
Insights into iodoplumbate complex evolution of precursor solutions for perovskite solar cells: from aging to degradation
Bo
Li
a,
Qilin
Dai
 *b,
Sining
Yun
*c and
Jianjun
Tian
*a
*b,
Sining
Yun
*c and
Jianjun
Tian
*a
aInstitute of Advanced Materials and Technology, University of Science and Technology Beijing, 100083, China. E-mail: tianjianjun@mater.ustb.edu.cn
bDepartment of Chemistry, Physics, and Atmospheric Sciences, Jackson State University, Jackson, MS 39217, USA
cFunctional Materials Laboratory (FML), School of Materials Science and Engineering, Xi'an University of Architecture and Technology, Xi'an, Shaanxi 710055, China
First published on 15th February 2021
Abstract
The power conversion efficiency (PCE) of solution-processed perovskite solar cells (PSCs) is significantly influenced by the characteristics of the perovskite precursor solutions. Iodoplumbate complexes such as PbI2, PbI3−, and PbI42− present in perovskite precursor solution in equilibria dictate the quality of perovskite films and the corresponding optoelectrical performance of PSCs. Meanwhile, the solvent and additive molecules compete with I− to coordinate with Pb2+ and shift the chemical equilibria based on their coordination capabilities. The proportion and species of iodoplumbate complexes have a time-dependent influence on the subsequent perovskite crystallization and the quality of perovskite films. This review presents the impact of the evolution of the characteristics of a perovskite precursor solution driven by the components of the precursor solution, aging, degradation, and humidity on the physical properties of perovskite films and the photovoltaic performance of PSCs. A brief outlook on further improving the reproducibility window for PSCs fabricated by the solution process is provided based on the current research progress in terms of perovskite precursor evolution in PSCs.
 Bo Li | Bo Li earned his PhD degree from the University of Science and Technology Beijing in 2019 under the supervision of Prof. Jianjun Tian. His study was focused on the fabrication of highly efficient perovskite solar cells in the Laboratory of Optoelectronic Materials and Devices. From 2019 to 2020, he worked as a postdoctoral researcher at Jackson State University USA. His research focuses on perovskite precursor solution chemistry. |
 Qilin Dai | Dr Qilin Dai is an assistant professor of physics at Jackson State University, USA. He earned his PhD degree in Condensed Matter Physics from the Chinese Academy of Sciences in 2009, and then served as a postdoctoral research associate in the USA at Florida State University and the University of Wyoming. His research interests include (1) perovskite solar cells with high stability and high efficiency and (2) nanosized metal oxide synthesis for energy storage devices. |
 Sining Yun | Sining Yun is a Professor in the School of Materials & Mineral Resources, Xi'an University of Architecture and Technology, China. He obtained his PhD degree from Xi'an Jiaotong University in 2007 and was a postdoctoral fellow at Yonsei University (Korea). He is a visiting professor at the State Key Laboratory of Fine Chemicals (China), University of Reading (UK), and Prof. Michael Grätzel and Prof. Anders Hagfeldt's laboratory at EPFL (Switzerland). He is currently a Research Director and Group Leader of the Functional Materials Laboratory and the Key Laboratory of Nanomaterials and Nanotechnology of Shaanxi Province (China). His research focuses on solar energy and biomass energy. Details can be found at: http://xy.xauat.edu.cn/gnclyjs/listyjsgk.asp?id=262%26bh=2080. |
 Jianjun Tian | Jianjun Tian is presently a professor at the Institute for Advanced Materials and Technology, University of Science and Technology Beijing. His current research is focused on the development of colloidal quantum dots and novel perovskite materials for the optoelectronic applications including the next generation solar cells, light-emitting diodes, and photodetectors. |
1. Introduction
Since the discovery of methylammonium lead halides (MAPbX3, X = Cl−, Br−, and I−) in 1978, the excellent optical and electronic properties of organic–inorganic hybrid metal halide perovskite materials have been intensively investigated for several decades.1–4 Polycrystalline MAPbI3 exhibits a direct bandgap (∼1.55 eV),5 high optical absorption coefficient (1.5 × 104 cm−1 at 550 nm),6 low Urbach energy (∼15 meV),7 small exciton binding energy (2–75 meV),8,9 high charge carrier mobility (10–24 cm2 V−1 S−1),9,10 long carrier diffusion length (up to 1 μm),11 low trap density (1015 to 1016 cm3),9,12,13 and low crystallization activation energy (97.3 kJ mol−1),14 making it a promising semiconductor material for light-energy conversion.15 The pioneering work on hybrid lead halide perovskite sensitized solar cells was reported by Miyasaka and co-workers in 2009;16 the devices showed poor stability due to the rapid dissolution of perovskite nanocrystalline particles in the liquid electrolyte that was commonly used in dye-sensitized solar cells.17 Interest in photovoltaics has increased sharply since the emergence of solid-state perovskite solar cells (PSCs) in 2012.6,18–24 So far, highly efficient PSCs have achieved a certified power conversion efficiency (PCE) of 25.5%.25 However, large PCE differences were reported among worldwide research groups, even adopting the same device architecture and solution process.26–29 There is an increasing consensus that the characteristics of the perovskite precursor solution are predominately responsible for the PCE differences.Recently, perovskite precursor solution chemistry has received intensive attention concerning its relationship with the physical properties of perovskite films.30–33 Perovskite precursor solutions are known as colloidal dispersions on the mesoscale. The inorganic component PbI2 undergoes complexation with the organic component CH3NH3I (MAI) to form several iodoplumbate complexes, which serve as precursors in the solution. Yan and co-workers first revealed that MAI could coordinate with the trigonal PbI2 colloid to form tetragonal iodoplumbate coordination complexes, or even corner-sharing and individual PbI64− octahedral units with a maximum coordination number, depending on the content of MAI. It indicates that the colloidal framework of iodoplumbate coordination complexes could be structurally tuned by the stoichiometric ratio of PbI2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MAI, which will determine the final perovskite morphology and crystallinity.30 Besides, the solvent–solute and additive-solute interactions also affect the species of iodoplumbate complexes in the precursor solutions and play a critical role in the subsequent perovskite crystallization,34–36 since the solvent and additive molecules are prone to compete with I− for coordination sites around Pb2+ according to their coordination capabilities, and thus control the perovskite formation.37 For example, the incorporation of 10 mM I3− into the perovskite precursor solution was reported to produce smaller high-valent clusters, which were closely related to the enhanced crystallinity, homogeneous chemical composition, and less defect concentration of the resulting perovskite films, as well as the improved efficiency of PSCs.29 Moreover, the status of perovskite precursor solutions changes during storage. High-quality perovskite films can only be achieved within the optimal precursor solution aging time because the perovskite precursor solutions suffer from degeneration during long-term aging.38–40 It is noteworthy that many different variations of solution-based deposition techniques of perovskite films have been investigated over the years, such as spin coating,41 spray coating,42 slot-die coating,43 doctor-blade coating,44,45 and inkjet printing;46,47 the optimal precursor solution concentration, stoichiometry, and composition may vary to meet desired crystallization kinetics in each case. However, for solution-processed PSCs, the characteristics of the perovskite precursor solution have been demonstrated to constantly evolve until reaching chemical equilibria.38,39,48 Although several review articles have described the fundamental understanding of the nucleation and growth mechanism and microstructural evolution of perovskite from the precursor solution to a crystalline solid phase,32,33,49 only a few reports focused on the time-dependent influence of the characteristics of the perovskite precursor solution on the subsequent perovskite film quality and the corresponding device performance.
:MAI, which will determine the final perovskite morphology and crystallinity.30 Besides, the solvent–solute and additive-solute interactions also affect the species of iodoplumbate complexes in the precursor solutions and play a critical role in the subsequent perovskite crystallization,34–36 since the solvent and additive molecules are prone to compete with I− for coordination sites around Pb2+ according to their coordination capabilities, and thus control the perovskite formation.37 For example, the incorporation of 10 mM I3− into the perovskite precursor solution was reported to produce smaller high-valent clusters, which were closely related to the enhanced crystallinity, homogeneous chemical composition, and less defect concentration of the resulting perovskite films, as well as the improved efficiency of PSCs.29 Moreover, the status of perovskite precursor solutions changes during storage. High-quality perovskite films can only be achieved within the optimal precursor solution aging time because the perovskite precursor solutions suffer from degeneration during long-term aging.38–40 It is noteworthy that many different variations of solution-based deposition techniques of perovskite films have been investigated over the years, such as spin coating,41 spray coating,42 slot-die coating,43 doctor-blade coating,44,45 and inkjet printing;46,47 the optimal precursor solution concentration, stoichiometry, and composition may vary to meet desired crystallization kinetics in each case. However, for solution-processed PSCs, the characteristics of the perovskite precursor solution have been demonstrated to constantly evolve until reaching chemical equilibria.38,39,48 Although several review articles have described the fundamental understanding of the nucleation and growth mechanism and microstructural evolution of perovskite from the precursor solution to a crystalline solid phase,32,33,49 only a few reports focused on the time-dependent influence of the characteristics of the perovskite precursor solution on the subsequent perovskite film quality and the corresponding device performance.
In this review, we explore the impact of the characteristics of perovskite precursors on the photovoltaic performance of PSCs. A fundamental understanding of the intrinsic relationships between the perovskite precursor solution chemistry and the physical properties of the perovskite films is presented. Then we summarize the evolution of iodoplumbate coordination complexes in perovskite precursor solutions driven by aging, degradation, and humidity with the consideration of their influence on the perovskite film quality. Finally, we provide a brief outlook on further improving the reproducibility window for PSCs fabricated by the solution process.
2. Perovskite precursor solution chemistry
2.1 Evolution of iodoplumbate complexes
The studies on the coordination chemistry of lead(II) have been well established over the past decade. The 6 s2 electron pair of lead(II) is stereochemically active and plays a role in the complexation with halide ions and solvents.50,51 In 1940, Lanford and co-workers found that the solubility of lead iodide (PbI2) in the solution increased in the presence of iodide ions, which could be explained by the formation of the complexes of PbI3− and PbI42−.52 Similar complexation between PbI2 and halide ions occurs in perovskite precursor solutions. Fig. 1(a) shows absorption spectra of a PbI2 solution with gradual increase of the MAI concentration; the appearance of two new concentration-dependent absorption bands with maxima at 370 and 425 nm confirms the presence of two separate complexes of PbI3− and PbI42−, and the equilibria between PbI2, PbI3−, and PbI42− can be described as eqn (1) and (2)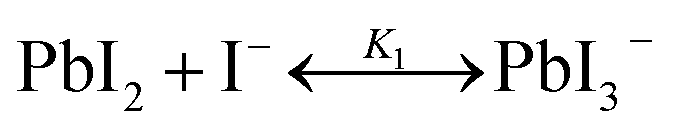 | (1) |
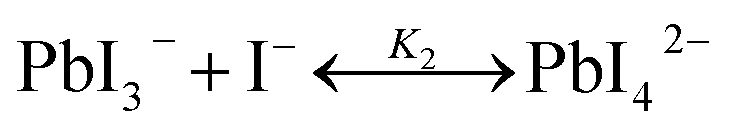 | (2) |
:MAI = 1:1, indicating that Pb2+ is not fully coordinated with six I− ions in the standard perovskite precursor solution. Excess MAI or other iodide sources are needed to achieve the full coordination of Pb2+. The results are consistent with the fact that solvent molecules can compete with I− for the coordination sites of Pb2+, depending on their coordinating capabilities, which inhibit the formation of high order iodoplumbate complexes. Based on density functional theory (DFT) calculations, the computational absorption spectra based on the optimized geometries of iodoplumbate complexes, including PbI2DMF3, [PbI3DMF]−, and PbI42− (Fig. 1(c)), show characteristic absorption at 311 nm, 375 nm, and 416 nm, respectively, which closely match the experimental results as shown in Fig. 1(a).55 The result further confirms the presence of solvated iodoplumbate complexes in the precursor solution.
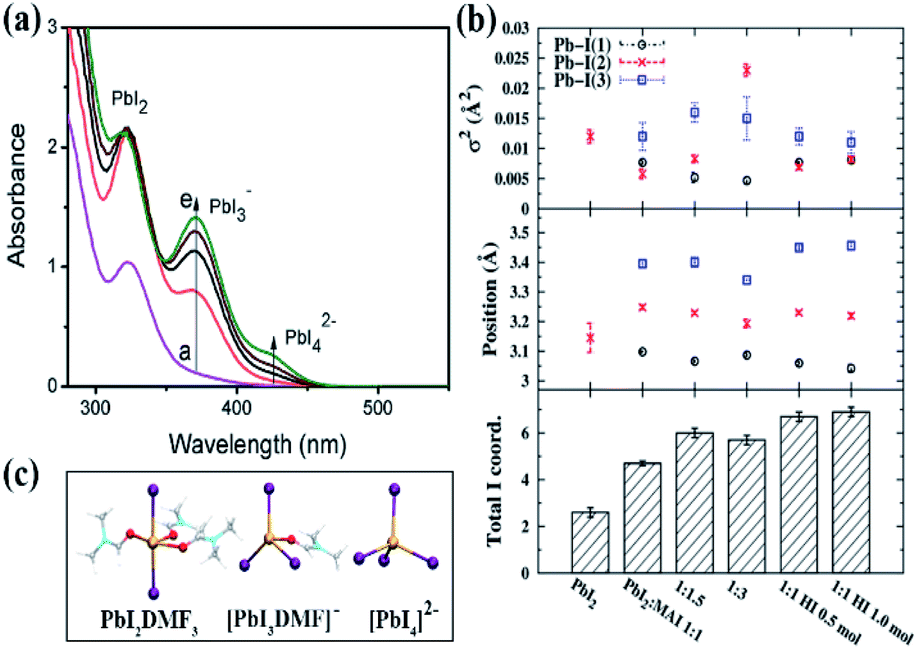 | ||
| Fig. 1 (a) Absorption spectra of 250 μM PbI2 solution in dimethylformamide with increasing concentration of MAI from 6 mM to 24 mM. Reprinted from ref. 53 with permission from the Royal Society of Chemistry. (b) Real-space extended X-ray absorption fine structure (EXAFS) fit parameters. The top panel plots the correlated Debye–Waller factor (σ2), the middle panel the Pb–I components' positions from the absorbing Pb atom, and the bottom panel the total iodide coordination number of the Pb atoms in solution. Note that the disorder increases as the value of σ2 increases and the Pb atoms in both PbI2 and MAPbI3 exhibit octahedral iodide coordination (PbI6). Reprinted from ref. 54 with permission from the American Chemical Society. (c) Optimized geometries for selected [PbImXn]2−m iodoplumbate complexes in DMF. Reprinted from ref. 55 with permission from the American Chemical Society. | ||
To establish the atomic origin and the evolution of these iodoplumbate complexes, Ahlawat and co-workers employed well-tempered metadynamics simulations to reveal the time evolution of iodoplumbate complexes in a perovskite precursor solution with γ-butyrolactone (GBL) as the solvent.56 In a well-equilibrated solution where the MA+, Pb2+, and I− ions are initially homogeneously distributed in GBL, the iodoplumbate complexes of PbI2, PbI3−, and PbI42− emerge spontaneously. The increased coordination of Pb2+ with I− ions contributes to the formation of face- or edge-sharing PbI64− octahedral clusters after ∼130–140 ns (Fig. 2). Then MA+ ions surround these clusters to balance the negative charge. The edge-sharing PbI64− octahedra start to transform into a PbI2-like structure by sharing at least three edges with neighboring PbI64− octahedra after 150 ns. As a result, the open structure enables the diffusion of MA+ ions in these spaces and eventually transform into PbI42− tetrahedra. Around 180 ns, this tetrahedral structure rearranges into corner-sharing PbI64− octahedra with the MA+ ions in the center. These simulation results are in general agreement with the experimental observation of PbI64− octahedral cage nanoparticles during perovskite crystallization.57 Finally, the amorphous clusters partially transform into the perovskite structure accompanied by the growth of perovskite crystals by the arrangement of ions around the initial nucleus. Based on the discussion above, we can now conclude that the perovskite precursor solution consists of solvated iodoplumbate complexes in a polar organic solvent or mixed solvents, and the species of iodoplumbate complexes evolve with the stoichiometric ratio or concentration of precursors until reaching equilibria.
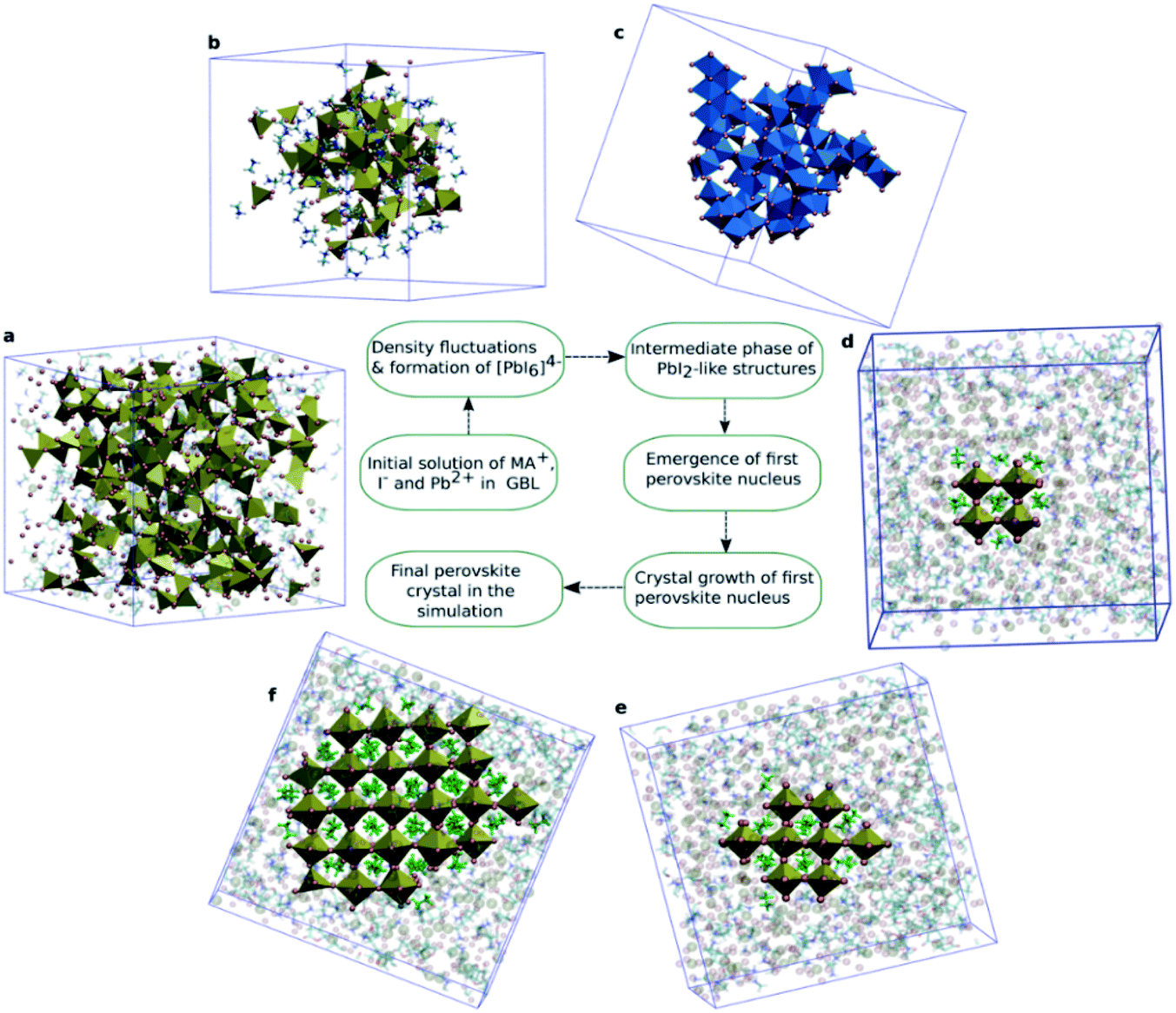 | ||
| Fig. 2 (a) Initial solution of MA+, I−, and Pb2+ in GBL. MA+ and GBL are shown semitransparent to visualize the random distribution of Pb2+ and I− in solution. (b) Initial cluster formation of Pb2+ and I− surrounded by MA+ ions. (c) Edge-sharing [PbI6]4− octahedra. (d) First perovskite nucleus observed in the solution. (e) Growth of the initial nucleus. (f) The largest perovskite crystal in the simulations. Reprinted from ref. 56 with permission from the American Chemical Society. | ||
2.2 Relationships with the physical properties of perovskite films
With the in-depth understanding of the perovskite precursor solution chemistry, there is an increasing consensus that the subtle changes of precursor solution parameters, including the precursor solution concentration, stoichiometry, temperature, and aging time, can result in a substantial variation in the physical properties of perovskite films. Fig. 3 summarizes the variability that often occurs in the physical properties of perovskite films induced by the characteristics of precursor solutions, such as the phase transition, morphology, crystallinity, component, and defect density.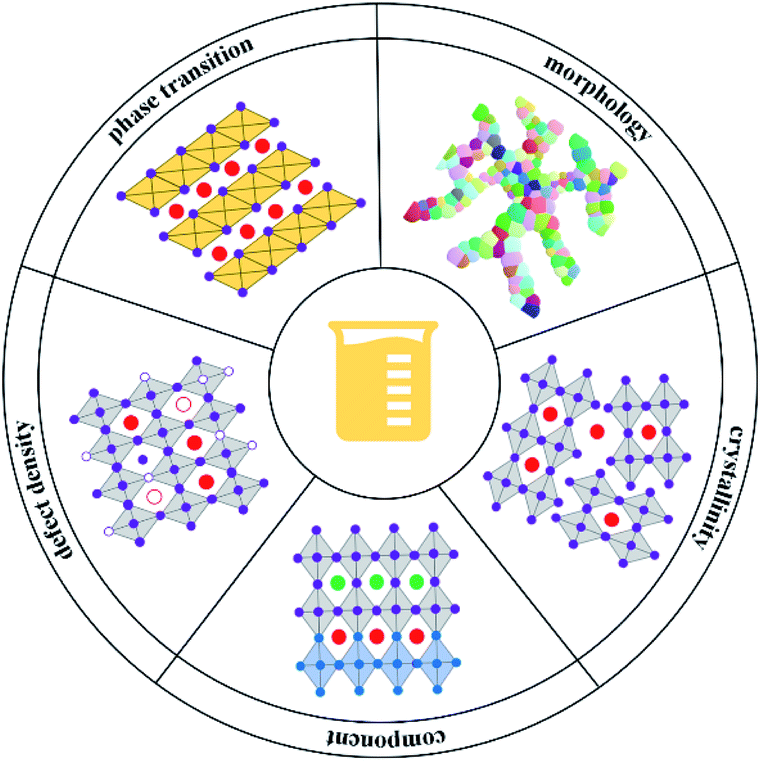 | ||
| Fig. 3 Schematic representation of the influence of the perovskite precursor solution on the phase transition, morphology, crystallinity, component, and defect density of perovskite films. | ||
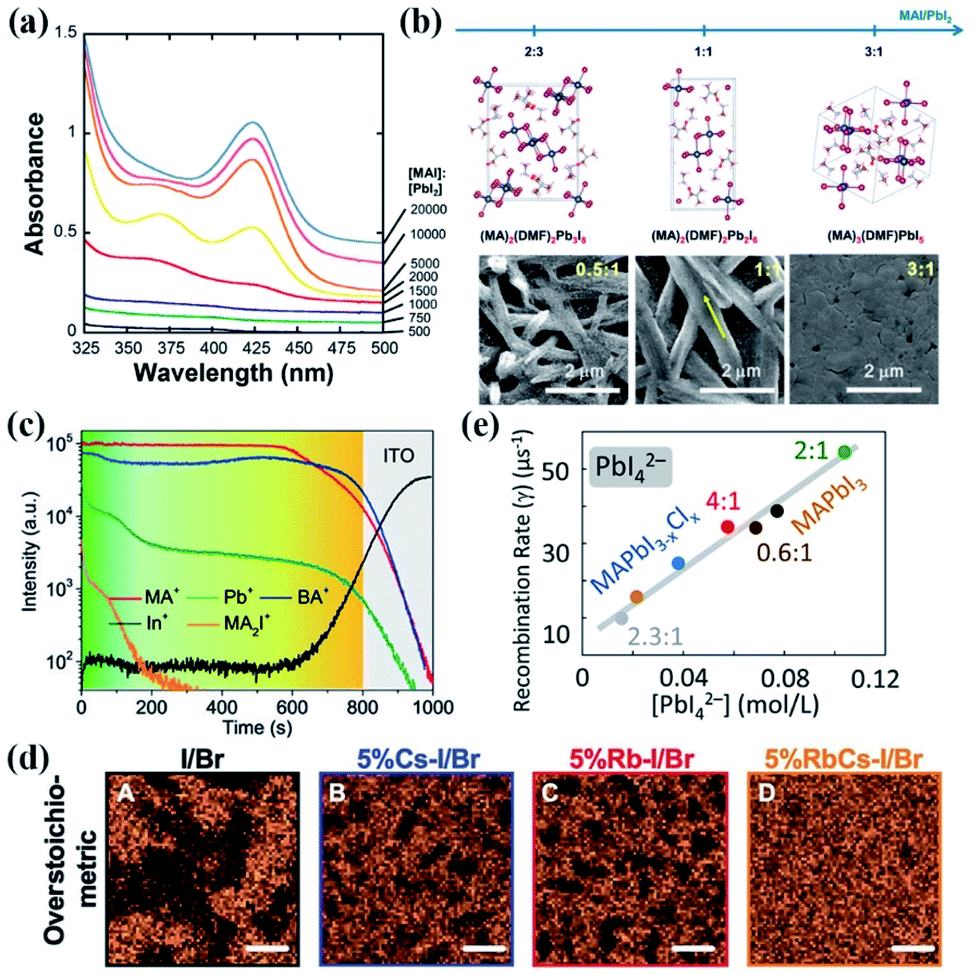 | ||
| Fig. 4 (a) Absorbance of 0.05 mM PbI2 with increasing [MAI] in DMPU. Spectral features corresponding to PbI3− at 370 nm and PbI42− at 425 nm are evident only at [MAI]:[PbI2] equal to or exceeding 1500. For [MAI]:[PbI2] less than 1500, the absorbances of PbI3− and PbI42− are negligible. Reprinted from ref. 37 with permission from the American Chemical Society. (b) Crystal structure motifs of three intermediate phases: PbI2-rich (MA)2(DMF)2Pb3I8, stoichiometric (MA)2(DMF)2Pb2I6, and MAI-rich (MA)3(DMF)PbI5 and the corresponding SEM micrographs of calcined thin films with CH3NH3I coordination engineering. Reprinted from ref. 68 with permission from the American Chemical Society. (c) ToF-SIMS patterns of BA2MA3Pb4I13 films prepared at 70 °C. Reprinted from ref. 90 with permission from Wiley. (d) X-ray fluorescence mapping indicates the heterogeneous distribution of Br as a function of alkali metal stoichiometric composition of the perovskite films. Scale bars, 2 mm. Reprinted from ref. 91 with permission from the American Association for the Advancement of Science. (e) Correlation of iodoplumbate and iodide species present in precursor solutions versus charge recombination rates measured in the perovskite films deposited from the corresponding solutions. Reprinted from ref. 96 with permission from the American Chemical Society. | ||
Solvent engineering and additive strategies have been successfully developed to modulate the precursor coordination and achieve controlled perovskite nucleation and crystal growth.64,69–71 To date, dimethyl sulfoxide (DMSO) as the dominant coordinating molecule is used to retard the fast interaction between MAI and PbI2, leading to the formation of a PbI2·MAI·DMSO intermediate phase.58,59,72 By controlling the mole ratio of DMSO:PbI2 to 10:1 in perovskite precursor solutions, a pure (MA)2Pb3I8(DMSO)2 intermediate phase was obtained.73 This induced the up-growth feature of perovskite films without horizontal grain boundaries. However, this retarding perovskite crystallization strategy often needs the extra steps of anti-solvent extraction,58 gas-quenching,74 or vacuum-assisted drying19 to rapidly remove the solvents from wet films, which increases the complexity of the film deposition process and is detrimental to device reproducibility. By the integration of solvent engineering and additive strategies, a spontaneously supersaturated nucleation strategy was devised to fabricate high-quality perovskite films without any additional steps.75 The incorporation of strong coordination additives into perovskite precursor solutions with a weak coordination solvent of 2-methoxyethanol (2-ME) could suppress the interaction of Pb2+ with I− and solvent, leading to the precipitation of PbI2-like coordination complexes in solutions. Volatile methylamine ethanol was further employed as a second solvent to dissolve these PbI2-like coordination complexes. As a result, the rapid dissolution-precipitation behavior of the precursor films could be triggered by the volatilization of MA ethanol, thereby achieving perovskite films with a uniform distribution of small-sized intermediate phase grains. During annealing, the growth of densely packed nanometer-sized intermediate phase grains results in mirror-like and compact large-area perovskite films (10 cm × 10 cm) with enhanced crystallinity. Finally, an average PCE of 19.4% was achieved for small area PSCs (0.1 cm2) due to the good reproducibility of high-quality perovskite films.
In addition, we found that the introduction of methylammonium chloride (MACl) into the perovskite precursor solution significantly increased the colloidal cluster size through coordination interaction.13 The large colloidal clusters were orderly arranged on the substrate and grown to form large grains with an average size of 3 μm after annealing. Similar results were observed for the precursor solution with the addition of methylammonium thiocyanate (MASCN).79 Han and co-workers demonstrated that the perovskite grain size in the final films strongly depends on the sizes of iodoplumbate complexes in precursor solutions. The grain sizes of the corresponding perovskite films increased from 100 nm to 2 μm for 0–50% MASCN additive levels.
Apart from the A site cation-initiated phase segregation, the halide anions are reported to form some anion-poor/rich phases. In our recent work, the Cl-rich phase-initiated precipitation behavior of the precursor solution was observed in the perovskite precursor solution with a weak coordination solvent of 2-methoxyenthanol (2-ME), leading to the formation of a Cl-rich intermediate phase in the precursor films.75 With the help of synchrotron-based nano-X-ray fluorescence, Correa-Baena and co-workers reported that although enhanced efficiency can be achieved in the case of perovskite films prepared from over-stoichiometric PbX2 and AX (X = halide anion) precursor solutions, large clusters of lower Br content with sizes of 6–8 μm on the perovskite films are observed, while the area fraction reduces to 32%, 29%, and only ∼6% upon the addition of RbI, CsI, and a mixture of RbI and CsI (Fig. 4(d)), respectively.91 However, the incorporation of alkali metals with a higher concentration produces optoelectronically inactive, current blocking, and possibly recombination-active clusters on perovskite films, which is detrimental to the PSC performance. It is noteworthy that the segregation of highly concentrated Rb clusters was also observed on the perovskite films obtained from the precursor solution even with added 1% Rb. Therefore, careful attention to the composition difference in the mixed cation or anion perovskite films is required and will benefit the development of highly reproducible optoelectronic devices.
3. Perovskite precursor solution aging effect
As discussed above, the perovskite precursor solution is composed of several iodoplumbate complexes in a polar aprotic organic solvent or mixed solvents, in which I− ions and solvent molecules are prone to coordinate with the Pb2+ ions. The incorporation of additives is expected to shift the equilibria position depending on their coordination capabilities, thus changing the distribution proportion and species of iodoplumbate complexes until reaching new chemical equilibria. Therefore, the evolution of iodoplumbate complexes during precursor solution aging will have a time-dependent influence on the subsequent perovskite crystallization and the quality of perovskite films.3.1 Perovskite precursor aging
It was reported that single precursor chemicals, such as FAI, MABr, PbI2, and PbBr2, could not be fully dissolved in a mixture of DMF/DMSO solvents within 2 h at room temperature.97 In this regard, the perovskite precursor solution needs a certain aging treatment to form a homogeneous solution, and this will have a time-dependent impact on the perovskite film quality as well. For example, a three-week-aged CsPbIBr2 precursor solution was reported to allow the fabrication of pure-phase and full-coverage CsPbIBr2 films with a lower formation temperature, and the PCE increased to 6.55% compared to only 3.36% for the PSCs derived from the nonaged precursor solution.98In a recent study, Su and co-workers investigated the aging effect of perovskite precursor solutions stored at different temperatures on the photovoltaic performance of PSCs.38 As shown in Fig. 5(a–c), for the precursor solution aged at 4 °C, the average PCE increased from 16.48% to 17.29% after 24 h aging treatment, and then it decreased to 15.73% upon further aging for 96 h. The aging effect was increased in the case of the precursor solution aged at room temperature. 2–4 h aging yielded a higher PCE of ∼17.5% compared to 16.48% for the PSCs obtained from the fresh solution. A more increased aging effect was achieved upon aging the precursor solutions at an elevated temperature of 65 °C. The highest PCE of ∼17.1% was obtained from the solution aged for only 1.5 or 2 h. As expected, the optimal aging time is shortened as the aging temperature increases. Fig. 5(d) shows the UV-vis absorption spectra of the precursor solution at different aging times. The intensity of the distinct absorbance peak of PbI2[S]4 at 292 nm increased after aging for 1.5 h, and then it decreased for a longer aging of 3 h, indicating that the species of iodoplumbate complexes evolve with the precursor solution aging time. Since strong coordination solvent molecules can cause insufficient coordination between Pb2+ and I− ions as mentioned in Section 2.1.1, an excess PbI2 or MAI was introduced as an iodide source to induce the formation of sufficient PbI2[S]4 in precursor solutions, and in turn compensate for the aging effect. This is confirmed by little dependence of the intensity on the aging time from the UV-vis absorption spectra shown in Fig. 5(e and f). So, the proportion of iodoplumbate complexes in the precursor solutions is sensitive to the temperature and precursor stoichiometry and will impact the corresponding perovskite film quality.99,100 The optimal operating window for perovskite precursor solutions under different storage conditions should be carefully adjusted.
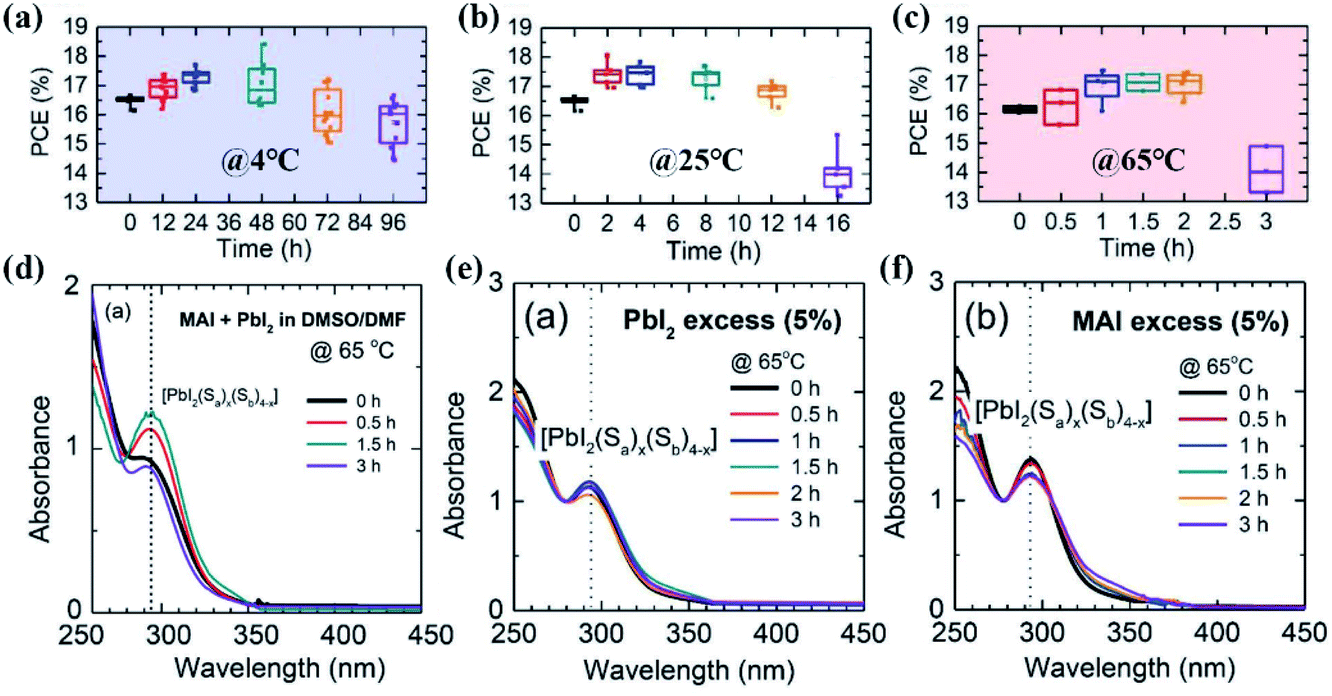 | ||
| Fig. 5 PCE of mesoscopic normal structured perovskite solar cells employing MAPbI3 films aged at (a) 4 °C, (b) room temperature (RT), and (c) 65 °C. The aging time was varied from 0 to 96 h at 4 °C, 16 h at RT, and 3 h at 65 °C. UV-vis absorption spectra of 10−4 M (MAI + PbI2) precursor solution dissolved in DMSO/DMF without (d) and with excess PbI2 (e) or excess MAI (f). Reprinted from ref. 38 with permission from Wiley. | ||
3.2 Additive induced aging
The incorporation of additives into perovskite precursor solution has been demonstrated to control the crystallization kinetics and improve the perovskite film quality.36,58,101–103 It is recognized that the additive induced aging effect is a critical parameter to affect the nucleation and growth of perovskite. For example, as shown in Fig. 6(a–e), the average grain-like size of perovskite films and PCE of the corresponding PSCs were found to increase with the precursor aging time for the precursor solution with the additive of methylammonium hydrochloride (MACl), accompanied by the increased colloidal size of iodoplumbate complexes in the precursor solution.39 The result is consistent with the finding that the perovskite film grain size strongly depends on the precursor aggregate size in precursor solutions.13,79 The authors speculated that the presence of nucleating iodoplumbate complexes and their size in the progressively aged precursor solution are directly correlated with the grain size in perovskite films. Based on the LaMer diagram, the nucleation of the iodoplumbate complexes induced by additives can be divided into three distinct processes: (1) prenucleation: a rapid increase in the concentration of free intermediates by dissolving the precursors and additive. (2) Burst nucleation: fast nucleation of iodoplumbate complex clusters coordinated with the additive. (3) Growth by diffusion: a decrease in the concentration of iodoplumbate complex clusters by forming larger crystallites. In parallel to the additive induced aging effect on the nucleation and growth of iodoplumbate complexes, the time-dependent reduction in the colloid size was reported for the precursor solution with a mixture of hydroiodic (HI) and hydrobromic (HBr) as additives (Fig. 6(f)).40 The authors demonstrated that these iodoplumbate complexes would serve as nucleation centers in the formation of perovskite films. Therefore, the density of nucleation sites of the precursor film would be reduced with the solution aging time for the solutions with the additives of HI and HBr, so that the growth of perovskite crystals would likely be dominant during perovskite formation. As a result, larger polycrystalline grain domains with preferred orientation and higher mobility were obtained from the aged precursor solution (Fig. 6(g)).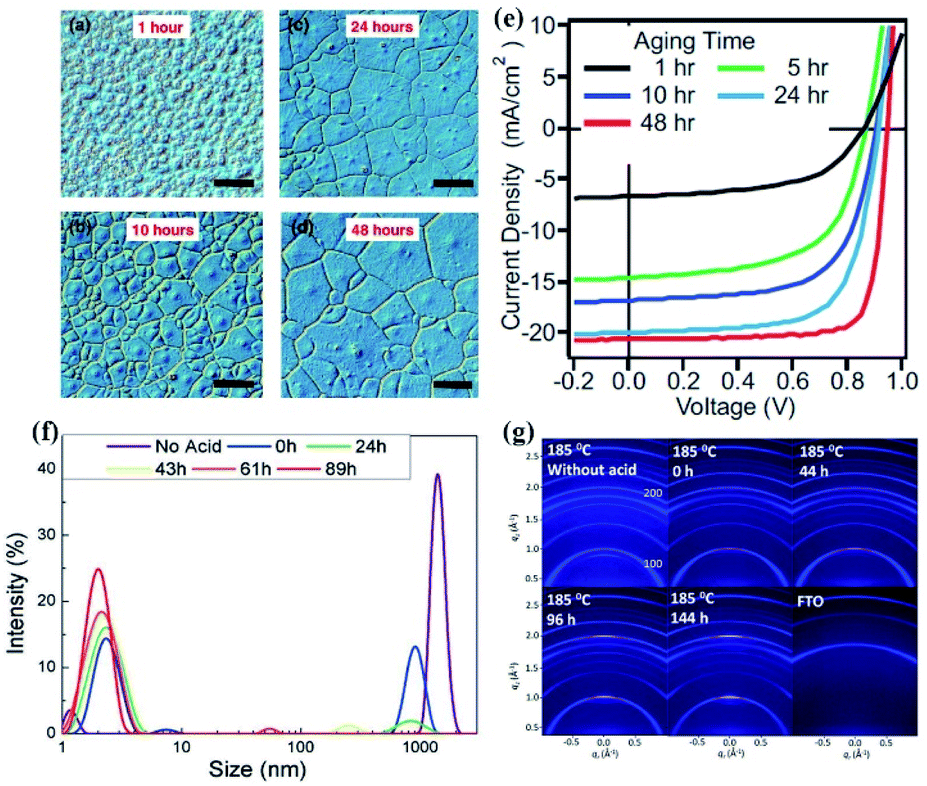 | ||
| Fig. 6 Optical microscope images of perovskite thin films produced by hot casting. The precursors are aged for different time periods: (a) 1 h, (b) 10 h, (c) 24 h, and (d) 48 h. Scale bar: 50 μm. (e) Average grain-size with the error bar determined for 1–48 h aging time. Reprinted from ref. 39 with permission from Wiley. (f) Colloidal hydrodynamic size distribution via dynamic light scattering of various perovskite solutions. (g) A series of 2D X-ray diffraction patterns of the corresponding thin film after the annealing step. Reprinted from ref. 40 with permission from Wiley. | ||
As discussed above, the optimal precursor solution aging time ranges from several hours to a few tens of hours. It is reasonable to speculate that the coordination capabilities of I− ions, solvent molecules, and additive molecules to Pb2+ may be comparable to each other, so it is difficult to overcome the energy barrier for the reconstruction of iodoplumbate complexes. Recently, a single perovskite crystal was reported as a solute to prepare the perovskite precursor solution, and high-quality perovskite films with reduced trap state density were obtained, as well as an impressive efficiency for PSCs.104,105 We speculate that the single perovskite crystal in the precursor solution may reduce the energy barrier and suppress the structural defects in both gel precursor films and perovskite films. So, this strategy may further mitigate the precursor solution aging effect and improve the reproducibility of high-quality perovskite films.
4. Degradation of perovskite precursor solutions
So far, we have discussed the beneficial effect of precursor solution aging on the performance of PSCs. However, the perovskite precursor solution suffers from degeneration during long-term storage due to the chemical reactions between the precursor solution's components and the environmental condition induced degradation, which drastically decreases the photovoltaic efficiency of the corresponding PSCs.4.1 Chemical reactions between components
Several studies focused on the chemical reactions between the solute and solvent occurring in perovskite precursor solutions. For example, proton transfer from MA+ to DMSO has been demonstrated in the perovskite precursor solution containing the MAI component and DMSO, in which the MA+ induces proton transfer to DMSO to form methylamine (CH3NH2), followed by methyl group transfer between CH3NH2 and residual MA+, resulting in ammonium (NH4+) and dimethylammonium (DMA+) (Fig. 7(a)).106 Meanwhile, the transmethylation from DMSO to MA+ leads to the formation of DMA+ and methylsulfonic acid (CH3SOH). Then CH3SOH can react with DMSO quickly to form methylsulfinic acid (CH3SOOH) and dimethylsulfide (CH3SCH3). Finally, the produced CH3SOOH undergoes further reaction with DMSO to form methylsulfonic acid (CH3SO3H) and another equivalent of CH3SCH3. The deprotonation of MA+ and the subsequent methyl group transfer represent the dominant pathway and introduces the cation impurities of NH4+ and DMA+ into the perovskite precursor solutions.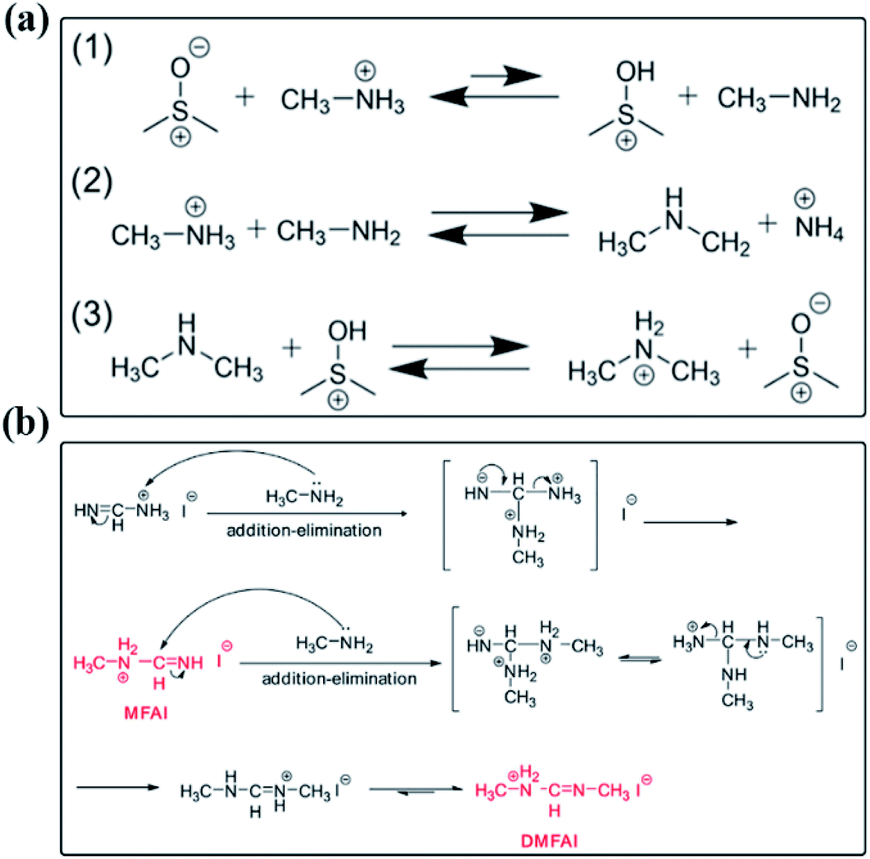 | ||
| Fig. 7 Possible reaction mechanism in perovskite precursor solutions. (a) Formation of DMA+ and NH4+ in DMSO. Reprinted from ref. 106 with permission from the American Chemical Society. (b) The addition–elimination reactions between MA and FAI in the perovskite precursor solution. Reprinted from ref. 107 with permission from Elsevier Inc. | ||
Except for the solute–solvent reactions in precursor solutions, the MAI and FAI contents in a mixed organic cation perovskite precursor solution were found to reduce sharply along with the aging time.107 In contrast, little changes occurred in the pure MAPbI3 precursor solution. It implies that there are potential chemical reactions between the two organic cations. As shown in Fig. 7(b), Wang and co-workers proposed the possible reactions in a co-existing perovskite precursor solution containing MAI and FAI. MAI is known to suffer from the deprotonation reaction and result in CH3NH2 in the solution. Due to the nucleophilicity of lone-pair electrons of the N atom in CH3NH2 and the active electrophilic imine bond in FAI, if the produced CH3NH2 could not volatilize, it would be constantly consumed by FAI through an addition–elimination reaction to form the condensation products of N-methyl FAI (MFAI). The formed MAFI also has an imine bond and is prone to induce a second addition–elimination reaction with CH3NH2 to produce N,N′-dimethyl FAI (DMFAI). Thus, the degradation products from the chemical reactions between the solute and solvent should be responsible for the low stability of perovskite precursor solutions.
4.2 Environmental factors
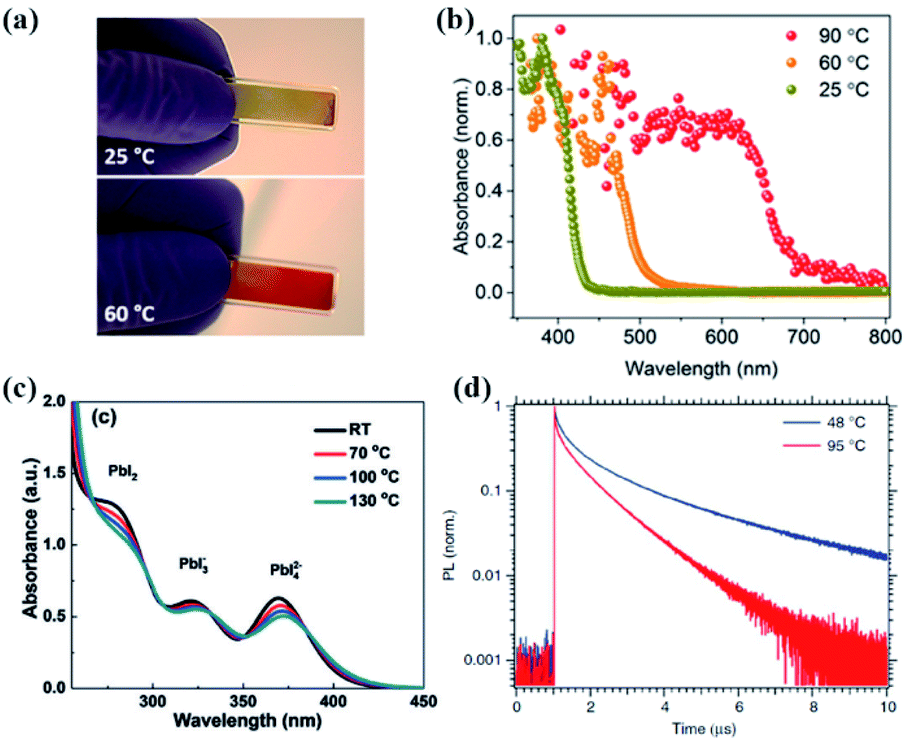 | ||
| Fig. 8 Picture (a) and absorption spectra (b) of a TC prototype after annealing at different temperatures: 25 °C yellow, 60 °C orange, and 90 °C red. Reprinted from ref. 112 with permission from the American Chemical Society. (c) Absorption spectra of a precursor solution with a MAI:PbI2 ratio of 1.7 at different temperatures. Reprinted from ref. 100 with permission from Wiley. (d) Photoluminescence decay of MAPbI3 single crystals grown at 48 °C and 95 °C. Reprinted from ref. 108 with permission from the Nature Publishing group. | ||
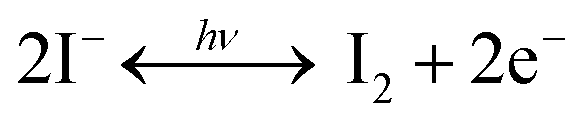 | (3) |
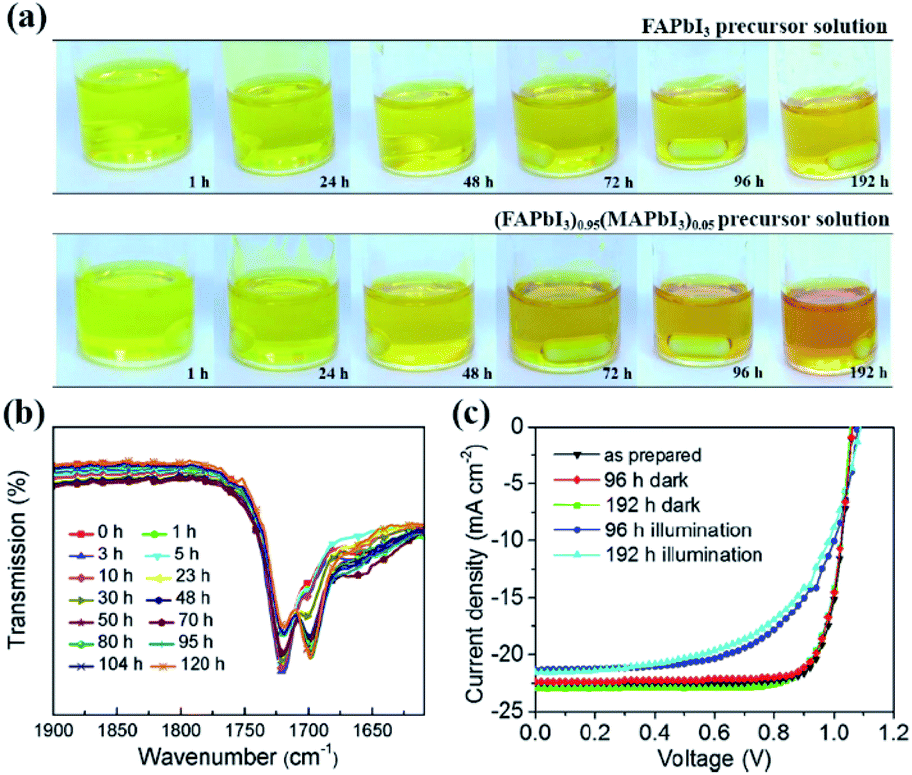 | ||
| Fig. 9 (a) Perovskite precursor solution color change depending on time. FAPbI3 black powder is dissolved in a mixed solvent of DMF/DMSO (top), a conventional perovskite precursor solution, wherein both FAPbI3 and MAPbBr3 powders are dissolved in the mixed solvent (bottom). Reprinted from ref. 48 with permission from Wiley. (b) FTIR spectra of FAPbBr3/DMSO solution after light soaking for different periods. (c) Current density–voltage (J–V) characteristics of mixed cation perovskite devices with different perovskite solution aging history. Reprinted from ref. 122 with permission from the American Chemical Society. | ||
Besides, light-induced FA+ ion degradation in perovskite precursor solutions is also reported. Wei and co-workers found that a redox reaction between the –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH bonds in FA+ and PbBr2 in a Br− ion based precursor solution took place under illumination.122 As a result, Pb2+ ions were reduced to Pb metal, and –CHNH bonds were oxidized to –CHO bonds as irreversible degradation (Fig. 9(b)). In contrast, the MA+ ions were much more stable in the precursor solutions due to the absence of more reactive –CNNH double bonds. However, under I− ion abundant conditions, photoinduced decomposition of PbI2 was observed in both MA+ and mixed Cs/MA+/FA+ containing precursor solutions, leading to the generation of iodine, which changed the color of the precursor solutions to dark red after 5 h, and then the produced iodine would further serve as a catalyst in the redox reaction to activate the transformation of –CHNH double bonds into –CHO bonds in FA+. Note that no such oxidization of FA+ ions in solid films was observed. As expected, the PCE of MAPbI3 based PSCs dropped from 19.6 to 15.6% due to the iodine interstitials or iodine vacancies in perovskite films after light soaking the precursor solution for 96 h, while it reduced from 19.5 to 14.3% for mixed Cs/MA+/FA+ based PSCs under a synergy effect between generated iodine and degraded –CO molecules (Fig. 9(c)).
NH bonds in FA+ and PbBr2 in a Br− ion based precursor solution took place under illumination.122 As a result, Pb2+ ions were reduced to Pb metal, and –CHNH bonds were oxidized to –CHO bonds as irreversible degradation (Fig. 9(b)). In contrast, the MA+ ions were much more stable in the precursor solutions due to the absence of more reactive –CNNH double bonds. However, under I− ion abundant conditions, photoinduced decomposition of PbI2 was observed in both MA+ and mixed Cs/MA+/FA+ containing precursor solutions, leading to the generation of iodine, which changed the color of the precursor solutions to dark red after 5 h, and then the produced iodine would further serve as a catalyst in the redox reaction to activate the transformation of –CHNH double bonds into –CHO bonds in FA+. Note that no such oxidization of FA+ ions in solid films was observed. As expected, the PCE of MAPbI3 based PSCs dropped from 19.6 to 15.6% due to the iodine interstitials or iodine vacancies in perovskite films after light soaking the precursor solution for 96 h, while it reduced from 19.5 to 14.3% for mixed Cs/MA+/FA+ based PSCs under a synergy effect between generated iodine and degraded –CO molecules (Fig. 9(c)).
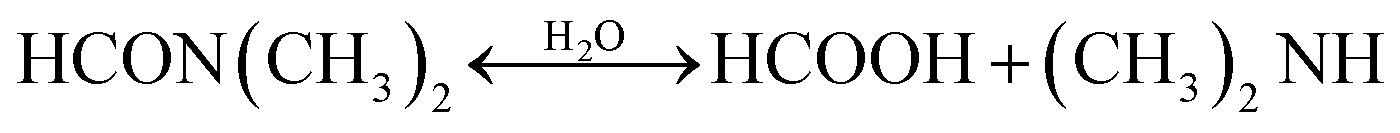 | (4) |
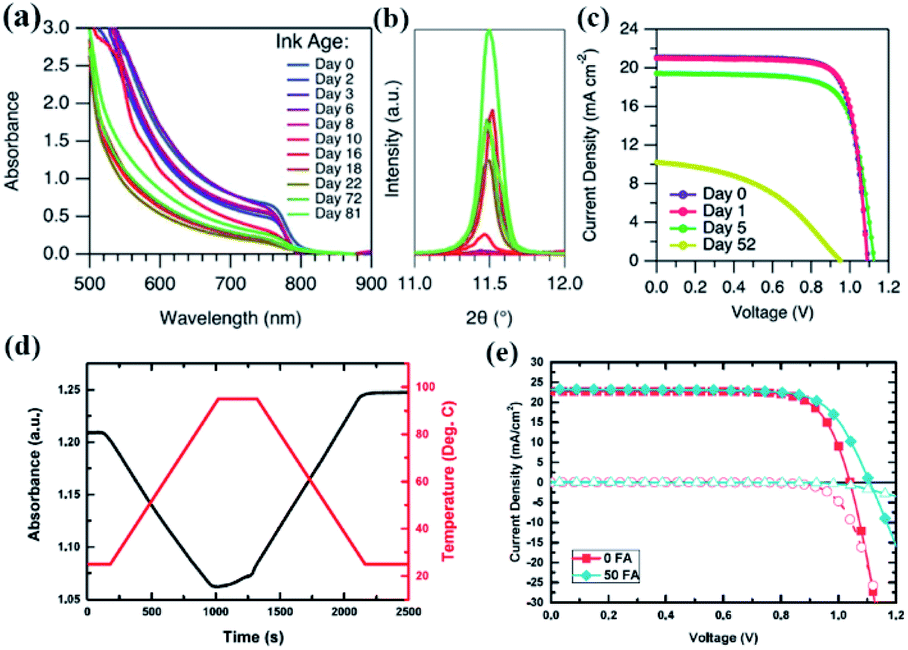 | ||
| Fig. 10 (a) Absorption spectra and (b) selected XRD patterns (not normalized) of FAMAC films fabricated from FAMAC ink that was stored for different days in the dark in N2. (c) Open-circuit voltage to short-circuit current scanned (reserve scan) J–V curves for the PSCs fabricated with FAMAC ink stored for different times: fresh ink and 1, 5, and 52 days. Reprinted from ref. 126 with permission from the American Chemical Society. (d) Evolution of the absorbance of thymol blue in DMF at 402 nm with time and temperature. The change in absorbance is depicted by the solid black line and the corresponding temperature curve is shown by the dashed red line. (e) Current–voltage curves of solar cells fabricated using neat DMF as a solvent, as well as DMF with 5 vol% of formic acid added. Solid symbols represent the light JV measurement, while hollow symbols show the dark JV measurement. Reprinted from ref. 129 with permission from Elsevier Inc. | ||
Moreover, the hydrolysis products change the pH of the precursor solution, which has been proven as one of the crucial parameters that significantly impacts the perovskite crystallization kinetics and optoelectronic properties of the perovskite films.128 Anhydrous DMF is a relatively neutral solvent with a pH of 6.3. Considering that the hydrolysis product of HCOOH is an acid and DMA is a base, both of which present in similar concentrations, the pH of hydrolyzed DMF solution depends on the pKa of the products; the lower pKa of DMA indicates that DMA is a stronger base and has a larger influence of the pH value. So, the hydrolyzed DMF tends to more basic. As shown in Fig. 10(d), by using thymol blue as a pH indicator in DMF, Noel and co-workers investigated the changes in pH during DMF hydrolysis at different temperatures and identified their impacts on the chemical properties of the perovskite precursor solution.129 When increasing the solution temperature to 95 °C, the equilibria of the hydrolysis reaction moved to the product side, and a decreased basicity was observed due to the encouraging dissociation of HCOOH at elevated temperature. Then gradually cooled the solution back down to room temperature, the hydrolyzed DMF solution became increasingly basic and even more than in its initial pH value. The authors suggested that the produced DMA through hydrolysis can increase the solvating power of DMF to solvate the perovskite precursor salts, leading to an enhanced perovskite film quality. By employing formic acid (FA) to artificially tune the age of the DMF, an increased PCE was achieved compared to the device fabricated from the neat precursor solution (Fig. 10(e)).
4.3 Effects on the perovskite film quality
The degradation of perovskite precursor solutions not only reduces the reproducibility of film fabrication but also deteriorates the perovskite film performance. Fig. 11(a–e) summarizes the effects of the degradation of precursor solutions on the performance of the corresponding films and PSCs. Firstly, changes in the crystal phase transition were commonly observed. It can be seen that the black mixed organic cation perovskite films gradually faded out to light yellow with the precursor solution aging time.130 The appearance of the diffraction peak at 11.7° with increasing aging time confirmed the formation of the hexagonal δ-phase of FAPbI3 induced by the stored precursor solution. The phase transition of δ-FAPbI3 to α-FAPbI3 is known to occur in the case of FA+ with a mixture of a certain amount of MA+; the reverse transition suggests the reduced content of MA+, and this may be related to the loss of MA+ through the deprotonation of MA+ to volatile CH3NH2 or the substitution of hydrolysis products of DMF in the perovskite structure discussed above. In addition, as shown in Fig. 11(c), PL quenching for perovskite films obtained from the stored precursor solutions was also observed, consistent with the formation of photoinactive δ-FAPbI3. Secondly, the reaction byproducts with a comparable molecule size from solute–solvent and solute–solute reactions and the hydrolysis of DMF were found to incorporate into the perovskite structure as A-site impurities and resulted in some impurity phases, which have significant effects on the stoichiometry and optical properties of the perovskite films. For example, Fig. 11(d) exhibits the ternary phase diagram of the alloying of the solute–solvent reaction products (NH4+ and DMA+) and MA+ in the perovskite structure, and perovskite films with different components and optical properties can be obtained.106 Moreover, the incorporated NH4+ and DMA+ in the perovskite structure were found to induce a significant density of occupied states in the band gap of MAPbI3 and limit the excursion of the Fermi level at the perovskite surface.113 Finally, the PCEs of devices derived from the precursor solution show a rapid reduction to 75% of the initial value (20.82%) after storing for 72 h (Fig. 11(e)).48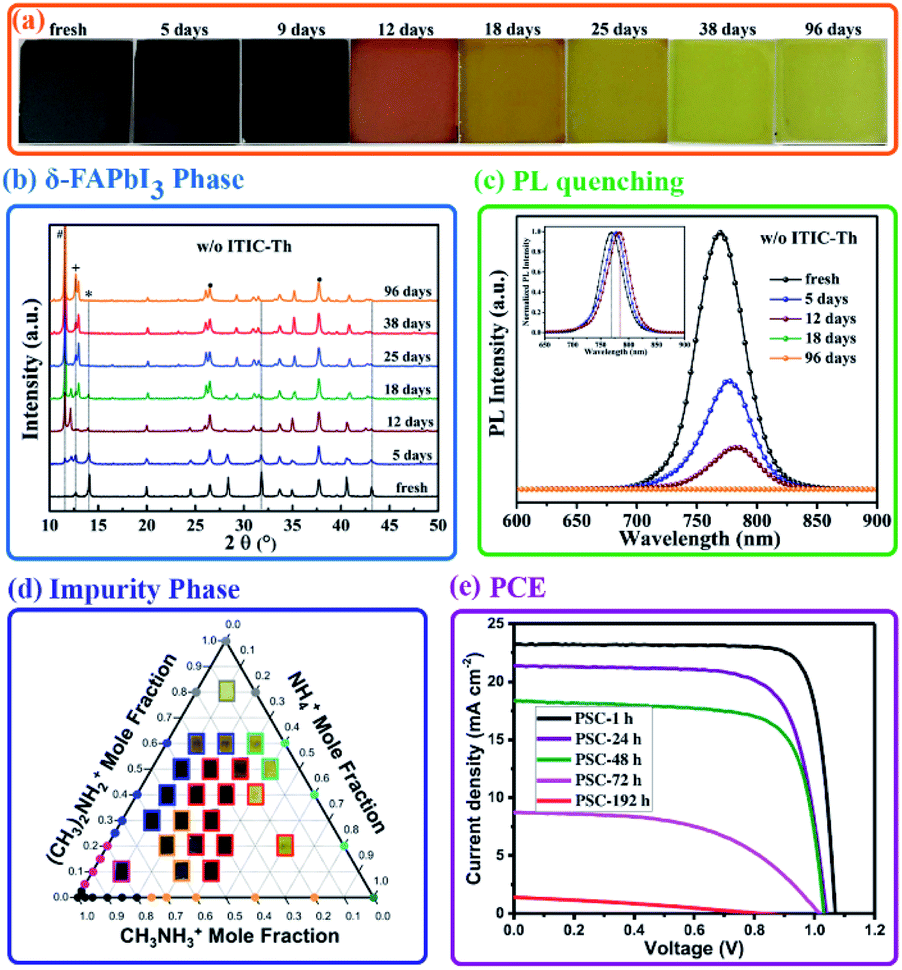 | ||
| Fig. 11 (a) A series of photographs of perovskite films fabricated from perovskite precursor solutions aged for different amounts of time. The corresponding XRD (b) and PL spectra (c) of the perovskite films. Reprinted from ref. 130 with permission from Wiley. (d) Partial ternary phase diagram of [DMA]x(NH4)yMA1−x−yPbI3. Photographs of triple organic cation films are overlaid on the corresponding data points in the center of the phase diagram. Reprinted from ref. 106 with permission from the American Chemical Society. (e) I–V curves and distribution of the PCE of PSCs fabricated as a function of the aging time of the precursor solution. Reprinted from ref. 48 with permission from Wiley. | ||
4.4 Stabilization of the perovskite precursor solution
A few studies have tried to enhance the interaction between perovskite precursors and additives, to inhibit the chemical reactions between the solute and solvent mentioned above. The organic molecules of 3,9-bis(2-methylene-(3-(1,1-dicyanomethylene)-indanone))-5,5,11,11-tetrakis(5-hexylthienyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene (ITIC-Th) and 1,4,7,10,13,16-hexaoxacyclooctadecane (18C6) have been incorporated into mixed cation perovskite precursor solutions to stabilize the perovskite precursor solution, in which the S in ITIC-Th and O in 18C6 were confirmed to form coordination or covalent bonds with Pb2+, respectively.130,131 Such bonding resulted in a more stable colloidal [PbI6]4− framework and suppressed the formation of δ-FAPbI3, which significantly prolonged the lifetime of precursor solutions for more than one month.To further suppress the reaction byproducts originating from the deprotonation of MAI as impurities in the perovskite structure, the stabilizer triethyl borate (TEB) with electron-withdrawing capability has been demonstrated to form a strong interaction with I− of MAI and reduce the electron cloud density of I−, making it hard to capture the proton in MA+ and efficiently inhibit the deprotonation of MAI.107 As a result, the applied window period of the precursor solution was widened for more than 24 h, and only 4% FAI was confirmed to form byproduct MFAI after aging for 7 days, compared to that of 75% for the normal precursor solution. A similar interaction strategy was reported by incorporating elemental sulfur into precursor solutions.48 The elemental sulfur exists as stable S8 rings in the precursor solution and forms a complex with MA+ by chemically interacting with the amine group. The complex could inhibit the volatilization of CH3NH2 and drastically improve the stability of the perovskite precursor solutions; the α-FAPbI3 phase was retained and no visible δ-FAPbI3 could be observed in the perovskite films prepared with the sulfur-containing precursor solution even after more than one week. Nevertheless, an alternative way is to decrease the MAI concentration or replace cesium or FAI in the DMSO and MAI containing perovskite precursor solution, which is suggested to essentially suppress the detrimental impact on the quality of perovskite films.113
Except for the suppression of chemical reactions in precursor solutions, the precursor solution degradation induced by environmental factors should also be avoided. For example, various additives, including hypophosphorous acid (HPA),121 methylamine (CH3NH2),132 formamidine acetate (FAAc),128 and elemental sulfur (S8),48 have been developed to eliminate the oxidized I2 in the precursor solutions. It was reported that the addition of CH3NH2 into the precursor solution effectively avoided the formation of oxidized I2 and the disturbance of Pb/I stoichiometry, and the defect density of the perovskite film derived from the suppressed I2 precursor solution decreased from 1.38 × 1016 to 1.5 × 1015 cm3, and a PCE of 20.02% with a stabilized output efficiency of 19.01% was achieved for the PSCs based on the thick perovskite film with a thickness of ∼650 nm. Besides, a solvent-less storage strategy was reported to avoid the modulation of the composition and crystal phase of the resulting perovskite films by the hydrolysis of DMF.126 By utilizing a ball mill to uniformly mix the precursor salts into a mixed perovskite powder with desired stoichiometry, it can be dissolved in solvents immediately before use. Perovskite films prepared from the powder show no significant changes in either the structure or the optical properties after storing for 31 days.
5. Humidity effect on perovskite formation
Although the chemical species in perovskite precursor solutions plays a critical role in perovskite formation, as discussed in Section 2.2.1, solvated gel complexes precipitated from the perovskite precursor solution during solidification are directly related to the subsequent perovskite crystallization as well as the quality of perovskite films.133,134 However, the gel complexes are metastable and sensitive to humidity since H2O has a stronger bond affinity towards PbI2 compared to the solvents in gel complexes, making it easy to coordinate with Pb2+ and suppress the formation of higher iodoplumbate complexes required for perovskite formation.135 As a result, the stoichiometric ratio of PbI2 and DMSO in precursor solutions needs to be carefully adjusted to achieve perovskite films with a desired morphology under different relative humidity conditions. Although a suitable amount of H2O was reported as an additive to improve the quality of perovskite films,136–138 unintentional moisture incursion can mediate the chemical species of the gel complexes and change the perovskite crystallization kinetics. In addition, the H2O molecule has a strong hydrogen bonding interaction with DMF or DMSO. It can break the Pb–O bond in solvated complexes and attenuate the complexation of Pb2+ in gel complexes by reducing the availability of solvents.139 Therefore, it is important to understand the underlying mechanism of the humidity effect on the chemical species of the gel complexes and further improve the reproducibility window for the future industrial fabrication of PSCs. In a very recent study shown in Fig. 12(a), the calculated and experimental results showed that one dimensional (1D) PbI2·2DMSO is quickly formed due to the strong coordination of DMSO to Pb2+ after mixing MAI/PbI2 in the DMF/DMSO mixed solution, and then they combine together and further convert to Pb2I4·2DMSO and MA2Pb3I8·2DMSO, driven by the concentration of the precursor.140 However, H2O will interact with DMSO in these lead complexes and reduce their relative electronic energies in a way (Fig. 12(b)), thus facilitating the conversion from the 1D H2O·PbI2·2DMSO lead complex to 1D MA2Pb3I8·2DMSO with much longer needle-like fibers in the morphology than those grown in the water-free environment as confirmed by XRD patterns in Fig. 12(c). This is consistent with the reported moisture-induced crystal-growth anisotropy phenomenon under ambient conditions, where porous perovskite films can even be fabricated by an anti-solvent assisted method (Fig. 12(d)).139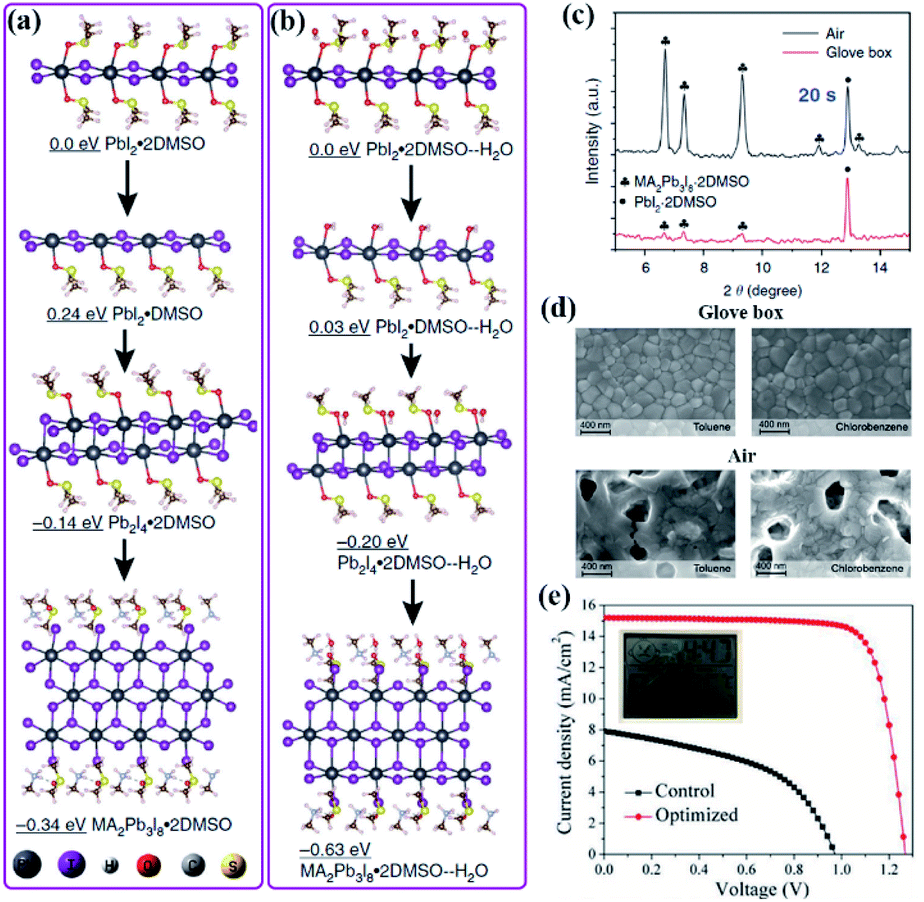 | ||
| Fig. 12 Calculated structures and electronic energies of lead complexes without (a) and with H2O (b). (c) The X-ray diffraction patterns (XRD) of Pb-complex films fabricated in ambient air and in a glove box with a sample spinning time of 20 s. Reprinted from ref. 140 with permission from the Nature Publishing group. (d) SEM images of perovskite films fabricated with different antisolvents—the commonly used are toluene and chlorobenzene. Reprinted from ref. 139 with permission from Wiley. (e) J–V curves of the control and optimized CsPbI2Br PSCs prepared in air with a high RH of 91% and temperature of 36 °C. Reprinted from ref. 141 with permission from Wiley. | ||
Except for the humidity effect on hybrid perovskites, the fabrication of high-efficiency inorganic PSCs is known highly sensitive to moisture and is generally limited in an inert-filled glovebox or dry air atmosphere. Very recently, to suppress the effects of humidity and achieve the air fabrication process of inorganic PSCs, a new precursor system by mixing HCOOCs, HPbI3, and HPbBr3 was developed to replace the traditional precursor solutions commonly used for the fabrication of inorganic PSCs.141 A new complex HCOOH–Cs+ present in the precursor system was confirmed to prevent moisture incursion and protect the gel precursor films from an unwanted phase transition in ambient air. Finally, as shown in Fig. 12(e), an impressive PCE of 15.1% was achieved for inorganic PSCs in the atmosphere with 91% relative humidity. These results suggest that tailored iodoplumbate complexes in perovskite precursor solutions can be obtained through precursor engineering, which can achieve desired perovskite crystallization under various fabrication conditions and further improve the PSC performance.
6. Conclusions and outlook
In summary, we presented insights into the evolution of the characteristics of a perovskite precursor solution driven by the components of the precursor solution, aging effect, degradation effect, and humidity effect and revealed their impact on the physical properties of perovskite films and the PSC performance. Iodoplumbate complexes, such as PbI2, PbI3−, and PbI42−, are present in the perovskite precursor solution in equilibria and serve as precursors that dictate the quality of perovskite films and the optoelectrical performance of PSCs. The solvent and additive molecules are prone to compete with I− to coordinate with Pb2+ due to their coordination capabilities, shifting the precursor equilibria to iodoplumbate complexes with a smaller number of I− ions, thus controlling perovskite formation. As a result, the perovskite precursor solutions often need a special aging time, ranging from a few to several tens of hours to achieve the optimal performance of PSCs. The status of the precursor solution continuously evolves and will have a time-dependent influence on the quality of perovskite films until the solution reaches new equilibria. Then the efficiency of PSCs drops rapidly due to the degradation of the perovskite precursor solution induced by the chemical reactions between components or exposure to external stimuli. In addition, the PCEs of PSCs are also sensitive to the fabrication atmosphere since moisture can interact with either Pb2+ ions in gel complexes or solvent molecules to modify the subsequent perovskite crystallization kinetics, which further reduces the reproducibility of highly efficient PSCs. Based on the discussion above, we present the following suggestions to further improve the reproducibility of highly efficient PSCs:(1) Increase the iodide coordination number in the coordination complexes present in the precursor solutions or gel precursor films before perovskite formation.
(2) Carefully adjust the optimal aging window for perovskite precursor solutions under different storage conditions.
(3) Avoid the degradation of precursor solutions induced by chemical reactions between the components and environmental conditions.
(4) Prevent unintentional moisture incursion during perovskite formation.
(5) Tailor iodoplumbate complexes in perovskite precursor solutions through precursor engineering to meet the various fabrication conditions.
Although excellent progress has been made in understanding the relationships between the characteristics of the perovskite precursor solution and the reproducibility of high-quality perovskite films and improvement of the efficiency of PSCs, the effect of precursor aging on the reproducibility of highly efficient PSCs is intractable, which needs to be carefully considered. The aging of the perovskite precursor could benefit the quality of the resultant perovskite film. However, long-term storage (or exposure to external stimuli) may lead to the degradation of the precursor and deteriorate the perovskite film. Efforts towards stabilizing the perovskite precursor solution and in-depth understanding of the degradation mechanism of the precursor are still required. Tailoring the perovskite precursor solution with weak coordination solvents may promote the complexation of Pb2+ and I− to form sufficient high order iodoplumbate complexes that are required for perovskite formation, which may suppress the effect of the precursor solution aging to facilitate the formation of highly reproducible perovskite films.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2017YFE0119700), National Natural Science Foundation of China (51961135107, 51774034, 51772026, and 51672208), Beijing Natural Science Foundation (2182039), and Open Foundation Project of Key Laboratory of Plateau Green Building and Ecological Community of Qinghai Province (KLKF-2019-002).References
- D. Weber, Z. Naturforsch. B, 1978, 33, 1443–1445 Search PubMed.
- D. B. Mitzi, J. Mater. Chem., 2004, 14, 2355–2365 RSC.
- M. I. Saidaminov, K. Williams, M. Wei, A. Johnston, R. Quintero-Bermudez, M. Vafaie, J. M. Pina, A. H. Proppe, Y. Hou, G. Walters, S. O. Kelley, W. A. Tisdale and E. H. Sargent, Nat. Mater., 2020, 19, 412–418 CrossRef CAS.
- E. M. Hutter, M. C. Gelvez-Rueda, A. Osherov, V. Bulovic, F. C. Grozema, S. D. Stranks and T. J. Savenije, Nat. Mater., 2017, 16, 115–120 CrossRef CAS.
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. I. Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS.
- H. S. Kim, C. R. Lee, J. H. Im, K. B. Lee, T. Moehl, A. Marchioro, S. J. Moon, R. Humphry-Baker, J. H. Yum, J. E. Moser, M. Gratzel and N. G. Park, Sci. Rep., 2012, 2, 591 CrossRef.
- S. De Wolf, J. Holovsky, S.-J. Moon, P. Löper, B. Niesen, M. Ledinsky, F.-J. Haug, J.-H. Yum and C. Ballif, J. Phys. Chem. Lett., 2014, 5, 1035–1039 CrossRef CAS.
- V. D'Innocenzo, G. Grancini, M. J. Alcocer, A. R. Kandada, S. D. Stranks, M. M. Lee, G. Lanzani, H. J. Snaith and A. Petrozza, Nat. Commun., 2014, 5, 3586 CrossRef.
- J. Huang, Y. Yuan, Y. Shao and Y. Yan, Nat. Rev. Mater., 2017, 2, 17042 CrossRef CAS.
- M. Li, B. Li, G. Cao and J. Tian, J. Mater. Chem. A, 2017, 5, 21313–21319 RSC.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341–344 CrossRef CAS.
- M. Wang, B. Li, P. Siffalovic, L.-C. Chen, G. Cao and J. Tian, J. Mater. Chem. A, 2018, 6, 15386–15394 RSC.
- B. Li, M. J. Li, C. B. Fei, G. Z. Cao and J. J. Tian, J. Mater. Chem. A, 2017, 5, 24168–24177 RSC.
- D. T. Moore, H. Sai, K. W. Tan, D. M. Smilgies, W. Zhang, H. J. Snaith, U. Wiesner and L. A. Estroff, J. Am. Chem. Soc., 2015, 137, 2350–2358 CrossRef CAS.
- S. Yun, Y. Zhang, Q. Xu, J. Liu and Y. Qin, Nano Energy, 2019, 60, 600–619 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS.
- S. Yun, N. Vlachopoulos, A. Qurashi, S. Ahmad and A. Hagfeldt, Chem. Soc. Rev., 2019, 48, 3705–3722 RSC.
- W. S. Yang, J. H. Noh, N. J. Jeon, Y. C. Kim, S. Ryu, J. Seo and S. I. Seok, Science, 2015, 348, 1234–1237 CrossRef CAS.
- X. Li, D. Bi, C. Yi, J.-D. Décoppet, J. Luo, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Science, 2016, aaf8060 Search PubMed.
- H. Tan, A. Jain, O. Voznyy, X. Lan, F. P. Garcia de Arquer, J. Z. Fan, R. Quintero-Bermudez, M. Yuan, B. Zhang, Y. Zhao, F. Fan, P. Li, L. N. Quan, Y. Zhao, Z. H. Lu, Z. Yang, S. Hoogland and E. H. Sargent, Science, 2017, 355, 722–726 CrossRef CAS.
- E. H. Jung, N. J. Jeon, E. Y. Park, C. S. Moon, T. J. Shin, T.-Y. Yang, J. H. Noh and J. Seo, Nature, 2019, 567, 511–515 CrossRef CAS.
- S. Yang, S. Chen, E. Mosconi, Y. Fang, X. Xiao, C. Wang, Y. Zhou, Z. Yu, J. Zhao, Y. Gao, F. De Angelis and J. Huang, Science, 2019, 365, 473–478 CrossRef CAS.
- H. Lu, Y. Liu, P. Ahlawat, A. Mishra, W. R. Tress, F. T. Eickemeyer, Y. Yang, F. Fu, Z. Wang, C. E. Avalos, B. I. Carlsen, A. Agarwalla, X. Zhang, X. Li, Y. Zhan, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, L. Zheng, A. Hagfeldt and M. Gratzel, Science, 2020, 370, eabb8985 CrossRef CAS.
- S. Yun, Y. Qin, A. R. Uhl, N. Vlachopoulos, M. Yin, D. Li, X. Han and A. Hagfeldt, Energy Environ. Sci., 2018, 11, 476–526 RSC.
- NREL, https://www.nrel.gov/pv/assets/images/efficiency-chart.png, accessed, January 2021.
- C. Fei, B. Li, R. Zhang, H. Fu, J. Tian and G. Cao, Adv. Energy Mater., 2017, 7, 1602017 CrossRef.
- M. Yang, Z. Li, M. O. Reese, O. G. Reid, D. H. Kim, S. Siol, T. R. Klein, Y. Yan, J. J. Berry, M. F. A. M. van Hest and K. Zhu, Nat. Energy, 2017, 2, 17038 CrossRef CAS.
- L. Li, N. Liu, Z. Xu, Q. Chen, X. Wang and H. Zhou, ACS Nano, 2017, 11, 8804–8813 CrossRef CAS.
- J. Kim, B. W. Park, J. Baek, J. S. Yun, H. W. Kwon, J. Seidel, H. Min, S. Coelho, S. Lim, S. Huang, K. Gaus, M. A. Green, T. J. Shin, A. W. Y. Ho-Baillie, M. G. Kim and S. I. Seok, J. Am. Chem. Soc., 2020, 142, 6251–6260 CrossRef CAS.
- K. Yan, M. Long, T. Zhang, Z. Wei, H. Chen, S. Yang and J. Xu, J. Am. Chem. Soc., 2015, 137, 4460–4468 CrossRef CAS.
- J. S. Manser, M. I. Saidaminov, J. A. Christians, O. M. Bakr and P. V. Kamat, Acc. Chem. Res., 2016, 49, 330–338 CrossRef CAS.
- B. Li, D. Binks, G. Cao and J. Tian, Small, 2019, 15, e1903613 CrossRef.
- M. Jung, S. G. Ji, G. Kim and S. I. Seok, Chem. Soc. Rev., 2019, 48, 2011–2038 RSC.
- Y. Guo, K. Shoyama, W. Sato, Y. Matsuo, K. Inoue, K. Harano, C. Liu, H. Tanaka and E. Nakamura, J. Am. Chem. Soc., 2015, 137, 15907–15914 CrossRef CAS.
- S. A. Fateev, A. A. Petrov, V. N. Khrustalev, P. V. Dorovatovskii, Y. V. Zubavichus, E. A. Goodilin and A. B. Tarasov, Chem. Mater., 2018, 30, 5237–5244 CrossRef CAS.
- X. Huang, R. Chen, G. Deng, F. Han, P. Ruan, F. Cheng, J. Yin, B. Wu and N. Zheng, J. Am. Chem. Soc., 2020, 142, 6149–6157 CrossRef CAS.
- J. C. Hamill, J. Schwartz and Y.-L. Loo, ACS Energy Lett., 2017, 3, 92–97 CrossRef.
- G. S. Shin, S. G. Kim, Y. Zhang and N. G. Park, Small Methods, 2019, 4, 1900398 CrossRef.
- H. Tsai, W. Nie, Y.-H. Lin, J. C. Blancon, S. Tretiak, J. Even, G. Gupta, P. M. Ajayan and A. D. Mohite, Adv. Energy Mater., 2017, 7, 1602159 CrossRef.
- D. P. McMeekin, Z. Wang, W. Rehman, F. Pulvirenti, J. B. Patel, N. K. Noel, M. B. Johnston, S. R. Marder, L. M. Herz and H. J. Snaith, Adv. Mater., 2017, 29, 1607039 CrossRef.
- M. Xiao, F. Huang, W. Huang, Y. Dkhissi, Y. Zhu, J. Etheridge, A. Gray-Weale, U. Bach, Y. B. Cheng and L. Spiccia, Angew. Chem., Int. Ed., 2014, 53, 10056–10061 CrossRef.
- F. Li, C. Bao, H. Gao, W. Zhu, T. Yu, J. Yang, G. Fu, X. Zhou and Z. Zou, Mater. Lett., 2015, 157, 38–41 CrossRef CAS.
- S. H. Huang, C. K. Guan, P. H. Lee, H. C. Huang, C. F. Li, Y. C. Huang and W. F. Su, Adv. Energy Mater., 2020, 10, 2001567 CrossRef CAS.
- Y. Zhong, R. Munir, J. Li, M.-C. Tang, M. R. Niazi, D.-M. Smilgies, K. Zhao and A. Amassian, ACS Energy Lett., 2018, 3, 1078–1085 CrossRef CAS.
- C. Li, J. Yin, R. Chen, X. Lv, X. Feng, Y. Wu and J. Cao, J. Am. Chem. Soc., 2019, 141, 6345–6351 CrossRef CAS.
- Z. Wei, H. Chen, K. Yan and S. Yang, Angew. Chem., Int. Ed., 2014, 53, 13239–13243 CrossRef CAS.
- F. Mathies, T. Abzieher, A. Hochstuhl, K. Glaser, A. Colsmann, U. W. Paetzold, G. Hernandez-Sosa, U. Lemmer and A. Quintilla, J. Mater. Chem. A, 2016, 4, 19207–19213 RSC.
- H. Min, G. Kim, M. J. Paik, S. Lee, W. S. Yang, M. Jung and S. I. Seok, Adv. Energy Mater., 2019, 9, 1803476 CrossRef.
- Y. Zhou, O. S. Game, S. Pang and N. P. Padture, J. Phys. Chem. Lett., 2015, 6, 4827–4839 CrossRef CAS.
- H. Miyamae, H. Toriyama, T. Abe, G. Hihara and M. Nagata, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 1559–1562 CrossRef.
- J. Parr, Polyhedron, 1997, 16, 551–566 CrossRef CAS.
- O. E. Lanford and S. J. Kiehl, J. Am. Chem. Soc., 1941, 63, 667–669 CrossRef CAS.
- K. G. Stamplecoskie, J. S. Manser and P. V. Kamat, Energy Environ. Sci., 2015, 8, 208–215 RSC.
- A. Sharenko, C. Mackeen, L. Jewell, F. Bridges and M. F. Toney, Chem. Mater., 2017, 29, 1315–1320 CrossRef CAS.
- E. Radicchi, E. Mosconi, F. Elisei, F. Nunzi and F. De Angelis, ACS Appl. Energy Mater., 2019, 2, 3400–3409 CrossRef CAS.
- P. Ahlawat, M. I. Dar, P. Piaggi, M. Grätzel, M. Parrinello and U. Rothlisberger, Chem. Mater., 2019, 32, 529–536 CrossRef.
- Q. Hu, L. Zhao, J. Wu, K. Gao, D. Luo, Y. Jiang, Z. Zhang, C. Zhu, E. Schaible, A. Hexemer, C. Wang, Y. Liu, W. Zhang, M. Gratzel, F. Liu, T. P. Russell, R. Zhu and Q. Gong, Nat. Commun., 2017, 8, 15688 CrossRef CAS.
- N. Ahn, D. Y. Son, I. H. Jang, S. M. Kang, M. Choi and N. G. Park, J. Am. Chem. Soc., 2015, 137, 8696–8699 CrossRef CAS.
- N. J. Jeon, J. H. Noh, Y. C. Kim, W. S. Yang, S. Ryu and S. I. Seok, Nat. Mater., 2014, 13, 897–903 CrossRef CAS.
- F. Cai, Y. Yan, J. Yao, P. Wang, H. Wang, R. S. Gurney, D. Liu and T. Wang, Adv. Funct. Mater., 2018, 28, 1801985 CrossRef.
- J. Kim, M. I. Saidaminov, H. Tan, Y. Zhao, Y. Kim, J. Choi, J. W. Jo, J. Fan, R. Quintero-Bermudez, Z. Yang, L. N. Quan, M. Wei, O. Voznyy and E. H. Sargent, Adv. Mater., 2018, 30, e1706275 CrossRef.
- J.-Y. Seo, T. Matsui, J. Luo, J.-P. Correa-Baena, F. Giordano, M. Saliba, K. Schenk, A. Ummadisingu, K. Domanski, M. Hadadian, A. Hagfeldt, S. M. Zakeeruddin, U. Steiner, M. Grätzel and A. Abate, Adv. Energy Mater., 2016, 6, 1600767 CrossRef.
- Y. Rong, Z. Tang, Y. Zhao, X. Zhong, S. Venkatesan, H. Graham, M. Patton, Y. Jing, A. M. Guloy and Y. Yao, Nanoscale, 2015, 7, 10595–10599 RSC.
- L. Chao, Y. Xia, B. Li, G. Xing, Y. Chen and W. Huang, Chem, 2019, 5, 995–1006 CAS.
- S. Rahimnejad, A. Kovalenko, S. M. Fores, C. Aranda and A. Guerrero, ChemPhysChem, 2016, 17, 2795–2798 CrossRef CAS.
- X. Guo, C. McCleese, C. Kolodziej, A. C. S. Samia, Y. Zhao and C. Burda, Dalton Trans., 2016, 45, 3806–3813 RSC.
- S. Bae, S. J. Han, T. J. Shin and W. H. Jo, J. Mater. Chem. A, 2015, 3, 23964–23972 RSC.
- A. A. Petrov, I. P. Sokolova, N. A. Belich, G. S. Peters, P. V. Dorovatovskii, Y. V. Zubavichus, V. N. Khrustalev, A. V. Petrov, M. Grätzel, E. A. Goodilin and A. B. Tarasov, J. Phys. Chem. C, 2017, 121, 20739–20743 CrossRef CAS.
- K. H. Hendriks, J. J. van Franeker, B. J. Bruijnaers, J. A. Anta, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2017, 5, 2346–2354 RSC.
- Y. Xia, C. Ran, Y. Chen, Q. Li, N. Jiang, C. Li, Y. Pan, T. Li, J. Wang and W. Huang, J. Mater. Chem. A, 2017, 5, 3193–3202 RSC.
- Y. Wu, F. Xie, H. Chen, X. Yang, H. Su, M. Cai, Z. Zhou, T. Noda and L. Han, Adv. Mater., 2017, 29, 1701073 CrossRef.
- L. Zhang, B. Li, J. Yuan, M. Wang, T. Shen, F. Huang, W. Wen, G. Cao and J. Tian, J. Phys. Chem. Lett., 2018, 9, 3646–3653 CrossRef CAS.
- Y. Bai, S. Xiao, C. Hu, T. Zhang, X. Meng, Q. Li, Y. Yang, K. S. Wong, H. Chen and S. Yang, Nano Energy, 2017, 34, 58–68 CrossRef CAS.
- F. Huang, Y. Dkhissi, W. Huang, M. Xiao, I. Benesperi, S. Rubanov, Y. Zhu, X. Lin, L. Jiang, Y. Zhou, A. Gray-Weale, J. Etheridge, C. R. McNeill, R. A. Caruso, U. Bach, L. Spiccia and Y.-B. Cheng, Nano Energy, 2014, 10, 10–18 CrossRef CAS.
- B. Li, Q. Zhang, S. Zhang, Z. Ahmad, T. Chidanguro, A. Hunter Davis, Y. C. Simon, X. Gu, W. Zheng, N. Pradhan and Q. Dai, Chem. Eng. J., 2021, 405, 126998 CrossRef CAS.
- Y. Li, Z. Zhao, F. Lin, X. Cao, X. Cui and J. Wei, Small, 2017, 13, 1604125 CrossRef.
- Y. C. Zheng, S. Yang, X. Chen, Y. Chen, Y. Hou and H. G. Yang, Chem. Mater., 2015, 27, 5116–5121 CrossRef CAS.
- B. Li, C. Fei, K. Zheng, X. Qu, T. Pullerits, G. Cao and J. Tian, J. Mater. Chem. A, 2016, 4, 17018–17024 RSC.
- Q. Han, Y. Bai, J. Liu, K.-z. Du, T. Li, D. Ji, Y. Zhou, C. Cao, D. Shin, J. Ding, A. D. Franklin, J. T. Glass, J. Hu, M. J. Therien, J. Liu and D. B. Mitzi, Energy Environ. Sci., 2017, 10, 2365–2371 RSC.
- Z. Xu, Z. Liu, N. Li, G. Tang, G. Zheng, C. Zhu, Y. Chen, L. Wang, Y. Huang, L. Li, N. Zhou, J. Hong, Q. Chen and H. Zhou, Adv. Mater., 2019, 31, e1900390 CrossRef.
- Z. Wang, Y. Zhou, S. Pang, Z. Xiao, J. Zhang, W. Chai, H. Xu, Z. Liu, N. P. Padture and G. Cui, Chem. Mater., 2015, 27, 7149–7155 CrossRef CAS.
- T. Bu, X. Liu, Y. Zhou, J. Yi, X. Huang, L. Luo, J. Xiao, Z. Ku, Y. Peng, F. Huang, Y.-B. Cheng and J. Zhong, Energy Environ. Sci., 2017, 10, 2509–2515 RSC.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- M. Abdi-Jalebi, Z. Andaji-Garmaroudi, S. Cacovich, C. Stavrakas, B. Philippe, J. M. Richter, M. Alsari, E. P. Booker, E. M. Hutter, A. J. Pearson, S. Lilliu, T. J. Savenije, H. Rensmo, G. Divitini, C. Ducati, R. H. Friend and S. D. Stranks, Nature, 2018, 555, 497–501 CrossRef CAS.
- D. Y. Son, S. G. Kim, J. Y. Seo, S. H. Lee, H. Shin, D. Lee and N. G. Park, J. Am. Chem. Soc., 2018, 140, 1358–1364 CrossRef CAS.
- C. Liang, H. Gu, Y. Xia, Z. Wang, X. Liu, J. Xia, S. Zuo, Y. Hu, X. Gao, W. Hui, L. Chao, T. Niu, M. Fang, H. Lu, H. Dong, H. Yu, S. Chen, X. Ran, L. Song, B. Li, J. Zhang, Y. Peng, G. Shao, J. Wang, Y. Chen, G. Xing and W. Huang, Nat. Energy, 2020, 6, 38–45 CrossRef.
- H. Tsai, W. Nie, J. C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk and M. G. Kanatzidis, Nature, 2016, 536, 312–316 CrossRef CAS.
- E. Shi, Y. Gao, B. P. Finkenauer, Akriti, A. H. Coffey and L. Dou, Chem. Soc. Rev., 2018, 47, 6046–6072 RSC.
- J. Hu, I. W. H. Oswald, S. J. Stuard, M. M. Nahid, N. Zhou, O. F. Williams, Z. Guo, L. Yan, H. Hu, Z. Chen, X. Xiao, Y. Lin, Z. Yang, J. Huang, A. M. Moran, H. Ade, J. R. Neilson and W. You, Nat. Commun., 2019, 10, 1276 CrossRef.
- L. Liu, Y. Bai, X. Zhang, Y. Shang, C. Wang, H. Wang, C. Zhu, C. Hu, J. Wu, H. Zhou, Y. Li, S. Yang, Z. Ning and Q. Chen, Angew. Chem., Int. Ed., 2020, 59, 5979–5987 CrossRef CAS.
- J. P. Correa-Baena, Y. Luo, T. M. Brenner, J. Snaider, S. Sun, X. Li, M. A. Jensen, N. T. P. Hartono, L. Nienhaus, S. Wieghold, J. R. Poindexter, S. Wang, Y. S. Meng, T. Wang, B. Lai, M. V. Holt, Z. Cai, M. G. Bawendi, L. Huang, T. Buonassisi and D. P. Fenning, Science, 2019, 363, 627–631 CrossRef CAS.
- S. Yun, X. Zhou, J. Even and A. Hagfeldt, Angew. Chem., Int. Ed., 2017, 56, 15806–15817 CrossRef CAS.
- D. Luo, R. Su, W. Zhang, Q. Gong and R. Zhu, Nat. Rev. Mater., 2019, 5, 44–60 CrossRef.
- J. M. Ball and A. Petrozza, Nat. Energy, 2016, 1, 16149 CrossRef CAS.
- W. S. Yang, B. W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS.
- R. J. Stewart, C. Grieco, A. V. Larsen, G. S. Doucette and J. B. Asbury, J. Phys. Chem. C, 2016, 120, 12392–12402 CrossRef CAS.
- P. Boonmongkolras, D. Kim, E. M. Alhabshi, I. Gereige and B. Shin, RSC Adv., 2018, 8, 21551–21557 RSC.
- W. Zhu, Q. Zhang, C. Zhang, Z. Zhang, D. Chen, Z. Lin, J. Chang, J. Zhang and Y. Hao, ACS Appl. Energy Mater., 2018, 1, 4991–4997 CrossRef CAS.
- J. S. Manser, B. Reid and P. V. Kamat, J. Phys. Chem. C, 2015, 119, 17065–17073 CrossRef CAS.
- B. Li, F. H. Isikgor, H. Coskun and J. Ouyang, Angew. Chem., Int. Ed., 2017, 56, 16073–16076 CrossRef CAS.
- T. Li, Y. Pan, Z. Wang, Y. Xia, Y. Chen and W. Huang, J. Mater. Chem. A, 2017, 5, 12602–12652 RSC.
- Z. Huang, X. Hu, C. Liu, L. Tan and Y. Chen, Adv. Funct. Mater., 2017, 27, 1703061 CrossRef.
- C. Fei, L. Guo, B. Li, R. Zhang, H. Fu, J. Tian and G. Cao, Nano Energy, 2016, 27, 17–26 CrossRef CAS.
- B.-X. Chen, W.-G. Li, H.-S. Rao, Y.-F. Xu, D.-B. Kuang and C.-Y. Su, Nano Energy, 2017, 34, 264–270 CrossRef CAS.
- J. Wang, F. Meng, R. Li, S. Chen, X. Huang, J. Xu, X. Lin, R. Chen, H. Wu and H.-L. Wang, Sol. RRL, 2020, 4, 2000091 CrossRef CAS.
- J. C. Hamill, J. C. Sorli, I. Pelczer, J. Schwartz and Y.-L. Loo, Chem. Mater., 2019, 31, 2114–2120 CrossRef CAS.
- X. Wang, Y. Fan, L. Wang, C. Chen, Z. Li, R. Liu, H. Meng, Z. Shao, X. Du, H. Zhang, G. Cui and S. Pang, Chem, 2020, 6, 1369–1378 CAS.
- P. K. Nayak, D. T. Moore, B. Wenger, S. Nayak, A. A. Haghighirad, A. Fineberg, N. K. Noel, O. G. Reid, G. Rumbles, P. Kukura, K. A. Vincent and H. J. Snaith, Nat. Commun., 2016, 7, 13303 CrossRef CAS.
- M. I. Saidaminov, A. L. Abdelhady, B. Murali, E. Alarousu, V. M. Burlakov, W. Peng, I. Dursun, L. Wang, Y. He, G. Maculan, A. Goriely, T. Wu, O. F. Mohammed and O. M. Bakr, Nat. Commun., 2015, 6, 7586 CrossRef.
- X. Cheng, S. Yang, B. Cao, X. Tao and Z. Chen, Adv. Funct. Mater., 2019, 30, 1905021 CrossRef.
- Y. Chen, M. He, J. Peng, Y. Sun and Z. Liang, Adv. Sci., 2016, 3, 1500392 CrossRef.
- M. De Bastiani, M. I. Saidaminov, I. Dursun, L. Sinatra, W. Peng, U. Buttner, O. F. Mohammed and O. M. Bakr, Chem. Mater., 2017, 29, 3367–3370 CrossRef CAS.
- F. Zhang, J. C. Hamill Jr, Y. L. Loo and A. Kahn, Adv. Mater., 2020, 2, e2003482 CrossRef.
- D. Bryant, N. Aristidou, S. Pont, I. Sanchez-Molina, T. Chotchunangatchaval, S. Wheeler, J. R. Durrant and S. A. Haque, Energy Environ. Sci., 2016, 9, 1655–1660 RSC.
- Y. Li, X. Xu, C. Wang, B. Ecker, J. Yang, J. Huang and Y. Gao, J. Phys. Chem. C, 2017, 121, 3904–3910 CrossRef CAS.
- D. W. deQuilettes, W. Zhang, V. M. Burlakov, D. J. Graham, T. Leijtens, A. Osherov, V. Bulovic, H. J. Snaith, D. S. Ginger and S. D. Stranks, Nat. Commun., 2016, 7, 11683 CrossRef CAS.
- Y. C. Zhao, W. K. Zhou, X. Zhou, K. H. Liu, D. P. Yu and Q. Zhao, Light: Sci. Appl., 2017, 6, e16243 CrossRef CAS.
- A. Ummadisingu, L. Steier, J. Y. Seo, T. Matsui, A. Abate, W. Tress and M. Gratzel, Nature, 2017, 545, 208–212 CrossRef CAS.
- G. Niu, W. Li, F. Meng, L. Wang, H. Dong and Y. Qiu, J. Mater. Chem. A, 2014, 2, 705–710 RSC.
- Z. Wang, Z. Shi, T. Li, Y. Chen and W. Huang, Angew. Chem., Int. Ed., 2017, 56, 1190–1212 CrossRef CAS.
- W. Zhang, S. Pathak, N. Sakai, T. Stergiopoulos, P. K. Nayak, N. K. Noel, A. A. Haghighirad, V. M. Burlakov, D. W. deQuilettes, A. Sadhanala, W. Li, L. Wang, D. S. Ginger, R. H. Friend and H. J. Snaith, Nat. Commun., 2015, 6, 10030 CrossRef CAS.
- H. Wei, S. Chen, J. Zhao, Z. Yu and J. Huang, Chem. Mater., 2020, 32, 2501–2507 CrossRef CAS.
- R. S. Brown, A. J. Bennet and H. Slebocka-Tilk, Acc. Chem. Res., 2002, 25, 481–488 CrossRef.
- D. Zahn, J. Phys. Chem. B, 2003, 107, 12303–12306 CrossRef CAS.
- T. Cottineau, M. Richard-Plouet, J.-Y. Mevellec and L. Brohan, J. Phys. Chem. C, 2011, 115, 12269–12274 CrossRef CAS.
- B. Dou, L. M. Wheeler, J. A. Christians, D. T. Moore, S. P. Harvey, J. J. Berry, F. S. Barnes, S. E. Shaheen and M. F. A. M. van Hest, ACS Energy Lett., 2018, 3, 979–985 CrossRef CAS.
- M. V. Lee, S. R. Raga, Y. Kato, M. R. Leyden, L. K. Ono, S. Wang and Y. Qi, J. Mater. Res., 2016, 32, 45–55 CrossRef.
- Y. Chen, N. Li, L. Wang, L. Li, Z. Xu, H. Jiao, P. Liu, C. Zhu, H. Zai, M. Sun, W. Zou, S. Zhang, G. Xing, X. Liu, J. Wang, D. Li, B. Huang, Q. Chen and H. Zhou, Nat. Commun., 2019, 10, 1112 CrossRef.
- N. K. Noel, M. Congiu, A. J. Ramadan, S. Fearn, D. P. McMeekin, J. B. Patel, M. B. Johnston, B. Wenger and H. J. Snaith, Joule, 2017, 1, 328–343 CrossRef CAS.
- M. Qin, J. Cao, T. Zhang, J. Mai, T.-K. Lau, S. Zhou, Y. Zhou, J. Wang, Y.-J. Hsu, N. Zhao, J. Xu, X. Zhan and X. Lu, Adv. Energy Mater., 2018, 8, 1703399 CrossRef.
- X. Wu, Y. Jiang, C. Chen, J. Guo, X. Kong, Y. Feng, S. Wu, X. Gao, X. Lu, Q. Wang, G. Zhou, Y. Chen, J. M. Liu, K. Kempa and J. Gao, Adv. Funct. Mater., 2019, 30, 1908613 CrossRef.
- Z. Liu, J. Hu, H. Jiao, L. Li, G. Zheng, Y. Chen, Y. Huang, Q. Zhang, C. Shen, Q. Chen and H. Zhou, Adv. Mater., 2017, 29, 1606774 CrossRef.
- K. Meng, L. Wu, Z. Liu, X. Wang, Q. Xu, Y. Hu, S. He, X. Li, T. Li and G. Chen, Adv. Mater., 2018, 30, 1706401 CrossRef.
- R. Munir, A. D. Sheikh, M. Abdelsamie, H. Hu, L. Yu, K. Zhao, T. Kim, O. E. Tall, R. Li, D. M. Smilgies and A. Amassian, Adv. Mater., 2017, 29, 1604113 CrossRef.
- C. Aranda, C. Cristobal, L. Shooshtari, C. Li, S. Huettner and A. Guerrero, Sustainable Energy Fuels, 2017, 1, 540–547 RSC.
- C.-H. Chiang, M. K. Nazeeruddin, M. Grätzel and C.-G. Wu, Energy Environ. Sci., 2017, 10, 808–817 RSC.
- X. Gong, M. Li, X.-B. Shi, H. Ma, Z.-K. Wang and L.-S. Liao, Adv. Funct. Mater., 2015, 25, 6671–6678 CrossRef CAS.
- J. Huang, S. Tan, P. D. Lund and H. Zhou, Energy Environ. Sci., 2017, 10, 2284–2311 RSC.
- D. Angmo, X. Peng, A. Seeber, C. Zuo, M. Gao, Q. Hou, J. Yuan, Q. Zhang, Y. B. Cheng and D. Vak, Small, 2019, 15, e1904422 CrossRef.
- K. Zhang, Z. Wang, G. Wang, J. Wang, Y. Li, W. Qian, S. Zheng, S. Xiao and S. Yang, Nat. Commun., 2020, 11, 1006 CrossRef CAS.
- C. Duan, J. Cui, M. Zhang, Y. Han, S. Yang, H. Zhao, H. Bian, J. Yao, K. Zhao, Z. Liu and S. Liu, Adv. Energy Mater., 2020, 10, 2000691 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |