
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Capture of toxic gases in MOFs: SO2, H2S, NH3 and NOx
Eva
Martínez-Ahumada
 a,
Mariana L.
Díaz-Ramírez
b,
Miriam de J.
Velásquez-Hernández
c,
Vojtech
Jancik
*de and
Ilich A.
Ibarra
*a
a,
Mariana L.
Díaz-Ramírez
b,
Miriam de J.
Velásquez-Hernández
c,
Vojtech
Jancik
*de and
Ilich A.
Ibarra
*a
aLaboratorio de Fisicoquímica y Reactividad de Superficies (LaFReS), Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Circuito Exterior s/n, CU, Del. Coyoacán, 04510, Ciudad de México, Mexico. E-mail: argel@unam.mx; Fax: +52(55) 5622-4595
bDepartment of Chemistry, Mississippi State University, Box 9573, Mississippi 39762, USA
cInstitute of Physical and Theoretical Chemistry, Graz University of Technology, 8010 Graz, Austria
dUniversidad Nacional Autónoma de México, Instituto de Química, Ciudad Universitaria, Ciudad de México, Mexico. E-mail: vjancik@unam.mx
eCentro Conjunto de Investigación en Química Sustentable UAEM-UNAM, Carr. Toluca-Atlacomulco Km 14.5, Toluca, Estado de México 50200, Mexico
First published on 28th April 2021
Abstract
MOFs are promising candidates for the capture of toxic gases since their adsorption properties can be tuned as a function of the topology and chemical composition of the pores. Although the main drawback of MOFs is their vulnerability to these highly corrosive gases which can compromise their chemical stability, remarkable examples have demonstrated high chemical stability to SO2, H2S, NH3 and NOx. Understanding the role of different chemical functionalities, within the pores of MOFs, is the key for accomplishing superior captures of these toxic gases. Thus, the interactions of such functional groups (coordinatively unsaturated metal sites, μ-OH groups, defective sites and halogen groups) with these toxic molecules, not only determines the capture properties of MOFs, but also can provide a guideline for the desigh of new multi-functionalised MOF materials. Thus, this perspective aims to provide valuable information on the significant progress on this environmental-remediation field, which could inspire more investigators to provide more and novel research on such challenging task.
1. Introduction
The accelerated growth of our modern society demands huge amounts of energy. Unwittingly, in order to provide these high energetic levels, an indiscriminate combustion of large volumes of fossil fuels occurs, leading to an incommensurable release of toxic pollutants to the atmosphere. Emissions of anthropogenic air pollutants generate a vast range of health complications (e.g., premature death and morbidity). Additionally, these air pollutants are also responsible for the reduction of the biodiversity, crop damages and acidification of soils and waters.1 As an example of the adverse impacts to humans, the World Health Organization (WHO) recently announced that air pollution was responsible for the premature mortality of approximately 4.2 million people in 2016 alone.2 In fact, air pollution is now the single largest environmental health risk worldwide since it is responsible for one in eight premature global deaths.3 For example, PM2.5 (fine inhalable particles, with diameters that are generally 2.5 micrometres and smaller) are responsible for approximately half of the deaths related to air pollution.4 Thus, PM2.5 have been classified as the air pollutant with the highest impact in premature mortality,5 and a reduction of their emissions is crucial. Such reduction can be achieved in two steps: (i) mitigation of primary PM2.5 emissions and (ii) extenuation of secondary inorganic aerosols (SIA); which are oxidised from precursor emissions such as sulphur dioxide (SO2), nitrogen oxides (NOx), ammonia (NH3) and volatile organic compounds (VOCs).6 Thus, in order to extenuate these emissions, different actions have been taken, such as the shutdown of some coal-fired power plants and their replacement by thermoelectric power plants, importing electricity from other countries with strict restrictions on the electricity production and the use of more environmentally friendly energy sources.7 In addition to these actions, the development of efficient technologies for the capture of toxic gases (e.g., NOx, SO2, NH3 and H2S) from static and mobile sources is necessary, in order to achieve a cleaner environment.8Porous metal–organic frameworks (MOFs) or porous coordination polymers (PCPs) are amongst the most promising candidates for the capture of these toxic gases since their sorption selectivity is directly tuneable as a function of the topology and chemical composition of the pores.9 Undoubtedly, for NOx, SO2, NH3 and H2S capture, there is a substantial emphasis on optimising the interactions between MOF materials and these toxic molecules, leading to the discovery of new functional porous materials with enhanced gas adsorption properties.10 Although MOF materials have shown very promising capabilities for the capture of these toxic gases, their main drawback is their vulnerability to these highly corrosive molecules capable of compromising the chemical stability of the MOFs. NOx, SO2, NH3 and H2S can disrupt the coordination bonds between the organic ligands (e.g., carboxylate) and the metal centres, occasioning the breakdown of the MOF structure. Therefore, chemical stability of the MOFs is a fundamental requirement for the capture of these toxic gases. The present contribution aims to provide a useful reference in the field of capturing toxic gases in MOFs, emphasising on their chemical stability and the role of the functional groups to enhance such captures. We hope to encourage more research groups to explore the exciting frontiers of science in environmental-remediation applications for MOFs.
2. Sulphur dioxide
The anthropogenic release of SO2 into the atmosphere is mainly due to the combustion of fossil fuels (e.g., electric power generation). One of the first environmental problems related to SO2 was observed during the last century in the acidic deposition.11 To avoid this, since 1990 many European countries have considerably reduced their SO2 emissions.12 Nevertheless, the rapid industrialisation of some developing countries has caused a constant presence of SO2 in the atmosphere,13 risking the health of millions of people (vide supra).SO2 is a colourless, non-flammable and corrosive gas with high solubility (120 g l−1) in water. Among the technologies for SO2 capture, wet and dry flue-gas desulfurization (FGD) processes are commonly used,14 where the cost and recyclability depend on the used technology. Among them, alkaline solutions, activated carbons, zeolites and silica have typically showed low SO2 adsorption capacities, accompanied by the corrosion of pipelines and high energy regeneration overheads.15 Thus, the design of new materials is extremely necessary in order to meet all the challenges that the capture of SO2 signifies. MOF materials are an exciting alternative for the capture of SO2 as they have demonstrated promising results for the SO2 capture assignment, mainly due to their specific chemical functionality and pore dimensions.16 The following section focuses on the highlights of remarkable MOF materials stable towards SO2, with the emphasis not only on their capture performance, but also on showing the role of different chemical functionalities which are the key for the SO2 capture.
2.1 MOFs for SO2 capture
The chemical nature of the SO2 molecule and its main binding modes: oxygen, an electron-rich atom that can act as a Lewis base; and sulphur that can be a Lewis acid site, have been exploited to improve its adsorption on the porous surface of the MOFs. Due to the specific functionalisation of these MOFs, interesting SO2 capture results have been achieved. Such functionalisation can be classified in four main categories: (i) coordinatively unsaturated metal sites (open metal sites), (ii) μ-OH groups, (iii) defective sites and (iv) halogen functionalisation.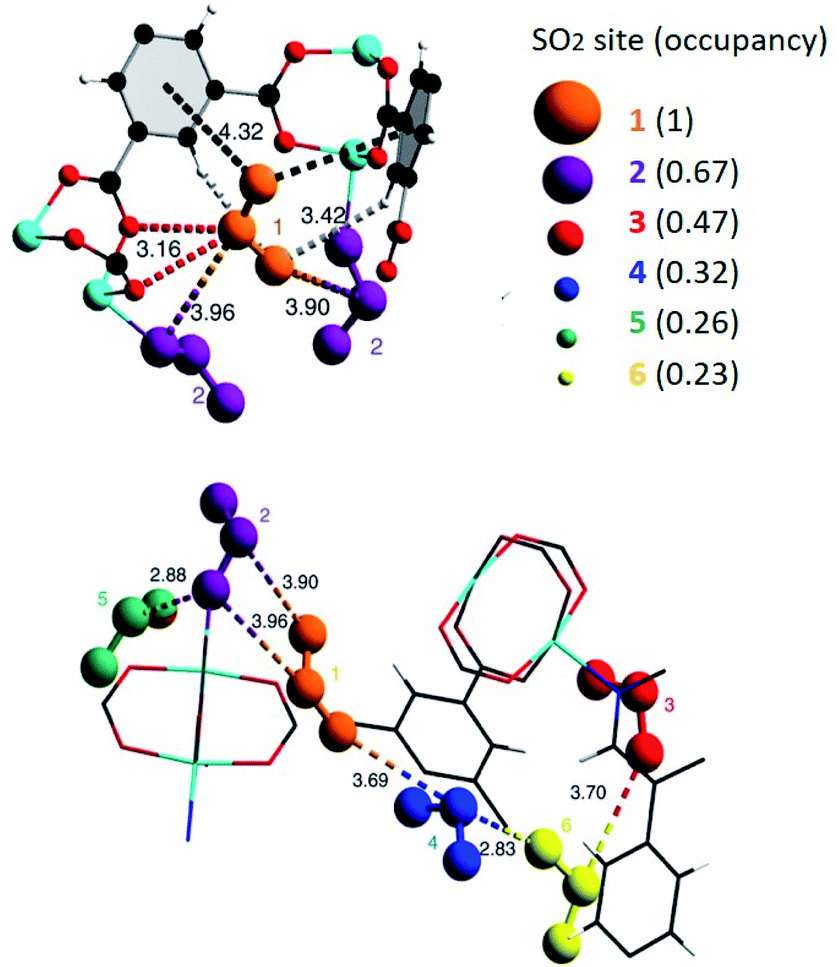 | ||
| Fig. 1 SO2 positions within MFM-170 pores obtained from in situ single-crystal X-ray diffraction. Purple SO2 molecule is located at the thermodynamically strongest binding site. Cu, cyan; C, black; O, red; N, blue. Reprinted (adapted) with permission from ref. 19. Copyright (2019) Springer Nature. | ||
Our group reported a partially fluorinated version of MIL-101(Cr) named MIL-101(Cr)-4F(1%).20 The particularity of MIL-101(Cr)-4F(1%) is the different chemical character of the Cr3+ metal centres, in comparison to MIL-101(Cr). The incorporation of fluorine in MIL-101(Cr) promoted a higher acidity of some of the Cr3+ metal centres (open metal sites), due to the capability of fluorine to attract electrons.20 Such difference afforded a total SO2 capture of 18.4 mmol g−1 at 298 K and up to 1 bar, chemical stability towards dry and humid SO2 and an exceptional cycling performance with facile regeneration.20
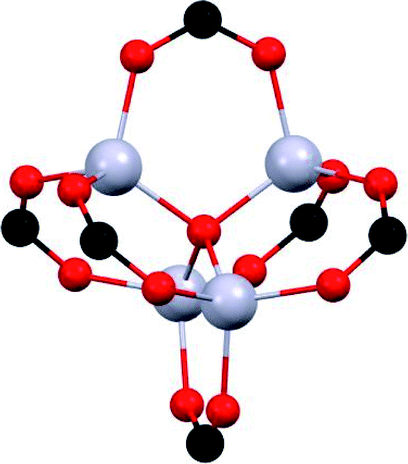
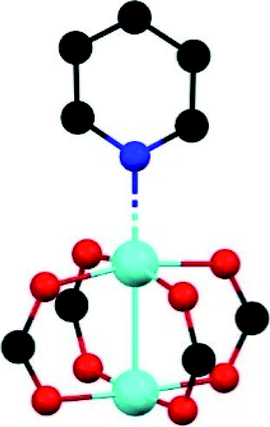
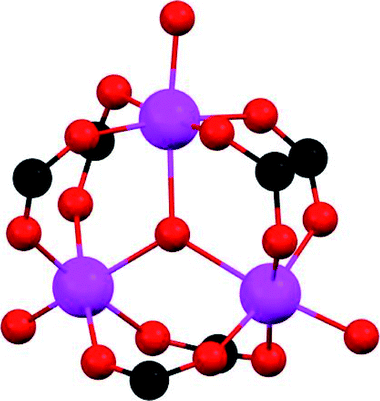
This uptake represents the highest SO2 capture for a structurally stable MOF material. In addition, in situ DRIFT spectroscopy upon the adsorption of CO demonstrated the efficient packing of SO2 molecules within MIL-101(Cr)-4F(1%). Thus, the SO2 uptake mechanism takes place in three stages: (i) adsorption at acid (Lewis) Cr3+ sites with a relatively high heat of adsorption for SO2; (ii) adsorption at both acid (Lewis and Brønsted) sites of MIL-101(Cr)-4F(1%) and (iii) adsorption within the cavities of this MOF material. Up to this point, we have presented three examples (MOF-177, MFM-170 and MIL-101(Cr)-4F(1%)) of MOF materials with coordinatively unsaturated metal sites that demonstrated the highest SO2 captures for any MOF. One material (MOF-177) demonstrated poor structural stability towards SO2versus the high structural stability of MFM-170 and MIL-101(Cr)-4F(1%) in the presence of SO2 and humid SO2. Unwittingly, the fundamental question arises: why is MOF-177 unstable while MFM-170 and MIL-101(Cr)-4F(1%) are stable to SO2? In order to answer this question, their comparison is presented in Table 1, allowing to propose a stability hypothesis for these materials.
| Material | MOF-177 | MFM-170 | MIL-101(Cr)-4F(1%) |
|---|---|---|---|
| Ligand | H3BTB | H4L = 4′,4′′′-(pyridine-3,5-diyl)bis([1,1′-biphenyl]-3,5-dicarboxylic acid) | |
| H2BDC/H2BDC-4F | |||
| SBU | [Zn4(μ4-O)(O2CR)6] | [Cu2(O2CR)4] | [Cr3(μ3-O)(O2CR)6] |
|
|
|
|
| BET surface area [m2 g−1] | 4100 | 2408 | 2176 |
| Pore volume [cm3 g−1] | 1.51 | 0.88 | 1.19 |
| SO2 adsorption [mmol g−1] | 25.7 | 17.5 | 18.4 |
| Packing density [cm3 g−1] | 1.09 | 1.27 | 1.00 |
| Stability under SO2 | Partial destruction after SO2 exposure | Stable after 50 cycles | Stable after 50 cycles |
The design of stable frameworks for capture, confinement and release of corrosive gases uses tools centred in the robustness of the metal-linker bond, the use of inert or higher-valent metals and, the use of poly-nuclear secondary building units (SBUs).21 In the case of MOF-177, MFM-170 and MIL-101(Cr)-4F(1%), these exhibit at least one of such design tools. These three MOFs are composed of carboxylate ligands and robust SBUs, this multinuclearity gives, in theory, a better thermal and chemical stability. Nonetheless, in the case of MOF-177,18 that possess the highest BET surface area, pore volume and the record of SO2 capture, the use of robust polynuclear cluster [Zn4(μ4-O)(O2CR)6] (see comparative Table 1), is not enough to achieve a reversible SO2 adsorption and the framework suffers a partial decomposition.
This result is most likely due to the propensity of the Zn–O bond to bind SO2 and form SO32−. Similar behaviour was observed on a ZnO surface exposed to SO2 gas, that reacts with the oxygen atoms from the Zn–O bonds forming the SO32− and SO42− species.22,23 In fact, other Zn-based MOFs that have been studied for SO2 adsorption have shown similar stability problems. This is also the case of Zn-MOF-74 (ref. 24) with a rod-like SBU and coordinatively unsaturated zinc sites, where a Zn⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO interaction leads to a chemisorption. Additionally, materials with paddlewheel SBUs, such as Zn2+-BDC (MOF-2) where the catalytic oxidation of SO2 under humid conditions was confirmed by the presence of sulphate species.25 Conversely, when the coordinatively unsaturated zinc site is blocked (i.e., pillared MOFs), direct interaction of adsorbate with the metal centre is not observed, as for example in Zn(bdc)(ted)0.5 (ref. 26) and Zn-DMOF.27 These comparative results suggest that in the case of Zn-based materials, the robustness of the SBU plays a secondary role in chemical stability against SO2, and the fact that they are constructed from a divalent metal that forms labile bonds,28 makes them more susceptible to collapse.
SO interaction leads to a chemisorption. Additionally, materials with paddlewheel SBUs, such as Zn2+-BDC (MOF-2) where the catalytic oxidation of SO2 under humid conditions was confirmed by the presence of sulphate species.25 Conversely, when the coordinatively unsaturated zinc site is blocked (i.e., pillared MOFs), direct interaction of adsorbate with the metal centre is not observed, as for example in Zn(bdc)(ted)0.5 (ref. 26) and Zn-DMOF.27 These comparative results suggest that in the case of Zn-based materials, the robustness of the SBU plays a secondary role in chemical stability against SO2, and the fact that they are constructed from a divalent metal that forms labile bonds,28 makes them more susceptible to collapse.
Although MFM-170 contains the Cu2+ paddlewheel SBU, a cluster frequently instable under harsh conditions such as corrosive or acidic gases,29,30 it is stable even after 50 adsorption–desorption cycles of SO2.19 This extraordinary chemical stability is mainly due to two important factors: (i) the unusual paddlewheel SBU formed in the framework and (ii) additional intermolecular interactions formed between SO2 and the pore-walls of the framework. The di-cooper paddlewheel SBU shows the particularity of only one of the Cu2+ centres axially coordinated to a nitrogen atom from the pyridyl-based ligand, leaving the other Cu2+ metal centre coordinatively unsaturated and pointing toward the centre of the pore (Table 1). The effect of this structural arrangement is evident on the high SO2 uptake, where the Cu2+ sites are able to fix SO2 by forming a strong Cu–O bond without ligand displacement. Such behaviour was previously observed in a Cu2+-BDC MOF material.25 It is significant that this interaction does not represent the main SO2 binding site due to the steric hindrance generated by a neighbouring SO2 molecule. This latter molecule has several interactions: (i) with the C–H portion of the ligand, (ii) with the oxygen atoms in the paddlewheel SBU, and (iii) with other SO2 molecules. All these interactions promote an efficient packing within the pore surface, maximising the adsorbate–adsorbent bonds and presumably avoids the frame destruction. Both materials reviewed above, MOF-170 and MFM-177, are formed by first-row transition metals Zn2+ and Cu2+, respectively, and follow the stability order of the Irving–Williams series for divalent metals: Mn < Fe < Co < Ni < Cu > Zn.31 Thus, the high SO2 capture can be attributed to the large BET surface area and pore size rather than to the local pore environment, while structural stability is associated with the type of metal, and the interactions adsorbate–adsorbent.
Additionally, the use of higher-valent metal ions such as Al3+, Sc3+, Cr3+ or Zr4+ allows the formation of stronger M–O bonds with carboxylate ligands compared to divalent ions.21 Thus, the lability of metal–ligand bonding is higher in Cu2+ or Zn2+-based MOFs that Cr3+ MOFs. As a consequence, MOFs constructed from divalent metals such Cu2+ and Zn2+, typically show partial or total degradation under harsh conditions.32 Due to the inertness of the Cr–O bond, Cr3+-based MOFs highly resistant to the attack of acid and base guests were reported.33,34 As described above, the third material, MIL-101(Cr)-4F(1%), has the highest SO2 adsorption among SO2-stable MOFs.20 This framework poses a robust trinuclear metal cluster with coordinatively unsaturated chromium sites after the activation process (Table 1). When comparing MIL-101(Cr)-4F(1%) to MFM-170, both materials stable towards SO2, we can observe that the Cr3+ is harder than Cu2+ ions according to the Pearson acid–base concept35 and therefore the coordinated bond with carboxylic ligands is stronger for the trivalent cation than for the divalent cation. However, the Cu2+ paddlewheel increases its stability when the N-containing ligand coordinates to one of the Cu2+ ions.36
| Metal center (M) | BET Surface area [m2 g−1] | Pore volume [cm3 g−1] | SO2 adsorption [mmol g−1] | Packing density [g cm−3] | Q st [kJ mol−1] | Stability |
|---|---|---|---|---|---|---|
| a Adsorption near the zero-coverage calculated by GCMC. | ||||||
| Al | 1370 | 0.375 | 7.1 | 1.213 | 31.25a37e | Stable over 4 years of exposure |
| In | 1071 | 0.419 | 8.28 | 1.266 | 34.5 | Stable in dry and humid conditions |
| Ga | 1045 | 0.62 | — | — | — | — |
| Sc | 1360 | 0.56 | 9.4 | 1.075 | 36.2 | Stable in dry and humid conditions/over 10 cycles |
The first MFM-300 material investigated for the capture of SO2 was MFM-300(Al).37a Although the SO2 capture was not remarkably high (see Table 2), the identification of the preferential SO2 adsorption sites was achieved by sophisticated in situ INS and PXRD experiments, revealing that the μ-OH groups bind SO2 molecules through the formation of OSO(δ−)⋯H(δ+)–O hydrogen bonds, reinforced by weak supramolecular interactions with C–H atoms from the aromatic rings of the framework (Fig. 2).37a In the case of MFM-300(In) (also known as InOF-1 (ref. 38)) the SO2 uptake was slightly higher than for MFM-300(Al) (see Table 2), and in situ INS and PXRD experiments corroborated the same preferential adsorption site for SO2 (see Fig. 3) with exceptional chemical stability for SO2 capture under both dry and humid conditions.37b In fact, Eddaoudi and Salama fabricated an advanced chemical capacitive sensor, using MFM-300(In), for the detection of very low concentrations of SO2 (≈5 ppb) at room temperature.39
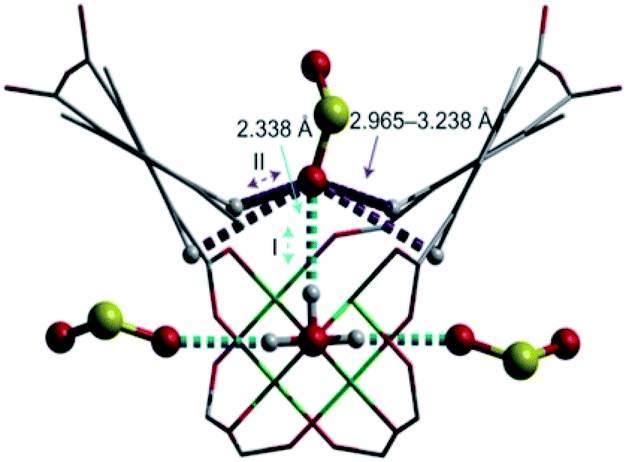 | ||
| Fig. 2 Preferred binding sites for SO2 molecules inside MFM-300(Al) determined by in situ PXRD. (I) Moderated hydrogen bond. (II) Weak hydrogen bond. Reprinted (adapted) with permission from ref. 37a. Copyright (2012) Springer Nature. | ||
 | ||
| Fig. 3 Binding sites for SO2 molecules within MFM-300(In) determined by single-crystal X-ray diffraction (a), and DFT calculations (b). Reprinted (adapted) with permission from ref. 37b. Copyright (2016) John Wiley & Sons. | ||
MFM-300(Sc) showed the highest SO2 uptake of the MFM-300(M) materials (see Table 2), Grand Canonical Monte Carlo (GCMC) simulations demonstrated the same preferential adsorption sites (μ-OH) and by confining small amounts of EtOH (2.6 wt%) the SO2 capture increased by 40% (see Fig. 4).37c The only MFM-300 remaining to be investigated in the capture of SO2 is MFM-300(Ga),37d where we anticipate, based on the pore volume, performance similar to MFM-300(Sc).
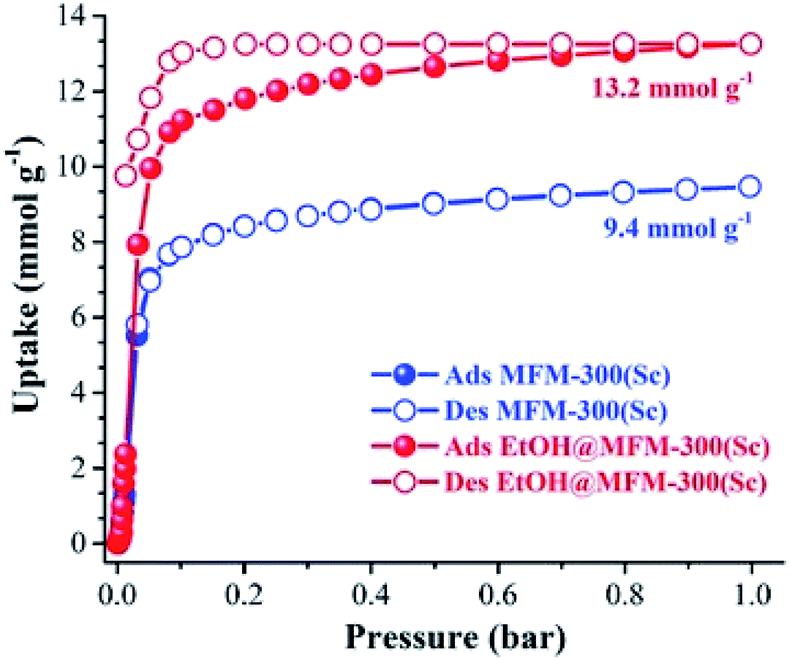 | ||
| Fig. 4 SO2 adsorption isotherm of scandium-based MFM-300 (blue isotherm) and after confinement of 2.8% EtOH (red isotherm). Reprinted with permission from ref. 37c. Published (2019) by The Royal Society of Chemistry. | ||
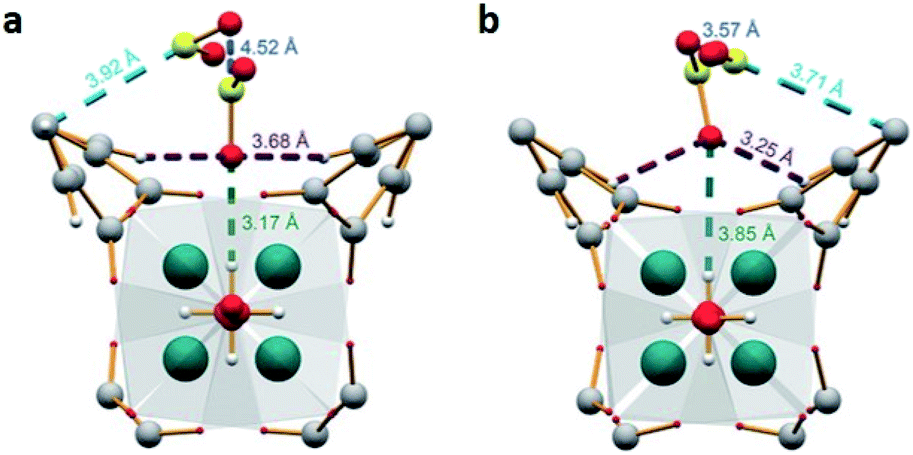
Thus, the MFM-300(M) MOF materials have shown the relevance of the hydroxo-functionalisation to the capture and detection of SO2 with high chemical stability and excellent cyclability involving a remarkably facile regeneration. The key for all of such outstanding properties is the strength of the coordination bonds between the oxygen atoms form the carboxylic ligand (biphenyl-3,3′,5,5′-tetracarboxylic acid) and the M3+ metal centres. The ionic radii of Al3+, In3+ and Sc3+ cations, in octahedral coordination, are 0.675, 0.940 and 0.885 Å, respectively.40a
This leads to a higher surface charge density for the Al3+ metal centre and, therefore, the M–O bond strength decreases in this order: Al3+ > Sc3+ > In3+. Thus, a higher ligand exchange can occur in the In-based complex.40b However, MFM-300(In) does not suffer apparent displacement of carboxylate ligands after SO2 exposure. In the MFM-300(M) family all the metal cations are nonmagnetic because of all paired electrons,41 and the comparison between octahedral complexes formed by trivalent cations of group 13 such as Al3+, Ga3+ and In3+, with the transition-metal analogue Sc3+, has shown that in the group 13 metal complexes the d orbitals are not involved in the metal–linker bonding due to their high energy (3d10 and 4d10 configuration). In the case of Sc3+, a complex with 12e−, d orbitals form part of the M–O bonds (Fig. 5a). On the other hand, these types of complexes are considered as electron-rich hypervalent species with 7-center-12-electron bonding pattern (Fig. 5b).40c This type of binding confers to the group 13 complexes thermal stability and in this particular case, the kinetic stability is comparable to that of the transition metal analogues. At this point, we can corroborate that frameworks constructed with trivalent cations are more stable than MOFs based on divalent cations (considering the stability of Irving–Williams series discussed above), as well that carboxylate ligands form with M3+ cations stronger bonds than divalent cations with N-based ligands.32
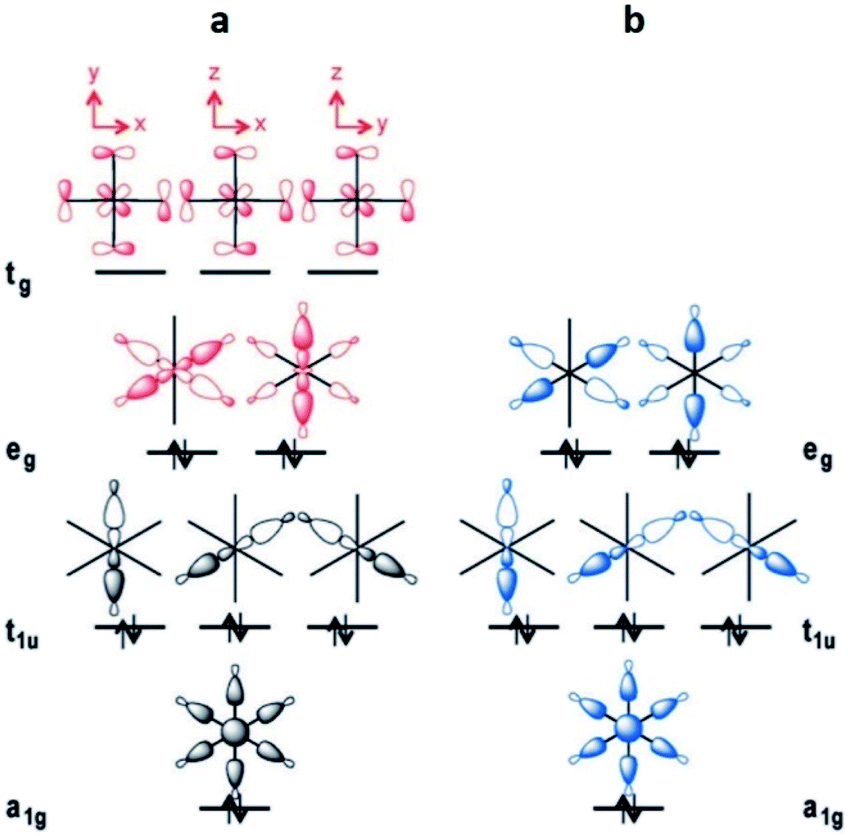 | ||
| Fig. 5 Schematic MO diagrams for an Oh-symmetric 12e− complex with d orbitals (a, red) and the MO diagram without d orbitals in 7c-12e− pattern of MO (b, blue). Reprinted (adapted) with permission from ref. 40c. Copyright (2015) John Wiley & Sons. | ||
Thus, the post-synthetic treatment of the [Ni8(OH)4(H2O)2(BDP_X)6] (H2BDP_X = 1,4-bis(pyrazol-4-yl) benzene-4-X with X = H (1), OH (2), NH2 (3)) systems with ethanolic solutions of potassium hydroxide leads to the formation of defective K[Ni8(OH)3(EtO)3(BDP_X)5.5] (1@KOH, 3@KOH) and K3[Ni8(OH)3(EtO)(BDP_O)5] (2@KOH).46 Exposing the 1@KOH–3@KOH materials to aqueous Ba(NO3)2 solutions incorporates barium cations into the solution, yielding the defective ion exchanged Ba0.5[Ni8(OH)3(EtO)3(BDP_X)5.5] (1@Ba(OH)2, X = H; 3@Ba(OH)2, X = NH2), and Ba1.5[Ni8(OH)3(EtO)(BDP_O)5] (2@Ba(OH)2) systems.45 Interestingly, density functional theory (DFT) calculations located the extra-framework cations close to the crystal defect sites (Fig. 6a). In addition, DFT results demonstrated that the average 3D structure of the MOF framework is preserved.45 SO2 dynamic adsorption experiments confirmed the beneficial influence of the deliberate introduction of defects by a subsequent K+ to Ba2+ ion exchange process on the SO2 capture.45 The authors demonstrated that the pre-synthetic introduction of amino and hydroxyl functional groups on the organic linkers and the post-synthetic modifications, synergistically increase the SO2 capture capacity of these materials. In addition, chemisorption of SO2 was observed in all the systems: after all available sites for SO2 chemisorption were occupied, during the first exposure, reversible adsorption takes place in a steady way evidencing the stability of the materials upon continuous SO2 exposure. This high affinity of these MOF materials for SO2 was attributed to the increased basicity of the nickel hydroxide clusters after the introduction of defects. Lewis acid–base interactions with SO2 molecules can afford the formation of HSO3− and SO32− species according to eqn (1):
| [Ni8(OH)6(OH)2]10+ + SO2 → [Ni8(OH)5(HSO3)]10+ → [Ni8(OH)4(SO3)(H2O)]10+ | (1) |
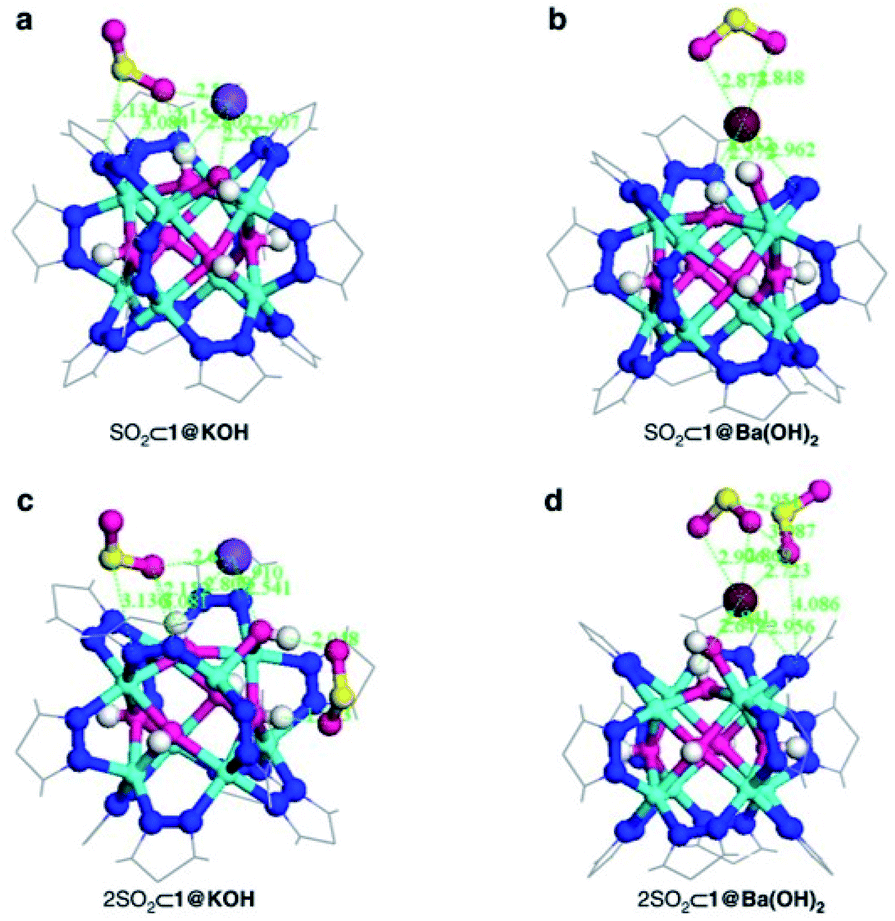 | ||
| Fig. 6 DFT structure minimization of molecular configuration of one (a and b) and two (c and d) adsorbed SO2 molecules on 1@KOH (left) and 1@Ba(OH)2 (right) materials. For sake of clarity, only the region around the metal cluster is shown. Ni (cyan); K (purple); Ba (wine); C (grey); N (blue); O (magenta); H (white); S (yellow). Reprinted (adapted) with permission from ref. 45. Copyright (2017) Springer Nature. | ||
Furthermore, the existence of extra-framework cations located close to defects sites promotes the formation of more stable MSO3 (M = Ba, 2 K) sulphite species according to eqn (2):
| M[Ni8(OH)4(SO3)(H2O)(BDPX)5.5]+ → (MSO3)[Ni8(OH)4(H2O)(BDP_X)5.5]+ | (2) |
These chemisorption mechanisms were supported by the DFT simulations (Fig. 6), showing that 1@Ba(OH)2 establishes stronger interactions with SO2 molecules than 1@KOH due to the specific interactions with extra-framework barium cations.
Thus, this sophisticated and comprehensive study, by Navarro et al.,45 carefully incorporated essential variables to improve the SO2 capture performance of these MOF materials. Such variables play together to increase the interaction with SO2 and can be summarised as (i) enhanced basicity of metal hydroxide clusters as a result of additional hydroxide anions replacing the missing linkers defects; (ii) affinity of extra-framework Ba2+ ions for SO2 sequestration; (iii) higher pore accessibility of the 3-D structure due to missing linker defects and (iv) the fine-tuning of the pore surface polarity by the benzene functional groups.
The post-synthetic inclusion of Ba2+ cations to improve the SO2 adsorption was later validated by Navarro and co-workers47 in the Ni-based MOF material Ni22+{Ni42+[Cu42+–(Me3mpba)2]3}·54H2O (1). In this case, the exchange of the Ni2+ cations for Ba2+ was accomplished by soaking 1 in an aqueous solution of Ba(NO3)2 for two days to finally obtain a novel crystalline phase of heterotrimetallic oxamato-based MOF with the formula [Ba2+(H2O)4]1.5[Ba2+(H2O)5]0.5 {Ni42+[Cu22+–(Me3mpba)2]3}·57.5H2O (2). On this occasion, the presence of the hydrated Ba2+ counterions within the MOF's pores allowed the SO2 molecules to interact with the network reversibly via Cu2+⋯OSO2 and OH2O⋯OSO2 interactions. The amount of SO2 adsorbed in 2 increased from 2.0 to 2.5 mmol g−1 in comparison to 1, respectively. This reversibility was conserved over 10 adsorption–desorption cycles. Theoretical calculations demonstrated that the principal binding site of SO2 molecules is with coordinated water molecules, and it showed moderate adsorption energy of −65.5 kJ mol−1, otherwise, the direct interaction of SO2 with Ba2+ cations would induce a higher adsorption energy (>100 kJ mol−1) due to the formation of an irreversible bond. Thus, this research group has elegantly proved that the formation of defective sites and the incorporation of large cations such as Ba2+ is a good and feasible post-synthetic method for modulating the adsorptive capacities of MOF materials.
Yang and Xing incorporated inorganic hexafluorosilicate (SiF62−, SIFSIX) anions (as pillars) into a series of MOF materials (SIFSIX-1-Cu, SIFSIX-2-Cu (2 = 4,4′-dipyridylacetylene), SIFSIX-2-Cu-i, SIFSIX-3-Zn (3 = pyrazine), and SIFSIX-3-Ni) and investigated the SO2 adsorption properties of these materials.51 The remarkable highly efficient removal of SO2 from other gases, particularly at a very low SO2 concentrations, and excellent SO2/CO2 and SO2/N2 selectivities were attributed to the strong electrostatic interactions between the SO2 molecules and the SiF62− anions (Sδ+⋯Fδ−), assisted by dipole–dipole interactions with the ligand (Oδ−⋯Hδ+).51 The authors identified the interactions between the SO2 molecule and SIFSIX materials by modelling studies using first-principles DFT-D (dispersion-corrected density functional theory) calculations (see Fig. 7).51 These computational calculations were experimentally corroborated by Rietveld refinement of the powder X-ray diffraction patterns of SO2-loaded samples to locate the adsorbed positions of the SO2 molecules in the crystal structure of SIFSIX materials.51 Thus, this strategy highlighted the relevance on creating the multiple binding sites (anionic and aromatic linkers) which can originate the specific recognition of SO2 to optimise its capture.
 | ||
| Fig. 7 Binding sites for SO2 molecules adsorbed within SIFSIX-1-Cu determined by DFT-D calculations. Interaction Sδ+⋯Fδ− (a), and dipole–dipole interactions with the ligand Oδ−⋯Hδ+ (b). Cu (dark teal); F (red); Si (light blue); C (grey); H (light grey); N (sky blue); O (orange); S (sea green). Reprinted (adapted) with permission from ref. 51. Copyright (2017) John Wiley & Sons. | ||
Similarly, Salama and Eddaoudi demonstrated how fluorinated MOFs (KAUST-7 and KAUST-8) can be formidable candidates for sensing SO2 from flue gas and air (250 ppm to 7% of SO2).52 SCXRD data collected on the SO2-loaded KAUST-7 and KAUST-8 identified the preferential adsorption sites for SO2 revealing, comparable to SIFSIX materials, strong electrostatic interactions between the SO2 molecules and the fluorinated pillars (Sδ+⋯Fδ−), supported by dipole–dipole interactions with the pyrazine ligand (Oδ−⋯Hδ+), see Fig. 8.52 These interactions were also corroborated by DFT calculations.
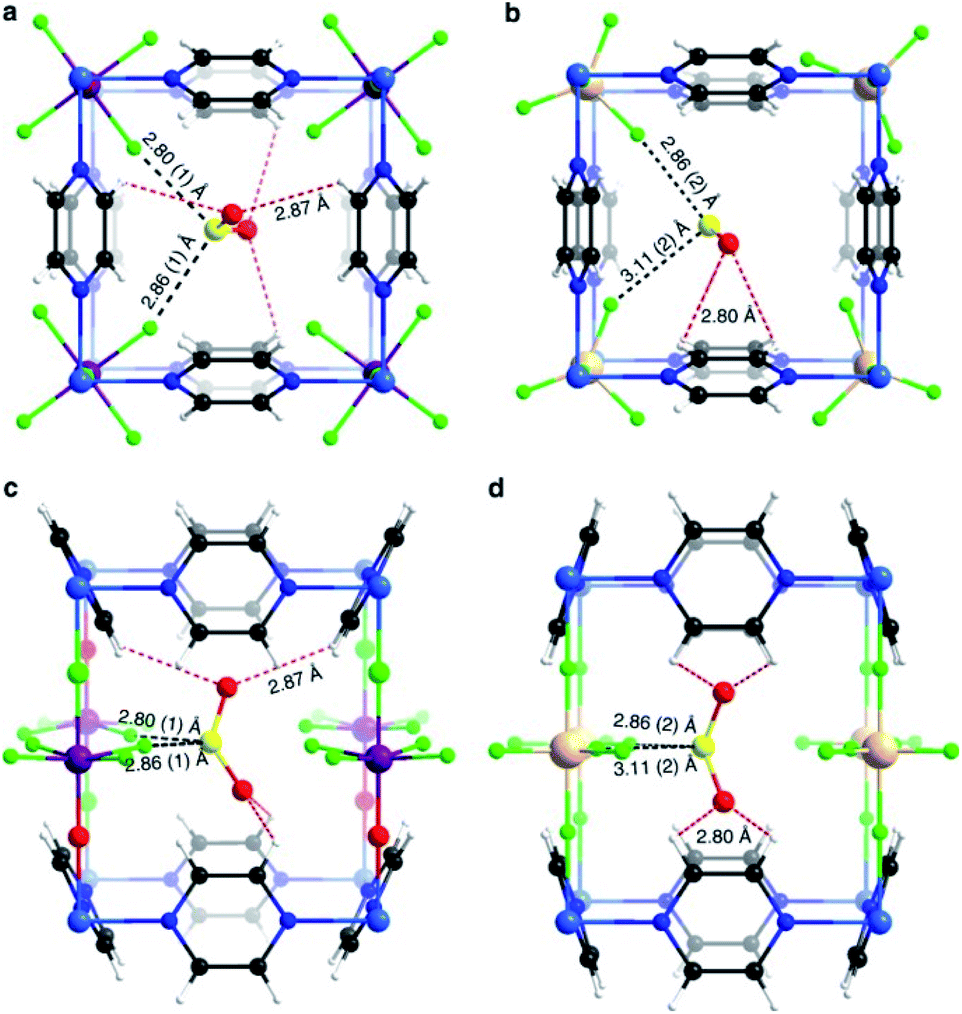 | ||
| Fig. 8 SO2 interactions within KAUST-7 (a and c), and KAUST-8 (b and d). Ni (blue light); Nb (purple); Al (beige); F (green); C (black); N (blue); O (red); H (white); S (yellow). Reprinted (adapted) with permission from ref. 52. Copyright (2019) Springer Nature. | ||
Another remarkable halogen functionalisation was presented by Janiak and co-workers,53 where MOF-801 was modified by reacting zirconium halides (ZrX4; X = Cl, Br, I) in water with acetylenedicarboxylic acid. The HX addition and material construction occurred in a one-pot reaction yielding three microporous HHU-2-X MOFs (X = Cl, Br, I). The material HHU-2-Cl showed in increased SO2 uptake (by 21%) in comparison to the nonhalogenated MOF-801.53 Therefore, as previously demonstrated by the SIFSIX materials, KAUST-7, and KAUST-8, the electrostatic interactions between the SO2 molecules and the Cl (Sδ+⋯Clδ−), might be responsible for such SO2 capture enhancement.
3. Hydrogen sulphide
H2S is released to the environment via natural events such as volcanic eruptions, gas streams, hot springs, decomposition of organic matter and bacterial reduction.54 Additionally, H2S is emitted by some chemical industries, e.g., oil desulphurisation processes at oil refineries, burning fossil fuels and mass transportation.55 H2S is a main air pollutant which negative impacts the environmental as it is one of the main sources of acid rain,55 and it is highly toxic to humans (concentrations above 700 ppm in the air can cause death).56H2S is a colourless, flammable gas with a characteristic rotten egg odour. Typical strategies to capture H2S comprise alkanolamines and ionic liquids, adsorption in solid materials (i.e., zeolites, activated carbons and metal oxides), membrane separation and cryogenic distillation.57 However, these techniques have major drawbacks, such as low H2S capture, corrosion of pipelines and large cost of expenditure and recovery.58 Thus, the development of new technologies for a dry adsorption process (avoiding solvents/water consumption is essential to reduce waste generation), for removal and capture of H2S signifies a technological challenge which could allow not only the capture of H2S from the main source, but also its use as feedstock, similarly to the CO2 technologies (CCS).59 MOFs have been visualised for the capture of H2S; however, most of them have shown poor chemical stability in its presence.60 Nonetheless, chemically stable MOF materials have demonstrated promising results in the reversible capture of H2S.61 This section highlights outstanding MOF materials that have shown high H2S capture performances and in situ H2S transformations, as well as biomedical applications.
3.1 MOFs for H2S capture
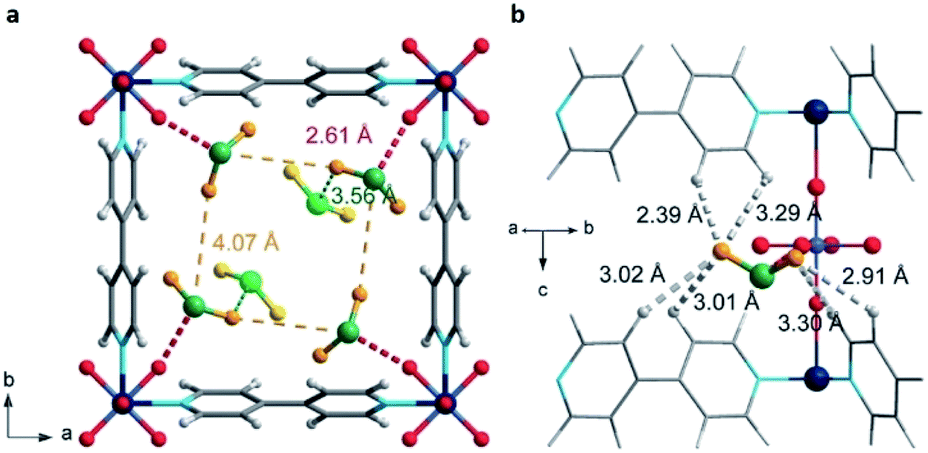
Distinctive investigations reported by De Weireld,62 Zou63 and Eddaoudi64 reveal that the majority of MOF materials experience structural decomposition upon adsorption of H2S or the desorption of it resulted complicated, due to relatively strong host–guest binding in the pores (strong physisorption or chemisorption). In such cases, the reactivation of the materials involves a large energy penalty or is not feasible. Thus, the identification of new MOFs capable of capturing H2S under industrially practical pressure-swing desorption conditions,65 represents a very important challenge to solve. This perspective focused on (i) high and reversible H2S capture in MOFs and (ii) chemical transformation of H2S within the pores of MOFs: formation of polysulphides.Humphrey and Maurin showed the H2S capture on Mg-CUK-1.68 This MOF is assembled from a 2,4-pyridinecarboxylate ligand and Mg2+ octahedral centres, connected into infinite chains of [Mg3(μ3-OH)]5+ clusters by μ3-OH groups. Although the total H2S capture of Mg-CUK-1 was relatively low (3.1 mmol g−1), this material demonstrated to be chemically stable to H2S and a remarkably easy regeneration after the H2S capture using temperature swing re-activation (TSR). Most importantly, GCMC simulations showed how H2S molecules interact with the hydroxo functional groups (by moderate hydrogen bonds μ3-O–H⋯SH2), see Fig. 9.68 Such moderate H2S/Mg-CUK-1 interaction was found to be consistent with easy regeneration of the material after H2S exposure.
 | ||
| Fig. 9 H2S adsorption preferred site of magnesium-based CUK-1 material. And after confinement of 2.8% EtOH (red isotherm). Reprinted (adapted) with permission from ref. 68. Published (2018) by The Royal Society of Chemistry. | ||
This work demonstrated experimentally and in good correlation with advanced computational calculations, the chemical structure integrity of a hydroxo-functionalised MOF material to H2S, and the critical role of a moderate interaction (hydrogen bonding) to facilitate its cyclability. The study of more μ-OH functionalised MOFs for the efficient and cyclable capture of H2S initiated.
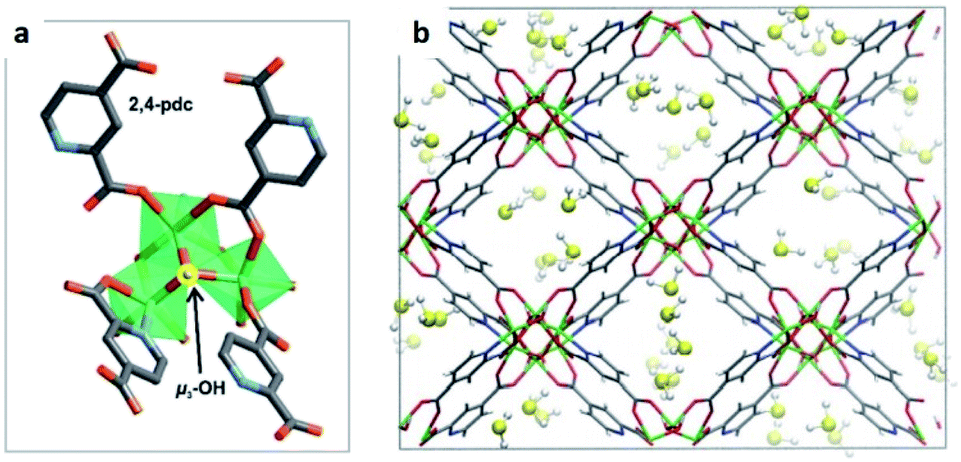
Maurin and Gutiérrez-Alejandre investigated another μ-OH functionalised MOF (MIL-53(Al)-TDC) for the capture of H2S.69 This material, an Al3+-based constructed with a carboxylate ligand (TDC = 2,5-thiophenedicarboxylate) which contains [AlO4-trans-(μ-OH)2] octahedra where the Al3+ centre coordinates to two μ-OH groups and six oxygen atoms from the TDC ligands, has established the highest H2S adsorption (18.1 mmol g−1 at room temperature) reported for a microporous material.69 Structural stability of MIL-53(Al)-TDC, after the H2S capture experiment was corroborated by PXRD and SEM analyses. TGA experiments demonstrated the complete desorption of H2S molecules at 65 °C. H2S sorption–desorption cycles (five cycles with a value of 18.5 ± 0.7 mmol g−1) demonstrated high H2S regeneration capacity. This cyclability confirmed the full regeneration of the material by only increasing the temperature to 65 °C, showing the low energy requirement to fully desorb H2S. This suitable cyclability indicated weak interactions between H2S molecules and the pores of the material. In situ DRIFT experiments investigated the interactions between H2S and MIL-53(Al)-TDC, showing (i) the formation of hydrogen bonds between H2S molecules themselves confined in the pores of MIL-53(Al)-TDC; (ii) relatively weak interaction between H2S and the μ-OH groups and (iii) H2S molecules interact with the other functionality of the MOF: the thiophene ring from the TDC ligand.69 To corroborate these interpretations from the DRIFT experiments, Monte Carlo simulations were carried out for different loadings corresponding to the experimental discoveries. These calculations corroborated that at low loading, H2S interacts via its S-atom with the H-atom of the μ-OH group (see Fig. 10), representing the preferential adsorption site. In addition, it was shown that H2S also interacts with the thiophene ligand, and, upon increasing the H2S loading, hydrogen bonds between the H2S molecules were also identified (Fig. 10).69
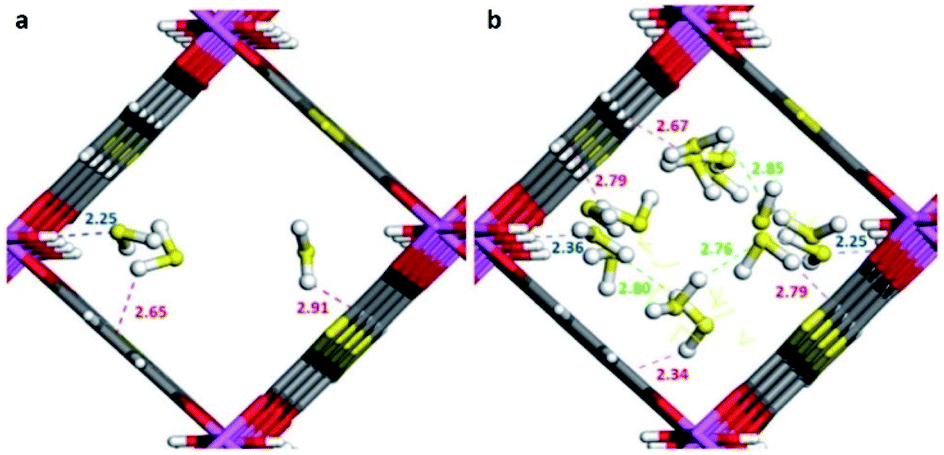 | ||
| Fig. 10 Illustrative arrangements of H2S in the pores of MIL-53-TDC generated from the GCMC simulations at (a) 0.5 mmol g−1 and (b) 18.5 mmol g−1. The distances are reported in Å. (Al, pink; O, red; S, yellow; C, grey; H, white). Reprinted (adapted) with permission from ref. 69. Published (2019) by The Royal Society of Chemistry. | ||
These theoretical findings revealed the fundamental role of the thiophene ligand. Experimentally speaking, this M–OH hydroxo group containing functional material shows an extraordinarily facile re-activation leading to an easy H2S cyclability, which should not be anticipated due to the dominant interaction between the μ-OH group and H2S reenforced further by the interaction of the H2S molecule with the thiophene unit as demonstrated by in situ DRIFT measurements. However, computational calculations postulate that the interaction of H2S with the thiophene ring weakens the interaction with the preferential adsorption site (μ-OH group), favouring the easy displacement of the H2S molecule when the material is re-activated.
As previously discussed, (vide supra) for the MFM-300(M) family, the extraordinary chemical stability arises from the strength of the coordination bonds between the oxygen atoms form the carboxylic ligand and the M3+ metal centres. Thus, MIL-53(Al)-TDC is very similar to MFM-300(Al) since it is also constructed with Al3+. Therefore, the chemical stability of MIL-53(Al)-TDC towards H2S is envisaged based on the same arguments used for MFM-300(Al).
First, MFM-300(Sc) exhibited a H2S uptake of 16.5 mmol g−1 (at 25 °C), which is comparable to the H2S uptake of MIL-53(Al)-TDC (18.1 mmol g−1).69 Upon an inspection of the structural integrity of the material after the H2S experiment, PXRD experiments revealed the retention of the crystalline structure. Interestingly, when investigating the porosity of MFM-300(Sc) after the H2S uptake experiment, the pore volume was reduced by 34% from 0.56 to 0.37 cm3 g−1 indicating that some remaining species are still present in the pores of the material. After confirming the retention of the crystallinity and a reduction of the intrinsic porosity of MFM-300(Sc) after the first H2S cycle, additional H2S cycling experiments were performed. Over the second cycle, the H2S adsorption capacity decreased by 39% to 10.08 mmol g−1, which is in a good agreement with the reduction of the pore volume by about 34%. The average H2S capture in cycles 2 to 5 was 10.22 mmol g−1. After the fifth cycle, PXRD experiments corroborated the retention of the crystalline structure of MFM-300(Sc), while a N2 adsorption experiment showed that the pore retained its reduced volume of 0.37 cm3 g−1. In an attempt to remove the remaining sulphur species formed inside the pores of MFM-300(Sc), the sample after the first H2S cycle was activated at 250 °C. After this thermal treatment, the pore volume did not change (0.37 cm3 g−1) confirming that the sulphur species were irreversibly adsorbed within the MFM-300(Sc) framework.70
The identification of these sulphur species was first approached by TGA and diffuse-reflectance infrared Fourier-transform spectroscopy (DRIFT) experiments which corroborated strong interactions between the guest species and the pore-walls of MFM-300(Sc). Raman spectroscopy, complemented with elemental analysis (EDX) and conventional elemental analysis, postulated the first indication of the possible nature of the remaining (irreversibly adsorbed) sulphur species within the pores, i.e., polysulphides (see Fig. 11).70 Even though the redox properties of polysulphides are highly complex, electrochemical experiments were the key to fully identify them. An electrochemical cell (MFM-300(Sc))-CSP/PVDF/1 M LiTFSI in triglyme/Li0 (PVDF = poly(vinylidene difluoride), LiTFSI = LiN[SO2CF3]2, triglyme = MeO[CH2CH2O]3Me) was examined (for 20 h) through open-circuit potential (OCP) measurements and analysed by cyclic voltammetry. Then, this electrochemical cell showed an initial potential of 2.29 V (vs. Li0/Li+) for MFM-300(Sc) and after reaching equilibrium, this potential was equal to 2.15 V (vs. Li0/Li+).
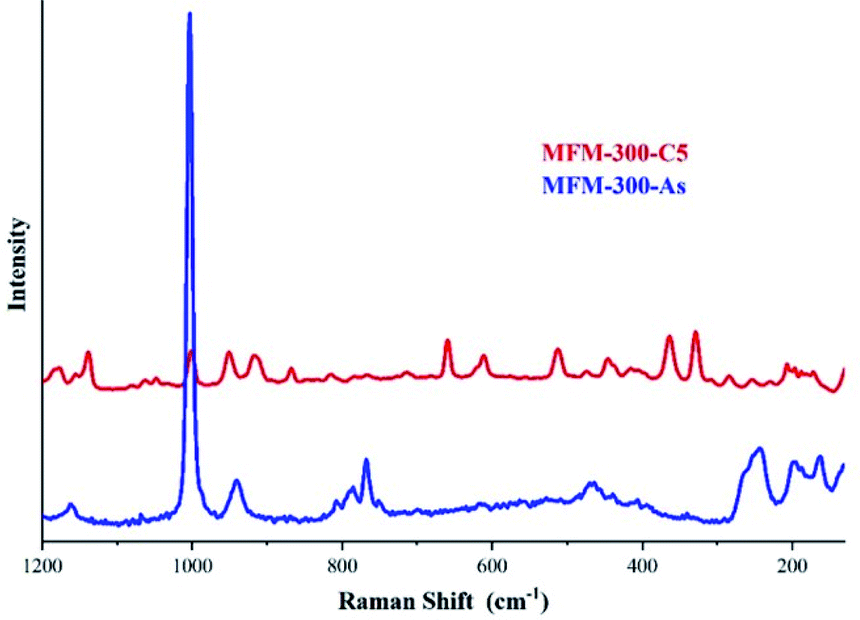 | ||
| Fig. 11 Raman spectra of MFM-300(Sc) after 5 cycles of H2S (red) and as synthesised (blue). The peaks observed in the 350–520 cm−1 region are associated to the S–S stretching vibrations modes of several polysulphides with various chain lengths. Reprinted with permission from ref. 70. Copyright (2020) American Chemical Society. | ||
Later, a H2S exposed sample of MFM-300(Sc) (H2S@MFM-300(Sc)) was used to construct a different electrochemical cell showing a different behaviour on the variation of the potentials (the potential increased from 1.90 to 2.33 V (vs. Li0/Li+)). According to Mikhaylik and Akridge,71 electrochemical potentials lower than 2.10 V correspond to low-order polysulphides (Sn2−), (n = 2). Thus, the initial potential for H2S@MFM-300(Sc) of 1.90 V suggested the formation of such low-order S22− polysulphides. The formation of these low-order species arises from the strong mutual H2S⋯H2S hydrogen bond interactions with a characteristic H(H2S)⋯S(H2S) distance of 2.91 Å, as demonstrated in the corresponding RDF plot from the Monte Carlo simulations.
In the case of MFM-300(In) by constructing the equivalent electrochemical cell (MFM-300(In))-CSP/PVDF/1 M LiTFSI in triglyme/Li0 and same operation conditions (OCP measurements and analysed by cyclic voltammetry for 20 h), the electrochemical cell exhibited an initial potential of 2.65 V (vs. Li0/Li+) for MFM-300(In) and after reaching equilibrium, this potential was equal to 2.15 V (vs. Li0/Li+). The difference of 0.36 V, from the initial potentials for MFM-300(Sc) and MFM-300(In), was attributed to the difference in the electronegativity of the metal centres (Sc3+ and In3+) in both materials. After the adsorption of H2S, H2S@MFM-300(In), MFM-300(In) showed a different trend than MFM-300(Sc): the potential decreased from 2.50 to 2.15 V (vs. Li0/Li+). Such a trend difference for both stabilisation potentials and the potential values indicated that the chemical composition of the sulphur species in both materials (MFM-300(Sc) and MFM-300(In)) were different. Again, Mikhaylik and Akridge indicate,71 that electrochemical potentials higher than 2.10 V correspond to high-order polysulphides (Sn2−), (n = 6). Such large polysulphides block completely the pores of MFM-300(In), as corroborated by the loss of the pore volume in H2S@MFM-300(In) (0.02 cm3 g−1) and Raman spectroscopy. Interestingly, although these two materials are isostructural, a small difference in the pore size (8.1 and 7.5 Å for MFM-300(Sc) and MFM-300(In), respectively) contributes along with the different electrochemical potential of both materials (different electron densities) to the formation of distinct polysulphide species.
Although there is a hypothesis that justifies the polysulphide formation in both materials (based on the “disproportionation” type reaction,72 were the protons of H2S play the role of an oxidant and the sulphide plays the role of a reducing agent), there is a great opportunity to deeply investigate this phenomenon. For example, can the polysulphide formation be a consequence of reversible metal–ligand bonding upon the adsorption of H2S? Recently Brozek and co-workers73 elegantly demonstrated metal–ligand dynamics for carboxylate benchmark MOFs, via variable-temperature diffuse reflectance infrared Fourier transform spectroscopy (VT-DRIFTS) coupled with ab initio plane wave density functional theory. Thus, new exciting horizons can be discovered by taking a different approach when H2S is adsorbed in MFM-300(M), which could explain such fascinating polysulphide formation.
The last MOF material, to date, that demonstrated the chemical transformation of H2S to polysulphides is SU-101.74
This bioinspired material was synthesised (by Inge et al.74), using ellagic acid, a common natural antioxidant, and bismuth (Bi2O(H2O)2(C14H2O8)·nH2O), see Fig. 12. Then, the resultant breakthrough H2S experiment led to a gas uptake of 15.95 mmol g−1, representing one of the highest H2S uptakes reported and even more interesting since the BET surface area of SU-101 (412 m2 g−1) is considerably lower than top H2S capture materials (e.g., MIL-53(Al)-TDC; BET = 1150 m2 g−1). On a second H2S adsorption cycle, the initially observed capacity and surface area were reduced to only 0.2 mmol g−1 and 15 m2 g−1, respectively, even though the crystallinity was retained as confirmed by PXRD. Raman spectra of SU-101 (before and after H2S adsorption) confirmed the presence of low-order polysulphides (n = 2), S42−. Thus, the electrochemical potential of SU-101 should be similar to the one for MFM-300(Sc) (2.29 V),70 considering that S42− species were only found for MFM-300(Sc). Future investigations are anticipated to continue in order to verify the electrochemical potential of SU-101 and any possible reversible metal–ligand bonding upon the adsorption of H2S.
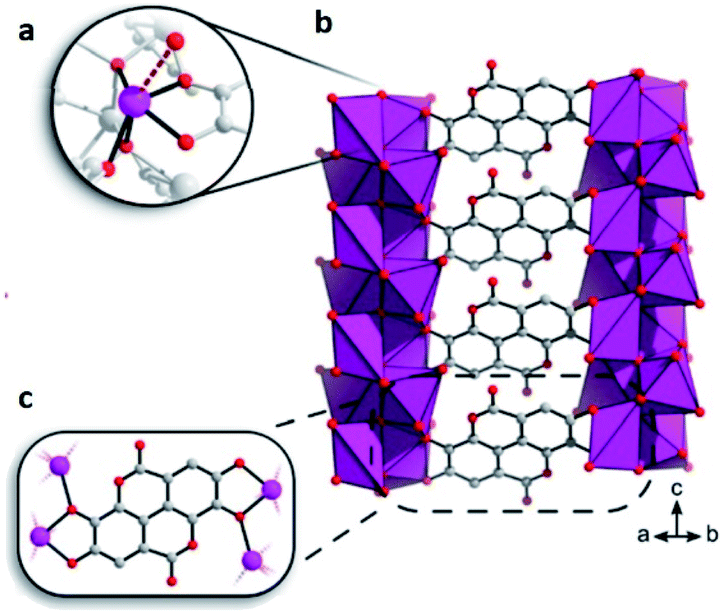 | ||
| Fig. 12 (a) The coordination environment around Bi3+. The bond to a coordinated water molecule is represented as a dashed line. (b and c) Chelation of ellagate ligand toward the bismuth oxo rods. Reprinted (adapted) with permission from ref. 74. Copyright (2020) American Chemical Society. | ||
A variety of MOFs, as well as classic materials such as zeolites, activated carbon and metal oxides, have been studied for the adsorption of these toxic gases, where some of these are summarized in Table 3. Noteworthy, the comparison of materials for H2S capture is sometimes challenging due to the difference in the experimental conditions (e.g., flue gas concentration).
| MOF | BET Surface area [m2 g−1] | H2S adsorption [mmol g−1] | Ref. |
|---|---|---|---|
a 15% vol H2S.
b GCMC simulations.
c Graphite-oxide composite.
d 1000 ppm H2S in moist air.
e H2S/H2/CO2/H2O (600 ppm![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25%:7.5%:1%).
f 10% vol H2S.
g 80 ppmv H2S.
h 4000 ppmv of H2S and 20 vol% of H2 in N2. :25%:7.5%:1%).
f 10% vol H2S.
g 80 ppmv H2S.
h 4000 ppmv of H2S and 20 vol% of H2 in N2.
|
|||
| MIL-47(V) | 930 | 14.6 | 67 |
| MIL-53(Cr) | 1946 | 12.0 | 67 |
| MIL-53(Al)-BDC | 1103 | 11.7 | 67 |
| Ni-CPO-27 | 1547 | 12.0 | 85 |
| Mg-CUK-1 | 604 | 3.1a | 68 |
| MIL-53(Al)-TDC | 1150 | 18.5 | 69 |
| MIL-100(Fe) | 2000 | 16.7 | 67 |
| MIL-101(Cr) | 2916 | 30.1 | 34 |
| MIL-101(Cr)-4F(2%) | 2176 | 36.9 | 34 |
| MFM-300(Sc) | 1350 | 16.5 | 70 |
| Cu-BTC | 1590 | 1.1 | 63 |
| SU-101 | 412 | 15.95 | 74 |
| PAF-302 | 5600 | 51.94 | 75 |
| COF-102 | 3621 | 35.57b | 75 |
| HKUST-1c | — | 2.7d | 60 |
| 10Mn–45Zn–45Ti–Oe | — | 0.0005 | 76 |
| 4A molecular sieve zeolite | 49.5 | 0.0002f | 77 |
| 13X zeolite | 440 | 0.007g | 78 |
| SBA-15 | 950 | 1.98h | 79 |
| PEI(50)/SBA-15(MBS-2) | 80 | 0.0002h | 80 |
| Wood-based AC | 1400 | 0.008 | 81 |
3.2 Bio-compatible MOFs for H2S detection and controlled delivery
Although H2S is catalogued as highly toxic, paradoxically, it can also be crucial as an endogenous biological mediator.82 Endogenous pathways for H2S have been found to have wide extending activities in vivo, such as in the cardiovascular system.83 For example, H2S executes a crucial activity in the regulation of blood pressure, neurotransmission, anti-inflammatory mechanism, anti-oxidation, angiogenesis and apoptosis.84 H2S can be biosynthesised by enzymatic reactions via a sequence of endogenous processes,85 and in the biological medium, the H2S concentration can fluctuate from nano-to micromolar levels.86 However, at higher concentrations of H2S in the bloodstream can cause severe physiological disorders such as diabetes, Alzheimer's disease, cirrhosis, different types of cancer and Down's syndrome.87Therefore, in order to recognise the specific role of H2S in these processes, the detection (spatial and temporal information) of H2S levels in living cells and organisms is crucial. However, due to the high reactivity, volatility and diffusible properties of H2S, traditional detection technologies (chromatography, high performance liquid chromatography, electrochemical analysis and colorimetric)88 are unsuitable for the identification of H2S in living cells because they result in destruction of the cells and tissues and difficulties associated to real-time detection.89 Fluorometric detection techniques have emerged as highly significant alternatives since their non-destructive characteristics, fast response, high selectivity and sensitivity, fast response, high spatial sampling capability, real-time monitoring and easy sample preparation.90 Turn-on type fluorescence probes for H2S detection have been recently investigated and these are commonly based on the reactivity characteristics of H2S (e.g., copper sulphide precipitation, dual nucleophilic addition, H2S-mediated hydroxyl amide and nitro/azide reduction).91 Such characteristic reactivity of H2S can efficiently distinguish it from other biological species; however, many of these detectors do not fulfil basic requirements such as rapid detection and selectivity. Thus, the investigation of new H2S probes is still very attractive due of the complexity of different molecular events involved in signalling transduction, and other complications related to the probes. Therefore, bio-compatible MOFs have been postulated as promising probes for the detection of H2S.
The first concept of a MOF material in in vitro H2S delivery was demonstrated by Morris and co-workers who used CPO-27 for this purpose.92 H2S can be released by exposing the material to H2O since CPO-27 is an open metal site system thus, the water molecule replaces the hydrogen sulphide molecule at the metal site. The authors analysed the H2S delivery in two isostructural materials (Zn-CPO-27 and Ni-CPO-27) finding that the Ni2+ material exhibited higher delivery capacity than the Zn2+ material: after 30 min 1.8 mmol g−1 of H2S was released from Ni-CPO and only 0.5 mmol g−1 from the Zn-CPO. After 1 h, the H2S release for both materials was completed. The amount of H2S released by each material corresponds to approximately 30% of the chemisorbed gas. Later, the release of H2S (stored in Zn-CPO-27 and Ni-CPO-27) was investigated under physiological conditions (endothelium-intact ring of pig coronary artery), finding a substantial vasodilatory action (relaxation) with only a short induction period of approximately 5 min.92 These results represented a very significant progress in the field of MOFs and biological applications, since such H2S release mechanisms offered new protocols to better understand the effects of H2S on this particular vascular system.
Later, the first fluorescence probe example based on a MOF for the detection of H2S was presented by Wang and co-workers.93 Through a post-synthetic modification of ZIF-90 with malononitrile (N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–CH2–CN), they obtained MN-ZIF-90 which undergoes a specific reaction with H2S, achieving an enhancement of photoluminescence, constituting the base for the detection of H2S (see Fig. 13). Then, the malononitrile moieties conjugated to the host (ZIF-90) through double bonds aided as quenchers of the host fluorescence through intramolecular photoinduced electron transfer. The detection mechanism was based on the α,β-unsaturated bond of the malononitrile which is susceptible to thiol compounds, leading to the breaking of the double bond and thus, the fluorescence of MN-ZIF-90 was recovered once H2S was introduced into the system. In addition, the probe exhibited favourable biocompatibility.93
C–CH2–CN), they obtained MN-ZIF-90 which undergoes a specific reaction with H2S, achieving an enhancement of photoluminescence, constituting the base for the detection of H2S (see Fig. 13). Then, the malononitrile moieties conjugated to the host (ZIF-90) through double bonds aided as quenchers of the host fluorescence through intramolecular photoinduced electron transfer. The detection mechanism was based on the α,β-unsaturated bond of the malononitrile which is susceptible to thiol compounds, leading to the breaking of the double bond and thus, the fluorescence of MN-ZIF-90 was recovered once H2S was introduced into the system. In addition, the probe exhibited favourable biocompatibility.93
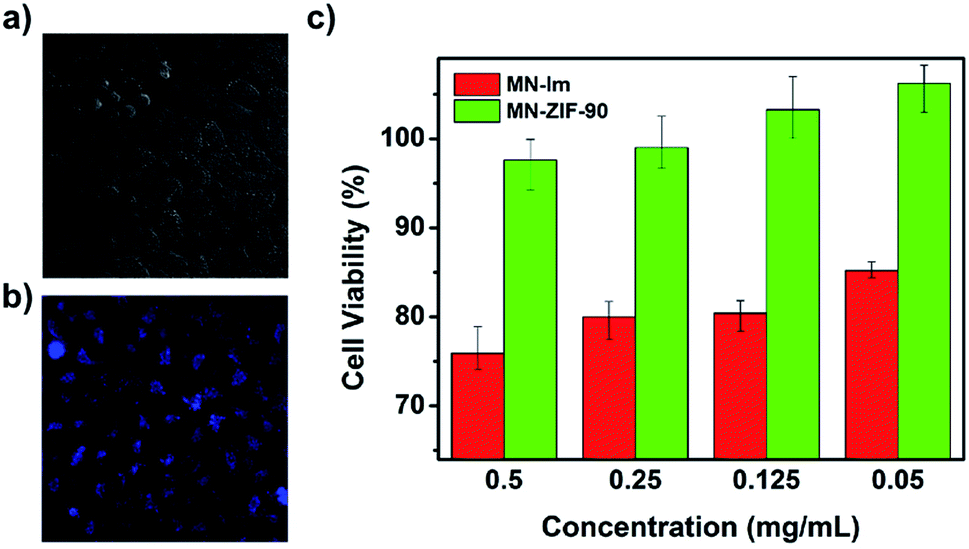 | ||
| Fig. 13 (a, b) Bright field image and fluorescence image after treating the HeLa cells with MN-ZIF-90 (0.05 mg mL−1) after 48 h of incubation. (c) In vitro HeLa cell viabilities after 48 h of incubation with MN-IM and MN-ZIF-90 at different concentrations. Reprinted with permission from ref. 93. Copyright (2014) Springer Nature. | ||
Taking a different approach but keeping the fundamental idea of introducing a reactive site for H2S, Tang et al.,94 demonstrated the use of a porphyrin-based MOF for the fluorescent detection of H2S. By introducing reactive Cu2+ metal centres into a Al3+ MOF material {CuL[AlOH]2}n (H6L = mesotetrakis(4-carboxylphenyl) porphyrin) the detection of H2S was selectively followed by fluorescence under physiological pH (see Fig. 14). Additionally, they successfully demonstrated the capability of the probe to detect exogenous H2S in living cells, while the probe showed low toxicity and high biocompatibility.94
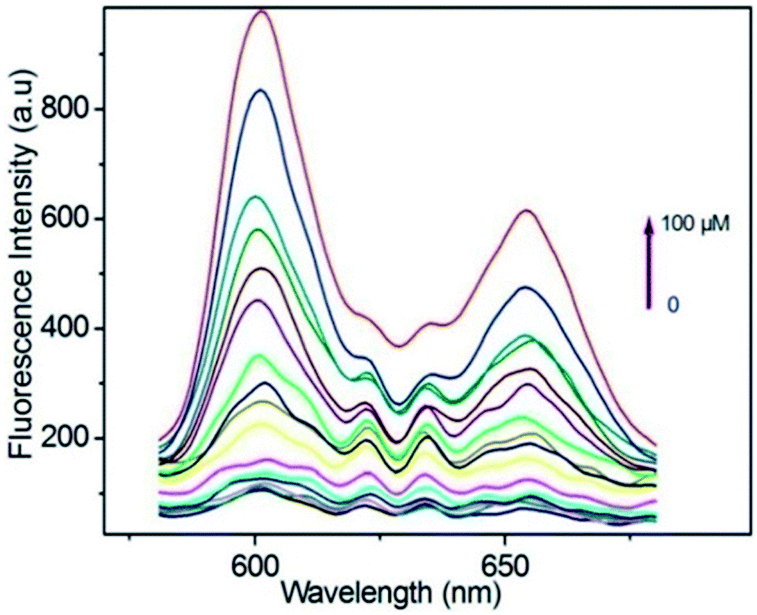 | ||
| Fig. 14 Fluorescence spectra of porphyrin ligand (10 μM) in BBS butter (pH = 7.40, 20 mM) after titration with HS− from 0–100 μM, λex = 419. Reprinted with permission from ref. 94. Copyright (2014) American Chemical Society. | ||
Later, Biswas and co-workers demonstrated, on a dinitro-functionalised UiO-66 (UiO-66-(NO2)2), fluorescence turn-on behaviour towards H2S in simulated biological medium (HEPES buffer, pH = 7.4).95 Remarkably, UiO-66-(NO2)2 exhibited highly sensitive fluorometric H2S sensing while also showing a visually detectable colorimetric change to H2S in daylight (see Fig. 15). In addition, the high selectivity of this functionalised MOF material to H2S was preserved even when several other biological species were present in the detecting medium. Finally, fluorescence microscopy studies on J774A.1 cells demonstrated the effectiveness of UiO-66-(NO2)2 for H2S imaging in living cells, detection of H2S in human blood plasma (HBP) and monitoring of the sulphide concentration in real water samples.95 These results emphasised the biocompatibility of UiO-66-(NO2)2. The reaction mechanism of UiO-66-(NO2)2 and H2S is caused by the reduction of the nitro groups to electron donating amine groups causing an increase in the fluorescence intensity that can be registered.
 | ||
| Fig. 15 Colorimetric detection behaviour of the HEPES suspension of UiO-66-(NO2)2 with increasing concentration of Na2S in daylight. Reprinted with permission from ref. 95. Published (2018) by The Royal Society of Chemistry. | ||
Eu3+/Ag+@UiO-66-(COOH)2 (EAUC) composites were reported by Li and Qian as biomarkers for the for potential diagnosis of asthma.96 The decrease of the H2S production in the lung has been identified as an early detection biomarker for asthma. Thus, fluorescent experiments showed that EAUC exhibited high selectivity and sensitivity with a limit of a real-time H2S detection of 23.53 μM. The detection of H2S by EAUC was based on the introduction of active metal centre (Ag+, H2S-responding site) to the Eu3+@UiO-66-(COOH)2 as the lanthanide-luminescence sensitiser.96 MTT assay and cell viability analysis in PC12 cells demonstrated relatively low cytotoxicity for EAUC and biocompatibility. Finally, the determination of H2S was tested in diluted fetal bovine and human serum samples demonstrating that EAUC can detect H2S in real biological samples. Zhang and co-workers97 covalently modified (via click chemistry) PCN-58 with target-responsive two-photon (TP) organic moieties to fluorescently detect H2S. These TP-MOF probes showed good photostability, high selectivity, minor cytotoxicity, and excellent H2S sensing performance in live cells. These modified PCN-58 materials also showed intracellular sensing and depth imaging capabilities (penetration depths up to 130 μm).97
Up to this point we have presented remarkable examples of MOF materials for the detection of H2S (biomarkers). All of these have been constructed with the fundamental principle of incorporating into these MOFs “the right chemical functionality” which can react with H2S. Remarkably, Wang and Xie98 took a step forward and very recently showed how an endogenous MOF-biomarker-triggered “turn-on” strategy was capable to produce therapeutic agents in situ, as a promising example in nanomedicine for the precise treatment of colon cancer. HKUST-1 was used as an endogenous H2S-activated copper MOF which demonstrated to synergistically mediate H2S-activated near-infrared photothermal therapy and chemodynamic therapy. As a proof of principle, the protocol worked as follows: in normal tissues, the photoactivity of as-synthesised HKUST-1 nanoparticles was in the “OFF” state, and no obvious adsorption within the NIR region was observed. On the other hand, when HKUST-1 nanoparticles with high overexpression of H2S reached the colon tumour tissues, these nanoparticles were activated to the “ON” state by reacting with endogenous H2S to produce in situ photoactive copper sulphide with stronger NIR absorption, which is viable for the subsequent photothermal therapy and corresponding thermal imaging (see Fig. 16). In addition, besides the in situ formation of the photothermal agent, HKUST-1 nanoparticles also showed a horseradish peroxidase (HRP)-mimicking activity to efficiently convert overexpressed H2O2 within cancer cells into more toxic ˙OH radicals for chemodynamic therapy. Thus, this promising H2S-triggered “turn-on” strategy based on the endogenous biomarker from the tumour microenvironment can provide a precise diagnosis, marginal invasion, and possibly clinical translation.98
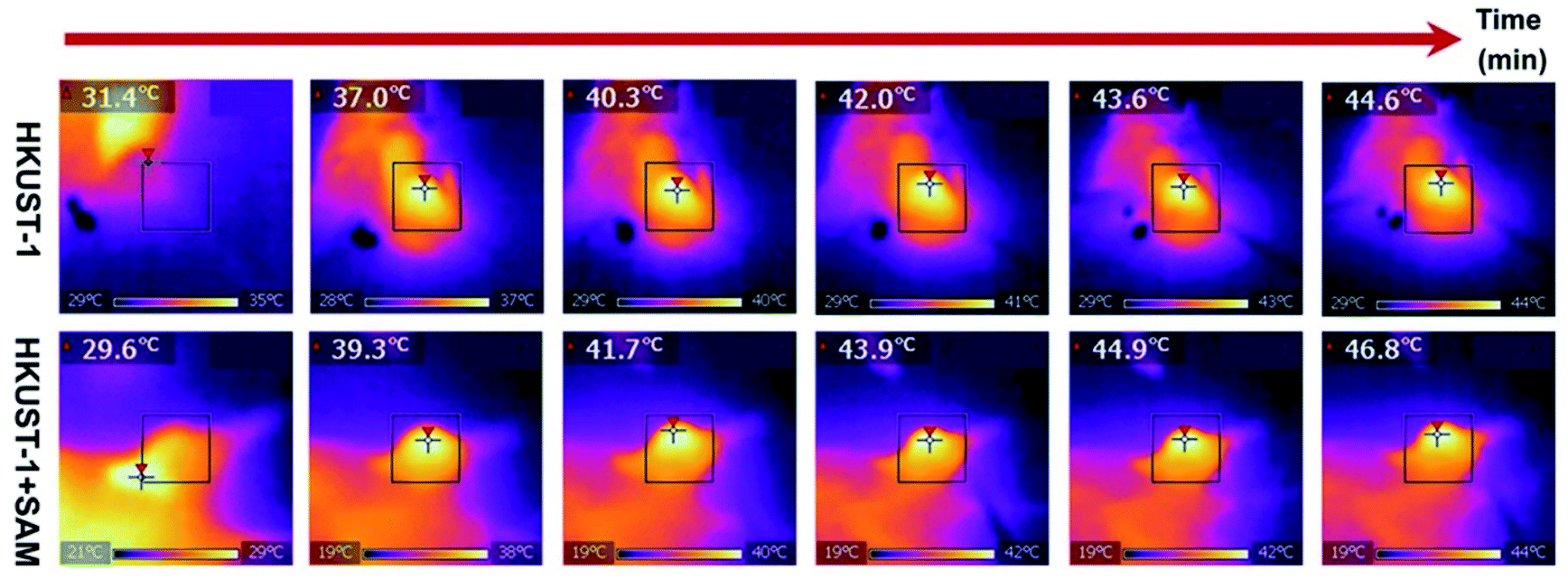 | ||
| Fig. 16 In vivo antitumour efficacy of HKUST-1. Photothermal images of the tumour site after 4 h of intratumoural injection of HKUST-1 and HKUST-1 + SAM exposure to laser irradiation (808 nm, 1 W cm−2) at the 2 min interval. Reprinted with permission from ref. 98. Copyright (2020) American Chemical Society. | ||
4. Ammonia
NH3 is one of the most important chemicals in the world since it is an irreplaceable feedstock for global agriculture and industry (e.g., fertilizers, drug production, heat pumps and fuel cells).99 Although fundamental to our diet supply and economy, NH3 is a highly toxic (even in small concentrations) and corrosive gas (i.e., difficult to handle and store). Thus, considerably large emissions of NH3 from livestock breeding, industrial production, fertiliser application and public transportation can have serious consequences on the environment (highly unfavourable to air quality and aquatic life), and human health.100 In addition, NH3 reacts with NOx and SO2 to form PM2.5, seriously contributing to the increase of air pollution.NH3 is a colourless gas with a characteristic pungent smell which is typically liquefied and confined in metal tanks in order to be efficiently stored and transported.101 This implies high pressures (approximately 18 bar) and constant corrosion of pipelines and containers. Thus, new sorbent technologies capable of efficiently removing or storing NH3 are highly desirable for air remediation and separation of NH3 from N2 and H2 since NH3 is considered an interesting energy intermediate.102 It is clear that capture and recycling of NH3 implies a dual connotation in the fields of environment and energy. Classic materials such as activated carbons, zeolites, silicas and even organic polymers have been investigated for the capture and separation of NH3 showing low uptakes, poor selectivity and, in some cases, irreversible storages.103,104 MOFs have been postulated as a promising option for NH3 capture, due to the access to a wide range of chemical functionality (e.g., Lewis or Brønsted acid sites which provide higher affinity to the basic NH3 molecule).104 However, the main problem that most MOF materials exhibit poor chemical stability towards NH3.105 The following section describes chemically stable MOFs with interesting NH3 capture performances.
4.1 MOFs for NH3 capture
The NH3 molecule is a Lewis and Brønsted base which is the key to its adsorption on the porous surface of chemically stable MOFs. Thus, the incorporation of acidic functional groups within the MOFs is required to achieve relevant NH3 capture results. These functionalisations can be organised in three main types: (i) coordinatively unsaturated metal sites (open metal sites), (ii) μ-OH groups and (iii) defective sites.Later, the Dincă's group reported another series of microporous triazolate MOFs (containing open metal sites; Cu2+, Co2+, and Ni2+, see Fig. 17), that exhibited remarkable static and dynamic NH3 capacities (up to 19.79 mmol g−1, at 1 bar and 298 K).108 These isoreticular analogues to the bisbenzenetriazolate examples,107 are constructed from smaller triazolate ligands and therefore, they exhibited smaller pore windows (see Fig. 17). Interestingly, these microporous MOFs showed higher NH3 captures than their mesoporous analogues.107 This remarkable property was not only accounted by the increase in the density of open metal sites, in addition cooperative proximity effects resulted very important. NH3 breakthrough experiments on these microporous materials showed the potential applicability for both personal protection and gas separations (NH3 capacities of 8.56 mmol g−1). Finally, once again, the superior chemical stability of these triazolate MOFs arises, as previously described, from the coordination of the N atom to metal centres (vide supra).
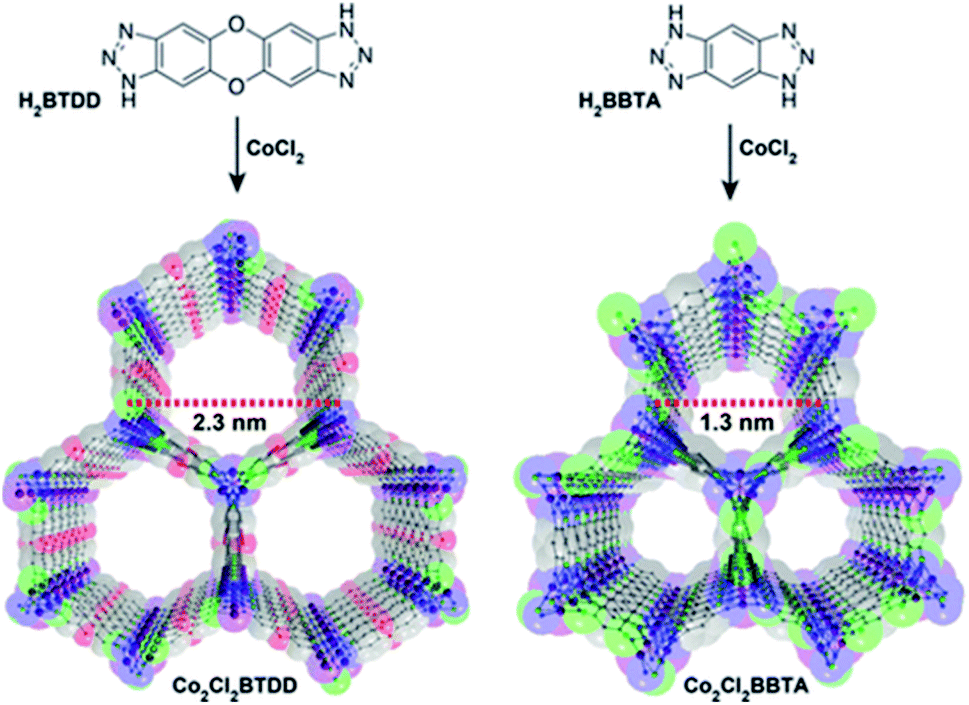 | ||
| Fig. 17 Structure of Co2Cl2 BTDD (left) and Co2Cl2 BBTA (right). C, gray; O, red; N, blue; Cl, green; Co, purple. Hydrogen atoms have been omitted for clarity. Reprinted with permission from ref. 108. Copyright (2018) American Chemical Society. | ||
Very recently, Hong and co-workers recently presented the NH3 adsorption properties of M2(dobpdc) MOFs (M = Mg2+, Mn2+, Co2+, Ni2+, and Zn2+; dobpdc4− = 4,4-dioxidobiphenyl-3,3-dicarboxylate), which after activation exhibited open metal sites (see Fig. 18).109 The NH3 uptake of Mg2(dobpdc) at 298 K and 1 bar was 23.9 mmol g−1 at 1 bar and 8.25 mmol g−1 at 570 ppm, representing the NH3 record high capacities at both pressures among existing porous adsorbents.109 Mg2(dobpdc) remarkably demonstrated chemical stability to not only dry NH3, but also to wet NH3 (NH3·H2O vapour). Three consecutive NH3 sorption isotherms (activation included only application of vacuum) demonstrated the cyclability of Mg2(dobpdc). To evaluate the affinity of the NH3 molecule to the open Mg2+ sites in Mg2(dobpdc), NH3-TPD curves demonstrated (based on the Redhead analysis) the activation energy for NH3 desorption of 146.7 kJ mol−1. The NH3 adsorption mechanism of Mg2(dobpdc) was investigated by in situ FTIR experiments, indicating that NH3 was preferentially adsorbed, by coordination bonds, to the open metal sites of Mg2(dobpdc). As the MOF sample was exposed to atmosphere the adsorbed NH3 in Mg2(dobpdc) was converted to NH4+, due to its reaction with atmospheric moisture. Dynamic NH3 breakthrough curves under dry conditions (0% relative humidity (RH) and 298 K) showed a NH3 capacity of 8.37 mmol g−1 for Mg2(dobpdc). When performing these NH3 breakthrough experiments under 80% RH conditions, the NH3 capacity of Mg2(dobpdc) decreased to 6.14 mmol g−1, due to the competitive adsorption between NH3 and H2O. Additionally, this NH3 capacity remained constant over five breakthrough wet cycles.109 Thus, Mg2(dobpdc) is an exceptional MOF material that can be easily synthesised (i.e., microwave-assisted), highly recyclable (even after exposure to a humid NH3) without structural degradation or NH3 capacity loss and holds the record for the highest NH3 uptake at room temperature and atmospheric pressure.
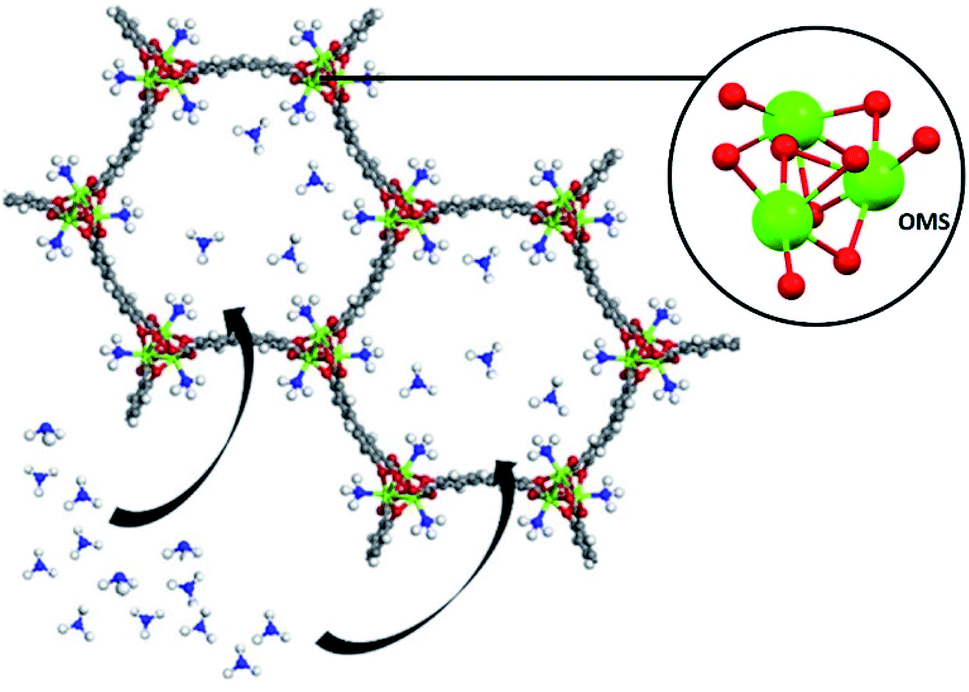 | ||
| Fig. 18 Atomic structure of M2(dobpdc) complexes with one-dimensional hexagonal channels. It is possible to observe that each coordinatively unsaturated metal site points towards the channel Reprinted (adapted) with permission from ref. 109. Copyright (2020) John Wiley & Sons. | ||
All of these relevant properties are possible due to the exceptional chemical stability of this Mg2+-based MOF material. But why is it so stable towards NH3? The authors proposed that such chemical stability was due to the higher affinity of Mg2+ to oxygen atoms than nitrogen atoms, as confirmed by van der Waals (vdW)-corrected density functional theory (DFT) calculations. They further investigated the origin of such remarkable chemical stability finding that the oxygen adjacent to the carboxylate participates in the coordination of the ligand to the Mg2+ cation acting as a tetratopic linker, similar to a chelate effect, increasing the stability of the cluster. This strategy is widely used to prevent pore collapse due to the activation energy barrier to ligand reorganisation or removal, which is increased by the higher linker connectivity.21
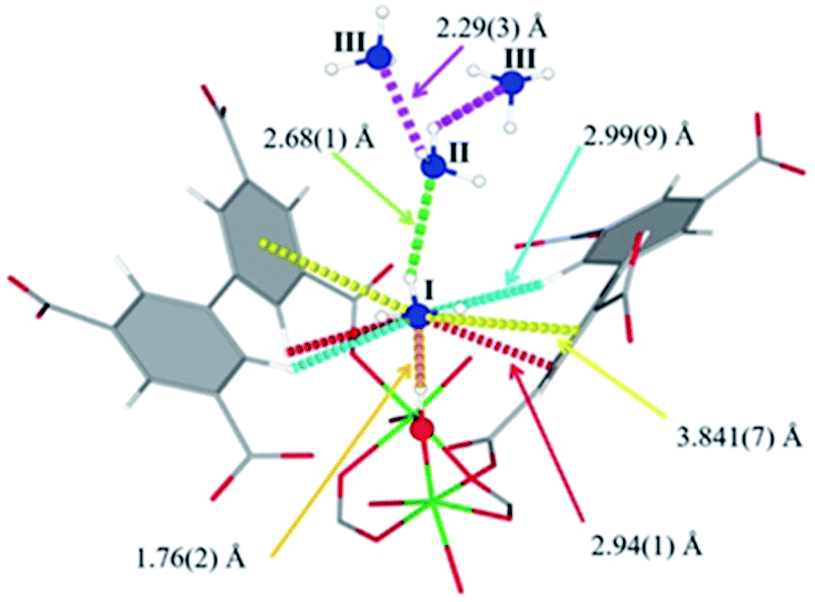 | ||
| Fig. 19 Binding sites for ND3 molecules adsorbed within MFM-300(Al) determined by in situ NPD studies. Reprinted with permission from ref. 110. Copyright (2018) John Wiley & Sons. | ||
Refinement of the NPD data for ND3-loaded MFM-300(Al) demonstrated that on increasing the loading and thus the ND3/μ2-OH ratio, the hydrogen atoms of the hydroxo functional groups experienced a reversible site exchange with the deuterium from the guest ND3 molecules residing at site I in the pore to resulting in the formation of μ2-OD moieties.110 Interestingly, this H–D reversible exchange did not lead to any detectable structural degradation of the long-range order of the MOF material. Thus, the adsorption of ND3 in MFM-300(Al) showed a new type of adsorption mechanism where adsorbent and adsorbate experienced a rapid site-exchange via reversible formation and cleavage of O–H and O–D chemical bonds, in other words a pseudo-chemisorption binding mechanism.
Very recently, Yang and Schröder expanded the investigation on more MFM-300(M) (M = Fe2+, V4+, Cr3+, In3+) materials and NH3.111 MFM-300(M) (M = Fe2+, V3+, Cr3+) demonstrated fully reversible NH3 capture for over 20 adsorption–desorption cycles, under pressure-swing conditions, reaching capacities of 16.1, 15.6, and 14.0 mmol g−1, respectively, at 273 K and 1 bar. In the case of MFM-300(In), a significant loss of NH3 capture capacity over repeated NH3 cycles was shown which corroborated its chemical instability. On the other hand, MFM-300(V4+) exhibited the highest NH3 uptake (17.3 mmol g−1) among the MFM-300(M) family. Interestingly, the NH3 desorption phase for MFM-300(V4+), with pore dimensions of approximately 6.7 × 6.7 Å2, showed a hysteresis loop. Although this could indicate a characteristic capillary NH3 condensation (e.g., in mesopores and/or due to a broad distribution of pore size and shape), the authors demonstrated, taking into account the pore dimensions of this MOF, a specific and potentially strong host–guest charge transfer upon the adsorption of NH3. In addition, MFM-300(V4+) showed an increase of both NH3 capacity and residue within the first 18 cycles, which was not observed for the rest of the MFM-300(M) materials. Such residual amount of NH3 left within MFM-300(V4+) upon regeneration (pressure-swing) gradually increased from 8 to 20% along these cycles, indicating an increase of strongly bound NH-derived species in MFM-300(V4+). This NH-derived residue, which was not desorbed by only reducing the pressure, was completely removed by increasing the temperature under dynamic vacuum, although some structural degradation of MFM-300(V4+) was observed. The chemical stability of these materials to NH3 under humid conditions was investigated by PXRD, confirming the retention of their crystallinity for both MFM-300(M) (M = Cr3+, V3+), while MFM-300(M) (M = Fe3+, V4+) showed some structural degradation.111
Neutron powder diffraction data for ND3-loaded MFM-300(M) combined with Rietveld refinements, showed the preferential binding sites for ND3 (Fig. 20). Two binding sites for ND3 were clearly identified for MFM-300(M) (M = In3+, V3+), while MFM-300(Fe) has an additional binding site for ND3. Site I showed the highest occupancy, with hydrogen bonding between the hydroxo functional group and the ND3 molecule (μ2-OH⋯ND3) (1.411–1.978 Å), complemented by additional hydrogen bonding from the organic ligand (Haromatic⋯ND3 = 2.738–3.174 Å; ND⋯Oligand = 3.078–3.179 Å) and electrostatic interactions (ND3⋯aromatic rings = 2.946–3.132 Å) (Fig. 20). Similar to MFM-300(Al),110 hydrogen/deuterium site exchange was also observed between the adsorbed ND3 and the μ2-OH group on for MFM-300(M) (M = In3+, Fe3+, V3+). Site II is located toward the centre of the pore (fixed by hydrogen-bonding interactions ND3⋯Oligand = 2.284–3.065 Å). Site III in MFM-300(Fe) is constructed by electrostatic interactions (ND3⋯aromatic rings = 3.146 Å). Additionally, in MFM-300(Fe), intermolecular hydrogen bonds between ND3 molecules (2.327 Å) were identified, propagating along the 1D channel to form a cooperative {ND3}∞ network.
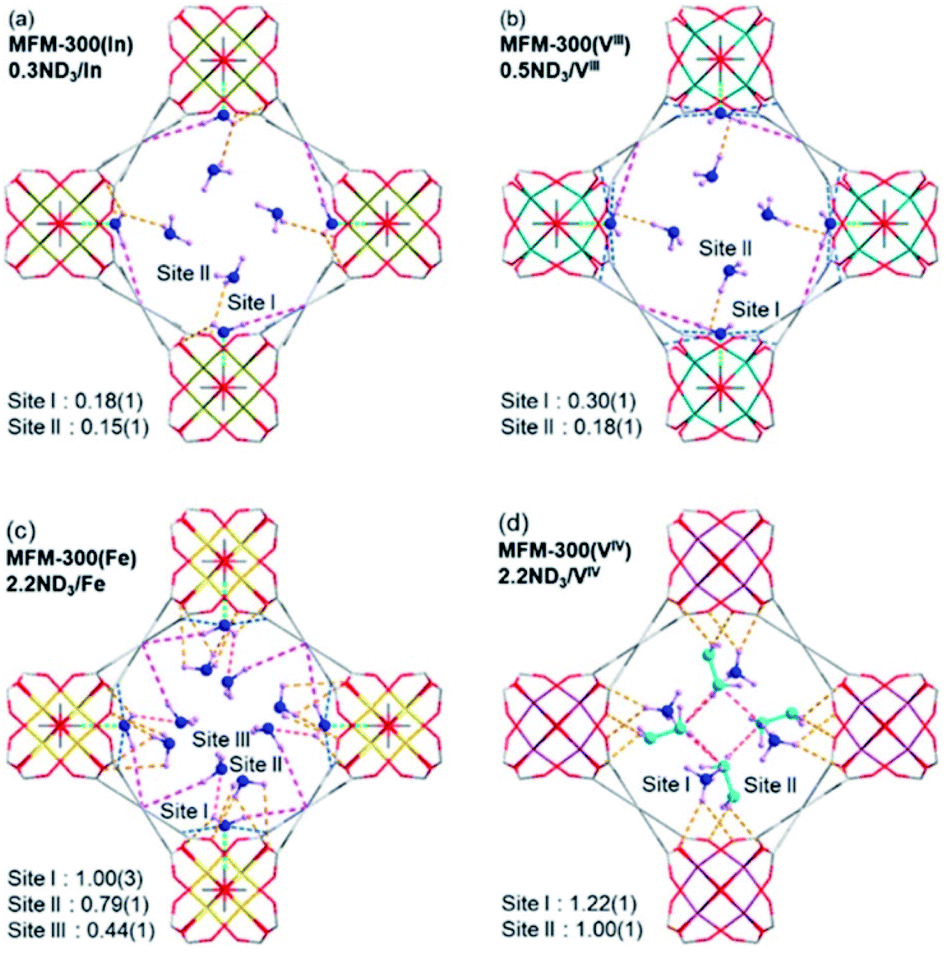 | ||
| Fig. 20 Preferential ND3 binding sites in MFM-300(M) family (M = Cr, Fe, V3+, V4+) determined by NPD at 10 K. Views along c axis. Reprinted with permission from ref. 111. Copyright (2021) American Chemical Society. | ||
The preferential ND3 binding sites for MFM-300(V4+) were identified and these were different to the previous examples. MFM-300(V4+) is not constructed with μ2-OH groups. It shows bridging oxo centres to balance the charge of the oxidised V centre. Thus, ND3 molecules are located in the centre of the pore. This particular arrangement implicates a very interesting situation: chemical reaction between the ND3 molecules: a N2D4 molecule was formed and located at site II with a ND3 molecule at site I (being partially protonated to ND4+). Then, both sites are stabilised via hydrogen bonding (ND3⋯Oligand = 2.529–3.092 Å), and the amount of N2D4 at site II was estimated as 0.5 N2H4 per V centre.
The formation of N2H4 was explained in terms of the redox activity of MFM-300(V4+) which promotes a host–guest charge transfer between the material and the adsorbed NH3 molecules, promoting the reduction of the V4+ centres and oxidation of NH3 to N2H4.111 These host–guest charge transfer results were experimentally determined by EPR and corroborated by bond valence sum (BVS) calculations, finding as well that the charge transfer between adsorbed NH3 molecules and the V4+ centre can only occur when the loading of NH3 is sufficiently high so that a predominant occupancy of the N site, which is located close to the metal chain, is reached to initiate the redox reaction. Finally, such mechanisms could also be investigated by the approach of metal–ligand dynamics as recently proposed by Brozek and co-workers,73 to fully understand the interaction of NH3 and the hydroxo functional group in the MFM-300(M) family.
Then, defective UiO-67 (non-monodisperse pore structure created by missing ligand defects) was synthesised thanks to the high connectivity of the Zr6 oxoclusters, which help to retain the overall crystal structures even with some ligands are missing. UiO-67 involves uniform trigonal windows with a diameter of 11 Å that lead to interconnected tetrahedral and octahedral pores inside the structures. In the presence of missing ligand defects, the trigonal windows surrounding the defects in UiO-bpydc are fused into lozenge windows with a dynamic size larger than 14 Å. NH3 adsorption–desorption isotherms at 298 K and up to 1 bar were performed for both materials: UiO-67 and UiO-bpydc. In the case of Ui-O-67, the adsorption isotherm showed “step-like shape” with two events (see Fig. 21, bottom). This characteristic isotherm shape is caused in MOFs by a gate-opening phenomenon due to the interaction between guest NH3 molecules and pore walls, or a pore filling process.113 Thus, at the beginning of the adsorption, the rapid and sudden increase in NH3 adsorption at approximately 30 mbar (1.70 mmol g−1) (position I) suggests the presence of strong adsorption sites inside the framework, e.g., strong interaction of the NH3 molecules with the hydroxo (μ3-OH) functional groups. Later, the NH3 uptake increased from 2.40 to 4.40 mmol g−1 at 250 mbar (position II) and from 5.60 to 8.40 mmol g−1 at 650 mbar (position III, Fig. 21). The desorption phase exhibited large, opened hysteresis, indicating that the NH3 molecules strongly interact with the pore walls of the material.112 These NH3 molecules were fully desorbed by heating up to 423 K for 1 h under dynamic vacuum. Later, the chemical stability of UiO-67 towards NH3 was investigated by conducting three NH3 adsorption–desorption cycles and finally exposing the material to NH3 vapour for 1 week. PXRD experiments corroborated the stability of UiO-67 to dry and humid NH3. Then, in order to identify the preferential NH3 adsorption sites in UiO-67, in situ high-resolution neutron powder diffraction (NPD) and synchrotron powder X-ray diffraction (SPXRD) experiments were carried out. The refined structures of UiO-67 at different ND3 pressures, reviled five such adsorption preferential sites (Fig. 21).112 At site I, the ND3 was found close to the μ3-OH with a OH⋯NI distance of 1.96(1) Å, and O⋯NI distance of 2.80(1) Å (see Fig. 21). This distance suggested the formation of a relatively strong H-bonding interaction between the μ3-OH and ND3, similar to the one reported by Yang and Schröder in the MFM-300(M) family.110,111 ND3 molecules at site II were situated close to the walls of the trigonal windows suggesting interactions with the MOF organic ligands (Hligand⋯NII = 2.58(1) and 2.68(1) Å), Fig. 21. On increasing the amount of ND3, these molecules were located near to the tetragonal pore (sites III and IV). At site III, the ND3 molecule filled the shallow pore positions with NII⋯NIII bond distances among these ND3 sites in the range of 2.33(1) and 2.57(1) Å, creating a H-bonded network of these ND3 molecules (Fig. 21).
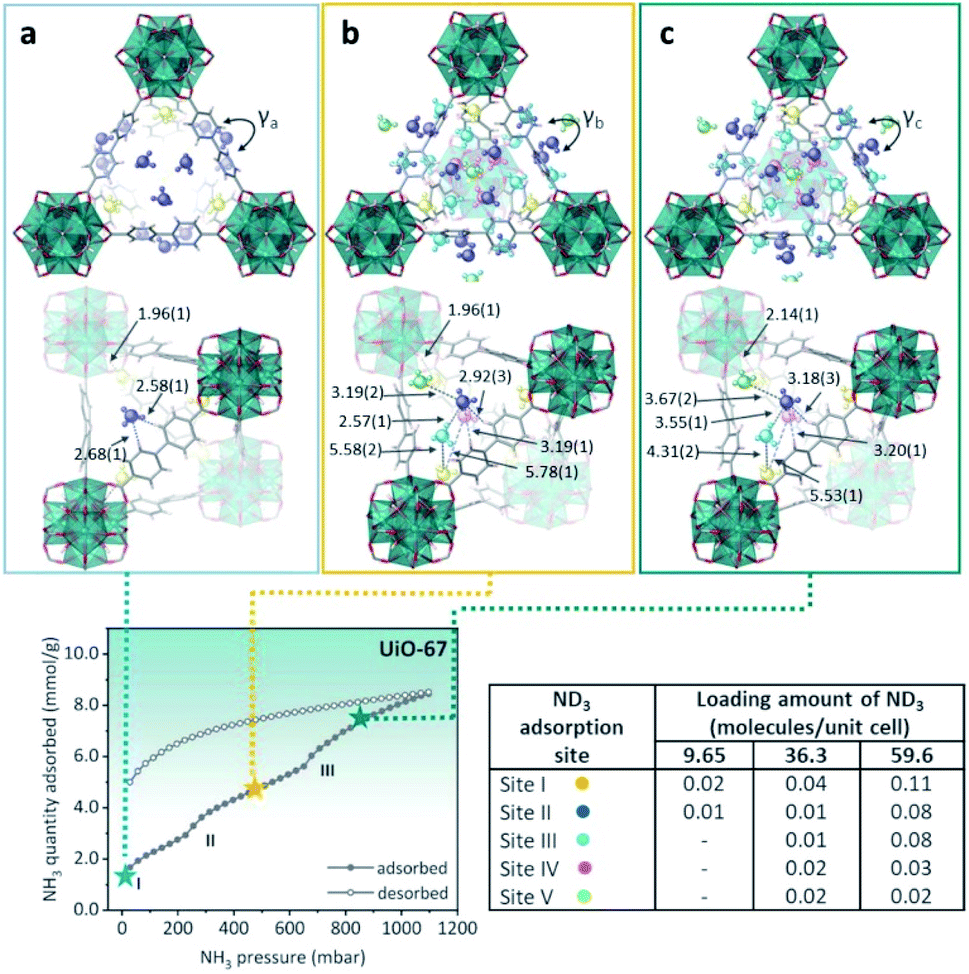 | ||
| Fig. 21 (Top) Refined ND3 positions in the UiO-67 structure at ND3 loadings of (a) 1.23, (b) 4.63, and (c) 7.60 mmol g−1 obtained from NPD data at 300 K. ND3 molecules at different binding sites: site I: yellow, site II: blue, site III: light blue, site IV: pink, and site V: green. (Bottom) NH3 sorption isotherm at 298 K and up to 1100 mbar; adsorption: closed circles; desorption: open circles reprinted with permission from ref. 112. Copyright (2021) American Chemical Society. | ||
In the case of UiO-bpydc, the NH3 adsorption isotherm exhibited an uptake of 8.4 mmol g−1 at 298 K and 1 bar (see Fig. 22). Conversely to UiO-67, UiO-bpydc showed only one large and sharper transition step (position II′) for the NH3 adsorption phase (Fig. 22). Then, in situ NPD experiments and followed by Rietveld refinements corroborated the existence of missing ligand defects. Similarly to UiO-67, the preferential NH3 adsorption sites within UiO-bpydc were also identified by in situ NPD experiments (replacing NH3 by ND3). Thus, two independent preferential sites of ND3 were found close to the μ3-OH functional group (site I′) and to the organic ligands (site II′, bipyridine ligand), Fig. 22. This strongly suggested that the adsorption of ND3 mainly takes place at site II′. For site I′, the OH⋯NI bond distance was estimated to be 2.10(2) Å, which is similar to that found in the isostructural UiO-67. Interestingly, the ND3–bipyridine ligand interaction was estimated to be stronger than the ND3–biphenyl ligand interaction with longer distances (Hligand⋯NII = 2.68(1), 3.19(1), and 3.20(1) Å, respectively) (see Fig. 22). Increasing the dosing of ND3 molecules, increased the H-bonding between the ND3 molecules giving rise to a network of six ND3 molecules with the three bipyridine ligand located around the trigonal window. This particular situation demonstrated the gate-opening/closing behaviour via ligand flipping of UiO-bpydc upon ND3 adsorption.112
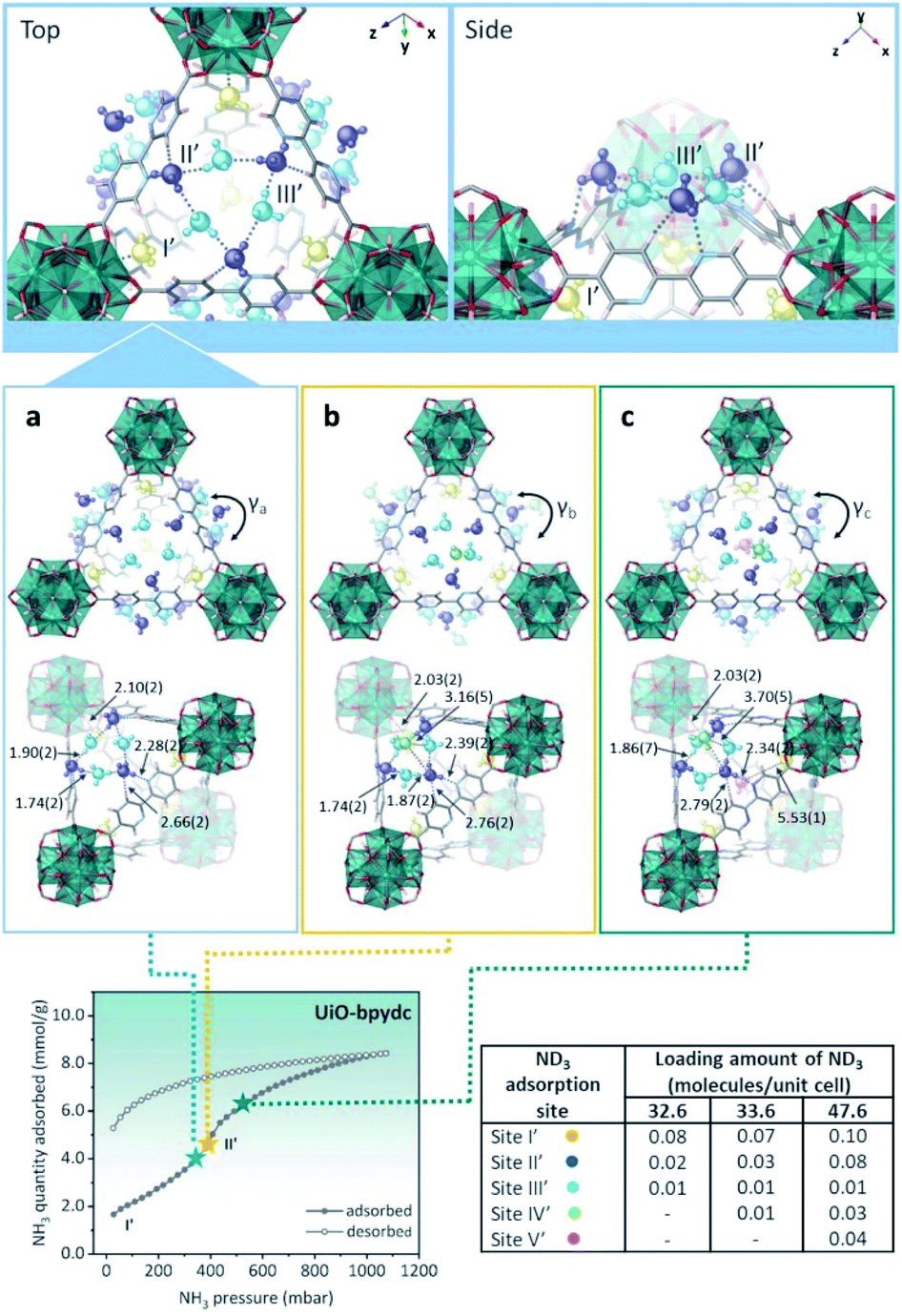 | ||
| Fig. 22 (Top) Refined ND3 positions in the UiO-bpydc structure at ND3 loadings of (a) 4.15, (b) 4.27, and (c) 6.06 mmol g−1 obtained from NPD data at 300 K. (Bottom) ND3 molecules at different binding sites: site I: yellow, site II: blue, site III: light blue, site IV: green, and site V: pink. NH3 sorption isotherm at 298 K and up to 1100 mbar; adsorption: closed circles; desorption: open circles reprinted with permission from ref. 112. Copyright (2021) American Chemical Society. | ||
This phenomenon was further investigated by DFT calculations to understand the role of the bipyridine ligands of UiO-bpydc in forming the H-bonding network with NH3 molecules, in comparison with the biphenyl ligands of UiO-67. Thus, the DFT-optimised structures demonstrated the progressive distortion of the aligned bipyridine ligands with γ changed from 20.34° (in good agreement with the NPD refined desolvated structure of UiO-bpydc) to 19.47°, 10.83°, 9.49°, and 6.33° by increasing the number of NH3 molecules at the trigonal window. Finally, these remarkable results demonstrated that the different pore openings (windows) induced by missing ligands can introduce stepped NH3 sorption with a strong hysteresis into the UiO type MOFs (i.e., biphenyl ligands are replaced by pyridine ligands).112
5. NOx
Nitrogen oxides (NOx = N2O, NO, N2O3, NO2 N2O4 and N2O5) are considered one of the major air pollutants generated by anthropogenic activities, particularly those that involve fuel combustion from stationary and mobile sources such as thermal power plants and vehicles.114 It has been estimated that fossil fuel combustion generates around 5% of NO2, and 95% NO.114 Nevertheless, once NO is released into the atmosphere, it rapidly reacts with O2 to form NO2. The latter is a poisonous red-orange gas responsible for the reddish-brown colour of the smog, although at lower temperatures it can dimerize into a colourless N2O4 dimer. Due to the highly reactive nature of NOx, the uncontrolled emissions of such gases into the atmosphere are associated with a series of health and environmental issues.114–116 For instance, the presence of high concentrations of NO2 has been directly related to the formation of tropospheric ozone O3,109,116 which not only is considered an important greenhouse gas; but is also associated with pulmonary and chronic respiratory diseases.115,116 The adverse effects caused by atmospheric NO2 have motivated an intense research for the development of NO2 abatement technologies.114,117One of the most studied methods to mitigate the atmospheric NO2 is the development of porous materials for the selective capture of this pollutant. The traditional adsorbent materials used for NO2 removal include zeolites,118 calcium-based adsorbents,119 and activated carbon.120 However, such materials are typically affected by the reactive oxidative nature of NO2, which hampers the fully reversible desorption of the guest molecules and limits the material regeneration.117
5.1 MOFs for NOx capture
Current investigations in NOx sorption have pointed out that MOFs represent an attractive alternative for storage, selective separation, and/or catalytic transformation of NO2.121 Such materials not only display a large surface area, but they also exhibit tuneable pore functionalities122 allowing for the stabilisation of guest molecules through the formation of supramolecular host–guest interactions. For instance, a study reported by Peterson et al.123 points out the key role of the organic linker for the capture of NO2. In this work, the authors conducted a comparative study bout the NO2 sorption performance of UiO-66 and UiO-66-NH2.The gas sorption experiments were performed under dry and controlled humid conditions. The results obtained from the microbreakthrough experiments reveal that UiO-66-NH2 exhibits a higher capacity for the NO2 sorption than UiO-66 (20.3 mmol g−1vs. 8.8 mmol g−1; respectively). Under humid conditions (80% RH), UiO-66-NH2 exhibits higher uptake of NO2 than under dry conditions (31.2 mmol g−1vs. 20.3 mmol g−1; respectively), and it produces a significantly lower amount of NO as a by-product than its analogous UiO-66 (4.5%, 9.5%; respectively).
Such differences were explained in terms of the higher capability of UiO-66-NH2 to adsorb H2O vapours. As the H2O co-adsorbed within the pore network might enhance the stabilization of NO2 molecules through the formation of supramolecular interactions and it facilitates the preferential formation of nitrous acid as a by-product. The complementary characterization of UiO-66-NH2 upon NO2 adsorption revealed that although the crystallinity of UiO-66-NH2 remains intact, the high reactivity of the adsorbate leads to a series of reactions with the organic linker, which ends up in the post functionalization of the phenyl group (Fig. 23).123
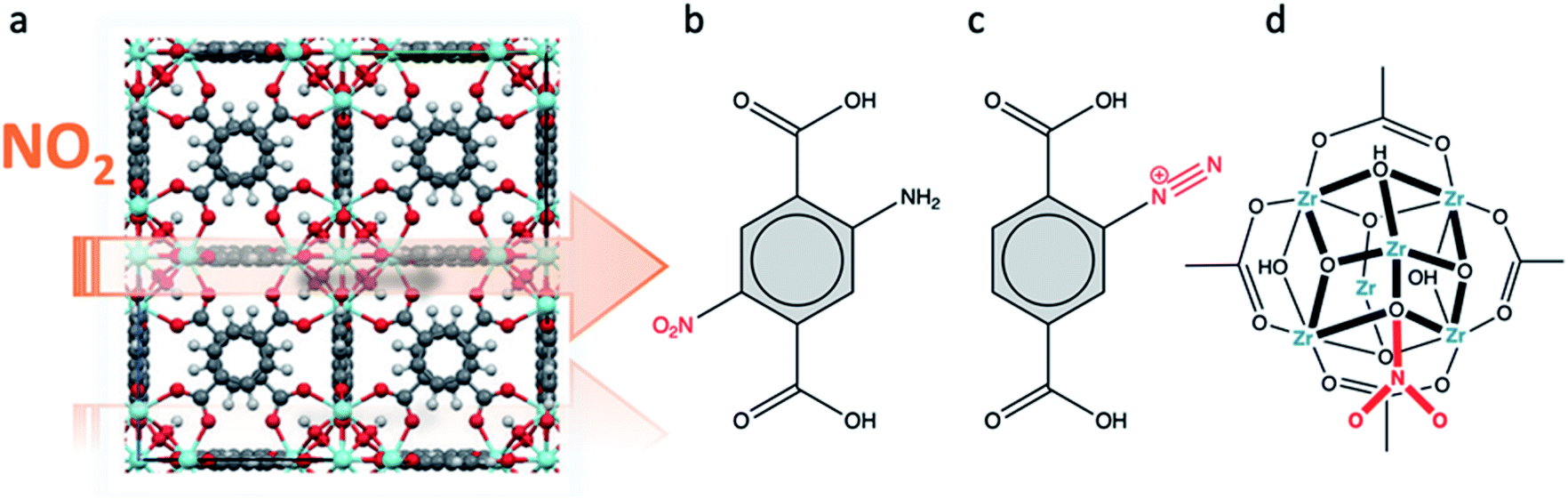 | ||
| Fig. 23 (a) Structure of UiO-66 analogues. Post functionalization of phenyl ring modifications upon NO2 sorption: (b) nitration of the phenyl ring, (c) diazonium ion formation. (d) Nitration of the bridging hydroxyl group at the SBU. Reprinted (adapted) with permission from ref. 123. Copyright (2016) John Wiley & Sons. | ||
The main transformation that suffered the organic linker upon NO2 uptake were the nitration of the aromatic ring and the formation of the diazonium ion at the amino group, while in the inorganic SBU the terminal –OH group is replaced by the NO3− ion. More recently, a study conducted by Schröder and Yang,124 demonstrated that the synergistic effect between the organic ligand and the inorganic building block in MFM-300(Al) allows for the stabilisation of highly reactive NO2 species within the pore network (Fig. 24a–c). The authors reported that under ambient conditions MFM-300(Al) exhibits a fully reversible NO2 isotherm uptake of 14.1 mmol g−1 at 298 K. Moreover, the host material retains its crystallinity and sorption capacity after five cycles of NO2 adsorption/desorption. Remarkably, MFM-300(Al) displays outstanding performance for the selective removal of low-concentration of NO2 (5000 to <1 ppm) from gas mixtures (Fig. 24d). The optimal uptake and selectivity of MFM-300(Al) for NO2 was attributed to the existence of both host–guest and guest–guest interactions. The former involves mainly the hydrogen bonding interaction between NOx and the –OH pendant group of the inorganic node, while the latter refers to the supramolecular interactions between the guest molecules in their monomeric (NO2) and dimeric (N2O4) form. Such interactions give rise to a one-dimensional helical chain arrangement comprised of alternating monomer-dimer molecules (NO2·N2O4)∞ running along the channel of MFM-300(Al) (Fig. 24d).
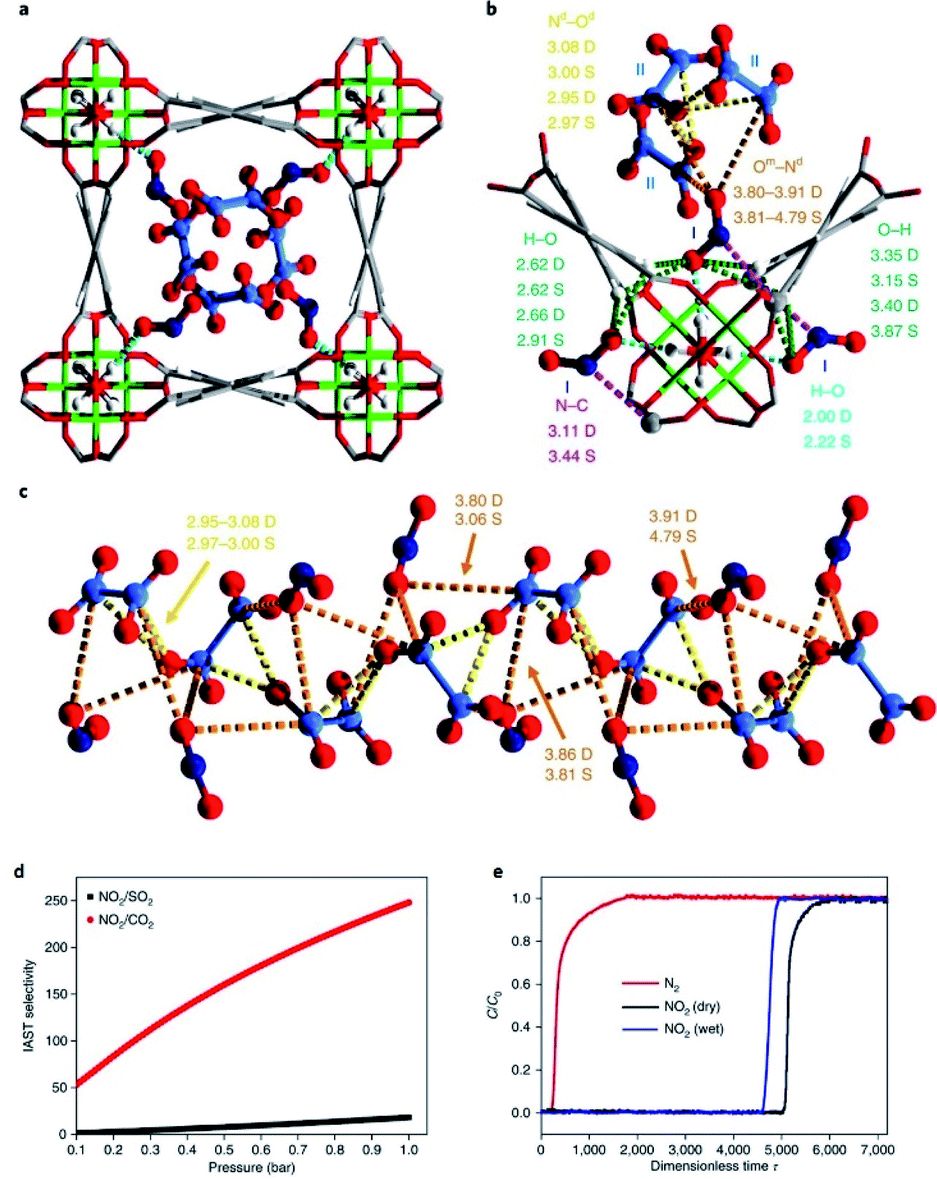 | ||
| Fig. 24 Views of the structural model of MFM-300(Al)·(NO2)(N2O4)2. Al-green, C-grey, O-red, H-white, N-blue. (a) and (b) host–guest supramolecular interactions. (c) Structural view of 1D helical chain (NO2, N2O4)∞ within the channel of MFM-300(Al). (d) Comparison of IAST selectivities for equimolar mixtures of NO2/SO2 and NO2/CO2 at 0.1–1.0 bar for MFM-300(Al) at 298 K. (e) Dimensionless breakthrough curve of 0.5% NO2 (5000 ppm) diluted in He/N2 under both dry and wet conditions through a fixed bed packed with MFM-300(Al) at 298 K and 1 bar. Reprinted (adapted) with permission from ref. 124. Copyright (2018) Springer Nature. | ||
The cooperative supramolecular interactions between NO2 and N2O4, confined within the pore network, allows for the stabilization of the highly reactive NO2 molecules and inhibits the guest–host electron transfer. This affords an unusual, fully reversible desorption of NO2 without altering the framework structure of the host material. The structural versatility of MOFs not only permits the stabilization of highly reactive NOx species within the pore channels but also opens the possibility for the catalytic conversion of NOx into more valuable and/or less abrasive compounds. In this regard, Schröder and Yang reported the adsorption and catalytic transformation of NO2 by using a redox-active MOF,125 termed MFM-300(V) [V2(OH)2(C16H6O8)]. This system not only exhibits high adsorption capacity for NO2 (13 mmol g−1 at 298 K and 1.0 bar), but it also allows for the catalytic reduction of NO2 into NO. The structural analysis reveals that upon gas sorption, NO2 molecules get primarily anchored to the pore walls through a hydrogen-bonding interaction with the bridging hydroxyl groups of the inorganic nodes. Further stabilization is reached by forming of 8-fold supramolecular interactions with the aromatic ligand (Fig. 25). Then, those interactions give rise to the host–guest charge transfer process and the formation of NO (through the oxidation of the metal centre from V3+ to V4+), and water, which is produced by the deprotonation of the bridging –OH groups anchored to the SBU.
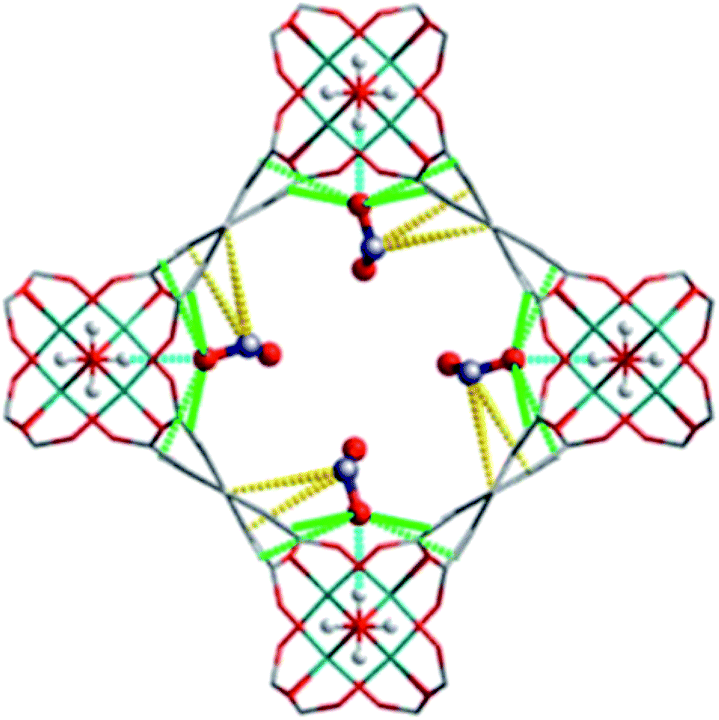 | ||
| Fig. 25 Rapid scan RS model showing the packing of guest molecules within MFM-300(V3+). Reprinted with permission from ref. 125. Copyright (2020) American Chemical Society. | ||
Recent research conducted by Dincă and co-workers highlights the capacity of M-MFU-4l (M = Cu, Zn) MOFs for NO capture and on-demand desorption of guest molecules by a thermal treatment.126 In this study, the authors demonstrated that the gas-sorption properties of the host material can be easily adjusted by modifying the cation identity and the oxidation state of the inorganic building block. To this aim, three different systems were selected as host materials (i) Zn5Cl4(BTDD)3; (ii) Zn3CuII2Cl4(BTDD)3; and (iii) Zn3CuI2Cl2(BTDD)3 (H2BTDD = bis(1H-1,2,3-triazolate[4,5-b;4′,5′-i])dibenzo[1,4] dioxin). The first material Zn5Cl4(BTDD)3 (Zn-MFU-4l) displayed the lowest capacity for NO sorption (0.51 mmol g−1 at 750 torr). The second system, Zn3CuII2Cl4(BTDD)3 (CuII-MFU-4l), was obtained by substituting two Zn2+ atoms from Zn-MFU-4l by Cu2+ ions. This subtle modification resulted in a significant increase in the NO sorption capacity (1.43 mmol g−1 at 750 torr).
These findings were explained in terms of the differences in the tetrahedral coordination environment around the metal centre N3M–Cl (M = Zn2+, Cu2+). In the first case, the saturated coordination environment around Zn2+ prevents the formation of stronger metal–NO interactions, whereas in the second system, the N3Cu–Cl geometry is sufficiently distorted to allow the NO molecules to approach closer to the metal centre leading to stronger Cu–NO interactions. The formation of Cu2+–nitrosyl species upon NO adsorption was corroborated by diffuse-reflectance infrared Fourier-transform spectroscopy (DRIFT) study. Finally, the third system (Cu+-MFU-4l) was obtained by the reduction of Cu2+ to Cu+ accompanied by the concomitant loss of chloride. This system displays a significant improvement in the low-pressure NO uptake (1.24 mmol g−1 below 1.4 torr). This value is three times the amount of NO adsorbed by Cu2+-MFU-4l (0.0008 mmol g−1) and Zn2+-MFU-4l (0.0002 mmol g−1) under the same conditions. The DRIFT analysis suggests that the metal centre in Cu1+-MFU-4l allows for the formation of Cu+–NO complex even when the material is exposed to low concentrations of NO in Ar (10 ppm). This interaction favours the NO disproportionation 3 NO → NO2 + N2O and the oxidation of the metal centre to Cu2+ which strongly binds to the NO2 product (Fig. 26). Then, the Cu2+–nitrite bond can be cleaved by exposing Cu2+-MFU-4l–(NO2) to high temperatures, releasing the anchored NO2 and thereby regenerating the adsorbent Cu+-MFU-4l. According to the authors, this catalytic system represents a potentially attractive scheme for cold-start NO capture. The design of porous materials for storage and controlled release of NO have gained relevance in biomedicine, as it has been shown that the exposure to controlled doses of NO induces wound healing and prevents the formation of blood clots. Moreover, NO is a potent antimicrobial agent; therefore, the development of materials for the local release of NO could be highly advantageous to reduce the risk of infections.
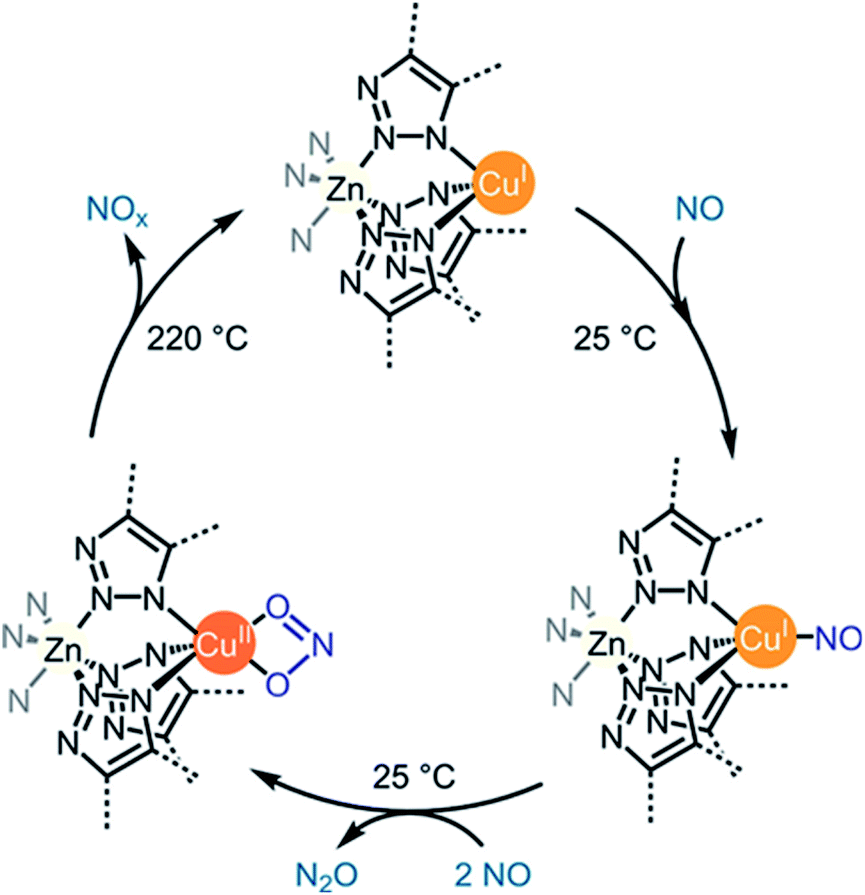 | ||
| Fig. 26 Proposed NO Disproportionation Cycle Using CuI-MFU-4l. During the thermal decomposition of Cu-MFU-4l(NO2), NO is the major NOx species detected. Reprinted with permission from ref. 126. Copyright (2021) American Chemical Society. | ||
One of the first reports about the use of MOFs for the on-demand delivery of NO was published in 2007 by Morris and co-workers.127 In this work, the authors used HKUST-1 as a reservoir for NO. The gravimetric adsorption experiments performed at 196 K and 298 K display significant isotherm hysteresis with an adsorption capacity of ca. 9 mmol g−1 and 3 mmol g−1 at one bar, respectively. Upon gas desorption processes, both isotherms exhibit a remaining amount of NO trapped within the pore network (2 mmol g−1). The IR analysis of NO-loaded material reveals that the presence of open copper sites in the walls of HKUST-1 framework allows for the formation of NO⋯Cu2+ coordination adduct; thereby leading to the irreversible adsorption of NO. However, NO molecules trapped within the pore network can be released on demand upon exposure of the NO-loaded HKUST-1 to water vapours, as H2O acts as a nucleophile replacing the NO molecules coordinated to the open copper sites.
More recently, the same research group reported the development of MOF-based composites for the controlled delivery of NO.128 The selected host material was CPO-27-Ni, as this MOF affords highly efficient adsorption, storage, and release cyclable profile for NO.129 Moreover, this material exhibits high stability towards adsorbed water, which allows for the controlled release of NO when exposed to moisture, without altering the structural arrangement of the host material.
The optimal MOF films were obtained by varying the amount of CPO-27-Ni (wt%) integrated within a polyurethane matrix. The composite films exhibited homogeneous distribution of the MOF within the polymer matrix. The release of NO was triggered by exposing the composite films to 11% or relative humidity. Such conditions allow for the controlled release of the NO adsorbed without affecting the polymer properties. Finally, the antimicrobial properties of the resultant MOF-based composite demonstrated that the controlled release of biologically active levels of NO provides a bactericidal effect against Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus).
6. Selectivity of trace gases
One important aspect of evaluating MOF materials is their selectivity of adsorbing one gas preferentially over other molecules that are present in the industrial flue gas streams, as these are mainly composed of CO2 (10–15% [v]), N2 (70–75% [v]), and H2O (∼10–18% [v]),130 whereas gases such as SO2 (0.00325% [v], 500–3000 ppm), H2S (13.5% [v]; 1000–4000 ppm), NH3 (300–800 ppm) and NOx (200–800 ppm) are present in trace concentrations.131 For this reason, it is highly desirable that the study of the capture of trace gases by MOFs should be accompanied by a selectivity study in the presence of other gases in order to have a complete profile of their potential applications at industrial levels.Table 4 lists the most promising MOF materials for the separation of SO2 and H2S gases, with a good selectivity performance.
| Gas target | MOF | Gas adsorption [mmol g−1] | Flue gas concentration | Selectivity | Ref. | |||
|---|---|---|---|---|---|---|---|---|
| CO2 | N2 | CH4 | Other gases | |||||
| a Breakthrough experimental result. b Obtained from ratio of slopes of initial adsorption isotherm plot at a low pressure region. c GCMC simulations at 303 K and 10 bar. d Membrane composite measured at 35 °C. e Composite materials. f Pellets. g GCMC simulations at 273 K. | ||||||||
| SO2 | MIL-160 | 7.2 | SO2/CO2 (10:90–50:50 v/v) |
124–128 (IAST) | 31.2537e | — | — | 18 |
| MIL-125(Ti)-NH2 | 10.8 | SO2/CO2 (10:90–50:50 v/v) |
42–55 (IAST) | — | — | — | 18 | |
| MFM-170 | 17.5 | Equimolar SO2/N2 | 35 | 944 | 260 | 203 (CO) | 19 | |
| MFM-300(Al) | 7.1 | 50–350 mbarb | — | 6522 | 3620 | 105 (H2) | 37a | |
| 3105(CO) | ||||||||
| 4974 (O2) | ||||||||
| MFM-300(In) | 8.28 | At 50:50 mixture of each gas |
60 | 5000 | 425 | — | 37b | |
| MFM-601 | 12.3 | SO2/CO2, SO2/N2 (50:50 to 10:90) |
32 | 255 | — | — | 132 | |
| ELM-12 | 2.73 | 10:90 mixture at 298 K and 1 bar |
30 | 4064 | 871 | — | 133 | |
| 1@Ba(OH)2 | 4.0a | N2/CO2/SO2 (83.5:14:2.5 v/v) |
990 | — | — | — | 45 | |
| SIFSIX-1-Cu | 11.01 | At 10:90 mixture of each gas |
70.7 | 3145.7 | 1241.4 | — | 51 | |
| SIFSIX-2-Cu-i | 6.9 | At 10:90 mixture of each gas |
87.1 | 3103.2 | 1017.1 | — | 51 | |
| SIFSIX-3-Zn | 2.1 | At 10:90 mixture of each gas |
— | 506.7 | 276 | — | 51 | |
| SIFSIX-3-Ni | 2.74 | At 10:90 mixture of each gas |
— | 701.8 | 371.6 | — | 51 | |
| KAUST-7 | 1.4a | SO2/CO2/N2 (4%:4%:balance) and (500 ppm:10%:balance) |
1 | — | — | — | 52 | |
| KAUST-8 | 1.6a | SO2/CO2/N2: 0.05/10/89.95 | 66 | — | — | — | 52 | |
| ECUT-77 | 8.0 | SO2/CO2 (1:99 v/v; 2000 ppm SO2) |
44–36 | — | — | — | 134 | |
| ECUT-100 | 4.95 | At 1:99 mixture of each gas |
27.5–26.9 | — | 2302 | — | 135 | |
| ECUT-111 | 11.56 | SO2/CO2, SO2/N2 (1:99 v/v) |
22.2–25.2 | 860.9 | — | — | 136 | |
| H2S | MIL-101(Cr) | 0.419a | H2S/CO2/He (1:10:89 v/v) |
5 | — | — | — | 63 |
| HKUST-1 | ∼1a | H2S/CO2/He (1:10:89 v/v) |
∼40 | — | — | — | 63 | |
| KAUST-7 | — | H2S/CO2/CH4 (20:20:60) |
— | — | 20.7d | — | 139 | |
| KAUST-8 | — | H2S/CO2/CH4 (20:20:60) |
— | — | 18.6d | — | 139 | |
| KAUST-8 | — | H2S/CO2/CH4 (5:5:90) |
1 | — | — | — | 137 | |
| SIFSIX-2-Ni-i | — | H2S/CO2/CH4 (5:5:90) |
3.3 | — | — | — | 137 | |
| MIL-125(Ti)-NH2 | — | H2S 0.001 | — | — | 70c | — | 138 | |
| Ga-soc-MOF-1a | 4.4 | CO2/H2S/CH4 (5%:5%:90%) |
7 | — | — | — | 64 | |
| Y-fum-fcu-MOF | 1.1a | CO2/H2S/CH4 (5%:5%:90%) |
6.4 | — | — | — | 139 | |
| [BMIM][Cl]/Cu-TDPATe | — | H2S 1000 ppm | — | — | 1302 | — | 140 | |
| Cu-TDPATe | — | H2S 1000 ppm | — | — | 141 | — | 140 | |
| MIL-53(Al)f | — | CH4/CO2/H2S (60%:39.9%:0.1%) |
— | — | 23–34 | — | 141 | |
| MIL-47(V)-Brg | — | H2S/CH4 (5:95) 0.1 MPa |
— | 278 | 68 | — | 142 | |
| MIL-47(V)g | — | H2S/CH4 (5:95) 0.1 MPa |
— | 179 | 223 | — | 142 | |
| H2S/CH4 (1:99) 0.1 MPa |
51 | |||||||
| H2S/N2 (50:50) |
||||||||
7. Stability and reusability of MOFs
As previously shown, there is a wide variety of MOFs with high structural stability towards corrosive gases (SO2, H2S, NH3 and NOx). The stability, as well as the reusability of the MOFs, is required for tangible industrial applications on the adsorption field. Considering that the CSD currently contains near to 70000 MOF structures,143 a selection criterion for MOFs that could be potentially useful in the adsorption of toxic gases is essential, as are the guidelines for generating new materials focused on this target.144 Although the nature of MOFs is very diverse, MOFs stable in the presence of corrosive gases feature some common characteristics: (i) the strength of the ligand–metal bond; (ii) oxidation state of the metal centre; (iii) robustness of the metallic cluster; and (iv) the formation of supramolecular host–guest interactions. The presence of at least one of these characteristics is responsible for the stabilization of the framework under such harsh conditions. For example, when the strongest binding site corresponds to a free metal site, the metal–ligand bond should be strong enough to avoid linker displacement and subsequent structure degradation. The combination of ligands with strong donor groups/atoms and late transition metals, or the use of metals with a higher oxidation state together with carboxylate ligands are good synthetic strategies to improve the strength of the metal–ligand bonds and, therefore, increase the overall stability of the framework.21,40a,145 For example, MOFs formed by triazolate ligands such as the M2Cl2BTDD family (M = Ni2+, Co2+, Mn2+, and Cu2+)107 with uncoordinated metal sites are stable even after several sorption and desorption cycles of NH3. Such stability increases according to the Irving–Williams series, being the Ni2+ framework more stable than the Cu2+. In this case, the presence of heterolytic metal–ligand bonds and a less labile metal centre, enhances the MOF's stability. In MFM-170,19 a MOF formed by the Cu2+ paddlewheel SBU, the coordination of one of the Cu2+ centres to a N-donating ligand, allowed reversible SO2 adsorption, even if one of the Cu2+ atoms presented weak contacts with SO2. Additionally, the interactions between SO2 and the surface of the framework contributed to the reversibility of the adsorption process. This research represents an example of enhancing the chemical stability of a MOF by increasing the metal–ligand bond strength, a highly effective strategy to increase the stability of MOFs with labile metals.
On the other hand, the use of small metal cations confers stability against chemical degradation, as shown for several MOFs constructed with trivalent and tetravalent cations such as Al3+, In3+, Sc3+, and Cr3+ and Zr4+, V4+, and Ti4+, respectively. These metal centres tend to form robust clusters, increasing the stability of the framework towards corrosive gases. For example, MIL-101(Cr) and MIL-101(Cr)-4F(1%) formed by [Cr3(μ3-O)(O2CR)6] clusters, are stable to SO2 and H2S;20,34 Mg-CUK-1 with [Mg3(μ3-OH)]5+ clusters is stable to H2S;68 MIL-53(Al)-TDC formed by trans-corner-sharing [Al(μ2-OH)(p-BDC)] polyhedra is stable to H2S.69 Additionally, the MFM-300 family formed by infinite chains of [M2(μ2-OH)2(C16O8H6)] (M = Al3+, In3+, Sc3+, V3+), has demonstrated to be stable to SO2, H2S, NH3, and NO2.37,70,110,111,125 This family represents an excellent example of how the non-covalent interactions formed between the framework surface and the host molecules provide to the additional structural stability and adsorption reversibility in almost all the studied cases. The principal interaction involves the μ2-OH motif as hydrogen bond donor and the gas molecule as an acceptor, followed by guest–ligand and guest–guest interactions. This last aspect has a direct impact on the recyclability of the MOF, where most of the materials with good cyclability exhibit similar host–guest interactions such as hydrogen bonds (strong and weak), electrostatic interactions inside the pore framework, and metal–guest interactions. For example: MFM-170,19 MIL-101(Cr)-4F(1%),20 are stable even after 50 SO2 adsorption–desorption cycles while the following MOFs are stable for different numbers of SO2 cycles [Ni8(OH)4(H2O)2(BDP_X)6] (10 cycles),45 SIFSIX-1-Cu (4–6 cycles),51 and MFM-300(Sc) (10 cycles).37c Furthermore, Mg-CUK-1,68 MIL-53(Al)-TDC,69 and MFM-300(Sc)70 are stable for at least five adsorption–desorption cycles of H2S while Mg2(dobpdc),109 Co2Cl2 BBTA and Co2Cl2 BTDD,108 MFM-300(Al) (50 cycles),111 and UiO-67.112 Finally, MFM-300(Al) is not affected even by five cycles of NO2.124
8. Conclusions and future perspectives
Air pollution due to the emissions of toxic gases is the single largest environmental health risk around the world since it is responsible for one in eight premature global deaths, reduction of the biodiversity, crop damages and acidification of soils and waters. Thus, the development of efficient technologies for the capture of toxic gases (i.e., NOx, SO2, NH3 and H2S) from static and mobile sources is essential, in order to achieve a cleaner environment. MOFs are amongst the most promising candidates for the capture of these toxic gases since their sorption selectivity is directly tuneable as a function of the topology and chemical composition of the pores, which can optimise the interactions between MOFs and these toxic molecules, affording enhanced gas adsorption properties. Although the main drawback of MOFs is their vulnerability to these highly corrosive gases which can compromise their chemical stability, remarkable examples have demonstrated high chemical stability to NOx, SO2, NH3 and H2S.Chemically stable MOFs for the capture of SO2 have provided promising advances in the field and the understanding of the role of different chemical functionalities, is the key for accomplishing superior SO2 captures. Coordinatively unsaturated metal sites (open metal sites) incorporated into chemical stable MOFs (e.g., MFM-170 and MIL-101(Cr)-4F(1%)) demonstrated the relevance of the interaction of these open metal sites and SO2 molecules. The design of new functionalised MOF materials with coordinatively unsaturated metal sites signifies an interesting alternative to obtain superior SO2 captures and therefore, the challenge is to be able to also incorporate chemical stability to these MOFs. Thus, high SO2 uptakes are related to large BET surface area, pore size and the open metal sites incorporation into MOFs, while structural (chemical) stability is associated with the type of metal, and the interactions adsorbate–adsorbent inside the pore.
Hydroxo-functionalisation (μ-OH groups) in structural stable MOFs (e.g., MFM-300(M)) have shown excellent capabilities for the capture and detection of SO2 with high chemical stability and excellent cyclability involving a remarkably facile regeneration. The key for all of such outstanding properties is the preferential interaction between the functional group (μ-OH) and SO2, in which the μ-OH groups binds SO2 molecules through the formation of OSO(δ−)⋯H(δ+)–O hydrogen bonds, reinforced by weak supramolecular interactions with C–H atoms from the aromatic rings of the framework. The chemical stability of the MFM-300(M) family relies on the strength of the coordination bonds between the oxygen atoms form the carboxylic ligand (biphenyl-3,3′,5,5′-tetracarboxylic acid) and the M3+ metal centres.
Defective sites a.k.a., crystal irregularities, composition inhomogeneities or defects in MOFs have exhibited increased SO2 adsorption capacities, energies, and SO2/CO2 selectivity. For example, in nickel pyrazolate MOFs it was demonstrated that the pre-synthetic introduction of amino and hydroxyl functional groups on the organic linkers and the post-synthetic modifications, synergistically increase the SO2 capture capacity of these MOF materials.
Halogen functionalised MOF materials (e.g., hexafluorosilicate (SiF62−) incorporation in SIFSIX MOFs) showed highly efficient removal of SO2 from other gases, at a very low SO2 concentrations, and excellent SO2/CO2 and SO2/N2 selectivities. These properties were attributed to the strong electrostatic interactions between the SO2 molecules and the SiF62− anions (Sδ+⋯Fδ−), assisted by dipole–dipole interactions with the ligand (Oδ−⋯Hδ+).
The identification of MOFs capable of capturing H2S under industrially practical pressure-swing desorption conditions, symbolises a very promising application which is based on the chemical stability of these materials. For example, stable MOF materials constructed with μ-OH functional groups (e.g., MIL-53(Al)-TDC) exhibited high and fully reversible H2S capture where the formation of hydrogen bonds between H2S and the μ-OH functional group (a relatively weak interaction) facilitates the H2S cyclability.
The chemical transformation of H2S within the pores of MOFs to produce, in situ, polysulphides is a new and exciting discovery in MFM-300(M) and SU-101 MOF materials. In addition to the promising application as novel MOF-lithium/sulphur batteries, the chemical investigation behind them, provided addition information on the electrochemical potential of MOFs which could guide new interesting properties to be discovered. Then, although the polysulphide formation was explained in terms of a “disproportionation” type reaction (protons of H2S play the role of an oxidant and the sulphide plays the role of a reducing agent), other hypotheses need to be explored. What if the polysulphide formation is a consequence of reversible metal–ligand bonding upon the adsorption of H2S? We anticipate new perspectives will be shortly investigated.
Paradoxically, the role of H2S as an endogenous biological mediator in the human body is crucial. For example, H2S executes a vital activity in the regulation of blood pressure, neurotransmission, anti-inflammatory mechanism, anti-oxidation, angiogenesis and apoptosis. Thus, the delivery and detection of H2S levels in living cells and organisms is an outstanding application for MOF materials. For example, Ni-CPO-27 showed a promising release of H2S (under physiological conditions) with a short induction period of approximately 5 min. In the field of H2S detection, for example, a post-synthetic modification of ZIF-90 with malononitrile (NC–CH2–CN), exhibited a specific reaction with H2S completing an enhancement of photoluminescence, which constitutes the base for the detection of H2S. Thus, the development of this research is expanding by the fundamental principle of introducing a reactive site for H2S within MOF material (monitored by fluorescence), which can work under physiological pH, to detect exogenous H2S in living cells, while the MOF-probe shows low toxicity and high biocompatibility.
Although most MOF materials have exhibited poor chemical stability towards NH3, chemically stable MOFs constructed with different chemical functionalities have shown interesting NH3 capture performances.
Coordinatively unsaturated metal sites incorporated, for example, in bisbenzenetriazolate and triazolate MOFs, showed high and reversible NH3 uptakes with strong interactions between the open metal sites and NH3. The chemical stability of these MOF materials was attributed to the strength of the N atom coordinated to divalent metal centres. Mg2(dobpdc) demonstrated high NH3 capture and chemical stability to not only dry NH3, but also to wet NH3. Interestingly, it was experimentally and computationally demonstrated that NH3 was preferentially adsorbed, by coordination bonds, to the coordinatively unsaturated metal sites of Mg2(dobpdc). The exceptional chemical stability of this Mg2+-based MOF material credited to the higher affinity of Mg2+ to oxygen atoms than nitrogen atoms, as confirmed by van der Waals (vdW)-corrected density functional theory (DFT) calculations.
The μ-OH functionalised MFM-300(M) family demonstrated remarkable NH3 adsorption properties, high chemical stability, high uptakes and attractive cyclabilities. Not surprisingly, as in the case of SO2 and H2S, the preferential adsorption sites of the MFM-300(M) materials were found at the hydroxo functional group. For example, in the case of MFM-300(Al) the adsorption of NH3 showed an atypical adsorption mechanism where adsorbent and adsorbate experienced a rapid site-exchange via reversible formation and cleavage of O–H and O–H(from NH3) chemical bonds, in other words a pseudo-chemisorption binding mechanism.
As previously described in the SO2 capture section, crystal irregularities can provide very particular physical and chemical properties. Then, defects in MOF materials also demonstrated remarkable NH3 adsorption properties. For example, UiO-67 and UiO-bpydc containing 4,4′-biphenyl dicarboxylate and 4,4′-(2,2′-bipyridine) dicarboxylate ligands, despite their structural similarities demonstrated a drastic difference in the NH3 adsorption properties when the biphenyl groups in the organic ligand were replaced by the bipyridine moieties. Such replacement can confer flexibility to the framework in the context of mainly ligand “flipping” but without significant pore volume alteration.
Current investigations in NOx capture have pointed out that chemical stable MOFs represent an attractive alternative for storage, selective separation, and/or catalytic transformation of NO2. For example, the synergistic effect between the organic ligand and the inorganic building block in MFM-300(Al) showed the stabilisation of highly reactive NO2 species within the micropores. The optimal uptake and selectivity of MFM-300(Al) for NO2 was attributed to the existence of both host–guest and guest–guest interactions. The former involves mainly the hydrogen bonding interaction between NOx and the hydroxo functional group, while the latter refers to the supramolecular interactions between the guest molecules in their monomeric (NO2) and dimeric (N2O4) form.
Interestingly, the design of MOF materials for storage and controlled release of NO have gained relevance in biomedicine, as it has been shown that the exposure to controlled doses of NO induces wound healing and prevents the formation of blood clots. For example, CPO-27-Ni exhibited highly efficient adsorption, storage, and release cyclable profile for NO with a high stability to water, allowing a controlled release of NO when exposed to moisture.
Despite the significant progress in the capture of NOx, SO2, NH3 and H2S pollutants by MOF-based technology, further research in this field is required to overcome some of the main challenges: chemical stability, reusability, and suitable functionalisation. Considering the large number of MOF materials synthesised to date (over 70000 structures reported in the CCD), it is crucial to understand what makes the above-reported MOFs so chemically stable. Thus, this perspective summarises their most important characteristics (metal centre, strength of the metal–ligand bond and functional groups) that should be further investigated in order to be able to design tailor-made MOFs for the capture of corrosive gases. Additionally, an important aspect to consider is the study of breakthrough experiments and selectivity calculations, where more realistic conditions for industrial applications are investigated, providing a direct performance comparison of MOFs to classic materials such as activated carbon, zeolites and metal oxides. On the other hand, the synthesis scalability of MOFs is one of the weak points to contemplate for corrosive gas adsorption applications, as only a few materials can be synthesised on an industrial scale. We desire that this perspective can provide useful information on the significant progress of this field and inspire new investigations to be carried out.
Author contributions
E. M.-A., M. L. D.-R., and M. J. V.-H. conducted the literature research and drafted the manuscript. V. J. and I. A. I. conceived the topic and structure of the article. Outlined, drafted and supervised the completion of the manuscript. All authors reviewed and contributed to this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank PAPIIT-UNAM (Grant IN202820), Mexico for financial support. E. M.-A. thanks to CONACYT for the PhD fellowship (770954). M. L. D.-R. thanks Mississippi State University for the financial support. Thanks to U. Winnberg (Pharma View Consulting SC) for scientific discussions and G. Ibarra–Winnberg for conceptualising the design of this contribution.Notes and references
- R. A. Silva, J. J. West, J.-F. Lamarque, D. T. Shindell, W. J. Collins, G. Faluvegi, G. A. Folberth, L. W. Horowitz, T. Nagashima, V. Naik, S. T. Rumbold, K. Sudo, T. Takemura, D. Bergmann, P. Cameron-Smith, R. M. Doherty, B. Josse, I. A. MacKenzie, D. S. Stevenson and G. Zeng, Nat. Clim. Change, 2017, 7, 647–651 CrossRef PubMed.
- World Health Organization, https://www.who.int/en/news-room/fact-sheets/detail/ambient-(outdoor)-air-quality-and-health, accessed December 2020.
- K. Balakrishnan, M. Brauer, N. Bruce, F. Forastiere, N. C. Gouveia, R. M. Harrison, F. J. Kelly, P. L. Kinney, M. Lippmann, S. Mehta, B. Ostro, A. Peters, P. H. N. Saldiva, J. M. Samet , M. J. Utell, M. Zuk, World Health Organization, Air quality guidelines. Global update 2005. Particulate matter, ozone, nitrogen dioxide and sulfur dioxide, WHO Regional Office for Europe, Copenhagen, 2006 Search PubMed.
- G. Shaddick, M. L. Thomas, P. Mudu, G. Ruggeri and S. Gumy, npj Clim. Atmos. Sci., 2020, 3, 1–5 CrossRef.
- J. Lelieveld, J. S. Evans, M. Fnais, D. Giannadaki and A. Pozzer, Nature, 2015, 525, 367–371 CrossRef CAS PubMed.
- B. Chu, X. Zhang, Y. Liu, H. He, Y. Sun, J. Jiang, J. Li and J. Hao, Atmos. Chem. Phys., 2016, 16, 14219–14230 CrossRef CAS.
- (a) Y. Wang, P. K. Hopke, D. C. Chalupa and M. J. Utell, Aerosol Sci. Technol., 45, 1245–1249 Search PubMed; (b) R. Figueiredo, P. Nunes, M. Meireles, M. Madaleno and M. C. Brito, J. Cleaner Prod., 2019, 222, 129–142 CrossRef; (c) X. Kan, F. Hedenus and L. Reichenberg, Energy, 2020, 195, 117434 CrossRef.
- (a) M. Sharifzadeh, G. Triulzi and C. L. Magee, Energy Environ. Sci., 2019, 12, 2789–2805 RSC; (b) J. Guo, W. Jiao, G. Qi, Z. Yuan and Y. Liu, Chin. J. Chem. Eng., 2019, 27, 1361–1373 CrossRef CAS.
- (a) E. Martínez-Ahumada, A. López-Olvera, V. Jancik, J. E. Sánchez-Bautista, E. González-Zamora, V. Martis, D. R. Williams and I. A. Ibarra, Organometallics, 2020, 39, 883–915 CrossRef; (b) T. Islamoglu, Z. Chen, M. C. Wasson, C. T. Buru, K. O. Kirlikovali, U. Afrin, M. R. Mian and O. K. Farha, Chem. Rev., 2020, 120(16), 8130–8160 CrossRef CAS PubMed.
- (a) K. Dedecker, E. Dumas, B. Lavédrine, N. Steunou and C. Serre, in Metal-Organic Frameworks (MOFs) for Environmental Applications, ed. S. K. Ghosh, Elsevier, Amsterdam, 2019, ch. 5, pp. 141–178 Search PubMed; (b) A. J. Rieth, A. M. Wright and M. Dincă, Nat. Rev. Mater., 2019, 4, 708–725 CrossRef CAS.
- F. C. Men and H. M. Seip, Environ. Sci. Policy, 2004, 7, 253–265 CrossRef.
- (a) M. R. Taylor, E. S. Rubin and D. A. Hounshell, Technol. Forecast. Soc. Change, 2005, 72, 697–718 CrossRef; (b) S. Reis, P. Grennfelt, Z. Klimont, M. Amann, H. ApSimon, J.-P. Hettelingh, M. Holland, A.-C. LeGall, R. Mass, M. Posch, T. Spranger, M. A. Sutton and M. Williams, Science, 2012, 338, 1153–1154 CrossRef CAS PubMed.
- T. Larssen, E. Lydersen, D. Tang, Y. He, J. Gao, H. Liu, L. Dual, H. M. Seip, R. D. Vogt, J. Mulder, M. Shao, Y. Wang, H. Shang, X. Zhang, S. Solberg, W. Aas, T. Okland, O. Eilertsen, V. Angell, Q. Li, D. Zhao, R. Xiang, J. Xiao and J. Luo, Environ. Sci. Technol., 2006, 40, 418–425 CrossRef CAS PubMed.
- R. K. Srivastava, W. Jozewicz and C. Singer, Environ. Prog., 2001, 20, 219–228 CrossRef CAS.
- (a) F. Rezaei, A. A. Rownaghi, S. Monjezi, R. P. Lively and C. W. Jones, Energy Fuels, 2015, 29(9), 5467–5486 CrossRef CAS; (b) Y. Sun, E. Zwolińska and A. G. Chmielewski, Crit. Rev. Environ. Sci. Technol., 2016, 46, 119–142 CrossRef CAS.
- X. Han, S. Yang and M. Schröder, Nat. Rev. Chem., 2019, 3, 108–118 CrossRef CAS.
- (a) W. P. Mounfield III, C. Han, S. H. Pang, U. Tumuluri, Y. Jiao, S. Bhattacharyya, M. R. Dutzer, S. Nair, Z. Wu, R. P. Lively, D. S. Sholl and K. S. Walton, J. Phys. Chem. C, 2016, 120(48), 27230–27240 CrossRef; (b) S. Bhattacharyya, S. H. Pang, M. R. Dutzer, R. P. Lively, K. S. Walton, D. S. Sholl and S. Nair, J. Phys. Chem. C, 2016, 120(48), 27221–27229 CrossRef CAS; (c) S. Glomb, D. Woschko, G. Makhloufi and C. Janiak, ACS Appl. Mater. Interfaces, 2017, 9, 37419–37434 CrossRef CAS PubMed.
- P. Brandt, A. Nuhnen, M. Lange, J. Möllmer, O. Weingart and C. Janiak, ACS Appl. Mater. Interfaces, 2019, 11, 17350–17358 CrossRef CAS PubMed.
- G. L. Smith, J. E. Eyley, X. Han, X. Zhang, J. Li, N. M. Jacques, H. G. W. Godfrey, S. P. Argent, L. J. M. McPherson, S. J. Teat, Y. Cheng, M. D. Frogley, G. Cinque, S. J. Day, C. C. Tang, T. L. Easun, S. Rudić, A. J. Ramirez-Cuesta, S. Yang and M. Schröder, Nat. Mater., 2019, 18, 1358–1365 CrossRef CAS PubMed.
- E. Martínez-Ahumada, M. L. Díaz-Ramírez, H. A. Lara-García, D. R. Williams, V. Martis, V. Jancik, E. Lima and I. A. Ibarra, J. Mater. Chem. A, 2020, 8, 11515–11520 RSC.
- A. J. Rieth, A. M. Wright and M. Dincă, Nat. Rev. Mater., 2019, 4, 708–725 CrossRef CAS.
- S. Luo, J. Liu and Z. Wu, J. Phys. Chem. C, 2019, 123, 11772–11780 CrossRef CAS.
- S. Chaturvedi, J. A. Rodríguez, T. Jirsak and J. Hrbek, J. Phys. Chem. B, 1998, 102(36), 7033–7043 CrossRef CAS.
- (a) D. Britt, D. Tranchemontagne and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11623–11627 CrossRef CAS PubMed; (b) T. G. Glover, G. W. Peterson, B. J. Schindler, D. Britt and O. Yaghi, Chem. Eng. Sci., 2011, 66, 163–170 CrossRef; (c) K. Tan, S. Zuluaga, H. Wang, P. Canepa, K. Soliman, J. Cure, J. Li, T. Thonhauser and Y. J. Chabal, Chem. Mater., 2017, 29, 4227–4235 CrossRef CAS.
- A. C. Elder, S. Bhattacharyya, S. Nair and T. M. Orlando, J. Phys. Chem. C, 2018, 122, 10413–10422 CrossRef CAS.
- K. Tan, P. Canepa, Q. Gong, J. Liu, D. H. Johnson, A. Dyevoich, P. K. Thallapally, T. Thonhauser, J. Li and Y. J. Chabal, Chem. Mater., 2013, 25, 4653–4662 CrossRef CAS.
- J. Hungerford, S. Bhattacharyya, U. Tumuluri, S. Nair, Z. Wu and K. S. Walton, J. Phys. Chem. C, 2018, 122, 23493–23500 CrossRef CAS.
- M. Eigen, Pure Appl. Chem., 1963, 6, 97–115 CAS.
- C. Petit, B. Levasseur, B. Mendoza and T. J. Bandosz, Microporous Mesoporous Mater., 2012, 154, 107–112 CrossRef CAS.
- C. Petit, B. Mendoza and T. J. Bandosz, ChemPhysChem, 2010, 11(17), 3678–3684 CrossRef CAS PubMed.
- H. Irving and J. P. Williams, J. Chem. Soc., 1953, 3192–3210 RSC.
- L. Feng, K.-Y. Wang, G. S. Day, M. R. Ryder and H.-C. Zhou, Chem. Rev., 2020, 120(23), 13087–13133 CrossRef CAS PubMed.
- L. Hamon, C. Serre, T. Devic, T. Loiseau, F. Millange, G. Férey and G. De Weireld, J. Am. Chem. Soc., 2009, 131, 8775–8777 CrossRef CAS PubMed.
- M. L. Díaz-Ramírez, E. Sanchez-González, J. R. Álvarez, G. A. González-Martínez, S. Horike, K. Kadota, K. Sumida, E. González-Zamora, M. A. Springuel-Huet, A. Gutiérrez-Alejandre, V. Jancik, S. Furukawa, S. Kitagawa, I. A. Ibarra and E. Lima, J. Mater. Chem. A, 2019, 7, 15101–15112 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85(22), 3533–3539 CrossRef CAS.
- M. Bosch, M. Zhang and H.-C. Zhou, Adv. Chem., 2014, 2014, 1155 Search PubMed.
- (a) S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. F. David, D. P. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schröder, Nat. Chem., 2012, 4, 887–894 CrossRef CAS PubMed; (b) M. Savage, Y. Cheng, T. L. Easun, J. E. Eyley, S. P. Argent, M. R. Warren, W. Lewis, C. Murray, C. C. Tang, M. D. Frogley, G. Cinque, J. Sun, S. Rudić, R. T. Murden, M. J. Benham, A. N. Fitch, A. J. Blake, A. J. Ramirez-Cuesta, S. Yang and M. Schröder, Adv. Mater., 2016, 28, 8705–8711 CrossRef CAS PubMed; (c) J. A. Zárate, E. Sánchez-González, D. R. Williams, E. González-Zamora, V. Martis, A. Martínez, J. Balmaseda, G. Maurin and I. A. Ibarra, J. Mater. Chem. A, 2019, 7, 15580–15584 RSC; (d) C. P. Krap, R. Newby, A. Dhakshinamoorthy, H. García, I. Cebula, T. E. Easun, M. Savage, J. E. Eyley, S. Gao, A. J. Blake, W. Lewis, P. H. Beton, M. R. Warren, D. R. Allan, M. D. Frogley, C. C. Tang, G. Cinque, S. Yang and M. Schröder, Inorg. Chem., 2016, 55, 1076 CrossRef CAS PubMed; (e) J.-X. Liu, J. Li, W.-Q. Tao and Z. Li, Fluid Phase Equilib., 2021, 536, 112963 CrossRef CAS.
- I. A. Ibarra, S. Yang, X. Lin, A. J. Blake, P. J. Rizkallah, H. Nowell, D. R. Allan, N. R. Champness, P. Hubberstey and M. Schröder, Chem. Commun., 2011, 47, 8304–8306 RSC.
- V. Chernikova, O. Yassine, O. Shekhah, M. Eddaoudi and K. N. Salama, J. Mater. Chem. A, 2018, 6, 5550–5554 RSC.
- (a) R. D. Shannon, Acta Crystallogr. A, 1976, 32, 751–767 CrossRef; (b) L.-H. Schilling, H. Reinsch and N. Stock, in The Chemistry of Metal-Organic Frameworks. Synthesis, Characterization, and Applications, ed. S. Kaskel, WILEY-VCH Verlag GmbH & Co. KGaA, Germany, 2016, vol. 1, ch. 5, pp. 106–108 Search PubMed; (c) M. G. Goesten, C. F. Guerra, F. Kapteijn, J. Gascon and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2015, 54, 12034–12038 CrossRef CAS PubMed.
- S. Kancharlapalli, S. Natarajan and T. K. Ghanty, J. Phys. Chem. C, 2019, 123(45), 27531–27541 CrossRef CAS.
- (a) S. Dissegna, K. Epp, W. R. Heinz, G. Kieslich and R. A. Fischer, Adv. Mater., 2018, 30, 1704501–1717045 CrossRef PubMed; (b) W. Xiang, Y. Zhang, Y. Chen, C.-J. Liu and X. Tu, J. Mater. Chem. A, 2020, 8, 21526–21546 RSC.
- (a) O. Kozachuk, I. Luz, F. X. Llabrés i Xamena, H. Noei, M. Kauer, H. B. Albadia, E. D. Bloch, B. Marley, Y. Wang, M. Muhler and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7058–7062 CrossRef CAS PubMed; (b) J. Canivet, M. Vandichel and D. Farrusseng, Dalton Trans.,45, 4090–4099 Search PubMed; (c) J. Wang, L. Liu, C. Chen, X. Dong, Q. Wang, L. Alfilfil, M. R. AlAlouni, K. Yao, J. Huang, D. Zhang and Y. Han, J. Mater. Chem. A, 2020, 8, 4464–4472 RSC; (d) R. S. Forgan, Chem. Sci., 2020, 11, 4546–4562 RSC.
- E. López-Maya, C. Montoro, L. M. Rodríguez-Albelo, S. D. A. Cervantes, A. A. Lozano-Pérez, J. L. Cenís, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2015, 54, 6790–6794 CrossRef PubMed.
- L. M. Rodriguez-Albelo, E. Lopez-Maya, S. Hamad, A. R. Ruiz-Salvador, S. Calero and J. A. R. Navarro, Nat. Commun., 2017, 8, 14457 CrossRef CAS PubMed.
- E. López-Maya, C. Montoro, V. Colombo, E. Barea and J. A. R. Navarro, Adv. Funct. Mater., 2014, 24, 6130–6135 CrossRef.
- M. Mon, E. Tiburcio, J. Ferrando-Soria, R. G. San Millan, J. A. R. Navarro, D. Armentano and E. Pardo, Chem. Commun., 2018, 54, 9063–9066 RSC.
- (a) Y. Yuan, J. Li, X. Sun, G. Li, Y. Liu, G. Verma and S. Ma, Chem. Mater., 2019, 31, 1084–1091 CrossRef CAS; (b) J. Hou, M. L. R. Gómez, A. Krajnc, A. McCaul, S. Li, A. M. Bumstead, A. F. Sapnik, Z. Deng, R. Lin, P. A. Chater, D. S. Keeble, D. A. Keen, D. Appadoo, B. Chan, V. Chen, G. Mali and T. D. Bennett, J. Am. Chem. Soc., 2020, 142(8), 3880–3890 CrossRef CAS PubMed; (c) L.-M. Yang, G.-Y. Fang, J. Ma, R. Pushpa and E. Ganz, Phys. Chem. Chem. Phys., 2016, 18, 32319–32330 RSC; (d) M. Kalaj, M. R. Momeni, K. C. Bentz, K. S. Barcus, J. M. Palomba, F. Paesani and S. M. Cohen, Chem. Commun., 2019, 55, 3481–3484 RSC.
- A. Karmakar, A. V. Desai and S. K. Ghosh, Coord. Chem. Rev., 2016, 307, 313–341 CrossRef CAS.
- X. Suo, Y. Yu, S. Qian, L. Zhou, X. Cui and H. Xing, Angew. Chem., Int. Ed., 2021, 60, 1–7 CrossRef PubMed.
- X. Cui, Q. Yang, L. Yang, R. Krishna, Z. Zhang, Z. Bao, H. Wu, Q. Ren, W. Zhou, B. Chen and H. Xing, Adv. Mater., 2017, 29, 1606929 CrossRef PubMed.
- M. R. Tchalala, P. M. Bhatt, K. N. Chappanda, S. R. Tavares, K. Adil, Y. Belmabkhout, A. Shkurenko, A. Cadiau, N. Heymans, G. De Weireld, G. Maurin, K. N. Salama and M. Eddaoudi, Nat. Commun., 2019, 10, 1328 CrossRef CAS PubMed.
- T. M. M. Ntep, H. Breitzke, L. Schmolke, C. Schlüsener, B. Moll, S. Millan, N. Tannert, I. E. Aita, G. Buntkowsky and C. Janiak, Chem. Mater., 2019, 31, 8629–8638 CrossRef.
- W. Jaeschke, H. Claude and J. Herrmann, J. Geophys. Res., 1980, 85, 5639–5644 CrossRef CAS.
- (a) O. A. Habeeb, R. Kanthasamy, G. A. M. Ali, S. Sethupathi and R. B. M. Yunus, Rev. Chem. Eng., 2018, 34, 837–854 CrossRef CAS; (b) G. E. Likens, R. F. Wright, J. N. Galloway and T. J. Butler, Sci. Am., 1979, 241, 43–51 CrossRef CAS.
- R. O. Beauchamp, J. S. Bus, J. A. Popp, C. J. Boreiko, D. A. Andjelkovich and P. Leber, CRC Crit. Rev. Toxicol., 1984, 13, 25–97 CrossRef CAS PubMed.
- M. S. Shah, M. Tsapatsis and J. I. Siepmann, Chem. Rev., 2017, 117, 9755–9803 CrossRef CAS PubMed.
- A. Ros, M. A. Montes-Moran, E. Fuente, D. M. Nevskaia and M. J. Martin, Environ. Sci. Technol., 2006, 40, 302–309 CrossRef CAS PubMed.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- C. Petit, B. Mendoza and T. J. Bandosz, ChemPhysChem, 2010, 11, 3678–3684 CrossRef CAS PubMed.
- M. Khabazipour and M. Anbia, Ind. Eng. Chem. Res., 2019, 58, 22133–22164 CrossRef CAS.
- L. Hamon, C. Serre, T. Devic, T. Loiseau, F. Millange, G. Férey and G. De Weireld, J. Am. Chem. Soc., 2009, 131, 8775–8777 CrossRef CAS PubMed.
- J. Liu, Y. Wei, P. Li, Y. Zhao and R. Zou, J. Phys. Chem. C, 2017, 121, 13249–13255 CrossRef CAS.
- Y. Belmabkhout, R. S. Pillai, D. Alezi, O. Shekhah, P. M. Bhatt, Z. Chen, K. Adil, S. Vaesen, G. De Weireld, M. Pan, M. Suetin, A. J. Cairns, V. Solovyeva, A. Shkurenko, O. E. Tall, G. Maurin and M. Eddaoudi, J. Mater. Chem. A, 2017, 5, 3293–3303 RSC.
- (a) J. Izumi, T. Morimoto, H. Tsutaya and K. Araki, Stud. Surf. Sci. Catal., 1993, 80, 293–299 CrossRef CAS; (b) P. B. S. Rallapalli, K. Cho, S. H. Kim, J.-N. Kim and H. C. Yoon, Fuel, 2020, 281, 118985 CrossRef CAS.
- P. Lyn and G. Maurin, ACS Appl. Mater. Interfaces, 2021, 13, 4813–4822 CrossRef PubMed.
- L. Hamon, C. Serre, T. Devic, T. Loiseau, F. Millange, G. Férey and G. De Weireld, J. Am. Chem. Soc., 2009, 131, 8775–8777 CrossRef CAS PubMed.
- E. Sánchez-González, P. G. M. Mileo, M. Sagastuy-Breña, J. R. Álvarez, J. E. Reynolds III, A. Villareal, A. Gutiérrez-Alejandre, J. Ramírez, J. Balmaseda, E. González-Zamora, G. Maurin, S. M. Humphrey and I. A. Ibarra, J. Mater. Chem. A, 2018, 6, 16900–16909 RSC.
- J. A. Zárate, E. Sánchez-González, T. Jurado-Vázquez, A. Gutiérrez-Alejandre, E. González-Zamora, I. Castillo, G. Maurin and I. A. Ibarra, Chem. Commun., 2019, 55, 3049–3052 RSC.
- J. G. Flores, J. A. Zárate-Colín, E. Sánchez-González, J. R. Valenzuela, J. A. Gutiérrez-Alejandre, J. Ramírez, V. Jancik, J. Aguilar-pliego, M. C. Zorrilla, H. A. Lara-García, E. González-Zamora, G. Guzmán-González, I. González, G. Maurin and I. A. Ibarra, ACS Appl. Mater. Interfaces, 2020, 12, 18885–18892 CrossRef CAS PubMed.
- Y. V. Mikhaylik and J. R. Akridge, J. Electrochem. Soc., 2004, 151, A1969–A1976 CrossRef CAS.
- D. Moy, A. Manivannan and S. R. Narayanan, J. Electrochem. Soc., 2015, 162, A1–A7 CrossRef CAS.
- A. B. Andreeva, K. N. Le, L. Chen, M. E. Kellman, C. H. Hendon and C. K. Brozek, J. Am. Chem. Soc., 2020, 142, 19291–19299 CrossRef CAS PubMed.
- E. S. Grape, J. G. Flores, T. Hidalgo, E. Martínez-Ahumada, A. Hautier, D. R. Williams, M. O. Kee, O. Lars, T. Willhammar, P. Horcajada, I. A. Ibarra and A. K. Inge, J. Am. Chem. Soc., 2020, 142, 16795–16804 CrossRef CAS PubMed.
- H. Wang, X. Zeng, W. Wang and D. Cao, Chem. Eng. Sci., 2015, 135, 373–380 CrossRef CAS.
- K. Polychronopoulou, J. L. G. Fierro and A. M. Efstathiou, Appl. Catal., B, 2005, 57, 125–137 CrossRef CAS.
- X. Liu and R. Wang, J. Hazard. Mater., 2017, 326, 157–164 CrossRef CAS PubMed.
- L. Sigot, G. Ducom and P. Germain, Chem. Eng. J., 2016, 287, 47–53 CrossRef CAS.
- X. Wang, X. Ma, X. Xu, L. Sun and C. Song, Top. Catal., 2008, 49, 108–117 CrossRef CAS.
- X. Ma, X. Wang and C. Song, J. Am. Chem. Soc., 2009, 131, 5777–5783 CrossRef CAS PubMed.
- F. Adib, A. Bagreev and T. J. Bandosz, Langmuir, 2000, 16(4), 1980–1986 CrossRef CAS.
- H. Dong, Q. Zhou, L. Zhang and Y. Tian, Angew. Chem., Int. Ed., 2019, 58, 13948–13953 CrossRef CAS PubMed.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- G. Yang, L. Wu, B. Jiang, W. Yang, J. Qi, K. Cao, Q. Meng, A. K. Mustafa, W. Mu, S. Zhang, S. H. Snyder and R. Wang, Science, 2008, 322, 587–590 CrossRef CAS PubMed.
- D. J. Polhemus and D. J. Lefer, Circ. Res., 2014, 114, 730–737 CrossRef CAS PubMed.
- M. N. Hughes, M. N. Centelles and K. P. Moore, Free Radical Biol. Med., 2009, 47, 1346–1353 CrossRef CAS PubMed.
- C. Szabo and A. Papapetropoulos, Pharmacol. Rev., 2017, 69, 497–564 CrossRef CAS PubMed.
- H. A. Henthorn and M. D. Pluth, J. Am. Chem. Soc., 2015, 137(48), 15330–15336 CrossRef CAS PubMed.
- D.-W. Li, L.-L. Qu, K. Hu, Y.-T. Long and H. Tian, Angew. Chem., 2015, 127, 12949–12952 CrossRef.
- C. Yang, X. Yin, S.-Y. Huan, L. L. Chen, X.-X. Hu, M.-Y. Xiong, K. Chen and X.-B. Zhang, Anal. Chem., 2018, 90, 3118–3123 CrossRef CAS PubMed.
- J. Yang and Y. W. Yang, Small, 2020, 16, 1906846 CrossRef CAS PubMed.
- P. K. Allan, P. S. Wheatley, D. Aldous, M. Infas, C. Tang, J. A. Hriljac, I. L. Megson, K. W. Chapman, G. D. Weireld, S. Vaesen and R. E. Morris, Dalton Trans., 2012, 41, 4060–4066 RSC.
- H. Li, X. Feng, Y. Guo, D. Chen, R. Li, X. Ren, X. Jiang, Y. Dong and B. Wang, Sci. Rep., 2014, 4, 4366 CrossRef PubMed.
- Y. Ma, H. Su, X. Kuang, X. Li, T. Zhang and B. Tang, Anal. Chem., 2014, 86, 11459–11463 CrossRef CAS PubMed.
- S. Nandi, S. Banesh, V. Trivedi and S. Biswas, Analyst, 2018, 143, 1482–1491 RSC.
- X. Zhang, L. Fang, K. Jiang, H. He, Y. Yang, Y. Cui, B. Li and G. Qian, Biosens. Bioelectron., 2019, 130, 65–72 CrossRef CAS PubMed.
- C. Yang, K. Chen, M. Chen, X. Hu, S. Y. Huan, L. Chen, G. Song and X. B. Zhang, Anal. Chem., 2019, 91, 2727–2733 CrossRef CAS PubMed.
- Y. Li, J. Zhou, L. Wang and Z. Xie, ACS Appl. Mater. Interfaces, 2020, 12, 30213–30220 CrossRef CAS PubMed.
- C. D. Demirhan, W. W. Tso, J. B. Powell and E. N. Pistikopoulos, AIChE J., 2019, 65, e16498 CrossRef.
- (a) A. Fangmaier, A. Hadwiger-Fangmaier, L. Van der Eerden and H. J- Jäger, Environ. Pollut., 1994, 86, 43–82 CrossRef; (b) S. N. Behera, M. Sharma, V. P. Aneja and R. Balasubramanian, Environ. Sci. Pollut. Res., 2013, 20, 8092–8131 CrossRef CAS.
- R. Metkemeijer and P. Achard, J. Power Sources, 1994, 49, 271–282 CrossRef CAS.
- Z. Tamainot-Telto, S. J. Metcalf, R. E. Critoph, Y. Zhong and R. Thorpe, Int. J. Refrig., 2009, 32, 1212–1229 CrossRef CAS.
- (a) C. Petit, B. Mendoza and T. J. Bandosz, Langmuir, 2010, 26, 15302–15309 CrossRef CAS PubMed; (b) M. J. Katz, A. J. Howarth, P. Z. Moghadam, J. B. DeCoste, R. Q. Snurr, J. T. Hupp and O. K. Farha, Dalton Trans., 2016, 45, 4150–4153 RSC.
- D. W. Kang, S. E. Ju, D. W. Kim, M. Kang, H. Kim and C. S. Hong, Adv. Sci., 2020, 16, 2002142 CrossRef.
- J. Helminen, J. Helenius, E. Paatero and I. Turunen, J. Chem. Eng. Data, 2001, 46, 391–399 CrossRef CAS.
- (a) C. Petit and T. J. Bandosz, Adv. Funct. Mater., 2010, 20, 111–118 CrossRef CAS; (b) D. Britt, D. Tranchemontagne and O. M. Yagui, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11623–11627 CrossRef CAS PubMed.
- A. J. Rieth, Y. Tulchinsky and M. Dincă, J. Am. Chem. Soc., 2016, 138, 9401–9404 CrossRef CAS PubMed.
- A. J. Rieth and M. Dincă, J. Am. Chem. Soc., 2018, 140, 3461–3466 CrossRef CAS PubMed.
- D. W. Kim, D. W. Kang, M. Kang, J.-H. Lee, J. H. Choe, Y. S. Chae, D. S. Choi, H. Yun and C. S. Hong, Angew. Chem., 2020, 59, 22531–22536 CrossRef CAS PubMed.
- H. G. W. Godfrey, I. da Silva, L. Briggs, J. H. Carter, C. G. Morris, M. Savage, T. L. Easun, P. Manuel, C. A. Murray, C. C. Tang, M. D. Frogley, G. Cinque, S. Yang and M. Schröder, Angew. Chem., Int. Ed., 2018, 57, 14778–14781 CrossRef CAS PubMed.
- X. Han, W. Lu, Y. Chen, I. Silva, J. Li, L. Lin, W. Li, A. M. Sheveleva, H. G. W. Godfrey, Z. Lu, F. Tuna, E. J. L. Mcinnes, Y. Cheng, L. L. Daemen, L. J. M. McPherson, S. J. Teat, M. D. Frogley, S. Rudic, P. Manuel, A. J. Ramirez-cuesta, S. Yang and M. Schröder, J. Am. Chem. Soc., 2021, 143(8), 3153–3161 CrossRef CAS PubMed.
- T. Yoskamtorn, P. Zhao, X.-P. Wu, K. Purchase, F. Orlandi, P. Manuel, J. Taylor, Y. Li, S. Day, L. Ye, C. C. Tang, Y. Zhao and S. C. E. Tsang, J. Am. Chem. Soc., 2021, 143(8), 3205–3218 CrossRef CAS PubMed.
- P. Zhao, H. Fang, S. Mukhopadhyay, A. Li, S. Rudic, I. J. McPherson, C. C. Tang, D. Fairen-Jimienez, S. C. E. Tsang and S. A. T. Redfern, Nat. Commun., 2019, 10, 999 CrossRef CAS PubMed.
- F. Gholami, M. Tomas, Z. Gholami and M. Vakili, Sci. Total Environ., 2020, 714, 136712 CrossRef CAS PubMed.
- D. Krewski, M. Jerrett, R. T. Burnett, R. Ma, E. Hughes, Y. Shi, M. C. Turner, C. A. Pope, G. Thurston, E. E. Calle, M. J. Thun, B. Beckerman, P. DeLuca, N. Finkelstein, K. Ito, D. K. Moore, K. B. Newbold, T. Ramsay, Z. Ross, H. Shin and B. Tempalski, Res. Rep. Health Eff. Inst., 2009, 5–114 Search PubMed ; discussion 115–136.
- S. Safieddine, C. Clerbaux, M. George, J. Hadji-Lazaro, D. Hurtmans, P.-F. Coheur, C. Wespes, D. Loyola, P. Valks and N. Hao, J. Geophys. Res. Atmos., 2013, 118, 10555–10566 CAS.
- F. Rezaei, A. A. Rownaghi, S. Monjezi, R. P. Lively and C. W. Jones, Energy Fuels, 2015, 29, 5467–5486 CrossRef CAS.
- J. Despres, M. Koebel, O. Kröcher, M. Elsener and A. Wokaun, Microporous Mesoporous Mater., 2003, 58, 175–183 CrossRef CAS.
- X. Chang, G. Lu, Y. Guo, Y. Wang and Y. Guo, Microporous Mesoporous Mater., 2013, 165, 113–120 CrossRef CAS.
- W. J. Zhang, S. Rabiei, A. Bagreev, M. S. Zhuang and F. Rasouli, Appl. Catal., B, 2008, 83, 63–71 CrossRef CAS.
- N. Gargiulo, A. Peluso and D. Caputo, Processes, 2020, 8, 613 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- G. W. Peterson, J. J. Mahle, J. B. DeCoste, W. O. Gordon and J. A. Rossin, Angew. Chem., Int. Ed., 2016, 55, 6235–6238 CrossRef CAS PubMed.
- X. Han, H. G. W. Godfrey, L. Briggs, A. J. Davies, Y. Cheng, L. L. Daemen, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, J. Sun, C. Drathen, M. W. George, A. J. Ramirez-Cuesta, K. M. Thomas, S. Yang and M. Schröder, Nat. Mater., 2018, 17, 691–696 CrossRef CAS PubMed.
- X. Han, Y. Hong, Y. Ma, W. Lu, J. Li, L. Lin, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, C. Dejoie, J. Sun, S. Yang and M. Schröder, J. Am. Chem. Soc., 2020, 142, 15235–15239 CrossRef CAS PubMed.
- A. M. Wright, C. Sun and M. Dincă, J. Am. Chem. Soc., 2021, 143, 681–686 CrossRef CAS PubMed.
- B. Xiao, P. S. Wheatley, X. Zhao, A. J. Fletcher, S. Fox, A. G. Rossi, I. L. Megson, S. Bordiga, L. Regli, K. M. Thomas and R. E. Morris, J. Am. Chem. Soc., 2007, 129, 1203–1209 CrossRef CAS PubMed.
- S. M. Vornholt, M. J. Duncan, S. J. Warrender, R. Semino, N. A. Ramsahye, G. Maurin, M. W. Smith, J.-C. Tan, D. N. Miller and R. E. Morris, ACS Appl. Mater. Interfaces, 2020, 12, 58263–58276 CrossRef CAS PubMed.
- A. C. McKinlay, B. Xiao, D. S. Wragg, P. S. Wheatley, I. L. Megson and R. E. Morris, J. Am. Chem. Soc., 2008, 130, 10440–10444 CrossRef CAS PubMed.
- (a) M. O. Andreae and P. Merlet, Global Biogeochem. Cycles, 2001, 15, 955–966 CrossRef CAS; (b) J.-Y. Lee, T. C. Keener and Y. J. Yang, J. Air Waste Manage. Assoc., 2009, 59, 725–732 CrossRef CAS PubMed.
- (a) G. V. Last and M. T. Schmick, Identification and Selection of Major Carbon Dioxide Stream Compositions, US Department of Energy, Pacific Northwest National Laboratory, Washington U. S. A., 2011 CrossRef; (b) R. Zevenhoven and P. Kilpinen, in Control of Pollutants in Flue Gases and Fuel Gases, Helsinki Univ. of Technology, Otaniemi (Finland). Energy Engineering and Environmental Protection, Finland, 2005, ch. 2, pp. 2–4 Search PubMed.
- J. H. Carter, X. Han, F. Y. Moreau, I. da Silva, A. Nevin, H. G. W. Godfrey, C. C. Tang, S. Yang and M. Schröder, J. Am. Chem. Soc., 2018, 140, 15564–15567 CrossRef CAS PubMed.
- Y. Zhang, P. Zhang, W. Yu, J. Zhang, J. Huang, J. Wang, M. Xu, Q. Deng, Z. Zeng and S. Deng, ACS Appl. Mater. Interfaces, 2019, 11(11), 10680–10688 CrossRef CAS PubMed.
- Y. L. Fan, H. P. Zhang, M. J. Yin, R. Krishna, X. F. Feng, L. Wang, M. B. Luo and F. Luo, Inorg. Chem., 2021, 60(1), 4–8 CrossRef CAS PubMed.
- L. J. Gou, X. F. Feng, Z. Gao, R. Krishna and F. Luo, Inorg. Chem., 2021, 60(3), 1310–1314 CrossRef PubMed.
- M. J. Yin, X. H. Xiong, X. F. Feng, W. Y. Xu, R. Krishna and F. Luo, Inorg. Chem., 2021, 60(5), 3447–3451 CrossRef CAS PubMed.
- Y. Belmabkhout, P. M. Bhatt, K. Adil, R. S. Pillai, A. Cadiau, A. Shkurenko, G. Maurin, G. Liu, W. J. Koros and M. Eddaoudi, Nat. Energy, 2018, 3, 1059–1066 CrossRef CAS.
- S. Vaesen, V. Guillerm, Q. Yang, A. D. Wiersum, B. Marszalek, B. Gil, A. Vimont, M. Daturi, T. Devic, P. L. Llewellyn, C. Serre, G. Maurin and G. De Weireld, Chem. Commun., 2013, 49, 10082–10084 RSC.
- P. M. Bhatt, Y. Belmabkhout, A. H. Assen, L. J. Weselinski, H. Jiang, A. Cadiau, D.-X. Xue and M. Eddaoudi, Chem. Eng. J., 2017, 324, 392–396 CrossRef CAS.
- Z. Li, Y. Xiao, W. Xue, Q. Yang and C. Zhong, J. Phys. Chem. C, 2015, 119(7), 3674–3683 CrossRef CAS.
- E. N. H. S. Vaese and G. De Weireld, Microporous Mesoporous Mater., 2012, 154, 93–99 CrossRef.
- J. Xu, W. Xing, H. Wang, W. Xu, Q. Ding, L. Zhao, W. Guo and Z. Yan, J. Mater. Sci., 2016, 51, 2307–2319 CrossRef CAS.
- A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef.
- D. Ongari, L. Talirz and B. Smit, ACS Cent. Sci., 2020, 6(11), 1890–1900 CrossRef CAS PubMed.
- A. J. Howarth, Y. Liu, P. Li, Z. Li, T. C. Wang, T. T. Hupp and O. K. Farha, Nat. Rev. Mater., 2016, 1, 15018 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |