
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA switchable sensor and scavenger: detection and removal of fluorinated chemical species by a luminescent metal–organic framework†
Hua-Qing
Yin
 abc,
Kui
Tan
d,
Stephanie
Jensen
e,
Simon J.
Teat
f,
Saif
Ullah
e,
Xiuze
Hei
a,
Ever
Velasco
a,
Kolade
Oyekan
d,
Noah
Meyer
e,
Xin-Yao
Wang
b,
Timo
Thonhauser
e,
Xue-Bo
Yin
*b and
Jing
Li
*a
abc,
Kui
Tan
d,
Stephanie
Jensen
e,
Simon J.
Teat
f,
Saif
Ullah
e,
Xiuze
Hei
a,
Ever
Velasco
a,
Kolade
Oyekan
d,
Noah
Meyer
e,
Xin-Yao
Wang
b,
Timo
Thonhauser
e,
Xue-Bo
Yin
*b and
Jing
Li
*a
aDepartment of Chemistry and Chemical Biology, Rutgers University, 123 Bevier Road, Piscataway, NJ 08854, USA. E-mail: jingli@rutgers.edu
bState Key Laboratory of Medicinal Chemical Biology, Tianjin Key Laboratory of Biosensing and Molecular Recognition, College of Chemistry, Nankai University, 94 Weijin Road, Tianjin 300071, P. R. China. E-mail: xbyin@nankai.edu.cn
cInstitute for New Energy Materials and Low Carbon Technologies, School of Materials Science & Engineering, Tianjin University of Technology, 391 Bin Shui Xi Dao Road, Tianjin 300384, P. R. China
dMaterials Science and Engineering, The University of Texas at Dallas, 800 W. Campbell Road, Richardson, TX 75080, USA
eDepartment of Physics and Center for Functional Materials, Wake Forest University, 1834 Wake Forest Road, Winston–Salem, NC 27109, USA
fAdvanced Light Source, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, CA 94720, USA
First published on 30th September 2021
Abstract
Fluorosis has been regarded as a worldwide disease that seriously diminishes the quality of life through skeletal embrittlement and hepatic damage. Effective detection and removal of fluorinated chemical species such as fluoride ions (F−) and perfluorooctanoic acid (PFOA) from drinking water are of great importance for the sake of human health. Aiming to develop water-stable, highly selective and sensitive fluorine sensors, we have designed a new luminescent MOF In(tcpp) using a chromophore ligand 2,3,5,6-tetrakis(4-carboxyphenyl)pyrazine (H4tcpp). In(tcpp) exhibits high sensitivity and selectivity for turn-on detection of F− and turn-off detection of PFOA with a detection limit of 1.3 μg L−1 and 19 μg L−1, respectively. In(tcpp) also shows high recyclability and can be reused multiple times for F− detection. The mechanisms of interaction between In(tcpp) and the analytes are investigated by several experiments and DFT calculations. These studies reveal insightful information concerning the nature of F− and PFOA binding within the MOF structure. In addition, In(tcpp) also acts as an efficient adsorbent for the removal of F− (36.7 mg g−1) and PFOA (980.0 mg g−1). It is the first material that is not only capable of switchable sensing of F− and PFOA but also competent for removing the pollutants via different functional groups.
Introduction
Fluoride ion (F−) is a kind of essential element of the human body and promote the formation of fluorapatite on the dental and skeletal surfaces at low concentration.1,2 However, when F− intake is beyond 0.05 mg per kg per day, it will become one of the most toxic and dangerous elements.3–6 Excessive consumption of F− has been proven to be the main cause of dental and bone fluorosis.7–10 Chronic absorption of an excess amount of F− would cause gastric and kidney disorders and even death.11–13 F− can be easily absorbed by the human body when in contact with water, earth, and even air.14,15 As the industrialization process continues to accelerate, more and more areas are suffering from fluorine pollution.16,17 Perfluorooctanoic acid (PFOA), on the other hand, is a surfactant containing fifteen fluorine atoms. It has a high surface activity and is used in a variety of products, from surface protectants to polytetrafluoroethene and insecticides.18–20 Because of the high binding energy of carbon–fluorine (C–F) bonds, PFOA shows great resistance to natural biodegradation.21–25Via the food chain, PFOA can accumulate in the human body and shows immunological and neurological toxicity.26,27 Hence, PFOA is regarded as one of the emerging contaminants by various organizations.28–30 To avoid F− and PFOA from being ingested by the human body, their monitoring, identification, and removal from drinking water are essential yet remain challenging.31Some molecular compounds demonstrate turn-on/off and ratiometric luminescence detection for F−.4,32,33 While they show relatively higher sensitivity, complex procedures are usually required for the synthesis of these compounds. Metal–organic frameworks (MOFs), built by self-assembly processes, are regarded as an ideal platform for such sensing applications due to their facile synthesis, high porosity and surface tunability which conventional molecular sensors are usually lacking.34 To this date, there are only a few reports on F− detection and adsorption by MOFs. For example, an europium-based MOF, Eu-MOF-1, with dual-emission is investigated for ratiometric detection of F− ions with high selectivity and the limit of detection (LOD) is 2 μM.35 Another MOF, SION-105 with boron as the receptor site, shows a good response to F− with high selectivity and a LOD of 0.1 ppm.1 SION-105 was also used to develop an F− detection device for convenient use. A guest encapsulated MOF, NH2-MIL-101(Al)@DCF, reaches a lower LOD for F− (0.05 μM).36 However, the F− response of NH2-MIL-101(Al)@DCF requires addition of H2O2, making the process inconvenient. The dominant technique for PFOA analysis at the present time is HPLC-MS with high sensitivity, but it is time- and energy-consuming and must be carried out in professional laboratories with cumbersome instruments.37 Recent studies have shown that luminescent materials are promising candidates for convenient and real-time PFOA detection. For example, a photoluminescence-based sensor made of molecularly imprinted polymer-coated quantum dots has been used for the enrichment and determination of PFOA in water with the LOD of 11.8 nM.22 Being highly porous and luminescent, luminescent MOFs (LMOFs) have considerable potential for simultaneous pre-concentration, detection and removal of fluorine containing species. Often a single LMOF may be capable of detecting different kinds of pollutants with different response behaviors.
For solution-based sensing applications, LMOFs must possess high stability. The stability of MOFs is mainly determined by thermodynamic and kinetic factors.38 The thermodynamic stability is consistent with the hard/soft acid/base (HSAB) principle. Hard bases, such as carboxylate-based ligands, can react with high-valence (hard acid) metals (e.g. Ti4+, Zr4+, Al3+, Fe3+ and In3+) to form stable MOFs.39 Soft bases such as pyrazole can combine with low-valence (soft acid) metals such as Ni2+, Co2+ and Zn2+ to afford highly water stable MOFs.40 For example, Li and co-workers developed a series of BUT-32 and BUT-33 (BUT = Beijing University of Technology) MOFs with remarkable water stability as a result of combination of soft base pyrazole and soft acid Ni2+.41,42 By use of hard acid (Hf4+ and Zr4+) and hard base carboxylate ligands PCN MOFs developed by Zhou and co-workers also demonstrate high water and acid/base stability.43–45
This work presents, for the first time, a new water stable LMOF for the sensitive detection of F− and PFOA via a distinctly different signal change, as well as effective adsorption/removal of both species. The LMOF, simplified as In(tcpp) or LMOF-651, was prepared by reacting InCl3 and 2,3,5,6-tetrakis(4-carboxyphenyl)pyrazine (H4tcpp) under solvothermal conditions. In(tcpp) shows high porosity, water stability, and strong luminescence-based turn-on detection of F− and turn-off detection of PFOA in aqueous solutions. LODs of 1.3 μg L−1 and 19 μg L−1 were achieved for F− and PFOA, respectively (Scheme 1). The F− detection process was fully recyclable. Furthermore, In(tcpp) adsorbs a relatively large amount of F− (36.7 mg g−1) and PFOA (980.0 mg g−1), demonstrating its strong potential for treating drinking water. IR spectroscopic analysis and DFT calculations were carried out to explore the mechanism of F− and PFOA detection and adsorption.
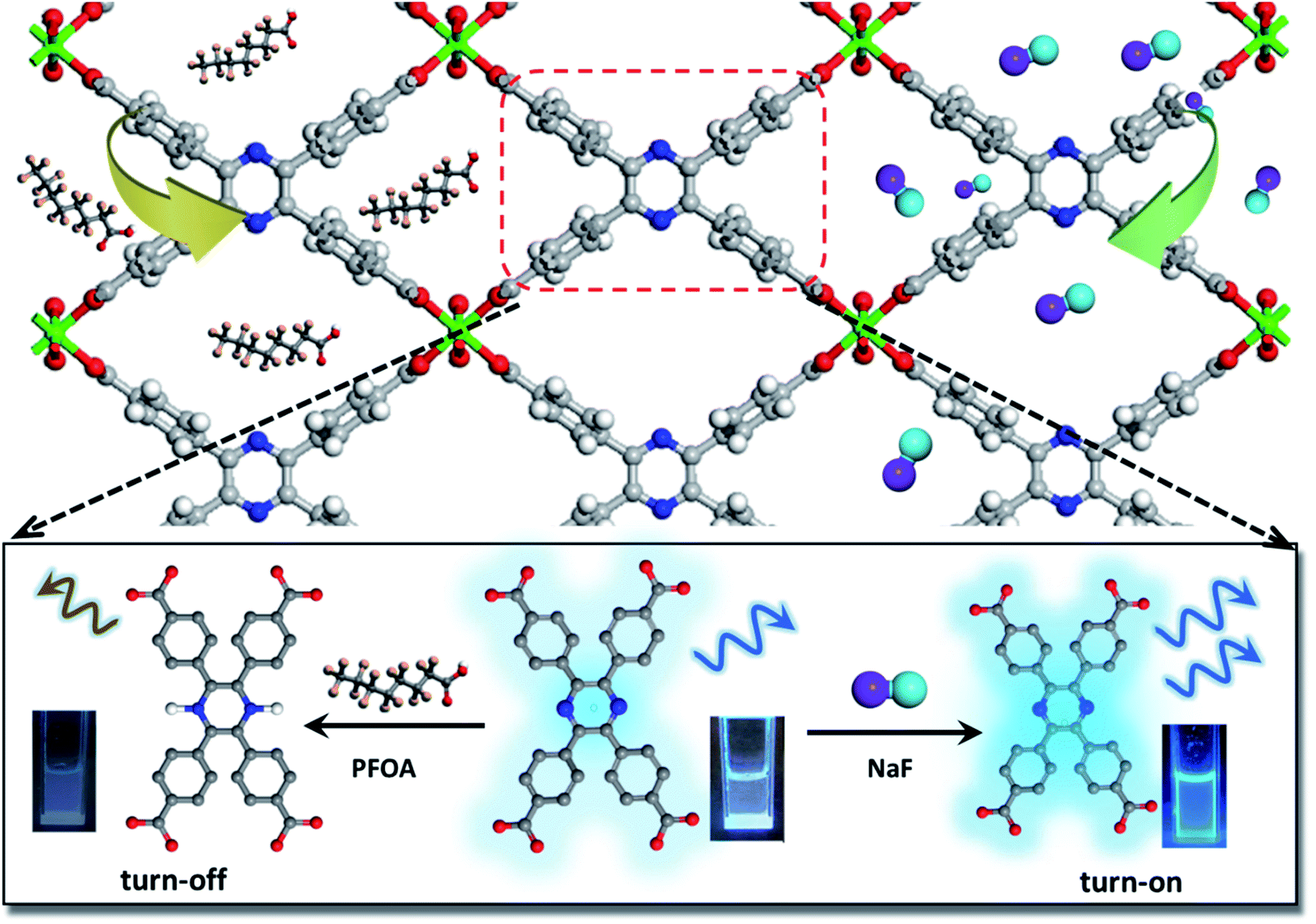 | ||
| Scheme 1 The structure of In(tcpp) (top) and schematic presentation of its luminescence switchable turn-off response toward PFOA (bottom left) and turn-on response toward F− ions (bottom right). Color codes: C grey; N blue; F light blue; O red; In green; Na purple; and H white. | ||
Results and discussion
Synthesis, crystal structure and characterization
H4tcpp was synthesized according to the former work.46 H4tcpp and InCl3 were used to prepare In(tcpp) by a solvothermal method in the presence of formic acid at 100 °C for 48 h (Fig. 1a and b). As a comparison, In(tcpb) or LMOF-652 was also constructed with InCl3 and 1,2,4,5-tetrakis(4-carboxyphenyl)benzene (H4tcpb) using the same synthetic method (ESI† for details). The two In-MOFs are isostructural and differ only by the two atoms in the central ring of the ligands, which allowed us to compare and understand their similarities and differences in the sensing behavior toward fluorine-based chemical species.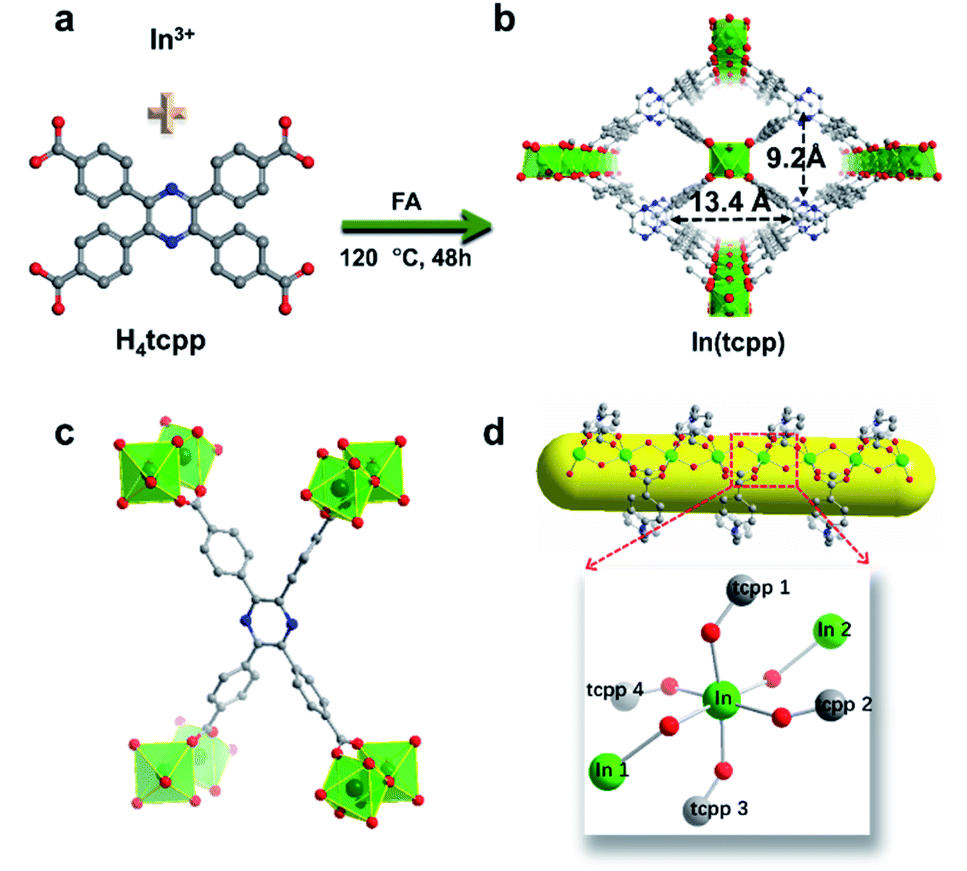 | ||
| Fig. 1 Representation of the crystal structure of In(tcpp). (a) The organic and inorganic building units of In(tcpp) and reaction conditions. (b) The one-dimensional channel in In(tcpp). (c) The coordination of the tcpp ligand. (d) The 6-coordinated In3+ ion and 1D In3+ chain along the a-axis. Color code: C grey; N blue; O red; In green; H atoms are omitted. | ||
The crystals of the resulting In(tcpp) and In(tcpb) are transparent, hexagonal-shaped rods (Fig. S1†). Single crystal X-ray diffraction analysis (Table S1†) reveals that In(tcpp) crystallizes in an orthorhombic space group of Cmmm (a = 7.2195(3) Å, b = 22.5222(10) Å, c = 15.7802(7) Å) with a chemical formula of [In2(tcpp)(OH)2]. Each tcpp is connected to eight In3+ ions through In–O bonds to form an ordered one-dimensional rhombic channel, with a cross-section of about 9.2 × 13.4 Å (Fig. 1b and c). The hexacoordinated In3+ ion bonds to four carboxylate oxygen atoms from different tcpp ligands, and two hydroxo oxygen atoms that connect the adjacent In3+ ions through the In–OH–In bonds (Fig. 1d).47,48 In3+ ions are linked to each other through the tcpp ligands and bridging oxygen atoms to form a 1D chain. The PXRD analysis confirmed the phase purity of In(tcpp) (Fig. S2†). A comparison of the single crystal and PXRD data of In(tcpb) verified that it is isoreticular to In(tcpp) with the same space group topology (Fig. S3 and Table S2†).
In(tcpp) samples were activated by exchanging the solvents with acetone and then evacuated at 60 °C under vacuum for 24 h. No obvious differences were observed in the PXRD patterns of the as-made and activated In(tcpp) samples, indicating high framework stability (Fig. S2†). Similar procedures were used to activate In(tcpb). Porosity characterization reveals type I adsorption behavior for both In(tcpp) and In(tcpb), illustrating their microporous structures (Fig. S4a and S5a†). The Brunauer–Emmett–Teller (BET) surface area of In(tcpp) and In(tcpb) was estimated to be 535 m2 g−1 and 534 m2 g−1, respectively. The pore size distribution analysis yielded a pore size ∼6 Å for both In(tcpp) and In(tcpb) (Fig. S4b and S5b†).
The thermal stability of both MOFs was evaluated. From the thermogravimetric analysis (TGA) curve, the as-made In(tcpp) shows 21 wt% mass loss at 200 °C, which comes from the uncoordinated solvent molecules (Fig. S6†). For the activated In(tcpp) sample, there was no weight loss until 200 °C, illustrating that the guest molecules were fully removed from the MOF channels. In(tcpb) showed 25 wt% loss of guest solvent molecules at 210 °C (Fig. S7†). Both compounds were stable up to 400 °C. Upon heating at 200 °C for 2 h, the characteristic diffraction peaks of In(tcpp) remained (Fig. S8†). The PXRD patterns kept unchanged after soaking the In(tcpp) sample in water, in pH 2 (HCl) and pH 12 (NaOH) aqueous solutions for 24 h, demonstrating its excellent chemical stability. Also, N2 adsorption isotherms were collected after the sample was heated at 200 °C or treated under different acidity conditions at pH 2 (HCl) and pH 12 (NaOH), and the porosity remains the same (Fig. S9†), which further confirms the high stability of In(tcpp). The thermal stability of In(tcpb) was found to be similar to that of In(tcpp). However, its resistance to the chemical environment was less than the former as it collapsed after being treated in pH 11 for 24 h (Fig. S10†).
The UV-vis spectra of In(tcpp) and H4tcpp ligand were collected at room temperature (Fig. S11†). H4tcpp and In(tcpp) show very similar absorption with a characteristic π → π* transition cantered at 325 nm. As a result, the emission of the In(tcpp) originates from the ligand. 3D luminescence spectra of In(tcpp) and H4tcpp were measured and the optimal excitation was observed at 280 nm (Fig. S12†). Under 280 nm excitation, In(tcpp) emits at 400 nm with 30 nm blue shift compared to the free H4tcpp (Fig. S13†). The blue shift of In(tcpp) was attributed to the different structural torsion of the free H4tcpp and tcpp fixed in the In(tcpp). As shown in Fig. S14a,† in the free H4tcpp, the dihedral angle between the benzene ring and pyrazine core is approximately 20°. However, the dihedral angle of tcpp increases to approximately 56° to cater the rigid structure of In(tcpp) (Fig. S14b†). The torsion of the benzene rings changes the π-conjugation of the whole ligand, resulting in the blue-shift emission.46 The optical properties of In(tcpb) were also measured and the results are shown in Fig. S15–S17.† The optimal excitation of 270 nm was selected to obtain the emission at 380 nm for In(tcpb).
Switchable detection and removal of F− and PFOA
Considering the interaction between guest analytes and functional groups of In(tcpp), activated In(tcpp) was selected for the detection experiments. In(tcpp) remained stable after being soaked in 1 mM NaF aqueous solution for 24 h (Fig. S18†), illustrating that it is sufficiently stable for F− detection. As shown in Fig. S19,† a drastic enhancement in the photoluminescence (PL) intensity was observed within ∼5 min upon addition of NaF solution.The results of the titration experiment are shown in Fig. 2a. Upon addition of F−, the PL intensity of In(tcpp) increased monotonically. The enhancement reached 250% at a concentration of 200 μM F−. The turn-on efficiency was calculated by the Stern–Volmer (SV) eqn (1):
| I0/I = Ksv[F−] + 1 | (1) |
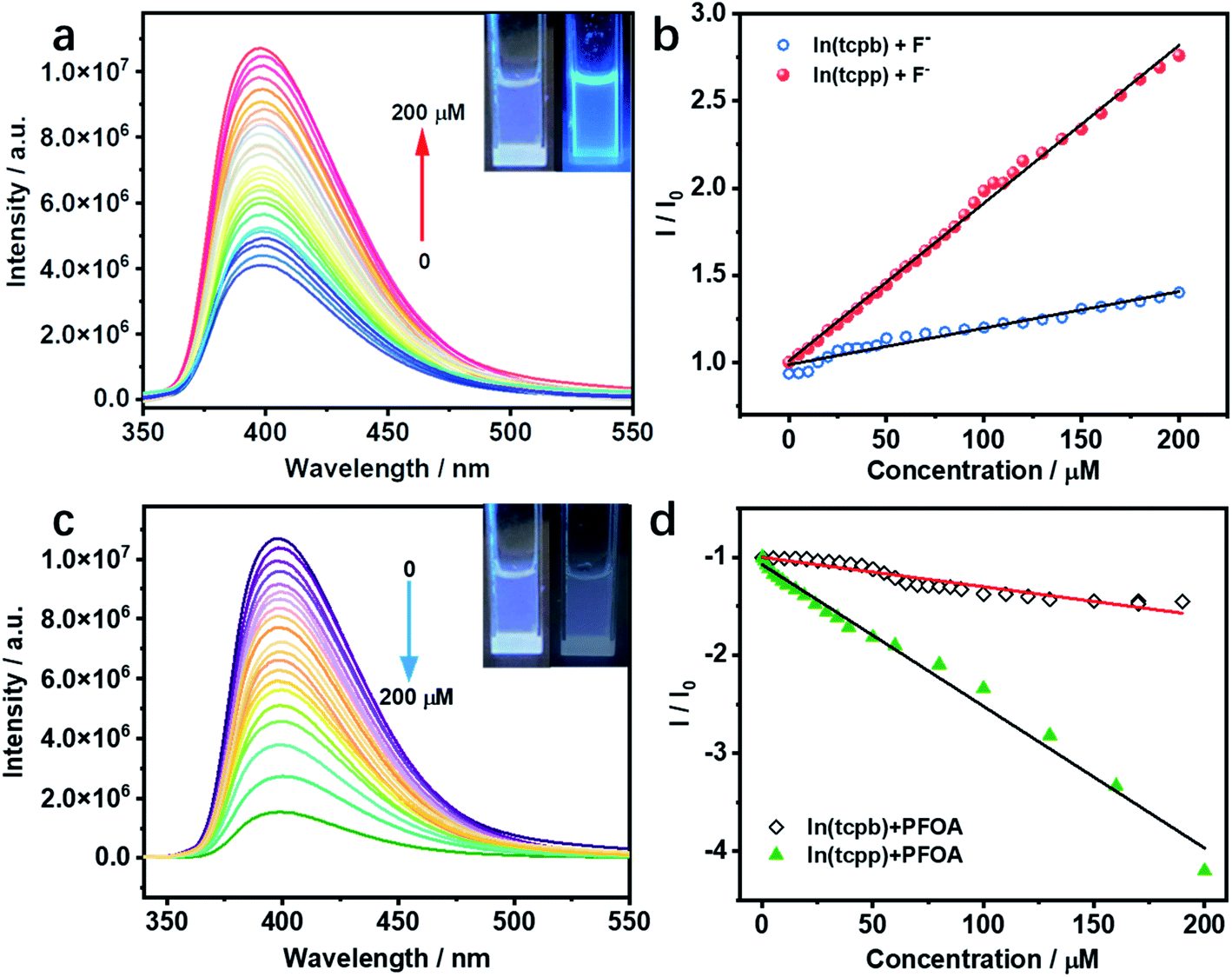 | ||
| Fig. 2 (a) In(tcpp) luminescence turn-on response to F− at various concentrations. (b) The concentration-dependent luminescence response of In(tcpp) and In(tcpb) to F−. (c) In(tcpp) luminescence turn-off response to PFOA at various concentrations. (d) The concentration-dependent luminescence response of In(tcpp) and In(tcpb) to PFOA. Insets in (a) and (c): images of the In(tcpp) solution samples before (left) and after (right) the addition of the analytes. The detection response time for (a)–(d) 5 min. | ||
In(tcpp) is stable in PFOA solution as there was no obvious change in its PXRD patterns before and after exposure to a 1 mM PFOA solution for 24 h (Fig. S21†). Interestingly, In(tcpp) shows monotonic quenching as a function of PFOA concentration (Fig. 2c). As depicted in Fig. 2d and S22,† In(tcpp) responds to PFOA at very low concentrations with the Ksv and LOD of 1.446 × 104 M−1 and 19 μg L−1, respectively.49 It shows a comparative sensitivity as a PFOA sensor (Table S4†). In addition, it can act as an excellent scavenger for PFOA removal as discussed in the following section. However, the isostructural In(tcpb) shows little PL response to F− and PFOA (Fig. 2b and d).
As stated earlier, excessive F− is regarded as a water contaminant, while some common light ions, such as Ca2+, Mg2+ and Cl−, are also found in drinking water and play important roles in biological processes. Therefore, selective detection of F− ions over other harmless ions is of great importance. As shown in Fig. 3a and b, In(tcpp) exhibits distinct selectivity toward F− over other cations and anions although their concentrations are higher than F−. The fast, selective, and turn-on response makes In(tcpp) a promising sensor for easy and convenient detection of F− ions. Further, the relative selectivity of In(tcpp) was tested in different pH solutions (Fig. S23 and S24†). We found that at higher pH values (e.g. pH 10), strong luminescence enhancement was observed with addition of F− and In(tcpp) exhibits good selectivity toward F−. While at lower pH (e.g. pH 3), F− gives rise to very little change in the luminescence intensity. Moreover, in pH 10 solution (Fig. S24†), the sensitivity of F− is similar to that in pure water.
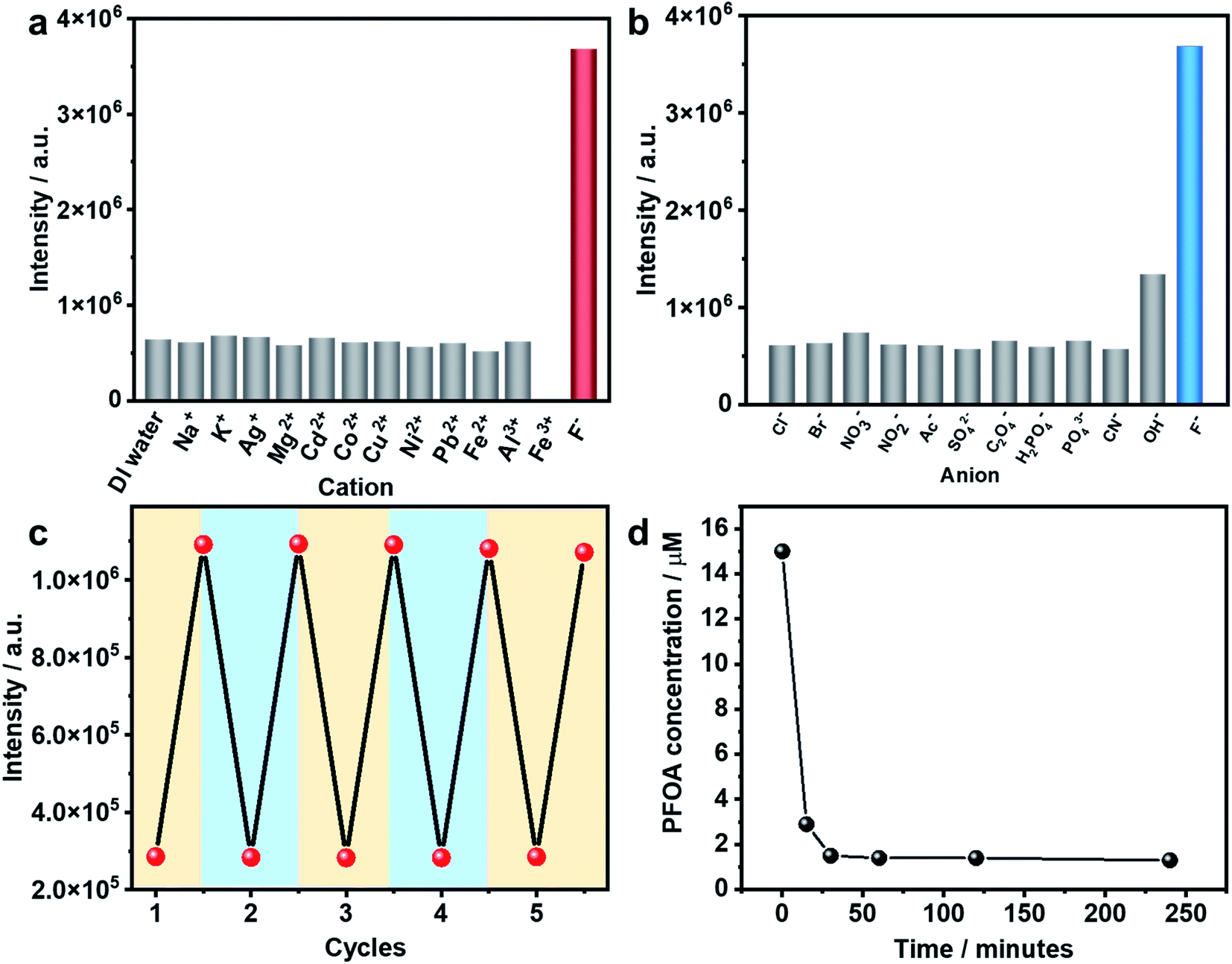 | ||
| Fig. 3 Selectivity of In(tcpp) for (a) cations and (b) anions. In(tcpp) 0.025 mg mL−1, the concentration of F− is 650 μM and other cations and anions are 1 mM. (c) The In(tcpp) luminescence turn-on and turn-off in 5 cycles. (d) The In(tcpp) PFOA removal efficiency in different pH solutions. | ||
Reusability is another important performance measure for a luminescence-based sensor. Considering the effective interaction between boric acid and F−,35 F−@In(tcpp) was dissolved in boric acid solutions of pH 3 to regenerate the sensor material. After being stirred and centrifuged three times, In(tcpp) was fully regenerated as its PL intensity reached the original level. The recovered In(tcpp) was then reused four times consecutively without apparent changes in its PL profile (Fig. 3c); the PXRD patterns show that In(tcpp) remained stable after 5 cycles (Fig. S25†), confirming its good recyclability. Taking into consideration its high porosity and stability, In(tcpp) was further used to remove F− ions. An uptake capacity of 36.7 mg g−1 (original concentration 100 ppb, remaining concentration 1.2 ppb) was achieved for this scavenger, a value that is higher than those of most of the F− removal materials, such as graphene, mesoporous alumina, and hydrous ferric oxide.50
The PFOA removal performance was assessed in different pH solutions. As shown in Fig. S26,† In(tcpp) adsorbs 980 mg g−1 PFOA in acidic solution (pH 3, original concentration 100 ppb, remaining concentration 2.14 ppb), which is higher than those of most of the porous materials (such as mesoporous silica, activated carbons, activated carbons, molecularly imprinted polymers, etc.) reported to date.51 We find that 90% of PFOA (at a concentration of 15 μM) can be removed within 30 min (Fig. 3d). Thus, strong evidence confirms that In(tcpp) is a good candidate not only for the switchable detection of PFOA and F−, but also for their capture and removal.
MOF–analyte interaction mechanism
To unveil the MOF–analyte interaction mechanism, we turned to DFT calculation and infrared/X-ray photoelectron spectroscopy to examine the binding of PFOA and NaF in In(tcpp). We calculated the removal efficiency based on XPS and HPLC-MS methods, which allow for the determination of the quantity of adsorbed F− and PFOA. The EPR and NMR experiments were carried out before and after adding NaF into the free ligand H4tcpp.52 As shown in Fig. S27 and S28,† there are no obvious differences before and after the NaF addition, suggesting that no charge/electron transfer interactions occurred between NaF and H4tcpp. Based on the UV-vis and PL spectra (Fig. S29†), small and gradual increase in the absorption intensity was observed as a function of time, while an appreciable enhancement in the PL intensity was noted after 30 min. These data indicate that the strong basicity of NaF promotes the dissolution of the ligand giving rise to the increases in the UV-vis and PL intensity.We then studied the configuration of F− in In(tcpp) via XPS. The content of F− is much higher than that of Na+ as shown from XPS results (Fig. S30 and Table S5†). We speculate that F− exists in the form of both NaF and HF within the sample, but the latter is the majority species. We thus analyzed the favorable binding site of HF within In(tcpp), which is near Ntcpp (Fig. S31a†). The H+ of HF is 1.733 Å away from the Ntcpp and the H–F bond length is 0.975 Å, which is slightly elongated compared to our calculated gas-phase bond length of 0.938 Å. The binding configuration predicted computationally is in good agreement with what we observed in the IR spectra (Fig. 4), namely, significant perturbations of the ring mode, e.g., blue shift of the ν(CN)pyrazine band is observed in IR spectra, indicating the direct interaction of HF with the tcpp linker. In addition, no amine deformation bands δ(NH)ip,oop were detected because H stays closer to F without protonating the N site on the organic linker, in perfect agreement with our theoretical predictions. Moreover, the blue shift of the ν(CN)pyrazine band as observed in our IR spectra is mimicked with the induced charge density calculations where a strong interaction of H@HF with the N@tcpp linker is clearly visible (Fig. S31b†).
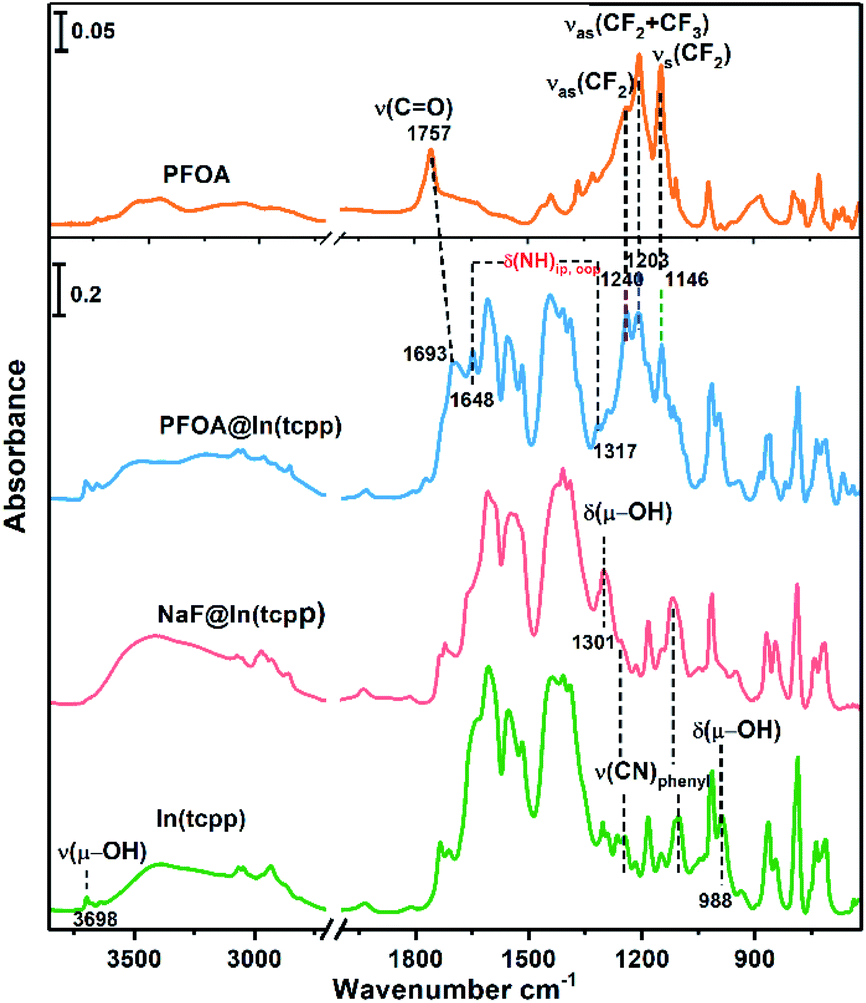 | ||
| Fig. 4 FTIR spectra of free PFOA, PFOA@In(tcpp), NaF@In(tcpp), and In(tcpp) (from top to bottom), referenced to the blank KBr pellet. | ||
We further assessed the possibility of the minority species NaF interacting directly with the N sites in In(tcpp), and found that they are unfavorable based on the estimated binding energetics (1.35 eV between Na+ and Ntcpp) (Fig. S32†). Further, numerous starting configurations of Na+, F−, and Na+F− interacting with the tcpp ligand or In node were considered. We found that after the F− adsorption, the coordination of F− at the axial position of In via μH–O displacement is energetically unfavourable. Instead, the most energetically favourable model for NaF binding is that Na+ interacts with oxygen on the In–Otcpp, and the F− interacts with both OH and Na, based on their charge rearrangement (Fig. 5a and b). This is consistent with the infrared spectroscopic results, which suggest that NaF most likely binds in close proximity to the COO and OH groups (Fig. 4). Firstly, the bridging OH stretching band ν(μ-OH) at 3698 cm−1 and bending band β(OH) at 983 cm−1 disappear. Additionally, a new band is observed at 1301 cm−1. DFT calculation has ascribed this new band to the shifted β(OH) vibration upon forming H-bonding interaction with F− (Table S6†). Furthermore, a decrease of the intensity of stretching bands ν(COO) is detected in the NaF@In(tcpp) spectrum, giving an indication that NaF interacts with the COO group. The photoluminescence enhancement was not captured, possibly due to exciton formation upon the photoexcitation.
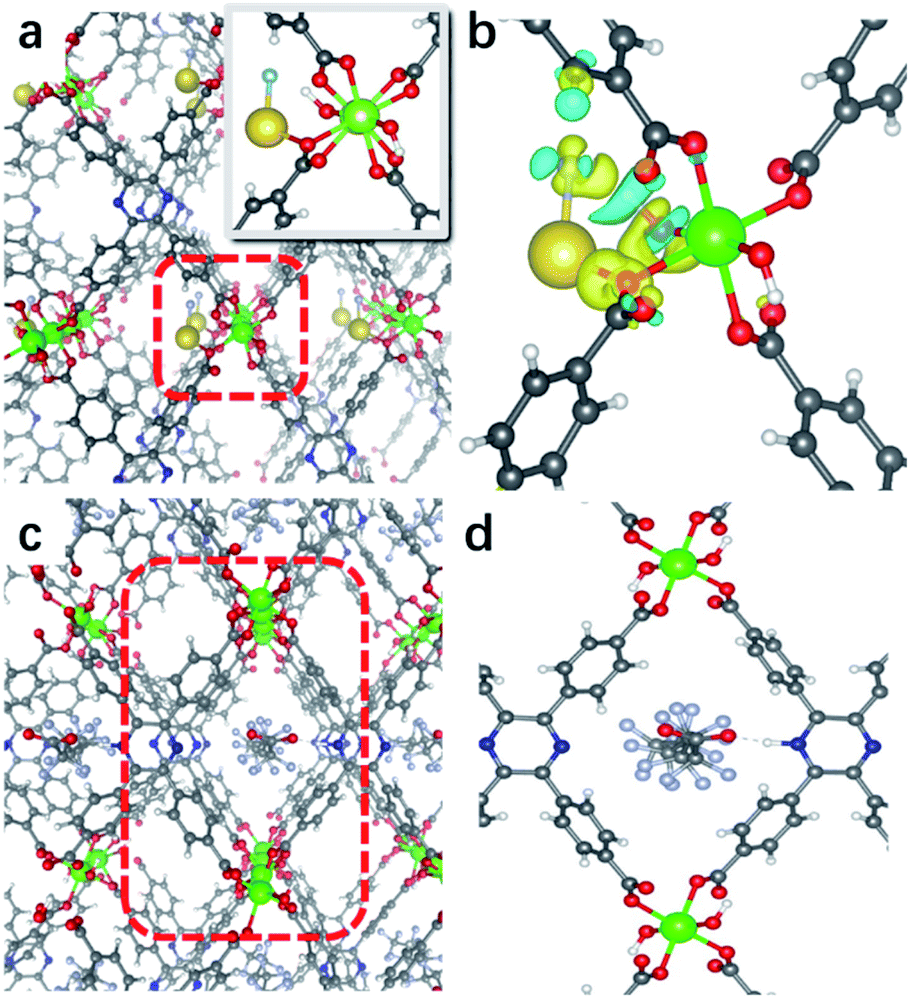 | ||
| Fig. 5 (a) Optimized structure of NaF@In(tcpp), highlighting Na+ and F− interactions. (b) Charge density rearrangement upon formation of the bond at an isolevel of 0.002 eV Å−3. (c) Binding of PFOA and In(tcpp) in PFOA@In(tcpp). (d) Configuration of PFOA-@In(tcpp) (atoms are colored as follows: C grey, O red, N blue, In green, F light blue, Na yellow, H white). | ||
Based on the titration PFOA turn-off results, we tested the model of PFOA binding to the nitrogen of the linker through its H+, forming PFOA−. Several other binding sites were also examined but were found to be less favorable and, therefore, are not part of this discussion. In the PFOA@In(tcpp), we found that hydrogen binds more favorably to the nitrogen of the linker, but is still close enough to the PFOA− molecule to form a hydrogen bond (Fig. 5c and d) with a strong binding energy of 1.57 eV. This is in accordance with an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of Ntcpp:PFOA, calculated based on our HPLC-MS results of 980.0 mg g−1 PFOA adsorption (see the characteristic absorption peak and standard curve of PFOA in Fig. S33†). The protonation of N is evidenced by the appearance of two new bands at 1648 and 1317 cm−1 as well as a red frequency shift of the ν(C
:1 ratio of Ntcpp:PFOA, calculated based on our HPLC-MS results of 980.0 mg g−1 PFOA adsorption (see the characteristic absorption peak and standard curve of PFOA in Fig. S33†). The protonation of N is evidenced by the appearance of two new bands at 1648 and 1317 cm−1 as well as a red frequency shift of the ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) band from 1757 cm−1 to 1693 cm−1 (Fig. S34†). Frequency modeling by DFT shows that these two bands at 1648 and 1317 cm−1 can be assigned to in-plane and out-of-plane deformation bands of the –NH group, respectively. The so formed –NHtcpp group can further interact with the PFOA's carboxylate group by hydrogen bonding interaction, resulting in an observed frequency shift of the ν(CO) band to 1693 cm−1 (Fig. 4). The protonation of N is evident from the Raman spectra (Fig. S35†). After interacting with PFOA, the v(N–H) band is observed at 1568 cm−1. Further, the hydrogen bonding between Ntcpp and PFOA blueshifts the v(C–O) from 1148 cm−1 to 1145 cm−1. Our theoretical and spectroscopic results show a distinct binding mechanism due to the presence of N in the organic component and the acid–base interaction between the PFOA molecule and In(tcpp) framework is the driving force for PFOA detection and adsorption (Fig. S36†). Using these models and comparing them to the pristine In(tcpp) MOF, we calculated the photoluminescence quenching of PFOA and PFOA− using our previously developed methodology.53,54 As can be seen in Fig. S37,† PFOA shows strong quenching of the photoluminescence in PFOA@In(tcpp) (92%).
O) band from 1757 cm−1 to 1693 cm−1 (Fig. S34†). Frequency modeling by DFT shows that these two bands at 1648 and 1317 cm−1 can be assigned to in-plane and out-of-plane deformation bands of the –NH group, respectively. The so formed –NHtcpp group can further interact with the PFOA's carboxylate group by hydrogen bonding interaction, resulting in an observed frequency shift of the ν(CO) band to 1693 cm−1 (Fig. 4). The protonation of N is evident from the Raman spectra (Fig. S35†). After interacting with PFOA, the v(N–H) band is observed at 1568 cm−1. Further, the hydrogen bonding between Ntcpp and PFOA blueshifts the v(C–O) from 1148 cm−1 to 1145 cm−1. Our theoretical and spectroscopic results show a distinct binding mechanism due to the presence of N in the organic component and the acid–base interaction between the PFOA molecule and In(tcpp) framework is the driving force for PFOA detection and adsorption (Fig. S36†). Using these models and comparing them to the pristine In(tcpp) MOF, we calculated the photoluminescence quenching of PFOA and PFOA− using our previously developed methodology.53,54 As can be seen in Fig. S37,† PFOA shows strong quenching of the photoluminescence in PFOA@In(tcpp) (92%).
Conclusions
In summary, we have designed and prepared a robust and porous LMOF, In(tcpp). In(tcpp) acts as a “switchable” sensor, showing a strong turn-on and turn-off PL signal when exposed to F− anions and PFOA. Its limits of detection for F− ions and PFOA are 1.3 μg L−1 and 19 μg L−1, respectively, indicating high sensitivity towards both chemical species. In(tcpp) also shows excellent selectivity for F− over other cations and anions commonly found in drinking water. The F− detection process is fully recyclable and the sensor material is reusable. Moreover, In(tcpp) can be used as an effective scavenger to capture and remove F− and PFOA from aqueous solution. FTIR analysis and DFT calculations were performed to explore the MOF–analyte interaction mechanism: in the case of PFOA, the Ntcpp is the site of interaction that is responsible for the quantitative adsorption of PFOA; the maximum loading amount is consistent with the number of available binding sites. The interaction between F− and In(tcpp) occurs primarily at the bridging –OH−, which is also consistent with the In/F ratio. This work demonstrates that switchable detection and removal of similar pollutants by the same MOF may be achieved and may serve as an effective approach in the future design of high performance MOF-based chemical sensors and scavengers.Experimental
Preparation of compounds
| LOD = kSb/a | (2) |
To estimate the PFOA removal efficiency of In(tcpp) in solutions of different pH, 55 mg In(tcpp) was added into 10 mL PFOA in different pH buffer (15 mM). The samples were sonicated to ensure good mixing. After 30 min, the concentration of supernatant PFOA was obtained after centrifugation.
Data availability
All experimental supporting data are provided in the ESI.†Author contributions
H. Yin and J. Li designed the experiments. H. Yin. performed the syntheses, measurements and detection on the compounds. K. Tan, S. Jensen, S. Ullah, K. Oyekan, N. Meyer and T. Thonhauser performed the FTIR measurements and DFT calculations. K. Tan organized the detection and removal mechanism. S. Teat performed single X-ray diffraction analyses. X. Hei, E. Velasco and X. Wang gave suggestions and helped in the whole process. H. Yin wrote the first draft of the manuscript, which was subsequently edited by J. Li. and X. Yin. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award No. DE-SC0019902 is gratefully acknowledged for support of the synthesis, structural characterization, adsorption and sensing studies, vibrational spectroscopy and all theoretical calculation work. This work was also supported partially by the National Natural Science Foundation of China (Grants No. 21874074). Hua-Qing Yin is grateful to the China Scholarship Council (20183101) for supporting her visit at Rutgers University. The Advanced Light Source (ALS) was supported by the Director, Office of Science, Office of Basic Energy Science, of the U.S. Department of Energy, under contract DE-AC02-05CH11231.Notes and references
- F. M. Ebrahim, T. N. Nguyen, S. Shyshkanov, A. Gladysiak, P. Favre, A. Zacharia, G. Itskos, P. J. Dyson and K. C. Stylianou, J. Am. Chem. Soc., 2019, 141, 3052–3058 CrossRef CAS PubMed.
- J. D. B. Featherstone, Community Dent. Oral Epidemiol., 1999, 27, 31–40 CrossRef CAS PubMed.
- Z. Kheradpisheh, M. Mirzaei, A. H. Mahvi, M. Mokhtari, R. Azizi, H. Fallahzadeh and M. H. Ehrampoush, Sci. Rep., 2018, 8, 2674 CrossRef PubMed.
- T. K. Mandal, Y. Hou, Z. Gao, H. Ning, W. Yang and M. Gao, Adv. Sci., 2016, 3, 1600217 CrossRef PubMed.
- O. Barbier, L. Arreola-Mendoza and L. M. Del Razo, Chem.-Biol. Interact., 2010, 188, 319–333 CrossRef CAS PubMed.
- A. Bontemps, L. Conquet, C. Elie, V. Magneron, C. Gloaguen, D. Kereselidze, K. Tack, O. C. Barbier and Y. Gueguen, Int. J. Environ. Res. Public Health, 2019, 16, 1136–1156 CrossRef CAS PubMed.
- Y. Liu, A. Jiang, Q. Jia, X. Zhai, L. Liu, L. Ma and J. Zhou, Chem. Sci., 2018, 9, 5242–5251 RSC.
- B. Ke, W. Chen, N. Ni, Y. Cheng, C. Dai, H. Dinh and B. Wang, Chem. Commun., 2013, 49, 2494–2496 RSC.
- S. Ayoob and A. K. Gupta, Crit. Rev. Environ. Sci. Technol., 2006, 36, 433–487 CrossRef CAS.
- S. Roy and G. Dass, Resour. Environ., 2013, 3, 53–58 Search PubMed.
- M.-L. Cittanova, B. Lelongt, M.-C. Verpont, M. Geniteau-Legendre, F. Wahbe, D. Prie, P. Coriat and P. M. Ronco, Anesthesiology, 1996, 84, 428–435 CrossRef CAS PubMed.
- M. I. Jimenez-Cordova, C. Gonzalez-Horta, J. C. Ayllon-Vergara, L. Arreola-Mendoza, G. Aguilar-Madrid, E. E. Villareal-Vega, A. Barrera-Hernandez, O. C. Barbier and L. M. Del Razo, Environ. Res., 2019, 169, 220–228 CrossRef CAS PubMed.
- Y. Tian, H. Che, J. Wang, D. Wang, L. Yang, L. Wang, Y. Nie and X. Tian, J. Hazard. Mater., 2021, 411, 125182 CrossRef CAS PubMed.
- A. Rasool, A. Farooqi, T. Xiao, W. Ali, S. Noor, O. Abiola, S. Ali and W. Nasim, Environ. Geochem. Health, 2018, 40, 1265–1281 CrossRef CAS PubMed.
- K. J. Barmao, J. K. Cherutoi, C. Y. Mitei, M. L. L. Were, A. Kiprop and O. G. Achieng', J. Environ. Manage., 2019, 8, 15–24 Search PubMed.
- Q. Zhang, S. Bolisetty, Y. Cao, S. Handschin, J. Adamcik, Q. Peng and R. Mezzenga, Angew. Chem., Int. Ed., 2019, 58, 6012–6016 CrossRef CAS PubMed.
- F. Ahmadijokani, H. Molavi, M. Rezakazemi, T. M. Aminabhavi and M. Arjmand, Coord. Chem. Rev., 2021, 445, 214037 CrossRef CAS.
- L. Zheng, Y. Zheng, Y. Liu, S. Long, L. Du, J. Liang, C. Huang, M. T. Swihart and K. Tan, Talanta, 2019, 194, 1–6 CrossRef CAS PubMed.
- K.-U. Goss, Environ. Sci. Technol., 2008, 42, 456–458 CrossRef CAS PubMed.
- Y. Wang, P. B. Groszewicz, S. Rosenfeldt, H. Schmidt, C. A. Volkert, P. Vana, T. Gutmann, G. Buntkowsky and K. Zhang, Adv. Mater., 2017, 29, 1702473 CrossRef PubMed.
- R. C. Buck, J. Franklin, U. Berger, J. M. Conder, I. T. Cousins, P. de Voogt, A. A. Jensen, K. Kannan, S. A. Mabury and S. P. van Leeuwen, Integr. Environ. Assess. Manage., 2011, 7, 513–541 CrossRef CAS PubMed.
- Z. Cheng, L. Du, P. Zhu, Q. Chen and K. Tan, Spectrochim. Acta, Part A, 2018, 201, 281–287 CrossRef CAS PubMed.
- C. Fang, X. Zhang, Z. Dong, L. Wang, M. Megharaj and R. Naidu, Chemosphere, 2018, 191, 381–388 CrossRef CAS PubMed.
- L. Liu, Y. Liu, N. Che, B. Gao and C. Li, J. Hazard. Mater., 2021, 416, 125866 CrossRef CAS PubMed.
- J. Wang, C. Cao, Y. Zhang, Y. Zhang and L. Zhu, Appl. Catal., B, 2021, 286, 119911 CrossRef CAS.
- X. Li, L. W. Y. Yeung, S. Taniyasu, M. Li, H. Zhang, D. Liu, P. K. S. Lam, N. Yamashita and J. Dai, Environ. Sci. Technol., 2008, 42, 7078–7083 CrossRef CAS PubMed.
- L. Xiang, L. Chen, L. Y. Yu, P. F. Yu, H. M. Zhao, C. H. Mo, Y. W. Li, H. Li, Q. Y. Cai, D. M. Zhou and M. H. Wong, Sci. Total Environ., 2018, 636, 999–1008 CrossRef CAS PubMed.
- F. Riva, S. Castiglioni, E. Fattore, A. Manenti, E. Davoli and E. Zuccato, Int. J. Hyg. Environ. Health, 2018, 221, 451–457 CrossRef CAS PubMed.
- J. M. Judge, P. Brown, J. G. Brody and S. Ryan, J. Health Soc. Behav., 2016, 57, 333–350 CrossRef PubMed.
- B. Vrana, F. Smedes, R. Prokeš, R. Loos, N. Mazzella, C. Miege, H. Budzinski, E. Vermeirssen, T. Ocelka, A. Gravell and S. Kaserzon, TrAC, Trends Anal. Chem., 2016, 76, 153–165 CrossRef CAS.
- J. Zhang, J. Qian, Q. Mei, L. Yang, L. He, S. Liu, C. Zhang and K. Zhang, Biosens. Bioelectron., 2019, 128, 61–67 CrossRef CAS PubMed.
- F. M. Hinterholzinger, B. Ruhle, S. Wuttke, K. Karaghiosoff and T. Bein, Sci. Rep., 2013, 3, 2562 CrossRef PubMed.
- M. Hirai and F. P. Gabbaï, Chem. Sci., 2014, 5, 1886–1893 RSC.
- B. Chen, L. Wang, F. Zapata, G. Qian and E. B. Lobkovsky, J. Am. Chem. Soc., 2008, 130, 6718–6719 CrossRef CAS PubMed.
- Z.-R. Yang, M.-M. Wang, X.-S. Wang and X.-B. Yin, Anal. Chem., 2017, 89, 1930–1936 CrossRef CAS PubMed.
- Y. Sun, X. Xu, Y. Zhao, H. Tan, Y. Li and J. Du, Talanta, 2020, 209, 120582 CrossRef CAS PubMed.
- P. Zhu, X. Ling, W. Liu, L. Kong and Y. Yao, J. Chromatogr. B, 2016, 1031, 61–67 CrossRef CAS PubMed.
- X. Liu, X. Wang and F. Kapteijn, Chem. Rev., 2020, 120, 8303–8377 CrossRef CAS PubMed.
- S. Yuan, L. Feng, K. Wang, J. Pang, M. Bosch, C. Lollar, Y. Sun, J. Qin, X. Yang, P. Zhang, Q. Wang, L. Zou, Y. Zhang, L. Zhang, Y. Fang, J. Li and H. C. Zhou, Adv. Mater., 2018, 30, 1704303 CrossRef PubMed.
- X. L. Lv, K. Wang, B. Wang, J. Su, X. Zou, Y. Xie, J. R. Li and H. C. Zhou, J. Am. Chem. Soc., 2017, 139, 211–217 CrossRef CAS PubMed.
- T. He, X. J. Kong, J. Zhou, C. Zhao, K. Wang, X. Q. Wu, X. L. Lv, G. R. Si, J. R. Li and Z. R. Nie, J. Am. Chem. Soc., 2021, 143, 9901–9911 CrossRef CAS PubMed.
- T. He, Z. Huang, S. Yuan, X. L. Lv, X. J. Kong, X. Zou, H. C. Zhou and J. R. Li, J. Am. Chem. Soc., 2020, 142, 13491–13499 CrossRef CAS PubMed.
- B. Wang, X. L. Lv, D. Feng, L. H. Xie, J. Zhang, M. Li, Y. Xie, J. R. Li and H. C. Zhou, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef CAS PubMed.
- B. Wang, P. Wang, L. H. Xie, R. B. Lin, J. Lv, J. R. Li and B. Chen, Nat. Commun., 2019, 10, 3861 CrossRef PubMed.
- W. Bin, L. Xiu-Liang, F. Dawei, X. Lin-Hua, Z. Jian, L. Ming, X. Yabo, L. Jian-Rong and Z. Hong-Cai, J. Am. Chem. Soc., 2016, 138, 6204–6216 CrossRef PubMed.
- H. Q. Yin, X. Y. Wang and X. B. Yin, J. Am. Chem. Soc., 2019, 141, 15166–15173 CrossRef CAS PubMed.
- M. D. DeFuria, M. Zeller and D. T. Genna, Cryst. Growth Des., 2016, 16, 3530–3534 CrossRef CAS.
- L. Liu, S.-M. Wang, Z.-B. Han, M. Ding, D.-Q. Yuan and H.-L. Jiang, Inorg. Chem., 2016, 55, 3558–3565 CrossRef CAS PubMed.
- A. H. Mahvi, M. A. Zazoli, M. Younecian, B. Nicpour and A. Babapour, J. Med. Sci., 2006, 6, 658–661 CrossRef CAS.
- G. K. Sarma and M. H. Rashid, J. Chem. Eng. Data, 2018, 63, 2957–2965 CrossRef CAS.
- Z. Du, S. Deng, Y. Bei, Q. Huang, B. Wang, J. Huang and G. Yu, J. Hazard. Mater., 2014, 274, 443–454 CrossRef CAS PubMed.
- H. C. Wentz, G. Skorupskii, A. B. Bonfim, J. L. Mancuso, C. H. Hendon, E. H. Oriel, G. T. Sazama and M. G. Campbell, Chem. Sci., 2019, 11, 1342–1346 RSC.
- S. Jensen, K. Tan, W. P. Lustig, D. S. Kilin, J. Li, Y. J. Chabal and T. Thonhauser, Chem. Mater., 2019, 31, 7933–7940 CrossRef CAS.
- S. Jensen, K. Tan, W. Lustig, D. Kilin, J. Li, Y. J. Chabal and T. Thonhauser, J. Mater. Chem. C, 2019, 7, 2625–2632 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, DFT calculation details, PXRD, BET, TG, UV-vis data and luminescence spectra, tables as well as crystal structures and crystallographic data. CCDC 2057106 and 2057107. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04070g |
| This journal is © The Royal Society of Chemistry 2021 |