
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColloidal nano-MOFs nucleate and stabilize ultra-small quantum dots of lead bromide perovskites†
Loredana
Protesescu‡
 a,
Joaquín
Calbo§
b,
Kristopher
Williams
c,
William
Tisdale
c,
Aron
Walsh
bd and
Mircea
Dincă
*a
a,
Joaquín
Calbo§
b,
Kristopher
Williams
c,
William
Tisdale
c,
Aron
Walsh
bd and
Mircea
Dincă
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA. E-mail: mdinca@mit.edu
bDepartment of Materials, Imperial College London, London SW7 2AZ, UK
cDepartment of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, Massachusetts 02139, USA
dDepartment of Materials Science and Engineering, Yonsei University, Seoul 03722, Korea
First published on 19th March 2021
Abstract
The development of synthetic routes to access stable, ultra-small (i.e. <5 nm) lead halide perovskite (LHP) quantum dots (QDs) is of fundamental and technological interest. The considerable challenges include the high solubility of the ionic LHPs in polar solvents and aggregation to form larger particles. Here, we demonstrate a simple and effective host–guest strategy for preparing ultra-small lead bromide perovskite QDs through the use of nano-sized MOFs that function as nucleating and host sites. Cr3O(OH)(H2O)2(terephthalate)3 (Cr-MIL-101), made of large mesopore-sized pseudo-spherical cages, allows fast and efficient diffusion of perovskite precursors within its pores, and promotes the formation of stable, ∼3 nm-wide lead bromide perovskite QDs. CsPbBr3, MAPbBr3 (MA+ = methylammonium), and (FA)PbBr3 (FA+ = formamidinium) QDs exhibit significantly blue-shifted emission maxima at 440 nm, 446 nm, and 450 nm, respectively, as expected for strongly confined perovskite QDs. Optical characterization and composite modelling confirm that the APbBr3 (A = Cs, MA, FA) QDs owe their stability within the MIL-101 nanocrystals to both short- and long-range interfacial interactions with the MOF pore walls.
Introduction
Stable lead halide perovskite (LHP) nanocrystals (NCs), with the composition APbX3 (A = Cs+, formamidinium FA+, methylammonium MA+; X = I−, Br−, Cl−)1–4 have emerged as superior materials for optoelectronic applications such as light emitting devices,5–8 lasers,9,10 or selective optical sensors.11 Aided by their intrinsic or surface defect-tolerant electronic structure, LHPs exhibit excellent optical properties that are tunable over the entire visible spectrum through modification of the halide composition. Thus far, the overwhelming majority of studies have focused on the weak confinement regime (i.e. NCs larger than 8 nm) because large NCs are easier to synthesize and stabilize. Nevertheless, smaller NCs are expected to show optical properties that diverge from their larger congeners. For instance, energy transfer between LHP and external acceptors (e.g. polycyclic aromatic hydrocarbons) only occurs for smaller (5–8 nm) NCs but not for weakly confined donors.12 However, synthesizing and stabilizing ultra-small LHP NCs (i.e. diameters below 4 nm) is extremely challenging owing to their highly ionic nature, labile surfaces, and metastable structures. Indeed, colloidal syntheses (such as hot injection method) are inefficient for the production of ultra-small NCs due to the fast reactivity of the precursors, which yields nanostructures such as nanoplatelets, nanosheets and nanocubes that are difficult to separate and isolate for further investigation.13 Templating agents such as porous silica,14 zeolites,15,16 and metal–organic frameworks (MOFs)7,15,17–20 have been more successful, but so far have also only produced large APbBr3 NCs with bulk-like emission maxima centered at 520–530 nm.21 Here, we demonstrate that Cr-MIL-101, a MOF exhibiting large mesoporous cages, is an effective platform for the synthesis and stabilization of highly monodisperse, ultra-small, 3 nm-wide LHP QDs when the MOF itself is in nanoparticle form.Results and discussions
Owing to their porosity and tunable composition, MOFs have attracted considerable interest for the development of heterostructures where the ordered porosity of the MOF, as host, can be combined with additional electrical, optical, or catalytic properties given by a guest nanoparticle.22–25 There are two well-established routes to immobilize nanoscale objects within MOFs: “ship in a bottle” and “bottle around the ship”. Whereas the latter is essentially an encapsulation process whereby pre-formed NCs are added to a solution containing the MOF precursors, the “ship in a bottle”26,27 strategy requires that the precursors for NCs penetrate the windows of the pre-synthesized MOFs and react within the pores to form the desired nanostructure. Ideally, this latter strategy uses the framework skeleton as a ligand shell to lower the surface energy of the nanoparticles and leads to the formation of highly monodisperse NCs whose size is defined by the pore of the MOF host.APbBr3 in bulk Cr-MIL-101
Cr-MIL-101 was chosen as a host for the synthesis of halide perovskite QDs because of its large, pseudo-spherical cages, enhanced chemical stability under challenging environments, and relatively well-developed encapsulation chemistry.28,29 It can be isolated as a green powder with micron-sized crystals upon reaction of Cr(NO3)3·9H2O with terephthalic acid (TA) for 8 hours at 220 °C in acidic media (see ESI† for full experimental details). After activation and drying, Cr-MIL-101 retains a zeolite-type MTN framework (a zeolite whose topology comprises two different kinds of subcages: a dodecahedral cage face-shared with four other adjacent cages, Fig. 1a) with large pseudo-spherical pores (2.9 nm-wide M cages and 3.4 nm-wide L cages) connected via 1.2 nm and 1.6 nm openings that we reasoned would allow for facile diffusion of LHP precursors into the cages. We found, however, that impregnating micron-sized crystals of Cr-MIL-101 with a solution of CsBr and PbBr2 in N-methylformamide (NMF) produced mostly bulk halide perovskites attached to the exterior surface of the MOF crystals, regardless of the reaction temperature. Fluorescence spectroscopy of these MOF/LHP composites revealed features dominated by bulk-like emission at 520 nm, suggestive of CsPbBr3 particles at least 10 nm in diameter (Fig. S1†). The same spectra nevertheless revealed a small contribution from confined emission at 440 nm, itself suggestive of small nanoparticles possibly formed within the MOF pores. Altogether, the emission data suggested that although the LHP precursors are able to penetrate within the MOF pores, a large concentration gradient is likely established within the relatively large MIL-101 crystals. Rapid nucleation of the perovskite causes blockage of the pore entrance with concomitant formation of predominantly bulk LHP at the exterior and only a small fraction of the desired pore-confined NCs, themselves likely localized near the MOF surface.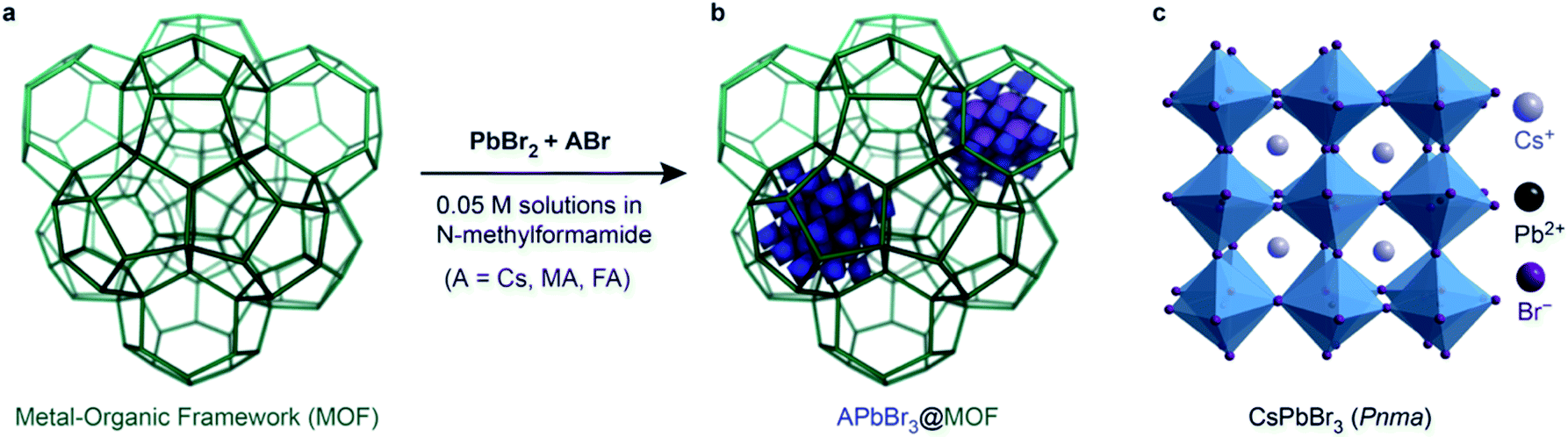 | ||
| Fig. 1 (a) A cartoon representing the 3D porosity of the MOFs before encapsulation of the LHP QDs and, (b) after, where the inclusions represent the LHP QDs trapped inside the pores, (c) crystal structure for γ-orthorhombic CsPbBr3 (Pnma). | ||
Synthesis of colloidal nano-Cr-MIL-101
Encouraged by the appearance of confined emission in bulk Cr-MIL-101, we sought to improve penetration and achieve even distribution of the LHP precursors throughout the host crystals by employing colloidal MOF nanoparticles. The synthesis of colloidal MOFs has been reviewed recently,30 and includes diverse strategies such as reverse addition of a co-solvent into the reaction mixture, microemulsion methodology,31 use of microwaves32 or ultrasound,33 and addition of a modulator such as a polymer or surfactant.34 Using a modified procedure described by the Edler group,34 we explored various modulators to synthesize colloidally stable, porous Cr-MIL-101 nanoparticles. The fundamental concept is to employ modulators that compete with the direct reaction between TA and Cr3+, the two main ingredients for Cr-MIL-101: amines that form acid–base adducts with TA, and monocarboxylic acids that bind Cr3+ and ideally provide colloidal stability to eventual MOF nanoparticles. A series of tests showed that benzoic, acetic, and hexanoic acids are ineffective as modulators for the production of colloidal Cr-MIL-101, all yielding either large crystals or samples with poor crystallinity. Similarly, using hexadecylamine or oleylamine as modulators produced only larger, highly polydisperse NPs with diameters exceeding 50 nm and low colloidal stability (Table S1†). However, addition of oleic acid (OA) to the MOF synthesis solution, in a molar OA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TA ratio of 1:12, produced colloidally stable 25 nm-wide Cr-MIL-101 particles with good crystallinity (nano-Cr-MIL-101, Fig. S2†). After the washing procedure, FTIR spectra (Fig. S3†) showed complete removal of the modulator since the band associated to free –COOH (from oleic acid) at 1700 cm−1 is no longer present. The vibrational spectrum of nano-Cr-MIL-101 differs from Cr-MIL-101 only due to the presence of NMF (νC–N at 1310 cm−1), with the NPs remaining colloidally suspended in the solvent of choice (acetone, NMF). The colloidal suspension is stable for over a year in acetone and NMF without presenting sedimentation while centrifuging the suspension at 4k rpm.
:TA ratio of 1:12, produced colloidally stable 25 nm-wide Cr-MIL-101 particles with good crystallinity (nano-Cr-MIL-101, Fig. S2†). After the washing procedure, FTIR spectra (Fig. S3†) showed complete removal of the modulator since the band associated to free –COOH (from oleic acid) at 1700 cm−1 is no longer present. The vibrational spectrum of nano-Cr-MIL-101 differs from Cr-MIL-101 only due to the presence of NMF (νC–N at 1310 cm−1), with the NPs remaining colloidally suspended in the solvent of choice (acetone, NMF). The colloidal suspension is stable for over a year in acetone and NMF without presenting sedimentation while centrifuging the suspension at 4k rpm.
APbX3 in nano-Cr-MIL-101
Solutions of ABr (A = Cs, MA, FA) and of PbBr2 in NMF were mixed with a stable, colloidal suspension of monodisperse nano-Cr-MIL-101 in NMF. After stirring these mixtures for 1 to 12 h at room temperature, the reaction mixture remained colloidal, a first sign that bulk LHP did not precipitate under these conditions. Reaction mixtures were subsequently drop- or spin-casted on quartz and glass substrates and dried by heating at 70 °C for 1 min multiple times to produce films with varying thickness between 100 to 300 nm (Fig. S4 and S5†). Optimized reactant ratios, 2:1 CsBr:PbBr2 for CsPbBr3 and 1:1 ABr:PbBr2 for FAPbBr3 and MAPbBr3 (Fig. S6 and S7†) produced films where the pXRD diffraction peaks pertaining to the nano-Cr-MIL-101 are broader, as expected for small NCs (Fig. S8†) but the crystallinity of the nano-MOF host is unaffected (Fig. 2). TEM images of the nano-MOF-encapsulated LHP nanocrystals revealed consistent sizes of approximately 3 nm, in line with the 2.9 nm and 3.4 nm cage sizes of the MOF (Fig. S9†). Furthermore, quantitative EDX mapping of the LHP NCs confirmed the ratio of 1:1:2.9 for CsPbBr3 (Fig. 2f and g).
 | ||
| Fig. 2 (a) View of the (111) plane in Cr-MIL-101, (b) view of the (110) plane in Cr-MIL-101, (c and d) TEM images for nano-Cr-MIL-101 (approx. 25 nm NPs), (e) TEM images for CsPbBr3@nano-Cr-MIL-101 (approx. 25 nm NPs), (f) STEM image for CsPbBr3@nano-Cr-MIL-101. (g) STEM EDX mapping for CsPbBr3@nano-Cr-MIL-101. | ||
As the 3 nm CsPbBr3 QDs have diffraction features that are too broad to be observed by PXRD (Fig. S8†), we confirmed their identity by Raman spectroscopy. The latter revealed the [PbBr6]4− vibration35 at 53 cm−1 along with unmodified bands from the MOF host (Fig. 3 and S10†).36 The vibrational band associated with the rattling of the Cs+ cation is weak in the bulk and undetectable for CsPbBr3@nano-Cr-MIL-101. We assign the 20 cm−1 shift of the [PbBr6]4− band relative to bulk37 to a larger contribution from the extended bonds of surface species, which in 3 nm particles represent a much larger proportion of all [PbBr6]4− species than in bulk CsPbBr3.
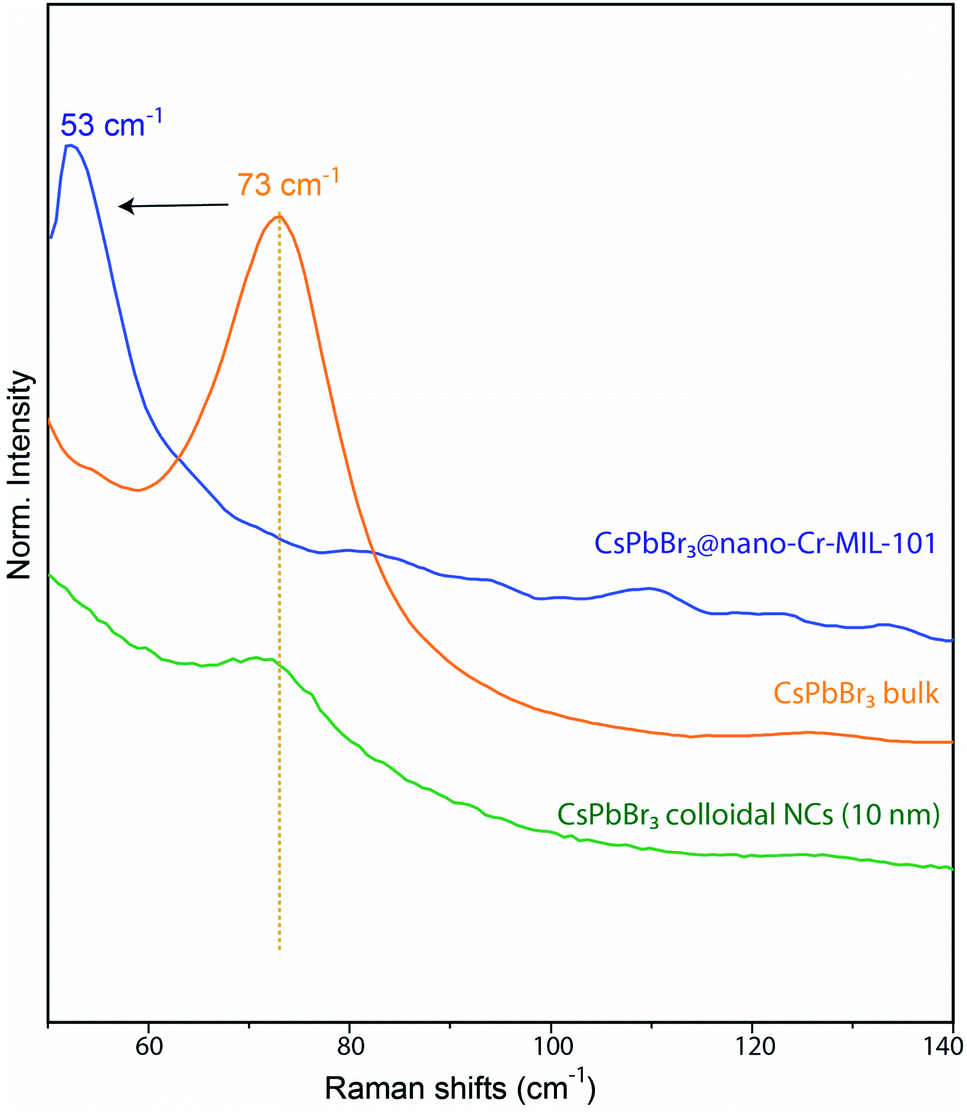 | ||
| Fig. 3 Comparison between Raman vibrational bands of bulk CsPbBr3, CsPbBr3@nano-Cr-MIL-101, and colloidal CsPbBr3 NCs (10 nm). | ||
Even when compared to a standard 10 nm colloidal suspension of CsPbBr3 NCs (79 cm−1),38 synthetized via hot injection method,1 the observed shift is an indication of the expansion of bond lengths35 in 3 nm CsPbBr3@nano-Cr-MIL-101 (Fig. 3 and S10†). As expected upon partial pore filling, N2 adsorption isotherms of CsPbBr3@nano-Cr-MIL-101 gave an apparent BET surface area of 1257 m2 g−1, a 20–25% decrease (on average, depending on sample) relative to bulk Cr-MIL-101 (Fig. S11†). Optical characterization provided important information on the nature of the LHP QDs within nano-Cr-MIL-101. Bare nano-Cr-MIL-101 shows two distinct features in the absorption spectrum: one at 447 nm attributed to π–π* transitions within the phenyl ring of the organic ligand, and one at 599 nm corresponding to the 4A2g–4T2g transition in Cr3+ (Fig. 4a). Importantly, the MOF itself is not photoluminescent (Fig. 4b). The representative features of the host persist in the absorption spectra after crystallization of APbBr3 within the pores. However, new well-defined excitonic peaks appear at 427 nm and at 432 nm for APbBr3@nano-Cr-MIL-101 (A = Cs, FA, respectively) (Fig. 4b). These correspond to 3 nm CsPbBr3 (440 nm) and FAPbBr3 (450 nm) photoluminescence peaks. For 3 nm MAPbBr3@nano-Cr-MIL-101, the emission peak maximum is at 446 nm (Fig. S7†). The lifetimes of the excited states are 1.5 ns (CsPbBr3) and 2 ns (FAPbBr3) at room temperature (Fig. S12†), in good agreement with predicted photoluminescence decay lifetimes for strongly confined CsPbBr3.39
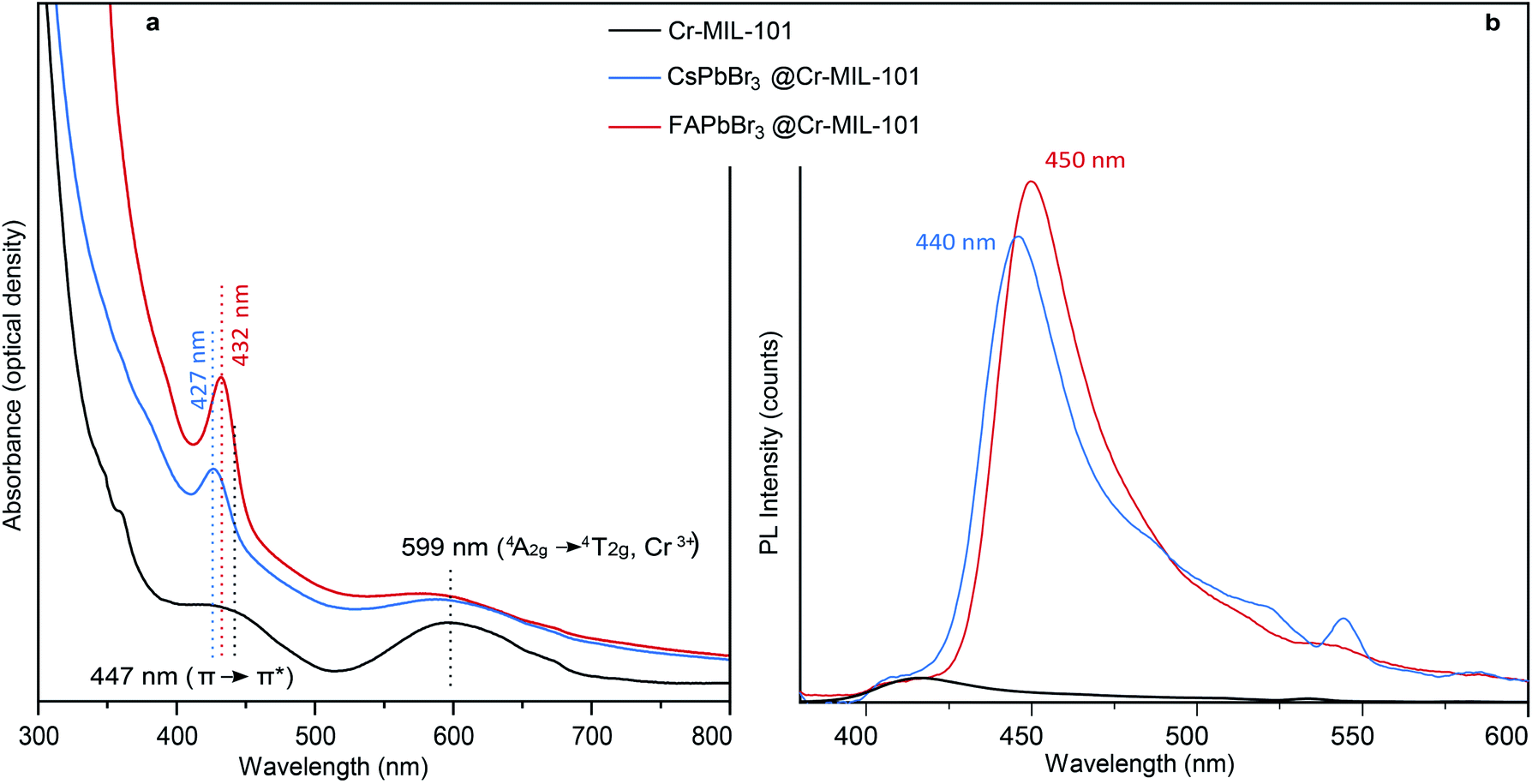 | ||
| Fig. 4 (a) Absorbance and (b) photoluminescence spectra for nano-Cr-MIL-101, CsPbBr3@nano-Cr-MIL-101, and FAPbBr3@nano-Cr-MIL-101. | ||
Tellingly, photoluminescence studies of CsPbBr3@nano-Cr-MIL-101 reveal an asymmetric peak defined by the morphology of the host itself. The ratio of 2.9 nm pores to 3.4 nm pores in pure Cr-MIL-101 is 2:1. Peak deconvolution in the emission spectrum revealed that the emission of QDs hosted in the 3.4 nm cages is red-shifted by 14 nm from the emission of the smaller QDs hosted in 2.9 cages (Fig. S13†).
Furthermore, variable-temperature (VT) experiments performed on films reveal energy transfer from the quantum dots hosted in the 2.9 nm cages to those in the larger cages upon raising the temperature from 80 K to 300 K (Fig. S14†). Energy transfer phenomena can only occur when the QDs are in close proximity, as might be expected in CsPbBr3@nano-Cr-MIL-101, and as reported for concentrated solutions of polydisperse NCs.40 Importantly, the position of the photoluminescence peak for CsPbBr3 did not change with temperature, confirming that the structure of the QDs remains constant over this temperature range and that QDs isolated within nano-Cr-MIL-101 retain the core structure of bulk CsPbBr3 (γ-orthorhombic, Pmna). Conversely, for FAPbBr3@nano-Cr-MIL-101, with emission centered at 2.755 eV at room temperature, clear evidence of the Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m structure of the 3 nm QDs is given by the abrupt change of the peak position while cooling the sample from 300 K to 80 K. Bulk FAPbBr3 itself transitions between 275 K and 250 K to a tetragonal P4/mbm phase, which further distorts to the orthorhombic Pnma phase between 150 K and 125 K.41 For the 3 nm FAPbBr3 QDs isolated within nano-Cr-MIL-101, the phase transition between cubic and tetragonal occurs at slightly lower temperature (∼250 K), as expected from the dramatic change in the material dimensions (from bulk to only 5–6 unit cells), while the orthorhombic Pnma is observed below 150 K.42
m structure of the 3 nm QDs is given by the abrupt change of the peak position while cooling the sample from 300 K to 80 K. Bulk FAPbBr3 itself transitions between 275 K and 250 K to a tetragonal P4/mbm phase, which further distorts to the orthorhombic Pnma phase between 150 K and 125 K.41 For the 3 nm FAPbBr3 QDs isolated within nano-Cr-MIL-101, the phase transition between cubic and tetragonal occurs at slightly lower temperature (∼250 K), as expected from the dramatic change in the material dimensions (from bulk to only 5–6 unit cells), while the orthorhombic Pnma is observed below 150 K.42
The invariance of the APbBr3 emission peak position over a wide range of temperatures informs on the influence of the MOF host on the optical properties of the perovskite QDs (Fig. 5). Typically, LHP NPs exhibit a blue shift with increasing temperature.43,44 This behavior is similar to that of Pb chalcogenide QDs and is thought to originate from the interplay between thermal expansion and electron–phonon coupling. With increasing temperature, thermal expansion becomes a dominating factor for CsPbBr3 because it decreases the interaction between the valence orbitals Pb 6s and I 5p. As a result, the forbidden gap increases, while the valence bandwidth narrows.44 The fact that in APbBr3@nano-Cr-MIL-101, the main PL peak position is invariant with temperature for both Cs and FA analogs (excluding the change in the crystal structure for FAPbBr3) confirm that the cages of the MOF constrain any lattice expansion with temperature (Fig. 5b). The PL quantum yield of all investigated samples was invariably below 5%, which suggests an active interaction of the MOF with the LHP QDs leading either to the creation of traps on the surface of the QDs or to non-radiative recombination via photon emission. Note that the samples of APbBr3@nano-Cr-MIL-101 were synthesized, processed into films, and stored for more than 6 months in ambient atmosphere without changes in their optical signatures. However, accelerated degradation studies under continuous laser beam irradiation at 405 nm (40% humidity and 20 °C) highlight the limitations of the MOF host under external stimuli: 3 minutes of exposure leads to the formation of the 520 nm emissive structures, which indicate the degradation of the matrix and the formation of large, non-uniform particles (Fig. S15†).
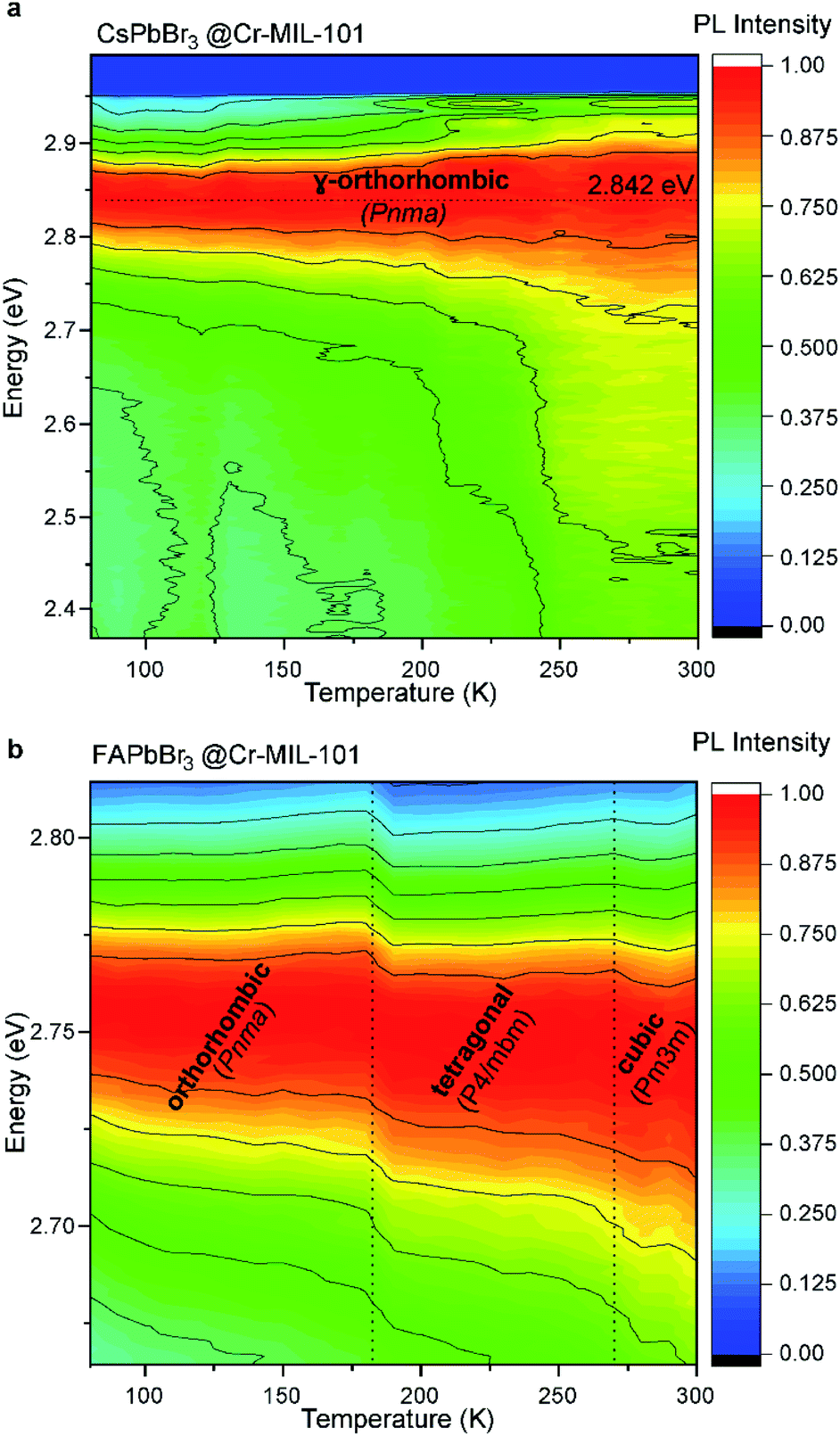 | ||
| Fig. 5 (a) Photoluminescence evolution over temperature (from 80 to 300 K) for CsPbBr3@nano-Cr-MIL-101. (b) Photoluminescence evolution over temperature (from 80 to 300 K) for FAPbBr3@nano-Cr-MIL-101. | ||
To gain a more thorough understanding on the unusual effects of the host MOF onto the structure and behavior of the QDs trapped within the MOF cages, we performed massively parallel density functional theory (DFT) calculations on pristine Cr-MIL-101 and CsPbBr3, as well as a full model of the CsPbBr3@nano-Cr-MIL-101 composite. The smaller pore M cage of Cr-MIL-101 and a CsPbBr3 NP of approximately 2.5 nm that perfectly fits into this cage were considered in a cluster approach (see the ESI† for full computational details). Geometries for the isolated systems were fully relaxed, and the density of states (DoS) for the minimum-energy structures of Cr-MIL-101 and CsPbBr3 are shown in Fig. 6. Analysis of the electronic structure indicates that the valence band (VB) of Cr-MIL-101 is localized on the inorganic cluster (primarily Cr d-electrons with additional contribution from the O and F p-electrons), whereas the conduction band (CB) is ligand-centered (delocalized frontier orbital over the terephthalate ligand; see Fig. S16†). The bandgap of Cr-MIL-101 in the spin-up channel (Cr-involved) is computed to be 2.51 eV (assigned to the experimental absorption band at 600 nm), and 3.17 eV in the spin-down channel (ligand-based, assigned to the experimental shoulder recorded at 447 nm) (Fig. 6a and S16a†). For the perovskite QDs, the VB is dominated by the Br p-orbital, whereas the CB belongs to the Pb p-orbital (Fig. 6b and S16b†). Notably, we predict a bandgap of 2.83 eV for the 2.5 nm CsPbBr3 QDs, which agrees well with the experimental absorption peak at 427 nm (2.90 eV). An embedding-inverted approach (nanoparticle atoms as monopoles) was used to analyze the effect of the perovskite nanoparticle onto the electronic structure of Cr-MIL-101 (see the ESI† for details). After taking into account for host–guest interactions through geometry relaxation, we find that both the VB and CB are slightly shifted to higher energy, with a negligible effect on the MOF bandgap itself (Table S2†). More importantly, the frontier energy levels of Cr-MIL-101 and CsPbBr3 in CsPbBr3@Cr-MIL-101 (Fig. S17†) are well aligned, which may explain the efficient energy transfer between the two moieties.
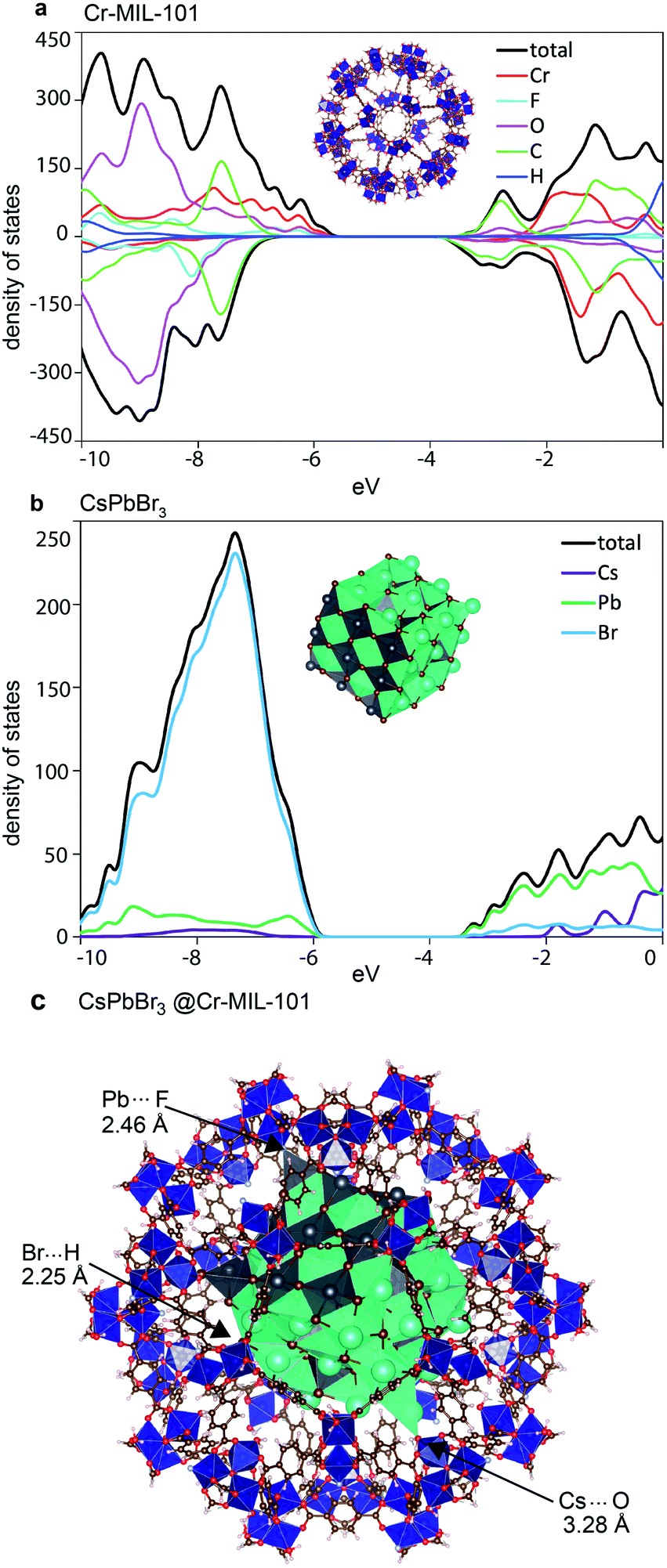 | ||
| Fig. 6 Electronic density of states calculated for (a) Cr-MIL-101 and (b) nano CsPbBr3. (c) Minimum-energy geometry for CsPbBr3@nano-Cr-MIL-101. | ||
Evaluating the binding energy in the model CsPbBr3@Cr-MIL-101 provided clues to the enhanced stability of the LHP QD within nano-Cr-MIL-101. Model optimization at the DFT level, including dispersion corrections, revealed strong interfacial interactions between the defectively-coordinated surface atoms of the perovskite nanoparticle and the inorganic coordination environment of the Cr-MIL-101 pore (Fig. 6c). For instance, Cs⋯O (H2O), Pb⋯F and Br⋯H (H2O) contacts with short distances of 3.28, 2.46 and 2.25 Å, respectively, are observed in the minimum-energy structure (Fig. 6c). These interactions are likely to be responsible for the low PL quantum yield observed for CsPbBr3 QDs inside the MOF. The binding energy, calculated as the energy difference between the minimum-energy of the composite and the constituting moieties, is predicted to be −232 kJ mol−1, with approximately 50% contribution from long-range van der Waals dispersion interactions and 50% from electrostatic forces and short-range contacts. This large and negative binding energy explains the unusually high stability of the CsPbBr3@nano-Cr-MIL-101 composite.
Conclusions
The foregoing results highlight the importance of nano-sizing the host itself in order to allow the crystallization/encapsulation and stabilization of ultra-small lead bromide perovskite QDs. Scaling down the particles of Cr-MIL-101 host to approximately 25 nm allows fast and efficient transport of LHP precursors within its pores and cages, which then enable the formation of 3 nm NPs of APbBr3. These ultra-small QDs exhibit significantly blue-shifted emission peaks at 440 nm (Cs+), 446 nm (MA+), and 450 nm (FA+). Variable-temperature photoluminescence measurements exposed strong interfacial host–guest interactions that were confirmed by computational studies and are likely responsible for the unusual stability of the QDs, whose optical properties remain unaltered after months under ambient atmosphere. Our approach enabled the formation of the smallest LHP QDs yet and establish a blueprint towards the exploration of other perovskite nanostructures whose quantum confinement effects have not yet been explored at this scale. Equally intriguing for future studies are explorations of different MOF hosts with a diminished effect on the electronic level of the guest QDs.Author contributions
L. P. and M. D. proposed the research project. L. P. prepared the materials and performed the structural and room temperature optical characterizations. K. W. performed low temperature photoluminescence measurements. J. C. conducted the DFT calculations. M. D., A. W., and W. T. provided critical insights for the analysis and discussion. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Army Research Office (grant number W911NF-17-1-0174). We thank Dr R. Day for SEM and Dr J. Gardener for assistance with STEM EDX mapping. L. P. acknowledges the Swiss NSF Postdoctoral Fellowship for support (Grant P2EXP2-172214). J. C. acknowledges the Generalitat Valenciana for the Postdoctoral APOSTD/2017/081 Fellowship. K. W. and W. A. T. were supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under award no. DE-SC0019345. This research was also supported by the National Research Foundation of Korea (NRF) funded by Ministry of Science and ICT (2018M3D1A1058536). Computational work used the ARCHER UK National Supercomputing Service (http://www.archer.ac.uk) via the HEC Materials Chemistry Consortium, which is funded by EPSRC (EP/L000202). We thank Mr G. Skorupskii for generating graphics for Fig. 1.References
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- M. V. Kovalenko, L. Protesescu and M. I. Bodnarchuk, Science, 2017, 358, 745–750 CrossRef CAS PubMed.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- S. Kumar, J. Jagielski, T. Tian, N. Kallikounis, W.-C. Lee and C.-J. Shih, ACS Energy Lett., 2019, 4, 118–125 CrossRef CAS.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- Z. Chen, Z.-G. Gu, W.-Q. Fu, F. Wang and J. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 28737–28742 CrossRef CAS PubMed.
- X. Zhang, B. Xu, J. Zhang, Y. Gao, Y. Zheng, K. Wang and X. W. Sun, Adv. Funct. Mater., 2016, 26, 4595–4600 CrossRef CAS.
- S. Yakunin, L. Protesescu, F. Krieg, M. I. Bodnarchuk, G. Nedelcu, M. Humer, G. De Luca, M. Fiebig, W. Heiss and M. V. Kovalenko, Nat. Commun., 2015, 6, 8056 CrossRef CAS PubMed.
- Y. Wang, X. Li, J. D. Song, L. Xiao, H. Zeng and H. Sun, Adv. Mater., 2015, 27, 7101–7108 CrossRef CAS PubMed.
- S. Yakunin, B. M. Benin, Y. Shynkarenko, O. Nazarenko, M. I. Bodnarchuk, D. N. Dirin, C. Hofer, S. Cattaneo and M. V. Kovalenko, Nat. Mater., 2019, 18, 846–852 CrossRef CAS PubMed.
- X. Luo, R. Lai, Y. Li, Y. Han, G. Liang, X. Liu, T. Ding, J. Wang and K. Wu, J. Am. Chem. Soc., 2019, 141, 4186–4190 CrossRef CAS PubMed.
- S. K. Yadav, G. K. Grandhi, D. P. Dubal, J. C. de Mello, M. Otyepka, R. Zbořil, R. A. Fischer and K. Jayaramulu, Small, 2020, 2004891 CrossRef CAS PubMed.
- D. N. Dirin, L. Protesescu, D. Trummer, I. V. Kochetygov, S. Yakunin, F. Krumeich, N. P. Stadie and M. V. Kovalenko, Nano Lett., 2016, 16, 5866–5874 CrossRef CAS PubMed.
- S. Ye, J.-Y. Sun, Y.-H. Han, Y.-Y. Zhou and Q.-Y. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 24656–24664 CrossRef CAS PubMed.
- Z. C. Kong, J. F. Liao, Y. J. Dong, Y. F. Xu, H. Y. Chen, D. B. Kuang and C. Y. Su, ACS Energy Lett., 2018, 3, 2656–2662 CrossRef CAS.
- H. He, Y. Cui, B. Li, B. Wang, C. Jin, J. Yu, L. Yao, Y. Yang, B. Chen and G. Qian, Adv. Mater., 2019, 31, 1806897 CrossRef PubMed.
- C. Zhang, B. Wang, W. Li, S. Huang, L. Kong, Z. Li and L. Li, Nat. Commun., 2017, 8, 1138 CrossRef PubMed.
- Q. Q. Zhang, H. S. Wu, W. Lin, J. N. Wang and Y. W. Chi, J. Solid State Chem., 2019, 272, 221–226 CrossRef CAS.
- X.-Y. Yang, L.-H. Chen, Y. Li, J. C. Rooke, C. Sanchez and B.-L. Su, Chem. Soc. Rev., 2017, 46, 481–558 RSC.
- C. Zhang, W. Li and L. Li, Angew. Chem., Int. Ed., 2021, 60, 7488–7501 CrossRef CAS PubMed.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- J. He, Z. Yan, J. Wang, J. Xie, L. Jiang, Y. Shi, F. Yuan, F. Yu and Y. Sun, Chem. Commun., 2013, 49, 6761–6763 RSC.
- J. Aguilera-Sigalat and D. Bradshaw, Coord. Chem. Rev., 2016, 307, 267–291 CrossRef CAS.
- S. Jin, H.-J. Son, O. K. Farha, G. P. Wiederrecht and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 955–958 CrossRef CAS PubMed.
- J. Liu, D. M. Strachan and P. K. Thallapally, Chem. Commun., 2014, 50, 466–468 RSC.
- S. Saha, G. Das, J. Thote and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 14845–14851 CrossRef CAS PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- D.-Y. Hong, Y. K. Hwang, C. Serre, G. Férey and J.-S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS.
- C. R. Marshall, S. A. Staudhammer and C. K. Brozek, Chem. Sci., 2019, 10, 9396–9408 RSC.
- W. Zheng, X. Hao, L. Zhao and W. Sun, Ind. Eng. Chem. Res., 2017, 56, 5899–5905 CrossRef CAS.
- S. Wuttke, A. Zimpel, T. Bein, S. Braig, K. Stoiber, A. Vollmar, D. Müller, K. Haastert-Talini, J. Schaeske, M. Stiesch, G. Zahn, A. Mohmeyer, P. Behrens, O. Eickelberg, D. A. Bölükbas and S. Meiners, Adv. Healthcare Mater., 2017, 6, 1600818 CrossRef PubMed.
- N. A. Khan and S. H. Jhung, Coord. Chem. Rev., 2015, 285, 11–23 CrossRef CAS.
- D. Jiang, A. D. Burrows and K. Edler, CrystEngComm, 2011, 13, 6916–6919 RSC.
- J.-H. Cha, J. H. Han, W. Yin, C. Park, Y. Park, T. K. Ahn, J. H. Cho and D.-Y. Jung, J. Phys. Chem. Lett., 2017, 8, 565–570 CrossRef CAS PubMed.
- O. Yaffe, Y. Guo, L. Z. Tan, D. A. Egger, T. Hull, C. C. Stoumpos, F. Zheng, T. F. Heinz, L. Kronik, M. G. Kanatzidis, J. S. Owen, A. M. Rappe, M. A. Pimenta and L. E. Brus, Phys. Rev. Lett., 2017, 118, 136001 CrossRef PubMed.
- Y. Gao, X. Zhao, P. Yin and F. Gao, Sci. Rep., 2016, 6, 20539 CrossRef CAS PubMed.
- M. Liao, B. Shan and M. Li, J. Phys. Chem. Lett., 2019, 10, 1217–1225 CrossRef CAS PubMed.
- M. A. Becker, R. Vaxenburg, G. Nedelcu, P. C. Sercel, A. Shabaev, M. J. Mehl, J. G. Michopoulos, S. G. Lambrakos, N. Bernstein, J. L. Lyons, T. Stöferle, R. F. Mahrt, M. V. Kovalenko, D. J. Norris, G. Rainò and A. L. Efros, Nature, 2018, 553, 189 CrossRef CAS PubMed.
- C. de Weerd, L. Gomez, H. Zhang, W. J. Buma, G. Nedelcu, M. V. Kovalenko and T. Gregorkiewicz, J. Phys. Chem. C, 2016, 120, 13310–13315 CrossRef CAS.
- E. C. Schueller, G. Laurita, D. H. Fabini, C. C. Stoumpos, M. G. Kanatzidis and R. Seshadri, Inorg. Chem., 2018, 57, 695–701 CrossRef CAS PubMed.
- P. Cottingham and R. L. Brutchey, Chem. Commun., 2016, 52, 5246–5249 RSC.
- G. Rainò, G. Nedelcu, L. Protesescu, M. I. Bodnarchuk, M. V. Kovalenko, R. F. Mahrt and T. Stöferle, ACS Nano, 2016, 10, 2485–2490 CrossRef PubMed.
- A. Shinde, R. Gahlaut and S. Mahamuni, J. Phys. Chem. C, 2017, 121, 14872–14878 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc00282a |
| ‡ Present address: Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, Netherlands. |
| § Present address: Instituto de Ciencia Molecular, Universidad de Valencia, 46890 Paterna, Spain. |
| This journal is © The Royal Society of Chemistry 2021 |