety at slightly higher energies. The electronic communication between the terminal electron-donor ZnP and the electron-acceptor C60 units is enhanced by the conjugated EDOTV-based spacer leading to photoinduced electron transfer over the distance >2 nm in the picosecond time domain. The involvement of the spacer in the electron transfer is confirmed by both photophysical measurements and theoretical calculations indicating the formation of intermediate ZnP–2EDOTVδ+–C60δ− charge-transfer states. Overall, the enhanced donor–acceptor electronic communication accelerates the photoinduced charge separation but also affects the charge recombination process.
Introduction
The challenge of using renewable energies instead of fossil fuels to fight the global warming is one of the main scientific tasks for the next few decades and the utilization of sunlight is the most promising alternative.1,2 A general approach is to design a system in which the energy of an excited state is converted to the energy of an electron–hole pair, or a cation–anion radical pair in the case of molecular system, which is used to generate electric current or to initiate a chemical reaction, respectively. In either case, it is important to achieve an as-long-as-possible charge separation (CS) to avoid fast charge recombination and allow efficient utilization of the charges or ions. In natural photosynthesis, this is achieved through multiple electron transfer reactions. The reaction centre consists of a series of redox active molecular entities placed close to each other such that the excitation of a primary electron donor (D) triggers a cascade of electron transfer (ET) reactions delivering an electron to a final electron acceptor (A) at a few nanometer distance from the primary donor.3–5 Natural photosynthesis utilizes chlorophylls and quinones as essential components,6 but numerous artificial systems were developed utilising porphyrins, the closest analogues of chlorophylls, and fullerenes, which are equivalent to quinones in their reduction potentials but have the advantage of a smaller reorganization energy associated with the ET.7–10 Although this strategy allows to design systems with almost unity quantum yield of the CS, it suffers from energy loss at each step of the ET and cumulative loss of energy from the excited state to the final CS state can be high, more than 50%. The ET distance for a single step of so-called through-space ET is limited by the fact that the electron must “jump” from a donor to an acceptor, and typically an increase of the distance by 1 Å reduces the ET time constant by factor of two.11An alternative approach is to design donor–bridge–acceptor (D–B–A) systems, in which the bridge plays the role of a molecular wire conducting an electron from the lowest-unoccupied molecular orbital (LUMO) of the donor to that of the acceptor.12,13 This requires that the LUMO of the bridge is located close to that of the donor, which decreases the ET rate sensitivity to the D–A distance by an order in magnitude or even more.13,14 The connection between the donor and acceptor, the bridge, therefore becomes an important player in the photoinduced ET of these systems. From this viewpoint, the design of efficient D–B–A systems required a careful adjustment of energies of all the three components in conjunction with each other. This in turn rises the requirements to the chemical engineering since the system cannot be considered as consisting of two relatively independent parts, the donor and the acceptor, but all three components must work coherently. Though this is a complication, the expected outcome is the longer distance of the CS with smaller energy losses.
To this end, porphyrins and fullerenes have been widely used as D and A, respectively, in D–B–A systems,15,16 whereas conjugated oligomers have been employed as bridges because they play an important role as semiconductors in molecular electronics.17 Porphyrin-B-C60 architectures using p-phenylenevinylene,18p-phenylene ethynylene,19 fluorene,20 thiophene21 or thienylenevinylene10,22 oligomers as bridges have been shown to exhibit efficient long-distance photoinduced electron transfer from the porphyrin to the fullerene.
In this work, the synthesis of the donor–spacer–acceptor ZnP–2EDOTV–C60 (1) triad, composed by a Zn–porphyrin as donor, a π-conjugated rigid oligomer formed by two units of 3,4-ethylenedioxythiophenevinylene (2EDOTV) as bridge and [60]fulleropyrrolidine as acceptor, is reported. Oligothienylenevinylenes (nTVs) have been reported to be one of the most planar and versatile conjugated oligomers described so far with a low attenuation factor.10,22,23 In addition, the inclusion of ethylenedioxy groups allows to obtain nEDOTVs, which, compared with nTVs, stabilize more efficiently positive charges.24 The electrochemical and photophysical properties of the new triad are studied in depth to get insight into the influence that the electronic coupling between the terminal ZnP donor and C60 acceptor units, through the 2EDOTV conjugated bridge, has over the photoinduced electron transfer. Theoretical calculations at the DFT level, including solvent effects, and detailed femtosecond transient absorption spectroscopic studies have been performed to investigate the electron transfer process.
Results and discussion
Synthesis
The synthesis of triad ZnP–2EDOTV–C60 (1) was achieved following a convergent methodology (Scheme 1). First, porphyrin 225 and bisaldehyde 326 were reacted by Witting–Horner olefination, under careful stoichiometric control, affording 4 in 42% yield. The 1H-NMR spectrum of 4 shows the H atom of the aldehyde at 9.83 ppm as a singlet (Fig. S1, ESI‡). Next, 1 was obtained by 1,3-dipolar cycloaddition of C60 and the azomethyne ylide generated “in situ” from 2 and N-methylglycine. The target compound 1 was isolated by column chromatography (silica gel, hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene 1:1), followed by gel permeation chromatography (Biobeads S-X1, toluene) to obtain a purple solid (44% yield). The MALDI-TOF MS spectrum shows the correct molecular ion at m/z = 1910.69 amu with a fragmentation peak at m/z = 720 amu due to the fullerene cage (Fig. S8, ESI‡). Triad 1 was characterized by 1H-NMR spectroscopy showing the signals corresponding to the porphyrin macrocycle between 8.7 and 9.0 ppm, and the vinyl signals of the oligomer as an AA′BB′ system between 6.9 and 7.5 ppm (Fig. S5, ESI‡). All double bonds are obtained as the E isomer as proved by the coupling constants (J = 16 and 16.2 Hz). The fingerprint of the fulleropyrrolidine ring appears as a singlet at 5.43 ppm and two doublets at 4.98 and 4.23 ppm. Synthetic details and NMR, FT-IR and MALDI-TOF MS data for molecules 1 and 4 are given in the Experimental section included in the ESI.‡
:toluene 1:1), followed by gel permeation chromatography (Biobeads S-X1, toluene) to obtain a purple solid (44% yield). The MALDI-TOF MS spectrum shows the correct molecular ion at m/z = 1910.69 amu with a fragmentation peak at m/z = 720 amu due to the fullerene cage (Fig. S8, ESI‡). Triad 1 was characterized by 1H-NMR spectroscopy showing the signals corresponding to the porphyrin macrocycle between 8.7 and 9.0 ppm, and the vinyl signals of the oligomer as an AA′BB′ system between 6.9 and 7.5 ppm (Fig. S5, ESI‡). All double bonds are obtained as the E isomer as proved by the coupling constants (J = 16 and 16.2 Hz). The fingerprint of the fulleropyrrolidine ring appears as a singlet at 5.43 ppm and two doublets at 4.98 and 4.23 ppm. Synthetic details and NMR, FT-IR and MALDI-TOF MS data for molecules 1 and 4 are given in the Experimental section included in the ESI.‡
 | ||
| Scheme 1 Synthesis of triad ZnP–2EDOTV–C60 (1). Reagents and conditions: (a) NaH in THF at 0 °C. (b) C60 and N-methylglycine in chlorobenzene at reflux. | ||
In addition, compounds 5 (Ph–2EDOTV), 6 (Ph–2EDOTV–C60), Zinc(II) 5,10,15,20-tetrakis(2,4,6-trimethylphenyl)porphyrin (ZnP) and N-methyl-[60]fulleropyrrolidine (NMFP) (Chart 1) were prepared as reference systems for comparison purposes (see the ESI‡).
 | ||
| Chart 1 Reference compounds Ph–2EDOTV (5), Ph–2EDOTV–C60 (6), ZnP and NMFP. | ||
Geometry and molecular orbitals
Theoretical calculations under the density functional theory (DFT) framework were performed for geometry and electronic structure analysis (see the ESI,‡ for full computational details). The minimum-energy structure of triad 1 (Fig. 1a) and its constituting derivatives 4, 5, 6, ZnP and NMFP (Fig. S26, ESI‡) were obtained after full geometry relaxation at the B3LYP/cc-pVDZ+LANL2DZ(Zn) level of theory including solvent effects (dichloromethane in a polarizable continuum model PCM approach). Triad ZnP–2EDOTV–C60 (1) shows a non-coplanar arrangement of the three constituting moieties (porphyrin, bridge and fullerene) with an almost orthogonal disposition (70°) of the π-conjugated 2EDOTV bridge with respect to the ZnP unit due to the internal rotation of the meso-phenyl ring (Fig. 1a). Among the different possible conformations of the 2EDOTV bridge, the one containing E double bonds with an anti orientation of the EDOT groups and sulphur atoms pointing inwards in the π-conjugated chain is found as the most stable arrangement (Fig. S26, ESI‡). | ||
| Fig. 1 (a) Minimum-energy structure (characteristic dihedral angles are indicated) and (b) frontier molecular orbital contours (isovalue = ±0.03) calculated for triad 1 at the B3LYP/cc-pVDZ+LANL2DZ(Zn) level including solvent effects. H and L denote HOMO and LUMO, respectively. | ||
Electronic structure calculations were performed on the minimum-energy geometries to unveil the nature of the frontier molecular orbitals in triad 1 (Fig. 1b) and relative compounds (Fig. S27 and S28, ESI‡). The highest-occupied molecular orbital (HOMO) of 1 is calculated at −4.83 eV and spreads over the π-conjugated bridge including the meso-phenyl ring (Fig. 1b). The HOMO shows a topology and an energy similar to those computed for the HOMOs of related Ph–2EDOTV (5), Ph–2EDOTV–C60 (6) and ZnP–2EDOTV (4) systems (Fig. S27 and S28, ESI‡). In contrast, the HOMO−1 and HOMO−2 of 1 are calculated at −5.21 and −5.43 eV, respectively, and are localized over the porphyrin moiety, in good analogy to that predicted for 4 and to the respective HOMO and HOMO−1 of ZnP (Fig. S27 and S28, ESI‡). These results suggest that the 2EDOTV bridge is a slightly stronger electron donor system compared to ZnP. The lowest-unoccupied molecular orbitals (LUMO, LUMO+1 and LUMO+2) of triad 1 are of C60 nature, in accordance with those computed for 6 and also for NMFP (Fig. S27, ESI‡), indicating the acceptor nature of the fullerene moiety. For 4, due to the absence of the C60 moiety, the LUMO is calculated high in energy at −2.59 eV and belongs to the 2EDOTV bridge, whereas the LUMO+1 and LUMO+2 are placed over the porphyrin unit (Fig. S27, ESI‡).
Electrochemical properties
The redox behaviour of triad 1 and of the reference compounds ZnP, NMFP, 5 and 6 was investigated by Oyster-Young square-wave voltammetry (OSWV), and the reversibility of each redox process was checked by cyclic voltammetry (CV). Table 1 collects the values measured for the first redox potentials.| E 2red | E 1red | E 1ox | E 2ox | |
|---|---|---|---|---|
|
a 10−3 M in o-dichlorobenzene:acetonitrile (4:1), Ag/AgNO3 (0.01 M) was used as reference electrode and checked against the (Fc/Fc+) couple, glassy carbon electrode, Pt counter electrode, 20 °C, 0.1 M Bu4NClO4, scan rate = 100 mV s−1.
b Irreversible process.
|
||||
| ZnP | −2.55 | −2.02 | 0.25 | 0.65 |
| NMFP | −1.59 | −1.16 | ||
| 5 | −2.37b | 0.16b | ||
| 6 | −1.55 | −1.15 | 0.19b | |
| 1 | −1.55 | −1.15 | 0.21 | 0.28 |
A complex cyclic voltammogram showing several reduction and oxidation processes was obtained for 1 (Fig. S33, ESI‡). The contribution of each electroactive moiety of the triad can be addressed by comparing the redox potentials measured for 1 with those of the reference compounds (Table 1). The reduction potentials of 1, registered at −1.15 and −1.55 V, are unequivocally assigned to the reduction of the C60 cage by comparison with those of NMFP and 6. In the anodic window, several waves corresponding to both the porphyrin moiety and the bridge were recorded. The first oxidation is attributed to the overlap of two waves at 0.21 and 0.28 V as can be deduced by careful deconvolution of the Oyster-Young voltammogram (Fig. S34, ESI‡). The similarity of the first oxidation potential of 1 (0.21 V) with that of 5 (0.16 V) and 6 (0.19 V) suggests that this process is centred on the oligomer bridge. Indeed, the shift to higher potentials of the oxidation of oligothienylenevinylenes linked to electron acceptors has been reported, and it is attributed to the presence of different rotameric conformations of the oligomer unit when attached (or not) to the bulky C60.27 The second oxidation peak of 1, at 0.28 V, is then attributed to the porphyrin macrocycle.
From these data, the energy of the radical ion pairs (ERIP)28 for ZnP–2EDOTV˙+–C60˙− and ZnP˙+–2EDOTV–C60˙− can be estimated as 1.36 and 1.44 V, respectively. These results suggest that the energy of both radical ion pairs is similar and that, in the eventual formation of a radical ion pair, both transient species would have almost the same energy.
Based on the results of the redox potential measurements, the energy diagram of HOMO and LUMO level relative positions can be drawn as presented in Fig. 2, though evaluations of the energies of the charge-separated (CS) states in the diagram do not account for the Coulomb potentials and can be taken as rough estimations only.
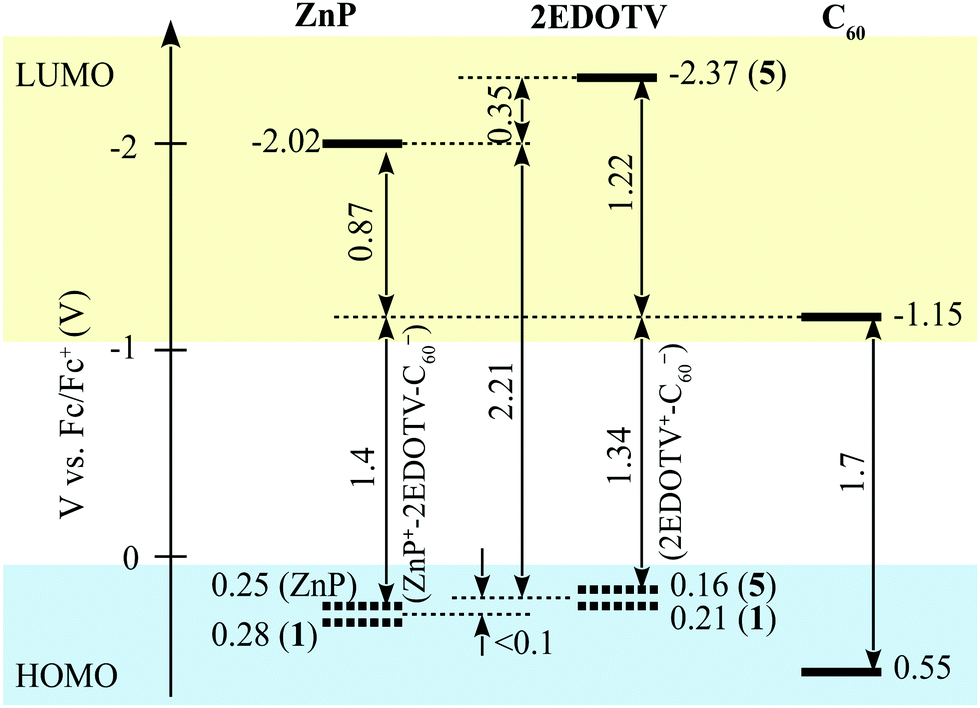 | ||
| Fig. 2 Relative energy positions of HOMO and LUMO levels of the chromophores constituting the Zn–2EDOTV–C60 triad. | ||
Theoretical calculations were performed on the charged species of triad 1 and reference compounds at the UB3LYP/cc-pVDZ+LANL2DZ(Zn)+PCM(dichloromethane) level to confirm the nature of the electrochemical processes recorded experimentally. Electron injection in the first and second reductions of 1 occurs on the electron-acceptor fullerene moiety, as indicated by the Mulliken net charge accumulated in C60 of −1.01e for the anion and of −1.99e for the dianion, to be compared with −0.04e in the neutral species (Table S1, ESI‡). These processes are predicted with a first and second adiabatic electron affinity of 3.34 and 2.62 eV, respectively. As shown in Fig. 3, the electron-unpaired spin-density contours computed for the anion and dianion species of 1 further support the exclusive participation of C60 in the first and second reductions. First ionization energies (IE1) calculated for the cation species indicate almost identical values for triad 1 and referable compounds 5 and 6 (IE1 of 4.67, 4.69 and 4.68 eV, respectively), in good correlation with HOMO energies (Fig. S28, ESI‡), suggesting a complete participation of the 2EDOTV bridge in the first oxidation process. On the other hand, the second oxidation of 1 is calculated with an IE2 = 5.27 eV, similar to the IE1 computed for referable ZnP (5.11 eV), indicating a main role of the porphyrin moiety in this oxidation process. Spin densities (Fig. 3), as well as accumulated Mulliken net charges (Table S1, ESI‡), further support that the first oxidation process takes place on the 2EDOTV bridge (cation in Fig. 3), whereas the second electron extraction occurs on the porphyrin unit (dication in Fig. 3), thus confirming the experimental assignments.
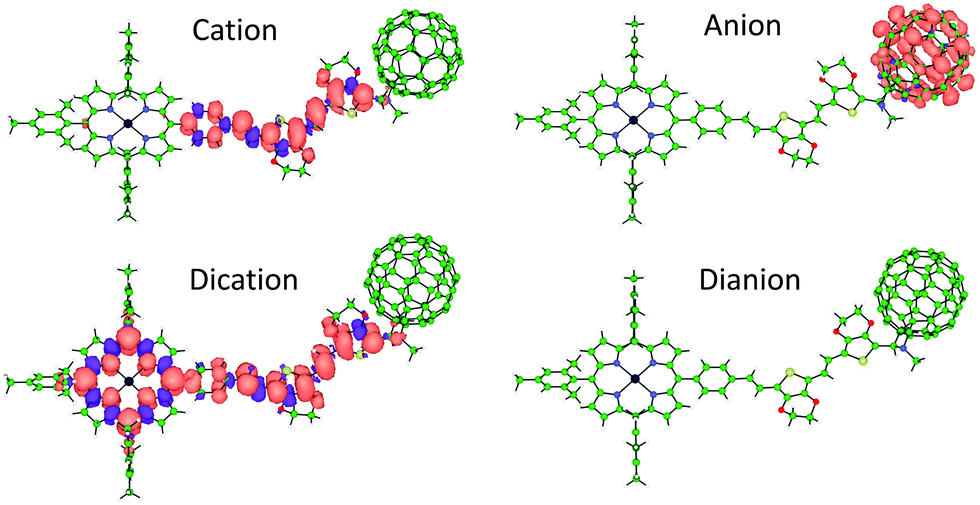 | ||
| Fig. 3 Spin densities calculated for the lowest-energy spin-configuration of the singly and doubly oxidized and reduced species of triad 1. Cation and anion: doublet state, dication: triplet state, dianion: closed-shell singlet state. | ||
Absorption and emission properties
To get insight into the optical properties of the D–B–A triad 1, the absorption spectrum of 1 and ZnP were recorded in CH2Cl2 and are shown in Fig. 4, whereas those of compounds 5 and 6 are displayed in Fig. S35 (ESI‡). Model compound Ph–2EDOTV (5) shows a structured absorption band with maximum at 424 nm, assigned to a π–π* transition, which red-shifts up to 435 nm in 6 owing to the interaction with the C60 cage. On the other hand, ZnP shows the characteristic Soret- and Q-band absorptions at 419 nm and 551 and 591 nm, respectively. The spectrum of compound 1 is the sum of the spectra of the three molecular components. Besides the characteristic broad absorption of the fullerene cage in the 300–400 nm region, the Soret band of the porphyrin moiety decreases in intensity and is slightly red-shifted (2 nm) and broadened compared to ZnP, probably due to the lower symmetry of the porphyrin macrocycle (Fig. 4). At lower energies, a broad band associated to the π–π* transition of the conjugated bridge is observed in the 450–500 nm region, and the components of the Q-band of ZnP appear at similar energies. | ||
| Fig. 4 UV-visible absorption spectra of ZnP–2EDOTV–C60 (1, black) and ZnP (blue) in dichloromethane solution. Concentration: 10−5 M. | ||
The absorption spectra of 1, ZnP, 5 and 6 were calculated at the TD-B3LYP/(cc-pVDZ+LANL2DZ(Zn))+PCM level using dichloromethane as solvent. Theoretical calculations predict the Q band of ZnP at 546 nm resulting from the S0 → S1, S2 degenerate excitations, which exhibit a small oscillator strength (f = 0.016) and are due to π–π* transitions centred over the porphyrin core (Fig. S36, ESI‡). The Soret band is predicted at 400 nm and is associated to a very intense doubly degenerate transition (S0 → S3, S4, f = 1.601 each) of π–π* nature. For 5 (Fig. S37, ESI‡), the S0 → S1 π–π* transition is calculated at 499 nm with high intensity (f = 1.491), slightly underestimating the energy of the experimental band around 450 nm (Fig. S35, ESI‡). This band red shifts to 516 nm in 6, in good agreement with the experiment. When moving to triad 1, several low-lying charge-transfer excitations are predicted in the low-energy range (Fig. S38 and Table S2, ESI‡). The first S0 → S1, S2, S3 transitions are calculated at 981, 902 and 765 nm, respectively, with small or negligible oscillator strengths (f = 0.002, 0.032 and 0.000, respectively). These transitions are described by monoelectronic excitations from the HOMO (2EDOTV) to the LUMO, LUMO+1 and LUMO+2 (C60), thus implying a charge transfer from the bridge to the fullerene ball (Fig. 1). Transitions S0 → S4, S5, …S10 computed below 700 nm mainly correspond to excitations from the porphyrin moiety to the C60 cage and present a negligible intensity (f < 0.001) due to the zero overlap between the molecular orbitals of the ZnP and C60 moieties (porphyrin-C60 centroid–centroid distance of 24.5 Å). Then, an intense CT transition from the 2EDOTV bridge to the porphyrin is predicted at 591 nm (S0 → S11, f = 0.767), and the characteristic π–π* excitation of the 2EDOTV bridge is calculated at 523 nm (S0 → S27, f = 1.520). These two intense transitions are calculated too low in energy and presumably give rise to the intense broad shoulder experimentally recorded in the 450–500 nm range (Fig. 4). The characteristic Q and Soret bands of the porphyrin moiety in triad 1 are predicted at similar energies as those calculated for ZnP in good agreement with the experimental data. The Soret band originates from transitions S0 → S53 (404 nm, f = 0.830) and S0 → S57 (399 nm, f = 1.121), which display lower intensities than for ZnP (f = 1.601) in good accord with the intensity decrease observed in Fig. 4 in passing from ZnP to 1. The Q band results from the S0 → S20, S22 transitions computed around 540 nm with intensities (f = 0.011 and 0.006, respectively) similar to ZnP.
The formation of the positively charged species was furthermore investigated by spectrochemistry measurements through stepwise addition of FeCl3. The UV-visible absorption spectra of the oxidized species of ZnP–2EDOTV–C60 (1) in dichloromethane are plotted in Fig. 5, and those of ZnP and Ph–2EDOTV–C60 (6) are displayed in Fig. S39 and S40 (ESI‡), respectively. For reference compounds ZnP and 6, clear oxidation processes are observed, with the formation of exclusively one cation species, as corroborated by the observation of well-defined isosbestic points. In contrast, two oxidation processes can be seen for 1 (Fig. 5). After the initial additions of FeCl3, the decrease of the band of the oligomer in the 450–500 range and the raise of a new band at 660 nm are observed. The new band is attributed to the formation of the cation ZnP–2EDOTV˙+–C60 as compared with the spectrochemical oxidation of Ph–2EDOTV–C60 (6) (Fig. S40, ESI‡). Theoretical calculations on the cation species of Ph–2EDOTV (5) and 6 further support this assignment by predicting an intense electronic excitation calculated at 621 nm (f = 1.444) for 5˙+ and 653 nm (f = 0.892) for 6˙+ (Fig. S37, ESI‡). Upon successive additions of FeCl3, the band at 421 nm decreases in intensity and two new bands appear at 442 and 646 nm (Fig. 5). Similar bands are observed in the spectrochemical oxidation of ZnP at 438 and 635 nm (Fig. S39, ESI‡), which are theoretically predicted at 470 and 668 nm for ZnP˙+ (Fig. S37, ESI‡), thus allowing the assignment of a radical located on the porphyrin moiety in the dication ZnP˙+–2EDOTV˙+–C60, as also suggested by theoretical calculations (see above). These findings support the easiness to oxidize both the oligomer bridge and the porphyrin moiety, and the preference to stabilize the first radical cation on the conjugated bridge.
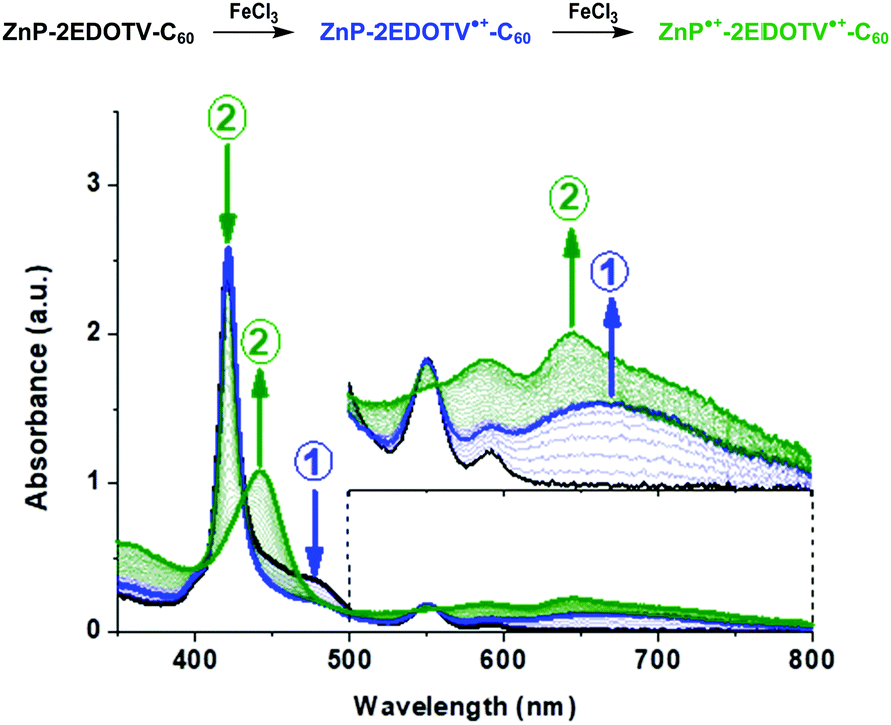 | ||
| Fig. 5 UV-Vis absorption spectra of neutral (black), cation (blue) and dication (green) species of ZnP–2EDOTV–C60 (1), in dichloromethane solution, generated upon successive additions of FeCl3. | ||
The photophysical properties of ZnP–2EDOTV–C60 were studied by steady-state emission spectroscopy in toluene and benzonitrile (Fig. 6). The spectra of compounds Ph–2EDOTV (5) and Ph–2EDOTV–C60 (6) are depicted, for comparison purposes, in Fig. S41 and S42 (ESI‡). For 1, the excitation wavelength was 426 nm, where mainly the porphyrin absorbs. In both solvents, a quenching of the fluorescence of the porphyrin by around 90% is observed.
 | ||
| Fig. 6 Steady-state fluorescence spectra of ZnP (purple line) and ZnP–2EDOTV–C60 (1, black line) in toluene (left) and benzonitrile (right). λexc = 426 nm. Concentration 10−6 M. | ||
Transient absorption spectra
The photo-induced reactions of the compounds were studied using transient absorption (TA) spectroscopy, namely a femtosecond pump–probe method with excitation (pump) by a short 100 fs pulse and monitoring the change (probe) in the visible and near-infrared (NIR) regions. Most of the measurements were carried out using two excitation wavelengths, 495 and 560 nm, which allows to excite predominantly either 2EDOTV or ZnP chromophores, respectively.The TA measurements on the Ph–2EDOTV (5) reference compound reveal that the singlet-excited state has a characteristic absorption band at 730 nm with a lifetime of 640 ps in PhCN (Fig. S43, ESI‡). The intersystem crossing efficiency of 5 is very low and the singlet state relaxes directly to the ground state leaving virtually no signal at delays longer than a few nanoseconds.
Excitation of dyad 4 (ZnP–2EDOTV) in toluene at 560 nm results in instant formation of the ZnP singlet-excited state, which decays to a state with a strong absorption band close to 610 nm, as presented in Fig. 7. This new band is formed with a time constant of 2.0 ± 0.2 ns, which is somewhat shorter than the lifetime of the ZnP singlet-excited state (2.4 ns). The spectrum of the state cannot be assigned neither to the ZnP triplet nor to the 2EDOTV singlet, and it is tentatively attributed to the 2EDOTV triplet, which can be reached in the case of rapid triplet–triplet energy transfer from ZnP to 2EDOTV, as will be discussed later. A tentative reaction scheme for ZnP–2EDOTV in toluene is
| ZnP–2EDOTV + hv → ZnPS–2EDOTV → ZnPT–2EDOTV → ZnP–2EDOTVT → ZnP–2EDOTV |
 | ||
| Fig. 7 Time-resolved TA spectra of ZnP–2EDOTV (4) in toluene excited at 560 nm. | ||
To ascertain the reaction scheme proposed above, the triplet excited states of ZnP–2EDOTV (4), ZnP and Ph–2EDOTV (5) were calculated at the TD-B3LYP/(cc-pVDZ+LANL2DZ(Zn))+PCM level using toluene as solvent. The lowest-lying triplet state T1 of ZnP–2EDOTV (4) is computed at 1.18 eV over the singlet ground state, and results from the HOMO → LUMO excitation associated to the 2EDOTV moiety (see Fig. S27, ESI‡). This is in line with the T1 state calculated for Ph–2EDOTV (5), which is predicted at a slightly higher energy (1.29 eV) due to a smaller π-conjugation compared to 4. The unpaired-electron spin-density computed for 4 after fully optimizing the geometry of the T1 state at the UB3LYP/(cc-pVDZ+LANL2DZ(Zn))+PCM(toluene) level spreads over the 2EDOTV bridge and is identical to that obtained for the T1 of 5 (Fig. S44, ESI‡). The T2 state of 4, corresponding to the excitation of the ZnP moiety (HOMO−1 → LUMO+1 excitation, Fig. S27, ESI‡), is calculated higher in energy at 1.57 eV, in good correlation with the lowest triplet state of ZnP (1.58 eV). Theoretical calculations therefore assign the lowest triplet of 4 to the ZnP–2EDOTVT species, and clearly support the reaction scheme described above with a downhill triplet–triplet energy transfer from ZnP to 2EDOTV.
In polar PhCN the TA response of the ZnP–2EDOTV dyad is more complex as presented in Fig. 8. Excitation at 560 nm results in an almost instant formation of a state with characteristic bleaching of the ZnP Q-bands (570 and 610 nm) and a deep at 670 nm which can be attributed to the ZnP stimulated emission typical of porphyrins. At the same time, a strong absorption band is formed in the red side with maximum close to 770 nm, which resembles the singlet state of Ph–2EDOTV. However, this band is much broader, exhibits a red shoulder extended to the NIR and differs significantly from the band of the Ph–2EDOTV singlet state (Fig. S43, ESI‡).
 | ||
| Fig. 8 Time-resolved TA spectra of ZnP–2EDOTV (4) in PhCN excited at 560 nm. | ||
One can notice that HOMO energies of ZnP and Ph–2EDOTV are very close to each other (Fig. 2 and Fig. S28 in the ESI‡), and that the HOMO of Ph–2EDOTV is only marginally higher than that of ZnP. In the singlet-excited state ZnPS, the HOMO of ZnP has one vacancy, which makes possible the hole transfer from the ZnP HOMO to the 2EDOTV HOMO, thus generating the charge-separated state ZnP−–2EDOTV+. However, we do not observe pure ZnP singlet-excited state with the time resolution of our instrument, roughly 200 fs. At 0.2 ps delay, the state with features of both ZnP and 2EDOTV excited states is seen instead. This is typical of the exciplex-like state, which involves both chromophores and is characterized by a partial electron density transfer, (ZnPδ−–2EDOTVδ+)*. Furthermore, one can speculate that this state is in equilibrium with the ZnPS locally excited state as indicated by a deep at 670 nm, which is typical of a ZnP stimulated emission feature. The global data fit shows that this state decays with time constant 160 ± 20 ps (Fig. S45, ESI‡). The relaxation is mainly seen as: (1) disappearance of the broad band absorption in the range from 700 nm to the NIR and (2) disappearance of the deep at 670 nm. The time-resolved spectrum at 500 ps delay is representative of this new intermediate state, it holds only features of the ZnP chromophore and is tentatively attributed to the ZnP triplet state. Unlike the triplet state of the isolated ZnP chromophore, the lifetime of this triplet in ZnP–2EDOTV is relatively short, and it does not relax directly to the ground state but decays with a time constant of 1.9 ± 0.6 ns to a long-lived state with the spectrum represented by the time-resolved spectrum at 6000 ps in Fig. 8. This spectrum can be assigned to the triplet state of 2EDOTV and the whole reaction scheme is
| ZnP–2EDOTV + hv → (ZnPδ−–2EDOTVδ+)* → ZnPT–2EDOTV → ZnP–2EDOTVT → ZnP–2EDOTV |
Switching the pump wavelength to 495 nm, which excites predominantly the 2EDOTV chromophore in the ZnP–2EDOTV dyad, does not change the TA response observed with 200 fs time resolution as presented in Fig. S43 and S45 (ESI‡). One can expect efficient energy transfer from the excited singlet state of 2EDOTV to ZnP, but the fact that we do not time-resolve this process can be interpreted in favor of a Dexter energy-transfer mechanism and a relatively strong electronic coupling between the two chromophores. This, the strong electronic coupling, explains relatively fast triplet state energy transfer taking approximately 2 ns.
The main difference between the photophysics in non-polar toluene and polar PhCN is the formation of the (ZnPδ−–2EDOTVδ+)* state, which is not observed in toluene. Since this state is characterized by a charge shift, its energy depends on the solvent polarity and it can be speculated that in toluene it is higher than that of the localized singlet state ZnPS–2EDOTV, and thus it is not observed. However, the energy transfer is rather insensitive to the solvent polarity and should be roughly the same in toluene and PhCN, or close to 2 ns, which is faster than intersystem crossing of ZnP. As the result, the triplet state of the ZnP chromophore is almost invisible in toluene as its relaxation rate (via energy transfer) is faster than the formation rate.
Excitation of the Ph–2EDOTV–C60 (6) dyad at 420 nm results in an instant formation of the 2EDOTVS excited state, which relaxes quickly by yielding the 2EDOTV+–C60− CS state followed by a rapid charge recombination. The three lowest-lying singlet states calculated for 6 below 1.55 eV actually correspond to 2EDOTV+–C60− CS states resulting from an electron transfer from the HOMO, located on the Ph–2EDOTV moiety, to the LUMO+1, LUMO+2 and LUMO+3 spreading over the C60 cage (Fig. S27, ESI‡). Fig. 9 presents the TA time-resolved spectra of the dyad in toluene. The spectrum at 0.2 ps delay time has one clear absorption maximum at 710 nm, which matches well that of the singlet-excited state of the reference compound Ph–2EDOTV (Fig. S43, ESI‡). Within few ps, a new absorption band develops at 1080 nm, which can be attributed to the fullerene anion C60−. At the same time, the band at 710 nm shifts to the blue (maximum close to 690 nm) and increases in intensity gradually. This band can be attributed to the cation 2EDOTV+, and correlates with the intense transition theoretically predicted for 6+ at 653 nm (f = 0.892). The CS state recombines with a time constant of 160 ± 10 ps according to the global fit results.
 | ||
| Fig. 9 Time resolved TA spectra of Ph–2EDOTV–C60 (6) in toluene excited at 420 nm. | ||
In PhCN, both charge separation and recombination are faster, which is typical for polar solvents as compared to non-polar (Fig. S48, ESI‡). The estimated time constants for the charge separation and recombination in PhCN are 2.4 ± 0.3 and 4.1 ± 0.4 ps, respectively. This behavior agrees with the energy diagram presented in Fig. 2. showing a rather large driving force for the CS and a marginally higher driving force for the recombination, which makes the recombination a fast process as well.
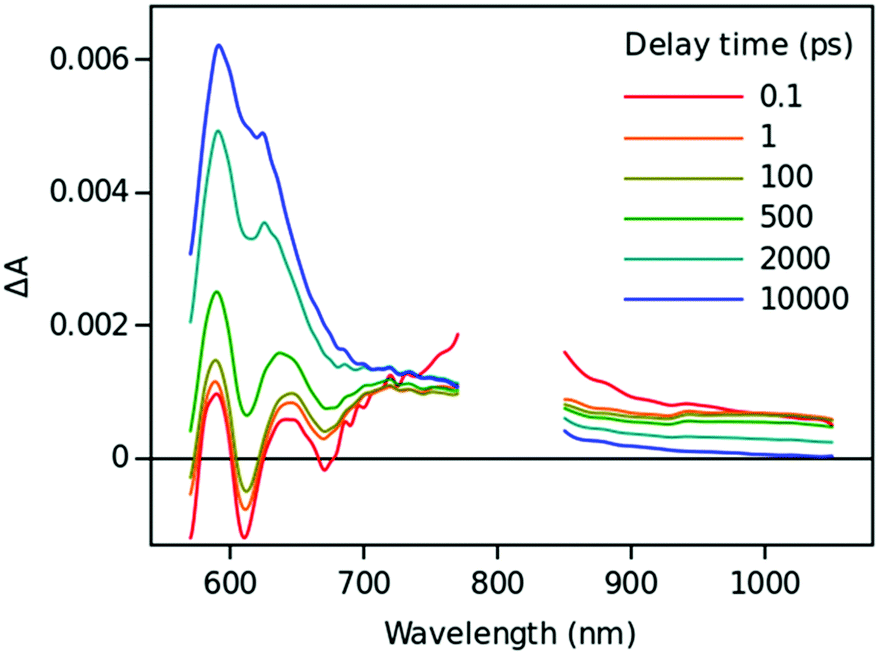
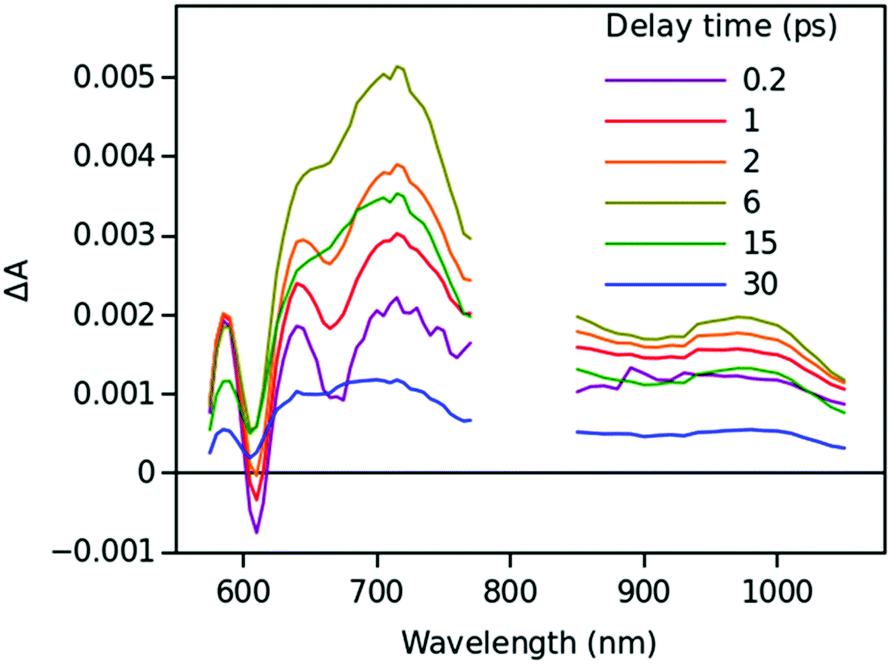
Time resolved TA spectra of the triad ZnP–2EDOTV–C60 (1) in toluene and PhCN obtained with excitation at 550 nm are presented in Fig. 10 and 11, respectively. The main difference between the responses in these two solvents is the time scale, the spectra shapes having many common features.
 | ||
| Fig. 10 Time resolved TA spectra of ZnP–2EDOTV–C60 (1) in toluene excited at 550 nm. | ||
 | ||
| Fig. 11 Time resolved TA spectra of ZnP–2EDOTV–C60 (1) in PhCN excited at 550 nm. | ||
At a short delay time (0.2 ps in Fig. 10), the time-resolved spectra resemble the singlet state ZnPS most closely. Further evolution of the spectrum is associated with the buildup of two broad absorption bands at roughly 720 and 970 nm and the disappearance of the deep at 670 nm. Characteristic spectra of this intermediate can be seen at roughly 6 and 40 ps delay time in PhCN and toluene, respectively. At least in PhCN, this spectrum resembles that of the ZnP+–C60− charge-separated state observed for numerous porphyrin–fullerene dyads though typical position of the fullerene anion radical band is at 1040 nm but not at 970 nm. However, in porphyrin–fullerene dyads with close proximity of the donor and acceptor moieties, an exciplex-like state is formed with the electron density spreading over both chromophores, ZnPδ+–C60δ−, instead of being localized completely on the acceptor. In this case, the fullerene anion-type band is shifted to 950–1000 nm and broadened.29,30 The exciplex-like state can be expected in the case of ZnP–2EDOTV–C60 compound as well, and it explains the spectrum shape in the NIR part.
There is a noticeable difference in the visible spectra between the sample in toluene and PhCN. The characteristic feature of the ZnP chromophore is the bleaching of the Q-band at 590 nm. This feature almost vanishes at 40 ps delay in toluene. At the same time, the rest of the spectrum resembles the spectrum of Ph–2EDOTV–C60 at similar delay time (20 ps in Fig. 9), which was assigned to the Ph–2EDOTV+–C60− CS state, though there is a perceptible difference between the spectra in the NIR. Therefore, the formation of an exciplex-like state between Ph–2EDOTV and C60, Ph–2EDOTVδ+–C60δ−, can be assumed. As mentioned above, the energies of the HOMO levels of ZnP and 2EDOTV are very close to each other and the energy difference between the states ZnP+–C60− and 2EDOTV+–C60−, or ZnPδ+–C60δ− and 2EDOTVδ+–C60δ−, must also be relatively low. This opens the possibility for the equilibrium
| ZnPδ+–2EDOTV–C60δ− ↔ ZnP–2EDOTVδ+–C60δ− |
| ZnP–2EDOTV–C60 + hv → (ZnPδ+–2EDOTV–C60δ− ↔ ZnP–2EDOTVδ+–C60δ−) → ZnP–2EDOTV–C60 |
Only two time constants are available from the TA measurements, 12 and 210 ps in toluene and 5 and 11 ps in PhCN. The first one corresponds to the charge separation and the establishment of the equilibrium, and the second one governs the relaxation to the ground state. The equilibrium rate constant is high in both cases and cannot be extracted from the TA measurements.
The charge transfer distance for the state ZnPδ+–2EDOTV–C60δ− can be estimated to be ∼2.45 nm and it is achieved in both solvents in a few picoseconds time. This is a long distance in the molecular scale, but this CS happens without the energy loss typically found for the more traditional design of molecular systems consisting of multiple donors and acceptors and implementing a multi-step ET,31 in which case the energy is lost at each sequential ET step. The long CS distance in ZnP–2EDOTV–C60 compound is achieved through enhanced electronic communication between the donor (ZnP) and the acceptor (C60) via the conjugated bridge (2EDOTV). This approach has been used in a number of D–B–A system designs leading to a one-step, long-distance CS via a molecular wire connecting the donor and acceptor.32,33 The time constants of the CS here reported are comparable to those systems with similar DA separation distance. A disadvantage of the DA electronic coupling enhanced by the molecular wire strategy is the accelerated charge recombination as well, which is also observed in the case of the ZnP–2EDOTV–C60 compound.
Conclusions
The design goal of the ZnP–2EDOTV–C60 triad is to provide relatively high electronic coupling between the electron donor (ZnP) and electron acceptor (C60) moieties through the π-conjugated bridge (2EDOTV), which was expected to accelerate the photoinduced electron transfer. The porphyrin–C60 centroid–centroid distance is estimated to be 24.5 Å for this compound and even in non-polar toluene the CS is as fast as 12 ps, and the goal of the fast CS is achieved. The observed equilibrium of hole shifting between ZnP and 2EDOTV in the triad (ZnPδ+–2EDOTV–C60δ− ↔ ZnP–2EDOTVδ+–C60δ−) can be considered as a drawback. However, we can envision that this problem can be solved by a more careful selection of the component energies. In particular, a conjugated bridge with a HOMO level lower in energy than that of the donor moiety in the D–B–A triad should help to avoid undesired intermediate states and to achieve a more complete charge separation with slower charge recombination.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors thank the Spanish Ministry of Science and Innovation (MICINN) (PID2019-105049RB-I00, PGC2018-099568-B-I00, RED2018-102815-T and Unidad de Excelencia María de Maeztu CEX2019-000919-M), the Junta de Comunidades de Castilla-La Mancha and European Social Fund (SBPLY/17/180501/000254), the Generalitat Valenciana (PROMETEO/2020/077 and SEJI/2018/035) and European FEDER funds (PGC2018-099568-B-I00) for financial support. J. A. acknowledges the MICINN for his “Ramón-y-Cajal” Fellowship (RyC-2017-23500).References
- R. E. Blankenship, Molecular Mechanisms of Photosynthesis, Wiley-Blackwell, Chichester, 2001 Search PubMed.
- I. McConnell, G. Li and G. W. Brudvig, Chem. Biol., 2010, 17, 434–447 CrossRef CAS PubMed.
- M. R. Wasielewski, Chem. Rev., 1992, 92, 435–461 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40–48 CrossRef CAS PubMed.
- M. E. El-Khouly, E. El-Moshnawy and S. Fukuzumi, J. Photochem. Photobiol., C, 2017, 31, 36–83 CrossRef CAS.
- M. Mamedov, Govindjee, V. Nadtochenko and A. Semenov, Photosynth. Res., 2015, 125, 51–63 CrossRef CAS PubMed.
- A. A. Strelnikov, A. S. Konev, O. V. Levin, A. F. Khlebnikov, A. Iwasaki, K. Yamanouchi and N. V. Tkachenko, J. Phys. Chem. B, 2020, 124, 10899–10912 CrossRef CAS PubMed.
- C. Schubert, J. T. Margraf, T. Clark and D. M. Guldi, Chem. Soc. Rev., 2002, 44(2015), 988–998 Search PubMed.
- T. Higashino, T. Yamada, M. Yamamoto, A. Furube, N. V. Tkachenko, T. Miura, Y. Kobori, R. Jono, K. Yamashita and H. Imahori, Angew. Chem., Int. Ed., 2016, 55, 629–633 CrossRef CAS.
- F. Oswald, D.-M. Shafiqul-Islam, Y. Araki, V. Troiani, R. Caballero, P. de la Cruz, O. Ito and F. Langa, Chem. Commun., 2007, 4498–4500 RSC.
- H. B. Gray and J. R. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3534–3539 CrossRef CAS.
- H. Oevering, M. N. Paddon-Row, M. Heppener, A. M. Oliver, E. Cotsaris, J. W. Verhoeven and N. S. Hush, J. Am. Chem. Soc., 1987, 109, 3258–3269 CrossRef CAS.
- O. S. Wenger, Acc. Chem. Res., 2011, 44, 25–35 CrossRef CAS.
- J. Sukegawa, C. Schubert, X. Zhu, H. Tsuji, D. M. Guldi and E. Nakamura, Nat. Chem., 2014, 6, 899–905 CrossRef CAS PubMed.
- D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22–36 RSC.
- H. Imahori and Y. Sakata, Adv. Mater., 1997, 9, 537–546 CrossRef CAS.
- J. M. Tour, Acc. Chem. Res., 2000, 33, 791–804 CrossRef CAS PubMed.
- G. de la Torre, F. Giacalone, J. L. Segura, N. Martín and D. M. Guldi, Chem. – Eur. J., 2005, 11, 1267–1280 CrossRef CAS PubMed.
- A. Lembo, P. Tagliatesta, D. M. Guldi, M. Wielopolski and M. Nuccetelli, J. Phys. Chem. A, 2009, 113(9), 1779–1793 CrossRef CAS PubMed.
- M. Wielopolski, G. de Miguel Rojas, C. van del Pol, L. Brinkhaus, G. Katsukis, M. R. Bryce, T. Clark and D. M. Guldi, ACS Nano, 2010, 4(11), 6449–6462 CrossRef CAS PubMed.
- J. Ikemoto, K. Takimiya, Y. Aso, T. Otsubo, M. Fujitsuka and O. Ito, Org. Lett., 2002, 4, 309–311 CrossRef CAS PubMed.
- F. Oswald, D.-M. Shafiqul Islam, M. E. El-Khouly, Y. Araki, R. Caballero, P. de la Cruz, O. Ito and F. Langa, Phys. Chem. Chem. Phys., 2014, 16, 2443–2451 RSC.
- C. Franco, P. Mayorga Berriezo, V. Lloveras, R. Caballero, I. Alcón, S. T. Bromley, M. Mas-Torrent, F. Langa, J. T. López-Navarrete, C. Rovira, J. Casado and J. Veciana, J. Am. Chem. Soc., 2017, 139, 686–692 CrossRef CAS PubMed.
- P. Mayorga-Burrezo, B. Pelado, R. Ponce-Ortiz, P. De la Cruz, J. T. López-Navarrete, F. Langa and J. Casado, Chem. – Eur. J., 2015, 21, 1713–1725 CrossRef CAS PubMed.
- A. Balakumar, A. B. Lysenko, C. Carcel, V. L. Malinovskii, D. T. Gryko, K.-H. Schweikart, R. S. Loewe, A. A. Yasseri, Z. Liu, D. F. Bocian and J. S. Lindsey, J. Org. Chem., 2004, 69, 1453–1460 CrossRef PubMed.
- B. Pelado, P. De La Cruz, V. González-Pedro, E. M. Barea and F. Langa, Tetrahedron Lett., 2012, 53, 6665–6669 CrossRef CAS.
- C. Martineau, P. Blanchard, D. Rondeau, J. Delaunay and J. Roncali, Adv. Mater., 2002, 14, 283–287 CrossRef CAS.
- The energy of each radical ion pair (RIP) is estimated from electrochemical measurements by applying the following equation (ERIP = Eox − Ered). Eox is 0.21 V for the radical ion pair ZnP–2EDOTV˙+–C60˙− and 0.28 V for ZnP˙+–2EDOTV–C60˙−.
- N. V. Tkachenko, H. Lemmetyinen, J. Sonoda, K. Ohkubo, T. Sato, H. Imahori and S. Fukuzumi, J. Phys. Chem. A, 2003, 107, 8834–8839 CrossRef CAS.
- V. Chukharev, N. V. Tkachenko, A. Efimov, D. M. Guldi, A. Hirsch, M. Scheloske and H. Lemmetyinen, J. Phys. Chem. B, 2004, 108, 16377–16385 CrossRef CAS.
- H. Imahori, Y. Sekiguchi, Y. Kashiwagi, T. Sato, Y. Araki, O. Ito, H. Yamada and S. Fukuzumi, Chem. – Eur. J., 2004, 10, 3184–3196 CrossRef CAS PubMed.
- W. B. Davis, M. A. Ratner and M. R. Wasielewski, J. Am. Chem. Soc., 2001, 123, 7877–7886 CrossRef CAS PubMed.
- J. Sukegawa, C. Schubert, X. Zhu, H. Tsuji, D. M. Guldi and E. Nakamura, Nat. Chem., 2014, 6, 899–905 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Jaume Veciana and Prof. Concepció Rovira on occasion of their 70 birthdays. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc01160j |
| This journal is © The Royal Society of Chemistry 2021 |