
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of the cobalt content in cobalt iron oxides on the electrocatalytic OER activity†
Sascha
Saddeler
a,
Georg
Bendt
 a,
Soma
Salamon
b,
Felix T.
Haase
c,
Joachim
Landers
b,
Janis
Timoshenko
c,
Clara
Rettenmaier
c,
Hyo Sang
Jeon
c,
Arno
Bergmann
c,
Heiko
Wende
b,
Beatriz
Roldan Cuenya
c and
Stephan
Schulz
*a
a,
Soma
Salamon
b,
Felix T.
Haase
c,
Joachim
Landers
b,
Janis
Timoshenko
c,
Clara
Rettenmaier
c,
Hyo Sang
Jeon
c,
Arno
Bergmann
c,
Heiko
Wende
b,
Beatriz
Roldan Cuenya
c and
Stephan
Schulz
*a
aInstitute for Inorganic Chemistry, Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Universitätsstr. 5-7, D-45117 Essen, Germany. E-mail: stephan.schulz@uni-due.de
bFaculty of Physics and Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Lotharstr. 1, D-47057 Duisburg, Germany
cDepartment of Interface Science, Fritz-Haber Institute of the Max Planck Society, Faradayweg 4-6, 14195 Berlin, Germany
First published on 15th October 2021
Abstract
Sub 10 nm cobalt ferrite CoxFe3−xO4 (x ≤ 1.75) nanoparticles and cobalt-rich wüstite (Cox/3Fe(1−x)/3)O nanoparticles (x ≥ 2) were synthesized in a solvothermal approach and characterized by powder X-ray diffraction (PXRD), selected area electron diffraction (SAED), transmission electron microscopy (TEM) as well as energy dispersive X-ray spectroscopy (EDX), IR, Raman, and 57Fe-Mössbauer spectroscopy. Their electrocatalytic activity in the oxygen evolution reaction (OER) was evaluated and the active state formation was tracked by operando X-ray absorption spectroscopy (XAS). Our studies demonstrate that the cobalt-rich wüstite (Cox/3Fe(1−x)/3)O nanoparticles underwent a phase-transformation into the spinels CoxFe3−xO4 (x ≥ 2) under the applied OER conditions. The overpotential η10 at 10 mA cm−2, serving as a benchmark for the OER activity of the cobalt ferrite nanoparticles in alkaline media, was lower than that of magnetite Fe3O4 even with low cobalt concentrations, reaching a minimum of 350 mV for Co2.25Fe0.75O4 with a Tafel slope of 50 mV dec−1. Finally, we identified that the catalytic activity is linked to the nanoparticle size as well as to the degree of Co redox activity and change in coordination during OER.
Introduction
The electrochemical water splitting reaction has been known for more than 200 years. This process offers easy access to the industrially important gases hydrogen and oxygen and is also becoming an important part of the modern energy economy.1 In contrast to the hydrogen evolution reaction (HER), the four-electron-transfer oxygen evolution reaction (OER) is kinetically hindered on most electrode surfaces, resulting in high overpotentials (theoretical potential = 1.23 V). Cobalt seems to be an important ingredient for the synthesis of highly active electrocatalysts for the oxygen evolution reaction (OER) in alkaline media and a variety of cobalt materials, i.e. layered double hydroxides,2,3 sulfides,4 phosphides,5,6 phosphates,7 borides,8,9 and spinel-type Co3O4,10–12 showed promising catalytic activities with low overpotentials. Moreover, surface modification of Co-based electrocatalysts by introducing either metals or organic groups or placing them on porous carbon-based materials has been demonstrated to substantially increase their OER activity.13–16 Unfortunately, the wide use of cobalt is not unproblematic due its toxic nature and the increasing global demand caused by the industrial production of batteries. The amount of cobalt in electrocatalysts should therefore be reduced as much as possible and replaced by non-toxic and earth abundant metals such as iron.17Spinel-type mixed-metal oxides AIIB2IIIO4 are particularly interesting as OER catalysts because the cationic sub-lattice is very tolerant to the substitution of other bivalent and trivalent cations, hence allowing a wide variation of the chemical composition and a fine-tuning of the catalytic properties. Moreover, they are very resistant to the harsh OER conditions as was recently shown for single CoFe2O4 nanoparticles with sub 5 nm size, which withstood OER conditions without changes in the morphology or crystal structure even at very high current densities in the range of several kA m−2.18,19 Spinel nanoparticles are accessible by a variety of synthetic routes, including hydrothermal,20 co-precipitation,21 sol–gel,22 and solid-state approaches.23 In solvothermal approaches, metal complexes are thermally decomposed in high-boiling solvents in the absence or presence of surfactants, so-called capping agents. This process not only allows the size- and morphology-controlled synthesis of spinel nanoparticles with precise control of the particle size down to 2–3 nm with narrow size distributions, but also gives access to hydrophobic24,25 and hydrophilic nanoparticles depending on the solvent and surfactant.26,27
Metal acetylacetonates M(acac)x are commercially available precursors for the synthesis of spinel-type nanoparticles with various compositions, including monometallic M3O4 (M = Fe,28 Co,29 Mn30), heterobimetallic MFe2O4 (M = Mg, Mn, Co, Ni, Cu, Zn)31,32 and even multimetallic materials such as Mn0.5Zn0.5Fe2O4.33 Moreover, thermal decomposition of various amounts of such precursors allowed for the synthesis of material series with systematically varied compositions as was demonstrated for the synthesis of a series of CoxFe3−xO4 nanoparticles, with x ranging from 0.5 to 2.34 This is important since the degree of inversion in cobalt ferrite spinels CoxFe3−xO4 is known to increase with the iron content.35 We recently reported on the OER activity of ternary spinels CoxNi1−xFe2O4 (0 ≤ x ≤ 1)36 and CoV2−xFexO4 nanoparticles (0 ≤ x ≤ 2),37 which showed much lower overpotentials and Tafel slopes compared to the binary spinels NiFe2O4 and CoFe2O4, respectively. Apart from the chemical composition, the electrocatalytic activity of spinel nanoparticles also largely depends on their size and surface termination. Spherical, cubic, and octahedral CoFe2O4 nanoparticles as well as hexagonal plates and nanofibers were synthesized,38–44 while we recently reported on the influence of the morphology and crystallographic surface termination on the electrocatalytic properties of Co3O4 nanoparticles.45
We herein became interested to investigate the influence of the cobalt content in spherical cobalt ferrite CoxFe3−xO4 nanoparticles on their electrocatalytic activity. Our attempted solvothermal synthesis of a series of cobalt ferrite CoxFe3−xO4 nanoparticles over the complete compositional range (x = 0–3, Δx = 0.25) yielded spherical spinel-type CoxFe3−xO4 nanoparticles with cobalt contents below x = 2, whereas higher amounts of the Co precursor (x ≥ 2) yielded biphasic mixtures of cobalt ferrite (CoxFe3−xO4) and cobalt–iron wüstite (Cox/3Fe(1−x)/3)O nanoparticles. The size of the nanoparticles was found to depend on the Co content and decreased from 9 nm (x = 0) to 3 nm (x = 2.5). In addition, the activity of the cobalt ferrite as well as cobalt–iron wüstite nanoparticles in the OER in alkaline media is reported.
Results and discussion
I Nanoparticle synthesis and pre-catalyst characterization
Thermal decomposition of varying amounts of Fe(acac)3 and Co(acac)2 in triethylene glycol (TEG) in the presence of polyethyleneimine (PEI) as capping agent at 250 °C yielded sub 10 nm nanoparticles. The elemental composition of the nanoparticles was determined by EDX analyses (Table S1†), both of single nanoparticles and larger nanoparticle aggregates, proving that the initial Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe precursor concentration is preserved in the resulting nanoparticles.
:Fe precursor concentration is preserved in the resulting nanoparticles.
Even though the XRD diffractograms show broad Bragg reflections due the nanosized nature of the oxide phases (Fig. 1), the crystalline materials were identified as phase-pure cubic spinels CoxFe3−xO4 in the case of cobalt contents x ≤ 1.75 on the basis of the Bragg peak positions (2θ values of 30.2, 35.6, 43.4, 57.2 and 62.6°), which correspond to the (220), (311), (400), (511), and (440) lattice planes (Fe3O4, PDF 01-077-1545, with a = 8.3970 Å). In contrast, higher concentrations of the cobalt precursor (x = 2.00–2.75) yielded biphasic mixtures of cobalt ferrite (CoxFe3−xO4) and cobalt–iron wüstite (Cox/3Fe(1−x)/3)O, resulting in a shoulder within the broad reflex at 43°. The fraction of the rock-salt phase as quantified by Rietveld refinement (Fig. S1 and Table S1†) was found to increase with increasing Co precursor concentration (x = 2.00, 30%; x = 2.25, 57%; x = 2.50, 90%; x = 2.75, 99%). The rock salt cobalt–iron wüstite phase was identified by Bragg reflections at 2θ = 36.5 and 34.4°, corresponding to the (111) and (200) lattice planes of this structure (CoO, PDF 01-071-1178, with a = 4.2631 Å). The Co-richest material within this series, iron-free cobalt(II) oxide CoO, formed a biphasic mixture of rock salt and wurtzite-type phases (CoO, PDF 01-089-2803). The phase transition from the cobalt ferrite (CoxFe3−xO4) to the cobalt–iron wüstite (Cox/3Fe(1−x)/3)O phase is also described in the Co–Fe–O phase diagram for cobalt-rich samples for low oxygen partial pressures.46
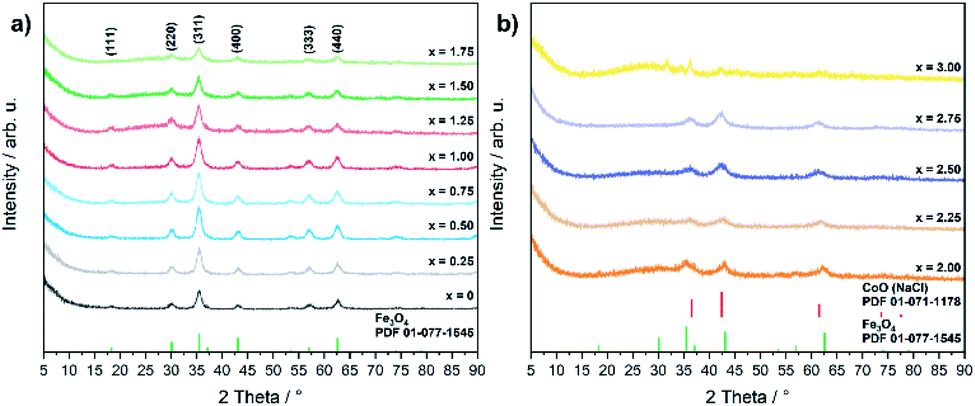 | ||
| Fig. 1 PXRD patterns of (a) CoxFe3−xO4 (x = 0–1.75) and (b) CoxFe3−xO4/(Cox/3Fe(1−x)/3)O (x = 2–3) with references for Fe3O4 (PDF 01-077-1545) and CoO (PDF 01-071-1178). | ||
Fig. 2 shows TEM images of CoxFe3−xO4 nanoparticles, proving the spherical morphology for all nanoparticles. The average particle size as-determined by analyzing 100 particles was found to decrease with increasing Co content from 9 nm (x = 0) to 3 nm (x = 2.50) (Fig. S2†) as was previously observed by Jalili and coworkers.47
 | ||
| Fig. 2 TEM images of CoxFe3−xO4 for x = 0 (a), 0.50 (b), 1.00 (c), 1.50 (d), 2.00 (e), 2.50 (f) and 3.00 (g) as well as SAED images for x = 1.00 (h) and 2.50 (i). | ||
The steadily decreasing size of the nanoparticles with increasing Co content was only observed for particles synthesized with PEI as surfactant, whereas analogous nanoparticles prepared in the absence of PEI showed constant particle sizes of 5–6 nm independent of the Co content, indicating that PEI differently binds to iron and cobalt. The binding strength of ligands to both the molecular metal precursor and the resulting nanoparticle surface is generally known to largely affect the nucleation and growth rates and consequently the final nanoparticle size.48 The low magnification micrograph shows well separated nanoparticles with a low tendency for agglomeration and a narrow size distribution except for the iron-free material (Fig. 2g). The average size of the CoFe2O4 nanoparticles is 8 nm, and the DF-STEM image (Fig. 2c) shows crystalline nanoparticles with a d-spacing of 2.53 Å corresponding to the (311)-lattice plane of CoFe2O4. In contrast, the DF-STEM image of biphasic nanoparticles (x = 2.50) (Fig. 2f) shows lattice fringes with a d-spacing of 2.46 Å, corresponding to the (111)-plane of the cubic CoO phase. The assignment of the crystalline phases CoFe2O4 and CoO is supported by the respective selected area electron diffraction (SAED) patterns (Fig. 2h and i).
The core-level XPS spectrum (Fig. S3†) for the Co 2p region shows the characteristic 2p3/2 and 2p1/2 peaks at ca. 779 and 795 eV, respectively. A satellite peak is visible for both peaks at higher binding energies of ca. 785 and 802 eV. These satellites are the signature for octahedrally coordinated high-spin Co2+ ions. Neither octahedrally coordinated low-spin Co3+ nor tetrahedrally coordinated Co2+ ions exhibits this feature. A high amount of Co2+ ions can be identified for x = 0.50 via peak shift to ca. 781 eV. Furthermore, the intensity of the satellite peak at ca. 785 eV decreases for higher amounts of Co from x = 1.00 to 1.50, indicating the presence of Co3+ ions.49,50 The octahedrally-coordinated CoO6 unit was found for sample x = 1.50 at ca. 779 eV, whereas the tetrahedrally-coordinated CoO4 unit is visible at ca. 781 eV.51 In contrast, the biphasic materials (x = 2.25, 2.50) show more pronounced satellites due to the presence of high amounts of Co2+, in accordance with the formation of the rock salt phase (CoxFe1−x)O, which exclusively contains bivalent Co. Cobalt-poor samples (x = 0.50, 1.00, 1.50) show peaks at ca. 711 and 724 eV in the Fe 2p region, which can be attributed to the Fe 2p3/2 and Fe 2p1/2 of Fe3O4.52 The satellite peak at 717 eV, which is characteristic for the presence of Fe2+ ions,53 increases with higher cobalt contents until only a weak satellite is visible in the sample with x = 1.75.
The nanoparticles were also characterized by FTIR and Raman spectroscopy. The spinel-type structure consists of tetrahedral MO4 and octahedral MO6 units (M = Co, Fe), which show characteristic signatures in the FTIR and Raman spectra. The spinel-type structure has five Raman active phonon modes (A1g, Eg, three T2g), while the A1g mode in inverse spinels such as CoFe2O4 splits into two modes, A1g and A1g*. Only two of them, A1g and T2g, are visible as broad modes in the Raman spectrum shown in Fig. S3a.† The A1g mode around 700 cm−1 most likely originates from the symmetric stretching of the MO4 tetrahedra and the T2g mode originates from the MO6 octahedra, even though this assignment is controversially discussed in the literature.54 According to Laguna-Bercero et al.55 the AO4 unit cannot be differentiated from the BO6 unit due to weaker A–O bonds compared to B–O bonds, which results in a mixture of both vibrations within the A1g mode. Both modes shift to lower wavenumbers with increasing cobalt content up to x = 1.75 due the incorporation of cobalt into the spinel-type structure as was previously reported.56 Starting at x = 2.0, the two bands not only shift back to higher wavenumbers but also become sharper with increasing value of x, and the Eg mode also becomes visible for x = 2.75 and x = 3.0, respectively. In contrast to the XRD results, only the spinel-type and not the wüstite-type phase was detected for high cobalt contents up to x = 3, most likely due to the ease with which CoO can be oxidized to Co3O4 even at moderate laser power.54 The influence of laser power on the transformation is shown in Fig. S3b,† and biphasic samples (x = 2–3) were measured at low laser power.
Cobalt ferrite not only shows five Raman active modes but also four IR-active modes (4 F1u). The FTIR spectra (Fig. S4†) show two major absorption bands at 386 and 579 cm−1 for the CoFe2O4 nanoparticles corresponding to the characteristic vibrations of octahedral MO6 (M = Co, Fe) unit and the tetrahedral MO4 unit, respectively.57 Both bands shift slightly, most likely due to a changing degree of inversion, but no clear trend is visible. Weak absorption bands around 1600, 1450, 1115 and 1059 cm−1 indicate the presence of small amounts of the organic capping agent (TEG, PEI) bonded to the nanoparticle surface.58,59
To further probe the composition-dependent structural properties using the underlying magnetic characteristics, CoxFe3−xOy nanoparticles with Co-contents of x = 1.25–2.50 were characterized via Mössbauer spectroscopy as well as magnetometry (Fig. 3).
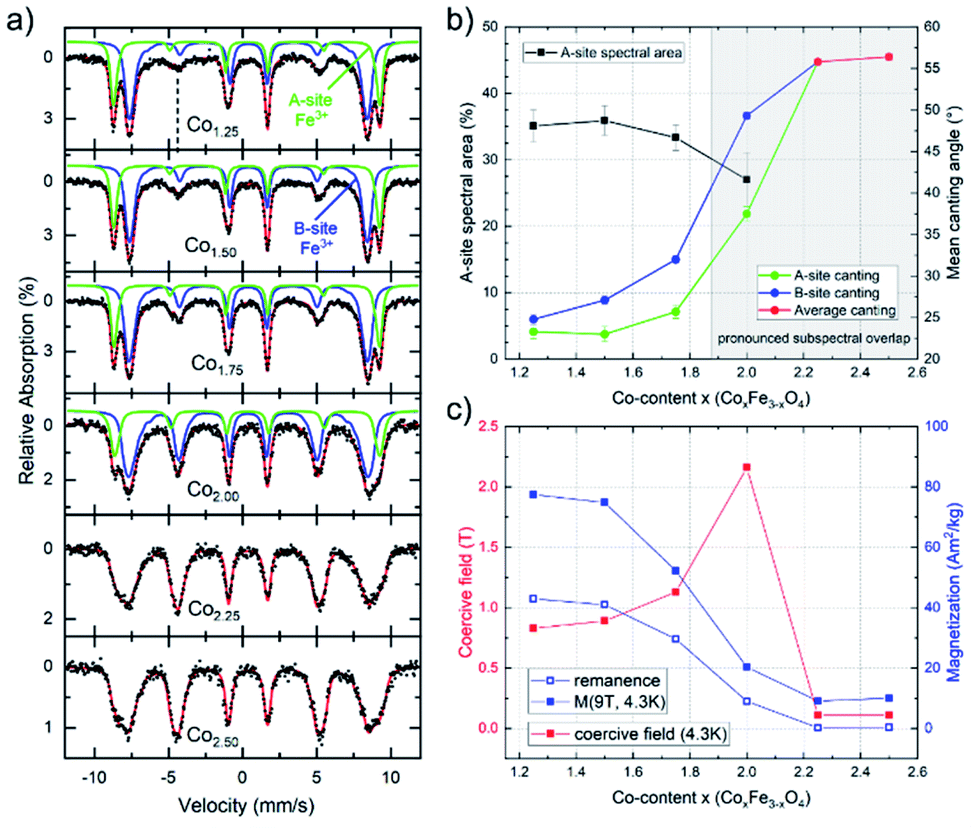 | ||
| Fig. 3 (a) In-field Mössbauer spectra of CoxFe3−xO4 (x = 1.25–2.5) nanoparticles, illustrating contributions of A- (green) and B-site (blue) subspectra. Spin canting angles were determined from the relative intensities of line 2 (marked by dashed line) and 5, strongly increasing upon rising Co-content. (b) Parameters extracted from in-field Mössbauer spectroscopy of CoxFe3−xO4 (x = 1.25–2.5) nanoparticles: The determination of the A-site spectral area (black) is hindered at x > 1.75 (shaded area) due to pronounced A- and B-subspectral overlap, spin canting angles of A- (green) and B-sites (blue) are provided up to x = 2.0. For higher Co-content the antiferromagnetic behavior prevents the resolution of the Fe-contributions on tetrahedral and octahedral positions and therefore, the average spin canting angles (red) are shown. (c) Remanent magnetization M(4.3 K,0 T) (blue, empty), high-field magnetization M(4.3 K,9 T) (blue, full) and coercive fields (red) determined at 4.3 K. | ||
Mössbauer spectra were recorded at 4.3 K in an applied field of 5 T (parallel to the γ-ray propagation direction) to determine the distribution of Fe3+ ions across different crystallographic sites, also allowing us to thereby (indirectly) extract the Co-ion site occupation. As shown in Fig. 3, samples with lower Co-content (x = 1.25–1.75) show a typical spinel spectrum with a clear separation between the sextet subspectra of the tetrahedrally coordinated A-sites (green) and octahedrally coordinated B-sites (blue). From the ratio of the spectral areas of these two sites, the degree of inversion can be calculated. As our samples deviate from the standard stoichiometry given above (AB2O4), a representation of the ion distribution via the inversion parameter would be uncommon. Thus, we present the ion site occupation by plotting the relative spectral area of Fe-ions on the A-site in Fig. 3a, which allows us to draw conclusions on the changing site preference upon increasing x. The displayed relative A-site spectral areas range from 33(1) % to 36(2) % for 1.25 ≤ x ≤ 1.75, which is slightly below the inversion parameter values commonly reported for pure bulk cobalt ferrite.60 Nonetheless, a more random placement is not unusual for nanoparticulate spinel systems, depending on the utilized synthesis procedure.
The finding from in-field Mössbauer spectroscopy regarding a rather random site-occupation is substantiated by the high magnetization values M(4.3 K,9 T) (Fig. 3c) and also illustrated in detail in the ESI.† The spectra with low Co-content are also characteristic in terms of revealing only a moderate degree of spin canting, which is shown by the intensities of lines 2 and 5 relative to lines 3 and 4, respectively. This indicates that the magnetic moments are predominantly aligned in the external field, with the small residual spin canting angle, as also given in Fig. 3b, presumably stemming from spin frustration effects at the surface. Upon increasing the Co-content (x ≥ 2.0) in these samples, two main effects can be observed. On the one hand, the average spin canting angles for both sites increase, as is visible by the increasing intensity of lines 2 and 5 in Fig. 3a. On the other hand, the two sextet subspectra increasingly overlap, making it impossible to accurately determine the contribution of the individual sites. Therefore, spectra were reproduced with only one distribution of effective magnetic fields for x > 2.0, where we observe a completely random orientation of the probed spins, as indicated by reaching the “magic angle” (≈54.7°). Such a behavior points towards a transition of the material from a ferrimagnetic state of the CFO endmember towards an increasingly antiferromagnetic state, as is typical for Co3O4, while the decreasing average particle diameter upon rising x will also likely result in stronger surface spin canting. Although X-ray diffractograms show the formation of a CoO phase for compositions with x > 2 (Fig. 1b), the comparatively high hyperfine fields and the isomer shifts of ca. 0.48 mm s−1 in the Mössbauer spectra point towards Fe3+ rather than the Fe2+ species one would expect for a Fe-fraction present within a wüstite-like CoO phase. For compositions up to x = 2.50, this can be resolved assuming a nearly Fe-free CoO main phase, while the increasingly minor Co3O4 phase (Table S2†) contains sufficient residual amounts of Fe, thus being much more strongly present in the Mössbauer spectra. As the CoO main phase is also antiferromagnetic,61 the low magnetization in the M(H) loops (Fig. S6b†) is consistent with this interpretation, as is the generally antiferromagnetic nature of the Mössbauer spectra for x > 2, preventing the resolution of individual subspectra due to a lack of orientation of magnetic moments relative to the applied magnetic field (Fig. 3a).
II OER activity
The electrochemical OER activity of as-prepared cobalt ferrite CoxFe3−xO4 (x = 0–1.75) and cobalt–iron wüstite (Cox/3Fe(1−x)/3)O (x ≥ 2) nanoparticles was determined using a rotating disk electrode. The nanoparticles were embedded in Nafion® on glassy carbon working electrodes in a standard three-electrode system with 1 M iron-free KOH as electrolyte and a Pt counter electrode at room temperature. To test the influence of solution of Pt ions from the counter electrode a linear sweep was performed with a graphite counter electrode (Fig. S8†). A catalyst loading of 100 μg cm−2 based on the geometrical surface area (0.196 cm2) was used for all experiments. Additionally, the OER activity and stability of CoFe2O4 (x = 1) was tested for catalyst loadings of 50 and 200 μg cm−2 (Fig. S9†). The working electrodes were activated by cyclic voltammetry (CV) with a scan rate of 100 mV s−1 in a potential window between 1 and 2 V vs. RHE until reproducible voltammograms were obtained and linear sweep voltammogram (LSV) measurements with a scan rate of 5 mV s−1 were recorded afterwards (1–2 V vs. RHE). The benchmark for evaluating electrocatalysts is the overpotential η10, which is necessary to deliver a geometric current density of 10 mA cm−2.62Fig. 4a and b display the polarization curves for the CoxFe3−xO4 (x = 0–1.75) and (Cox/3Fe(1−x)/3)O (x ≥ 2) nanoparticles. The cobalt-free Fe3O4 nanoparticles show only a very low electrocatalytic activity and the highest overpotential (699 mV) within this material series. The introduction of a small fraction of cobalt (x = 0.25) leads to a significant reduction of the overpotential by 250 mV, reaching a value that is comparable to the overpotential of Co3O4 (η = 440 mV).63 The overpotential further decreases monotonically with increasing cobalt content and reaches a minimum at η10 = 350 mV for the composition Co2.25Fe0.75O4. Increasing cobalt content (x > 2.25) then leads to an increase of the overpotential, which reaches a maximum for the pure cobalt oxide (CoO) at η10 = 410 mV.
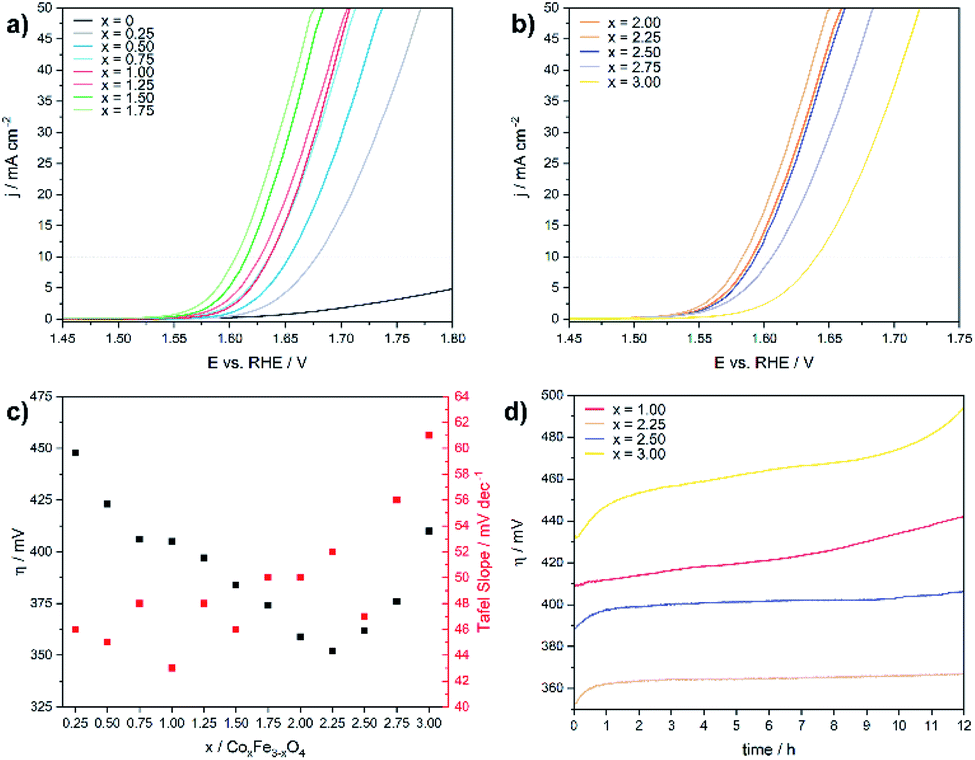 | ||
| Fig. 4 Linear sweep voltammograms for (a) CoxFe3−xO4 (x = 0–1.75) and (b) (Cox/3Fe(1−x)/3)O (x ≥ 2) in 1 M KOH electrolyte with a scan rate of 5 mV s−1, (c) overpotential and Tafel slope versus the cobalt content and (d) chronoamperometry measurements at 10 mA cm−2 (geometrical surface area). | ||
Fig. 4c shows the Tafel slopes for the CoxFe3−xO4 (x = 0–2) and (Cox/3Fe(1−x)/3)O (x ≥ 2) nanoparticles, and Fig. S10† shows the overpotential η is plotted against the logarithmic current density log (j). All electrocatalysts show Tafel slopes in a narrow range of 43–61 mV dec−1. A low Tafel slope indicates favourable OER reaction kinetics which can imply differences in the OER surface chemistry.64,65 The observed trend for the Tafel slope is comparable with the trend visible for a series of CoV2−xFexO4 (0 ≤ x ≤ 2)37 and Co1−xNixFe2O4 (0 ≤ x ≤ 1)36 nanoparticles. Fig. 4d displays chronoamperometry measurements at 10 mA cm−2 for CoFe2O4 (x = 1), which were performed to investigate the stability of the nanoparticles under these conditions. The increasing overpotential with increasing measurement time can be caused by several processes like nanoparticle growth, detachment, dissolution and compositional changes. For instance, leaching of Co2+ ions from the nanoparticle surface into the solution as was previously reported66 would result in cobalt vacancies or Fe accumulation on the nanoparticle surface and reduce the number of cobalt atoms on the surface. The reduced cobalt content should lead to a decrease of the OER activity as we observed in the LSV curves for spinel compounds with low amounts of cobalt. In contrast, the phase mixtures of spinel and rock salt type materials (x = 2.25, 2.50) show a constant overpotential over time. The observed rising overpotential for the CoO nanoparticles (x = 3.00) can be attributed to the oxidation of Co2+ to Co3+ under electrochemical conditions over the formation of Co(OH)2, CoO(OH) and finally Co3O4.67–69 The long-term stability in OER was also investigated by chronoamperometric measurement of CoFe2O4 (x = 1) at 1 mA cm−2 over 48 h (Fig. S11†) with a slight increase of the potential most likely due to leaching of Co into solution.
Electrochemical impedance spectra (EIS) were recorded to gain further insights in the electron transfer kinetics. Fig. 5 shows the Nyquist plots for the CoxFe3−xOy nanoparticles at an overpotential of 550 mV.
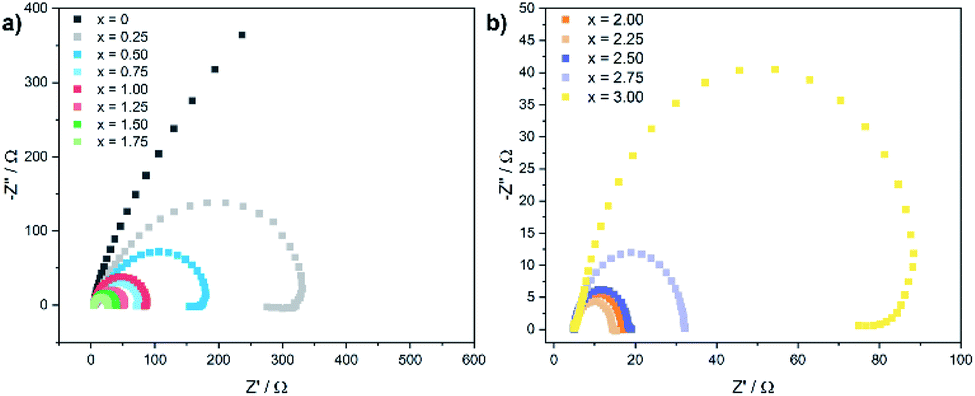 | ||
| Fig. 5 Electrochemical impedance spectra (EIS) for (a) CoxFe3−xO4 (x = 0–1.75) and (b) (Cox/3Fe(1−x)/3)O (x ≥ 2) nanoparticles recorded at an overpotential of 550 mV. | ||
With increasing cobalt content, the semicircular arc becomes narrower up to x = 2.25 and widens up again with further increase of x. The charge-transfer resistance Rct was extracted by fitting the Nyquist plot using a Randles circuit. The cobalt-free catalyst exhibits very high values for Rct with ∼2000 Ω (Fe3O4). Similarly to the behaviour of the overpotential and the Tafel slope, Rct also decreases rapidly with increasing cobalt content and reaches a minimum value of 10 Ω for x = 2.25, increasing slightly up to 90 Ω for x = 3.00. The substitution of iron by cobalt in the cationic sublattice of the spinel structure obviously leads to a significantly improved charge transport for nanoparticles with a cobalt-rich composition, which is in good agreement with the observed dependence of the Tafel slope and the overpotential on the elemental composition.
III Operando characterization
Operando X-ray absorption spectroscopy (XAS) measurements were conducted at the Co and Fe K-edges for four different nanoparticle compositions: Co2.25Fe0.75O4, CoFe2O4, Co0.25Fe2.75O4 and CoOX. Thus, the metal ion chemical state and local atomic structure could be determined for the most important cases of the composition–activity profile. The operando XAS data of the catalysts were recorded in the as-prepared state, after an electrochemical conditioning treatment (20 CVs, 1–1.8 VRHE, 50 mV s−1) in the OER active state at +1.8 VRHE as well as after OER in the electrolyte at +1.0 VRHE. All measurements were carried out in 0.1 M KOH. All spectra are provided in the ESI (Fig. S7–S13†) including a table with all fitting parameters (Table S6†).The X-ray absorption near edge structure (XANES) spectra of Co2.25Fe0.75O4, CoFe2O4, Co0.25Fe2.75O4 and CoOX at the Co K- and Fe K-edge are provided in Fig. 6a and b and S12a–e.† The corresponding oxidation states of Co and Fe were calculated by comparison of the absorption edge position with known reference compounds.70–72 The extracted oxidation states are displayed in Fig. 6c for Co and Fig. S12f† for Fe, with an uncertainty of ±0.1 and ±0.2 for Co and Fe, respectively. The average Co oxidation state increases for all samples during OER and this oxidation process is partially reversible as the Co oxidation state after OER exhibits a reduction. The average oxidation state of Fe is close to +3 for all samples and changes significantly less than that of Co under the different electrochemical conditions, especially considering the error bars. In detail, we identified that the average initial Co oxidation state ranges from +2.3 to +2.6, which increases with the percentage of Co within the sample. The initial Fe oxidation state as-prepared is +2.8 to +3, with no distinct trend with nanoparticle composition. During OER, the Co oxidation state increases from +2.4 to +3.0. The cobalt-rich CoOX and Co2.25Fe0.75O4 exhibit the highest Co oxidation state of +3.0, followed by CoFe2O4 with +2.9 and Co0.25Fe2.75O4 with +2.4. All samples possess an Fe oxidation state ranging from +2.9 to +3.0 during OER. After OER, the cobalt oxidation decreases to +2.5 and +2.7 for CoFe2O4 and Co2.25Fe0.75O4, respectively, but stays constant within error bars for CoOX and Co0.25Fe2.75O4. In this regard, the Co2.25Fe0.75O4 and CoFe2O4 nanoparticles reduce stronger after OER than the Co0.25Fe2.75O4 and CoOX nanoparticles, implying a larger redox activity of the Co ions. The Fe oxidation state after OER remains unchanged within the error. Interestingly, the so-called white line position (energy of the intensity maximum) of the Fe XANES profile for Co2.25Fe0.75O4 shifts reversibly from 7131.5 eV to 7132.5 eV, which indicates subtle changes in the electronic structure under OER conditions.
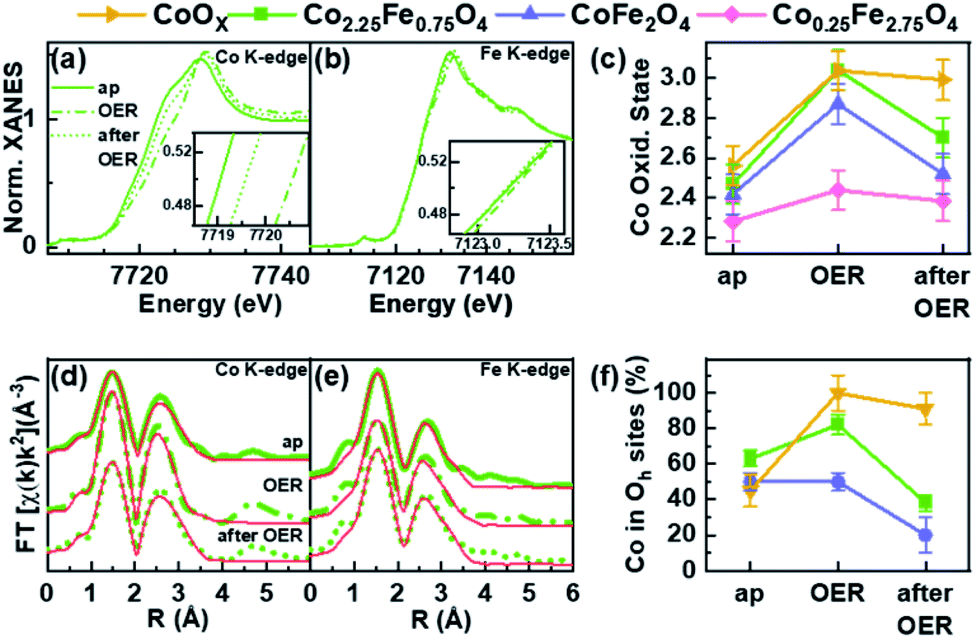 | ||
| Fig. 6 (a) Co and (b) Fe K-edge XANES of Co2.25Fe0.75O4 nanoparticles as-prepared (ap), during OER at 1.8 VRHE and after OER at OCV at ∼1 VRHE. (c) Co oxidation state ap, during and after OER of Co2.25Fe0.75O4, CoFe2O4, Co0.25Fe2.75O4 and CoOX nanoparticles. (d) Co and (e) Fe K-edge EXAFS of Co2.25Fe0.75O4 nanoparticles and (f) the fraction of Co ions occupying available octahedral sites. | ||
The extended X-ray absorption fine structure (EXAFS) analysis for bimetallic CoXFe2−XO4 nanoparticles was carried out based on a spinel-structure with a fixed 2:1 ratio of octahedral and tetrahedral site occupancy (see ESI† for further details). This analysis was carried out at both the Co and Fe K-edges for Co2.25Fe0.75O4 and CoFe2O4 nanoparticles. The Co0.25Fe2.75O4 nanoparticles were excluded from the former analysis due to insufficient data quality caused by the low Co content. For the CoOx nanoparticles, the same model was applied without a fixed 2:1 ratio to determine the fraction of octahedrally-coordinated Co ions in the presence of the wurtzite-CoO phase. The fits are displayed in Fig. 6d, e and S11a–e† for measurements carried out on the as-prepared samples, during OER and after OER. The first peak corresponds to the M–O coordination at ∼1.6 Å (phase uncorrected). The peak of the second coordination shell at 2.5 Å (phase uncorrected) corresponds to two di-μ-oxo bridged metal ions in octahedral sites. A third coordination shell peak at 3.1 Å (phase uncorrected) originates from mono-μ-oxo bridges between metal ions occupying tetrahedral and octahedral sites.73,74
As discussed above, the wüstite CoOX nanoparticles do not exhibit a spinel structure and the XANES can be best described as a linear combination of rs-CoO, w-CoO and CoOOH reference spectra. In the as-prepared state, the EXAFS spectra show a pronounced contribution of the second coordination shell due to the occupation of octahedral sites, and some contribution in the third coordination shell from tetrahedral sites as-prepared. During OER, a CoOOH-like structure with only octahedral sites occupied is formed, which is typical for cobalt oxides. After OER, the pure CoOx nanoparticles were found to restructure, as some tetrahedral sites are occupied with Co ions afterwards leading to a Co3O4-like structure.
Fig. 6f shows the fraction of Co located in available octahedrally coordinated sites in CoOX, Co2.25Fe0.75O4 and CoFe2O4 nanoparticles. As-prepared, the fraction is highest for Co2.25Fe0,75O4 and similar for CoOX and CoFe2O4. During OER, the fraction of Co occupying the octahedral sites is larger than before and/or after OER and thus, linked to the oxidative reaction conditions and an (irreversible) increase in the average Co oxidation state. This process is strongest yet irreversible for CoOX and lowest for CoFe2O4. It is not surprising that CoOX forms a CoOOH-like structure under OER conditions with all Co octahedrally coordinated. Remarkably during OER, the most active Co2.25Fe0.75O4 catalyst exhibits an increase in octahedral sites occupied by Co whereas the less-active CoFe2O4 is almost identically unchanged as compared to the state after OER. Recently, octahedral di-μ-oxo bridges have been identified as a common structure formed during OER independent of the initial crystal structure including Co3O4 spinel oxides.65 After OER we notice a decrease of octahedrally coordinated Co which arises as tetrahedrally coordinated sites of the third coordination shell. The EXAFS fits furthermore revealed that the identified reversible oxidation of the Co ions results in a contraction of the average Co–O bond during OER by 0.03–0.04 Å for all analysed samples as compared to after OER. The di-μ-oxo bridges and mono-μ-oxo bridges contract by 0.01–0.02 Å for Co ions. The Fe–M distances contract within ∼0.02 Å for second and third coordination shell.
In summary, XANES and EXAFS analyses suggest an increasing average Co oxidation state with increasing Co content in the as-prepared state. The most active Co2.25Fe0.75O4 shows pronounced (reversible) changes in the Co oxidation state as well as di-μ-oxo bridges during OER, which is similar to pure CoOX nanoparticles. This contrasts with CoFe2O4 nanoparticles which show a strong Co oxidation during OER but miss the transformation during OER. Co0.25Fe2.75O4 nanoparticles do not show changes, underlining their low reactivity. In general, the Fe ions do not undergo a pronounced redox transition during OER and the Fe K-edge data do not show strong Fe oxidation or Fe–O contraction. It must be noted that based on the operando XAS results, the Co ions participate significantly stronger in the redox chemistry and the degree of average structural change during OER, as referred to the post-catalysis state is linked to the catalytic activity.
IV Post-catalyst characterization
The OER is performed in strong alkaline media under high potentials and current densities which can lead to structural and chemical changes of the electrocatalyst. We therefore performed a post-catalysis characterization of the nanoparticles. EDX analyses of the bulk CoFe2O4 spinel nanoparticles show no change of their chemical composition after catalysis.The crystalline structure of the spinel nanoparticles (x < 2) was unaffected by OER conditions according to XRD and SAED analyses. This agrees with the reversible structural transformation determined by operando XAS and previous reports.14 However, the formation of very small nanoparticles (1–2 nm) was observed for CoFe2O4 (x = 1) (Fig. 7b), which could not be analyzed in more detail due to their small size. Additionally, the elemental composition becomes less homogeneous in a single particle after electrochemical treatment (Fig. 7c and d). However, these data should be regarded with care since only a single particle was analysed. In contrast, the cobalt–iron wüstite nanoparticles underwent major structural changes during OER and were transformed into the spinel-type structure as was proven by SAED (x = 2.25, 2.50, Fig. 7e and f) and GIXRD (x = 2.50, Fig. 8) with no residues of rock salt type material. The characteristic reflections due to the iron–cobalt wüstite phase disappeared, whereas reflections due to the spinel structure are present. These findings agree with the results obtained by operando XAS.
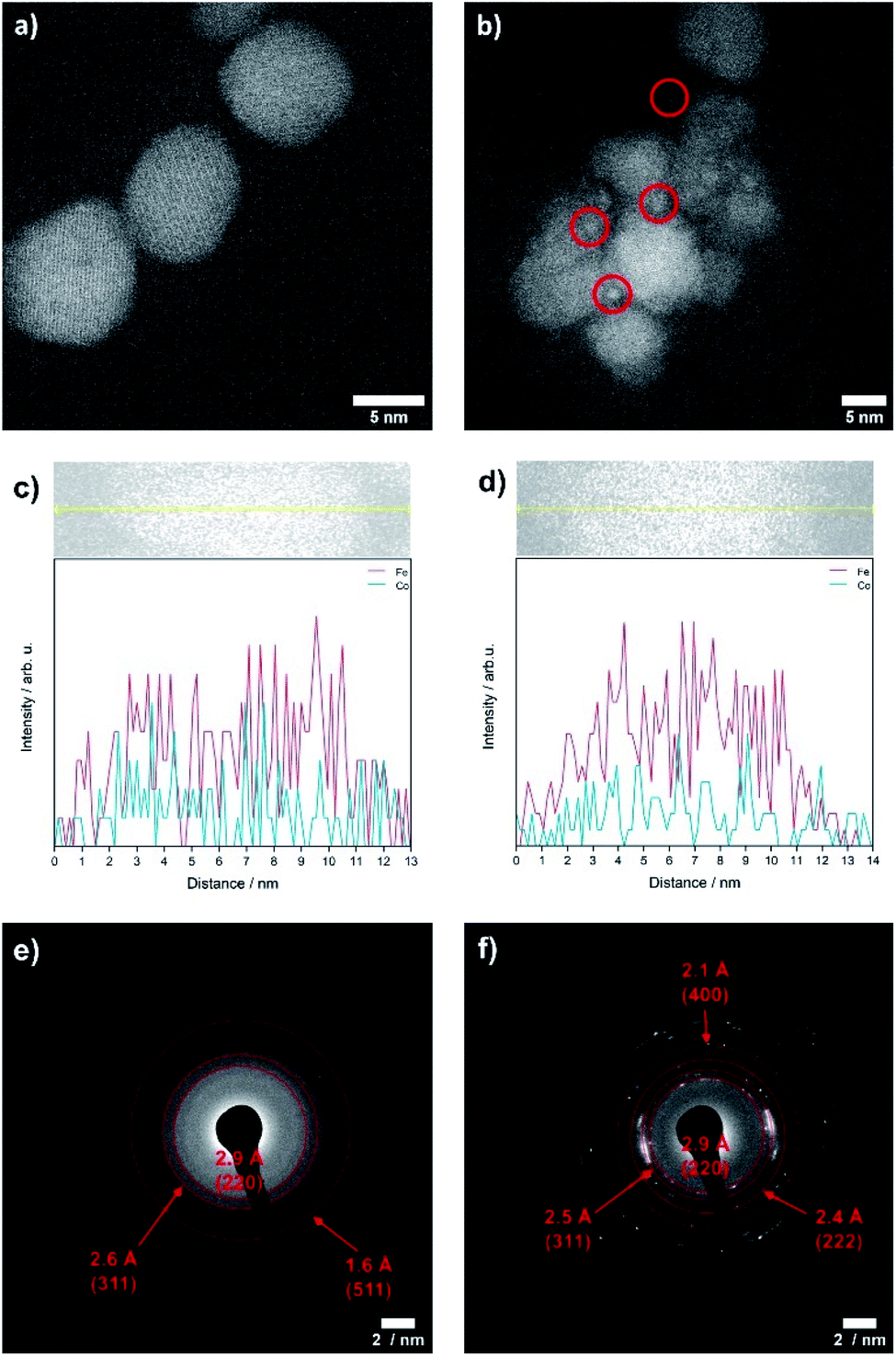 | ||
| Fig. 7 TEM images of CoFe2O4 (x = 1) nanoparticles before (a) and after OER (b), TEM-EDX linescans on a single particle for CoFe2O4 (x = 1) before (c) and after electrochemical treatment (d) and SAED patterns after OER measurements for CoxFe3−xO4 with x = 2.25 (e) and 2.50 (f). | ||
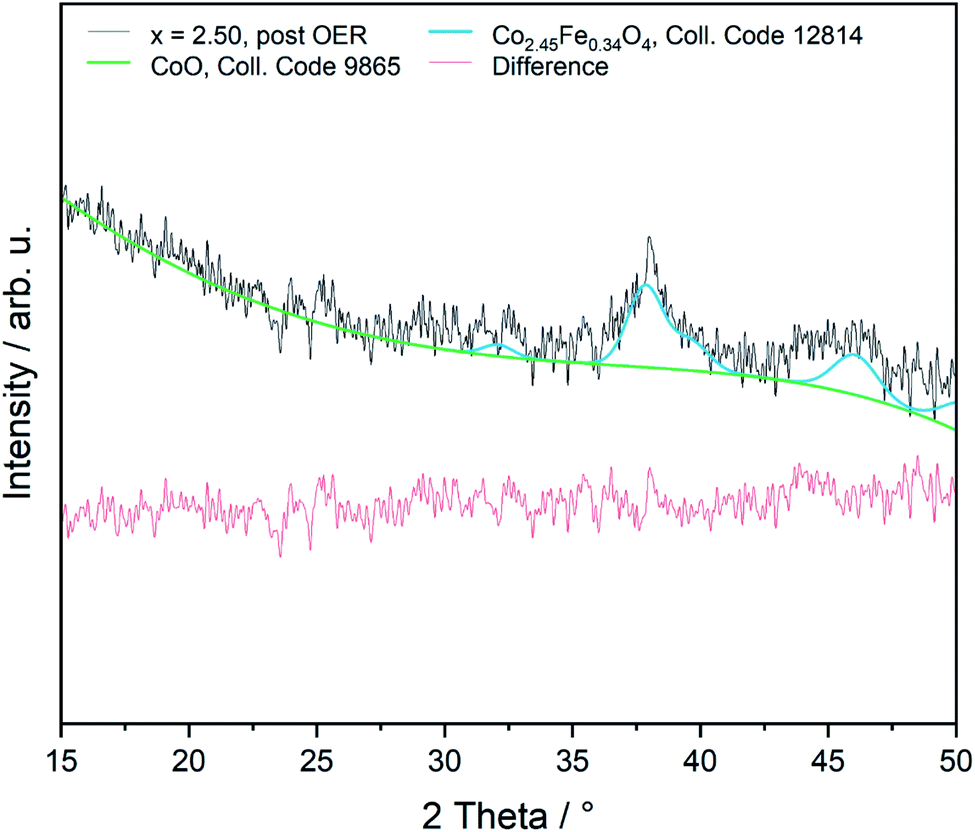 | ||
| Fig. 8 Rietveld refinement of sample x = 2.50 on Si after OER measurement (GIXRD, 1°). | ||
Conclusions
A series of sub 10 nm PEG/PEI stabilized phase-pure cobalt ferrite CoxFe3−xO4 (x ≤ 1.75) and cobalt-rich cobalt–iron wüstite (Cox/3Fe(1−x)/3)O nanoparticles (x ≥ 2) were synthesized and characterized by EDX, XRD, TEM, IR and Raman spectroscopy. A rather random ion site-occupation with a minor Co B-site preference was found by in-field Mössbauer spectroscopy, in agreement with magnetometry results. The nanoparticles were tested as OER catalyst in alkaline media, and operando XAS measurements revealed a correlation of the content of di-μ-oxo bridged cobalt–metal sites and the extent of reversible Co oxidation state change during OER with the reactivity. Post catalysis GIXRD and SAED analyses revealed the phase transition of iron–cobalt wüstite (Cox/3Fe(1−x)/3)O (x ≥ 2) nanoparticles into the spinel phase. The introduction of cobalt into the spinel structure improves the OER reaction kinetics and the charge transport of the nanoparticles, resulting in a reduction of the onset potential and the overpotential η10 for the OER compared to the cobalt-free Fe3O4. A maximum OER activity is reached for x = 2.25, whereas a further increase of the Co content results in a steady increase of the overpotential.Experimental
Synthetic procedures
:1 mixture of acetone and ethyl acetate resulted in precipitation of the CoxFe3−xOy nanoparticles, which were isolated by centrifugation (3000 rpm, 10 min), washed two times with acetone and dried at ambient temperature.
Methods
:1) and 10 μL of a 5 wt% Nafion 117 solution by ultrasonication for 30 min. 3.92 μL of the catalyst suspension was drop-coated onto the polished glassy carbon electrode and dried in air at room temperature resulting in a mass loading of 0.10 mg cm−2. Prior to the OER measurements, modified electrodes were subjected to continuous potential cycling in the potential window of 1 V to 2 V vs. RHE with a scan rate of 100 mV s−1 until reproducible voltammograms were obtained and LSV measurements for OER activity were recorded with a scan rate of 5 mV s−1 in a potential window of 1 V to 2 V vs. RHE. Electrochemical impedance spectroscopy (EIS) was performed at frequencies between 100 kHz and 0.1 Hz with an overpotential of 550 mV. The resistance of the solution was determined from the resulting Nyquist plot, and then later used for ohmic drop correction according to the relation, Ec = Em − iR, where Ec is the corrected potential and Em is the applied potential. All reported current densities were calculated using the geometric surface area of the electrode.
Author contributions
S. Saddeler and G. Bendt performed the synthesis and characterization (XRD, EDX, IR, Raman, CV) of the nanoparticles. Analysis of magnetic properties and Mössbauer spectroscopy measurements were performed by S. Salamon and J. Landers. Operando X-ray absorption spectroscopy (XAS) measurements were performed by F. T. Haase, J. Timoshenko, C. Rettenmaier, H. S. Jeon and A. Bergmann. The work was supervised by B. Roldan Cuenya, H. Wende and S. Schulz. The manuscript was written by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) under Project 388390466 with the collaborative research center/transregio TRR 247 “Heterogeneous Oxidation Catalysis in the Liquid Phase”; subprojects B2 (HW), A4 (BRC) and C3 (StS). We are also thankful to Dr Heidelmann (DFG core facility ICAN, University of Duisburg-Essen) for TEM characterization. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III and we would like to thank Dr Vadim Murzin and Dr Wolfgang Caliebe for using the P64 beamline.Notes and references
- S. Trasatti, J. Electroanal. Chem., 1999, 476, 90 CrossRef CAS
.
- X. Xu, Z. Zhong, X. Yan, L. Kang and J. Yao, J. Mater. Chem. A, 2018, 6, 5999 RSC
- F. Yang, K. Sliozberg, I. Sinev, H. Antoni, A. Bähr, K. Ollegott, W. Xia, J. Masa, W. Grünert, B. Roldan Cuenya, W. Schuhmann and M. Muhler, ChemSusChem, 2017, 10, 156 CrossRef CAS PubMed
- Y. R. Hong, S. Mhin, K. M. Kim, W. S. Han, H. Choi, G. Ali, K. Y. Chung, H. J. Lee, S. I. Moon, S. Dutta, S. Sun, Y.-G. Jung, T. Song and H. Han, J. Mater. Chem. A, 2019, 7, 3592 RSC
- P. Mei, Y. Yamauchi, M. Pramanik, A. Fatehmulla, A. M. Adhafiri, W. A. Farooq, Y. Bando, M. J. A. Shiddiky, Y. V. Kaneti, J. Lin and Y. Kim, Electrochem. Commun., 2019, 104, 106476 CrossRef CAS
- J. Masa, S. Barwe, C. Andronescu, I. Sinev, A. Ruff, J. Kolleboyina, K. Elumeeva, B. Konkena, B. Roldan Cuenya and W. Schuhmann, ACS Energy Lett., 2016, 1, 1192 CrossRef CAS
- Y. Shao, X. Xiao, Y. P. Zhu and T. Y. Ma, Angew. Chem., Int. Ed., 2019, 58, 14599 CrossRef CAS PubMed
- J. Masa, I. Sinev, H. Mistry, E. Ventosa, M. de la Mata, J. Arbiol, M. Muhler, B. Roldan Cuenya and W. Schuhmann, Adv. Energy Mater., 2017, 7, 1700381 CrossRef
- J. Masa, C. Andronescu, H. Antoni, S. Seisel, K. Elumeeva, S. Barwe, S. Marti-Sanchez, J. Arbiol, I. Sinev, B. Roldan Cuenya, M. Muhler and W. Schuhmann, ChemElectroChem, 2019, 6, 235 CrossRef CAS
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277 CrossRef CAS PubMed
- X. Zhou, X. Shen, Z. Xia, Z. Zhang, J. Li, Y. Ma and Y. Qu, ACS Appl. Mater. Interfaces, 2015, 7, 20322 CrossRef CAS PubMed
- R. Li, D. Zhou, J. Luo, W. Xu, J. Li, S. Li, P. Cheng and D. Yuan, J. Power Sources, 2017, 341, 250 CrossRef CAS
- C.-F. Li, J.-W. Zhao, L.-J. Xie, J.-Q. Wu, Q. Ren, Y. Wang and G.-R. Li, Angew. Chem., Int. Ed., 2021, 60, 18129 CrossRef CAS PubMed
- J.-W. Zhao, C.-F. Li, Z.-X. Shi, J.-L. Guan and G.-R. Li, Research, 2020, 6961578 CAS
- H. Xu, Z.-X. Shi, Y.-X. Tong and G.-R. Li, Adv. Mater., 2018, 30, 1705442 CrossRef PubMed
- S.-H. Ye, Z.-X. Shi, J.-X. Feng, Y.-X. Tong and G.-R. Li, Angew. Chem., Int. Ed., 2018, 57, 2672 CrossRef CAS PubMed
- S. Han, S. Liu, S. Yin, L. Chen and Z. He, Electrochim. Acta, 2016, 210, 942 CrossRef CAS
- A. El Arrassi, Z. Liu, M. V. Evers, N. Blanc, G. Bendt, S. Saddeler, D. Tetzlaff, D. Pohl, C. Damm, S. Schulz and K. Tschulik, J. Am. Chem. Soc., 2019, 141, 9197 CrossRef PubMed
- T. Quast, H. B. Aiyappa, S. Saddeler, P. Wilde, Y.-T. Chen, S. Schulz and W. Schuhmann, Angew. Chem., Int. Ed., 2021, 60, 3576 CrossRef CAS PubMed
- J. Huo and M. Wei, Mater. Lett., 2009, 63, 1183 CrossRef CAS
- K. F. Ortega, S. Anke, S. Salamon, F. Özcan, J. Heese, C. Andronescu, J. Landers, H. Wende, W. Schuhmann, M. Muhler, T. Lunkenbein and M. Behrens, Chem.–Eur. J., 2017, 23, 12443 CrossRef CAS PubMed
- A. Hajalilou, S. A. Mazlan, M. Abbasi and H. Lavvafi, RSC Adv., 2016, 6, 89510 RSC
- P. C. R. Varma, R. S. Manna, D. Banerjee, M. R. Varma, K. G. Suresh and A. K. Nigam, J. Alloys Compd., 2008, 453, 298 CrossRef CAS
- V. Georgiadou, C. Kokotidou, B. Le Droumaguet, B. Carbonnier, T. Choli-Papadopoulou and C. Dendrinou-Samara, Dalton Trans., 2014, 43, 6377 RSC
- Y. Eom, M. Abbas, H. Noh and C. Kim, RSC Adv., 2016, 6, 15861 RSC
- N. V. Long, Y. Yang, T. Teranishi, C. M. Thi, Y. Cao and M. Nogami, RSC Adv., 2015, 5, 56560 RSC
- C. An, Y. Wang, Y. Huang, Y. Xu, C. Xu, L. Jiao and H. Yuan, CrystEngComm, 2014, 16, 385 RSC
- S. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS PubMed
- K. M. Nam, J. H. Shim, D. W. Han, H. S. Kwon, Y. M. Kang, Y. Li, H. Song, W. S. Seo and J. T. Park, Chem. Mater., 2010, 22, 4446 CrossRef CAS
- R. Bussamara, W. W. M. Melo, J. D. Scholten, P. Migowski, G. Marin, M. J. M. Zapata, G. Machado, S. R. Teixeira, M. A. Novak and J. Dupont, Dalton Trans., 2013, 42, 14473 RSC
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273 CrossRef CAS PubMed
- K. M. Nam, J. H. Shim, D. W. Han, H. S. Kwon, Y. M. Kang, Y. Li, H. Song, W. S. Seo and J. T. Park, Chem. Mater., 2010, 22, 4446 CrossRef CAS
- K. Parekh, R. V. Upadhyay, L. Belova and K. V. Rao, Nanotechnology, 2006, 17, 5970 CrossRef CAS
- L. Ajroudi, N. Mliki, L. Bessais, V. Madigou, S. Villain and C. Leroux, Mater. Res.
Bull., 2014, 59, 49 CrossRef CAS
- L. Kampermann, J. Klein, J. Korte, O. Kowollik, O. Pfingsten, T. Smola, S. Saddeler, T. H. Piotrowiak, S. Salamon, J. Landers, H. Wende, A. Ludwig, S. Schulz and G. Bacher, J. Phys. Chem. C, 2021, 125, 14356 CrossRef CAS
- K. Chakrapani, G. Bendt, H. Hajiyani, I. Schwarzrock, T. Lunkenbein, S. Salamon, J. Landers, H. Wende, R. Schlögl, R. Pentcheva, M. Behrens and S. Schulz, ChemCatChem, 2017, 9, 2988 CrossRef CAS
- K. Chakrapani, G. Bendt, H. Hajiyani, T. Lunkenbein, M. T. Greiner, L. Masliuk, S. Salamon, J. Landers, R. Schlögl, H. Wende, R. Pentcheva, S. Schulz and M. Behrens, ACS Catal., 2018, 8, 1259 CrossRef CAS
- Q. Song and Z. J. Zhang, J. Am. Chem. Soc., 2004, 126, 6164 CrossRef CAS PubMed
- K. M. Nam, J. H. Shim, D. W. Han, H. S. Kwon, Y. M. Kang, Y. Li, H. Song, W. S. Seo and J. T. Park, Chem. Mater., 2010, 22, 4446 CrossRef CAS
- L. T. Lu, N. T. Dung, L. D. Tung, C. T. Thanh, O. K. Quy, N. V. Chuc, S. Maenosono and N. T. K. Thanh, Nanoscale, 2015, 7, 19596 RSC
- Y. Eom, M. Abbas, H. Y. Noh and C. G. Kim, RSC Adv., 2016, 6, 15861 RSC
- Y. Kumar, A. Sharma and P. M. Shirage, RSC Adv., 2017, 7, 55778 RSC
- Y. Kumar, A. Sharma, M. A. Ahmed, S. S. Mali, C. K. Hong and P. M. Shirage, New J. Chem., 2018, 42, 15793 RSC
- M. Hennous, E. V. Ramana, D. M. Tobaldi, B. F. O. Costa, M. A. Valente, J. Labrincha and M. Karmaoui, New J. Chem., 2019, 43, 10259 RSC
- S. Saddeler, U. Hagemann and S. Schulz, Inorg. Chem., 2020, 59, 10013 CrossRef CAS PubMed
- I.-H. Jung, S. A. Decterov, A. D. Pelton, H.-M. Kim and Y.-B. Kang, Acta Mater., 2004, 5, 507 CrossRef
- H. Jalili, B. Aslibeiki, A. Ghotbi Varzaneh and V. A. Chernenko, Beilstein J. Nanotechnol., 2019, 10, 1348 CrossRef CAS PubMed
- S. Mozaffari, W. Li, C. Thompson, S. Ivanov, S. Seifert, B. Lee, L. Kovarik and A. M. Karim, Nanoscale, 2017, 9, 13772–13785 RSC
- L. Lukashuk, N. Yigit, H. Li, J. Bernardi, K. Fottinger and G. Rupprechter, Catal. Today, 2019, 336, 139 CrossRef CAS
- Y. G. Borod'ko, S. I. Vetchinkin, S. L. Zimont, I. N. Ivleva and Y. M. Shul'ga, Chem. Phys. Lett., 1976, 42, 264 CrossRef
- R. Ji, C. Cao, Z. Chen, H. Zhai and J. Bai, J. Mater. Chem. C, 2014, 2, 5944 RSC
- W. Zhang, X. Li, R. Zou, H. Wu, H. Shi, S. Yu and Y. Liu, Sci. Rep., 2015, 5, 11129 CrossRef PubMed
- L. Ouyang, L. Zhu, J. Jiang and H. Tang, Anal. Chim. Acta, 2014, 816, 41 CrossRef CAS PubMed
- B. Rivas-Murias and V. Salgueiriño, J. Raman Spectrosc., 2017, 48, 837 CrossRef CAS
- M. A. Laguna-Bercero, M. L. Sanjuán and R. I. Merino, J. Phys.: Condens. Matter, 2007, 19, 186217 CrossRef CAS PubMed
- N. Bahlawane, P. H. T. Ngamou, V. Vannier, T. Kottke, J. Heberle and K. Kohse-Höinghaus, Phys. Chem. Chem. Phys., 2009, 11, 9224 RSC
- A. V. Raut, R. S. Barkule, D. R. Shengule and K. M. Jadhav, J. Magn. Magn. Mater., 2014, 358–359, 87 CrossRef CAS
- Y. Hao, X. Wang and L. Li, Nanoscale, 2014, 6, 7940 RSC
- A. Kasprzak, M. Popławska, M. Bystrzejewski, O. Łabędź and I. P. Grudziński, RSC Adv., 2015, 5, 85556 RSC
- G. Concas, G. Spano, C. Cannas, A. Musinu, D. Peddis and G. Piccaluga, J. Magn. Magn. Mater., 2009, 321, 1893 CrossRef CAS
- N. Fontaíña-Troitiño, S. Liébana-Viñas, B. Rodríguez-González, Z.-A. Li, M. Spasova, M. Farle and V. Salgueiriño, Nano Lett., 2014, 14, 640 CrossRef PubMed
- C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef CAS PubMed
- M. S. Ahmed, B. Choi and Y.-B. Kim, Sci. Rep., 2018, 8, 2543 CrossRef PubMed
- H. N. Nong, L. J. Falling, A. Bergmann, M. Klingenhof, H. P. Tran, C. Spöri, J. Timoshenko, G. Zichittella, A. Knop-Gericke, S. Piccinin, J. Pérez-Ramírez, B. Roldan Cuenya, R. Schlögl, P. Strasser, D. Teschner and T. E. Jones, Nature, 2020, 587, 408 CrossRef CAS PubMed
- A. Bergmann, T. E. Jones, E. Martinez Moreno, D. Teschner, P. Chernev, M. Gliech, T. Reier, H. Dau and P. Strasser, Nat. Catal., 2018, 1, 711 CrossRef CAS
- E. A. Schultz-Sikma, H. M. Joshi, Q. Ma, K. W. Macrenaris, A. L. Eckermann, V. P. Dravid and T. J. Meade, Chem. Mater., 2011, 23, 2657 CrossRef CAS PubMed
- J. Fester, M. Garcia-Melchor, A. S. Walton, M. Bajdich, Z. Li, L. Lammich, A. Vojvodic and J. V. Lauritsen, Nat. Commun., 2017, 8, 14169 CrossRef CAS PubMed
- J. Kim, W. H. Doh, Y. Kim, K.-J. Kim and J. Y. Park, ACS Appl. Energy Mater., 2019, 2, 8580 CrossRef CAS
- P. F. Liu, S. Yang, L. R. Zheng, B. Zhang and H. G. Yang, J. Mater. Chem. A, 2016, 4, 9578 RSC
-
J. Dittmer, L. Iuzzolino, W. Dörner, H.-F. Nolting, W. Meyer-Klaucke and H. Dau, Photosynthesis: Mechanisms and Effects, 1998, p. 1339 Search PubMed
- H. Dau, P. Liebisch and M. Haumann, Anal. Bioanal. Chem., 2003, 376, 562 CrossRef CAS PubMed
- M. Risch, K. Klingan, F. Ringleb, P. Chernev, I. Zaharieva, A. Fischer and H. Dau, ChemSusChem, 2012, 5, 542 CrossRef CAS PubMed
- A. Bergmann, E. Martinez-Moreno, D. Teschner, P. Chernev, M. Gliech, J. F. de Araújo, T. Reier, H. Dau and P. Strasser, Nat. Commun., 2015, 6, 8625 CrossRef CAS PubMed
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Additional analytical data including PXRD, TEM, Mössbauer, XANES, EXAFS, magnetization curves. See DOI: 10.1039/d1ta06568h |
| This journal is © The Royal Society of Chemistry 2021 |