DOI:
10.1039/D1NR02872C
(Paper)
Nanoscale, 2021,
13, 16525-16532
Improving the knock-in efficiency of the MOF-encapsulated CRISPR/Cas9 system through controllable embedding structures†
Received
5th May 2021
, Accepted 18th August 2021
First published on 1st September 2021
Abstract
Appropriate tuning of robust artificial coatings can not only enhance intracellular delivery but also preserve the biological functions of genetic molecules in gene based therapies. Here, we report a strategy to synthesize controllable nanostructures in situ by encapsulating CRISPR/Cas9 plasmids into metal–organic frameworks (MOFs) via biomimetic mineralization. The structure-functionality relationship studies indicate that MOF-coated nanostructures dramatically impact the biological features of the contained plasmids through different embedding structures. The plasmids are homogeneously distributed within the heterogeneous nanoarchitecture and protected from enzymatic degradation. In addition, the plasmid–MOF structure exhibits excellent loading capability, pH-responsive release, and affinity for plasmid binding. Through in vitro assays it was found that the superior MOF vector can greatly enhance cellular endocytosis and endo/lysosomal escape of sheltered plasmids, resulting in successful knock-in of GFP-tagged paxillin genomic sequences in cancer cell lines with high transfection potency compared to our previous studies. Thus, the development of new cost-effective approaches for MOF-based intracellular delivery systems offers an attractive option for overcoming the physiological barriers to CRISPR/Cas9 delivery, which shows great potential for investigating paxillin-associated focal adhesions and signal regulation.
1. Introduction
Gene editing technologies have pioneered new areas of research for the treatment of various diseases, especially for cancer, and have emerged as a revolutionary technique in the field of gene therapeutics.1 Among the different gene editing tools, the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein-9 nuclease (Cas9) system represents the most promising option at present, which utilizes single guide RNA (sgRNA) for site identification and can repair DNA double-strand breaks (DSBs) by non-homologous end joining (NHEJ) and homologous directed repair (HDR) through cleavage by Cas9 protein.2,3 In contrast to NHEJ that generates knockout epitopes by introducing mutable insertions or deletions (indels) into DSBs, the HDR pathway can create exact deletions, base substitutions, or insertions into target-coding genes.4,5 Thus, the HDR pathway can be used to foster correction of diseased genes, insertion of epitope labels or fluoro-reporters, and over-expression of genes of interest in a site-specific way.6,7 Unlike advanced gene knockout, it remains a great challenge to improve the efficiency of accurate CRISPR/Cas9-mediated gene editing or HDR-mediated gene knock-in.8–10 In addition to the low efficiency of CRISPR/Cas9-mediated HDR-induced knock-in itself, effective delivery is an important constraint to the success of this approach. Therefore, the exploitation of safe and efficient delivery vectors is critical for CRISPR/Cas9 based gene therapy. Although traditional viral systems11 such as adenoviruses and lentiviruses are versatile carriers with great transfection efficiency, their clinical utility raises significant concerns about cytotoxicity, pathogenicity, off-targeting, DNA carrying capability, and potential side-effects.12 Considering the shortcomings of viral vectors, non-viral delivery systems have shown better safety, less immunogenicity, cheaper production and greater carrying capacity, and easier functional modification.13 Therefore, developing novel non-viral vectors to improve the biocompatibility and to better protect and target editing of CRISPR/Cas9 will greatly promote the application of HDR-mediated gene knock-in.14,15
Metal organic frameworks (MOFs)16 comprising metal ions/clusters with organic ligand linkages have become a prospective platform for medical applications owing to their high porosity, versatility, good biocompatibility and biodegradability.17–20 To date, zeolitic imidazole frameworks (ZIFs)21 are the most extensively studied MOFs for accommodating biomacromolecules (including proteins, DNA, and enzymes) that can be in situ entrapped into the growing ZIFs by biomimetic mineralization, which provides biocompatible synthesis conditions that shield the biomolecules from denaturation in the formation process.22–24 During biomimetic mineralization, biomolecules can effectively induce the formation of ZIFs and manipulate the structures of the resulting porous structures, and the resulting ZIF structures vary widely in terms of retention and protection of biomolecular functions.25 To promote the biological function of a MOF–biomolecule complex, a better comprehension of the structure–function relationship between the MOF structure and the guest biomolecule is needed.
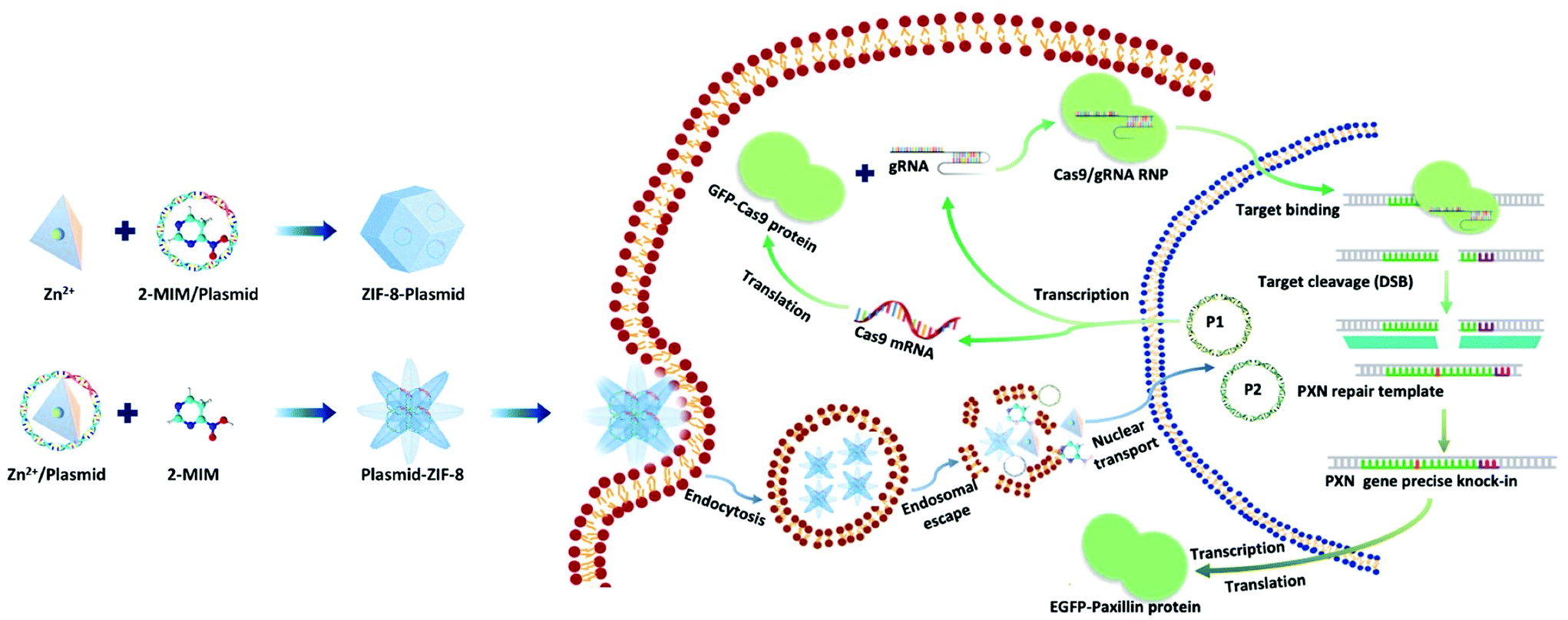
Here, we present a biomimetic strategy that allows in situ tailored ZIF-8 biocomposites to control the nanostructures. As shown in Scheme 1, by adding the plasmids in different parts of the precursor, the formed ZIF-8 coatings can be changed from being homogeneous structures to heterogeneous structures. The obtained ZIF-8-plasmid (ZP) tends to enlarge homogeneously by slow precipitation in premixed plasmid/organic linker solutions due to the competitive coordination effect of plasmids with 2-methylimidazole (2-MIM) ligands. While in the premixed plasmid/metal solution, the plasmid is enriched with metal ions via electrostatic interactions and rapidly triggers nucleation,26,27 which significantly promotes the formation of heterostructured plasmid-ZIF-8 (PZ). We then evaluated the effects of both embedding strategies of nanostructures on plasmid biofunctionality and showed that the heterostructured nanoparticles could enhance the retention and protect the hosted plasmid. Based on the above synthetic strategy, we explored the intracellular delivery capability of the CRISPR/Cas9 PZ system for Paxillin (PXN) knock-in via the HDR pathway. Paxillin28 is the earliest protein to be recruited to nascent adhesions and plays a critical role in mediating signalling events between the topical tumor microenvironment and tumor cells, which can influence both tumor spread and metastasis.29,30 After encapsulation of both different plasmids that cleave the PXN gene (GFP-Cas9-paxillin_gRNA, P1, 9288 bp) and repair (AICSDP-1: PXN-EGFP, P2, 5448 bp), the nanocarriers can facilitate cellular endocytosis and endo/lysosomal escape and subsequently initiate GFP-tagged PXN gene sequence-specific knock-in. The study showed that these proof-of-concept functionalized MOF nanovectors can overcome different physical barriers to efficiently deliver CRISPR/Cas9 plasmids to cells, thereby effectively recovering the edited genes via HDR.
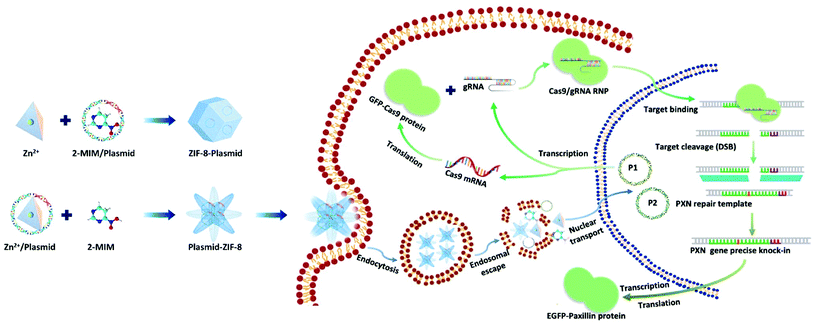 |
| | Scheme 1 Schematic illustration of the ZIF-8-based nanocarrier for the delivery of CRISPR/Cas9 plasmids: preparation of plasmid loaded nanostructures, intracellular delivery of the two kinds of plasmids to the nucleus for paxillin gene knock-in. | |
2. Results and discussion
2.1. Plasmid-directed morphological evolution of MOF biomimetic mineralization
Plasmids are ring-shaped double-stranded DNA (dsDNA) with intact genes on the sequence, which mostly exist in bacteria and can be expressed in mammalian cells by transfection.31,32 P1 and P2 plasmids were chosen for this proof-of-concept knock-in research due to their broad properties, easy availability, and routine use in cell and molecular biological studies. The plasmids themselves are nonfluorescent but can emit intense green fluorescence when P1 transcribes and translates the GFP-tagged Cas9 protein and when P2 knocks-in successfully and sequentially expresses the EGFP-tagged paxillin protein (as in Scheme 1). To the best of our knowledge, there has been little exploration of CRISPR/Cas9 plasmid-encapsulated MOF vectors for intracellular gene delivery and knock-in via biomimetic mineralization synthesis.
Here, P1 was chosen as a model plasmid, and in a typical biomineralization process, different amounts of P1 (from 1 μl to 6 μl lyophilized) were dispersed in an aqueous Zn2+ solution and then added to a 2-MIM solution, forming P1-ZIF-8 (P1Z) nanostructures. Alternatively, the plasmid was distributed in an aqueous solution containing 2-MIM, followed by the addition of Zn2+ solution to form ZIF-8-P1 (ZP1) nanostructures. The evolution of the embedding structure of the composites was studied by transmission electron microscopy (TEM), dynamic light scattering (DLS) and zeta potential, respectively (Fig. 1). The mechanism of biomineralization that occurs during natural processes is extensively ascribed to the particular ability of amino acids, peptide segments and more complicated biological entities to concentrate inorganic cations (e.g. Ca2+ and Zn2+) into the seed biominerals. Thus, when P1 was first introduced in a Zn2+ solution, plasmids with abundant phosphate groups tended to condense Zn2+ and promoted the formation of the P1Z complexes. As shown in Fig. 1a, when the biomineralization was modulated with 1 to 3 μl of the plasmid, the complex formed a homogeneous rhombohedral dodecahedron with a diameter of about 100 nm, the same as the ZIF-8 nanoparticles without plasmid in Fig. S1† during the synthesis. Upon adding more plasmids (4 to 5 μl), the nanostructures gradually evolved into an irregular polyhedron with spiky surfaces (Fig. 1a, 4 and 5 μL). A further increase in plasmid dose (to 6 μl) resulted in morphological evolution to decussation of approximately 200 nm size (Fig. 1a, 6 μL). Further raising the plasmid dose lead to the assembly into a 2D layered architecture until the crystal structure disappeared as shown in Fig. S2.† Therefore, the local concentration of Zn2+ cations was increased to promote the pre-nuclear clustering of ZIF-8 surrounding the plasmid, thus controlling the crystal formation. From the DLS and zeta potential measurements of P1Z, as shown in Fig. S3a,† the surface charges are quite stable in this system, although the hydrated size of the nanostructure is slightly larger than that shown in TEM. However, in the ZP1 system (Fig. 1b), the nucleation rates of a series of nanoparticles were much slower due to the coordination competition effect between the plasmid and the 2-MIM ligand, in which the morphologies of the nanoparticles remained uniformly rhombohedron dodecahedron, but the size of the ZP1 nanostructures increased from 100 nm to greater than 200 nm, with the doping of the plasmid from 1 to 6 μl, and the hydrated sizes even reached 600 nm as shown in Fig. S3b.† As can be seen from the time-dependent growth rates of ZIF-8, ZP1, and P1Z shown in Fig. 2a–c, this morphological evolution is believed to be a result of the competition between the plasmid and the ligand for coordination. After 30 min, the ZP1 nanoparticles increased in size as the continuous growth slowed down and the nanoparticles precipitated out, while the other two solutions remained homogeneous with small sized nanoparticles. To further confirm the distribution of P1 in ZIF-8, P1 was labelled with DAPI, the ZIF-8 precursor was mixed with sulfo-Cyanine5.5 NHS ester (Cy5.5), and a similar encapsulation process was carried out. Three-dimensional confocal laser scanning microscopy (CLSM) images showed that P1 was uniformly dispersed within P1Z and heavily aggregated in the ZP1 nanostructure (Fig. 2d and e). Together, these results demonstrate the formation of different plasmid–MOF embedding structures with the expected controllable structure.
 |
| | Fig. 1 TEM images showing the morphologies of P1Z (a) and ZP1 (b) composites formed with different amounts of plasmid. | |
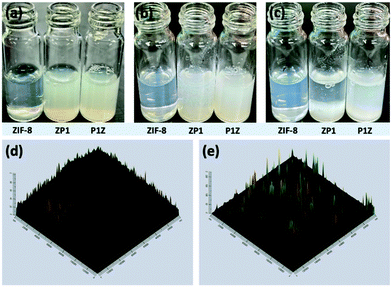 |
| | Fig. 2 Time-dependent characterization of ZIF-8, ZP1, and P1Z nanostructures that illustrates the growth progress of the as-obtained composites at each investigated time point: 10 s (a), 10 min (b), and 30 min (c), respectively. The 3D-CLSM profiles of the distribution of plasmid (green fluorescence) and ZIF-8 (red fluorescence) in P1Z (d) and ZP1 (e). | |
2.2. The effect of MOF nanostructures on plasmid functionality
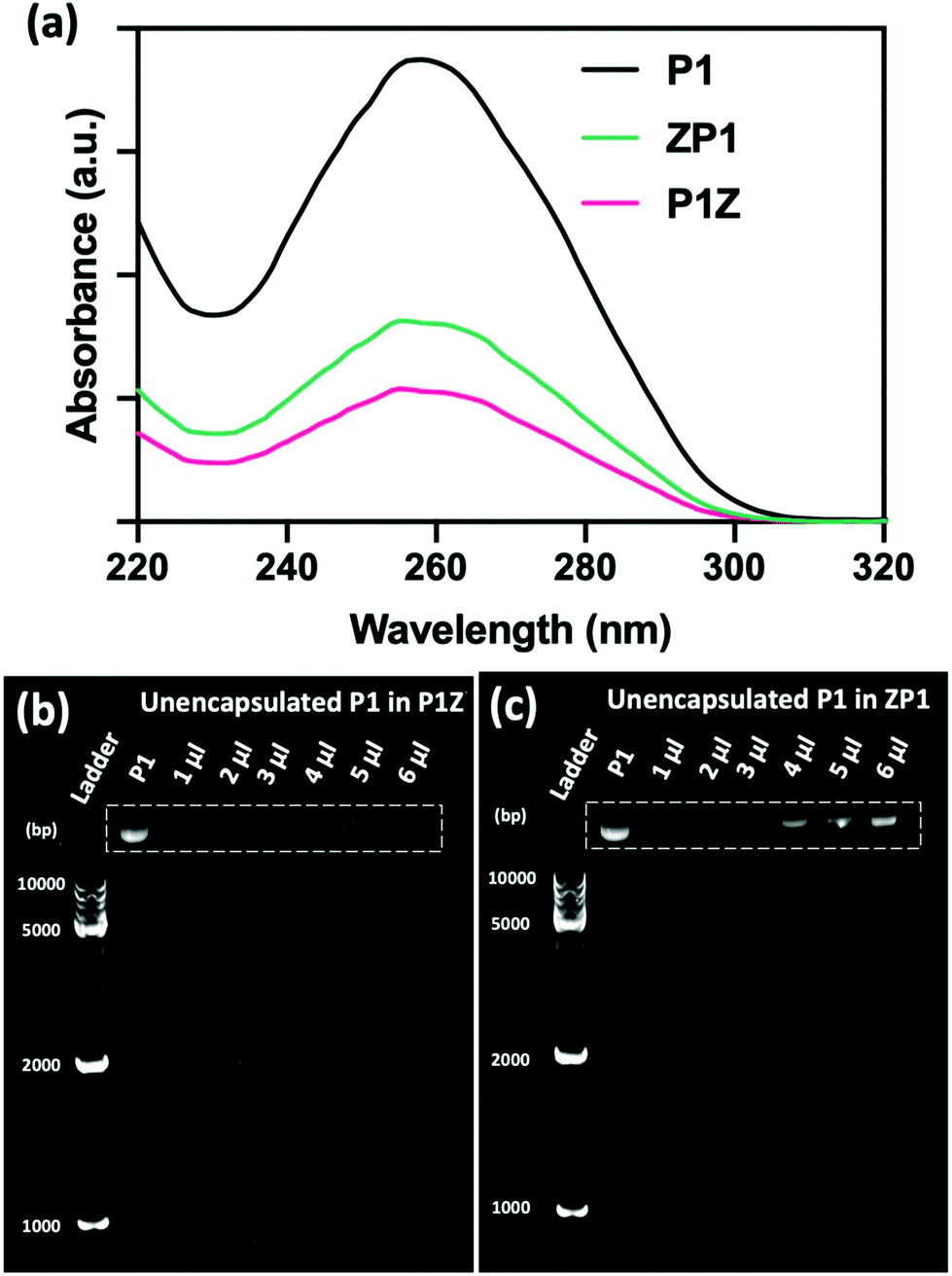
The loading ability and binding affinity of the carrier to the plasmid are essential elements to obtain the required gene therapy outcome.33 The loading capacity of P1Z and ZP1 nanostructures was measured by UV-vis absorption spectroscopy to determine the difference of P1 concentration in the supernatant before and after encapsulation, as shown in Fig. 3a. The P1 concentration was determined from the supernatant after centrifugation, the encapsulation capacity of P1 in different embedding structures are listed in Table S1,† the maximum loading of P1 was calculated as 2.0 μg (80% in 5 μl) of P1Z and 1.2 μg (60% in 4 μl) of ZP1 nanostructures in 10 synthesis solutions, respectively. Agarose gel electrophoresis was also performed to check the loading capacities of P1Z and ZP1 nanostructures (in Fig. 3b and c). As the dose of P1 increased, only very faint bands were seen for the supernatants of P1Z with a high amount of P1. However, the intensity of the bands gradually became apparent for the supernatant of ZP1, which is attributed to the high loading capacities of the P1Z vector on the CRISPR/Cas9 plasmid. The increased loading capacity of P1Z compared to ZP1 nanostructures was ascribed to the rapid nucleation due to the stronger electrostatic binding of the metal ions and plasmid. The binding affinity of P1 in the P1Z and ZP1 nanostructures was investigated via agarose gel electrophoresis. Bare P1 with complete electrophoretic displacement was observed in both P1 lanes in Fig. S4,† appearing as a bright primary band in the highly abundant P1 supercoiled form and as a faint secondary band in the other circular form. P1 was fully bound within the P1Z nanostructures at amounts of P1 ranging from 1 to 6 μl (Fig. S4a†). In contrast, a faint supercoiled P1 band was observed in the agarose gel of the ZP1 nanostructure (Fig. S4b†). This suggests that in the ZP1 embedding structure, some P1 cannot be entirely bound under the presence of an electric field, which may lead to the discharge of some P1 molecules attached to the ZIF surface. The results reveal that in the P1Z embedding structure, the binding affinity is enhanced by premixing the plasmid with metal ions.
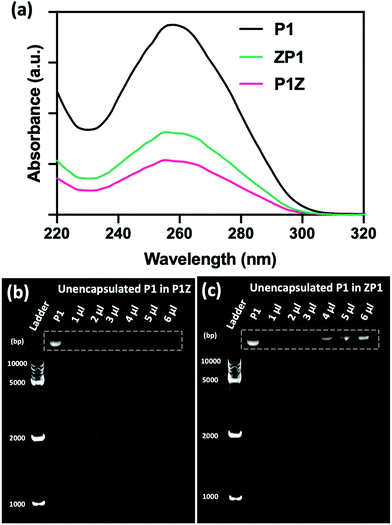 |
| | Fig. 3 (a) UV-vis spectra of original P1 before encapsulation and the residual P1 in supernatant after encapsulated by P1Z and ZP1 nanostructures. The agarose gel shifts of the supernatants from P1Z (b) and ZP1 (c) nanostructures with the increase of plasmid dosage of P1. | |
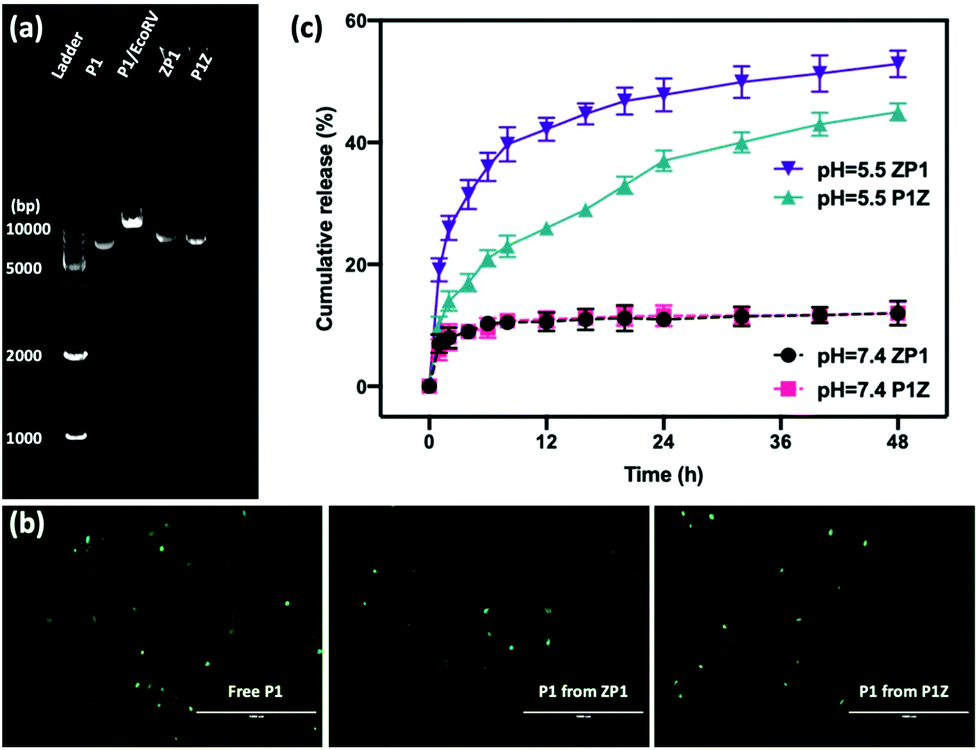
Moreover, the protection of CRISPR/Cas9 plasmids from restriction enzyme degradation by the carrier is also a crucial issue of gene delivery. The stability of P1 against Eco32I (EcoRV) degradation was investigated in two structures by gel electrophoresis. As shown in Fig. 4a, free P1 severely cleaved into a linearized DNA conformation and left a lower band in P1/EcoRV. P1 was extracted from both ZP1 and P1Z nanostructures after EcoRV treatment, where the DNA backbone was not digested and was found to be in the same form as free P1. Moreover, the intact P1 in P1Z accounted for 82.3% as found via a gel electrophoresis analysis upon EcoRV processing, which was better than that in ZP1 (64.5%), displaying better conservation of the P1Z structure. To demonstrate that the encapsulated plasmids remain intact in terms of functionality, transfection experiments were performed by using released plasmids with a regular transfection medium (Lipofectamine 2000). Human osteosarcoma epithelial cells (U2OS) were transfected with P1, P1Z and ZP1. Any considerable impairment of the plasmid will prevent effective gene expression and therefore the cells will not display fluorescence. Lipid-based standard cells transfected with P1, P1 released from P1Z, and ZP1, all showed fluorescence (Fig. 4b). That means the plasmid does not suffer any kind of damage in the encapsulation process that would have negative effects on the functional activity. Furthermore, the pH-responsive release behaviour of P1 in both structures was investigated by immersing them in PBS solutions with different pH values and their cumulative release from nanocarriers was detected by UV-visible spectrophotometry. Under normal physiological conditions (pH = 7.4), both P1Z and ZP1 nanostructures were stable as shown in Fig. 4c and no significant release of P1 (10%) was detected. In an acidic environment (pH = 5.5), 52% of P1 was unleashed from ZP1 nanostructures in 48 h. The more controlled pH-dependent release was less explosive for 12 h and slower (40% after 48 h) due to the fast nucleation of P1Z nanoparticles. These results suggest that P1Z-based nanocarriers exhibit efficient pH-responsive release, which is a more optimal feature for gene delivery and expression.
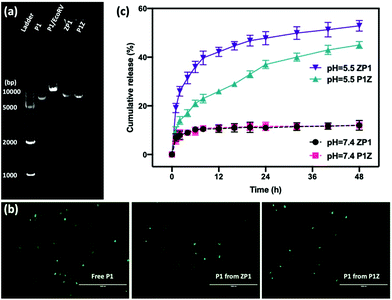 |
| | Fig. 4 (a) The agarose gel electrophoresis image of the EcoRV digestion assay: ladder, naked P1, EcoRV-treated P1, the releasing P1 from ZP1 and P1Z after EcoRV treatment. (b) DNA remains functionally intact during encapsulation and release. Fluorescence microscopy images of U2OS cells transfected with P1, with P1 previously encapsulated in ZP1 and P1Z, the scale bar = 1000 μm. (c) The cumulative release profile of P1 from P1Z and ZP1 nanocarriers at different pH values, (dash line pH = 7.4 and solid pH = 5.5). | |
2.3. Transfection study of plasmid-ZIF nanostructure in vitro
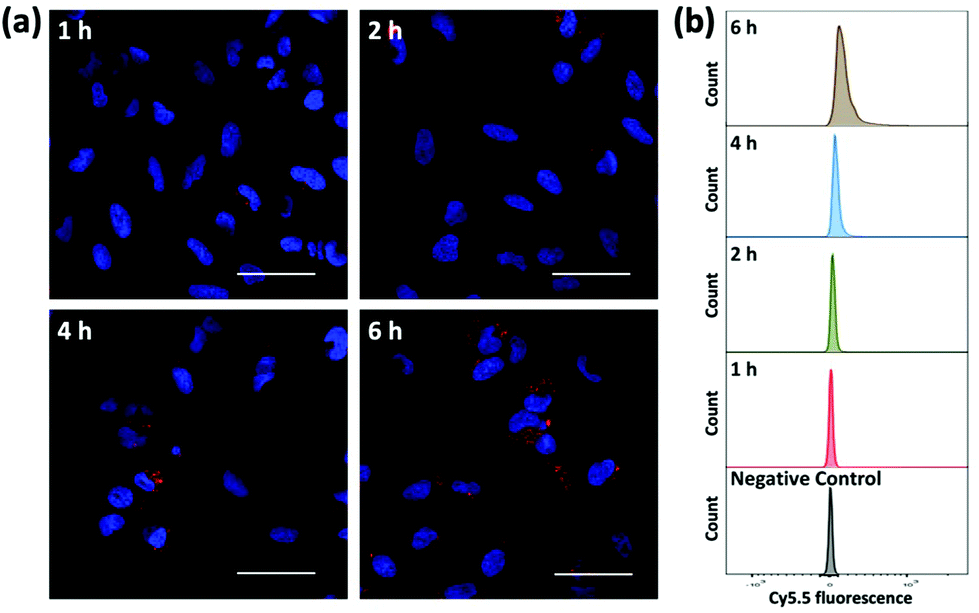
To evaluate the ability of plasmid-ZIF nanostructures for gene delivery, their effects on cell viability, endocytosis and transfection were studied. An essential feature of any biomaterial is low cellular toxicity and high biological compatibility. To characterize the effect of P1Z and P1P2-ZIF-8 (P1P2Z) on cell viability, a 48 h WST-1 assay was carried out on U2OS cells. As shown in Fig. S5,† there was no remarkable change in cell viability at different concentrations of P1Z and P1P2Z compared to the control based on the amount of P1 (up to 2000 ng mL−1). Efficient internalization of plasmid-ZIF into target cells is essential for valid CRISPR/Cas9-based gene editing. Depending on the cytotoxicity assay results, P1Z was incubated with U2OS cells for various time periods with certain concentrations of P1Z. The cellular uptake behaviour of P1Z (red fluorescence from Cy5.5) was evaluated by CLSM and flow cytometry. The red signal of P1Z gradually increased with time as illustrated in Fig. 5a. After 2 hours of incubation, significant uptake of P1Z was observed, demonstrating their rapid endocytosis into the cells. Similar results were seen in the median fluorescence intensity (MFI) from flow cytometry analysis (Fig. 5b). The results of the quantification analysis indicate that increased uptake can be derived from different time points. The endocytosis of different MOF nanoparticles (ZIF-8, P1Z and ZP1) was also compared, as shown in Fig. S6,† because of the difference in particle size; ZIF-8 has the smallest size and was more easily taken up by the cells and therefore the most efficient, followed by P1Z and then the largest sized ZP1, which indicates that cellular uptake is strongly dependent on particle size.34,35
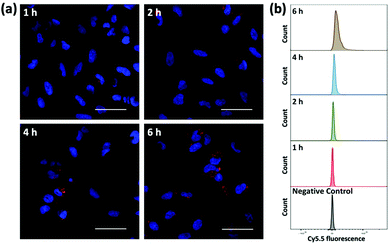 |
| | Fig. 5 Cellular uptake and intracellular distribution of P1Z nanocarriers after 1, 2, 4 and 6 h incubation detected by CLMS (a) and flow cytometry (b). The nanocarriers exhibit a red signal with Cy5.5, while the nuclei were stained with DAPI; the scale bar = 50 μm. | |
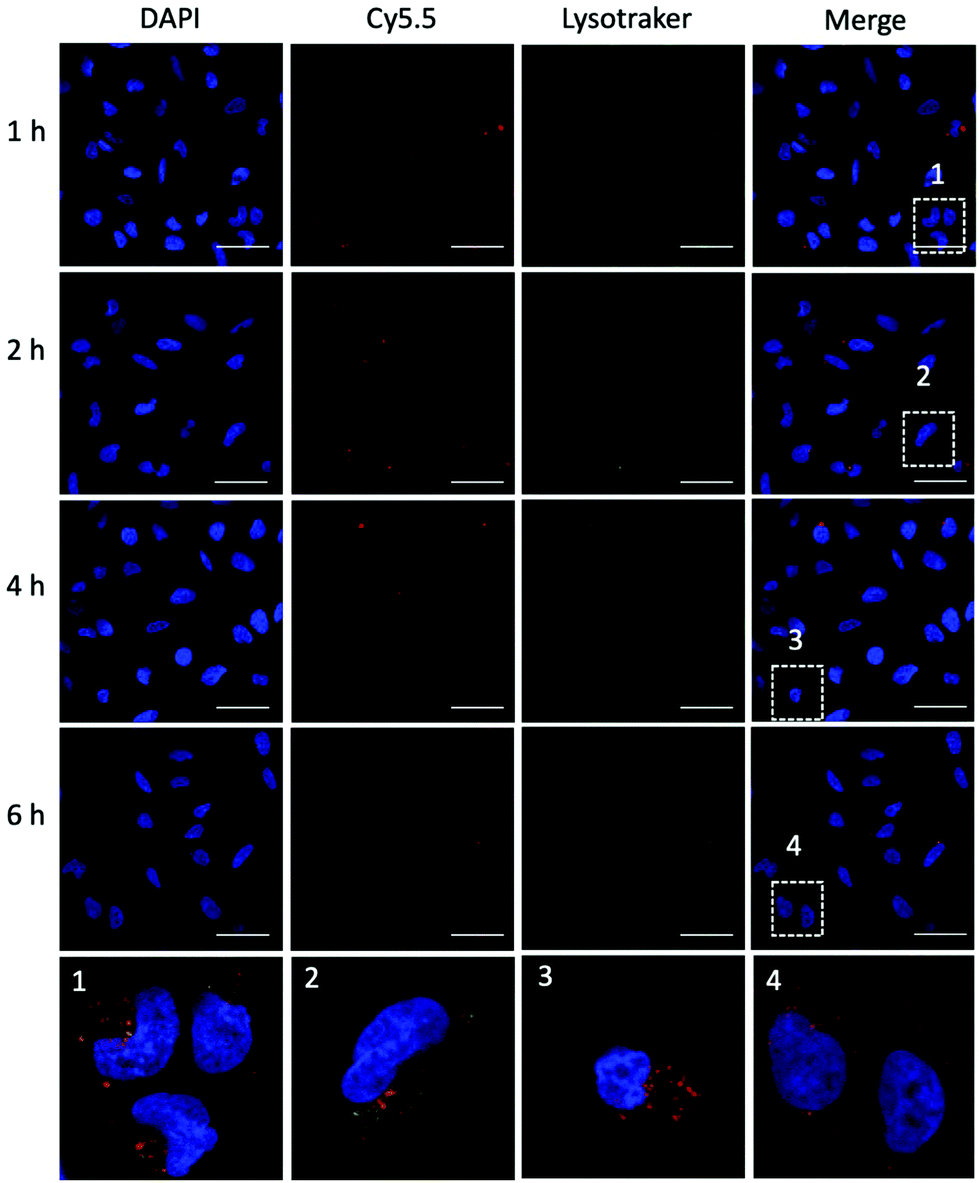
To achieve efficient gene editing, endo/lysosomal escape is a further pivotal feature of nanocarriers.36 To investigate the endo/lysosomal escape ability, U2OS cell lines were incubated with Cy5.5-labeled P1Z and then the cells were visualized with CLSM after 1, 2, 4 and 6 hours of incubation. Endo/lysosomal staining of cells was performed with LysoTracker@Green prior to confocal microscopy observation. As shown in Fig. 6, after 1 h of incubation, a clear overlap (yellow) between the red fluorescence of the nanocarriers and green fluorescence of the endo/lysosomes can be observed, indicating that P1Z can be localized in the endosomes after internalization into U2OS cells. After 6 h of incubation, in the absence of gene expression, the intracellular Cy5.5-labelled P1Z showed an increase in red fluorescence intensity and a greater overlap with the nucleus-specific blue fluorescence (Fig. 6, enlarged 4), while the green fluorescence became weaker as the escape of nanostructures led to a reduction in intracellular lysosomes. The overlapping fluorescence was also analysed with the ImageJ software, based on the data calculated from the Pearson correlation coefficient (PCC, see Fig. S7†), where 0 is no overlapping up to 100% for total colocalization. At 1 h, the red and green fluorescence largely overlapped together reaching 77% and then with time the red and green fluorescence gradually separated to 68% at 2 h and 50% at 4 h. At 6 h, they almost completely separated, and the overlap fluorescence ratio decreased to 11%, which means that most of the nanoparticles were released from the lysosomes at 6 h. Together, the data clearly demonstrated that P1Z can efficiently deliver plasmids into cells via the endocytosis approach, which depends on excellent escape due to the proton sponge effect37 from endo/lysosomes. This is essential to avoid plasmid degradation in cells and can guarantee the successful initiation of gene expression.
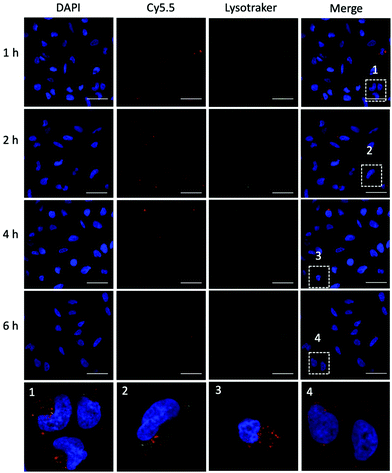 |
| | Fig. 6 CLSM images (63×) of U2OS cells after incubation with P1Z (red) for 1, 2, 4 and 6 h. Lysotracker@Green (green) was used to stain the acidic organelles (endosomes) and DAPI was used to stain the nucleus. Co-localization was indicated by yellow fluorescence; the scale bar = 50 μm. | |
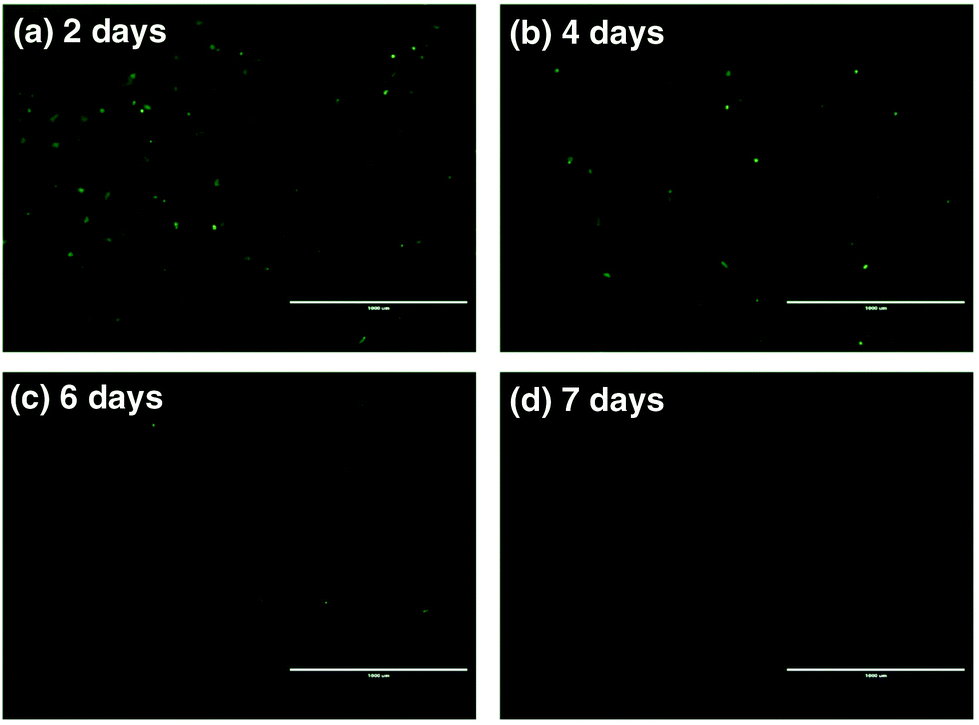
To further assess the ability of nanovectors for CRISPR/Cas9 plasmid delivery, we then investigated plasmid expression in target tumor cells. P1-encoded Cas9 and sgRNA scaffolds were fused with the downstream GFP expression sequence oligos, which can serve as fluoro-markers. To track the green signal at various time spots upon transfection, we cultured U2OS tumor cells with P1, ZIF-8 and P1Z and monitored the fluorescence after 2, 4, 6 and 7 days. A distinct blocky GFP signal could be observed in U2OS cells by fluorescence microscopy after a 2 day treatment with P1Z as illustrated in Fig. 7. In contrast, U2OS cells treated with free P1 or ZIF-8 did not display detectable GFP signals, which indicates the value of using the devised delivery platform for plasmid transfection. The transfection efficiency of P1Z was then evaluated, as shown in Fig. S8,† with Lipofectamine-2000 as a positive control and flow cytometry results after 48 hours of nanoparticle treatment showing that P1Z had a transfection efficiency of 8.02%, comparable to that of Lipofectamine-2000 at 9.08%, but P1Z has the great advantage of being less expensive and easy to prepare. Nevertheless, the GFP-positive cell population was drastically reduced due to the dilution of plasmids in cell proliferation. A lower expressing signal could be determined after 4 to 6 days of treatment and when cells were incubated for 7 days, the GFP signal almost disappeared.
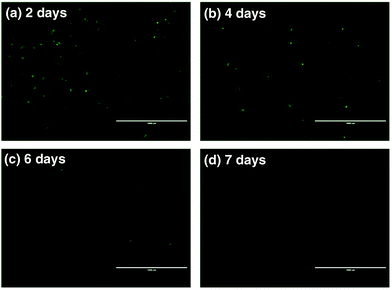 |
| | Fig. 7 Fluorescence microscopy images of U2OS cells treated with P1Z after 2 (a), 4 (b), 6 (c), and 7 day (d) incubation to confirm the in vitro transfection and GFP expression; the scale bar = 1000 μm. | |
2.4.
In vitro gene knock-in
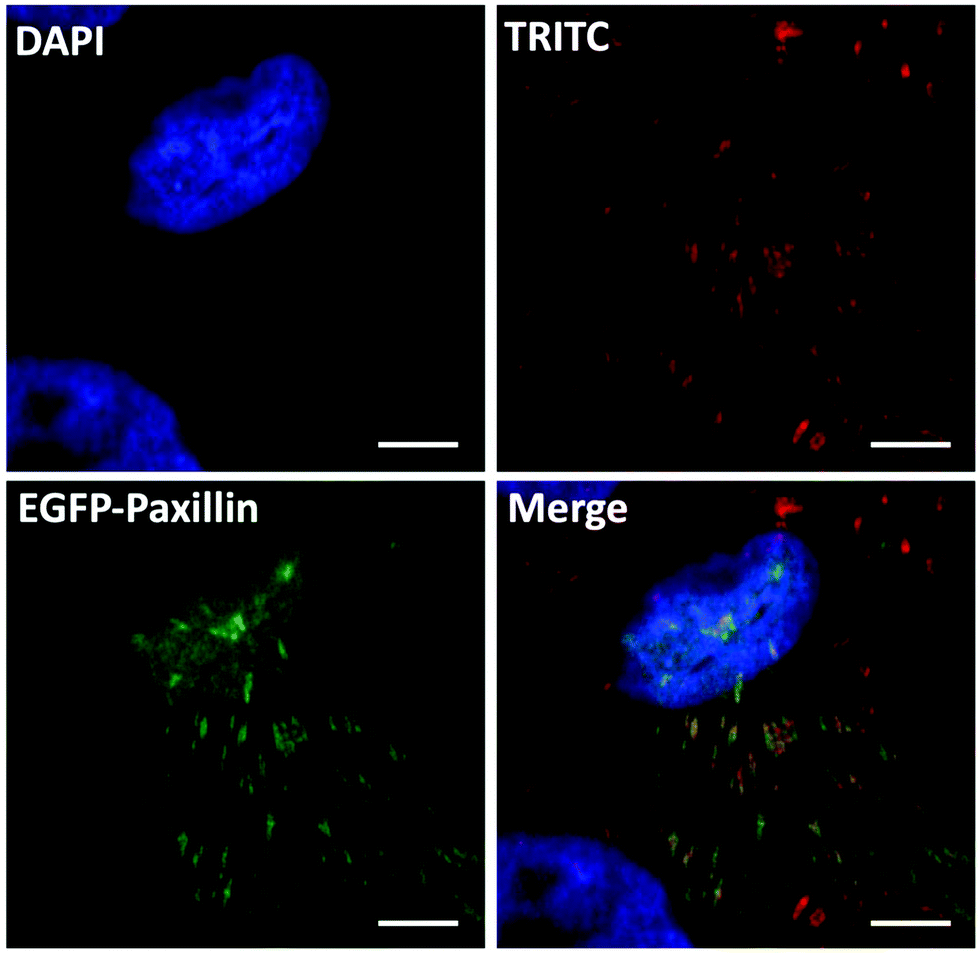
To verify the gene editing efficiency of the proposed nanovectors, we investigated the knock-in efficacy of PXN in vitro. In the assay, U2OS cells were transfected with nanovectors containing P1 and P2 in equal molar ratios. As the PXN sequence in plasmid P2 has been tagged with a GFP-linked expression cassette, a western blotting analysis was performed on whole-cell lysates from the edited U2OS cells after P1P2Z treatment for 96 and 120 hours38,39 (Fig. S9†). Immunoblotting with an antibody against the endogenous Paxillin protein yielded products with molecular weights that are consistent with the expected molecular weight of the unlabelled and EGFP-labelled protein. The expression of the fused EGFP-Paxillin protein gradually increased along with the gene knock-in and repair processes. Notably, the appropriate EGFP-Paxillin fusion protein product was obtained despite the low expression efficiency, indicating that the EGFP-Paxillin sequence of P2 was well inserted into the DSB of Paxillin cleaved by the Cas9/sgRNA ribonucleoprotein complexes expressed by P1 and successfully expressed the EGFP-Paxillin protein. According to the discussion above, green signals can be clearly separated from P1 expression disturbances, which can disappear up to 7 days because of transient expression. The gene editing cells could subsequently be checked by fluorescence microscopy.40 Therefore, the fluorescence study of gene editing behaviour on U2OS cells was initiated on the 8th-day after the treatment with P1P2Z. A distinct GFP luminescence could be visualized by fluorescence microscopy as expected and as shown in Fig. S10†. To validate the proper structural localization of GFP to Paxillin, focal adhesion and immunofluorescence colocalization of fibronectin staining have been investigated. Fibronectin is a ligand for more than a dozen integrin receptor family members that bind to paxillin and allows an environmental signal to transduce into the cell interior. After plating with fibronectin, the focal adhesion sites localized by U2OS cells showed a distribution of spots with a brilliant green fluorescence. A clear colocalization of distinct fluorescent channels (paxillin, red and GFP, green) was obtained when the edited cells were stained with monoclonal paxillin antibody and TRITC-conjugated goat anti-rabbit lgG (expected to label both GFP-tagged and untagged paxillin protein derivatives in the same cell), as observed in Fig. 8. Taken together, the results demonstrate that the GFP-tagged paxillin gene has been inserted into the proper location in U2OS cells, suggesting that the plasmid-ZIF-based nanovector is reliable and specialized for the delivery of the CRISPR/Cas9 editing system.
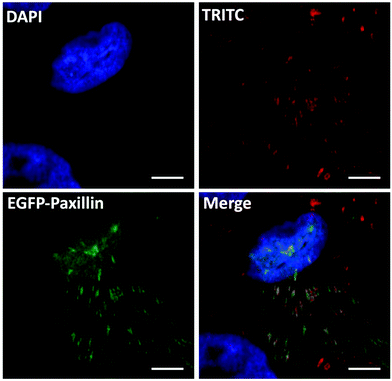 |
| | Fig. 8 Confocal immunofluorescent analysis of P1P2Z-treated U2OS cells using primary antibody (anti-paxillin antibody [Y113]) and secondary antibody (TRITC conjugated Goat Anti-Rabbit IgG (H + L)) showing colocalization with GFP signals; the scale bar = 5 μm. | |
3. Conclusions
In summary, we present a cytocompatible, plasmid-directed approach that is able to tailor MOF-encapsulated bioarchitecture into controllable nanostructures via biomimetic mineralization and emphasize the significance of MOF structures for upholding the biological functionality of the obtained nanostructures. It was found that plasmids can be well distributed throughout the plasmid-MOF embedding structure and benefit from efficient protection against enzymatic degradation. The CRISPR/Cas9 plasmid-MOF system showed good loading, proper protection against enzymatic degradation and pH-responsive release of the plasmid, exhibiting better cellular endocytosis and efficient endo/lysosomal escape properties, making it outstanding for gene knock-in. This work not only provides a fast, facile and low-cost method to load gene motifs in controlled nanostructures for efficient intracellular transfection but also sheds light on the potential of MOF-based non-viral vectors for use in a variety of gene therapies.
Author contributions
C. L., X. X. and H. Z. designed the project. O. K. prepared the samples for the plasmids. W. Z. prepared the samples for the gel electrophoresis. X. X. performed the CLSM measurements. G. J. supervised the cell experiments. Data collection and analysis were conducted by C. L., J. M. R., and H. Z. supervised the project. All the authors contributed to and commented on this paper.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
C. L. and X. X. contributed equally to this work. The authors are thankful to Prof. Johanna Ivaska for donating the plasmids used in this manuscript. The Cell Imaging and Cytometry Core facility (Turku Bioscience, University of Turku, Åbo Akademi University and Biocenter Finland) is acknowledged for its services, instrumentation, and expertise. Electron microscopy samples were processed and analyzed in the Electron Microscopy Laboratory, Institute of Biomedicine, the University of Turku, which receives financial support from Biocenter Finland. This work was supported by the National Natural Science Foundation of China (81871472), the Academy of Finland (grants no. 309374 and 328933) and the Sigrid Jusélius Foundation.
Notes and references
- A. S. Timin, A. R. Muslimov, K. V. Lepik, O. S. Epifanovskaya, A. I. Shakirova, U. Mock, K. Riecken, M. V. Okilova, V. S. Sergeev, B. V. Afanasyev, B. Fehse and G. B. Sukhorukov, Nanomedicine, 2018, 14, 97–108 CrossRef CAS PubMed
 .
.
- S. K. Alsaiari, S. Patil, M. Alyami, K. O. Alamoudi, F. A. Aleisa, J. S. Merzaban, M. Li and N. M. Khashab, J. Am. Chem. Soc., 2018, 140, 143–146 CrossRef CAS PubMed .
- L. Cong, F. A. Ran, D. Cox, S. L. Lin, R. Barretto, N. Habib, P. D. Hsu, X. B. Wu, W. Y. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed .
- H. X. Wang, M. Li, C. M. Lee, S. Chakraborty, H. W. Kim, G. Bao and K. W. Leong, Chem. Rev., 2017, 117, 9874–9906 CrossRef CAS PubMed .
- K. Yoshimi, Y. Oka, Y. Miyasaka, Y. Kotani, M. Yasumura, Y. Uno, K. Hattori, A. Tanigawa, M. Sato, M. Oya, K. Nakamura, N. Matsushita, K. Kobayashi and T. Mashimo, Hum. Genet., 2021, 140, 277–287 CrossRef CAS PubMed .
- L. Li, L. J. Song, X. W. Liu, X. Yang, X. Li, T. He, N. Wang, S. L. X. Yang, C. Yu, T. Yin, Y. Z. Wen, Z. Y. He, X. W. Wei, W. J. Su, Q. J. Wu, S. H. Yao, C. Y. Gong and Y. Q. Wei, ACS Nano, 2017, 11, 95–111 CrossRef CAS PubMed .
- J. P. Zhang, X. L. Li, G. H. Li, W. Chen, C. Arakaki, G. D. Botimer, D. Baylink, L. Zhang, W. Wen, Y. W. Fu, J. Xu, N. Chun, W. Yuan, T. Cheng and X. B. Zhang, Genome Biol., 2017, 18, 35 CrossRef PubMed .
- C. Z. Long, J. R. McAnally, J. M. Shelton, A. A. Mireault, R. Bassel-Duby and E. N. Olson, Science, 2014, 345, 1184–1188 CrossRef CAS PubMed .
- D. Heck, M. S. Kowalczyk, D. Yudovich, R. Belizaire, R. V. Puram, M. E. McConkey, A. Thielke, J. C. Aster, A. Regev and B. L. Ebert, Nat. Biotechnol., 2014, 32, 941–946 CrossRef PubMed .
- X. J. He, C. L. Tan, F. Wang, Y. F. Wang, R. Zhou, D. X. Cui, W. X. You, H. Zhao, J. W. Ren and B. Feng, Nucleic Acids Res., 2016, 44, e85 CrossRef PubMed .
- N. Yang, Int. J. Pharm. Invest., 2015, 5, 179–181 CrossRef CAS PubMed .
- F. Chen, M. Alphonse and Q. Liu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2020, 12, e1609 Search PubMed .
- S. Chira, C. S. Jackson, I. Oprea, F. Ozturk, M. S. Pepper, I. Diaconu, C. Braicu, L. Z. Raduly, G. A. Calin and I. Berindan-Neagoe, Oncotarget, 2015, 6, 30675–30703 CrossRef PubMed .
- J. R. Ohlfest, A. B. Freese and D. A. Largaespada, Curr. Gene Ther., 2005, 5, 629–641 CrossRef CAS PubMed .
- W. Walther, I. Fichtner, D. Kobelt, R. Siegel, P. Schlag and U. Stein, Xenotransplantation, 2008, 15, 297–297 CrossRef .
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed .
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed .
- D. D. Wang, D. L. Jana and Y. L. Zhao, Acc. Chem. Res., 2020, 53, 1389–1400 CrossRef CAS PubMed .
- C. Doonan, R. Ricco, K. Liang, D. Bradshaw and P. Falcaro, Acc. Chem. Res., 2017, 50, 1423–1432 CrossRef CAS PubMed .
- C. B. He, D. M. Liu and W. B. Lin, Chem. Rev., 2015, 115, 11079–11108 CrossRef CAS PubMed .
- K. S. Park, Z. Ni, A. P. Cote, J. Y. Choi, R. D. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed .
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 1–8 Search PubMed .
- X. L. Pei, Y. F. Wu, J. P. Wang, Z. J. Chen, W. Liu, W. K. Su and F. M. Liu, Nanoscale, 2020, 12, 967–972 RSC .
- X. Z. Lian, Y. Fang, E. Joseph, Q. Wang, J. L. Li, S. Banerjee, C. Lollar, X. Wang and H. C. Zhou, Chem. Soc. Rev., 2017, 46, 3386–3401 RSC .
- G. S. Chen, S. M. Huang, X. X. Kou, F. Zhu and G. F. Ouyang, Angew. Chem., 2020, 59, 13947–13954 CrossRef CAS PubMed .
- B. Seoane, S. Castellanos, A. Dikhtiarenko, F. Kapteijn and J. Gascon, Coord. Chem. Rev., 2016, 307, 147–187 CrossRef CAS .
- G. S. Chen, S. M. Huang, X. X. Kou, S. B. Wei, S. Y. Huang, S. Q. Jiang, J. Shen, F. Zhu and G. F. Ouyang, Angew. Chem., Int. Ed., 2019, 58, 1463–1467 CrossRef CAS PubMed .
- N. O. Deakin, J. Pignatelli and C. E. Turner, Genes Cancer, 2012, 3, 362–370 CrossRef PubMed .
- C. E. Turner, Nat. Cell Biol., 2000, 2, E231–E236 CrossRef CAS PubMed .
- M. R. Bai, J. Xie, X. Y. Liu, X. Chen, W. J. Liu, F. Z. Wu, D. Chen, Y. M. Sun, X. Li, C. L. Wang and L. Ye, ACS Appl. Mater. Interfaces, 2018, 10, 26917–26927 CrossRef CAS PubMed .
- J. Kroll, S. Klinter, C. Schneider, I. Voss and A. Steinbuchel, Microb. Biotechnol., 2010, 3, 634–657 CrossRef CAS PubMed .
- S. M. Deane and D. E. Rawlings, J. Bacteriol., 2004, 186, 2123–2133 CrossRef CAS PubMed .
- W. Cheng, J. P. Nie, L. Xu, C. Y. Liang, Y. Peng, G. Liu, T. Wang, L. Mei, L. Q. Huang and X. W. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 18462–18473 CrossRef CAS PubMed .
- S. L. Zhang, J. Li, G. Lykotrafitis, G. Bao and S. Suresh, Adv. Mater., 2009, 21, 419–424 CrossRef CAS PubMed .
- R. H. Pan, G. Q. Liu, Y. S. Li, Y. Wei, S. R. Li and L. Tao, Nanoscale, 2018, 10, 8269–8274 RSC .
- X. D. Xu, J. Wu, Y. L. Liu, M. Y. Yu, L. L. Zhao, X. Zhu, S. Bhasin, Q. Li, E. Ha, J. J. Shi and O. C. Farokhzad, Angew. Chem., Int. Ed., 2016, 55, 7091–7094 CrossRef CAS PubMed .
- M. Li, Y. Tao, Y. L. Shu, J. R. LaRochelle, A. Steinauer, D. Thompson, A. Schepartz, Z. Y. Chen and D. R. Liu, J. Am. Chem. Soc., 2015, 137, 14084–14093 CrossRef CAS PubMed .
- A. Stubb, C. Guzman, E. Narva, J. Aaron, T. L. Chew, M. Saari, M. Miihkinen, G. Jacquemet and J. Ivaska, Nat. Commun., 2019, 10, 1–15 CrossRef CAS PubMed .
- A. Stubb, R. F. Laine, M. Miihkinen, H. Hamidi, C. Guzman, R. Henriques, G. Jacquemet and J. Ivaska, Nano Lett., 2020, 20, 2230–2245 CrossRef CAS PubMed .
- F. A. Ran, P. D. Hsu, J. Wright, V. Agarwala, D. A. Scott and F. Zhang, Nat. Protoc., 2013, 8, 2281–2308 CrossRef CAS PubMed .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr02872c |
|
| This journal is © The Royal Society of Chemistry 2021 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *abf
*abf