
Kinetic stability modulation of polymeric nanoparticles for enhanced detection of influenza virus via penetration of viral fusion peptides†
Chaewon
Park
a,
Jong-Woo
Lim
a,
Geunseon
Park
a,
Hyun-Ouk
Kim
 b,
Sojeong
Lee
a,
Yuri H.
Kwon
a,
Seong-Eun
Kim
c,
Minjoo
Yeom
d,
Woonsung
Na
ef,
Daesub
Song
d,
Eunjung
Kim
*g and
Seungjoo
Haam
*a
b,
Sojeong
Lee
a,
Yuri H.
Kwon
a,
Seong-Eun
Kim
c,
Minjoo
Yeom
d,
Woonsung
Na
ef,
Daesub
Song
d,
Eunjung
Kim
*g and
Seungjoo
Haam
*a
aDepartment of Chemical and Biomolecular Engineering, Yonsei University, Seoul 03722, Republic of Korea. E-mail: haam@yonsei.ac.kr
bDivision of Chemical Engineering and Bioengineering College of Art, Kangwon National University, Chuncheon 24341, Republic of Korea
cHuvet Bio Inc, Seoul 05836, Republic of Korea
dCollege of Pharmacy, Korea University, Sejong 30019, Republic of Korea
eCollege of Veterinary Medicine, Chonnam National University, Gwangju 61186, Republic of Korea
fAnimal Medical Institute, Chonnam National University, Gwangju 61186, Republic of Korea
gDivision of Bioengineering, Incheon National University, Incheon 22012, Republic of Korea. E-mail: e.kim@inu.ac.kr
First published on 14th October 2021
Abstract
Specific interactions between viruses and host cells provide essential insights into material science-based strategies to combat emerging viral diseases. pH-triggered viral fusion is ubiquitous to multiple viral families and is important for understanding the viral infection cycle. Inspired by this process, virus detection has been achieved using nanomaterials with host-mimetic membranes, enabling interactions with amphiphilic hemagglutinin fusion peptides of viruses. Most research has been on designing functional nanoparticles with fusogenic capability for virus detection, and there has been little exploitation of the kinetic stability to alter the ability of nanoparticles to interact with viral membranes and improve their sensing performance. In this study, a homogeneous fluorescent assay using self-assembled polymeric nanoparticles (PNPs) with tunable responsiveness to external stimuli is developed for rapid and straightforward detection of an activated influenza A virus. Dissociation of PNPs induced by virus insertion can be readily controlled by varying the fraction of hydrophilic segments in copolymers constituting PNPs, giving rise to fluorescence signals within 30 min and detection of various influenza viruses, including H9N2, CA04(H1N1), H4N6, and H6N8. Therefore, the designs demonstrated in this study propose underlying approaches for utilizing engineered PNPs through modulation of their kinetic stability for direct and sensitive identification of infectious viruses.
Introduction
Viruses are nanosized pathogens that are entirely reliant on the host cell biosynthetic machinery to promote defined stages of their life cycles. The viruses display remarkable specific interactions with host cells, including attachment to cell surface receptors and fusion of their envelope lipid bilayers to target membranes, leading to subsequent viral replication pathways to cause infections.1–3 A representative example includes influenza virus infection, where the hemagglutinin (HA) surface glycoprotein mediates virus attachment and entry to target cells. In order for the influenza virus to replicate, HA protein first binds to sialic acid-presenting glycolipids or glycoproteins on the host membrane and mediates fusion of the host and viral membranes in acidic endosomal compartments. Notably, the fusion potential of HA is activated by cellular proteases and acidic pH, in which an inactive monomer of HA (HA0) is cleaved into a complex of heterodimers of HA1 (HA receptor-binding subunit) and HA2 (fusion subunit). The liberation of a fusion peptide sequestered in a pocket of HA2 at its N-terminus initiates a conformational transition of the HA1–HA2 complex, leading to the insertion of fusion peptides into the host membrane.4 This action brings contacts between the two opposing membranes and instigates the opening of a fusion pore, allowing the transfer of the viral genome into the host cell.5On the basis of the interplay between viruses and host cells, many attempts have been made to utilize nanomaterials that resemble such interactions to induce signals in the monitoring or diagnostic systems.6–10 Rationally designed nanomaterials offer versatile tools for highly efficient surface interactions with viruses in that they can serve as a host cell-mimicking system with unique features and the possibility of modulations such as modifying receptors,11–13 organizing valency and spacing of ligands on the nanoparticle surface,14–17 and tuning their size,18,19 and surface roughness.20–23 Their nanoscale size and high surface-to-volume ratio further strengthen the efficiency of nanomaterial-based approaches against viral infection due to enhanced surface interactions with viral receptors or lipid membranes.24
Notably, there have been reports on exploiting host cell membrane-mimicking nanomaterials for the direct detection of fusogenic viruses in real-time.25–27 These mimics include liposomes, polymeric vesicles, and natural cell membrane-decorated nanoparticles, presenting no sialic acid groups on their surface but enabling efficient virus trapping. This is because the fusion peptide forms amphiphilic helices at low pH and easily inserts its hydrophobic residues into the membrane structures.28 Their sensing approaches primarily rely on fluorescence signal changes derived from the penetration of viral fusion peptides into fluorophore-entrapped nanoparticles upon being exposed to specific conditions (e.g., low pH, enzymatic cleavage). Although various nanosystems to decoy fusogenic viruses have been developed for virus detection applications, they are limited to mimicking cellular membrane structures and compositions and lack of studying mutual interactions between viruses and nanoparticles to correlate the kinetic stability of nanoparticles with their sensing capability. Indeed, membrane destabilization due to the insertion of fusion peptides is a process that overcomes the substantial energy barriers arising from perturbing thermodynamically stably self-assembled structures, further imposing the importance of modulating the disassociation of nanoparticles.29–33 With this purpose, comprehensive analysis and control of the kinetic stability of nanoparticles and their behavior to withstand different stimuli that disrupt their membrane integrity are necessary to improve the detection performance of virus decoying-based nanosensors.
In this study, we developed a rapid and straightforward sensing system of an influenza A virus (IAV) using polymeric nanoparticles (PNPs) mimicking the host cell membranes. We chose IAV as a model pathogen that involves a pH-sensitive fusion process and sought to screen the resistance of PNPs against membrane disrupting agents to evaluate their kinetic stability. We modulated the morphology and membrane thickness of PNPs by varying the compositions of a polymer backbone and analyzed their structural integrity using a solubilization assay and atomic force microscopy (AFM)-based nanoindentation tests. We performed the detection assay by interacting self-assembled PNPs with activated IAVs under trypsin treatment at the reduced pH, promoting disaggregation of the polymer chains with the release of the entrapped fluorescent dye molecules and decreasing their Förster resonance energy transfer (FRET) signals (Fig. 1). We demonstrate that the PNP stability can be leveraged to enhance the penetration of amphiphilic fusion peptides, leading to highly amplified signals for the direct detection of viruses. The PNPs showing a susceptible membrane structure against the activated IAVs produced an enhanced detection signal compared to the PNPs with more resistant membranes. Therefore, analysis of the responses of PNPs to structure-disrupting stimuli provides insight into their kinetic stability, governing their sensitivity in the development of a virus detection system.
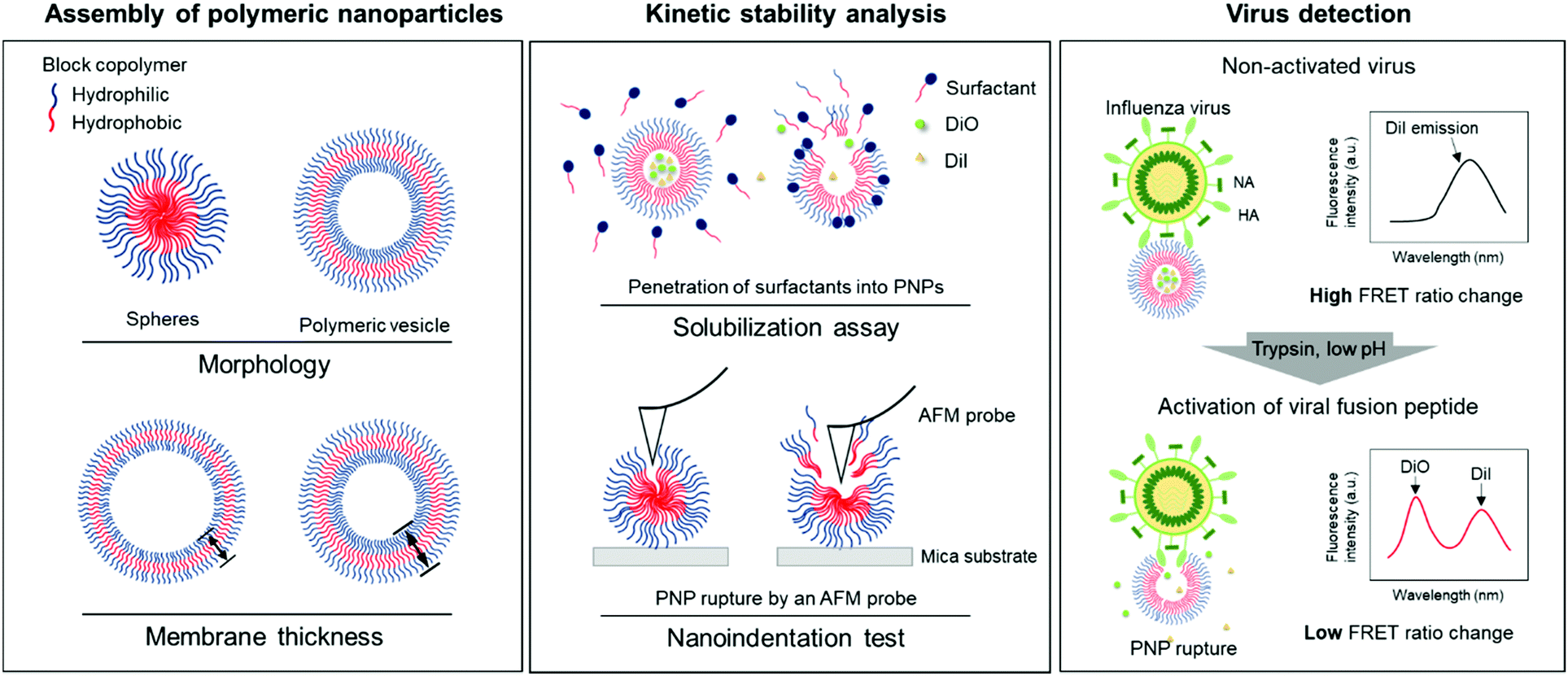 | ||
| Fig. 1 Schematic illustration of the assembly of polymeric nanoparticles (PNPs) and their kinetic stability analysis for virus detection. HA; hemagglutinin, NA; neuraminidase, FRET; Förster resonance energy transfer, PNP; polymeric nanoparticle, AFM; atomic force microscopy. | ||
Experimental
Materials
Methoxypolyethylene glycol amine with molecular weights of 1 kDa and 2 kDa was purchased from Laysan Bio (Arab, AL, USA). D,L-Leucine, triphosgene, anhydrous tetrahydrofuran (THF), anhydrous N,N-dimethylformamide (DMF), anhydrous chloroform, chloroform-d, trypsin from bovine pancreas, sodium acetate buffer solution (pH 5.2), and Triton™ X-100 were obtained from Sigma-Aldrich (Saint Louis, MO, USA). A cellulose ester (CE) membrane (Spectra/Por® Biotech CE Tubing, MWCO: 10 kDa) was purchased from Spectrum Labs (Los Angeles, CA, USA), and 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other chemicals and reagents were of analytical grade and used without further purification unless otherwise stated. 400-Mesh copper grids with formvar and carbon support films and the highest grade V1 AFM mica discs were purchased from Ted Pella, Inc. (Redding, CA, USA). The AFM cantilever (SD-R30-CONT) was purchased from Park Systems (Gyeonggi-do, South Korea).Synthesis of leucine-N-carboxyanhydride
Leucine-N-carboxyanhydride (Leu-NCA) was synthesized using triphosgene, as previously reported.42D,L-Leucine (3 g, 22.9 mmol) was dispersed in 80 mL of anhydrous tetrahydrofuran (THF) in a 250 mL three-necked round-bottom flask. Triphosgene (3.7 g, 12.6 mmol) was dissolved in 10 mL of THF, followed by addition to the D,L-leucine solution through dropwise injection using a syringe. The resulting suspension was reacted at 50 °C for 45 min under nitrogen and then cooled to room temperature. After the reaction, the THF was removed using a rotary vacuum evaporator, and the concentrated reaction solution was filtered with a 0.45 μm syringe filter, precipitated in 1 L of cold n-hexane, and stored at −20 °C overnight. The resulting solid precipitates were obtained by filtration through a cellulose filter paper with a pore size of 7 μm (ADVANTEC®, Japan) under reduced pressure, dried in a high vacuum chamber for 24 h, and stored at −20 °C for future use.Synthesis of methoxypolyethylene glycol-block-polyleucine copolymers
Methoxypolyethylene glycol-block-polyleucine (mPEG-b-pLeu) block copolymers were synthesized by ring-opening polymerization of Leu-NCA monomers with amine-terminated mPEG as a macroinitiator. To vary the hydrophilic fraction (f-values) in the copolymers, various molar ratios of Leu-NCA to mPEG (Leu-NCA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mPEG = 43.7, 22.5, and 12.6) were used. For the synthesis, mPEG (300 mg, 0.2 mmol) was dissolved in 5 mL of anhydrous N,N-dimethylformamide (DMF) in a 150 mL three-necked round-bottom flask and degassed by purging with nitrogen. The requisite amounts of Leu-NCA (6.6, 3.4, and 1.9 mmol) were dissolved in 5 mL of DMF and added to the mPEG solution dropwise using a syringe. Polymerization was carried out with a reflux condenser at 40 °C for 72 h under nitrogen. After completing the reaction, the mixture was precipitated in 1 L of cold diethyl ether, stored at −20 °C overnight, and isolated by removal of the solvent with filtration under reduced pressure. The polymer products were dried under a vacuum for 24 h and stored at −20 °C.
:mPEG = 43.7, 22.5, and 12.6) were used. For the synthesis, mPEG (300 mg, 0.2 mmol) was dissolved in 5 mL of anhydrous N,N-dimethylformamide (DMF) in a 150 mL three-necked round-bottom flask and degassed by purging with nitrogen. The requisite amounts of Leu-NCA (6.6, 3.4, and 1.9 mmol) were dissolved in 5 mL of DMF and added to the mPEG solution dropwise using a syringe. Polymerization was carried out with a reflux condenser at 40 °C for 72 h under nitrogen. After completing the reaction, the mixture was precipitated in 1 L of cold diethyl ether, stored at −20 °C overnight, and isolated by removal of the solvent with filtration under reduced pressure. The polymer products were dried under a vacuum for 24 h and stored at −20 °C.
Characterization of mPEG-b-pLeu
To analyze the chemical structure and molecular weight of each synthesized polymer, proton nuclear magnetic resonance (1H NMR) spectra were recorded on a 400 MHz spectrometer (Bruker AVANCETM III HD 400, Bruker BioSpin Corporation, Billerica, MA, USA) using chloroform-d as a solvent. The f-values of the polymers, defined as the ratio of the molecular weight of the hydrophilic PEG segments to the total molecular weight of the copolymer, were determined by NMR. The chemical shifts (δ) were given in ppm using tetramethyl silane as an internal standard. 1H NMR (400 MHz, CDCl3, δ, ppm): 0.92 (b, (CH3)2–CH–), 1.85 (b, (CH3)2–CHCH2–), 3.41 (t, –CH3), 3.55–3.80 (m, –OCH2CH2O–). Fourier transform infrared (FT-IR) spectra of the powder samples were recorded on a Spectrum TwoTM spectrometer (PerkinElmer, Inc., Waltham, MA, USA) using the attenuated total reflection method. Gel permeation chromatography (GPC) was performed using a YL9100 GPC System (Young-In Chromass, Gyeonggi-do, South Korea) equipped with a refractive index detector, autosampler, and C18-4D column (4.6 mm × 150 mm, 120 Å pore size). THF in HPLC grade was used as the mobile phase at a flow rate of 1 mL min−1. The columns were calibrated using commercial polystyrene standards. The polymer solutions (0.1 mg mL−1) prepared in THF were injected into the GPC system using an autosampler.Preparation of polymeric nanoparticles using mPEG-b-pLeu
Polymeric nanoparticles (PNPs) were prepared using a polymer thin film hydration method and tip sonication, as reported previously.43–45 Briefly, 10 mg of the mPEG-b-pLeu copolymer was dissolved in 1 mL of chloroform to prepare a 10 mg mL−1 polymer solution and mixed with 20 μL of DiO and DiI solutions (1 mg mL−1 in chloroform). The chloroform was eliminated using a vacuum rotary evaporator for 30 min to form thin films on the bottom of round-bottom flasks. The thin films were then entirely hydrated with 5 mL of deionized water by incubating at 60 °C for 6 h and magnetic stirring at 60 °C overnight. The dye-containing PNP dispersion was sonicated using a tip sonicator (VCX-750 Vibra Cell Processor, Sonics & Materials, Inc. Newtown, CT, USA) for 10 min to make them homogeneous and monodisperse. The prepared PNP solution was then placed into cellulose ester membrane tubes (molecular weight cutoff 5 kDa) and dialyzed against deionized water under mild stirring for 48 h to remove residual dyes.Characterization of PNPs
The hydrodynamic size of PNPs was measured by dynamic light scattering (DLS) at 25 °C using an ELSZ-2000ZS (Otsuka Electronics, Osaka, Japan). An average of three measurements was taken on 0.1 mg mL−1 PNP solution. The morphology of the PNPs was investigated by transmission electron microscopy (TEM) using a JEM-F200 (JEOL, Tokyo, Japan) at an accelerating voltage of 120 kV. The TEM samples were prepared by placing 10 μL of PNP dispersion on the formvar/carbon-coated 400-mesh copper grids (Ted Pella, USA). The samples were negatively stained using a 3% (w/v) aqueous neutral solution of phosphotungstic acid and dried overnight.Solubilization assay
The solubilization assay was performed using a non-ionic surfactant Triton X-100 (Tx-100). In this study, Tx-100 was used to destabilize the membrane or polymeric shell structures of a series of PNPs with various f-values. The assay was carried out by incubating the dye-incorporated PNPs (50 μL) with varying concentrations of Tx-100 (50 μL) from 0.01% to 10% (v/v) in deionized water for 20 min at room temperature to allow sufficient interaction between them. The fluorescence emission intensity spectra were recorded using a microplate reader (Spectra Max® i3x, Molecular Devices, LLC. Sunnyvale, CA, USA) with excitation at 475 nm. The FRET ratio is defined as the relative fluorescence intensity of an acceptor normalized to the total fluorescence intensity (FRET ratio = FIDiI/FItotal, where FItotal = FIDiO + FIDiI and FIDiO and FIDiI are the maximum fluorescence intensities of the DiO and DiI dyes at 504 and 574 nm, respectively). The normalized FRET ratio was calculated by defining the maximum and minimum FRET ratios as 100% and 0%, respectively. The critical molar ratios of Tx-100 to the polymer that initiate the rapid change of FRET signals were determined at the onset of PNP solubilization.Nanoindentation tests using atomic force microscopy
The local resistance of individual PNPs to the applied force by a cantilever tip at the nanoscale was assessed by atomic force microscopy (AFM)-based nanoindentation tests. For sample adsorption, the mica surfaces were freshly cleaved with Scotch tape. The PNP solution (1 mg mL−1) was deposited onto a mica surface and probed with an AFM microscope (NX-10, Park Systems, South Korea) using a silicon cantilever probe (SD-R30-CONT, Park Systems, South Korea) in a force–distance measurement mode. The approaching velocity of the tip was maintained at 500 nm s−1. For each PNP sample having different f-values, force–distance measurements were performed for more than 50 PNPs, and the critical force (Fc) required for the cantilever tip to rupture the assembled PNP structure was determined based on batch analysis of the obtained force–distance curves.Preparation of influenza viruses
The influenza A viruses (IAV), including three avian influenza H9N2 (A/Wild bird/Korea/JNU01-05/2012), H4N6 (A/Wild bird/Korea/JNU03-05/2015), H6N8 (A/Wild bird/Korea/JNU03-07/2015), and human influenza H1N1 (A/California/04/2009) were propagated in the allantoic cavity of 11 day-old embryonated chicken eggs. Each virus stock (100 μL) was inoculated into the allantoic cavities of chicken eggs. After 72 h incubation at 37 °C, eggs were chilled overnight at 4 °C. The allantoic fluid containing the propagated viruses was harvested and purified by centrifugation at 4000 rpm for 20 min. The supernatant was frozen at −80 °C for long-term storage and direct use in the assay.Detection of influenza a virus
The detection assay using FRET pairs-incorporated PNPs was carried out by adding the IAV solution with or without activation. Here, virus activation was performed by incubating 25 μL of viruses (102.026–103.833 TCID50 mL−1 of H9N2, 106.375 TCID50 mL−1 of CA04 (H1N1), 104.5 TCID50 mL−1 of H4N6, and 105.625 TCID50 mL−1 of H6N8) with 25 μL of trypsin (0.1 mg mL−1) for 20 min, to prepare for conformational changes under acidic conditions. The trypsin-treated viruses were then incubated with 25 μL of 300 mM sodium acetate buffer (pH 5.2) for an additional 20 min to induce conformational changes in the hemagglutinin of the activated fusion protein. For the assay, dye-containing PNPs (1 mg mL−1, 50 μL) were added to the activated virus solution (75 μL) in 60 mM sodium acetate buffer (pH 5.2) and incubated for 20 min at 37 °C. Inactivated virus samples and reaction buffer were used as controls. The fluorescence emission spectra were monitored using a microplate reader with excitation at 475 nm.The minimum FRET ratio (Rmin) was obtained from the FRET ratio of PNPs treated with 10% (v/v) Tx-100 (eqn (1)) and used to calculate the detection signal of each PNP. In eqn (1), Rx and Rdw refer to the FRET ratio changes of the PNPs under a given condition and in deionized water. As shown in eqn (2), the difference in FRET ratio changes of PNPs (N) between the activated and non-activated virus groups was defined as the detection signal (ΔN).
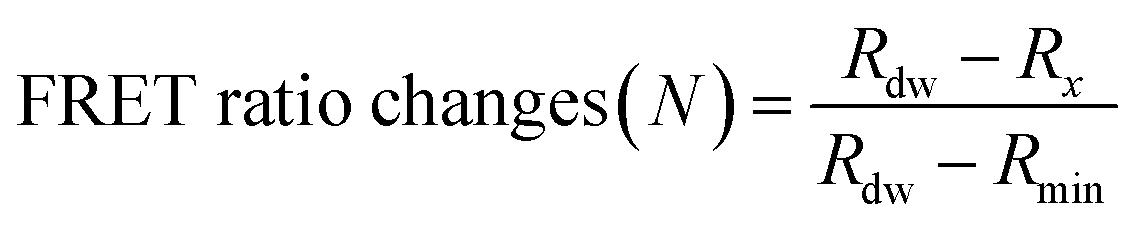 | (1) |
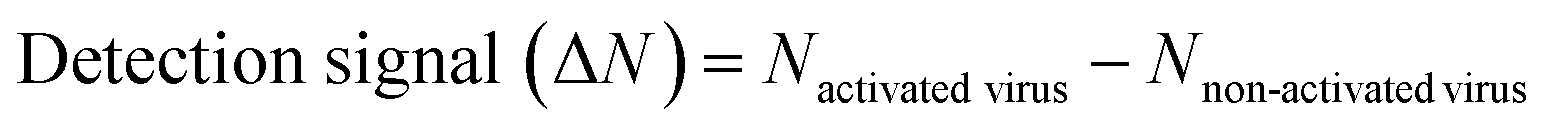 | (2) |
Results and discussion
Synthesis and characterization of mPEG-b-pLeu copolymers
The hydrophilic fraction (f-value) of the amphiphilic block copolymers, defined as the ratio of the molecular weight of the hydrophilic part to the total molecular weight, allows for determining the morphological features of the self-assembled polymeric nanoparticles (PNPs) in an aqueous solution.34–36 Previous studies have demonstrated that when the f-value of the amphiphiles is higher than 0.6 (i.e., the hydrophilic segments are dominant in copolymer chains), the polymers tend to form spherical nanoparticles. In contrast, when the relative portion of hydrophobic parts in the copolymers gradually increases and their f-value becomes less than 0.3, the nanoparticles spontaneously transform from spheres to bilayered vesicular structures with a hydrophilic aqueous core. The major driving force to self-assemble PNPs lies in the hydrophobic intra-particular interactions. This is a class of properties of nonpolar molecules (hydrophobic parts of amphiphilic molecules) and drives the molecules to constitute an anhydrous domain in an aqueous solution.37,38 Therefore, we hypothesized that amphiphilic copolymers with varying f-values or copolymer chain lengths would present sphere or vesicle-like structures with different thicknesses of brush-like polymeric shells or membranes, which would, in turn, modulate the kinetic stability of the resulting nanoparticles in the presence of surfactants or the response to the applied indentation force.39–41To obtain amphiphilic polymers with different f-values, methoxy poly(ethylene glycol)-block-polyleucine (mPEG-b-pLeu) copolymers were synthesized by ring-opening polymerization of N-carboxyanhydride (NCA), as described previously.42 We used mPEG with amino end groups as a hydrophilic macroinitiator for the polymerization of hydrophobic D,L-leucine monomers. Leucine-NCA (Leu-NCA) was first synthesized in the presence of triphosgene (Fig. S1, ESI†), and its subsequent polymerization reaction was then triggered by an mPEG initiator, giving rise to block copolymers with controlled repeating units of leucine molecules (Fig. S2a, ESI†). The reaction yield typically relies on various conditions, such as concentrations of macroinitiators and monomers, molecular weights of initiators, reaction time, and catalysts. To finely tune the chain length of the polymers, we chose 1 kDa and 2 kDa of mPEG and varied the relative molar ratios of monomers to initiators (Leu-NCA:mPEG = 3.3:1, 1.7:1, and 0.9:1). The chemical structures of the resulting copolymers were confirmed by Fourier transform-infrared (FT-IR) spectroscopy (Fig. S2b and e, ESI†). Their characteristic peaks appear at 3300 cm−1 and 1600–1650 cm−1, which is indicative of the formation of amide bonds driven by the elongation of leucine monomers. Gel permeation chromatography (GPC) elution profiles of mPEG and the synthesized copolymers were obtained with polystyrene calibrants to assess their polydispersity in molecular weights, suggesting that they show a good molecular weight distribution (<1.2 polydispersity index) (Fig. S2c and f, ESI†). The molecular weights of pLeu and mPEG-b-pLeu were determined by resolving the proton nuclear magnetic resonance (1H NMR) spectrum of mPEG-b-pLeu with the numerical integration of their characteristic proton peaks (i.e., δ = 3.55–3.80 ppm for methylene protons C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) – of PEG, and δ = 0.92 ppm for protons in methyl groups (C
– of PEG, and δ = 0.92 ppm for protons in methyl groups (C![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) )2– of the polyleucine chain).43–45 Three different molar ratios of Leu-NCA to mPEG (Leu-NCA:mPEG = 43.7, 22.5, and 12.6) were used to create copolymers with calculated f-values of 0.3, 0.45, and 0.6, respectively. Additionally, each mPEG-b-pLeu copolymer synthesized with 2 kDa of PEG exhibited approximately a two-fold larger molecular weight than that prepared with 1 kDa of PEG at the same f-value. Here, we refer to a series of synthesized block copolymers as mPEG-b-pLeu (molecular weight of mPEG, calculated f-value of PEG or fPEG), as summarized in Table 1.
)2– of the polyleucine chain).43–45 Three different molar ratios of Leu-NCA to mPEG (Leu-NCA:mPEG = 43.7, 22.5, and 12.6) were used to create copolymers with calculated f-values of 0.3, 0.45, and 0.6, respectively. Additionally, each mPEG-b-pLeu copolymer synthesized with 2 kDa of PEG exhibited approximately a two-fold larger molecular weight than that prepared with 1 kDa of PEG at the same f-value. Here, we refer to a series of synthesized block copolymers as mPEG-b-pLeu (molecular weight of mPEG, calculated f-value of PEG or fPEG), as summarized in Table 1.
| Block copolymer | MWa (Da) | f PEG | Polydispersity indexc (Đ) | Degree of polymerizationd |
|---|---|---|---|---|
| a Molecular weight of mPEG-b-pLeu copolymers obtained from 1H NMR spectra. b The hydrophilic fraction calculated by the molecular weight ratio of the PEG chain to the final copolymer, estimated from numerical integration of the proton peaks assigned to methylene (CH2–) and protons in methyl groups ((CH3)2–) in the 1H NMR spectra. c The polydispersity index of the copolymers confirmed by GPC. d The average degree of polymerization defined as the number of repeating leucine monomer units in the synthesized polymers and determined by 1H NMR spectra. | ||||
| mPEG-b-pLeu (1k, 0.3) | 3225.8 | 0.310 | 1.08 | 20 |
| mPEG-b-pLeu (1k, 0.45) | 2352.9 | 0.425 | 1.07 | 12 |
| mPEG-b-pLeu (1k, 0.6) | 1650.2 | 0.606 | 1.08 | 6 |
| mPEG-b-pLeu (2k, 0.3) | 6535.9 | 0.306 | 1.18 | 40 |
| mPEG-b-pLeu (2k, 0.45) | 4566.2 | 0.438 | 1.17 | 23 |
| mPEG-b-pLeu (2k, 0.6) | 3300.3 | 0.606 | 1.08 | 11 |
Preparation and characterization of PNPs
PNPs were prepared using polymer film hydration and tip sonication with slight modifications, as described previously.46,47 Briefly, a homogeneous thin film was prepared by evaporating the organic solvent (chloroform) containing amphiphilic mPEG-b-pLeu copolymers, hydrated with deionized water, and sonicated using a tip sonicator to enforce the self-assembly into homogeneous nanoparticles before purification with dialysis. We refer to the resulting PNPs along with the constituent block copolymer as PNP (molecular weight of mPEG, fPEG) as follows: PNP (1k, 0.3), PNP (1k, 0.45), PNP (1k, 0.6), PNP (2k, 0.3), PNP (2k, 0.45), and PNP (2k, 0.6). The sizes and morphologies of the prepared PNPs were analyzed using transmission electron microscopy (TEM) and dynamic light scattering (DLS). As shown in Fig. 2, the representative TEM images showed an appreciable difference in their morphology depending on the f-values of the copolymers, indicating that a low PEG fraction (∼30%) is required to assemble the corresponding polymers into vesicular structures. Indeed, a morphological transition of the PNPs was observed from unilamellar vesicles to spheres with an increasing fraction of PEG segments (ranging from ∼30% to ∼60%). Similar results were also found in the case of PNPs prepared using copolymers with 2 kDa of mPEG. Note that the TEM images showed the dehydrated structures of PNPs stained negatively with phosphotungstic acid, imposing that the observed contrasts did not perfectly represent their native structures in the working environment. These drying and staining procedures and imaging under high vacuum can affect the structure and morphology of the sample, thus necessitating extra caution in the interpretation of TEM images.48,49 Nonetheless, vesicular structures with hydrophobic bilayers were clearly seen in the TEM images of PNP (1k, 0.3) and PNP (2k, 0.3) (Fig. S2, ESI†). The formed vesicles or spheres showed a fairly monodisperse DLS intensity distribution (Fig. S3, ESI†). The average hydrodynamic size and polydispersity index (Đ) with the observed morphology from TEM images are summarized in Table 2. The PNPs analyzed by DLS in aqueous solution give hydrodynamic diameters that are slightly larger than number-based measurements with TEM because intensity-based DLS analysis weights large particles and shifts the average size towards a larger value than TEM-based measurements. This increase in size also indicates a significant hydration layer of the swollen state of PNPs in the aqueous phase.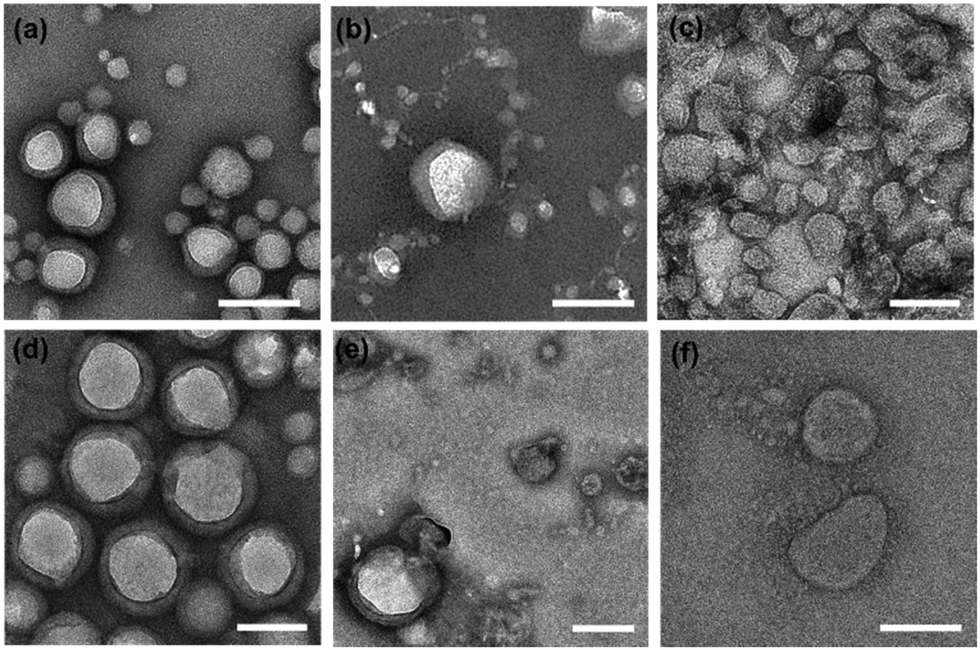 | ||
| Fig. 2 Representative TEM images of PNPs: (a) PNP (1k, 0.3), (b) PNP (1k, 0.45), (c) PNP (1k, 0.6), (d) PNP (2k, 0.3), (e) PNP (2k, 0.45), and (f) PNP (2k, 0.6). The samples were negatively stained with 3% (w/v) solution of neutral phosphotungstic acid (scale bar: 100 nm). | ||
| Type | Hydrodynamic diametera (nm) | Polydispersity indexa (Đ) | Morphologyb | Membrane thicknessc (nm) |
|---|---|---|---|---|
| a Hydrodynamic diameter determined by DLS with a polydispersity index (Đ). b Characteristic morphology observed from TEM images. c Thickness of bilayer membranes assessed from the TEM images. | ||||
| PNP (1k, 0.3) | 180.0 | 0.13 | Vesicles | 10.73 ± 3.38 |
| PNP (1k, 0.45) | 162.0 | 0.21 | Mixture of vesicles and spheres | — |
| PNP (1k, 0.6) | 149.0 | 0.24 | Spheres | — |
| PNP (2k, 0.3) | 246.5 | 0.16 | Vesicles | 19.21 ± 5.55 |
| PNP (2k, 0.45) | 206.9 | 0.23 | Mixture of vesicles and spheres | — |
| PNP (2k, 0.6) | 170.4 | 0.13 | Spheres | — |
Moreover, the structural changes seen in TEM imaging agree well with previous reports of the formulation of PNPs using PEG-based amphiphiles. The increase in the PEG fraction on the surface of vesicles caused an increase in the free energy of the vesicular membrane (mainly directed by hydrated, swollen PEG chains) and induced a transformation of vesicles to spheres that exhibit relatively large surface free energy. When assembling vesicles, their membrane thickness can be adjusted by varying the molecular weights of the copolymers.39–41 As shown in Fig. 2a and d, two types of vesicles, including PNP (1k, 0.3) and PNP (2k, 0.3), present unilamellar vesicular structures under TEM imaging. To investigate whether the molecular weight of the polymers alters the membrane thickness of the PNPs, we determined their average membrane thickness by direct measurement of the bilayer in TEM images of more than 30 PNPs. As a result, the membrane thickness of PNP (1k, 0.3) and PNP (2k, 0.3) appear to be approximately 10.7 ± 3.4 nm (n = 31) and 19.2 ± 5.6 nm (n = 31), respectively, showing a significant difference in their membrane thickness (Fig. S4, ESI†).
Evaluation of kinetic stability of PNPs using solubilization assay
We then explored the ability of the PNPs to withstand external amphiphilic stimuli that perturb the arrangement and packing of polymer chains constituting the nanoparticles, which is indicative of the kinetic stability of their structures. To do so, we employed a Triton X-100 (Tx-100)-based solubilization assay that measures the resistance of PNPs to a surfactant. Here, Tx-100 as a non-ionic surfactant is the most widely used detergent in membrane studies, which is classically applied to destabilize lipid membranes in biological compartments or liposomes because it can rapidly and favorably insert between lipid bilayers, causing membrane solubilization.50–52 The surfactant-driven solubilization has been extensively studied with a mixture of lipids and surfactants in an aqueous solution.53,54 The solubilization of lipid bilayered structures by surfactants can be explained by the three-stage model addressing common features of the kinetic changes in lipid bilayers with the stepwise addition of surfactants to lipid dispersions.55 As depicted in Fig. 3a, the surfactants are first inserted into the lipid bilayer when the concentration of surfactants is sufficiently low (stage I). As increasing the number of surfactants, the surfactant-saturated bilayer is disintegrated and begins to form micelles (stage II, at which solubilization starts to take place). Finally, complete solubilization of the bilayer into mixed micelles formed by both lipids and surfactants occurs with additional surfactants. This solubilization kinetics of lipid or polymer vesicles induced by surfactants can be investigated using fluorescence spectroscopy analysis.56–58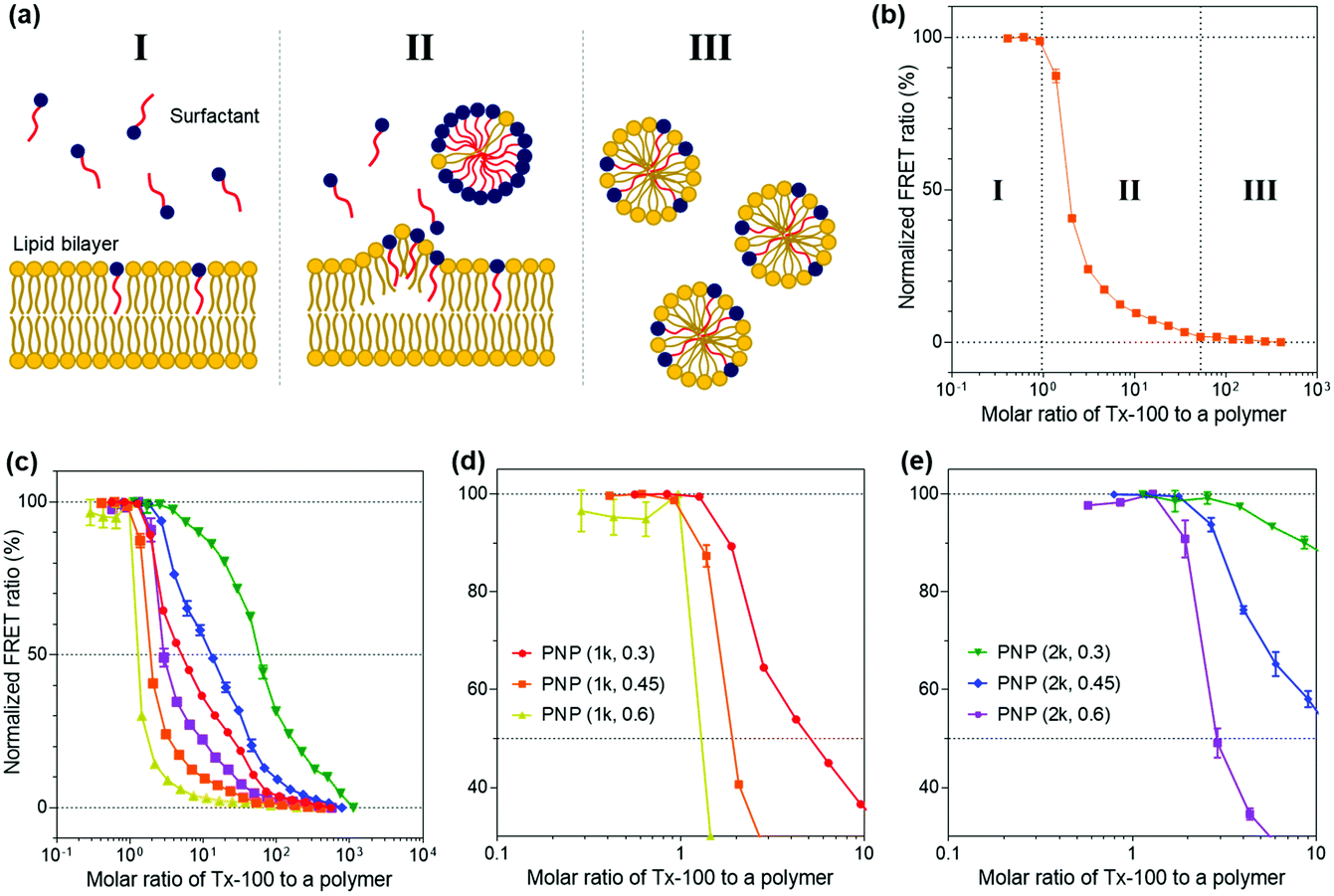 | ||
| Fig. 3 Evaluation of kinetic stability of various PNPs containing DiO and DiI FRET pairs using the solubilization assay. (a) Schematic of a three-stage model showing membrane solubilization by amphiphilic surfactants. (b) The changes in normalized FRET ratios in the corresponding stage. (c–e) Normalized FRET ratios of a series of PNPs with increasing amounts of Tx-100 after 20 min: FRET signal profiles of (c) all PNPs, and magnified profiles of (d) PNP (1k) and (e) PNP (2k) in the range of 0.1 to 10 molar ratio of Tx-100 to a polymer. The maximum and minimum FRET ratios were normalized to 100% and 0% FRET ratios, respectively. All fluorescence measurements were carried out after 20 min incubation with Tx-100 at room temperature. Data represent mean ± standard deviation (n = 3). | ||
In the context of our study, upon the addition of Tx-100, the integrity of PNP membranes or polymeric shells becomes weakened and solubilized. The Tx-100-induced solubilization can be assessed by FRET-based signal readouts that are mediated by a DiO (as a donor) and DiI (as an acceptor) pair encapsulated in PNPs.59,60 In other words, fewer FRET pairs remain within PNP constructs after treatment with an increasing amount of Tx-100, reducing the FRET ratio. Here, the FRET ratio is defined as the relative fluorescence intensity of an acceptor normalized to the total fluorescence intensity (FRET ratio = FIDiI/FItotal, where FItotal = FIDiO + FIDiI and FIDiO and FIDiI are the fluorescence intensity maxima of the DiO and DiI dyes at 504 and 574 nm with excitation at 475 nm, respectively) (Fig. 3b). We also sought to differentiate the extent of incorporation of Tx-100 into the PNPs by varying its concentration from 0.01% to 10% (v/v), which is below its critical micelle concentration (13.3% (v/v) or 0.22 mM in water).61 This study is likely to resemble the penetration of amphiphilic viral fusion peptides into the host cell's membrane and further allows us to examine their resistance capacity against the disruptive force of Tx-100. Membrane fusion of influenza A virus (IAV) as a model pathogen in our study is driven by low pH-triggered exposure of the hemagglutinin (HA) fusion peptides derived from the N-terminal region of the HA2 subunit. The fusion peptide is thought to form amphiphilic α-helical conformation upon binding to the membrane, penetrate the lipid bilayer, and destabilize the bilayer structure to induce fusion.28,62,63 Thus, we thought that solubilization of PNPs by Tx-100 surfactants is likely to feature the ability of amphiphilic helical fusion peptides to penetrate the host membrane.
We first tested the FRET ratios of PNPs after incubation with 10% (v/v) of Tx-100 for 20 and 40 min to ensure that the minimum FRET ratio was the lowest signal within this assay and to confirm whether the 20 and 40 min is enough to reach the equilibrium state of PNP disassociation. As shown in Fig. S5 (ESI†), the FRET ratios reached approximately ca. 0.2–0.3, and there were no significant changes in the FRET ratios over time. Therefore, we considered incubation with 10% (v/v) Tx-100 for 20 min as the optimal condition required for complete disintegration of the PNPs and consequently used the FRET signals at this condition as the minimum signal (0% FRET). We also used the maximum FRET signals with Tx-100 producing a negligible change in acceptor emission as the maximal signal (100% FRET) to normalize the obtained sample signals. Fig. S6a and b (ESI†) show the representative fluorescence emission spectral changes of PNP (1k, 0.3) after adding varied concentrations of Tx-100. With increasing amounts of Tx-100, the spectra show a remarkable increase in donor emission at 504 nm while the acceptor emission at 574 nm is reduced, confirming the disassociation of the FRET dyes due to Tx-100-triggered solubilization of PNPs.
Grounded on the three-stage model of lipid solubilization by surfactants, we could draw three apparent stages from the results of PNP's FRET signal changes as a function of Tx-100 concentration. Notably, the PNPs started to elute the encapsulated fluorescent dyes when Tx-100 reached the critical concentration, entering the solubilization stage (at which the polymer membrane or shell cannot accommodate more surfactants) and forming the mixed micelles of surfactants and polymers (stage II in Fig. 3b). Fig. 3c–e and Fig. S6c–e (ESI†) showed the solubilization assay results after 20 and 40 min incubation with the gradual addition of Tx-100 to PNPs, respectively. We further estimated the critical molar ratios of surfactants to the polymer that initiate the rapid change of FRET signals (defined as stage II in our results), allowing us to assess the capability of PNPs to endure the penetration of amphiphilic stimuli. The surfactant concentrations causing the onset of solubilization of individual PNPs were found to be 1.261 for PNP (1k, 0.3), 0.920 for PNP (1k, 0.45), 0.287 for PNP (1k, 0.6), 2.555 for PNP (2k, 0.3), 1.785 for PNP (2k, 0.45), and 1.290 for PNP (2k, 0.6). These results showed that the higher f-values the PNPs have, the lower critical surfactant ratios they obtained, implying sphere-like PNPs were more prone to surfactant penetration and solubilization than vesicle-like ones. Therefore, sphere-like PNPs showed reduced kinetic stability and sensitive deformation towards structure-disrupting agents than those with polymer membranes. From these data, it is expected for PNP spheres to have substantial interactions with viruses through the penetration of HA fusion peptides, which can further maximize their sensing performance. Another interesting point will be to explore the rate of fluorescence signal changes of PNPs over time depending on their polymer composition. This will ascertain how fast these PNPs can be disassembled against structure-disrupting amphiphilic molecules and further estimate how rapidly the viruses can be detected with various types of PNPs.
AFM-based nanoindentation tests
While the solubilization assay using surfactants suggests kinetic stability of PNPs in the presence of amphiphilic stimuli at the ensemble level (i.e., bulk measurements), atomic force microscopy (AFM)-based nanoindentation measurements enable us to extract the response of nanostructured polymer materials at the nanoscale.64–68 To characterize their deformability and compliance upon indentation of the nanosized edge of an AFM probe, we obtained force–distance curves for individual PNPs immobilized on a mica substrate using a silicon cantilever tip. In each trial, the cantilever tip approached a sample from a height of 3 μm at a velocity of 500 nm s−1. As depicted in Fig. 4a, consecutive linear force responses were obtained for a single PNP (1k, 0.3) upon indentation instead of a single linear regime.65 It is theorized that the mechanical response of the outer layer of the membrane (or shell) was initially associated, and the elastic deformation is then transmitted to the PNP that overcomes the intermolecular forces holding the polymer chains together. In the second stage, the AFM tip ruptured the PNP to the underlying mica substrate until the applied force reached a threshold value, followed by repulsion between the tip and mica substrate. In this force–distance curve, we can measure the critical force (Fc) required for the AFM tip to penetrate through the polymer layers or brushes of the PNP at a breakthrough point.68–70 In other words, the PNP is resistant to exterior mechanical force until the Fc is applied. Therefore, determining Fc for each PNPs enabled us to examine their resistance capacity against the disruptive force.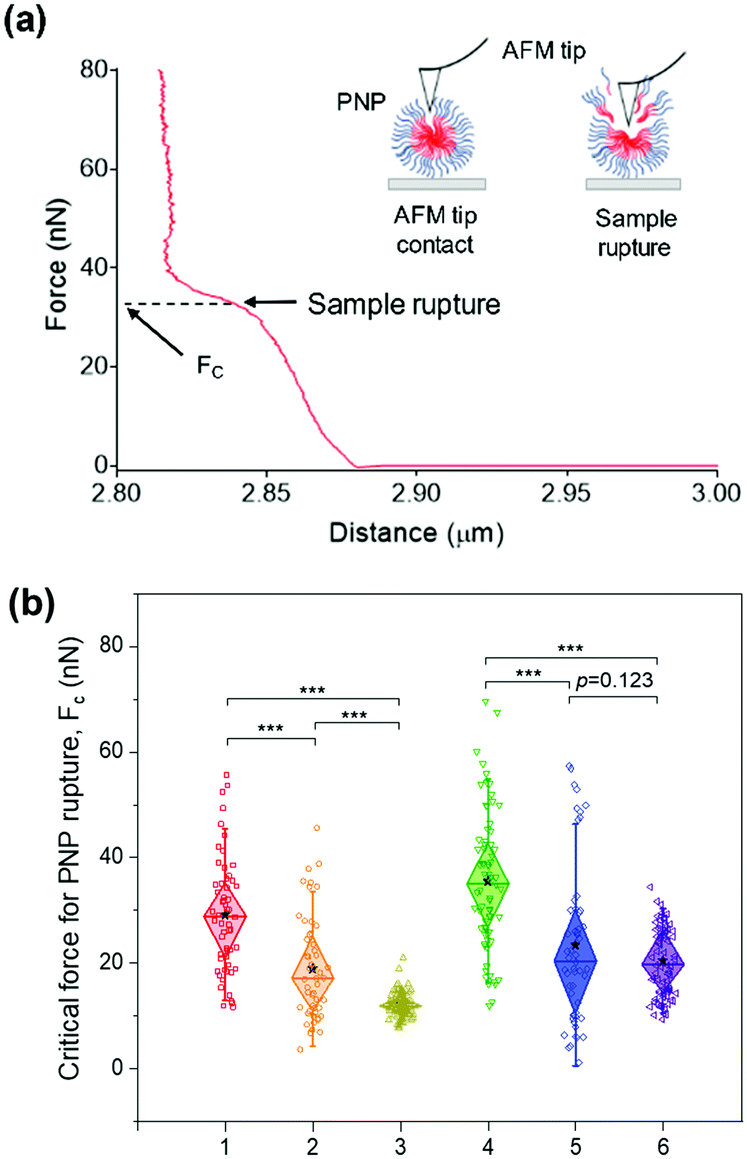 | ||
| Fig. 4 AFM-based nanoindentation tests of PNPs. (a) A representative force–distance curve to measure the critical force (Fc) required for the AFM tip to rupture a single PNP (1k, 0.3). (b) The average Fc values for each PNP formulation. Sample are: 1: PNP (1k, 0.3), 2: PNP (1k, 0.45), 3: PNP (1k, 0.6), 4: PNP (2k, 0.3), 5: PNP (2k, 0.45), and 6: PNP (2k, 0.6). Box and whisker plots represent median values (central lines), 25 and 75% quartile ranges around the median (diamond), mean values (asterisk), and standard deviation (whisker). Unpaired Student's t-test and one-way analysis of variance (ANOVA) were used for statistical analysis of the measured critical force between the groups. (***p < 0.001, n for each sample: n(1) = 57; n(2) = 51; n(3) = 108; n(4) = 73; n(5) = 52; n(6) = 80). | ||
The average Fc values for at least 50 PNPs per condition were determined to be 29.01 ± 10.76 nN for PNP (1k, 0.3), 18.74 ± 9.68 nN for PNP (1k, 0.45), 12.05 ± 2.19 nN for PNP (1k, 0.6), 35.35 ± 12.81 nN for PNP (2k, 0.3), 23.28 ± 15.12 nN for PNP (2k, 0.45), and 20.29 ± 6.56 nN for PNP (2k, 0.6) (Fig. 4b and Fig. S7, ESI†). Statistical analysis revealed that the difference between Fc of PNPs with f-values of 0.3 and 0.6 is significant (p-value < 0.001). Importantly, PNP (1k, 0.3) shows a 2.41-fold higher force response to rupture than PNP (1k, 0.6), which agrees well with the results obtained from the solubilization assay. Likewise, the PNPs with 2 kDa PEG showed an f-value dependence on the Fc magnitude, leading to PNP rupture. This substantial difference in the Fc of PNPs with different f-values and lengths of PEG chains makes it possible to predict the fate of PNPs when interacting with amphiphilic HA fusion peptides and relate their reactivity to the capability to detect IAVs.
Influenza virus detection with PNPs
Having successfully demonstrated the kinetic stability and mechanical responses of the prepared PNPs, we explored their application in influenza virus detection assays. The detection system relies on a low-pH- and enzyme-induced irreversible conformational change of a HA fusion protein of the IAV to the fusion-active state and the FRET signals of PNPs produced by their reduced kinetic stability upon interaction with IAV fusion peptides.71,72 It is important to note that the release of a fusion peptide is triggered by proteolytic cleavage of HA and reduced pH, not necessarily by binding with sialic acid receptors.25,73,74 In this regard, when the HA-containing molecules (e.g., full-length HA, influenza virus-like particles, etc.) were activated with trypsin and reduced pH, there was a remarkable increase in fusion interactions between HA and nanoparticles presenting no sialic acids or other ligands. This is because the fusion peptide forms amphiphilic helices and easily inserts its hydrophobic residues into the target membrane.Inspired by such a critical molecular step underlying virus fusion, we applied PNPs to the H9N2 IAV in the allantoic fluids of infected embryonated chicken eggs. For the preparation of activated viruses, the allantoic fluid samples were pretreated with trypsin (0.02 mg mL−1) and acidic buffer (60 mM sodium acetate buffer, pH 5.2), inducing insertion of the HA fusion peptide into the PNP membrane and releasing the FRET dyes via structural deformation of the PNPs. In this experiment, we obtained FRET ratio changes (N) by calculating the relative changes in FRET ratios of PNP-treated viruses and 10% (v/v) Tx-100 (as a minimum FRET ratio), normalized with respect to the maximal changes in FRET ratios (see Experimental Section for details of calculation). Here a 100% FRET ratio change (N = 100%) indicates complete disassembly of the PNPs. Fig. 5a shows the obtained FRET ratio changes of each PNP formulation after incubation with activated and non-activated viruses and reaction buffer containing trypsin and acidic buffer. The PNPs displayed significantly different levels of FRET signal changes when exposed to activated viruses presenting fusion peptides. In contrast, either non-activated viruses or enzyme- and pH-adjustments did not cause any marked increase in FRET signals of PNPs with the exception of PNP (1k, 0.6), which could be attributed to their lowest structural stability, as seen in Fig. 3c and 4b. Notably, the allantoic liquid collected from infected eggs presents a biological medium containing different proteins, indicating that our sensing design is capable of directly detecting IAV in a biologically relevant matrix with minimal sample preparation. The negative N values observed in reaction buffer conditions (negative controls) also indicate a larger FRET ratio of the PNPs than the PNPs dispersed in deionized water, implying hampered dispersion of the particles via the moderate ionic strength of the pH buffer.
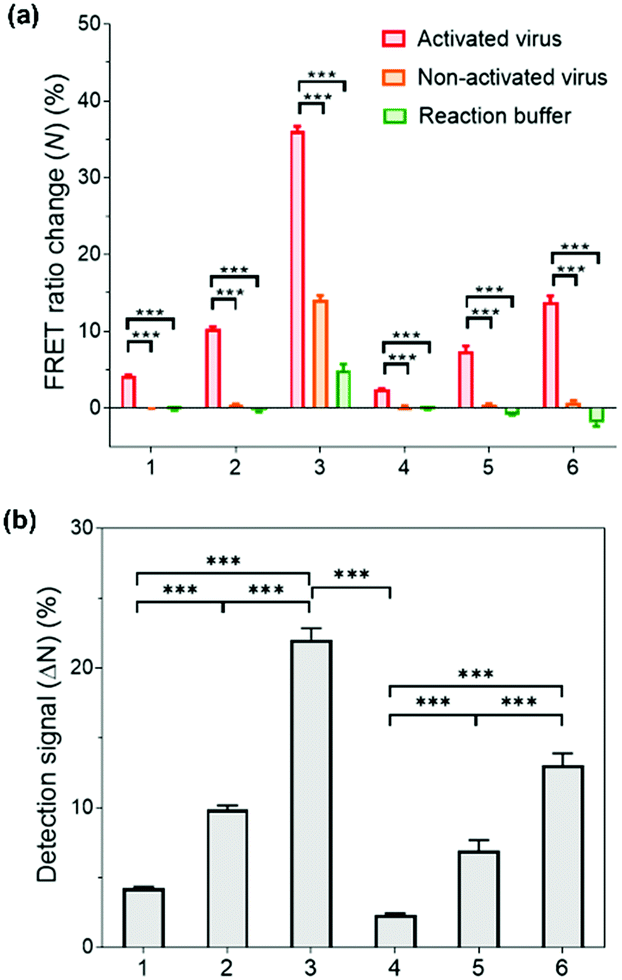 | ||
| Fig. 5 Influenza virus detection with PNPs. (a) FRET ratio chagnes (N) after incubation with H9N2 viruses (103.833 TCID50/mL). Each PNP was incubated with activated viruses in the presence of trypsin and acidic buffer (red), with non-activated viruses in the absence of trypsin and acidic buffer (orange), and in a solution containing trypsin and acidic reaction buffer (green). (b) Detection signals obtained from the PNPs with H9N2 viruses. The average detection signals (ΔN) were obtained by subtracting FRET ratio changes (N) of the activated groups from non-activated virus groups. Samples are: 1: PNP (1k, 0.3), 2: PNP (1k, 0.45), 3: PNP (1k, 0.6), 4: PNP (2k, 0.3), 5: PNP (2k, 0.45), and 6: PNP (2k, 0.6). Box plots represent mean ± standard deviation (n = 3). Student's t-test was used for statistical analysis of the measured FRET ratio changes and detection signals between the groups (***p < 0.0001). | ||
To compare the detection capacity of the PNPs in a reasonable manner, we refined the dataset of Fig. 5a by calculating the difference in FRET ratio changes between the activated and non-activated virus groups, defined as a detection signal (ΔN). As shown in Fig. 5b, PNP (1k, 0.6) and PNP (2k, 0.6) displayed a 5.22-fold and 5.69-fold higher detection signal than PNP (1k, 0.3) and PNP (2k, 0.3), respectively, implying that micelles are likely to be more deformable and produce more significant signals than vesicles. In other words, the PNPs with higher f-values and smaller lengths of PEG chains in the copolymer exhibit higher detection signals, suggesting that the kinetic stability of the PNPs is inversely related to the degree of disassembly of their structure upon reacting with activated IAVs. We note that these results clearly match previous results showing the mechanical responses of various PNPs to the addition of Tx-100 solution and indentation with AFM tips. For example, PNP (1k, 0.6) with more responsive micellar structures showed a ca. 9.6-fold higher detection signal for H9N2 IAV than PNP (2k, 0.3) with rigid and bilayered thick membranes. Therefore, modulating the unique features of nanosystems can lead to finely tuned, nanoparticle-based sensing platforms with virus-sensitive structures, providing an optimal selection of PNP structures (e.g., PNP (1k, 0.6)). The effectiveness of the developed detection system was further carried out with other subtypes of IAVs, including one human IAV (H1N1) and two avian IAVs (H4N6 and H6N8) (Fig. S8, ESI†). The observed detection signals are in line with the results obtained from H9N2 virus detection, demonstrating that our HA fusion-peptide-responsive nanosystem offers great potential in identifying a range of enveloped viruses with class I fusion proteins.
Sensitivity of IAV detection with PNPs
Finally, we tested the sensitivity of the detection assay using PNPs towards various concentrations of the H9N2 IAV. For the assay, the allantoic fluid containing the virus was pretreated with 0.1 mg mL−1 trypsin for 20 min to trigger the cleavage of HA proteins, followed by incubation in 300 mM sodium acetate buffer (pH 5.2) for an additional 20 min to expose the fusion peptides on the surface of the virus. Each PNP formulation (1 mg mL−1) with encapsulated FRET pairs was added to the activated virus samples at various virus titers (102.026–103.833 TCID50 mL−1). After incubation at 37 °C for 20 min, the fluorescence emission signals were recorded at 475 nm excitation on a microplate reader, and their detection signals (ΔN) were obtained by assessing the FRET ratios and FRET ratio changes (N), respectively. After fitting the detection signal data to the linear dose–response curve, the limit of detection (LOD) was determined based on the three-sigma (3σ) method, providing the lowest concentration of virus that yields a signal three times higher than the standard deviation (σ) of the control (Fig. 6 and Table 3). As expected, PNP (1k, 0.6), having the highest deformability among other PNPs, shows the most responsive behavior against the fusion peptides, offering a highly improved limit of detection of 102.082 TCID50 mL−1. These results confirm that the assay using FRET pair-incorporated PNPs allows simple, rapid detection of the presence of a fusion-activated virus with enhanced sensitivity within half an hour. In contrast to other virus detection methods such as polymerase chain reaction (PCR), our approach reduces complicated sample preparation and operation steps and shortens measurement time. It also provides a simple experimental workflow with a short detection time and comparable sensitivity compared to recently reported detection methods for influenza virus detection (Table S1, ESI†).75–77 More importantly, the insight gained into the mechanical properties and interaction of soft nanomaterials with viruses provides an important methodology for the systematic design of a nanomaterial-based sensing system.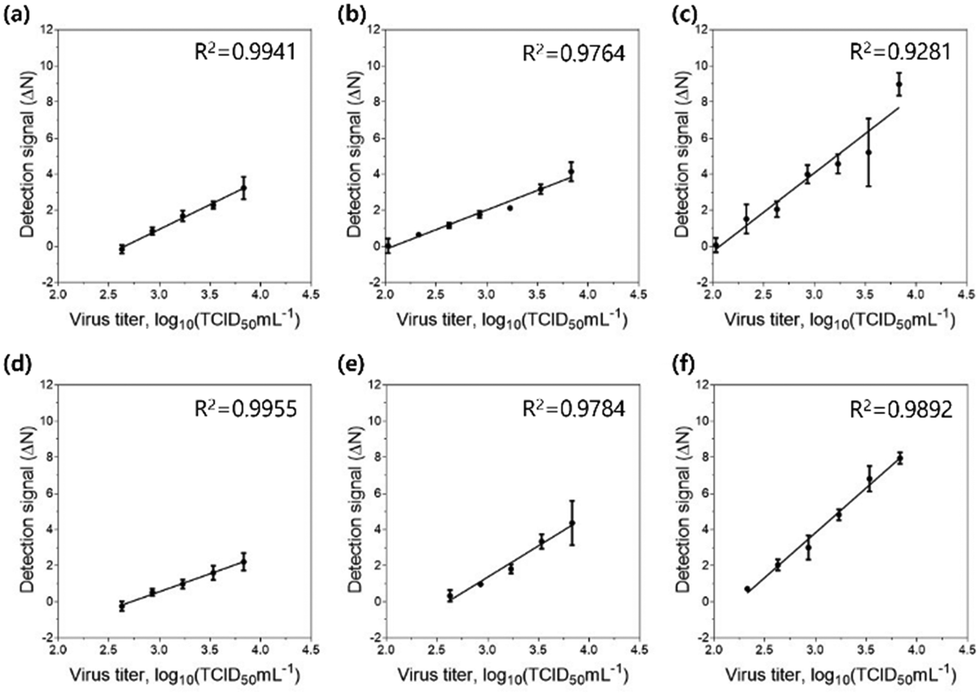 | ||
| Fig. 6 Detection of H9N2 influenza virus using PNPs. Assay sensitivity with (a) PNP (1k, 0.3), (b) PNP (1k, 0.45), (c) PNP (1k, 0.6), (d) PNP (2k, 0.3), (e) PNP (2k, 0.45), and (f) PNP (2k, 0.6) after incubation with various titers of H9N2 at 37 °C for 20 min. Data represent mean ± standard deviation (n = 3). | ||
Conclusions
A thorough understanding of the interactions of polymer-based nanomaterials with viruses as biological entities provides a valuable methodology for designing virus detection assays. In this study, we developed a host cell-mimetic fluorescence assay with the insightful use of PNPs for the detection of IAVs. Our sensing approach is based on the fluorescence signal readouts of FRET pairs released from disassembled PNPs that are driven by the insertion of the exposed fusion peptides of an activated IAV. This strategy utilizes the fusion mechanism of the IAV with host cells, where its HA fusion peptide acts partly as an amphiphilic helix and interacts with the target membrane to induce local structural disruption. By altering the hydrophilic fraction and molecular weight of the copolymers, we formulated a set of PNPs with varying morphology and membrane thickness and showed that their behavior against penetrating and disrupting forces could be effectively analyzed using the Tx-100-based solubilization assay and AFM-based nanoindentation studies. Thus, the PNPs having more flexible, monolayered membrane structures exhibited increased responsiveness to membrane-disrupting stimuli, leading to higher detection signals than relatively rigid and bilayered PNPs. We also demonstrated that the prepared PNPs generate positive signals for four types of IAVs, such as H9N2, CA04(H1N1), H4N6, and H6N8, activated by a short incubation with trypsin and reduced pH. We observed that the most responsive PNPs achieved a limit of detection of 102.082 TCID50 mL−1 using a laboratory microplate reader, which was approximately 4.95-fold lower than that of the most stable PNPs. These findings serve to guide the design and investigation of the effects of the kinetic stability of nanoparticles on nano-bio interaction. Therefore, our system has significant potential in the development of new diagnostic nanoplatforms by providing mechanistic and kinetic insights into responses associated with viral fusion peptides.Author contributions
C. Park, J.-W. Lim and G. Park developed the basic concept and analyzed the data. C. Park conducted the experiments and wrote the paper. H.-O. Kim, S. Lee, and S.-E. Kim directed and performed the analysis of experiments and quality of control samples. Y. H. Kwon assisted in conducting experiments and writing the paper. M. Yeom, W. Na, and D. Song advised the biological aspect of virus detection and provided stocks and information of influenza virus. E. Kim and S. Haam revised the paper, contributed to supervision and directed the project. All authors reviewed the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. K. acknowledges support from the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (no. 2020R1F1A1066247). S. H. acknowledges support from the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (no. 2017M3A7B4041798) and by Korea Environment Industry & Technology Institute(KEITI) funded by Korea Ministry of Environment(MOE)(RE202101004).References
- S. Belouzard, J. K. Millet, B. N. Licitra and G. R. Whittaker, Viruses, 2012, 4, 1011–1033 CrossRef CAS PubMed.
- M. E. Burlone and A. Budkowska, J. Gen. Virol., 2009, 90, 1055–1070 CrossRef CAS PubMed.
- Y. Matsuoka, H. Matsumae, M. Katoh, A. J. Eisfeld, G. Neumann, T. Hase, S. Ghosh, J. E. Shoemaker, T. J. Lopes, T. Watanabe, S. Watanabe, S. Fukuyama, H. Kitano and Y. Kawaoka, BMC Sys. Biol., 2013, 7, 1–18 CrossRef PubMed.
- M. Kielian, Annu. Rev. Virol., 2014, 1, 171–189 CrossRef PubMed.
- S. C. Harrison, Nat. Struct. Mol. Biol., 2008, 15, 690–698 CrossRef CAS PubMed.
- E. Vanderlinden and L. Naesens, Med. Res. Rev., 2014, 34, 301–339 CrossRef CAS PubMed.
- L. Rao, R. Tian and X. Chen, ACS Nano, 2020, 14, 2569–2574 CrossRef CAS PubMed.
- A. Knodler, J. Celli and B. B. Finlay, Nat. Rev. Mol. Cell Biol., 2001, 2, 578–588 CrossRef PubMed.
- D. A. Bricarello, M. A. Patel and A. N. Parikh, Trends Biotechnol., 2012, 30, 323–330 CrossRef CAS PubMed.
- B. Kong, S. Moon, Y. Kim, P. Heo, Y. Jung, S.-H. Yu, J. Chung, C. Ban, Y. H. Kim and P. Kim, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- S. Song, K. Ha, K. Guk, S.-G. Hwang, J. M. Choi, T. Kang, P. Bae, J. Jung and E.-K. Lim, RSC Adv., 2016, 6, 48566–48570 RSC.
- X. Wei, G. Zhang, D. Ran, N. Krishnan, R. H. Fang, W. Gao, S. A. Spector and L. Zhang, Adv. Mater., 2018, 30, 1802233 CrossRef PubMed.
- L. Rao, W. Wang, Q.-F. Meng, M. Tian, B. Cai, Y. Wang, A. Li, M. Zan, F. Xiao, L.-L. Bu, G. Li, A. Li, Y. Liu, S.-S. Guo, X.-Z. Zhao, T.-H. Wang, W. Liu and J. Wu, Nano Lett., 2018, 19, 2215–2222 CrossRef PubMed.
- C. Lee, J. Jeong, T. Lee, W. Zhang, L. Xu, J. E. Choi, J. H. Park, J. K. Song, S. Jang and C.-Y. Eom, Biomaterials, 2018, 183, 234–242 CrossRef CAS PubMed.
- J. Ingale, A. Stano, J. Guenaga, S. K. Sharma, D. Nemazee, M. B. Zwick and R. T. Wyatt, Cell Rep., 2016, 15, 1986–1999 CrossRef CAS PubMed.
- H. Chun, M. Yeom, H.-O. Kim, J.-W. Lim, W. Na, G. Park, C. Park, A. Kang, D. Yun and J. Kim, Polym. Chem., 2018, 9, 2116–2123 RSC.
- S.-J. Kwon, D. H. Na, J. H. Kwak, M. Douaisi, F. Zhang, E. J. Park, J.-H. Park, H. Youn, C.-S. Song, R. S. Kane, J. S. Dordick, K. B. Lee and R. J. Linhardt, Nat. Nanotechnol., 2017, 12, 48–54 CrossRef CAS PubMed.
- S. Gurunathan, M. Qasim, Y. Choi, J. T. Do, C. Park, K. Hong, J.-H. Kim and H. Song, Nanomaterials, 2020, 10, 1645 CrossRef CAS PubMed.
- Y. Mori, T. Ono, Y. Miyahira, V. Q. Nguyen, T. Matsui and M. Ishihara, Nanoscale Res. Lett., 2013, 8, 1–6 CrossRef PubMed.
- Y. Niu, M. Yu, S. B. Hartono, J. Yang, H. Xu, H. Zhang, J. Zhang, J. Zou, A. Dexter and W. Gu, Adv. Mater., 2013, 25, 6233–6237 CrossRef CAS PubMed.
- R. Y. Siddiquie, A. Agrawal and S. S. Joshi, Trans. Ind. Natl. Acad. Eng., 2021, 1–11 Search PubMed.
- A. Piloni, C. K. Wong, F. Chen, M. Lord, A. Walther and M. H. Stenzel, Nanoscale, 2019, 11, 23259–23267 RSC.
- Y. Niu, M. Yu, A. Meka, Y. Liu, J. Zhang, Y. Yang and C. Yu, J. Mater. Chem. B, 2016, 4, 212–219 RSC.
- F. Li, J. Lu, X. Kong, T. Hyeon and D. Ling, Adv. Mater., 2017, 29, 16505897 Search PubMed.
- H. O. Kim, W. Na, M. Yeom, J. Choi, J. Kim, J. W. Lim, D. Yun, H. Chun, G. Park and C. Park, Adv. Funct. Mater., 2018, 28, 1800960 CrossRef PubMed.
- D. M. Jones and S. Padilla-Parra, Sci. Rep., 2015, 5, 1–10 Search PubMed.
- L. Gui and K. K. Lee, Influenza Virus-Liposome Fusion Studies Using Fluorescence Dequenching and Cryo-electron Tomography. Influenza Virus, Springer, 2018, pp. 261–279 Search PubMed.
- S. T. Smrt, A. W. Draney and J. L. Lorieau, J. Biol. Chem., 2015, 290, 228–238 CrossRef CAS PubMed.
- P. Guo, D. Liu, K. Subramanyam, B. Wang, J. Yang, J. Huang, D. T. Auguste and M. A. Moses, Nat. Commun., 2018, 9, 1–21 CrossRef PubMed.
- Y. Hui, D. Wibowo, Y. Liu, R. Ran, H.-F. Wang, A. Seth, A. P. Middelberg and C.-X. Zhao, ACS Nano, 2018, 12, 2846–2857 CrossRef CAS PubMed.
- Kenry, T. Yeo, P. N. Manghnani, E. Middha, Y. Pan, H. Chen, C. T. Lim and B. Liu, ACS Nano, 2020, 14, 4509–4522 CrossRef CAS PubMed.
- Y. Hui, X. Yi, F. Hou, D. Wibowo, F. Zhang, D. Zhao, H. Gao and C.-X. Zhao, ACS Nano, 2019, 13, 7410–7424 CrossRef CAS PubMed.
- J. Sun, L. Zhang, J. Wang, Q. Feng, D. Liu, Q. Yin, D. Xu, Y. Wei, B. Ding and X. Shi, Adv. Mater., 2015, 27, 1402–1407 CrossRef CAS PubMed.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- A. Tandon, N. E. Rothfuss and M. D. Petters, Atmos. Chem. Phys., 2019, 19, 3325–3339 CrossRef CAS.
- Z.-Z. Tong, R.-Y. Wang, J. Huang, J.-T. Xu and Z.-Q. Fan, Polym. Chem., 2015, 6, 2214–2225 RSC.
- A. Sánchez-Iglesias, M. Grzelczak, T. Altantzis, B. Goris, J. Perez-Juste, S. Bals, G. Van Tendeloo, S. H. Donaldson Jr, B. F. Chmelka and J. N. Israelachvili, ACS Nano, 2012, 6, 11059–11065 CrossRef PubMed.
- D. Chandler, Nature, 2005, 437, 640–647 CrossRef CAS PubMed.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS PubMed.
- H. Bermudez, A. K. Brannan, D. A. Hammer, F. S. Bates and D. E. Discher, Macromolecules, 2002, 35, 8203–8208 CrossRef CAS.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- A. Nagai, D. Sato, J. Ishikawa, B. Ochiai, H. Kudo and T. Endo, Macromolecules, 2004, 37, 2332–2334 CrossRef CAS.
- R.-A. Miranda, E. Finocchio, J. Llorca, F. Medina, G. Ramis, J. E. Sueiras and A. M. Segarra, Phys. Chem. Chem. Phys., 2013, 15, 15645–15659 RSC.
- P. F. Gu, H. Xu, B. W. Sui, J. X. Gou, L. K. Meng, F. Sun, X. J. Wang, N. Qi, Y. Zhang and H. B. He, Int. J. Nanomed., 2012, 7, 109 CrossRef CAS PubMed.
- R. Wang, G.-T. Chen, F.-S. Du and Z.-C. Li, Colloids Surf., B, 2011, 85, 56–62 CrossRef CAS PubMed.
- J.-W. Lim, W. Na, H.-O. Kim, M. Yeom, A. Kang, G. Park, C. Park, J. Ki, S. Lee and B. Jung, J. Mater. Chem. B, 2020, 8, 5620–5626 RSC.
- M. Dionzou, A. Morère, C. Roux, B. Lonetti, J.-D. Marty, C. Mingotaud, P. Joseph, D. Goudounèche, B. Payré and M. Léonetti, Soft Matter, 2016, 12, 2166–2176 RSC.
- A.-L. Robson, P. C. Dastoor, J. Flynn, W. Palmer, A. Martin, D. W. Smith, A. Woldu and S. Hua, Front. Pharmacol., 2018, 9, 80 CrossRef PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- R. B. Brown and J. Audet, J. R. Soc., Interface, 2008, 5, S131–S138 CrossRef CAS PubMed.
- N. T. Tran, Z. Jia and M. J. Monteiro, Ind. Eng. Chem. Res., 2019, 58, 21003–21013 CrossRef CAS.
- N. Deo and P. Somasundaran, Langmuir, 2003, 19, 2007–2021 CrossRef CAS.
- H. Heerklotz, Q. Rev. Biophys., 2008, 41, 205–264 CrossRef CAS PubMed.
- D. Lichtenberg, H. Ahyayauch, A. Alonso and F. M. Goñi, Trends Biochem. Sci., 2013, 38, 85–93 CrossRef CAS PubMed.
- A. Helenius and K. Simons, Biochim. Biophys. Acta, Rev. Biomembr., 1975, 415, 29–79 CrossRef CAS.
- M. Ollivon, O. Eidelman, R. Blumenthal and A. Walter, Biochemistry, 1988, 27, 1695–1703 CrossRef CAS PubMed.
- A. Walter, G. Kuehl, K. Barnes and G. VanderWaerdt, Biochim. Biophys. Acta, Rev. Biomembr., 2000, 1508, 20–33 CrossRef CAS.
- J. Juan-Colás, L. Dresser, K. Morris, H. Lagadou, R. H. Ward, A. Burns, S. Tear, S. Johnson, M. C. Leake and S. D. Quinn, Langmuir, 2020, 36, 11499–11507 CrossRef PubMed.
- Y. Liu, G. Yang, S. Jin, R. Zhang, P. Chen, L. Wang, D. Chen, D. A. Weitz and C. X. Zhao, Angew. Chem., Int. Ed., 2020, 59, 20065–20074 CrossRef CAS PubMed.
- J. Gravier, L. Sancey, S. Hirsjärvi, E. Rustique, C. Passirani, J.-P. Benoît, J.-L. Coll and I. Texier, Mol. Pharm., 2014, 11, 3133–3144 CrossRef CAS PubMed.
- G. E. Tiller, T. J. Mueller, M. E. Dockter and W. G. Struve, Anal. Biochem., 1984, 141, 262–266 CrossRef CAS PubMed.
- K. J. Cross, W. A. Langley, R. J. Russell, J. J. Skehel and D. A. Steinhauer, Protein Pept. Lett., 2009, 16, 766–778 CrossRef CAS PubMed.
- M. Fuhrmans and S. J. Marrink, J. Am. Chem. Soc., 2012, 134, 1543–1552 CrossRef CAS PubMed.
- J. Song, R. Kahraman, D. W. Collinson, W. Xia, L. C. Brinson and S. Keten, Soft Matter, 2019, 15, 359–370 RSC.
- B. L. Micklavzina, S. Zhang, H. He and M. L. Longo, Langmuir, 2017, 33, 2122–2132 CrossRef CAS PubMed.
- A. M. Díez-Pascual, M. A. Gómez-Fatou, F. Ania and A. Flores, Prog. Mater. Sci., 2015, 67, 1–94 CrossRef.
- K. Jaskiewicz, M. Makowski, M. Kappl, K. Landfester and A. Kroeger, Langmuir, 2012, 28, 12629–12636 CrossRef CAS PubMed.
- S. Garcia-Manyes, L. Redondo-Morata, G. Oncins and F. Sanz, J. Am. Chem. Soc., 2010, 132, 12874–12886 CrossRef CAS PubMed.
- S. Garcia-Manyes and F. Sanz, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 741–749 CrossRef CAS PubMed.
- J. D. Unsay, K. Cosentino and A. J. García-Sáez, J. Vis. Exp., 2015, 101, e52867 Search PubMed.
- M. Peiris, K. Yuen, C. Leung, K. Chan, P. Ip, R. Lai, W. Orr and K. Shortridge, Lancet, 1999, 354, 916–917 CrossRef CAS.
- M. Ali, T. Yaqub, N. Mukhtar, M. Imran, A. Ghafoor, M. F. Shahid, M. Naeem, M. Iqbal, G. J. Smith and Y. C. Su, Emerging Infect. Dis., 2019, 25, 136 CrossRef CAS PubMed.
- K. K. Lee, A. Pessi, L. Gui, A. Santoprete, A. Talekar, A. Moscona and M. Porotto, J. Biol. Chem., 2011, 286, 42141–42149 CrossRef CAS PubMed.
- J. M. White, S. E. Delos, M. Brecher and K. Schornberg, Crit. Rev. Biochem. Mol. Biol., 2008, 43, 189–219 CrossRef CAS PubMed.
- L. Xu, D. Li, S. Ramadan, Y. Li and N. Klein, Biosens. Bioelectron., 2020, 170, 112673 CrossRef CAS PubMed.
- H. Zhang and B. L. Miller, Biosens. Bioelectron., 2019, 141, 111476 CrossRef CAS PubMed.
- N. Akkilic, S. Geschwindner and F. Hook, Biosens. Bioelectron., 2020, 151, 111944 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures. See DOI: 10.1039/d1tb01847g |
| This journal is © The Royal Society of Chemistry 2021 |