
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnesium–halobenzene bonding: mapping the halogen sigma-hole with a Lewis-acidic complex†
Alexander
Friedrich
a,
Jürgen
Pahl
a,
Jonathan
Eyselein
a,
Jens
Langer
 a,
Nico
van Eikema Hommes
bc,
Andreas
Görling
c and
Sjoerd
Harder
*a
a,
Nico
van Eikema Hommes
bc,
Andreas
Görling
c and
Sjoerd
Harder
*a
aInorganic and Organometallic Chemistry, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 1, 91058 Erlangen, Germany. E-mail: sjoerd.harder@fau.de
bComputer Chemistry Center, Friedrich-Alexander-Universität Erlangen-Nürnberg, Nägelsbachstraße 25, 91052 Erlangen, Germany
cTheoretical Chemistry, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany
First published on 16th December 2020
Abstract
Complexes of the Lewis base-free cations (MeBDI)Mg+ and (tBuBDI)Mg+ with Ph–X ligands (X = F, Cl, Br, I) have been studied (MeBDI = HC[C(Me)N-DIPP]2 and tBuBDI = HC[C(tBu)N-DIPP]2; DIPP = 2,6-diisopropylphenyl). For the smaller β-diketiminate ligand (MeBDI) only complexes with PhF could be isolated. Heavier Ph–X ligands could not compete with bonding of Mg to the weakly coordinating anion B(C6F5)4−. For the cations with the bulkier tBuBDI ligand, the full series of halobenzene complexes was structurally characterized. Crystal structures show that the Mg⋯X–Ph angle strongly decreases with the size of X: F 139.1°, Cl 101.4°, Br 97.7°, I 95.1°. This trend, which is supported by DFT calculations, can be explained with the σ-hole which increases from F to I. Charge calculation and Atoms-In-Molecules analyses show that Mg⋯F–Ph bonding originates from electrostatic attraction between Mg2+ and the very polar Cδ+–Fδ− bond. For the heavier halobenzenes, polarization of the halogen atom becomes increasingly important (Cl < Br < I). Complexation with Mg leads in all cases to significant Ph–X bond activation and elongation. This unusual coordination of halogenated species to early main group metals is therefore relevant to C–X bond breaking.
Introduction
Halogenated benzenes (Ph–X, X = F, Cl, Br, I) are key building blocks in organic syntheses and also often used as polar but weakly coordinating solvents. This last property explains the scarcity of metal⋯X–Ph complexes. There is only a handful of well-characterized metal⋯X–Ph complexes.1,2 Despite the availability of three lone-pairs of electrons, Ph–X is a very weak Lewis base and electron donor. This is illustrated by the fact that its oxidized form, Ph–X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, is only known for iodosobenzene (X = I) and even this compound is highly explosive.3 In the series Ph–X (X = F, Cl, Br, I), iodobenzene is likely the best electron pair donor especially for the softer, larger transition metals. It is, however, also most susceptible to C–X bond cleavage and indeed the formation of transition metal⋯X–Ph complexes is proposedly the first step towards oxidative addition. Metal–halobenzene complexes are crucial to C–X bond activation and catalysis4,5 and especially of great importance in the challenging activation of strong C–F bonds.2,6
O, is only known for iodosobenzene (X = I) and even this compound is highly explosive.3 In the series Ph–X (X = F, Cl, Br, I), iodobenzene is likely the best electron pair donor especially for the softer, larger transition metals. It is, however, also most susceptible to C–X bond cleavage and indeed the formation of transition metal⋯X–Ph complexes is proposedly the first step towards oxidative addition. Metal–halobenzene complexes are crucial to C–X bond activation and catalysis4,5 and especially of great importance in the challenging activation of strong C–F bonds.2,6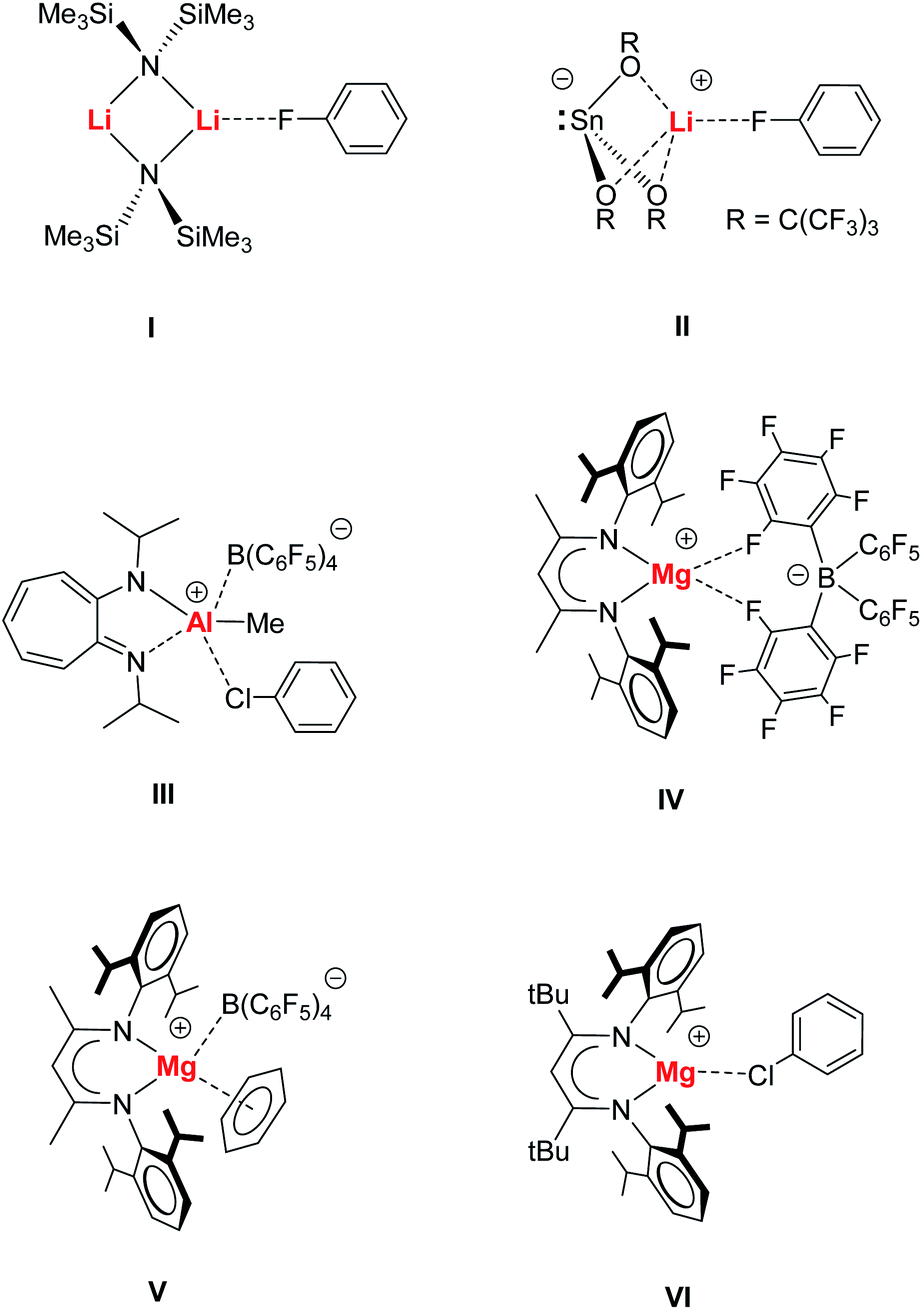
Main group metal⋯X–Ph complexes are even rarer and most examples deal with metal bonding of the most polar halobenzene Ph–F, e.g.I7 or II.8 Metal bonding of the heavier halobenzenes is much more uncommon. While metal⋯Cl–Ph complexes have been identified for Re, Zr and Rh,9a–d the cationic Al complex III represents a rare example for binding of a main group metal to a heavier halobenzene.9e Complexation of Ph–Br and Ph–I is limited to the softer metals Pd, Pt and Ag10 and to the best of our knowledge, main group complexes with such ligands are unknown.
We recently introduced syntheses and structures of cationic, Lewis base-free, alkaline earth metal complexes.11–16 The cationic β-diketiminate Mg complex [(MeBDI)Mg+][B(C6F5)4−] (IV) shows a chelating B(C6F5)4− anion but cation–anion association is weak and (MeBDI)Mg+ is more Lewis acidic than (MeBDI)AlMe+ or the benchmark Lewis acid B(C6F5)3.11 Highly Lewis acidic IV was shown to be able to form π-arene complexes (e.g.V),11,13–15 alkynes complexes11,16 and even a complex of the silyl ether O(SiMe3)2, that is normally fully inert to metal bonding, has been isolated.12
While Hill and co-workers reported a similar set of cationic complexes with the somewhat weaker coordinating anion Al(OC4F9)4−,17 we introduced a cationic β-diketiminate Mg complex with a much bulkier β-diketiminate ligand carrying tBu groups in the ligand's backbone. Restriction of the Mg coordination sphere weakens the Mg⋯B(C6F5)4− contacts and this allowed for isolation of a unique (tBuBDI)Mg+⋯Cl–Ph species (VI).15 Herein, we present extensive experimental and theoretical investigations on Mg–halobenzene complexes and discuss bonding and bond activation.
Results and discussion
Synthesis and solid state structures of cationic Mg⋯halobenzene complexes
In order to prepare complexes of the type [(BDI)Mg+·XPh][B(C6F5)4−] (X = F, Cl, Br, I) any coordinating ethereal solvent and aromatic solvents should be strictly avoided. Reaction of [(MeBDI)MgnPr]2 or (tBuBDI)MgnBu with excess [Ph3C+][B(C6F5)4−] in neat halobenzene generally led to a color change from dark orange to pale yellow, which indicated the reaction to be completed. For the cation with the smaller BDI ligand, (MeBDI)Mg+, only the PhF complex could be isolated (1); Scheme 1. The B(C6F5)4− anion is still weakly bound to Mg with two Mg⋯F contacts. Recrystallization from PhF led to [(MeBDI)Mg+·(FPh)3][B(C6F5)4−] (2) in which cation–anion contacts are fully cleaved. All attempts to isolate PhCl, PhBr or PhI complexes failed. These halobenzenes are clearly less polar than PhF and cannot compete with the Mg⋯B(C6F5)4− interaction in IV.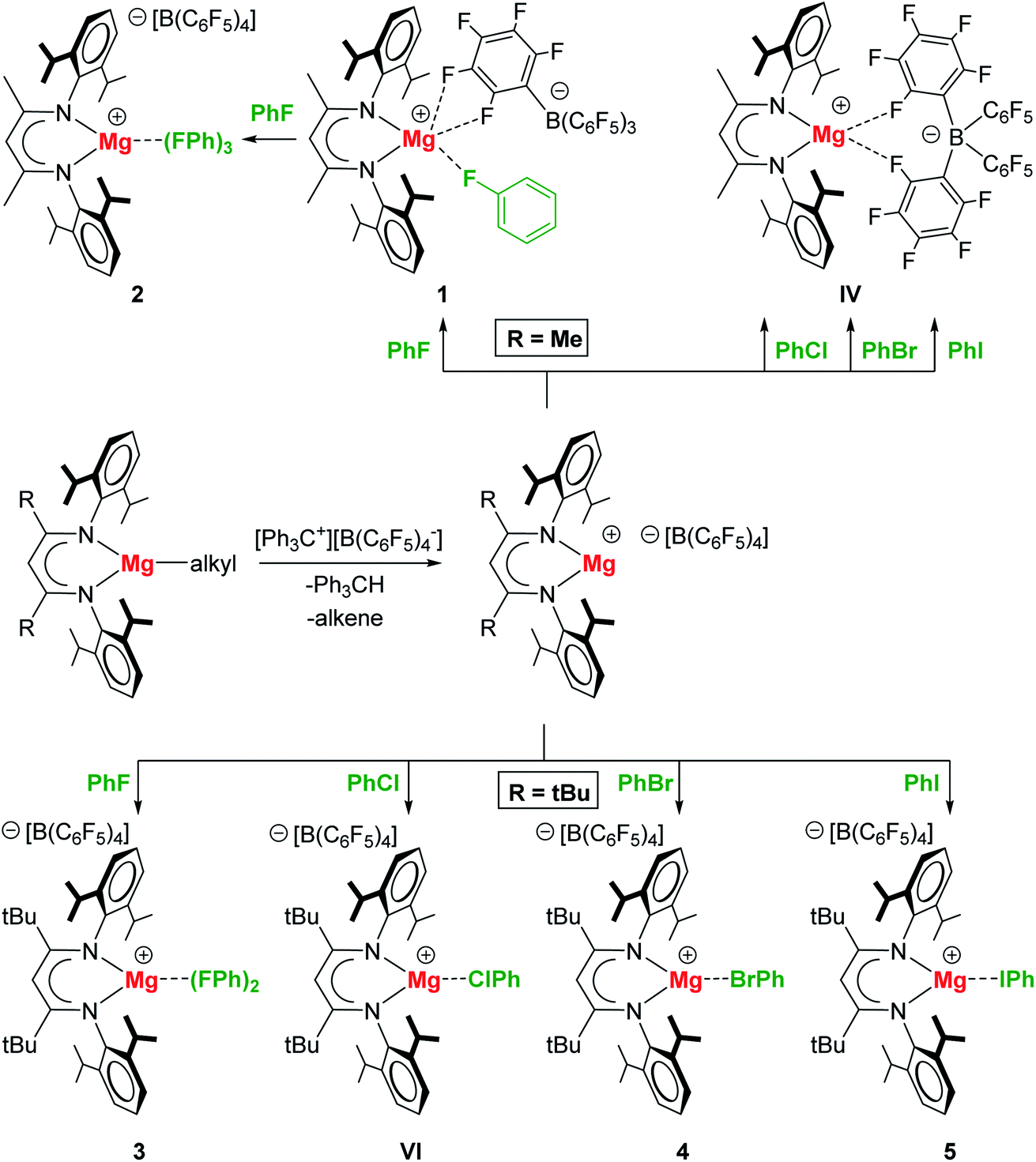 | ||
| Scheme 1 Syntheses of Mg⋯XPh complexes (X = F, Cl, Br, I). | ||
Switching to the cation with the bulkier tBuBDI ligand, however, weakens the Mg⋯B(C6F5)4− interaction and in this case we were able to isolate and structurally characterize the complete series of halobenzene complexes. The increased bulkiness of the BDI ligand led in all cases to cation–anion separation. For the most polar and least sterically encumbered PhF, a complex with two PhF ligands was isolated (3); Fig. 1 and Table 1. The geometry of the (tBuBDI)Mg+·(FPh)2 cation in 3 closely matches that of the cation in a similar complex with a different counter anion: [(CF3)3O]3Al–F–Al[O(CF3)3]3−, see Fig. S26.† This shows that the nature of the non-coordinating anion has no effect on Mg⋯FPh coordination. For the heavier, less polar, halobenzenes only complexes with one PhX ligand could be isolated: [(BDI)Mg+·XPh][B(C6F5)4−] (X = Cl, Br, I) (4–5, VI); Fig. 1 and Table 1. Complexes 4 and 5 are the first examples for the coordination of PhBr and PhI to a main group metal.
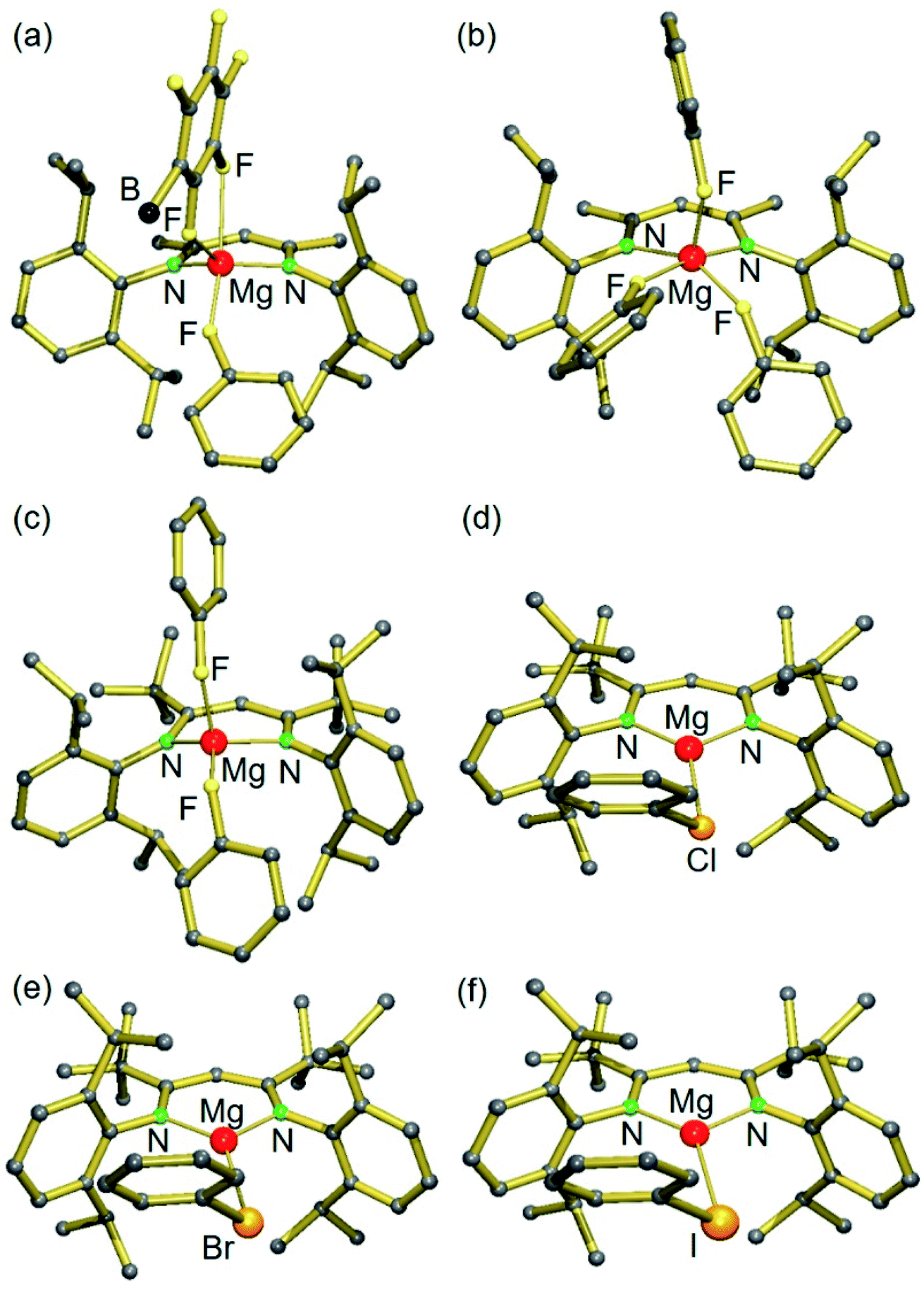 | ||
| Fig. 1 Crystal structures of (a) [(MeBDI)Mg+·FPh][B(C6F5)4−]; borate anion partially shown (1), (b) [(MeBDI)Mg+·(FPh)3] (2), (c) [(tBuBDI)Mg+·(FPh)2] (3), (d) [(tBuBDI)Mg+·ClPh] (VI),15 (e) [(tBuBDI)Mg+·BrPh] (4) and (f) [(tBuBDI)Mg+·IPh] (5). Anions and H atoms are not shown for clarity. | ||
| Mg⋯X(Ph) [Å] | Angle Mg–X–C [°] | (Ph)C–X [Å] | Free (Ph)C–X [Å]21 | |
|---|---|---|---|---|
| [(MeBDI)Mg+·FPh][B(C6F5)4−] (1) | 2.007 (2) | 151.9 (1) | 1.417 (3) | 1.364 (15) |
| [(MeBDI)Mg+·(FPh)3] (2) | 2.044 | 147.1 | 1.408 | |
| [(tBuBDI)Mg+·(FPh)2] (3) | 1.977 [1.961] | 139.1 [131.4] | 1.417 [1.431] | |
| [(tBuBDI)Mg+·ClPh] (VI) | 2.414 (1) [2.422] | 101.4(1) [101.2] | 1.788(2) [1.792] | 1.737 (5) |
| [(tBuBDI)Mg+·BrPh] (4) | 2.551 (1) [2.563] | 97.7(1) [97.9] | 1.938(1) [1.949] | 1.899 (12) |
| [(tBuBDI)Mg+·IPh] (5) | 2.748(1) [2.759] | 95.1(1) [93.8] | 2.118(2) [2.136] | 2.095 (15) |
While the Mg metal center in 1 and 2 is five-coordinate, that in the complexes with the bulkier tBuBDI ligand is four- (3) or three-coordinate (4–5, VI). The Mg⋯F(Ph) contacts range from 1.946(1) to 2.0533(14) Å and, although PhF is a neutral ligand that is known to be a weak donor, the shortest contact is just as short as the Mg2+–F− bonds in the F-bridged dimer [(MeBDI)Mg(μ-F) (THF)]2 (Mg–F av. 1.95 Å).18 Likewise, the distances Mg⋯Cl(Ph) and Mg⋯Br(Ph) of 2.414(1) Å and 2.551(1) Å, respectively, can be compared to the corresponding compounds containing X− anions: [(MeBDI)Mg(μ-Cl)]2 (Mg–Cl av. 2.3884 Å) and [(MeBDI)Mg(μ-Br)]2 (Mg–Br av. 2.539 Å).19 The Mg⋯I(Ph) contact of 2.748(1) Å in 5 is considerably longer than in monomeric (tBuBDI)MgI (Mg–I 2.597(1) Å)20 which is due to the terminal character of the Mg–I bond in the latter.
Interaction of the highly Lewis acidic (BDI)Mg+ cations with Ph–X causes substantial activation of the C–X bond, indicated by significant C–X bond lengthening. Compared to other metal⋯F(Ph) complexes the average C–F bond of 1.417 Å in 3 is elongated by 0.053 Å compared to that in free PhF (Table 1). Also the Mg complexes of PhCl, PhBr and PhI show a significant activation of the C–X bond. The extent of bond elongation decreases along the row C–F (0.053 Å) > C–Cl (0.051 Å) > C–Br (0.039 Å) > C–I (0.023 Å), i.e. with decreasing C–X bond polarity. There are only very few examples in literature in which a larger extent of C–X bond activation was observed. The elongation of the C–X bonds by cationic Mg complexes is of similar magnitude as C–X bond activation by transition metals like Zr, Re, Pd, Pt, Rh or Ag (see Table S2† for a detailed compilation of literature Ph–X⋯metal complexes).
Another interesting aspect of these Mg–halobenzene complexes is the Mg⋯X–Ph angle which steadily decreases with halogen size: F 139.1–151.9(1)° > Cl 101.4(1)° > Br 97.7(1)° > I 95.1(1)°. While the Ph–F⋯Mg angle is closest to linear, the Ph–I⋯Mg in 5 is nearly perpendicular. This trend is also found for the DFT calculated complexes and can be explained by an increasing σ-hole along the row F < Cl < Br < I (vide infra).
Cationic Mg⋯halobenzene complexes in solution
It is difficult to identify the Mg⋯halobenzene complexes in solution. They are insoluble in inert solvents like pentane or hexane and use of aromatic solvents would lead to Mg⋯π-arene complexes. Complexes were therefore characterized by NMR techniques in bromobenzene-d5. The existence of Mg⋯FPh interactions has been proven with 19F NMR. Complexes [(MeBDI)Mg+·FPh][B(C6F5)4−] (1) and [(tBuBDI)Mg+·(FPh)2] [B(C6F5)4−] (3) show signals at −120.9 ppm and −116.3 ppm, respectively. These are downfield shifted in respect to uncoordinated PhF in this solvent (−111.7 ppm). Concentration dependent NMR analysis gave upon dilution a shift of the 19F NMR resonance towards the value for uncoordinated PhF. The 1H NMR signals for the PhCl complex (VI) dissolved in bromobenzene-d5 overlap with the residual solvent signals and are not informative. For the PhBr complex (4) and PhI complex (5) exchange of the halobenzene with bromobenzene-d5 is expected. Indeed, the 1H NMR spectrum of 5 in bromobenzene-d5 only shows signals for free PhI.DFT calculations
DFT calculations were performed at the B3LYP/def2TZVP level of theory with the Grimme D3 dispersion correction using Becke–Johnson damping, unless indicated otherwise (see ESI† for further details). Preliminary studies on the interaction of very simple model systems (HMg+ and HC[C(H)NH]2Mg+) with Ph–X (X = F, Cl, Br, I) showed that, apart from Mg⋯X, also Mg⋯π-arene interactions contribute to their stability (see Fig. S29 and S30†). Although the latter interactions were not found in the Mg⋯F–Ph complexes, they become more prevalent along the row Mg⋯π-PhCl < Mg⋯π-PhBr < Mg⋯π-PhI, i.e. with increasing electron density in the ring π-system. This influence of the inductive effect of the X-substituent on metal⋯π-PhX interactions has been previously recognized experimentally and theoretically.22,23 Such metal⋯π-PhX interactions could be avoided by calculations on the full ligand system. Calculated structures for (tBuBDI)Mg+⋯XPh (X = F, Cl, Br, I) closely match the crystal structures (Fig. S31† and Table 1). Not only the Mg+⋯XPh distances but especially the Mg+⋯X–Ph angles are in excellent agreement with those found in the crystal structures (Table 1). Given the fact that angular distortions are generally very facile, this close match between experiment and theory is surprising.The gradually decreasing Mg⋯X–Ph angle along the row X = F > Cl > Br > I finds its origin in the concept of halogen bonding. Dating back to the first observation of I2⋯NH3,24 halogen bonding has been reviewed as early as 1968.25 Halogen bonding, generally defined as a noncovalent interaction between a halogen atom and a Lewis base,26 consists of different contributions including electrostatic interaction, dispersion, charge transfer and halogen atom polarization.27 The angular geometries of large numbers of halogen-bonded complexes have been statistically evaluated and were rationalized by the halogen's σ-hole.28 Previously dubbed as “polar flattening”,26 the σ-hole is a region of positive electrostatic potential on the halogen's surface (Fig. 2). Although halogen atoms are rich in free electron pairs, the surface of the atom opposite of the C–X bond is positively charged. This region increases with halogen size and is generally discussed to explain Lewis base-halogen interactions,29 however, less frequently is also taken into account when discussing Lewis acid-halogen interactions.30 Decreasing Mg⋯X–Ph angles can be explained by the gradually decreasing belt of electron density on the halogen with increasing halogen size (Fig. 2). An alternative way to look at increased bending of the Mg⋯X–Ph geometries along the series X = F < Cl < Br < I is the change in the halogen atom hybridization31 which could be seen as the origin of the σ-hole. Two of the free electron pairs on the halogen are in p-orbitals perpendicular to the C–X axis. The third unshared pair along the C–X axis is in a hybrid orbital of s-character and some degree of p-character which is strongly reduced with increasing halogen size (for F circa 25% and for I circa 8%) explaining that the σ-hole is particularly large for the heavier halogen atoms.28
 | ||
| Fig. 2 (Top) Schematic representation of the σ-hole and possible coordination sites for Mg. (Bottom) Electrostatic maps of PhF, PhCl, PhBr and PhI at an isosurface of 0.003 a.u. of the total electron density. | ||
In order to quantify the driving force for the bent Mg⋯X–Ph structures, the energies for linearization have been calculated for the full cation (tBuBDI)Mg+⋯XPh (X = F, Cl, Br, I) (Fig. S33†). While linearization of the Mg⋯F–Ph unit hardly costs any energy (ΔH = 2.0 kcal mol−1), linearization free energies strongly increase for PhCl (ΔH = 15.1 kcal mol−1), PhBr (ΔH = 18.8 kcal mol−1) and PhI (ΔH = 22.1 kcal mol−1).
Formation of the (tBuBDI)Mg+⋯XPh complexes is in all cases exothermic. The following ΔH values have been calculated for the gas phase interaction of (tBuBDI)Mg+ with PhX (kcal mol−1): PhF −21.6, PhCl −28.0, PhBr −29.3 and PhI −30.6. However, using a solvent correction (PCM = benzene) turns this order and gives lower, more realistic, values (kcal mol−1): PhF −12.8, PhCl −12.3, PhBr −11.8 and PhI −10.9. Although the complexation energies decrease from PhF to PhI, the difference within the series is very small. The strong Mg⋯FPh interaction can be attributed to the highly polar Cδ+–Fδ− bond which upon bonding with Mg2+ is even further polarized (see NPA charges in Fig. 3). The charge distribution also shows that there is hardly electron transfer from PhF to Mg. However, electron transfer from PhX to Mg increases along the row X = F < Cl < Br < I. This is likely related to decreasing electronegativity differences in the Mg⋯X bond along the same row which makes the Mg⋯I interaction partially covalent. The Mg⋯IPh bond is strong on account of polarization of the core electron density of the iodine atom. The latter has been visualized by an Atoms-In-Molecules (AIM) analysis.
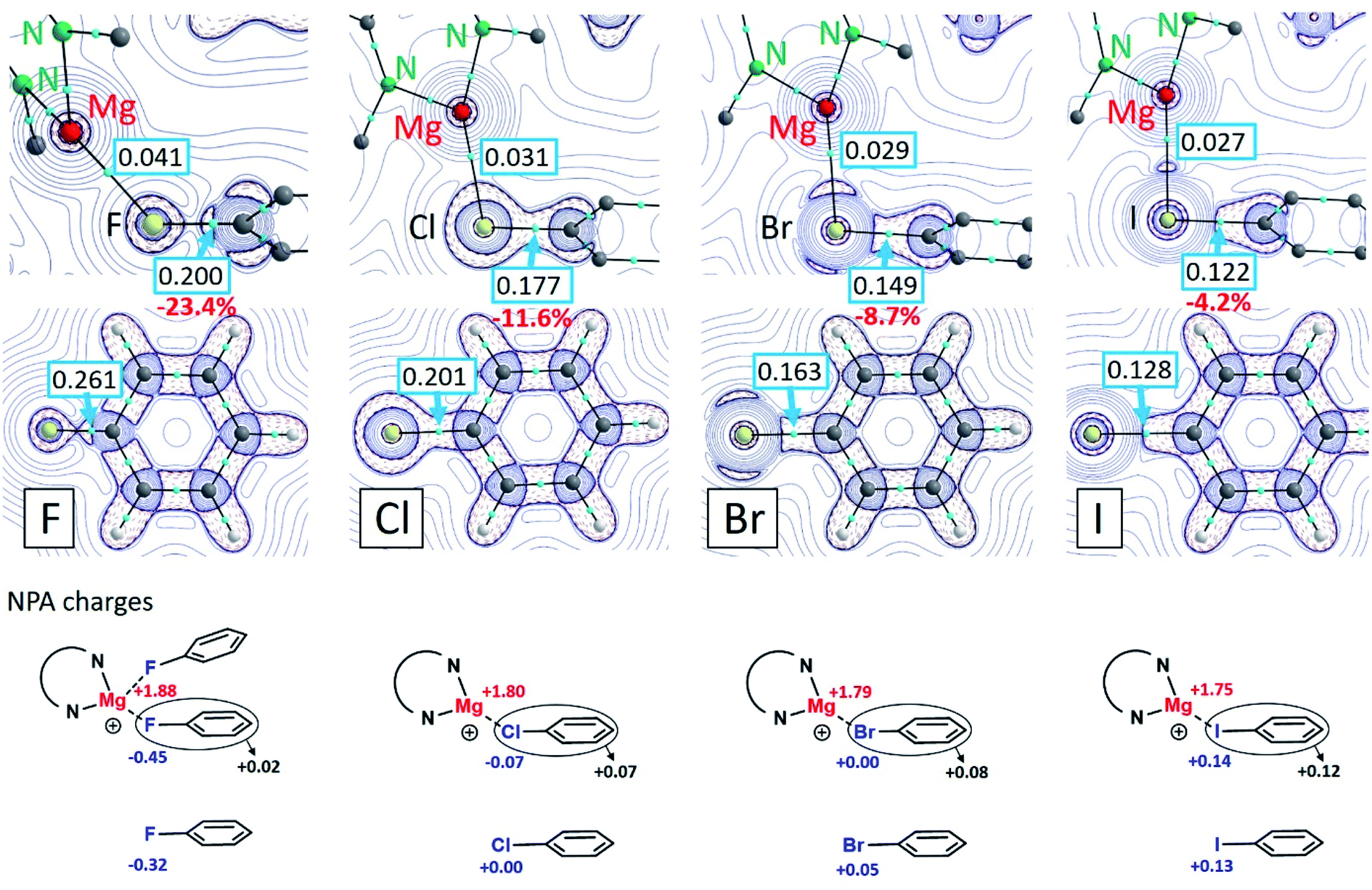 | ||
| Fig. 3 Contour plots of the negative Laplacian, −∇2ρ(r), for the cations (tBuBDI)Mg+⋯XPh (X = F, Cl, Br, I) in the Mg⋯X–C plane (top row) and for the free halobenzenes PhX in the molecule plane (middle row). Light-blue dots are bond-critical-points (BCP) and boxed numbers show the electron densities in these points. NPA charges are shown in the bottom part. | ||
Contour plots of the negative Laplacian, −∇2ρ(r), for the cations (tBuBDI)Mg+⋯XPh (X = F, Cl, Br, I) are shown in Fig. 3 which for comparison also shows the electron densities for the free halobenzene ligands. In all cases, distinct Mg⋯X bond paths have been found. The electron density in the bond-critical-point (BCP) decreases gradually from X = F to I. The electron density in the BCP on the C–X axis of the coordinated PhX ligands is always smaller when compared to the free PhX ligands. This is in line with the observed C–X bond activation by Mg coordination. Weakening of the C–X bond decreases along the row: C–F (−23.4%) > C–Cl (−11.6%) > C–Br (−8.7%) > C–I (−4.2%). This is in agreement with the experimentally observed elongations of the C–X bond (Table 1): C–F (+3.7%) > C–Cl (+2.9%) > C–Br (+2.0%) > C–I (+1.1%). Analysis of the Wiberg bond indices for the C–X bonds by the NBO method also shows that Mg coordination leads in all cases to C–X bond weakening (Fig. S32†). While the bonding interaction between Mg2+ and PhF can be explained by electrostatic interaction with the strongly polar Cδ+–Fδ− bond, for the heavier halogens mainly polarization of the halogen atom is important. This is clearly visible by accumulation of electron density on the Mg⋯X axis (Fig. 3).
Conclusion
Highly Lewis acidic cationic Mg complexes with “naked” Lewis base-free Mg centers have been the key to the isolation of the full series of halobenzene complexes: Mg⋯XPh (X = F, Cl, Br, I). Although bonds between PhF and PhCl and main group metals have been reported, complexation of the softer ligands, PhBr and PhI, by main group metals is unprecedented.Tuning the bulk of the β-diketiminate ligand (BDI) is crucial to the isolation of (BDI)Mg+⋯XPh complexes. For the smaller MeBDI ligands a competition between (MeBDI)Mg+⋯B(C6F5)4− and (MeBDI)Mg+⋯XPh interaction is observed and only complexes with the most polar halobenzene, PhF, could be isolated. Using the bulkier tBuBDI ligand shuts off cation–anion contacts and the full series (tBuBDI)Mg+⋯XPh (X = F, Cl, Br, I) could be structurally characterized.
Complexation of PhX with Mg leads to C–X bond activation. Elongation of the C–X bond is most extreme for PhF and decreases for the heavier halogens. Most remarkable is the Mg⋯XPh coordination mode: whereas the Mg⋯F–C angle tends to be large and linear, the Mg⋯I–C angle is close to perpendicular. The steadily decreasing Mg⋯X–C angle (X = F > Cl > Br > I) can be explained with the halogen σ-hole. This region of positive electrostatic potential on the halogen's surface, opposite of the C–X bond, increases with halogen size. Previous reports on the statistical examination of non-bonded interactions of arylhalides with neighboring molecules in crystal structures have shown the preferred bonding sites for electrophiles and nucleophiles.30 For the heavier arylhalides (Br, I), however, only scatter plots for interactions with nucleophiles have been identified. This is due to the lack of metal interactions with arylbromide and iodide species. Here we show mapping of the σ-hole down the full halogen group with a single Lewis acidic Mg cation.
The experimentally observed structures are surprisingly well modelled by DFT calculations. Since bending angles are generally subject to variation, there is clearly a distinct preference for the bent Mg⋯X–Ph coordination modes observed for the heavier halogens. Indeed, the halogen–metal bond is highly directional27 and the linearization energies strongly increase along the row X = F < Cl < Br < I.
Although weaker than a classical O–H⋯O hydrogen bond, the complexation enthalpies between 11–13 kcal mol−1 (ΔH) for (tBuBDI)Mg+⋯XPh formation are still considerable and relevant for physical interaction. Bonding between Mg and PhF is mainly dictated by the very polar Cδ+–Fδ− bond. Bonding between Mg and the heavier PhX ligands relies increasingly on halogen polarization. Evidence for electrostatic Mg⋯X–Ph interactions is mainly based on solid state structures. In solution there is competition with Mg⋯solvent bonding and dissociation equilibria lie largely on the side of free uncoordinated PhX. Despite their weakness, we embrace the idea that metal⋯X–Ph interactions can play an important role in halogen exchange reactions. Being stronger than typical van der Waals interactions, the preferred approach of early main group metal reagents and halobenzenes is defined by metal⋯X–Ph bonding. Since the interaction between a positively charged s-block metal and organic halides activates the C–X bond, they could be especially important in the C–X bond breaking process. Although the here presented Mg⋯XPh complexes are in solution short-living intermediates, their challenging isolation and structural characterization contribute to mechanistic insights in the emerging field of s-block metal catalysis.32
Experimental section
General considerations
All experiments were conducted under an inert nitrogen atmosphere using standard Schlenk and glovebox techniques (MBraun, Labmaster SP). All solvents were degassed with nitrogen, dried over activated aluminium oxide (Solvent Purification System: Pure Solv 400-4-MD, Innovative Technology) and stored over 3 Å molecular sieves unless noted otherwise. Fluorobenzene, chlorobenzene, bromobenzene and iodobenzene were dried over calcium hydride, distilled under N2 atmosphere and stored over molecular sieves 3Å. C6D6 and C6D5Br (99.6% D, Sigma Aldrich) were dried over 3Å molecular sieves. [CPh3+][B(C6F5)4−] (Boulder Scientific) was used as received. Complexes [(MeBDI)MgnPr]2,11 (tBuBDI)MgnBu,33 and [(tBuBDI)Mg+·(PhCl)][B(C6F5)4−]15 were synthesized according to literature procedures. NMR spectra were recorded with a Bruker Avance III HD 400 MHz or a Bruker Avance III HD 600 MHz spectrometer. The spectra were referenced to the respective residual signals of the deuterated solvents. Elemental analysis was performed with an Euro EA 3000 (Euro Vector) analyzer. All crystal structures have been measured on a SuperNova (Agilent) diffractometer with dual Cu and Mo microfocus sources and an Atlas S2 detector. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. 2042836 (1), 2042837 (2), 2042838 (3), 2042839 (4), 2042840 (5) and 2042841 for [(tBuBDI)Mg+·(FPh)2][{(CF3)3O}3Al–F–Al{O(CF3)3}3−].Author contributions
A. Friedrich: conceptualization, investigation, validation, formal analysis, writing – original draft, visualization. J. Pahl: conceptualization, investigation, validation, formal analysis. J. Eyselein: formal analysis, validation. J. Langer: formal analysis, validation. N. van Eikema Hommes: formal analysis, validation, supervision. A. Görling: validation, supervision. Sjoerd Harder: conceptualization, writing – original draft – review and editing, visualization, validation, supervision, project administration.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We acknowledge Mrs C. Wronna and A. Roth (University of Erlangen-Nürnberg) for CHN analyses and J. Schmidt and Dr C. Färber (University of Erlangen-Nürnberg) for assistance with the NMR analyses.References
- R. J. Kulawiec and R. H. Crabtree, Coord. Chem. Rev., 1990, 99, 89–115 CrossRef CAS.
- H. Plenio, Chem. Rev., 1997, 97, 3363–3384 CrossRef CAS.
- V. V. Zhdankin, Hypervalent Iodine Chemistry, John Wiley & Sons Ltd, Chichester, UK, 2013 Search PubMed.
- T. G. Richmond, Topics in Organometallic Chemistry – Activation of Unreactive Bonds and Organic Synthesis, Springer-Verlag, Berlin Heidelberg, 1999 Search PubMed.
- F. J. Fernandez-Alvarez, M. Iglesias, L. A. Oro and V. Passarelli, Comprehensive Inorganic Chemistry II – Bond Activation and Catalysis, Elsevier, Amsterdam, 2013 Search PubMed.
- U. Rosenthal, V. V. Burlakov, P. Arndt, A. Spannenberg, U. Jaeger-Fiedler, M. Klahn and M. Hapke, Activating Unreactive Substrates, Wiley-VCH, Weinheim, 2009 Search PubMed.
- P. G. Williard and Q. Y. Liu, J. Org. Chem., 1994, 59, 1596–1597 CrossRef CAS.
- A. V. Korolev, F. Delpech, S. Dagorne, I. A. Guzei and R. F. Jordan, Organometallics, 2001, 20, 3367–3369 CrossRef CAS.
- (a) X.-Y. Liu, K. Venkatesan, H. W. Schmalle and H. Berke, Organometallics, 2004, 23, 3153–3163 CrossRef CAS; (b) A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 18463–18478 CrossRef CAS; (c) O. J. Metters, S. J. K. Forrest, H. A. Sparkes, I. Manners and D. F. Wass, J. Am. Chem. Soc., 2016, 138, 1994–2003 CrossRef CAS; (d) P. Ren, S. D. Pike, I. Pernik, A. S. Weller and M. C. Willis, Organometallics, 2015, 34, 711–723 CrossRef CAS; (e) A. V. Korolev, E. Ihara, I. A. Guzei, V. G. Young and R. F. Jordan, J. Am. Chem. Soc., 2001, 123, 8291–8309 CrossRef CAS; (f) S. Balasubramaniam, S. Kumar, A. P. Andrews, B. Varghese, E. D. Jemmis and A. Venugopal, Eur. J. Inorg. Chem., 2019, 3265–3269 CrossRef CAS.
- (a) M. D. Butts, B. L. Scott and G. J. Kubas, J. Am. Chem. Soc., 1996, 118, 11831–11843 CrossRef CAS; (b) L. P. Press, B. J. McCulloch, W. Gu, C.-H. Chen, B. M. Foxman and O. V. Ozerov, Chem. Commun., 2015, 51, 14034–14037 RSC; (c) J. Powell, M. Horvath and A. Lough, J. Organomet. Chem., 1993, 456, 27–28 CrossRef; (d) J. Powell, M. J. Horvath, A. Lough, A. Phillips and J. Brunet, J. Chem. Soc., Dalton Trans., 1998, 637–646 RSC.
- J. Pahl, S. Brand, H. Elsen and S. Harder, Chem. Commun., 2018, 54, 8685–8688 RSC.
- J. Pahl, H. Elsen, A. Friedrich and S. Harder, Chem. Commun., 2018, 54, 7846–7849 RSC.
- J. Pahl, A. Friedrich, H. Elsen and S. Harder, Organometallics, 2018, 37, 2901–2909 CrossRef CAS.
- S. Brand, H. Elsen, J. Langer, S. Grams and S. Harder, Angew. Chem., Int. Ed., 2019, 58, 15496–15503 CrossRef CAS.
- A. Friedrich, J. Pahl, H. Elsen and S. Harder, Dalton Trans., 2019, 48, 5560–5568 RSC.
- J. Pahl, T. E. Stennett, M. Volland, D. M. Guldi and S. Harder, Chem.–Eur. J., 2019, 25, 2025–2034 CrossRef CAS.
- L. Garcia, M. D. Anker, M. F. Mahon, L. Maron and M. S. Hill, Dalton Trans., 2018, 47, 12684–12693 RSC.
- (a) H. Hao, H. W. Roesky, Y. Ding, C. Cui, M. Schormann, H. G. Schmidt, M. Noltemeyer and B. Žemva, J. Fluorine Chem., 2002, 115, 143–147 CrossRef CAS; (b) C. Bakewell, A. J. P. White and M. R. Crimmin, J. Am. Chem. Soc., 2016, 138, 12763–12766 CrossRef CAS.
- A. P. Dove, V. C. Gibson, P. Hormnirun, E. L. Marshall, J. A. Segal, A. J. P. White and D. J. Williams, Dalton Trans., 2003, 3088–3097 RSC.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem.–Eur. J., 2010, 16, 938–955 CrossRef CAS.
- Crystal structure of PhF: V. R. Thalladi, H. C. Weiss, D. Bläser, R. Boese, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1998, 120, 8702–8710 CrossRef CAS ; crystal structure of PhCl: N. P. Penionzhkevich, N. I. Sadova and L. V. Vilkov, Zh. Strukt. Khim., 1979, 20, 527–529 Search PubMed ; crystal structures of PhBr and PhI: F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, 1–19 RSC.
- J.-F. Gal, P.-C. Maria, M. Decouzon, O. Mó and M. Yáñez, Int. J. Mass Spectrom., 2002, 219, 445–456 CrossRef CAS.
- N. Hallowita, E. Udonkang, C. Ruan, C. E. Frieler and M. T. Rodgers, Int. J. Mass Spectrom., 2009, 283, 35–47 CrossRef CAS.
- F. Guthrie, J. Chem. Soc., 1863, 16, 239–244 RSC.
- H. A. Bent, Chem. Rev., 1968, 68, 587–648 CrossRef CAS.
- R. Sedlak, M. H. Kolár and P. Hobza, J. Chem. Theory Comput., 2015, 11, 4727–4732 CrossRef CAS.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- T. Clark, M. Henneman, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291–296 CrossRef CAS.
- (a) G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS; (b) J. Y. C. Lim and P. D. Beer, Chem, 2018, 4, 731–783 CrossRef CAS.
- N. Ramasubbu, R. Parthasarathy and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308–4314 CrossRef CAS.
- W. Kutzelnigg, Angew. Chem., Int. Ed. Engl., 1984, 23, 272–295 CrossRef.
- (a) S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS; (b) M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC; (c) Early Main Group Metal Catalysis – Concepts and Reactions, ed. S. Harder, Wiley-VCH, Weinheim, Germany, 2020 Search PubMed.
- V. Balasanthiran, M. H. Chisholm, K. Choojun, C. B. Durr and P. M. Wambua, Polyhedron, 2016, 103, 235–240 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic details including ORTEP plots, 1H, 11B, 19F and 13C NMR spectra, details for the DFT calculations including XYZ-files. CCDC 2042836–2042841. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06321e |
| This journal is © The Royal Society of Chemistry 2021 |