DOI:
10.1039/D1MH00773D
(Review Article)
Mater. Horiz., 2021,
8, 2964-3008
Progress on photocatalytic semiconductor hybrids for bacterial inactivation
Received
13th May 2021
, Accepted 8th September 2021
First published on 13th September 2021
Abstract
Due to its use of green and renewable energy and negligible bacterial resistance, photocatalytic bacterial inactivation is to be considered a promising sterilization process. Herein, we explore the relevant mechanisms of the photoinduced process on the active sites of semiconductors with an emphasis on the active sites of semiconductors, the photoexcited electron transfer, ROS-induced toxicity and interactions between semiconductors and bacteria. Pristine semiconductors such as metal oxides (TiO2 and ZnO) have been widely reported; however, they suffer some drawbacks such as narrow optical response and high photogenerated carrier recombination. Herein, some typical modification strategies will be discussed including noble metal doping, ion doping, hybrid heterojunctions and dye sensitization. Besides, the biosafety and biocompatibility issues of semiconductor materials are also considered for the evaluation of their potential for further biomedical applications. Furthermore, 2D materials have become promising candidates in recent years due to their wide optical response to NIR light, superior antibacterial activity and favorable biocompatibility. Besides, the current research limitations and challenges are illustrated to introduce the appealing directions and design considerations for the future development of photocatalytic semiconductors for antibacterial applications.

Jiayu Zeng
| Jiayu Zeng received her BS Degree in South China University of Technology in 2019. She is currently pursuing a Master's Degree in the State Key Laboratory of Bioelectronics of Southeast University under the supervision of Professor Xuemei Wang and Hui Jiang. Her research focuses on the design of novel bio-imaging probes for target bacteria theranostics and photodynamic therapy. |

Ziming Li
| Ziming Li received his BS Degree from South China University of Technology in 2019 and is now pursuing a Master's Degree in the Department of Polymer Science and Engineering, Zhejiang University under the supervision of Prof. Changyou Gao. He is currently working on the design of stimuli-responsive hydrogels for spinal cord injury (SCI) repair and regeneration. Besides, he is also interested in photocatalytic materials for antibacterial applications and tissue engineering. |

Hui Jiang
| Hui Jiang received his PhD (2005) from the University of Science and Technology of China. After completing postdoctoral research at Nanjing University, he joined the faculty of Biomedical Engineering at Southeast University in 2007, and now he is an Associate Professor. His research interests include nanocluster assembly and biological imaging, micro–nano biosensors and the research on POCT. |

Xuemei Wang
| Xuemei Wang received her PhD in Chemistry from Nanjing University in 1994 and then worked as a Lecturer in the Department of Chemistry in Nanjing University from 1995 to 1996. From 1996 to 1998 she studied in Germany with Alexander von Humboldt Fellowship. Then she came back to Nanjing and worked in the School of Biological Science and Medical Engineering as an Associate Professor in Southeast University. Since 2001 she has worked in Southeast University as a Full Professor. Her main research interests cover the design of novel bio-imaging probes including self-assembled biosynthesized functionalized nanoclusters/or smart nano-robots for target cancer theranostics, biosensors and nanomedicine. |
1. Introduction
1.1 The crisis of bacterial infections and the development of photocatalytic antibacterial applications
Bacterial infection has become a prominent underlying cause of numerous diseases threatening human life.1 In the past few decades, antibiotics have been widely used to combat bacteria.2 However, the off-label abuse of antibiotics due to insufficient surveillance has resulted in an emergent crisis of antibiotic resistance and even the creation of superbugs.3,4 Furthermore, the unsatisfactory input–output ratio and inadequate research activities have resulted in an equally alarming decline in the development of new antibiotics. Therefore, there is an urgent requirement for the development of novel antibacterial techniques to overcome the growing bacterial threats and address the issue of bacterial resistance.
Among the various exploited antibacterial techniques, photocatalytic disinfection as a green sustainable strategy has attracted extensive extension due to its rapid and high-efficient inactivation effect without notable drug resistance and side effects.5 As an exogenous antibacterial technique, it relies on the strong oxidation towards bacteria induced by photogenerated ROS, which contains no chemical additives or harmful inactivation by-products.6 Driven by solar energy, the antibacterial process is controllable, recyclable and low cost. Studies have shown that photocatalysis has broad-spectrum inactivation properties for many G-(+) and G-(−) bacteria including E. coli, S. aureus, P. aeruginosa and S. choleraesuis.7 The photocatalytic antibacterial technique can be applied for various occasions ranging from environmental purification to biomedical treatment of bacterial infections, including environmental water disinfection, medical equipment disinfection, food sterilization, human implant sterilization, bacteria-infected wound cure and even bacterial inflammation therapy.8,9 With the development of the photocatalytic technique, some commercial industrial applications have emerged such as self-cleaning antibacterial films, ceramic tiles, fabrics and glasses.10
1.2 The development of semiconductors for photocatalytic inactivation
In photocatalysis, the choice of suitable catalysts is crucial for antibacterial effects, and the photocatalysts are mainly semiconductor materials. Since Fujishima and co-workers11 discovered the catalytic decomposition of water on the surface of the TiO2 semiconductor with UV irradiation, photocatalysis has become a research hotspot for developing a novel field of semiconductor applications. Also, the use of photocatalytic bacterial inactivation was discovered by T Matsunaga's team,12 who reported the photoelectrochemical sterilization of S. cerevisiae with semiconductor TiO2/Pt powder as the catalyst. Inspired by this work, there have been numerous reports on photocatalytic semiconductor sterilization. N-type semiconductors such as n-TiO2 and n-ZnO were extensively reported initially; however, pristine semiconductors suffer from inherent drawbacks such as wide energy gap, which can only be excited under UV (less than 4% of the solar spectrum13,14), and high rate of photogenerated change carrier recombination,15 leading to unsatisfactory photocatalytic activity.
To overcome these drawbacks, researchers have explored the modification and reconstruction of pristine semiconductors.16 For example, it has been proven that noble metals (for example, Ag and Au NPs) loaded on the surface of TiO2 can effectively separate photo-generated electron–hole pairs.15,17 Also, anion and cation doping have been investigated18,19 to successfully restructure the bandgap of pristine semiconductors. Especially, 2D semiconductors such as g-C3N4, MoS2, and BPs have attracted extensive attention in the field of photocatalytic applications in recent years,20,21 owing to their appealing properties such as wide light response to NIR and layers-dependent band energy. However, there are few comprehensive reviews on pristine semiconductors and their modified hybrids for photocatalytic sterilization.
Herein, we comprehensively review the research progress on photocatalytic antibacterial semiconductors in recent years, including their energy band structures, synthetic techniques, photocatalytic mechanisms, ROS-induced antibacterial effects, and the influence of different modification strategies. Besides, the biosafety of the material such as cell compatibility and the evolution/degradation behaviors of semiconductors for further use in the biomedical field will be presented. Finally, some challenges and perspectives on semiconductor photocatalysts will be also discussed to spark appealing directions and design considerations for future semiconductors (Fig. 1).
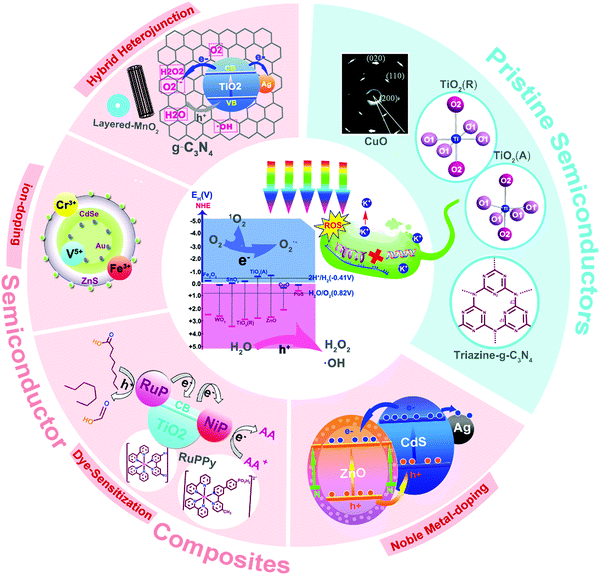 |
| | Fig. 1 Various photocatalytic semiconductor systems for photocatalytic bacterial inactivation. | |
2. Fundamentals of photocatalytic semiconductor hybrids for bacterial inactivation
2.1 Fundamentals of heterogeneous photocatalysis for bacterial inactivation
Knowledge on bacterial inactivation is crucial for the effective development of novel composite materials. Microbial disinfection driven by solar energy has attracted widespread attention as a sustainable green strategy, which achieves rapid disinfection through a powerful oxidation process without the requirement of additional chemical substances.6 The key to achieve rapid photocatalytic disinfection is ROS-induced toxicity.
ROS is mainly involved in cellular oxidative stress, and although its inactivation mechanisms are not very clear to date, its potential mechanisms are as follows: (1) cell wall or cell membrane damage may be induced by intracellular K+ leakage. For example, Lu and Thabet et al.22,23 found that in the process of cells killing, photocatalysis alters the cell membrane permeability and leads to intracellular K+ leakage. (2) Small enough engineered nanoparticles penetrate the cell membrane and photocatalysis occurs inside. NPs may indirectly attack intracellular substances by ROS or directly attack them to cause cell death. For example, it has been found that ultrafine TiO2 particles can break bacterial plasmid DNA molecules.24 (3) ROS causes intracellular acetyl-CoA oxidative/reductive injury, leading to cell respiratory system damage.25 (4) Photo-generated ROS may cause intracellular overproduction of ROS, which induces oxidative stress. For instance, Wu et al.26 reported that a red phosphorus/g-C3N4 composite system caused the sunlight-driven photogeneration of ROS, which could enter bacteria and then induce enhanced endogenous O2− and H2O2 production. Also, Wang et al. invented various methods for the detection and tracing of intracellular ROS or other related substances.27–33
2.2 The photocatalytic mechanisms of semiconductor hybrids in photocatalytic bacterial inactivation
2.2.1 The band structure of semiconductors based on density functional theory (DFT) calculation.
Generally, the energy band structure refers to the fact that the electronic states exhibited by electrons in a solid enable them to exist continuously in some energy ranges under the action of the potential field formed by the periodically distributed atomic nuclei, while the others are completely forbidden in other energy ranges. In a narrow sense, the electronic energy band structure refers to the dependence of the energy of a single electron state near the Fermi energy on the lattice wave vector k, which is also called the band diagram (band diagram). The electronic energy band structure discussed here covers the meaning of the above-mentioned two aspects.
Specifically, first-principles electrons based on Kohn–Sham (KS) density functional theory based on the local density approximation (LDA) or generalized gradient approximation (GGA) structural theory play an increasingly important role in semiconductor physics, especially in the study of band gaps. The latter reformulates the complex quantum multisystem problem as a formal single-system problem by establishing the mapping relationship between a real multi-electron interaction system and a virtual non-interaction system with the same ground state electron density. Due to its conceptual intuition, the energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is used to describe the band gap and is known as the Kohn–Sham band gap, which has been widely used to describe the electronic band structure properties of materials, i.e.,
Moreover, for the dependence of the single-electron state energy near the Fermi energy on the lattice wave vector
k, the band gaps with orbital energies corresponding to the same wave vector are called direct band gaps, whereas those with different wave vectors are called indirect band gaps. For many systems, LDA/GGA can give a qualitatively correct image of the electronic energy band structure, but because the pure ground state theory DFT ignores the consequences of excitation, the basic energy gap and optical transition can be clearly underestimated and revealed, and thus there is a systematic error in quantitative analysis. Compared to the LDA/GGA, the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional with range separation can usually reproduce the hybrid orbital of many semi-conductor band gaps more accurately. For more details, please refer to the literature.
34–38 Herein, we pay focus on whether the lowest excitation energy (whether direct or indirect) calculated by
eqn (1) can be excited by ordinary visible light or ultraviolet light, given that the size and properties of the band gap contribute greatly to photocatalysis.
2.2.2 The photogenerated charge carriers and ROS.
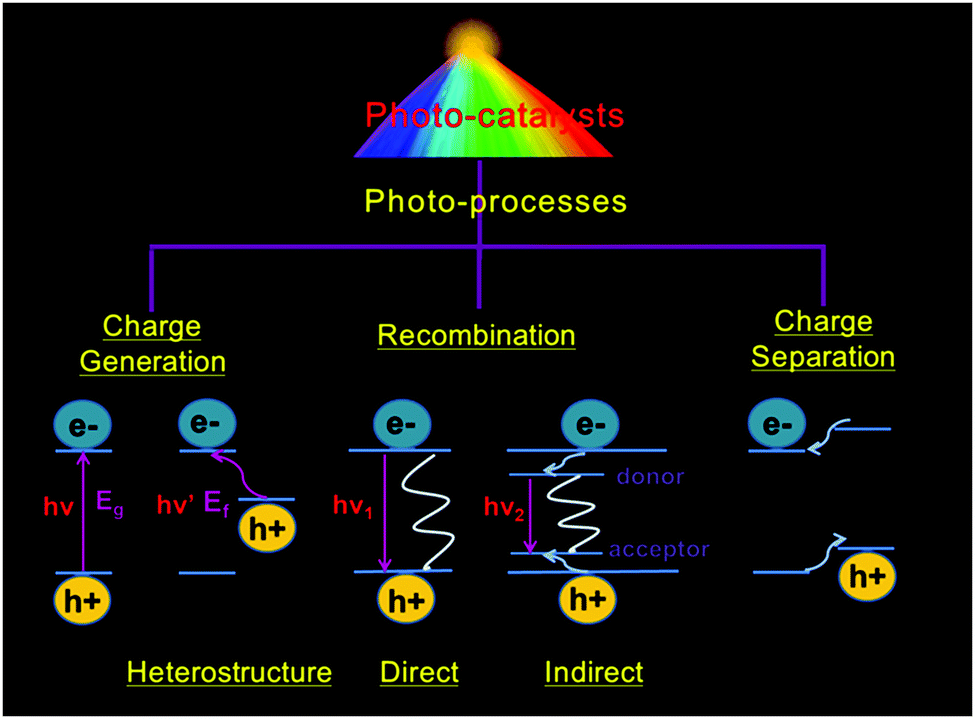
During photocatalysis, the photocatalytic material assimilates photon energy exactly equivalent to or larger than its own band gap. An electron with a negative charge (e−) in the valence band (VB) of the semiconductor jumps towards the conduction band (CB), leaving a void with a positive charge (h+) in the VB. The photogenerated h+ and e− diffusing on the surface of semiconductors act as strong oxidizing and reducing agents, respectively. They perform electrochemical redox reactions, which macroscopically presents the photoinduced chemical synthesis process. These photo-responsive and catalytic semiconductors are defined as photocatalysts. However, unsatisfactory recombination of charge carriers tends to occur, reducing their photocatalytic efficiency. The relaxed electrons will return to the conduction band directly or recombine with holes in traps.39,40 These mechanisms are illustrated in Fig. 2.
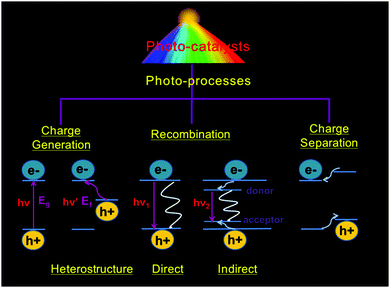 |
| | Fig. 2 Basic photo-process of photocatalysts. Eg: band gap of a semiconductor and Ef: Fermi energy of the immobilized noble metal particles. After photo-excitation, radiative and non-radiative recombination can occur, as depicted by the straight and wavy lines, respectively. | |
ROS, mainly consisting of superoxide radical (O2˙−), hydrogen peroxide (H2O2), hydroxyl radical (˙OH) and singlet oxygen (1O2), are also generated in the process of photocatalysis. When irradiated charge carriers are formed, O2˙− appears via the interaction between oxygen and electrons. Also, ˙OH will be formed through the oxidation of H2O or OH−. 1O2 can be generated from the subsequent reduction of the superoxide anion or directly generated from oxygen (O2) by utilizing the energy from recombination.41,42 These processes can be depicted by eqn (2)–(5) as follows:
It should be noted that the bottom of the CB of the photocatalytic material should be more negative than the potential of O
2/O
2˙
− or the top of VB should be more positive than the potential of H
2O/˙OH to produce redox reactions. The occurrence of redox reactions also requires certain thermodynamic conditions.
5
2.3 The current research on photocatalytic semiconductor hybrids for bacterial inactivation
Photocatalytic bacterial inactivation has become an appealing direction for the application of semiconductors. However, the photocatalytic antibacterial mechanisms remain unclear such as the surface interactions between bacteria and semiconductors and ROS-induced antibacterial actions, which will attempt to be solved in the following sections with emphasis on the reactive sites and specific antibacterial mechanisms of each semiconductor. Also, a unified measurement of antibacterial ability of semiconductors is lacking, given that the photocatalytic antibacterial effect is affected by various internal/external conditions, such as preparation method, material morphology, catalyst concentration, reaction environment (such as pH, temperature, solution, and light intensity),10 which will be summarized and discussed in the tables below. For many semiconductors, they mainly depend on visible-light-driven energy, which is restricted to disinfection in the external environment, but does not meet the medical requirements of antibacterial or anti-inflammatory in vivo, demanding deeper photon penetration.43 Also, cell compatibility and the evolution or degradation of materials in the physiological environment should be considered for further medical applications. 2D semiconductors seem to be more suitable in this regard, which will be discussed in detail.
3. Pristine semiconductors for bacterial inactivation
3.1 The classification of semiconductors
3.1.1 n-type semiconductor.
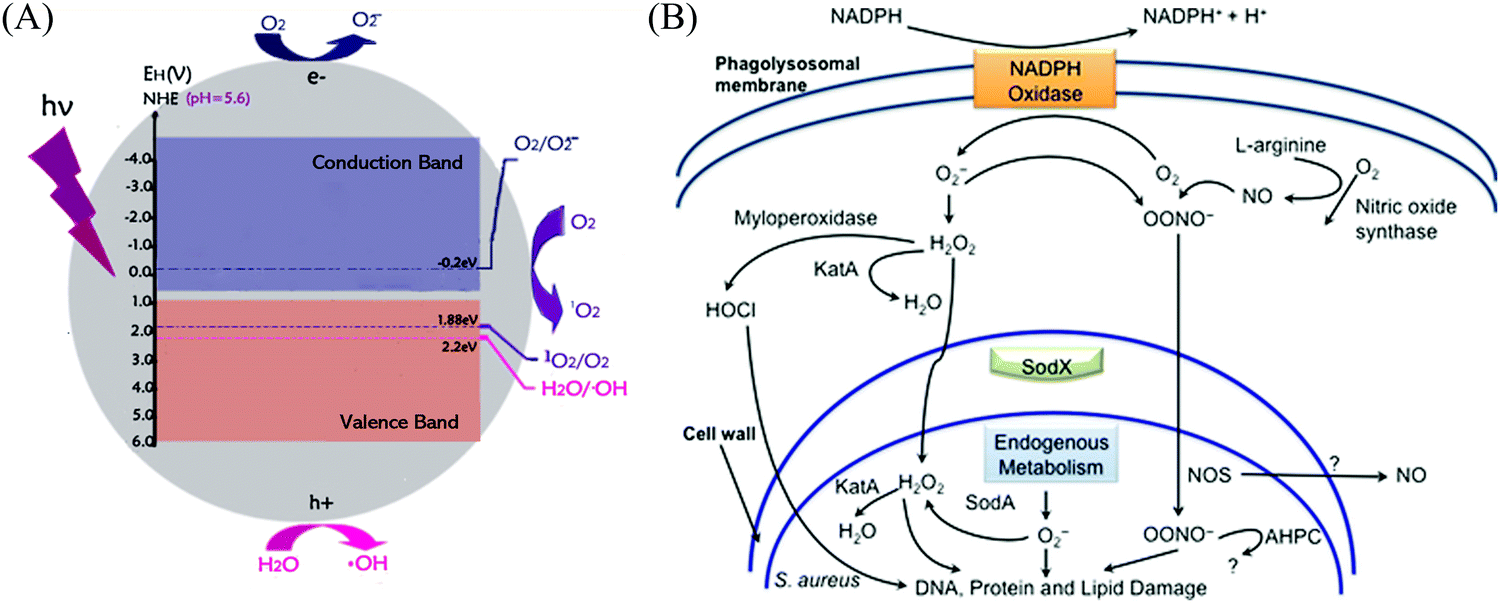
Semiconductors are currently classified into p-type and n-type semiconductors according to their different charge carriers.44 In n-type semiconductors, electrons serve as the main photo-generated carriers,45,46 whereas positive charge carriers (holes) are the majority in p-type semiconductors.47,48 Also, there is a significant difference in the mobility between them. It is widely recognized that n-type semiconductors possess a higher mobility due to the adequate electrons produced by oxygen vacancies,49,50 together with the unimpeded electron-transport paths mainly composed of spatially spread metal s orbitals in the conduction-band minimum (CBM).51 It should be noted that these spatially spread and isotropic orbitals result in a highly dispersed and delocalized CBM, facilitating a low electron effective mass, and therefore the photo-excited electron can move without limitation, making reduction reactions easy (Fig. 3).52
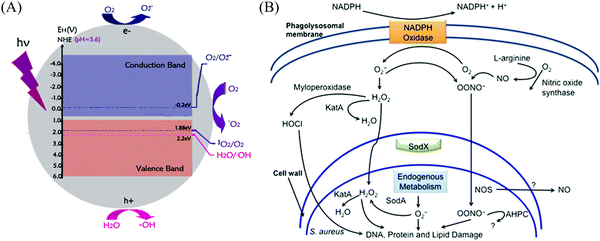 |
| | Fig. 3 (A) Edge locations of H2O and O2 in water at pH 5.6. The normal hydrogen electrode (NHE) is used as a reference. Reprinted from ref. 53. (B) Intracellular mechanism of ROS-induced bacterial inactivation. Reprinted from ref. 54. | |
The typical n-type TiO2 and ZnO as photocatalysts have been widely investigated; however, they still suffer from a wide bandgap from their O 2p orbitals to M 3d (M = Ti and Zn) orbitals,55,56 which can only be excited by light in the UV region with a wavelength below 390 nm57 and 370 nm,58 respectively. When irradiated by enough photon energy, photocatalysis will occur on their surface as follows (using TiO2 as an example):59–61
| | | TiO2 + hν (λ < 390 nm) → hvb+ + ecb− | (6) |
The photogenerated electron and/or hole react with the hydrated (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) TiIVOH) surface functionality of TiO2 to form [TiIIIOH] and [TiIVOH]+, as depicted in eqn (7) and (8), where TiIVOH represents the primary hydrated surface functionality of TiO2.
TiIVOH) surface functionality of TiO2 to form [TiIIIOH] and [TiIVOH]+, as depicted in eqn (7) and (8), where TiIVOH represents the primary hydrated surface functionality of TiO2.
| | | ecb− + TiIVOH → [TiIIIOH] | (7) |
| | | hvb+ + TiIVOH → [TiIVOH]+ | (8) |
[TiIIIOH] acts as a reducing agent to reduce oxygen into superoxide radicals, which can be subsequently reduced to 1O2 by [TiIIIOH], as shown in eqn (9) and (10). For eqn (11), water is oxidized to hydroxyl radicals by [TiIVOH]+, which functions as an oxidant agent.
| | | [TiIIIOH] + O2 → O2˙− + TiIVOH | (9) |
| | | O2˙− + [TiIIIOH] → 1O2 | (10) |
| | | [TiIVOH˙]+ + H2O → ˙OH + TiIVOH | (11) |
Serving as an h+-trapper and e−-trapper, [TiIVOH˙]+ and [TiIIIOH] contribute to the indirect recombination of charge carriers, respectively.
| | | ecb− + [TiIVOH˙]+ → TiIVOH | (12) |
| | | hvb+ + [TiIIIOH] → TiIVOH | (13) |
However, due to the low photoelectric conversion efficiency and high photogenerated charge carrier rate,
62 the effect of photogenic ROS is far from satisfactory. For example, Macyk
et al.63 reported the low efficiency of
1O
2 formation and sometimes even no generation of
1O
2 on pure TiO
2, whereas when the abundant OH
− groups on the surface of TiO
2 were substituted by other atoms such as [PtCl
6]
2−, the efficiency of the energy transfer pathway would be enhanced, and therefore generating
1O
2.
3.1.2 p-type semiconductors.
For p-type semiconductors, CuO is a representative one,64 which is attributed to the presence of negatively charged copper vacancies and interstitial oxygen.65 However, its conductivity mechanism is still under debate. It is widely acknowledged that CuO has an open 3d shell level (3d94s0), instead of a closed one (3d104s0), which can avoid absorption in the visible range from the d–d transition like Cu2O,66 endowing CuO with a direct and narrow band gap (about 1.2 eV in the bulk),67 enabling it to absorb near infrared light.67,68 According to Zhang,69 the optical absorption spectrum for nanostructured CuO can cover nearly the whole visible range depending on various process parameters and measurement techniques.
Actually, the antibacterial activity of copper and copper oxide have not been well investigated, partially due to the rapid oxidation of metallic copper nanostructures exposed to air70 and physical and chemical instability of copper oxides at room temperature, particularly in the presence of Cu2+.71 Akhavan72 reported the superiority of bacterial disinfection using Cu compared with CuO nanoparticles, attributing to the better electron accepting ability of Cu, as a good electron acceptor, which led to an enhancement in antibacterial activity via disruption of the wall membrane of the bacteria.
Also, the photocatalytic reaction and photogenerated ROS present on the surface of CuO are as follows:73,74
| | | CuO + hν (<660 nm) → CuO (ecb−, hvb+) | (14) |
The excited electron can either react (a) directly with O2, forming O2˙− or (b) reduce the Cu2+ lattice to Cu+, leading to the following reactions (15)–(17) with the formation of O2˙− radicals:
| | | CuO (ecb−) + O2 → CuO + O2˙− | (15) |
| | | CuO (ecb−) → CuO (Cu+) | (16) |
| | | CuO (Cu+) + O2 → CuO (Cu2+) + O2˙− | (17) |
Reaction (18) shows that the equilibrium between H+ and O2˙− leads to the formation of the HO2˙ radical. Also, H2O2 can be produced either by HO2˙ (reaction (19)) or by (reaction (20)) as follows:
| | | H+ + O2˙− → HO2˙ pKa = 4.8 | (18) |
| | | CuO (Cu+) + HO2˙ (partial O2˙−) + H+ → CuO (Cu2+) + H2O | (19) |
| | | 2Cu(I) + O2 + 2H+ → 2Cu(II) + H2O2 | (20) |
Notably, p-type semiconductors possess a low mobility, considering that the higher formation energy of the intrinsic acceptor and lower formation energy of the natural donor will harden the generation of holes.
75,76 Furthermore, owing to the anisotropy and O2p orbital with less overlap, the transfer of holes in the valence-band-maximum (VBM) is difficult, which reduces its mobility.
68 This low mobility of charge carriers hampers the development and further use of p-type semiconductors, remaining a challenge to researchers.
77
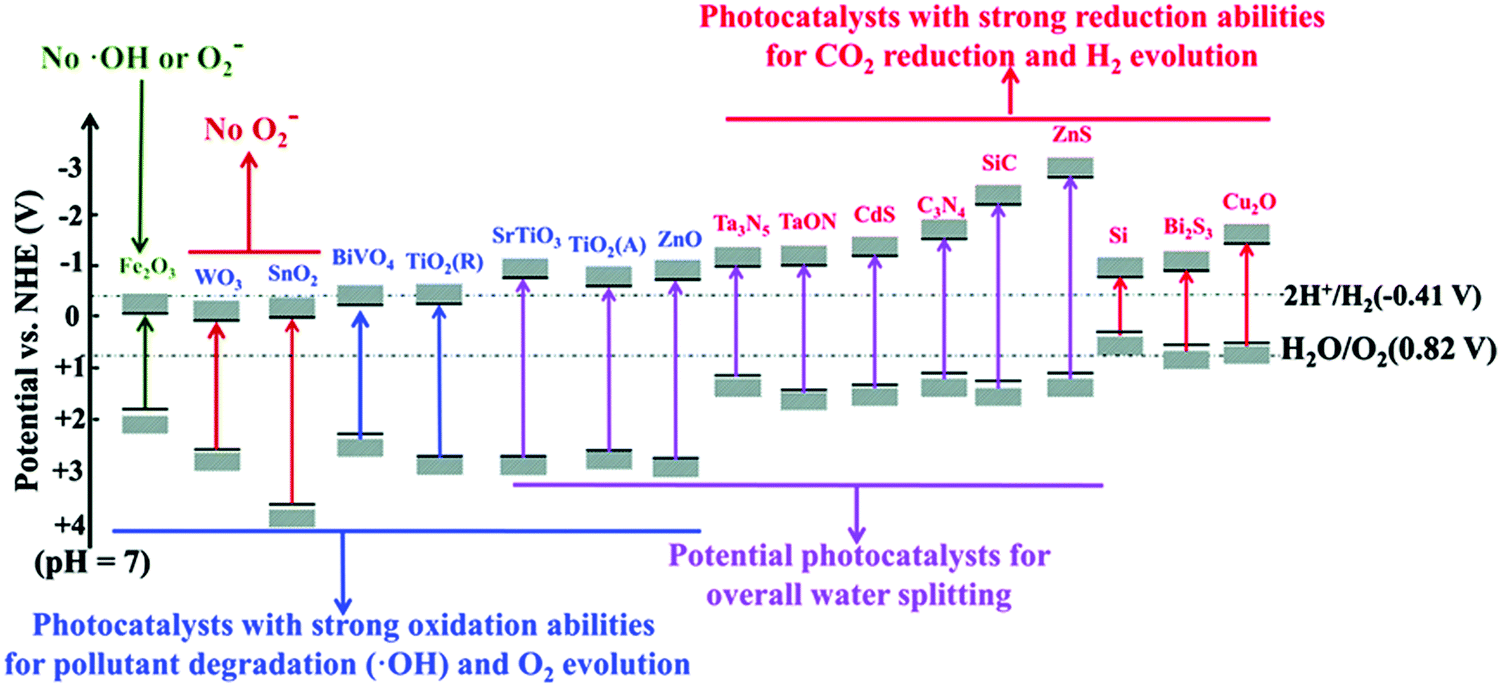
The bandgap of some typical pristine semiconductors including nano TiO2, ZnO, and CuO are illustrated in Fig. 4.
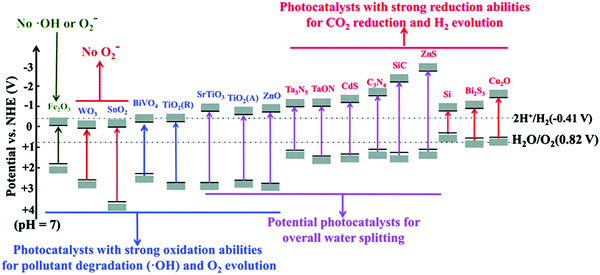 |
| | Fig. 4 Band gap of some typical pristine semiconductors (at pH = 7) and their potential applications. Reprinted from ref. 78. | |
3.2 The active sites and catalytic activity
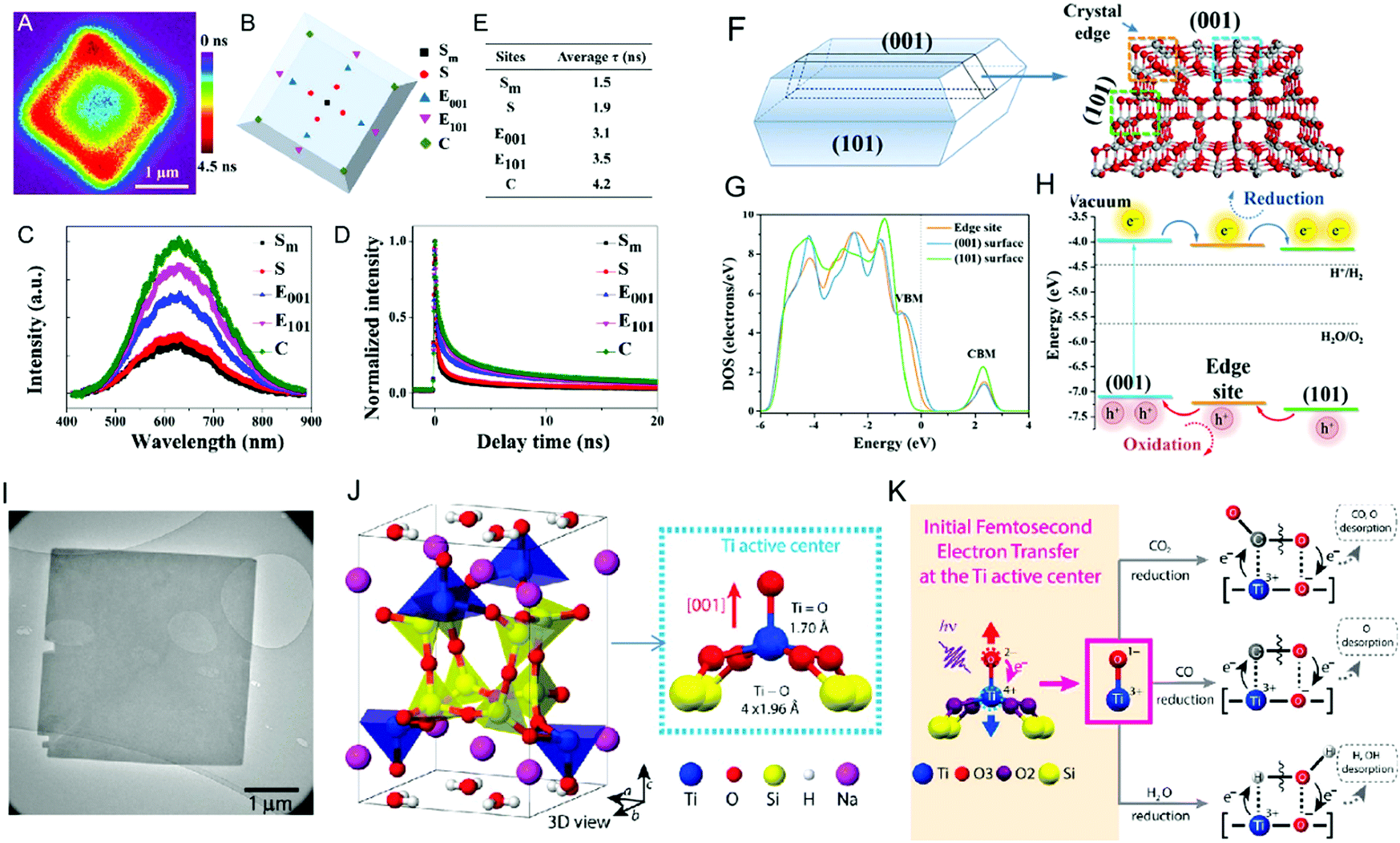
Photocatalytic reactions tend to occur at the active sites on the surface of the material, where clarification of the active site is beneficial to further understand the photocatalysis mechanism and improve the photoelectric conversion efficiency.79 The active sites are normally located at the edge, unsaturated steps, and/or corner atoms of semiconductors.80 The coordination of unsaturated transition metal (Ti, Cr, Fe, Mn, etc.) compounds are important in photocatalytic applications.81 Generally, the lower the coordination number, the higher the chemical activity.82 Fujitsuka et al.83 identified the photocatalytic active sites of 6-coordinated configurational-TiO2 at the single-molecule level, where they proposed that the surface heterojunction (edges and corners) formed by the (001) planes and (101) planes can act as active sites to accumulate photogenerated electrons and holes. Zewail et al.81 proposed the 5-coordinated Ba2Ti2Si2O8 photocatalyst, where photoexcited electrons are rapidly transferred from the apex oxygen to the central Ti atom, resulting in the separation of the two atoms and the transformation of the original Ti4+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O2− double bond state into a Ti3+–O1− single bond, which acts as the active site of the catalytic reaction (Fig. 5).
O2− double bond state into a Ti3+–O1− single bond, which acts as the active site of the catalytic reaction (Fig. 5).
It has been reported that the crystal structure also affects the photocatalytic activity of materials. Pang et al.84 discovered the shape-dependent antibacterial behavior of Cu2O, whose inactivation effect changed from broad-spectrum bacteriostasis to high selectivity with a morphology ranging from cubic to octahedral for different surface facets of its various crystal structures (such as (100) surfaces for cubes and (111) surfaces for octahedrons), exhibiting the respective adsorption and desorption behaviors towards bacteria.84 According to Prasanna85 and Gunder,86 intrinsic defects such as the shallow defect state formed by Zn interstitials (Zni) and deep defect state formed by oxygen vacancies (VO) can reduce the charge carrier recombination efficiency. The introduction of structural defects or disorder enables a reduction in the coordination number of adjacent sites and affects the electronic structure of the catalyst to adjust the activity of its active sites.87
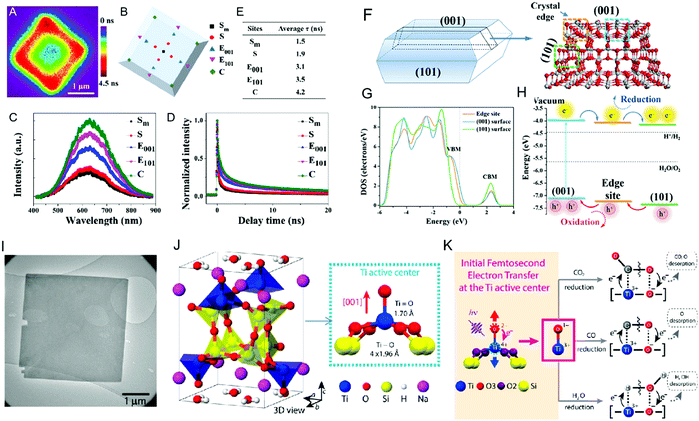 |
| | Fig. 5 Crystal structures and active sites of different coordination Ti (TiO2 and Ba2Ti2Si2O8). (A) PL lifetime mapping of a single anatase TiO2 particle shown with color bar. (B) Site distribution of a single anatase TiO2 particle and its PL spectra (C) and lifetime (D). (E) Delay time of each site. (F) Crystal and electronic model of anatase TiO2. (G) Different band structure characteristics between edge site and (001) and (101) surfaces. (H) Energy level structure of different photocatalytic sites and photogenerated carrier transfer process. (I) and (J) Bright-field image and schematic crystal structure of Ba2Ti2Si2O8. (K) Rapid electron transfer from O to central Ti when excited. (A)–(H) Reprinted from ref. 83. (I)–(K) Reprinted from ref. 81. | |
Besides, the interactions between the reactive states of a photocatalyst and the solvents around affect its photocatalytic activity. For example, Niu et al.88 reported the differences in the antibacterial effects of ZnO NPs in different media. It was shown that ZnO NPs in DI water exhibited a better effect on E. coli of than in other electrolyte solutions because less aggregation of the particles occurred, with the greater generation of ROS. Also, the nano ZnO was positively charged in DI water, which facilitated the attachment of the ZnO NPs to the negatively charged E. coli. Gaffney's team described the solvation kinetics around the catalytic active site, showing that the structure of the photocatalyst changes with the structure of its solvated shell.89 Muckerman90 demonstrated that the catalytic activity exhibited at the interface between the semiconductor and solvent is caused by a complicated series of physical and chemical steps.
3.3 An overview of 2D nanomaterials
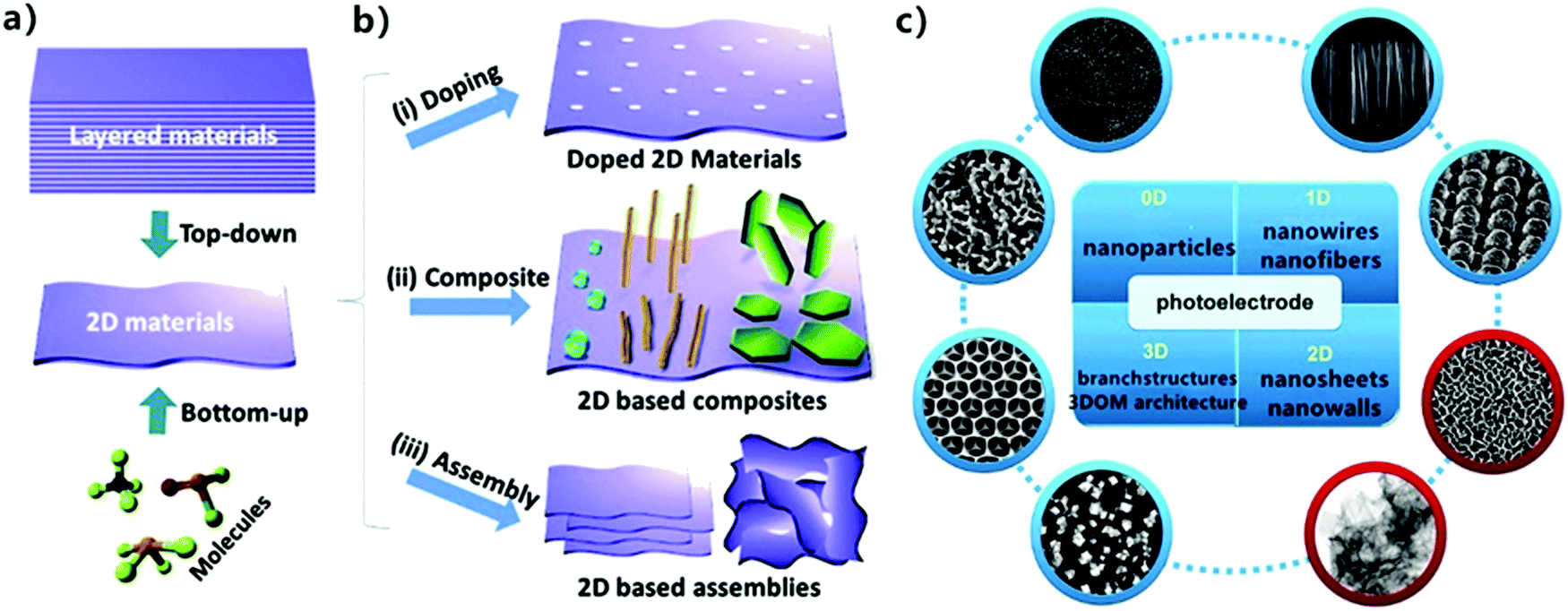
The scale of materials has a critical effect on their photocatalytic performance. For example, Lakshmi91 found that ZnO nanoparticles (NPs) exhibited stronger inactivation than ZnO microparticles (MPs) because of their high surface area and quantum size effect. Also, the 2D morphology of MoS2 exhibits better antibacterial activity compared with raw MoS2 powder (0D) due to its planar structure with more exposed active sites.92 Two-dimensional (2D) nanomaterials first appeared when Ferrari et al.93 successfully exfoliated mono-layered graphene from bulk graphite in 2014. Monolayer or thin-layer graphene is formed by a network of hexagonal enclosed carbons, which are stacked by van der Waals forces, with each layer spaced at 3.3 Å.94 Owing to their intriguing physicochemical properties such as limited quantum effect, atomic thickness, high specific surface area, adjustable electronic structure, low recombination of electrons and holes, and abundant surface active sites,95,96 2D nanomaterials have become promising candidates in photocatalytic applications, and thus various methods for the preparation of 2D materials have been developed.97,98 They mainly include two categories, one is the “top-down” exfoliation of layered materials and the other is the “bottom-up” synthesis of molecular precursors.99 It has also been reported that a controllable layer number (thickness), dye doping construction and heterostructure composition during the preparation of 2D materials are critical to their photocatalytic properties and applications (Fig. 6).
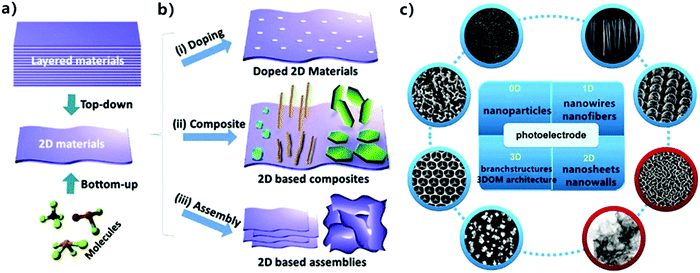 |
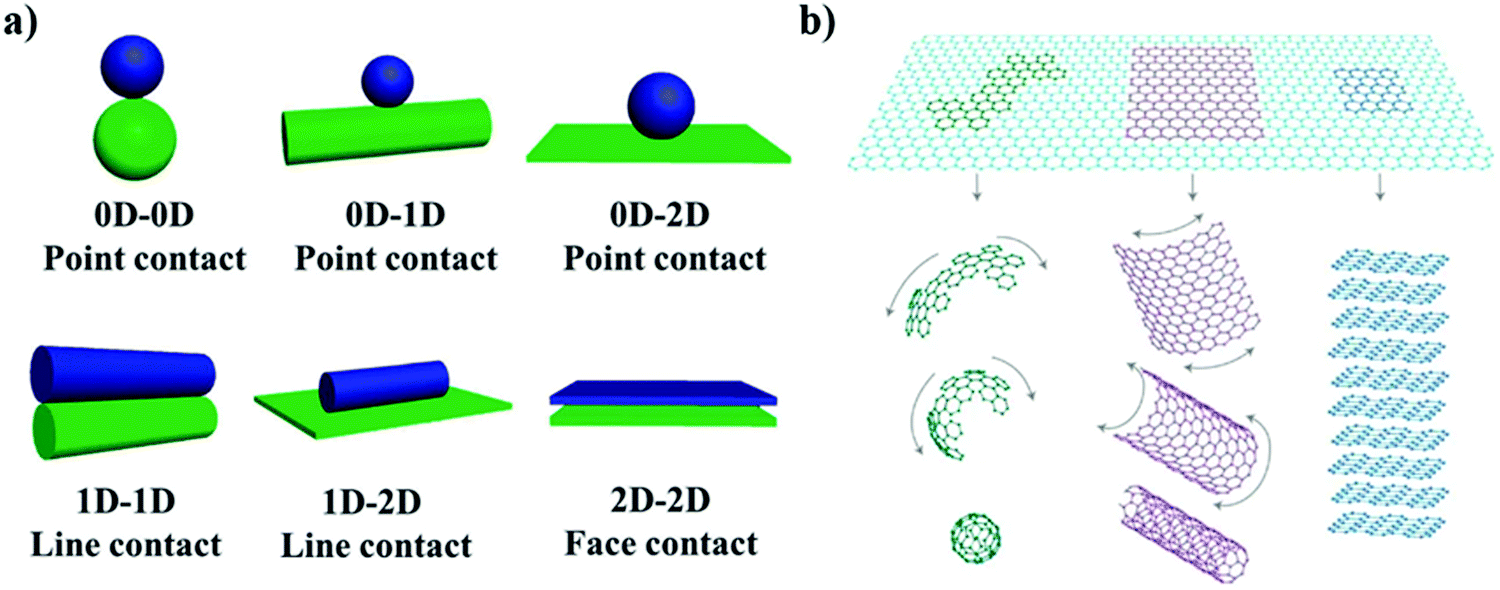
| | Fig. 6 Scheme of (a) “top-down” and “bottom-up” methods to synthesize 2D materials. (b) Modification of 2D materials as photocatalysts, consisting of (i) doping construction, (ii) compositing with other 0D/1D nanomaterials, and (iii) 3D structure assembled with 2D layers. Reprinted from ref. 100. (c) Basic geometrical motifs of different dimension materials. Reprinted from ref. 95. | |
3.3.1 The thickness-modulated energy gap and light absorption of 2D semiconductors.
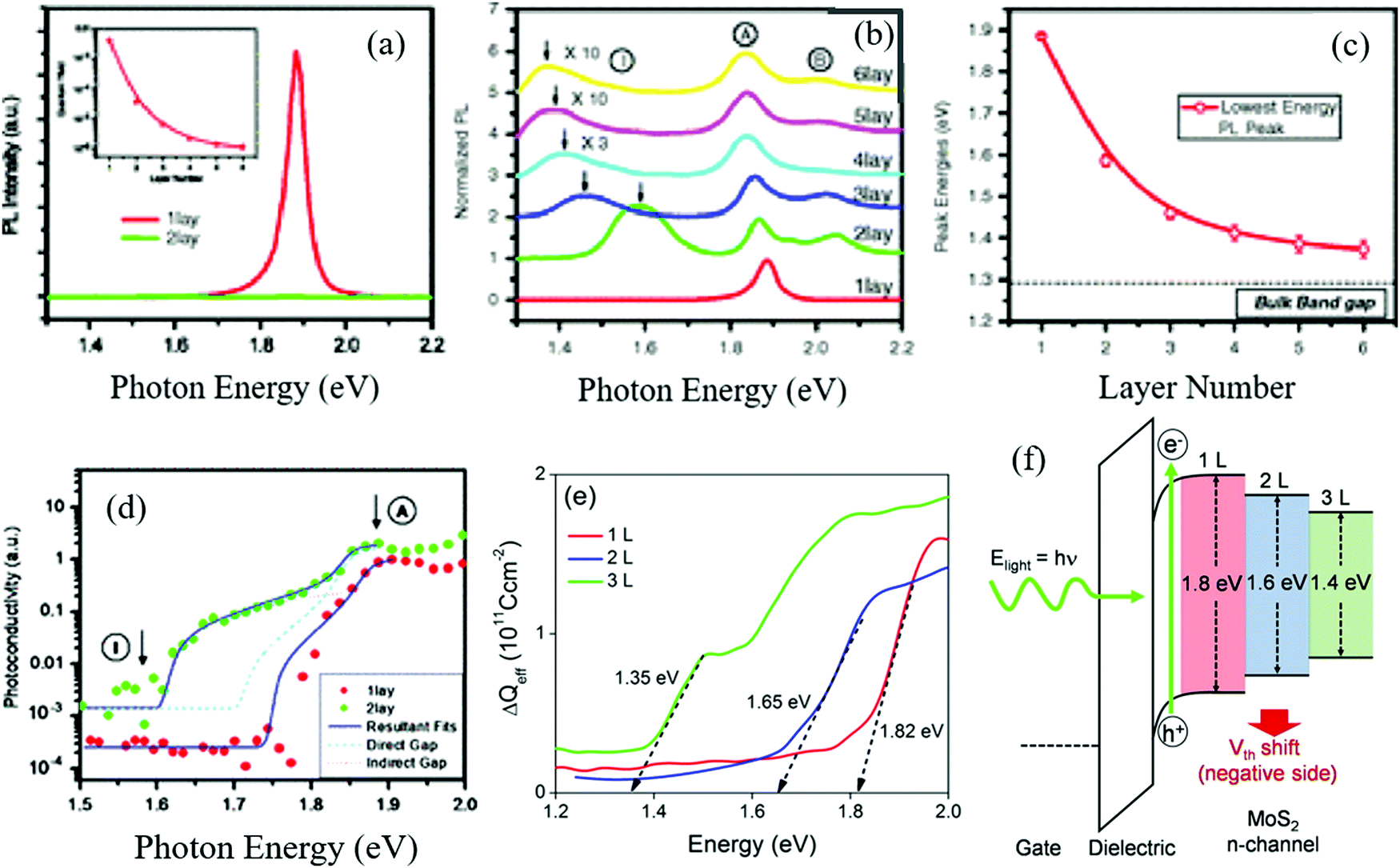
2D nanostructures materials are mainly monolayer or thin-layer exfoliated from bulk their counterpart,101 such as nanosheets33,102 and nanowalls.103,104 Also, the indirect energy gap of bulk materials differs from that of monolayer materials, which importantly exhibits a well-defined dependence on thickness.105 For example, monolayer MoS2 possesses a direct energy gap of about 1.8 eV, consisting of weak S–Mo–S coupling layers.106 As the number of layers increases, the indirect band gap of the bulk material shifts downwards monotonically until its threshold due to the perpendicular quantum confinement.105,107 Moreover, multiple-layer MoS2 conventionally stacks in a staggered arrangement of Mo atom layers and S atom layers,108 and the actual determining factor of band gap energy becomes the layer separation due to this arrangement109,110 (Fig. 7). Similar to MoS2, BP possesses a direct energy gap ranging from 2.0 to 0.3 eV (from bulk to monolayer), depending on the number of layers (or thickness111,112), endowing BP a tunable band gap width and light absorption and photo-excitation capacity by UV, visible or NIR light.113 It should also be noted that the charge transport anisotropy in BP dominantly relies on the number of layers,114,115 which provides a balance between the suppression of charge carrier recombination and wide light absorption range.
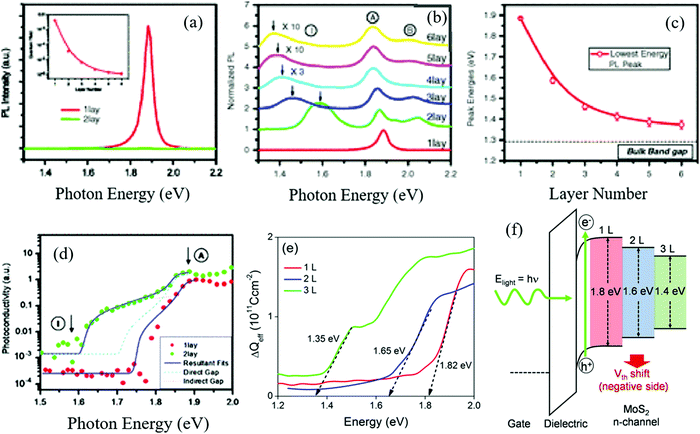 |
| | Fig. 7 (a) Photon energy of MoS2 with single and two layers. (b) Relative photon energy of different numbers of MoS2 layers. (c) Energy gap of different numbers of MoS2 layers. (d) Photoconductivity spectra for 1-layer and 2-layer samples, (e) measurements of their band gap energy, and (f) photoelectric effects of MoS2 with different layers, respectively. (a)–(d) Reprinted from ref. 116. (e) and (f) are Reprinted from ref. 117. | |
3.3.2 The photocatalytic active sites and disinfection performance.
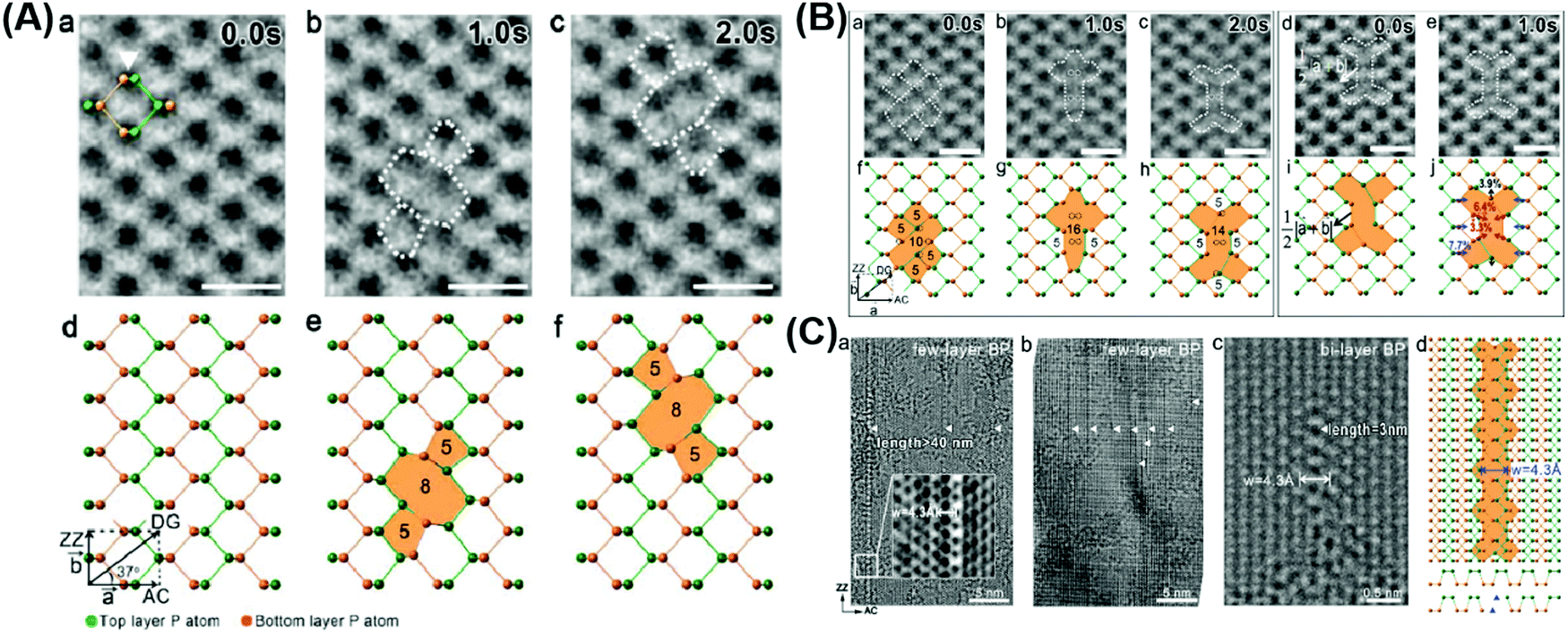
Increasing the specific surface area of materials remains a major strategy to proliferate their active sites due to the greater contact between semiconductors and reactive substrates and additional edges,118 which can be achieved by morphology regulation from bulk to 2D counterparts. It has been reported that numerous exposed surface atoms in ultra-thin 2D nanosheets tend to escape from the lattice, leading to the creation of defects.80 Structural defects and disorder will cause a decrease in the coordination number of the central atoms, affecting the electronic structure and the activity of the reaction sites (Fig. 8).119
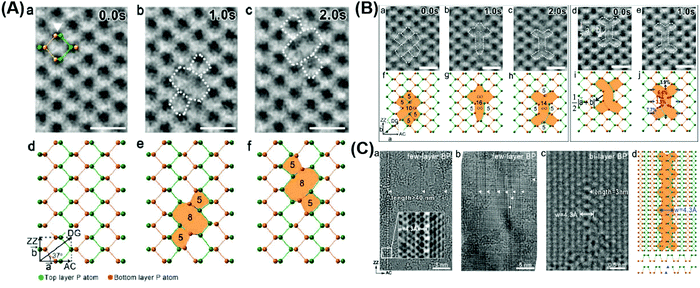 |
| | Fig. 8 Various vacancy defects of atomically thin BPs. (A) HRTEM images and the corresponding schematical atomic structures of the time-dependent formation and migration of binary vacancies (a–f). (B) HRTEM images and the corresponding schematic atomic structures of the time-dependent formation and migration of a tetra-vacancy (a–j). (C) TEM images of long vacancy line arrays existing in few-layer BP (a and b) and bi-layer BP (c), and (d) schematic crystal structure of the line vacancy corresponding to (c). Reprinted from ref. 120. | |
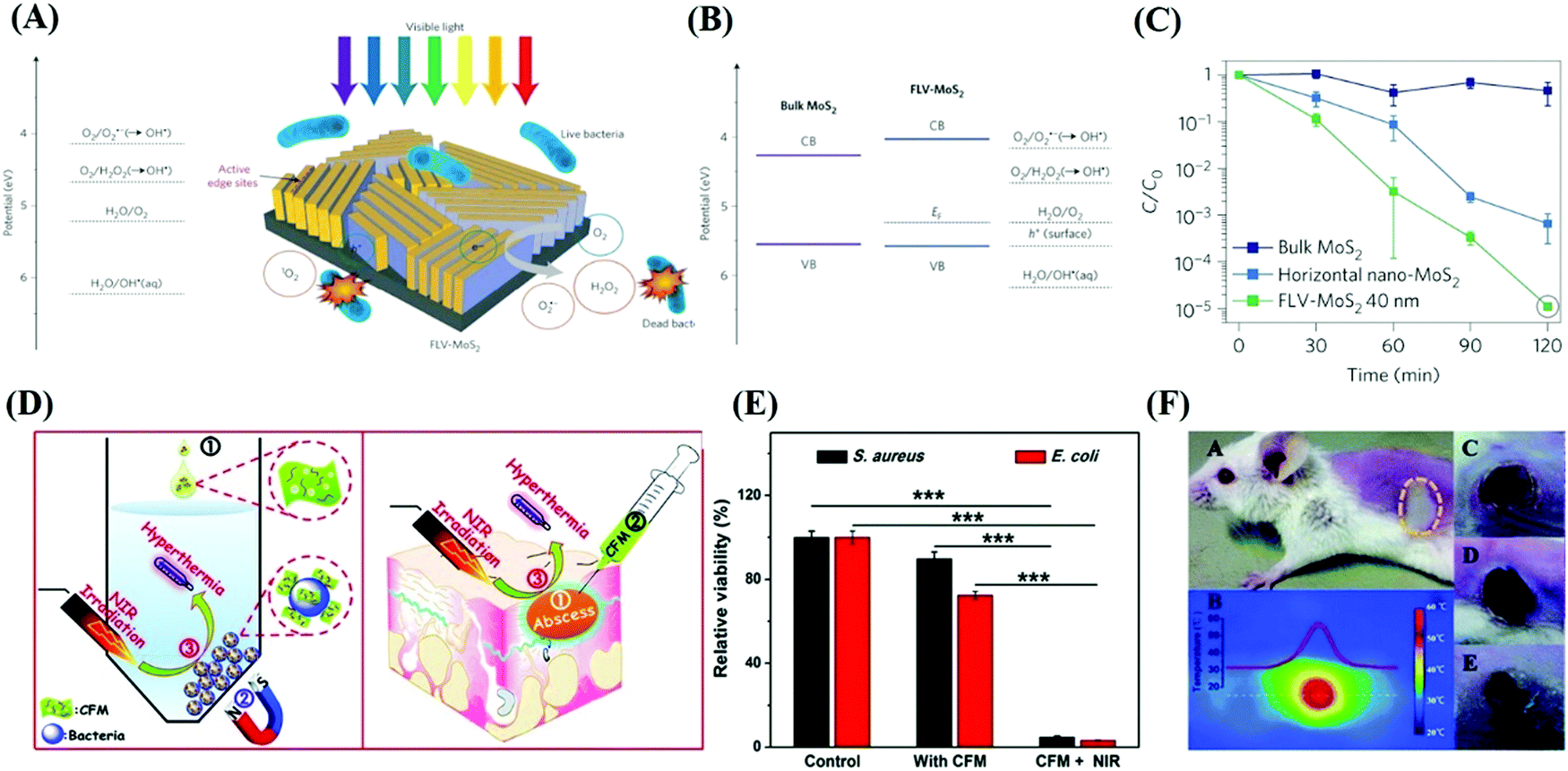
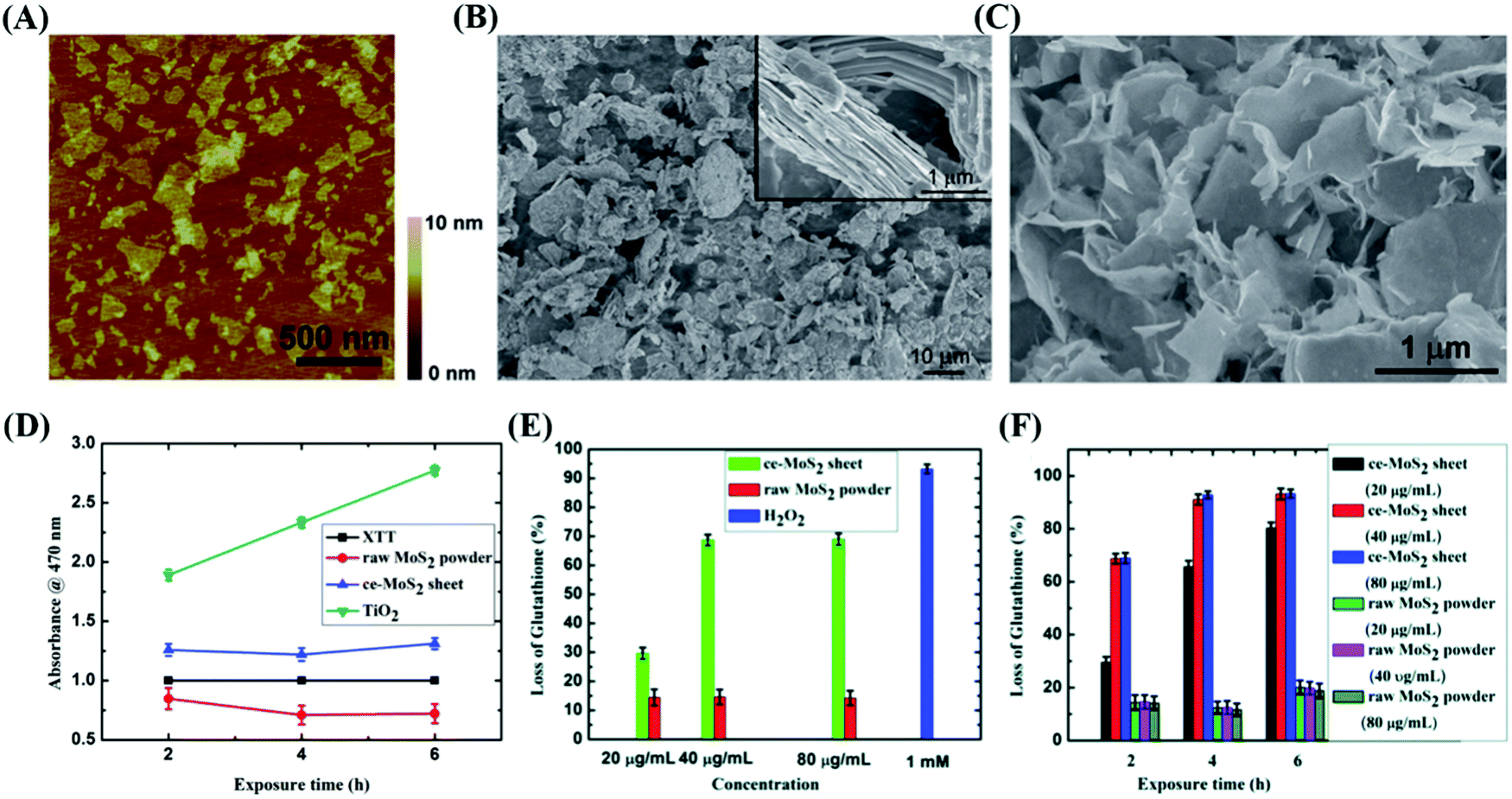
Due to their wide active edges, high charge carrier separation and special surface area, thin-layered nanosheets are capable of enhancing the photoproduction of multiple ROS, and ROS-induced oxidative stress has been proven to play a principle role in the disinfection mechanism.121 For example, 2D MoS2 nanosheets with few layers possess stronger inactivation behaviors in the presence of visible light compared with bulk MoS2,122 implying that the photocatalytic disinfection effects of MoS2 materials can be improved by reducing their layer thickness and lateral size. According to Chong,123 four types of ROS (O2˙−, 1O2, ˙OH and H2O2) were detected in MoS2-based materials (Fig. 9(A)), among which H2O2 possess the strongest antibacterial ability (Fig. 10).
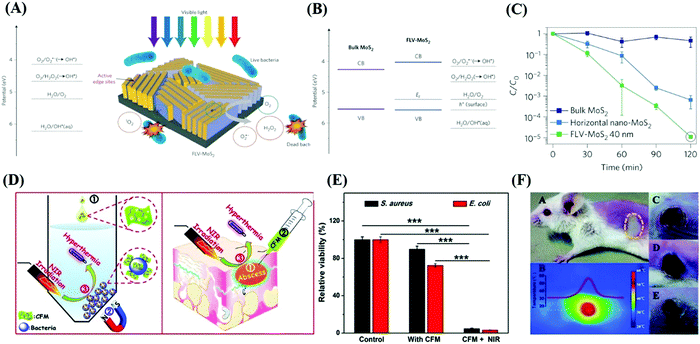 |
| | Fig. 9 Photocatalytic antibacterial effects of MoS2 in response to various wavelengths. (A) Potential of ROS referring to the vacuum level and schematic illustration of photogenerated ROS-induced bacteria death in the present of optical light. (B) Band edge of two different MoS2 with respect to the ROS formation potential. (C) Corresponding inactivation effects of three MoS2-based materials. (A)–(C) Reprinted from ref. 122. (D) Photothermal disinfection effects in vitro and local infection treatment in vivo by MoS2-based materials with NIR exposure. (E) Inactivation capability of CFM with or without NIR irradiation. (F) Serial characterization of photothermal therapy effects of CFM treatment in infected mouse in vivo. (D)–(F) Reprinted from ref. 124. | |
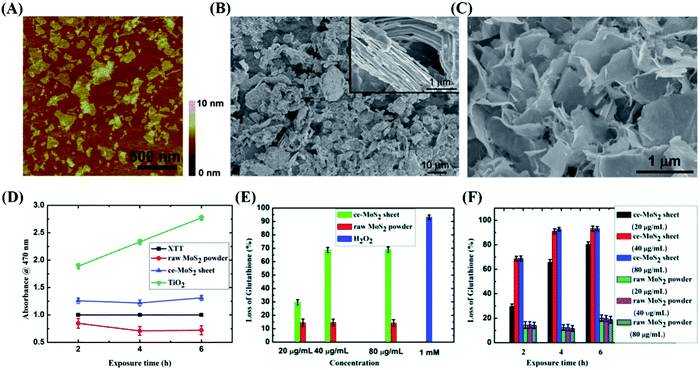 |
| | Fig. 10 Significant distinction in morphology and antibacterial properties between MoS2 particles and sheets. Reprinted from ref. 129. | |
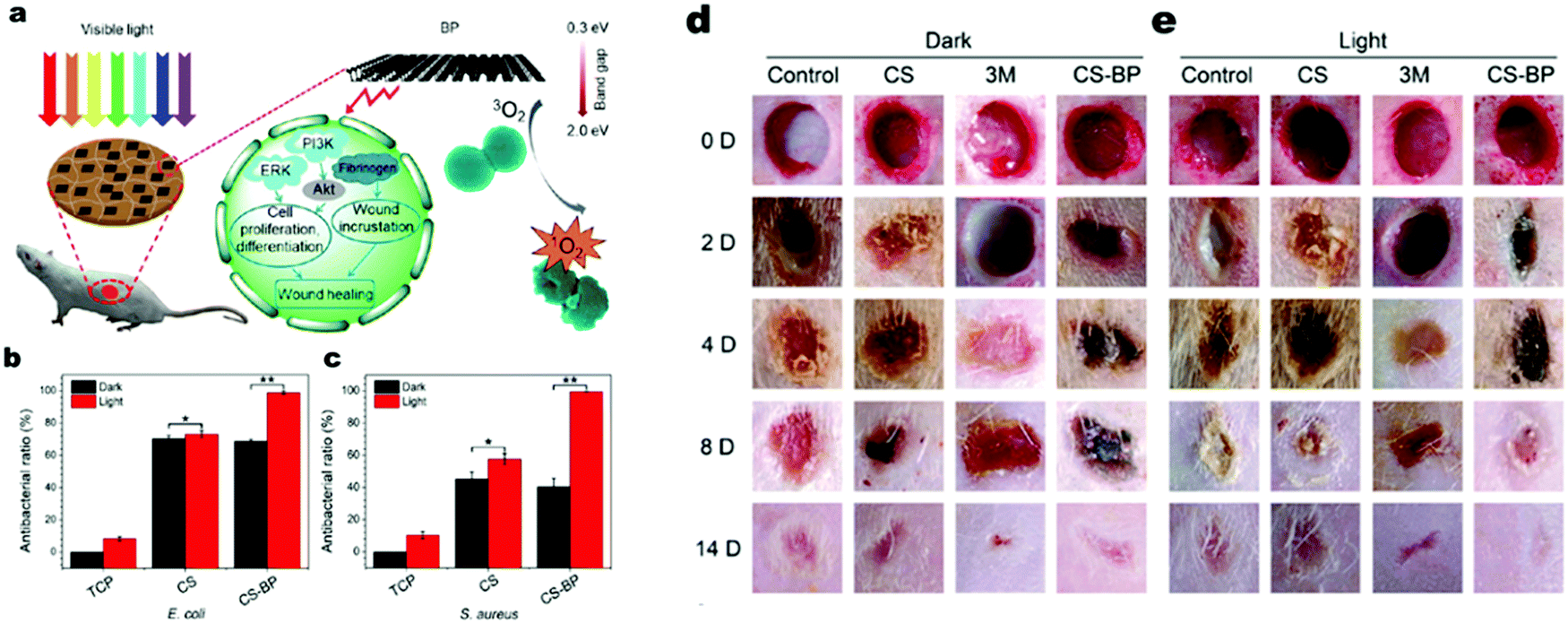
For their further application in the biomedical field, photocatalytic antibacterial agents should possess low cytotoxicity to avoid tissue inflammation or toxicity and high biodegradability with nontoxic decomposition products in vivo.125,126 The degradation product of BP exists in form of PxOyn−, which are non-toxic towards cells,127 endowing the material low cyto-toxicity. Besides, it was revealed that BP can somehow reduce inflammation by down-regulating inflammatory cytokines such as IL-6 and up-regulating regenerative cytokines such as IL-10.128 Besides, BP has good photo-thermal bacterial inactivation,128 which may exhibit a synergetic effect on disinfection. It should be noted that unlike other common photocatalysts, the photo-excitation process of BP mainly produces 1O2 upon excitation by O2 (triplet oxygen) rather than O2˙− or ˙OH to achieve photocatalytic activity (Fig. 11).127
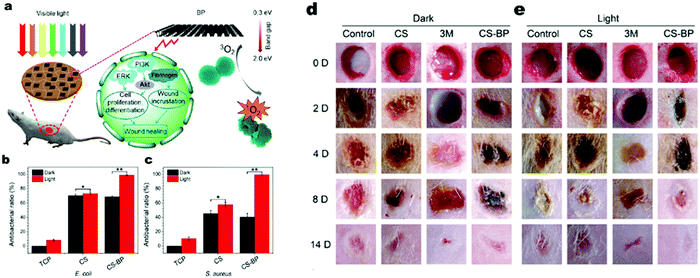 |
| | Fig. 11 Photocatalytic bacterial inactivation and would healing promotion by BP. (a) Schematic illustration of photocatalytic bacterial inactivation and wound healing by BP. (b) and (c) In vitro photocatalytic bacterial inactivation against two different bacteria. (d) and (e) In vivo wound healing process. Reprinted from ref. 137. | |
3.3.3 The deficiencies and limitations.
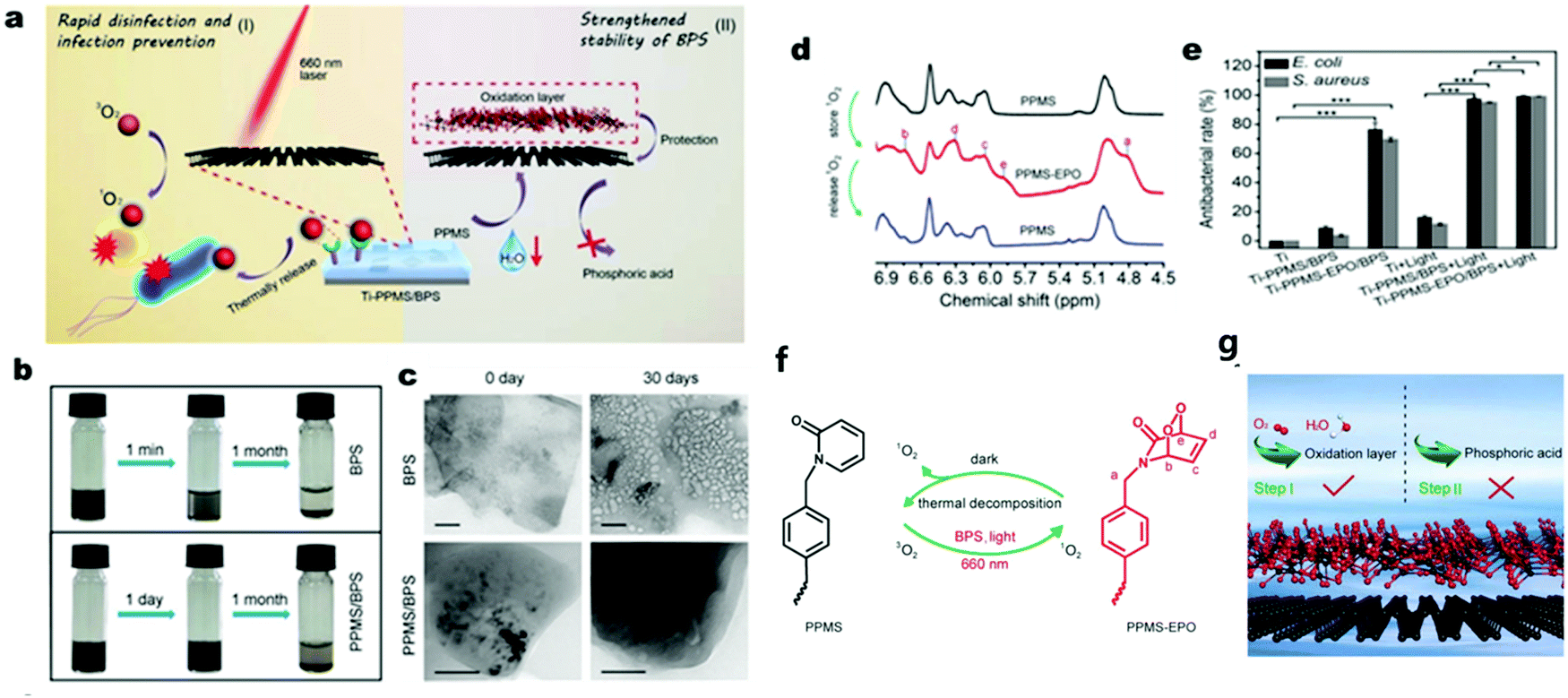
Although the above-mentioned 2D semiconductors possess very different merits for photocatalytic disinfection, they still have some limitations as pure components. For example, the zero band gap property of graphene (including graphene oxide) impedes its application as a photocatalyst given that it is unable to generate photo-induced electrons or holes.130,131 Besides, the carrier mobility MoS2 displays a strong temperature and density dependence at low temperature ranges,132,133 which suggests that it may not be an ideal photocatalyst. Also, BP nanosheets (BPS) suffer from poor chemical stability given that they tend to degrade in ambient oxygen and water, leading to unstable photocatalytic activity.134,135 Therefore, strategies have been investigated to protect BPS from being oxidized.135 For example, Wu et al.136 reported poly(4-pyridonemethylstyrene) (PPMS) wrapped in the external layer of BPS to strengthen chemical stability of BPS and realize the storage and thermal-responsive release of photo-generated 1O2 for on-demand antibacterial activity. Besides, PPMS was detected to capture some of the photo-generated 1O2via their reaction and later restore 1O2 in the form of PPMS endoperoxide (PPMS-EPO). The stored 1O2 was released through the reduction of PPMS-EPO to PPMS by thermal decomposition in the dark, which explained why the composite showed a disinfection effect even without irradiation (Fig. 12) and Table 1.
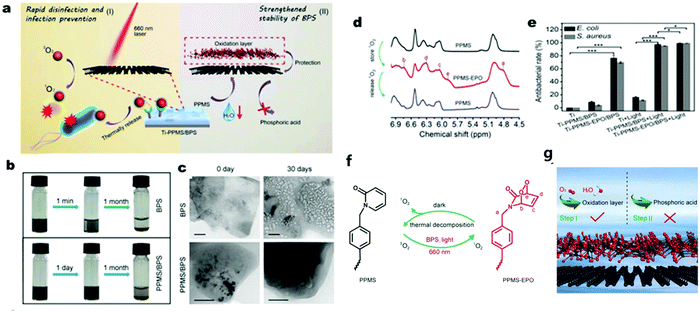 |
| | Fig. 12 Rapid bacterial inactivation and strengthened chemical stability of PPMS/BPS. (a) Schematic illustration of PPMS/BPS bacteria disinfection by singlet oxygen (1O2) with and without light, respectively. (b) Images of long-period dispersity and stability of PPMS/BPS in comparison to that of BPS. (c) Change in TEM characteristics of BPS and PPMS/BPS after one month. (d) 1H NMR spectra of PPMS and PPMS-EPO illustrating their reversible structure change. (e) Antibacterial rate results for two different bacteria. (f) Mechanism of the reversible 1O2 storage and release between PPMS and PPMS-EPO. (g) Graphical abstract of the protection of BPS with outer PPMS layers. Reprinted from ref. 136. | |
Table 1 Comparison of pristine semiconductors with different conditions for bacterial inactivation
| Semiconductor |
Type |
Morphology |
Preparation methods |
Model bacteria |
Initial condition 1 (CFU mL−1) 2 amount of photocatalyst |
Light condition |
Inactivation efficiency |
Radicals |
Ref. |
|
Λ (nm) |
LPD (mW cm−2) |
˙OH |
1O2 |
O2˙− |
H2O2 |
|
| “NA” stands for “not analyzed during the study”; “✓” means can be detected during the study of role of ROS; and “N” refers to “not detected” or “not responsible for the photocatalytic bacterial inactivation” as proven by experiments. |
| TiO2 |
n |
Nanoparticles |
P25, purchased from Degussa Co. |
E. coli
|
1: from 4.4 × 105 to 8.4 × 105 |
300 to 420 |
NA |
>99.9% for 2.25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log within 120 min log within 120 min |
✓ |
NA |
✓ |
✓ |
235
|
| 2: 1.0 g L−1 |
| TiO2 |
n |
Film layers |
Sol–gel approach |
Bacillus subtilis
|
1: 107 |
UV light 310–390 |
40 |
>99.9% within 30 min |
✓ |
NA |
✓ |
NA |
236
|
| 2: P25 (10 g L−1) |
| TiO2 |
n |
Nanosheets |
Hydrothermal method |
S. mutans
|
1: 1700 × g |
UV light at 365 nm |
2.5 |
30 min |
✓ |
NA |
✓ |
NA |
237
|
| 2: 0–0.4 mg mL−1 |
| ZnO |
n |
Nanoparticles |
Temperature and solvothermal synthesis |
S. aureus strain |
1: 1 × 108 (OD = 600 nm) |
300–400 |
NA |
99% within 6 h |
✓ |
NA |
✓ |
✓ |
238
|
| 2: 5 mM, 60–420 μL |
| ZnO |
n |
Flowers |
Co-precipitation process |
P. aeruginosa
|
1: 1 × 108 (OD = 600 nm) |
Visible light |
100 |
Completely inactive within 45 min |
✓ |
NA |
✓ |
NA |
239
|
| 2: 83.3 mM, 60 mL |
| Fe2O3 |
n |
Nanofilms |
Electrochemical anodization of steel |
P. aeruginosa
|
1: 2.0 × 107 |
Visible light ≥400 |
100 |
99.9% within 6 h |
✓ |
NA |
✓ |
NA |
240
|
| 2: 1 cm2 film |
| CuO |
p |
Powder |
Sol–gel method |
E. coli
|
1: 1 × 108 |
90%, ≥400 |
29.2 |
100% within 3 h |
✓ |
NA |
✓ |
✓ |
73
|
| 2: 6.15 μg |
| MoS2 |
p |
QDs |
Sonication and solvothermal treatment of bulk MoS2 |
E. coli, S. aureus subsp. |
1: 1 × 106 |
Visible light |
NA |
Less than 40% within 1 h |
✓ |
✓ |
✓ |
NA |
241
|
| 2: 50 μg mL−1 |
| MoS2 |
p |
a: few-layered nano film |
In situ growth |
E. coli
|
1: 1 × 106 |
Visible light |
NA |
a: >99.999% |
✓ |
✓ |
✓ |
✓ |
123
|
| b: bulk |
2: from 0.1 g L−1 to 2 g L−1 |
|
b: 54% within 120 min |
| Cu2O |
p |
Nanoparticles with 10 nm |
Chemical deposition method |
E. coli
|
1: 2 × 105 |
≥400 |
100 |
100% within 80 min |
✓ |
NA |
N |
✓ |
242
|
| 2: from 5 to 200 μg mL−1 |
| Cu2O |
p |
Films |
Electrochemical deposition |
E. coli
|
1: 2 × 107 |
≥400 |
1200 |
100% within 6 h |
N |
N |
N |
✓ |
243
|
| 2: NA |
| g-C3N4 |
n |
Mesoporous |
Self-condensation reaction of cyanamide |
E. coli K-12
|
1: 2.5 × 106 CFU mL−1, 100 mL |
Visible light (≤465 nm) |
NA |
100% at least 4 h |
N |
✓ |
N |
✓ |
244
|
| 2: 1 mg mL−1 |
| g-C3N4 |
n edges, hydrophilic groups |
Nanofilms |
Liquid-phase stripping method from bulk g-C3N4 powder |
S. aureus, E. coli |
1: 11.4 CFU cm−2 |
Visible light |
30 |
93.7% ± 2.7% was removed by 0.62 J dosage for 120 min |
✓ |
NA |
✓ |
✓ |
245
|
| 2: 10 mg mL−1 |
| RP |
p |
Nanosheets |
Hydrothermal purification |
E. coli
|
1: 0.5log10 |
Visible light |
193 |
Complete inactivation was obtained within 60 min |
✓ |
✓ |
✓ |
✓ |
246
|
| 2: 0.13 g mL−1 |
| BP |
p |
Nanosheets |
Basic solvent exfoliation from bulk BP |
E. coli and S. aureus |
1: 107 |
LED light irradiation |
5 |
>80% for all concentrations and 100% for 5 mg L−1 within 3 h |
N |
✓ |
N |
NA |
127
|
| 2: 0.01–5 mg L−1 |
4. Semiconductor hybrid composites for enhanced photocatalytic bacterial inactivation
4.1 Noble metal-doped hybrid composites
4.1.1 The electron transfer and plasmonic effects of metal-semiconductors.
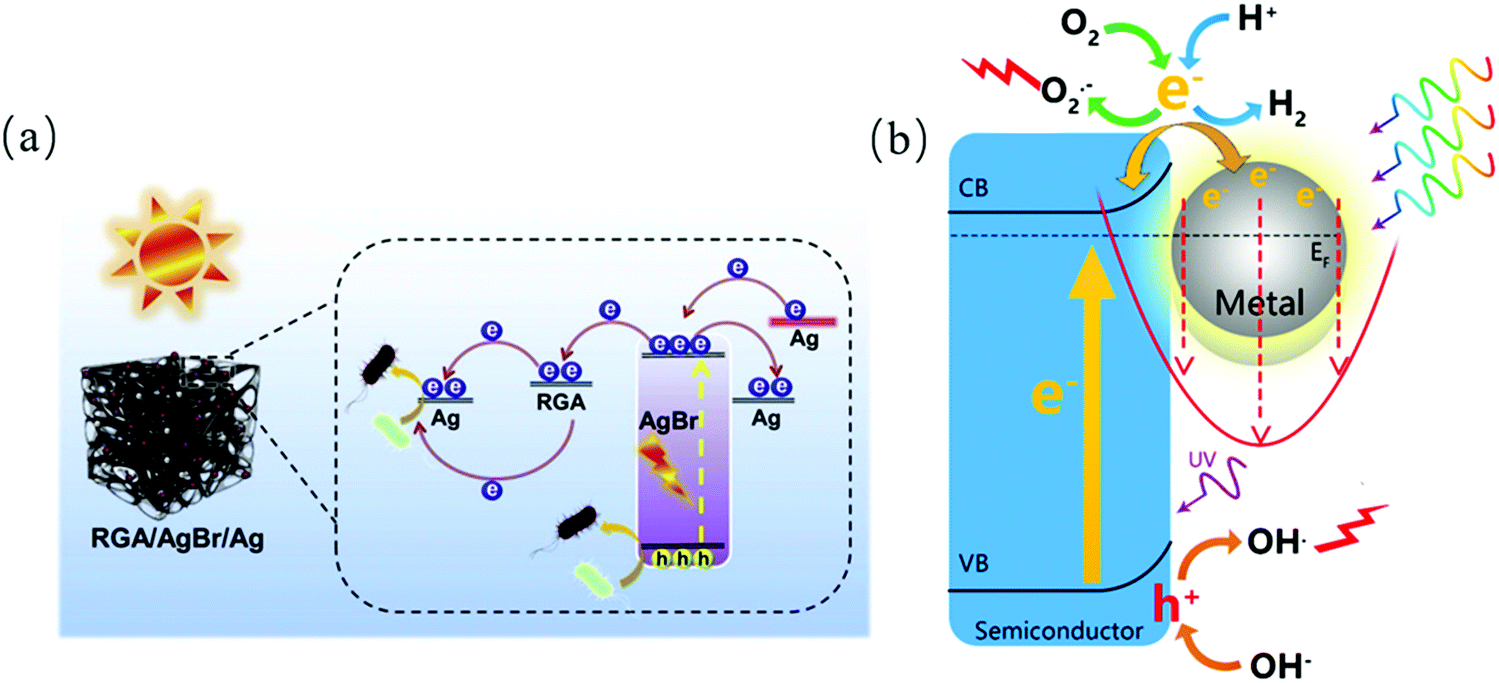
Noble metals (Au, Ag, Ru, Ir, Pt, etc.) refer to metals with strong chemical inertness.138 It has been proven that metals loaded on semiconductor materials as co-catalysts can significantly improve their photocatalytic performances by promoting the mobility of electrons,139,140 and thus recent research has been focused on modifying pristine semiconductors. For instance, Xin et al. confirmed that Ag nanoparticles deposited on a graphene oxide aerogel (RGA) can be applied as an electron delivery medium for improving the transfer of charge carriers.141 Baig's team142 used palladium-decorated silicon carbide (Pd–SiC) to inactivate sulfate-reducing bacteria (SRB). Pd–SiC showed superiority to SiC nanoparticles in photocatalytic disinfection, which can be attributed to the synthesis of Pd–SiC junctions, resulting in an increase in the separation of photo-generated charge carriers. Also, it exhibited a decrease in energy gap through the characterization of its diffuse reflectance spectra (DRS). Besides, some studies also indicate that more tight contact can be achieved between the atomic metal and semiconductors than that of metal clusters and semiconductors, which enhance the charge transfer for catalytic reactions.143,144 The localized surface plasmonic resonance (LSPR) of noble metals have proven to be inducible in solar photocatalysis. LSPR is defined as specific oscillations of electrons and electromagnetic fields,145 where a metal nanostructure strongly absorbs photon energy. Also, the plasmonic effect on photocatalysis can be classed as photon enhancement, hot electron injection and near-field enhancement.146,147 For example, Yu et al. designed a plasmonic nanostructure with three components(CdS/Au/SrTiO3),148 where hot injections were injected from Au to the neighboring CdS/SrTiO3, enabling a hot electron-assisted energy cascade for electron transfer, and thus photon absorption enhancement. The enhanced near-field amplitude effect applies to the case of insulation between a plasmonic metal and semiconductor. For instance, Wang et al. synthesized a plasmonic Au/TiO2/Au couple for electromagnetic enhancement (Fig. 13).148
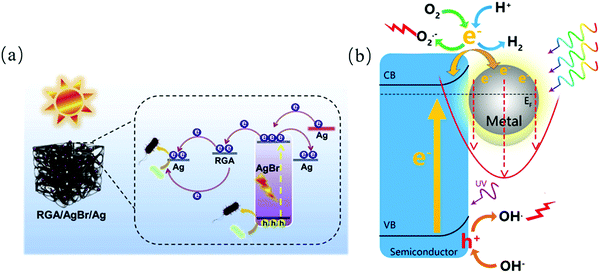 |
| | Fig. 13 (a) Schematic illustration of electron transfer in Ag-deposited RGO/AgBr photocatalyst141 and (b) plasmonic-mediated photocatalysis.147 | |
4.1.2 The active sites and optimal doping.
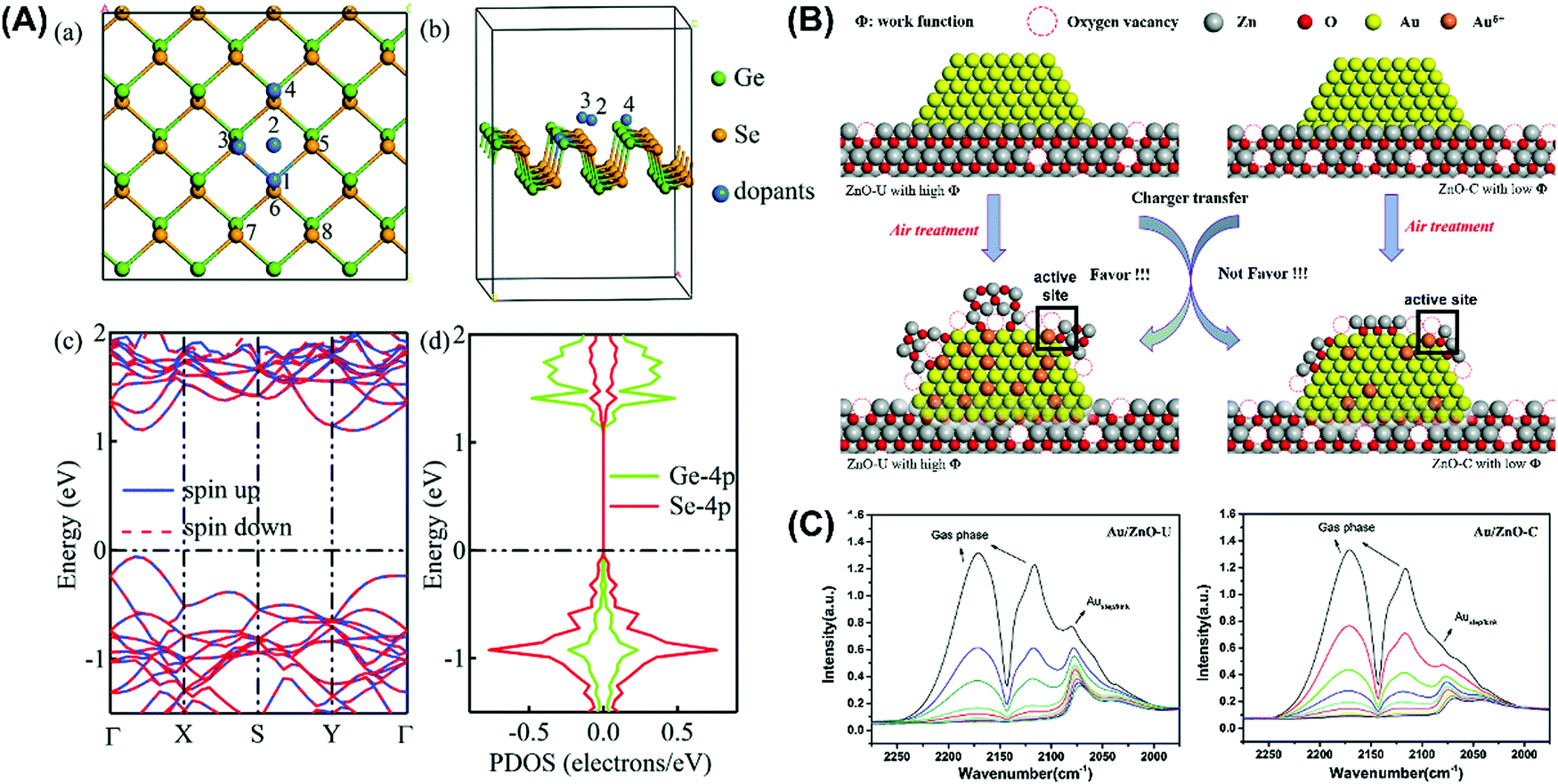
Given that photoinduced reactions mainly occur on the surface of nanoparticle catalysts and are dominated by surface atoms,143,149,150 the electron transfer between semiconductors and doped metal atoms may cause the surface metal atoms to act as electron-rich or electron-deficient active sites for the adsorption of reactants or desorption of products, and consequently enhance the catalytic performance.151 Huang et al.144 successfully synthesized monatomic silver chains from Ag particles via a simple thermal method. Also, this single-atom catalyst allowed the effective activation of lattice oxygen and oxygen molecules. Our team prepared a series of metal clusters that showed very surprising photochemical performances.152–157 It was found that the doped metal atoms can exist either as substitutes for the original metal atoms or adsorbed on the surface. For example, He et al.158 discovered that metal dopants were inclined to occupy the Ge sites rather than adsorb on the surface of GeSe (Fig. 14) and doping increased the oxidation of photogenic holes and/or the reduction of photogenic electrons. It was proven that the electron-deficient structure induced by black phosphorus-modified Pd nanoparticles improved the catalytic activity,151 and the redox cycle formed between Pd0 and Pd2+ also enhanced the activation of oxygen.159 Besides, the interface between dopants and the semiconductor is also worth studying. According to Li et al.,160 the insertion of Au into the ZnO lattice at the Au/ZnO interface leads to the formation of vacancies in the subsequently generated oxygen due to the charge mismatch, which increases the number of active sites on the catalyst surface. Nevertheless, it should be mentioned the strategy of metal doing has some disadvantages such as high cost, poor thermal stability and tightly controlled concentration of metals,161,162 where excess metal can act as recombination centres, leading to a decline in quantum efficiencies. It was reported an Au content in the range of 1 to 5 wt% shows efficiency within 3% deviation,162,163 which decreases once it exceeds 8 wt%.
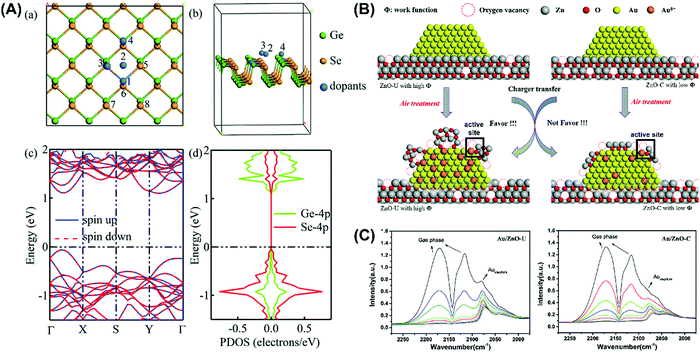 |
| | Fig. 14 Mechanism of dopant sites and creation of active sites for the introduction of noble metals. (A) Crystal lattice structure of noble metal-doped GeSe. (a) and (b) Location of doped metals with different perspectives. (c) Band structure and (d) partial density of states (PDOS) of monolayer GeSe. (B) Creation of active sites after Au doping with oxygen vacancy. (C) In situ FTIR spectra of CO adsorption for the study of adsorption sites and valence states of Au catalyst. (A) Reprinted from ref. 158. (B) and (C) reprinted from ref. 160. | |
4.1.3 ROS generation for antibacterial activity and biocompatibility.
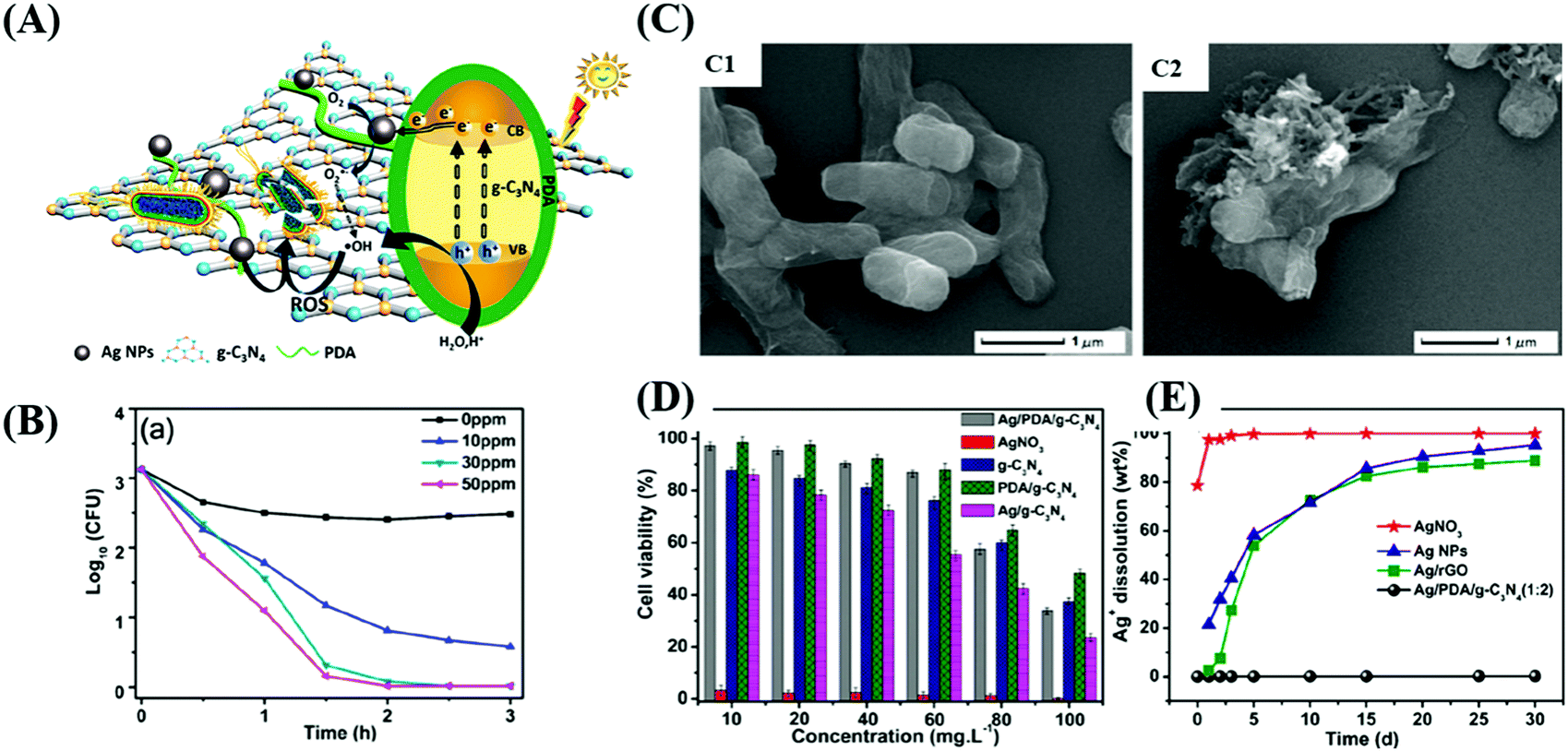
Studies on the use of metal-loaded semiconductors for bacterial disinfection have been widely reported.164–167 However, they rarely discussed biosecurity issues, where the release of metal ions is beneficial for synergistic disinfection, but leads to an increase in toxicity.168,169 Compared to other applications, photocatalytic sterilization demands higher requirements because photocatalysts have potential to be used in the disinfection of biological and medical devices.170,171 International supervisory agencies have created serial nanomaterial supervisions and regulations,172 through which the regulation, adaptability, biosafety and biocompatibility173,174 of photocatalytic materials will need to be considered,175 such as low cytotoxicity,176 the controlled release of drug or metallic ions,170 green synthesis of photocatalysts,175 and stability and reusability.174,177 It has been confirmed that cell death is probably due to the damage of the cell membrane integrity driven by photogenerated ROS, which can induce the release of components in bacteria, such as ions, phospholipids and proteins.178,179 As shown in Fig. 15, Wu's team180 synthesized a sterilized material silver/polydopamine/graphitic (Ag/PDA/g-C3N4), in which PDA/g-C3N4 was used as the carrier of silver to control the release of Ag+. The modified PDA not only could promote the dissolvability and dispersibility of the graphitic, but was beneficial for the deposition of small-sized and highly dispersive Ag NPs. The Ag NPs contributed to the promotion of the photo-excited capacity of the system by broadening the range of light absorption and narrowing the energy gap.181 It was indicated that ˙OH and O2˙− radicals are the dominant photoexcited radicals in disinfection by damaging the bacterial membrane. Also, the composite showed good cytocompatibility and hypotoxicity as evaluated by MTT assays and the detection of dissolved Ag+ (Table 2).
Table 2 Systematic review of the recent publications, showing the best doping of noble metals and ROS generation from different NPs
| Num. |
Materials |
Powder size in diameter |
Fabrication methods or main process |
Wavelength range of light |
Forms of noble metals |
The mechanism for bacteria death |
The (best) loading of metals |
ROS |
Ref. |
| ˙OH |
O2˙− |
1O2 |
H2O2 |
| “✓” indicates the corresponding type of ROS (˙OH, O2˙−, 1O2, and H2O2), “N” indicates no significant ROS detected, and “NA” means “not available” or not provided in the literature. |
| 1 |
Ag/PDA/g-C3N4 nanoparticles |
3.6 nm of AgNPs in Ag/PDA(1:4)-g-C3N4; 8.0 nm of AgNPs in Ag/PDA (1:2)-g-C3N4; 10.5 nm of AgNPs in Ag/PDA (1:1)-g-C3N4 |
Thermal treatment and in situ polymerization method |
Visible light |
Ag+/Ag0 |
Electrostatic adsorption, ROS generation; damage of E. coli bacteria cell membrane |
21.3 wt% Ag/PDA (1:2)-g-C3N4 |
✓ |
✓ |
NA |
✓ |
180
|
| 2 |
TiO2/ACF–Pt electrode |
<100 nm of Pt particles electrode |
Ion sputtering and coating operation |
UV light, <385 nm |
Pt0; Pt2+; Pt4+ |
ROS generation and ion release of Pt; damage to the plasma membrane P. expansum spores |
NA |
✓ |
✓ |
NA |
NA |
166
|
| 3 |
Ag/ZnO nanoparticle |
∼20 nm of Ag nanoparticle |
Preparation of ZnO via solution combustion method, and adding AgNO3 |
Under natural sunlight with 0.753 kW m−2 of average solar intensity |
Ag+/Ag0 |
ROS generation and ion release of Ag; disruption of cell membrane and invasion of DNA structure |
1 atom% |
✓ |
N |
NA |
✓ |
248
|
| 4 |
RGA/AgBr/Ag nanofilm |
80 nm of AgBr; too small to observe Ag in SEM |
Low-temperature chemical reduction method |
Visible light, λ > 400 nm |
Ag+/Ag0 |
ROS generation and ion release of Ag; E. coli rupture |
NA |
✓ |
✓ |
NA |
NA |
141
|
| 5 |
Bi2MoO6/Ag–AgCl microsphere |
NA |
AgCl sediment and Ag photoreduction on the surface of Bi2MoO6 |
Solar simulator 300 W Xe lamp |
Ag+/Ag0 |
Surface plasmon resonance of Ag, ROS generation; destruction of E. coli cells membrane, and leakage of intracellular components |
Mass ratio of AgCl to Bi2MoO6 is 15% |
✓ |
N |
NA |
NA |
249
|
| 6 |
Pb–BiFeO3/rGO nanoparticle |
3.1 nm of Pb–BiFeO3/rGO |
Hydrothermal method |
Visible-light irradiation (λ ≥ 400 nm) |
Pb2+, Fe3+ Bi2+/Bi3+ |
ROS generation, the release of Bi2+, Fe3+ and Pb2+; cell membrane damage, nucleic acid and protein leakage |
5 wt% rGO (PbBiFeO3–0.5% rGO) |
✓ |
N |
N |
N |
250
|
| 7 |
Ag–Au/ZnO |
4–100 nm of Ag–Au/ZnO |
Precipitation-decomposition method |
In nature visible light |
Ag+/Ag0 |
ROS generation membrane damage |
Au-1 wt% and Ag-5 wt% |
✓ |
✓ |
NA |
NA |
251
|
| 8 |
Ag/TiO2 nanofibers |
NA |
Mainly hydrothermal synthesis |
Visible lamps (Goodly, F8T5/D 8 W) |
Ag+/Ag0 |
ROS generation, the release of Ag+; damage the cellular membrane and cause osmotic pressure imbalance of S. aureus and E. coli |
5 mol% |
N |
✓ |
NA |
NA |
252
|
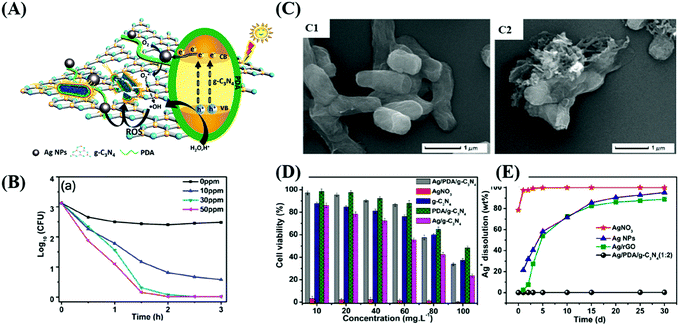 |
| | Fig. 15 Scheme showing the composition and biological performance of Ag/PDA/g-C3N4 photocatalysts. (A) Synthesis of the material via thermal treatment and in situ polymerization. (B) Disinfection curve of bacteria co-cultured with different concentrations of reagents. (C) Change in SEM image of E. coli after incubation with reagents. (D) Cytotoxicity of HUVEC cells treated with different materials. (E) Release of Ag+ from different materials. Reprinted from ref. 180. | |
4.2 Ion-doped hybrid semiconductors
Among the various photocatalysts, metal-oxide semiconductors have been widely applied due to their outstanding physicochemical properties.182 Nevertheless, most oxide-based semiconductors are not accessible for the production of H2 due to their very low CB edge position or optical light absorption due to high edge location of the VB, especially for d0 transition metal oxides, which mainly depend on the O2p orbitals (>3.0 eV).183,184 According to current research, doping has become one of the most effective strategies applied for the band structure modification, and therefore photocatalytic performance enhancement of semiconductors especially oxide-based semiconductors (Table 3).185,186
Table 3 Anion/cation dopedsemiconductors and their inactivation activity
| Doping type |
Photocatalyst |
Synthesis method or main process |
Doping mechanism |
Anion amount |
Band gap (eV) |
Photocatalytic antibacterial activity |
Antibacterial mechanism |
Ref. |
| “/” means not mentioned. |
| Cation |
Zr–Ag-co-doped TiO2 |
Sol–gel method |
Narrows the energy gap, inhibit charge carrier regeneration |
5% mol silver and 10% mol zirconium |
/ |
Complete inhibition of E. coli within 20 min under fluorescent light |
Zr4+ increases adhesion; the toxicity of Ag+; the photogenerated ROS |
196
|
| Cu-MOFs |
Hydration heating |
Introduced Cu2+ can serve as the trap of charges |
10 molar ratio Cu2+ |
/ |
99.71% S. aureus was inactivated within 20 min |
MOFs increase membrane permeation; synergistic effects of ROS and heat |
256
|
| Fe3+ and Pb2+ co-doped zinc oxide |
Microwave-assisted hydrothermal method |
Forms defects in the crystalline lattice of TiO2 |
8% Fe3+ and Pb2+, respectively |
3.15 → 3.06 |
microwave-assisted hydrothermal method |
Hydrophobicity of ZnO, electrostatic interaction |
257
|
| Cd/Fe-co-doped zinc oxide |
Sol–gel method |
Replaces Zn2+ ions by cations and create defects in the pure ZnO crystalline structure |
4% doping of (Cd, Fe) content |
/ |
High inhibition activity of P. aeruginosa and B. flexus |
Release of Zn2+ and ROS |
258
|
| Cu-doped zinc oxide |
Simple solvothermal route |
Narrows the band gap by electronic transition |
3.13 molar ratio of Cu ions |
3.20 → 2.74 |
Complete inhibition of E. coli under visible light for 4 h |
Release of Zn2+ and ROS, the rupture of cell membrane, inhibits growth. |
194
|
| Mg2+-doped TiO2 |
High-energy ball milling |
Forms defects in the crystalline lattice of TiO2 |
20% Mg2+ |
3.01 → 2.94 |
E. coli inhibition rate of 66.8%; S. aureus inhibition rate of 61.1% |
The toxicity of Mg2+; the presence of Ti3+ and Ti4+ enhances material photocatalysis; ROS |
259
|
| La+ doped-TiO2 |
High-energy ball milling |
Forms defects in the crystalline lattice of TiO2 |
10% La+ |
3.01 → 2.95 |
E. coli inhibition rate of 87.4%; S. aureus inhibition rate of 80.7% |
The toxicity of La+; Ti3+ and Ti4+ enhance material photocatalysis; the presence of ROS |
259
|
|
|
| Anion |
C–B co-doped TiO2 |
Sol–gel technology |
Narrows the band gap |
/ |
3.21 → 3.16 |
High inhibition activity of E. coli and S. aureus |
TiO2(B) phase and B2O3 phase benefit antibacterial effects; the photogenerated ˙OH |
260
|
|
|
F&N co-doped TiO2 |
Wet milling process |
Surface fluorination and interstitial doping |
F and N are (i) 7.45 at% and 2.83 at%, respectively |
/ |
Complete inhibition of E. coli (∼4.1 × 106 CFU mL−1) within 60 min; light source: 4.2 mW cm−2 |
The photogenerated ROS, especially ˙OH |
212
|
|
|
N-doped ZnO |
Self-assembly combustion |
Narrows energy gap with the blending of the 2p state of O |
1.04% nitrogen content |
/ |
Significant bacteriostatic zone with lamp illumination |
The introduction of nitrogen could significantly enhance the antibacterial performance of ZnO nanocrystals |
213
|
|
|
C-doped anatase-rutile nano-heterojunctions |
Microwave-assisted technique |
C 2p states benefit band gap narrowing |
/ |
3.16 → 2.90 |
C-doped nanoheterojunctions had a higher disinfection rate compared with the standard photocatalyst (0.0023 min−1 and −0.0081 min−1, respectively) |
Photogenerated ROS |
261
|
|
|
Cl-doped TiO2 |
Atomic layer deposition |
/ |
4% Cl content |
/ |
Reduction in CFU of C. albicans up to 59.5% after UV treatment |
Toxicity of Cl−, photocatalytic activity |
262
|
|
|
| Cation/anion co-doping |
Cu–N-co-doped TiO2 |
Sol–gel process |
Reduces the bandgap |
0.75% Cu–N content |
/ |
High inhibition activity of E. coli and S. aureus |
Toxicity of TiO2; the photogenerated ROS |
263
|
|
|
Mesoporous TiO2 with Ni–S co-doping |
Sol–gel method |
Ni2+: mingles in titanium dioxide lattice to suppress charge regeneration; sulfur: enters titanium dioxide lattice to decrease the energy gap |
0.75% of Ni–0.25% of S |
3.2 → 2.68 |
High inhibition activity of two types of bacteria |
Photogenerated ROS, e−/h+, O2− and ˙OH |
264
|
|
|
Ta/N–TiO2 |
Sol–gel method |
Narrows energy band gap by mixing of N 2p and O 2p, the new form of N 2p–Ta 5d hybrid |
The amount of Ta is 20 mol% |
3.1 → 2.20 |
High inhibition activity of three types of bacteria cultured with 300 μg mL−1 Ta/N–TiO2 powders for 2 h |
Photogenerated charge carriers |
221
|
|
|
B and Ce-co-doped TiO2 |
Sol–gel procedure |
Substitute B to occupy O sites |
/ |
/ |
Bacteria cultured with materials all have large inhibition zone within 120 min |
Smaller size of nanoparticle and higher specific area; broaden the range of visible-light response; surface hydroxyl groups; the toxicity produced by Ce3+/Ce4+ ions |
265
|
|
|
F,Cu-co-doped TiO2 |
Sol synthesis |
Charge compensation between F- and Ti4+ suppresses the regeneration of charge carriers |
F: 0.3–0.5 at%, Cu: / |
/ |
High antibacterial activity of the Cu-doped TiO2 with or without light |
Photogenerated ROS and copper ion toxicity, causing cell membrane rupture |
266
|
|
|
Co/F-co-doped ZnO |
Soft chemical method |
The introduction of Co and Co & F reduced the particle size; the form of defects in the crystalline lattice |
Co (1 at%) and F (10 at%) |
3.32 → 3.28 |
Induced big zone of inhibition for four types of bacteria due to the addition of dopants |
Due to the release of Zn2+ ions, the generation of ROS |
267
|
|
|
Sulfated and Eu-doped anatase |
Precipitation |
Formation of oxygen vacancy; Eu3+ dopants as an electron trap effect; lanthanoids reduce the titania band gap; increase surface area and pore volume |
/ |
/ |
Effective antimicrobial action of TiO2 nano-powders after exposure to UVA radiation within 60 min |
Inactivation of intracellular proteins; local damage of cell wall |
268
|
4.2.1 Cation-doped hybrid semiconductor photocatalysts.
Metal cation doping enables the promotion of the photocatalytic effects of semiconductors by reducing their photogenerated charge carrier recombination rates, extending their photon absorption to the visible range and enhancing the interfacial charge-transfer reactions.18,187 Choi et al.188 demonstrated that doping with some transition metal cations could significantly improve the photoactivity of TiO2, and a redshift occurred in the energy gap transition as the doping concentration increased, narrowing the band gap. However, they also discovered188 that the introduction of Co3+ and Al3+ reduced the photoactivity for some catalytic reactions due to the reduction in quantum yields obtained during continuous wave photolysis. It was shown that metal ions could be incorporated into the TiO2 lattice via substitution for certain Ti4+ ions, subsequently forming defects and oxygen vacancies.189 Also, the ions can function as charge carrier traps, resulting in the inhibition of their recombination.190,191
| Mn+ + ecb− → M(n−1)+ electron trap |
| Mn+ + hvb− → M(n+1)+ hole trap |
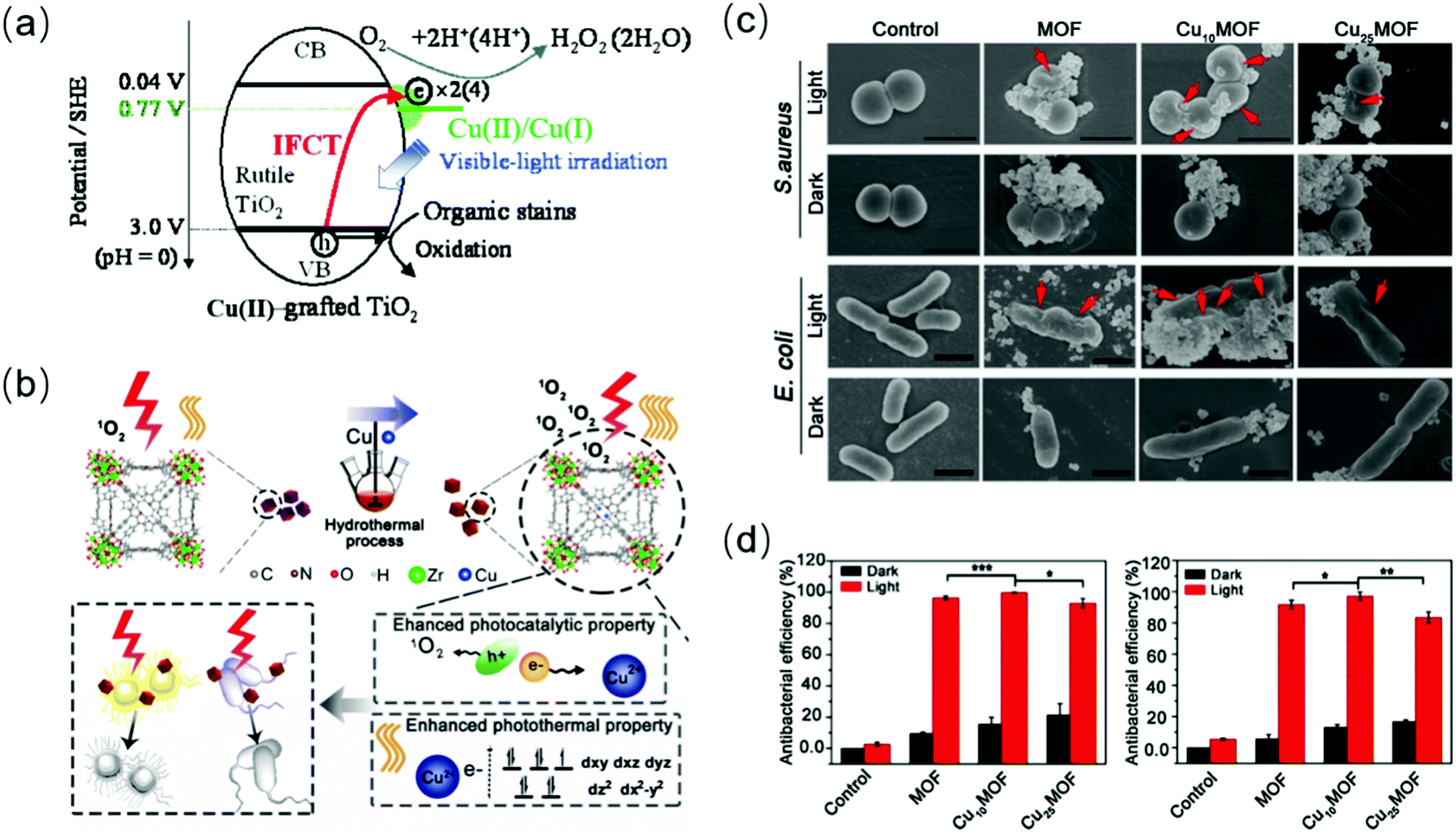
Especially, some metal dopants (such as Fe and Cu ions) enable the creation of additional energy levels near the TiO2 band, serving as charge traps.189,192 Also, their enhanced response to visible light is also attributed to photo-induced interface charge shift from the holes in the VB of the semiconductor to the adsorbed ions (such as Cu(II) species) on the surface and multi-electron reduction catalyzed by their reductive species (such as Cu(I) ions).189 For in situ Fe3+-doped BiOCl, the doped Fe3+ not only acts as an “electron transfer bridge” to capture and transport electrons, but the active center of catalytic reactions.193 Besides, the facet transition from the original BiOCl(001) to Fe–BiOCl(110) and (102) causes more exposure of the iron active sites. Various cation ion-doped hybrid semiconductors have been applied in the photocatalytic antibacterial field,194,195 for example, Ag+ (5% mol) and Zr4+ (10% mol) co-doped TiO2 showed complete inhibition of E. coli within 20 min,196 where Ag+ and Zr4+ functioned to narrow the energy gap and inhibit the regeneration of charge carriers, respectively. The presence of zirconium was beneficial to increase the adhesion between the Zr–Ag co-doped TiO2 particles and E. coli via electrostatic force. MOF doping with 10% Cu2+ (Cu10MOF) strengthened the antibacterial activity and photothermal effect for bacteria-infected wounds, mainly due to the photon absorption caused by the d–d transition of Cu2+197 and its capture of electrons (Fig. 16(b and c)). Cu10MOF also exhibited good biocompatibility in vitro and in vivo.198 However, an increase in the doping concentration of Cu2+ (Cu20MOF) resulted in an acceleration in electrons and holes regeneration and even a decline in their transfer velocity, which reduced the photocatalytic activity (Fig. 16(d)).
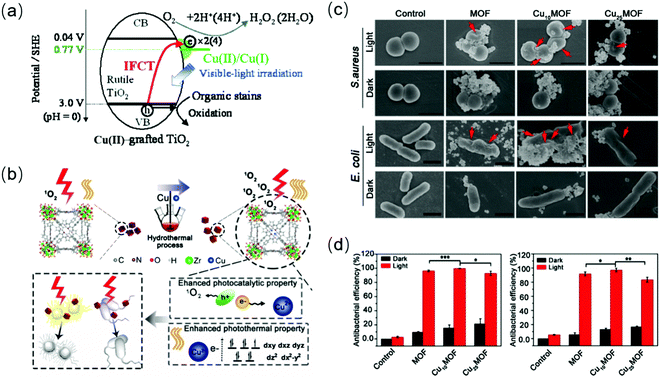 |
| | Fig. 16 Schematic illustration of the transition metal ion doping mechanism and its antibacterial effects upon exposure to light. (a) Possible photocatalytic mechanism of Cu(II)TiO2, including interfacial charge transfer (IFCT) and multielectron reduction processes. (b) Schematic diagram of the increase in photocatalytic and photothermal effects in the system. (c) SEM images showing the destruction of the morphology and structure of bacteria cultured with Cu2+-doped MOF under 660 nm light. (d) Evaluation of antibacterial action for different materials. (a) Reprinted from ref. 199. (b)–(d) Reprinted from ref. 198. | |
4.2.2 Anion-doped hybrid semiconductor photocatalysts.
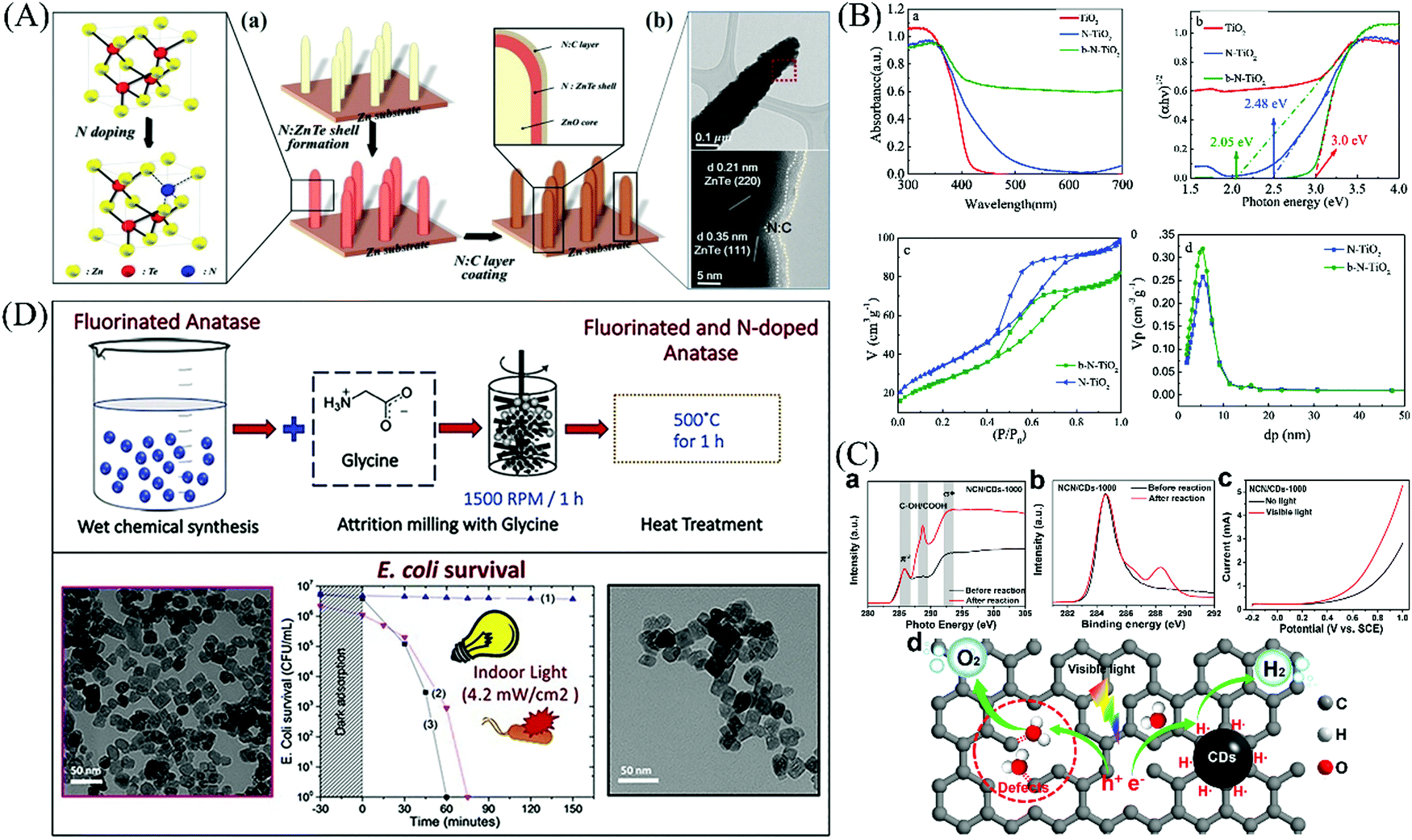
Vautier firstly found the that the introduction of nitrogen could strengthen the photo-degradation effects of TiO2 towards carmine dye in the present of visible light in 2001,200 followed by many other non-metal elements.201–206 It has been reported that non-metal dopants are superior to most of the metal ions with less generation of recombination centers.207 More vacancies or defects can be introduced into the original lattice after ion doping, which provide more active sites for photo-oxidation reactions.208 For example, in N-doped ZnTe with an N-doped carbon layer,209 the N atom inserted into the ZnTe lattice, leading to a slight reduction in the band gap and promotion of the charge transfer. Also, the N-doped carbon layers can be used as the active sites for subsequent reactions (Fig. 17(A)). In the case of N-doped TiO2,210 atom insertion similarly occurs in the lattice located above the conduction band and induces impurity levels with the introduction of Ti3+, further narrowing the band gap (Fig. 17(B)). The enhanced response to visible light can be attributed to the presence of oxygen vacancies, Ti3+ and N species. Defects can serve as active sites according to the previous discussion. Kang et al.211 designed a photocatalyst with inherent carbon defects (NCN/CDs) by removing N atoms, in which the defects bind to the adsorbed H2O as oxidation sites for the formation of O2 (Fig. 17(C)).
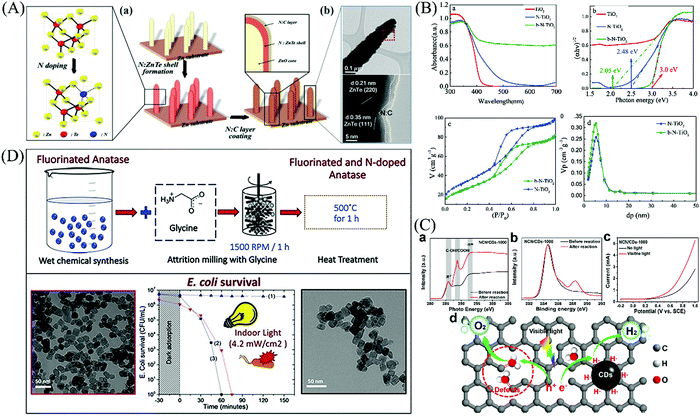 |
| | Fig. 17 Mechanism of anion-doped semiconductors. (A) Insertion of N atom in ZnTe lattice (a) and HRTEM images under low (upper) and high (below) magnification (b). Reprinted from ref. 209. (B) UV-visible solid absorption spectra (a) and band gap width (b) for N–TiO2 systems. N2 adsorption–desorption isotherm curves (c) and corresponding pore size distribution curve (d) of N–TiO2 systems. Reprinted from ref. 210. (C) Photoactivation mechanism of photocatalysts with rich carbon defects. Reprinted from ref. 211. (D) Preparation and characterization of F&N-doped TiO2 photocatalyst and its photocatalytic antibacterial effect. Reprinted from ref. 212. | |
Non-metal dopants have been well investigated for improving the antibacterial performance of materials. For example, Wu et al.213 fabricated N-doped ZnO nano-crystallites via a simple self-assembly combustion technique, which showed an obvious bacteriostatic circle of bacteria cultured with materials under lamp illumination, indicating that the incorporation of nitrogen into the ZnO lattice extended the light response and enhanced the antibacterial effect. Also, the inactivation ability of nanomaterials is mainly attributed to the photogeneration of ROS, where non-metal dopants are beneficial for the excitation of electrons through the overlap of non-metal the 2p levels and O 2p orbitals, leaving holes to produce the strongly oxidizing ˙OH.214 Milosevic et al.212 developed novel fluorine and nitrogen co-doped TiO2 nanoparticles, in which the amount of fluorine reached 7.45 at%. They showed that the F&N co-doped TiO2 was beneficial for disinfection due to its low agglomeration. It was further reported surface fluorination can help to induce the photogeneration of hydroxyl groups215 and the formation of superficial Ti3+ donor energy levels located near the CB,216 which accelerate the excitation of electrons, and therefore facilitate the continuous generation of ROS.
4.2.3 New strategy of co-doping hybrid with cations and anions.
Single-element doping causes a problem in the process of photocatalytic enhancement because the doping level will become the recombination center of photo-generated charges217 and even the impurity level formed by more discrete doping increases the migration and separation of light holes or electrons. Recent research results showed that by co-doping transition metals with non-metallic elements,218 such as N–La,219 S–La,220 and N–Ta,221 the co-doping defect level is passivated because the charge compensation of the anion and cation pairs is not likely to become a carrier recombination center.219,222 The synergistic effects of co-doping contribute to the enhancement of photocatalysis because of the extension of the range of light absorption and low regeneration efficiency.223
For example, Phattalun's team224 proposed a novel passivated co-doping method to produce vanadium–nitrogen (V–N) co-doped TiO2, in which the V–N pair led to a reduction in the energy gap of TiO2. V–N co-doping also has the potential to generate ˙OH and O2˙− due to the suitable band edges, which can serve as antibacterial agents.
4.3 Semiconductor hybrid heterojunctions
Although ion doping may serve as a solution to the rapid recombination of electrons and holes in semiconductor photocatalysts, it unavoidably reduces the oxidation and reduction potentials of the excited charge carriers and may be limited by the maximum doping ability, leading to an minor enhancement or even decrease in the photocatalytic bacterial inactivation.39 Since the CdS/TiO2 semiconductor hybrid system was firstly investigated and found to be able to transfer photo-generated electrons from the CB of CdS to TiO2 in 1984,225 it has become a hotspot for heterojunctions between two or three solid-state photocatalysts.226,227 It was found that this type of transfer process can serve as a pathway for charge separation, which contributes to reducing or slowing down the recombination of charges, and therefore enhances the photocatalytic efficiency.228,229 The past decades have witnessed the emergence of and subsequent expansion in the semiconductor hybrids for bacterial inactivation, which will be described in detail in the following sections. Besides, in the last Section 4.3.6, we discuss the heterojunctions of 2D materials, in which heterojunctions are defined and divided based on material dimensions rather than electron transport pathways, with emphasis on the interface contact effects between semiconductors with different dimensions.
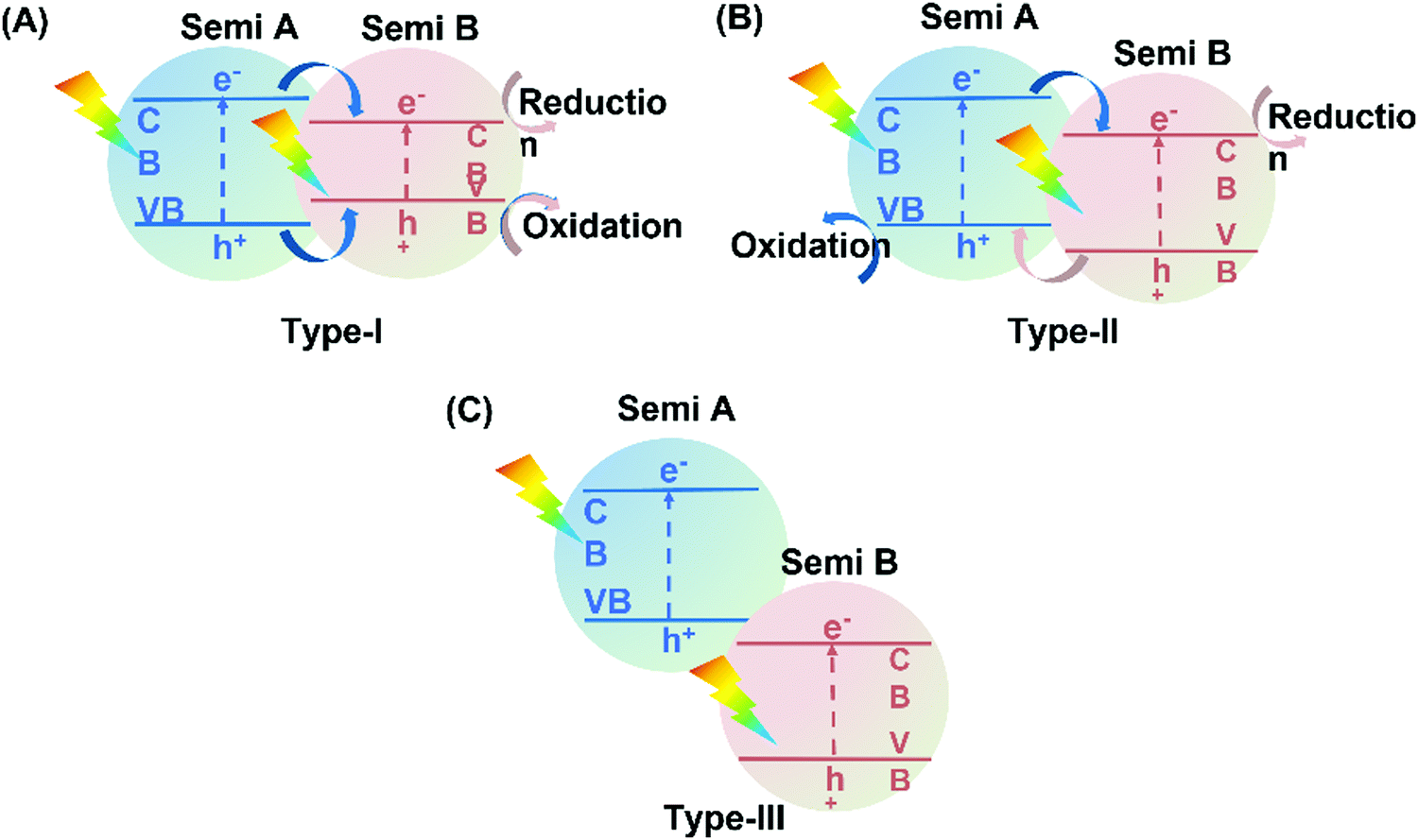
4.3.1 Type II heterojunction hybrids.
Typical heterojunction photocatalysts can be divided into three types depending on the band gap positions between two semiconductors (Fig. 18). According to Yu et al.,230 only type-II heterojunction is efficient to enhance photocatalytic activity due to its appropriate band structure for the spatial separation of charge carriers.
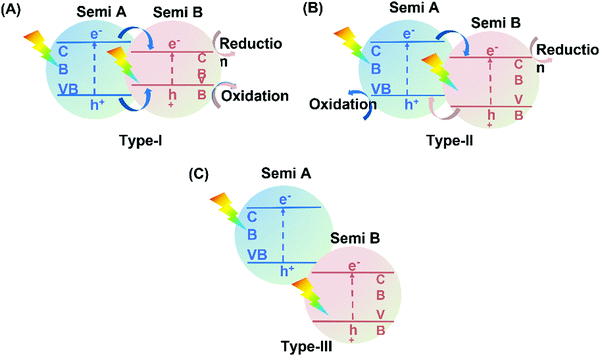 |
| | Fig. 18 Schematic illustration of electron transfer in the three different heterojunctions: (A) type-I, (B) type-II, and (C) type-III heterojunctions. Semi refers to semiconductor. | |
Type II heterojunctions have become common semiconductor photocatalysts used for bacterial inactivation,231,232 detailed information of which can be found in Table 4. Jia233 reported the synthesis of TiO2–Bi2WO6 bi-nanosheets to increase the antibacterial activity, and systematically investigated the underlying mechanism of inactivation. In the scavenger and EPR study, it was found that both the photo-excited electrons and holes contribute to the inactivation. However, h+ is the dominant factor. Also, as shown by the PCR and BCA assay, the components leaked were the genome and total protein. Moreover, the decrease in recombination of carriers was directly confirmed by the detection of the photocurrent, which makes it special among numerous studies. Ng234 reported a natural-light-responsive magnetic Fe2O3–AgBr for the photo-induced disinfection for E. coli and studied the factors influencing its activity, including temperature, dissolved ions and pH. As shown by their results, at 37 °C or/and pH = 7.67, the semiconductor hybrids exhibited the best photocatalytic bacterial inactivation, but a detailed explanation how temperature and pH affect the inactivation was not demonstrated. Moreover, the existence of Ca2+ would promote the inactivation, while anions such as SO42− would inhibit it given that a cation would offset the mutual repulsion between Fe2O3–AgBr and the bacterial membrane, both of which are negatively charged. The detailed inactivation data is shown in Table 4.
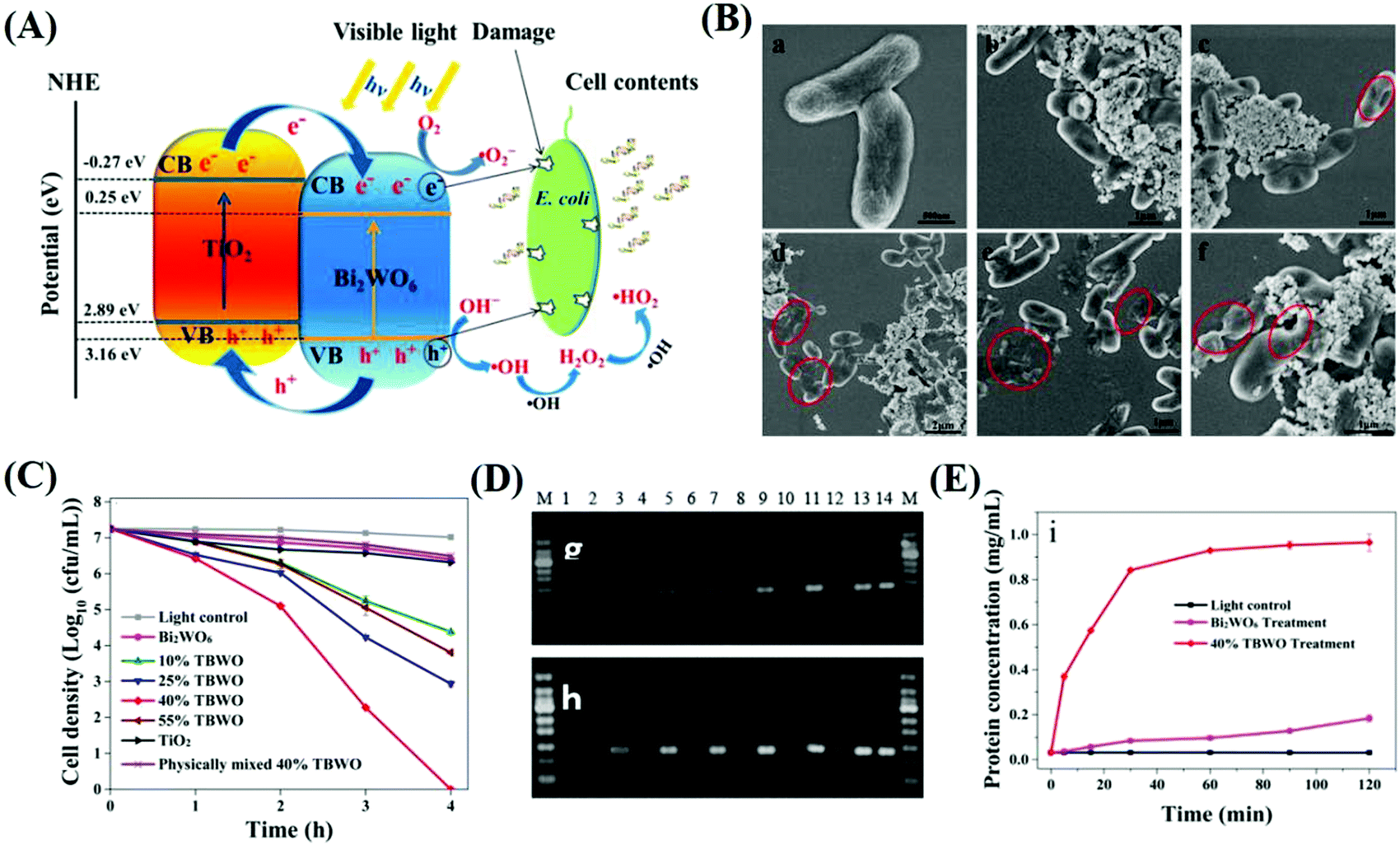
Generally, the geometrical structure of a heterojunction, especially its crystal plane, will greatly affect the charge redistribution at the heterointerface, and thus, change its photocatalytic efficiency. For example, Wu et al.26 reported a novel RP/ZnO heterojunction wafery film for solar or LED-driven rapid, efficient and safe disinfection and investigated the effect of the crystal plane. It was determined that in the RP/ZnO heterojunction, the RP(001) and ZnO(002) plane had the largest charge reallocation via density functional theory (DFT) calculations, and therefore, exhibited the greatest stability. In this study, photo-generated e− of the RP(001) plane jumped to the ZnO(002) plane, while h+ generated in ZnO would transfer to the VB of RP. Compared with RP, the RP/ZnO hybrid system can generate more ˙OH via the oxidation of water to enhance the antibacterial effect, whereas compared to ZnO, the hybrid system had an increased response to visible light and even NIR light and ensured sufficient carrier separation. Through the combined action of ˙OH, O2˙− and 1O2, a strong and rapid inactivation effect was achieved. This study showed that ROS could sterilize bacteria by triggering the overproduction of ROS in cells and causing oxidative stress to promote bacterial apoptosis, as confirmed by DCFH. S. aureus has a thick cell wall, which can efficiently slow down the penetration of ROS into its cells and rupture of its cell membrane, and thus excess ROS can be more easily neutralized. However, ROS-scavenging enzymes in E. coli were rapidly destroyed, and thus E. coli was quickly overwhelmed, which explains why the hybrids showed a better and faster inactivation towards E. coli(Fig. 19).
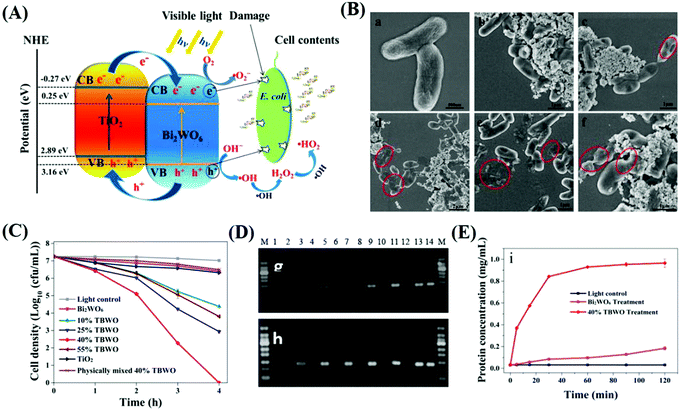 |
| | Fig. 19 Photocatalytic disinfection mechanism of a typical type II heterojunction, TiO2-Bi2WO6 nanosheets (TBWO). (A) Schematic illustration of the antibacterial process of TBWO. (B) Inactivation effect of E. coli at different times under visible light. (C) Bactericidal kinetic curve of TBWO with different concentrations. (D) Detection of leakage of bacterial 16rsDNA and (E) total protein in E. coli. Reprinted from ref. 231. | |
Although the type II heterojunction is ideal for separating electron–hole pairs in space with potential differences between the components, the enhanced electron–hole separation is still insufficient to overcome the ultra-fast electron–hole recombination on the semiconductor.247 Therefore, other types of heterojunctions have appeared to fill the gaps.
4.3.2 p–n heterojunction hybrids with noble metal nanoparticles.
p–n heterojunctions contribute to the migration or separation of photo-generated electron–hole pairs, and therefore the photocatalytic performance.253 Owing to their surface plasma resonance (SPR), high Fermi energy (Ef) and good antibacterial effect, noble metal nanoparticles, especially AgNPs, are widely used for the decoration of p–n semiconductor hybrids to improve their photocatalysis efficiency.
It is believed that AgNPs can be excited and generate electrons that transfer between other semiconductors. Jing et al. reported visible light-driven magnetic CoFe2O4/Ag/Ag3VO4254 and ZnFe2O4/Ag/Ag3VO4255 photocatalysts for highly efficient photocatalytic bacterial inactivation. In these systems, AgNPs generate plasma hot electrons, while Ag3VO4 and CoFe2O4 or ZnFe2O4 generate charge carriers. Due to the successive decline in the CB position, the electron would transfer from the AgNPs to Ag3VO4, and then to CoFe2O4 or ZnFe2O4. Meanwhile, the holes generated from CoFe2O4 or ZnFe2O4 would transfer to Ag3VO4, which greatly suppressed their recombination, and hence enhanced the photocatalytic antibacterial efficiency.
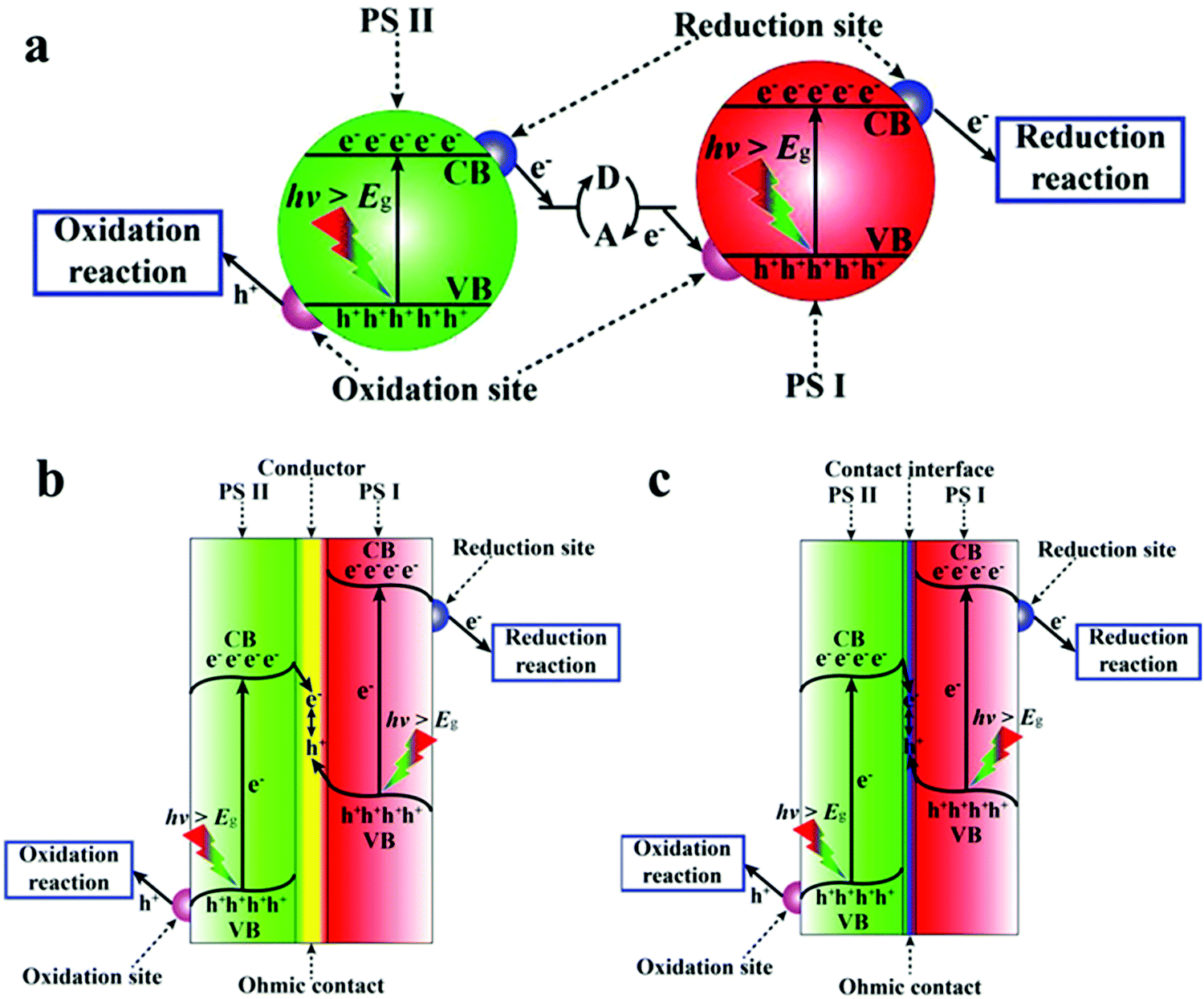
4.3.3 Z-scheme-type heterojunction hybrids.
Z-scheme type heterojunctions are named after the Z-like shape of the charge process.269 This type of heterojunction contains three parts, an electron mediator and two semiconductors labeled PS I and PS II. Generally, the semiconductor from which the photo-generated holes transfer is called PS I and the other is PS II. Z-scheme heterojunctions are currently divided into three types according to the electron mediator, including an acceptor/donor system (PS-A/D-PS system), an external conductor (PS–C–PS system) and a touch interface between (PS–PS system). Usually, in the latter two systems, the oxidation reaction occurs only in PS II and the reduction reaction only in PS I, whereas in the PS-A/D-PS system, the redox reaction can occur in both.269 A schematic diagram of the three types of Z-scheme heterojunction hybrids is shown in Fig. 20.
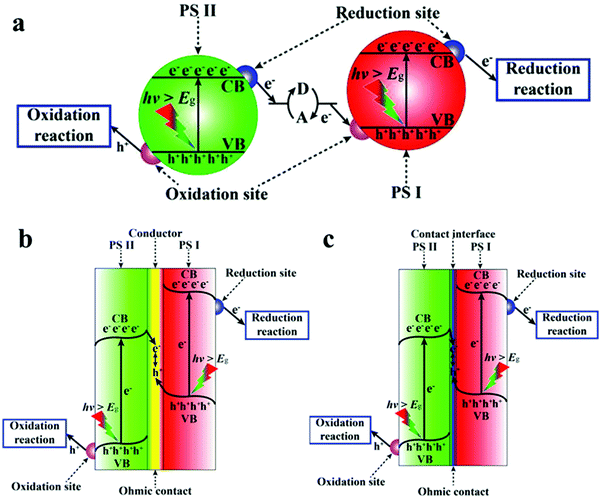 |
| | Fig. 20 Schematic diagram showing three types of Z-scheme heterojunctions: (a) PS-A/D-PS system; (b) PS–C–PS system, and (c) PS–PS system. Reprinted from ref. 269. | |
Xia et al.270 reported a novel g-C3N4/m-Bi2O4 PS–PS heterojunction system for enhancing the photocatalytic disinfection with a visible light source towards E. coli and investigated the different inactivation efficiencies caused by different ratios of g-C3N4 to m-Bi2O4, demonstrating that the optimal one was g-C3N4/m-Bi2O4 (1:0.5). The PL spectrum and photocurrent study further proved that the recombination process is mostly inhibited in g-C3N4/m-Bi2O4 (1:0.5), which directly improve its photocatalysis efficiency. Also, it is believed that the excess m-Bi2O4 can function as a recombination center of photo-generated electron–hole pairs to suppress the photo-induced activity when the m-Bi2O4 content is too high, corresponding to the higher efficiency of g-C3N4/m-Bi2O4 (1:1) compared to g-C3N4/m-Bi2O4 (1:0.5). Besides, the ROS production was also systematically investigated. Zeng et al.271 successfully synthesized a highly dispersed TiO2NC/WO3NR/rGO composite for enhanced bacterial inactivation, which can be classified as a PS–C–PS system. Due to its excellent electron conductivity and mobility, the original p–n heterojunction TiO2/WO3 photocatalysis system would be converted into a Z-scheme heterojunction hybrid once rGO is applied. After modification by rGO, the photogenerated electrons would no longer transfer from TiO2 to WO3, whose CB is at +0.4 V and more positive than −0.33 V, the redox potential for O2˙−/O2, in which case O2 could not be reduced to O2˙−. Instead, the electrons generated by TiO2 remained in its CB, the potential of which can thermodynamically produce both O2˙− and H2O2. Wu et al.272 synthesized a novel PS-A/D-PS system-ZnO/CDots/g-C3N4, in which CDots efficiently inhibited the recombination of photogenerated charge carriers, which served as electron mediators. This Z scheme heterojunction possessed photoresponsive antibacterial properties for synergistic effects by photoinduced ROS inactivation and photothermal therapy (CDots serve as photothermal material), resulting in the accelerated healing of infected wounds due to the release of Zn2+ with a promoting effect on cell proliferation (Fig. 21).
 |
| | Fig. 21 Schematic illustration of the band structure and photocatalytic antibacterial behaviors for 3 types of Z scheme heterojunctions. (A) and (B) Band structure and photoinduced behaviors of the CeO2/TiO2 direct PS–PS heterojunction. (C) Time-dependent antibacterial effects of CeO2/TiO2 materials under different conditions. (D) Band structure and photoinduced behaviors of TiO2/rGO/WO3 heterojunction (PS–C–PS system). (E1) Antibacterial dynamic curve and (E2) antibacterial plate experiments of TiO2/rGO/WO3 system for different times. (F) Band structure and photoinduced behaviors of ZnO/CDot/g-C3N4 heterojunction (PS-A/D-PS system). (G1) Photothermal curves with visible light irradiation and (G2) corresponding infrared thermal photos for different materials. (H1) Photos of S. aureus-infected wounds treated/not treated with ZnO/CDots/g-C3N4 for different times and (H2) corresponding size of the wound. (H3) Infections in wound after 2 days of treatment showed by Giemsa staining images. (H4) Inflammatory cell ratio calculated from H&E staining data. (I) Skin tissue infections showed by H&E staining images at 2 and 12 days. (A)–(C) Reprinted from ref. 273. (D) and (E) Reprinted from ref. 271. (F)–(I) Reprinted from ref. 272. | |
The other Z-scheme heterojunction hybrids and their detailed bacterial inactivation data are shown in Table 5.
Table 4 Type II heterojunction semiconductor hybrids and their inactivation process
| Semiconductor 1 |
Semiconductor 2 |
Methods |
Bacteria |
Light condition |
Inactivation efficiency |
Radicals |
Mechanism of inactivation |
Ref. |
|
λ (nm) |
LPD (mW cm−2) |
˙OH |
1O2 |
O2˙− |
H2O2 |
| “✓” represents that the radical was characterized and detected by ESR and the meanings of the other symbols are equivalent to that depicted in Table 1. |
| Bi2WO6 |
TiO2 |
Hydrothermal method |
E. coli
|
≥420 |
NA |
100% within 4 h (for 40% TiO2 doping) |
✓ |
NA |
✓ |
✓ |
cell wall rupture & cellular component leakage |
233
|
| Fe2O3 |
AgBr |
Electrospinning-calcination method |
E. coli K-12
|
480–680 |
100 |
100% within 4 h |
N |
NA |
N |
✓ |
Cell membrane rupture |
274
|
| TiO2 |
Cu2O |
Electrochemical deposition |
E. coli
|
≥400 |
82 |
100% within 20 min |
✓ |
NA |
NA |
NA |
Deformation of cell membrane |
275
|
| Bi2WO6 |
BiVO4 |
Hydrothermal method. |
P. aeruginosa
|
≥420 |
NA |
Almost all (99.99%) within 30 min |
✓ |
NA |
N |
NA |
Cell membrane rupture & cytoplasm leakage |
276
|
| AgBr |
ZnO |
Deposition–precipitation |
E. coli
|
Visible light region |
NA |
87.5% within 315 min |
✓ |
NA |
✓ |
NA |
Membrane degradation by h+, ˙OH and O2˙− |
277
|
| ZnFe2O4 |
ZnO |
Thermal decomposition of precursors |
E. coli
|
Visible light region |
NA |
100% for ZnFe2O4/ZnO with ratio 1:3 1:2 and 1:1 |
✓ |
NA |
✓ |
NA |
NA |
278
|
| TiO2 |
CNT |
Sol–gel method |
E. coli ATCC25922
|
≥400 |
110 |
48% within 1 h; (BC: 21%) |
✓ |
NA |
✓ |
NA |
NA |
279
|
| ZnO |
CNT |
Sol–gel method |
E. coli ATCC25922
|
NA |
110 |
100% within 10 min |
✓ |
NA |
✓ |
NA |
NA |
280
|
| TiO2 |
CeO2 |
Microemulsion method |
E. coli
|
350 |
1.5 kJ m−2 |
k = 98.4; k = 9.9 |
NA |
NA |
NA |
NA |
NA |
281
|
| 500 |
| TiO2 nanobelts |
Ag2O |
Deposition-precipitation |
E. coli ATCC15597
|
Visible light region |
NA |
Kill 4.7log of viable E. coli within 3 h |
N |
NA |
✓ |
✓ |
Cell membrane rupture |
282
|
| g-C3N4 |
α-Sulfur |
Modified heat-etching method |
E. coli K-12
|
UV (λ < 400 nm) |
193 |
Kill 6log of viable E. coli within 4 h |
✓ |
✓ |
NA |
✓ |
Cell membrane degradation |
283
|
Table 5 Z-scheme heterojunction hybrids and their inactivation
| PS I |
PS II |
Electron mediator |
Preparation method |
Model bacteria |
Initial condition 1: (CFU mL−1) 2: (mg) |
Light condition |
Inactivation efficiency |
Radicals |
Ref. |
|
λ (nm) |
LPD (mW cm−2) |
˙OH |
1O2 |
O2˙− |
H2O2 |
| p.s.: “—” represents that it is a PS–PS system, in which case the interface between two semiconductors serves as the electron mediator; “GI” stands for “greatly improved” compared to pure PS I or/and PS II, while “SI” means “slightly improved”; “#” represents a greatly enhanced production vs. pure PS I but reduced production in comparison to pure PS II as the blank control; and “✓” refers to be detected but not considering the PS II contribution. |
| g-C3N4 |
m-BiO4 |
— |
Hydrothermal |
E. coli K-12
|
1: 1× 106 |
≥400 |
193 |
100% within 1.5 h (for g-C3N4: Bi2O4 = 1:0.5) |
GI |
NA |
# |
GI |
270
|
| 2: 50 |
| AgI |
BiVO4 |
— |
Deposition-precipitation |
E. coli ATCC 25922
|
1: 7× 107 |
≥420 |
NA |
100% within 50 min (for 9.09% AgI contained) |
NA |
NA |
N |
N |
284
|
| 2: 20 |
| TiO2 |
WO3 |
rGO |
Hydrolysis-hydrothermal |
E. coli ATCC 11775
|
1: 2 × 103 |
Solar light |
NA |
97.3% within 80 min |
GI |
NA |
SI |
GI |
271
|
| 2: 30 |
| g-C3N4 |
Mg1.2Ti1.8O5 |
— |
Sol–gel and ionothermal method |
E. coli
|
1: 5 × 107 |
Solar light |
300 W |
100% over 100 min |
GI |
NA |
GI |
NA |
285
|
| 2: 0.01 mol |
| AgBr |
Ag3VO4 |
AgNPs |
Ionic liquid-assisted precipitation |
E. coli
|
1: 1 × 107 |
≥420 |
NA |
Almost all (99.99%) within 30 min |
✓ |
NA |
✓ |
NA |
286
|
| 2: NA |
| g-C3N4 |
ZnO |
CDots |
Thermal polymerization and one-pot method |
S. aureus and E. coli |
1: 1 × 107 |
300–800 nm |
NA |
99.97% and 99.99% against S. aureus and E. coli, respectively in visible light for 15 min |
✓ |
✓ |
NA |
NA |
272
|
| 2: 200 μg mL−1 |
| BiO2−x |
BiOBr |
— |
High temperature hydrothermal method |
S. aureus and E. coli |
1: 1 × 107 |
Visible light |
NA |
100% against S. aureus and E. coli in 20 min |
✓ |
NA |
✓ |
NA |
287
|
| 2: 20 |
| CeO2 |
AgBr |
— |
Simple sedimentation-precipitation method |
E. coli ATCC 25922
|
1: 4.7 × 107 |
Visible light, ≥400 |
300 W |
99.7% against E. coli in 180 min |
✓ |
NA |
✓ |
NA |
288
|
| 2: 0.4 mg mL−1 |
| g-C3N4 |
BiOCl |
— |
Hydrothermal method |
E. coli (BL21)
|
1: 1 × 106 |
Visible light |
300 W |
96.5% against E. coli in 40 min |
✓ |
NA |
✓ |
NA |
289
|
| 2: 0.4 mg mL−1 |
4.3.4 S-scheme heterojunction hybrids.
Type II heterojunctions separate photogenerated charges via the electric potential differences between their components because this type of heterostructure reduces the redox capacity of photogenerated electrons and holes.290 For p–n heterostructures, this also causes the redox capacity for the reduction and oxidation processes occurring on semiconductors to have lower reduction and oxidation potentials, respectively. The direct Z-scheme heterostructure can overcome the drawbacks of reduced photo-generated charge redox ability during the transfer process because electrons with a low reducing capacity tend to be quenched by holes with low oxidizability, while the longer transmission distance of the Z scheme due to the charge loss during the migration restricts its further application.291 To fill these gaps, Yu et al.292 proposed a novel of step-scheme heterojunction called S-scheme heterojunctions. This new type of heterojunction system mainly consists of two n-type semiconductor photocatalysts, which are used as an oxidation photocatalyst (OP) and reduction photocatalyst (RP), respectively.293,294 The internal electric field formed at the interface of the two photocatalysts provides the driving force to promote charge transfer, which is beneficial to reduce the transport distance of photogenerated carriers with the cooperative interface between the two components.229,295 As illustrated in Fig. 22(A), the photogenerated electrons from the lower CB of the OP will combine with the photogenerated holes on the VB of the RP, while strong photogenerated electrons and holes are retained in the CB of the RP and the VB of the OP, respectively, which causes a strong redox potential for the subsequent photochemical reaction.296,297
 |
| | Fig. 22 Photogenerated electron transfer and photocatalytic inactivation of S-scheme heterojunctions. (A) Photogenerated electron transport mechanism of S-scheme heterostructure (TaON/Bi2MoO6 as an example). (B) Electron transfer and photocatalytic inactivation effects and numerical calculation of CeO2/PCN with different conditions. (A) Reprinted from ref. 297. (B)–(D) Reprinted from ref. 298. | |
Due to the tight interface contact, large surface area and efficient separation of photoinduced electron–hole pairs,299 the S-type heterojunction exhibits favorable catalytic activity and retains powerful photogenerated carriers, making it suitable for photoactivated disinfection. Yu et al.298 reported that 0D/2D S-scheme heterojunction-CeO2 QDs and polymeric carbon nitride (CeO2/PCN) exhibited strong photocatalytic inactivation efficiency. The accumulated h+ at CeO as the oxidation site was involved in the creation of ˙OH, while the gathered e− at PCN was involved in the production of O2˙−. The S-scheme material exhibited high photocatalytic efficiency of 88.1% towards S. aureus with visible light, compared with 32.2% and 10.7% for single CeO and PCN, respectively. However, because of the novel ideas and the difficulty of construction of matched band structures, to date, there are only a few reports on photocatalytic disinfection using S-scheme heterojunctions.
Table 6 Typical 2D hybrid material-based composite photocatalysts and ROS generation from different NPs
| Dimensionality |
2D material-based composite |
Methods |
Bacteria and their concentration |
Wavelength range |
Inactivation efficiency |
ROS |
ROS radicals |
Ref. |
| ˙OH |
1O2 |
O2˙− |
H2O2 |
| “Y” indicates total ROS (˙OH, O2˙−, 1O2, H2O2, h+, and e−) were detected, “N” indicates no significant ROS were detected, “✓” indicates the corresponding type of ROS was detected, and NA means “not available” or not provided in the literature. |
| 0D/2D |
Ag–ZnO nanoparticle/g-C3N4 nanosheet |
One-pot hydrothermal process |
E. coli 106 CFU mL−1 |
Very low intensity UV light (2.2 W cm−2, 20%) |
100% under 3 h |
Y |
N |
N |
✓ |
NA |
300
|
| Bi2MoO6/Ag nanoparticle/g-C3N4 nanosheet |
Facile hydrothermal strategy |
E. coli and S. aureus |
500 W halogen lamp (λ > 400 nm) |
99.6% within 80 min under visible light |
Y |
✓ |
NA |
✓ |
✓ |
301
|
| Vanadium–TiO2 porous discrete/g-C3N4 nanosheet |
Facile calcination and ultrasonic dispersion facilitated hydrothermal techniques |
E. coli (G−) and S. aureus (G+) bacteria |
Visible light (384–440 nm) |
Efficiency over 99.5% for 60 min |
Y |
✓ |
NA |
✓ |
✓ |
302
|
| Ag nanoparticle/g-C3N4 nanosheet |
Hydrothermal as well as photo-assisted reduction |
S. aureus 107 CFU mL−1 |
500 W Xe lamp with 420 nm filter |
Inactivation time: 180 min |
Y |
N |
NA |
✓ |
N |
303
|
| Ag nanoparticle/g-C3N4 nanosheet |
Microemulsion technique |
E. coli 1337-H 8.9 × 109 CFU mL−1 |
UV light with 385 nm and visible light with 515 nm |
Inactivation time: 60 min |
Y |
✓ |
NA |
N |
N |
304
|
| m-Bi2O4 nanoparticle/g-C3N4 nanosheet |
One-pot hydrothermal method |
E. coli K-12 106 CFU mL−1 |
Xenon lamp (300 W) with UV light filter |
Completely inactivate E. coli within 1.5 h |
Y |
✓ |
NA |
✓ |
✓ |
270
|
| ZnO@stellerite nanoparticle/g-C3N4 nanosheet |
Coupled hydro thermal-thermal polymerization process |
S. aureus 107 CFU mL−1 |
Visible light irradiation (λ < 420 nm) |
Nearly 90% disinfection rate for 2 h |
Y |
N |
NA |
✓ |
✓ |
305
|
|
|
| 1D/2D |
PDA nanofibers/g-C3N4@Ag nanosheet |
30 mg of g-C3N4 powder, 40 mg of HAD, 60 mg mL−1 AgNO3 aqueous solution |
E. coli OD600 ≈ 1 |
Visible light |
100% under 3 h |
Y |
N |
✓ |
✓ |
NA |
306
|
| TNA (titanium nanotube array)/g-C3N4 nanosheet |
Electrochemical anodization |
E. coli
|
500 W Xe lamp |
Reached 100% for 60 min |
Y |
N |
NA |
✓ |
NA |
307
|
| Au nanorods (NR)/TiO2 nanobelts (NBs) |
TiO2 NBs were synthesized via a hydrothermal method, and then immersed in HAuCl4/NaOH aqueous solution by a photoreduction method to obtain Au NR/TiO2 NB |
E. coli, ATCC 25922 |
Visible source: a 300 W Xe arc lamp with a cutoff filter (λ < 400 nm) NIR light source: 250 W infrared lamp with a cutoff filter (λ < 760 nm) |
Reached 65% for 120 minutes with visible light; 49% for 120 min with NIR light |
Y |
✓ |
NA |
✓ |
NA |
308
|
| ZnO nanorod (NR)/RGO nanosheet |
GO sheet was synthesized from graphite flake via a modified Hummers’ method, then RGO–ZnO NR was synthesized via typical hydrothermal method |
E. coli (MTCC-4296) and S. aureus (MTCC-3196) |
Natural sunlight |
Almost 100% towards E. coli and S. aureus for 60 min |
Y |
✓ |
NA |
NA |
NA |
309
|
| TiO2 nanotube arrays (TiNT)/g-C3N4 layer |
One-step chemical vapor deposition (CVD) technique |
E. coli
|
Visible light at 420 nm |
∼84% for 3 h |
Y |
✓ |
NA |
NA |
✓ |
310
|
| ZnO nanoflowers/V2O5 nanowires (NWs) |
V2O5 NWs were synthesized by a hydrothermal method, and ZnO/V2O5 NWS were prepared via one-pot method |
S. aureus (ATCC25923) 106 CFU mL−1 |
Stimulated sunlight (300–2500 nm) with xenon lamp |
99.8% for 12 h |
Y |
NA |
NA |
NA |
NA |
311
|
|
|
| 2D/2D |
g-C3N4 nanosheet/red P hybrid nanosheets |
Ultrasonic-assisted wet chemical method |
E. coli 107 CFU mL−1 |
300 W Xe lamp |
Inactivation time: 120 min |
Y |
N |
NA |
✓ |
✓ |
312
|
| Mg1.2Ti1.8O5 (mT) nanoflakes/g-C3N4 (CN) nanosheets |
Sol–gel method and ionothermal method, respectively |
E. coli 5 × 107 CFU mL−1 |
300 W Xe lamp with an Am1.5 filter |
Total E. coli was eliminated by mT/CN within 100 min |
Y |
✓ |
NA |
✓ |
NA |
285
|
| RGO nanosheets/g-C3N4 (CN) sheets |
Modified heat-etching method |
E. coli K-12 2 × 106 CFU mL−1 |
300 W Xe lamp with UV light filter |
4 h of VL irradiation |
Y |
✓ |
NA |
✓ |
✓ |
283
|
| WO3–metal nanoporous sheet/g-C3N4 nanosheet (metal = Cu, Ag, Au) |
g-C3N4 nanosheets were deposited on WO3–metal nanoporous sheet by electrochemical method |
NA |
White light source |
Inactivation time: 120 min |
Y |
✓ |
NA |
✓ |
✓ |
313
|
| Bi2MoO6(BM) nanoplates/g-C3N4 sheets (CNN) |
In situ solvothermal method |
E. coli 2.5 × 107 CFU mL−1 |
300 W Xe lamp equipped with a UV cutoff filter (λ < 420 nm) |
99.99% within 4 h for 10%, 30%, 40%-BM/CNN; 99.99% within 2.5 h for 10%, 30%, 40%-BM/CNN |
Y |
✓ |
NA |
✓ |
NA |
314
|
| PEG-MoS2/rGO-streptomycin sulfate (SS) nanoflakes |
Traditional hydrothermal method was used to fabricate tPEG–MoS2/rGO nanoflowers, the as-prepared nanoflowers of PEG–MoS2/rGO were exfoliated into nanoflakes |
E. coli and S. aureus 108 CFU mL−1 |
NIR, 808 nm, 2.0 W cm−2 |
Beyond 90% within 4 h for PEG–MoS2/rGO–SS towards E. coli and S. aureus |
Y |
✓ |
NA |
✓ |
✓ |
315
|
| BiOI/BiOBr nanoflakes/g-C3N4 nanosheets |
g-C3N4 was synthesized by the thermal polycondensation of urea, the pure BiOBr, BiOI and g-C3N4/BiOI/BiOBr composite were prepared by chemical precipitation |
E. coli ATCC 15597 109 CFU mL−1 |
300 W xenon lamp with a UV cutoff filter (λ < 400 nm) |
Almost all E. coli cells were killed for 3 h |
Y |
✓ |
NA |
✓ |
NA |
316
|
| TiO2–Bi2WO6 binanosheet |
Two-step hydrothermal method |
E. coli 109 CFU mL−1 |
300 W xenon lamp with a UV cutoff (λ < 420 nm) |
The count of bacteria remained 10° with 40% TBWO (100 μg mL−1) for 4 h |
Y |
✓ |
NA |
✓ |
NA |
317
|
4.3.5 Dual heterojunction hybrids.
The definition of dual heterojunctions remains ambiguous, where some claim that a heterojunction with two semiconductors components can be called a (direct) dual heterojunction, such as the direct PS–PS system318 and InGaP/GaAs dual heterojunction.319 However, the majority consider that only ternary heterojunction photocatalysts can be classed as dual heterojunctions.320,321 The lifetime of surface photogenerated carriers in ternary systems can be effectively improved by electron transfer between interfaces.322 The most common type of dual heterojunction belongs Z-scheme dual heterojunctions.320,323 In the Z scheme ternary system (PS–A/D–PS), photoexcited charge transfer occurs through solid mediators located at the interface of PS–PS, which promotes the electron transfer and energy conversion. As shown in Fig. 23(A), Niu et al.324 reported a novel g-C3N4/Ti3C2 MXene/BP ternary system with a Z-scheme constructed via CN, Ti3C2 MXene and BP and another P-bridging effect between the N atom (CN with nitrogen deficiency) and P atom (BP nanosheet). The electron transfer from CN to the BP nanosheets and the shifter transfer of electrons can be attributed to the introduction of highly conductive MXene solid mediators. It was confirmed that O2˙− and 1O2 were the two major ROS species through scavenger and ESR experiments, which were created by the reaction between the photogenerated e− and dissolved O2 in the CB of the BP nanosheets, and the interaction between the accumulated h+ and dissolved O2 in the VB of CN, respectively. Zhou et al.325 synthesized a dual Z-scheme tandem heterojunction (WS2 QDs/MoS2@WO3−x) (Fig. 23(B)) because of the abundant edge active sites created by 2D MoS2 and better special separation and transfer of photo-produced charges due to the O dopants and tandem heterojunction formation, which exhibited favorable photocatalytic activity. Besides, a heterojunction is not necessary for two different components, it can also be formed by different facets of one semiconductor. The exposed [101] and [001] facets of anatase TiO2 are good catalytic sites for n–p heterojunction formation.326 There have been studies on n–p–n dual heterojunctions constructed by the [101] and [001] facets of anatase TiO2 and metal sulfide,327,328 in which the photoinduced electrons from TiO2 transfer to the [101] surface of TiO2 rather than MoS2, while the photoinduced holes from TiO2 migrate to MoS2, resulting in a higher separation efficiency (Fig. 23(C)).
 |
| | Fig. 23 Band structure and electron transfer of dual heterojunctions. (A) (a) and (b) Detection of photogenerated ROS species in g-C3N4/Ti3C2 MXene/BPs. (c) Band structure and redox reactions with visible light. (B) Band structure and ROS generation in WS2 QD/MoS2@WO3−x core–shell dual Z-scheme tandem heterojunctions. (C) Creation of n–p–n heterojunctions with the (101) and (001) facets of anatase TiO2 and SnS2. (A) Reprinted from ref. 324. (B) Reprinted from ref. 325. (C) Reprinted from ref. 328. | |
4.3.6 2D heterojunction hybrids.
4.3.6.1 The interface between different dimension hybrids.
As shown in (Fig. 30), the interfaces between different dimension materials are diverse. 0D materials refer to point contact, 1D materials refer to line contact, and their 2D counterparts refer to surface contact. As mentioned above, compared to their 0D and 1D counterparts, 2D layered materials show distinguished photoelectric and photocatalytic properties mainly due to their larger contact surface, and thus more superficial active sites and efficient interfacial charge transfer. Due to their atomic thickness (less than 5 nm), 2D nanomaterials show flexible grid structures. They can be rounded into 0D materials as nanoparticles, rolled into 1D materials as nanotubes and nanowires or stacked into 3D architectures (Fig. 24(b)).329
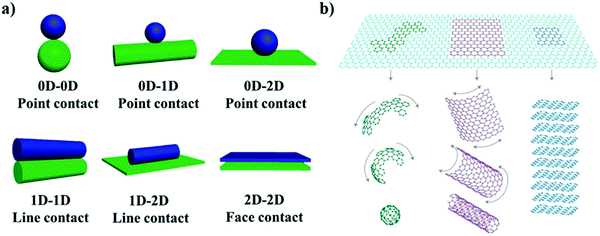 |
| | Fig. 24 (a) Scheme of the specific contact interface of 0D/0D, 0D/1D, 0D/2D, 1D/1D, 1D/2D and 2D/2D nanomaterials. Reprinted from ref. 330. (b) 2D graphene nanosheets can be circled to balls (0D material), rolled into tubes (1D material) or folded into 3D graphite. Reprinted from ref. 329. | |
In the last five years, research on 1D/2D heterostructure hybrids has become a hot topic and many 1D/2D materials have appeared, such as 1D CdS nanowires/2D BiVO4 nanosheets,332 1D Nb2O5 nanorods/2D g-C3N4 nanosheets,336 1D TiO2 nanofibers/2D MoS2 nanosheets,337 1D WO3−x nanowires/g-C3N4 nanosheets,338 1DNiCo2S4 nanowires and 2D layered-δ MnO2.339
(3) 2D/2D heterojunction hybrids.
As mentioned above, 2D semiconductors are promising photocatalysts due to their great properties.340 Thus, it can be predicted that 2D/2D heterojunction hybrids may have better photocatalysis efficiency. Cheng et al.341 developed 2D/2D heterojunction photocatalysts by coupling g-C3N4 (CN) nanosheets with oxygen vacancies confined in Bi20TiO32 (BTO) mesoporous nanosheets with exposed (001) facets via a facile hydrothermal process with the assistance of post-treatment.
Besides, the majority of the developed photocatalytic materials are at least one metal-based or metal-containing composites. However, these photocatalysts would release toxic metal ions, which pose a great threat to human health. Therefore, 2D/2D heterojunction hybrids for bacterial inactivation are in great demand. Zhang et al.,342 for the first time, fabricated a metal-free BP/g-C3N4 2D/2D heterojunction for visible light-driven water disinfection. In the in vitro antibacterial assay, BP/g-C3N4 exhibited around a seven-times inactivation efficiency comparing to bare g-C3N4 given that the photogenerated e− could transfer from g-C3N4 to BP, which greatly enhanced the charge carrier separation.
4.4 Dye-sensitized hybrid semiconductors
4.4.1 The basic principle of dye-sensitized hybrid semiconductors.
Dye-sensitization, which was first proposed by Grätzel et al.343 as a satisfying strategy to improve the activity of dye-sensitized solar cells (DSSCs), has been widely used to improve the photo-quantum efficiency and broaden the photo-absorption response of photocatalysts.344 The use of dye-sensitizers has matured considerably in photocatalytic applications such as photodynamic antimicrobial chemotherapy (PACT).345,346 PACT relies on the generation of ROS in the presence of photosensitizer (PS) molecules under illumination. The PS tends to be NIR absorbing dyes, such as phthalocyanines,347 porphyrins,348,349 and porphins,350 which are beneficial to reduce the interference of self-absorption and spontaneous fluorescence of tissue, and enhance the tissue penetration depth under illumination.346 Nevertheless, PSs suffer from weak hydrophilicity, strong aggregation, high doses, and low biocompatibility.351,352 It seems a feasible scheme to address these issues by using materials as suitable conjugates for PSs as delivery systems against microorganisms. For example, combination of dye and semiconductor (dye-sensitized semiconductors) has become an effective modification strategy for both dyes and semiconductors as photocatalysts, which effectively absorb visible or even NIR light, where efficient semiconductor stimulation can be an alternative approach of great use.353 As illustrated in Fig. 25, in a typical dye-sensitized photocatalyst, the CB value of the semiconductor is lower than the dye LUMO value, while higher than its HOMO,343,354 in which the dye serves as an antenna, enabling visible light to be received and resulting in the follow-up behaviors of photoinduced reactions via the injection of electrons into the CB of the semiconductor.355 This procedure is principally follows two steps: (1) the photogenerated electrons of the illuminated dye molecules transfer from the HOMO to the LUMO and (2) the transfer of electrons from the LUMO of the dye to the CB of the semiconductor. As reported by Zhang,356 besides the dye and semiconductor, the co-catalyst and sacrificial electron donor used for dye recycle also play dominant roles in photoactivity. Currently, noble metal-based (Pt, Ru, Rh, and Pd)357–361 and noble-metal-free362–365 co-catalysts have been comprehensively investigated to extract photogenerated carriers. Also, research has revealed that the electron donor species366–368 can influence the oxidation half reaction,369,370 which prominently impacts the overall photoactivity of dye-sensitized semiconductors.
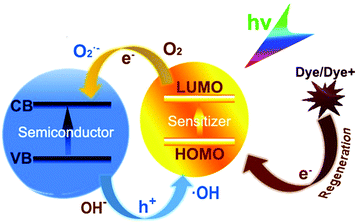 |
| | Fig. 25 Mechanism of the photocatalytic reaction on a dye-sensitized hybrid semiconductor. | |
4.4.2 Antibacterial activity of dye-sensitized hybrid semiconductors.
Dyes as photosensitizers can trigger photodynamic antibacterial behaviors (Fig. 26(A and B)), making them potential antibacterial materials acting on deeper issues in response to NIR light. In the dye-sensitized semiconductor system, dyes are loaded into semiconductors (as a drug delivery platform) via physical encapsulation, adsorption or chemical coupling, which further improves the drug delivery efficiency and avoids the inherent drawbacks of dyes (such as poor water solubility). TiO2 with a wide band gap has been functionalized by dye sensitization in recent years. For example, Insin et al.371 prepared FeOx–SiO2–TiO2–metalloporphyrin compounds with a wide light response (Fig. 26(C)), good dispersion in solution and high photoactivation effects. Giridhar et al.372 evaluated p-methyl red, an azobenzene compound, for the photocatalytic enhancement of TiO2, which could serve as photo switches when attached to TiO2, rendering the system photo responsive to external illumination. Besides, research on manipulating the surface of titanium natural dyes with biological safety and economic advantages has been well investigated to sensitize TiO2.373 Xing's team proved that three natural dyes (including alizarin red, fuchsin acid, and alizarin) as modified sensitizers of anatase could efficiently extend its optical absorption edge, and therefore increase its antimicrobial efficiency.374 For other semiconductors, anthocyanin (a type of natural molecule)-sensitized ZnO proved to increase the bacterial loss by ∼10% compared to naked ZnO nanoparticles under solar simulator light.375 It was proven that the 2D material g-C3N4 nanosheets sensitized with Cu diphenylamine complexes were accessible to eliminate pathogens, in which the layered structure with delocalized π-electrons of Cu–biphenylamine complexes serves well in the charge transfer process of photocatalysis.376 Moreover, some dyes possess favorable optothermal effects, and thus dye-sensitized systems can be used for bacterial inactivation with synergetic photothermal and photocatalytic therapy.377 Photogenerated ROS remains a principal antibacterial mechanism of dye-sensitized semiconductors, causing cell membrane rupture, and therefore intracellular DNA and protein leakage.178,179 The photogenerated ROS processes can be depicted in eqn (21)–(28) below:378| | | Dye + semiconductor → dye–semiconductor | (21) |
| | | trans dye–semiconductor → cis dye*–semiconductor | (22) |
| | | cis dye*–semiconductor → semiconductor + decomposed dye | (23) |
| | | Dye + semiconductor + O2 → semiconductor + decomposed dye + O2− | (24) |
| | | HO2˙ + H+ + e(CB–TiO2) → H2O2 | (26) |
| | | 2O2˙− + 2H+ → 2OH˙ + O2 | (27) |
| | | OH˙ + cell → damaged cell | (28) |
 |
| | Fig. 26 Photoinduced bacterial disinfection for dye-sensitized semiconductors. (A) Mechanism of photodynamic antibacterial therapy. Reprinted from ref. 379. (B) Photodynamic antibacterial effect of Ce6 modified with hollow SiO2/chitosan. Reprinted from ref. 380. (C) Synthesis of FeOx–SiO2–TiO2–metalloporphyrins and (D) their TEM images. (C) and (D) Reprinted from ref. 371. (E) Schematic illustration of the synthesis of BP@PEG/Ce6. (F) Photothermal evaluation and cytocompatibility (hemolysis test) of the as-prepared NPs in (E). (E) and (F) Reprinted from ref. 377. | |
4.4.3 Common types of dye-sensitizer hybrids and their photocatalytic inactivation application.
Research on manipulating the surface of photocatalysts with highly active, safe and inexpensive dyes has become a critical hotspot in photocatalytic disinfection recently.344 According to the current research, dye sensitizers principally include natural organic dyes such as chlorophyll,381,382 cytochrome c,383,384 anthocyanin,375,385 and alizarin red,386,387 as well as artificial composite dyes, such as porphyrin compounds,388,389 phthalocyanine compounds390 and polypyridyl complexes.391–393
4.4.3.1 Photocatalytic mechanism of porphyrin-based functional nano-systems for bacterial inactivation.
As the photosynthesis reaction center of green plants, porphyrin with a planar π-conjugated aromatic macrocycle394 benefits the enhancement of electron coupling and transfer of photogenerated carriers.395 Owing to its wide visible light absorption range (400–800 nm),396 porphyrin is frequently applied as a photon-trapping antenna molecule and co-catalyst in photocatalytic models.397 It has been reported that self-assembled molecular aggregates are candidate models for energy transfer processes,398 which resemble the photon-collected congeries in photosynthetic bacteria.399 Porphyrins naturally self-assemble into aggregates of π-conjugated molecules due to their easily modified porphyrin ring,400 and thus there are numerous porphyrin derivatives. For example, Wang's team401 synthesized supra-amphiphiles by combining a heteroditopic porphyrin amphiphile with zinc, which could be further used in controllable self-assembly applications. According to the report of Wang et al.,402 porphyrin J-aggregates could be self-assembled by two porphyrins with opposite ionic charges. In the case of porphyrin-sensitized semiconductors, Ghosh et al.403 well investigated the photoelectron-transfer mechanism in porphyrin aggregates, and observed an enhancement in charge carrier separation at the porphyrin/TiO2 interface (Fig. 27). According to Sorgues,404 the zinc tetracationic porphyrin [ZnTmePyP](4+) was successfully synthesized through electrostatic complexation of porphyrin and polyoxometalates (POm), and later the occurrence of electron jump from the porphyrin to the POm was observed in the presence of visible light.
 |
| | Fig. 27 (A) Distinction of absorption spectra between monomer- and J-aggregated porphyrin–TiO2 NPS with different delay times. (B) Mechanism of charge-carrier shift in the J-aggregate porphyrin–TiO2 system. Reprinted from ref. 403. | |
Furthermore, some porphyrin derivatives have been applied for photodynamic therapy (PDT). He et al.405 chose the second-generation photosensitizer hematoporphyrin monomethyl ether (HmmE) to sensitize TiO2 nanocomposites owing to its good photodynamic effects, low toxicity in the dark and fast removal rate.406 Also, the HmmE–TiO2 NCs showed strong ROS generation capability and inactivation effect of KB cells (type of cancer cells). Besides, 5,10,5,20-tetraphenyl-21H,23H-porphine nickel (TPPN) was applied to doped titanium dioxide via the sol–gel method. The TiO2/TPPN thin film was employed for the disinfection of the plant pathogen E. carotovora subsp. with a visible spectrum response, which showed over 90% bacteriostasis rate with white light (>400 nm) illumination for 60 min.406 As illustrated in Fig. 28(A), the synthesis of a 2D nanostructure diporphyrin honeycomb membrane consisting of TiO2, protoporphyrin IX, and hemin (TiO2/PPIX/Hem) was reported via dehumidifying with bidentate binding.407 The generation of an anodic photocurrent was observed on the surface of the PS/TiO2/PPIX/FTO electrode through electron transfer processes (Fig. 28(B)), and therefore proved its high photoactivity. Due to its inactivation effect, it showed the highest inhibition of Bacillus subtilis cultured with TiO2/PPIX/Hem under visible light compared with TiO2/PPIX and naked TiO2. Also, the TiO2/PPIX/Hem group further exhibited a much more rapid bacterial clearance rate in bactericidal kinetics characteristics. Furthermore, Wang et al. used water-soluble porphyrin–tetra sulphonatophenyl porphyrin (TSPP)–TiO2 nanowhiskers for the early diagnosis of inflammation together with bio-imaging and treatments of microbial infections.408–413
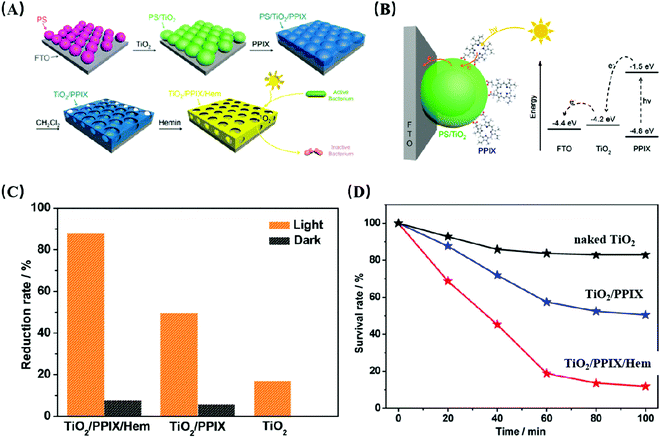 |
| | Fig. 28 (A) Schematic illustration of the synthesis of the TiO2/PPIX/Hem composite membrane. (B) Electron jump mechanism of the PS/TiO2/PPIX/FTO rheophore system under visible light. (C) Antibacterial activity toward Bacillus subtilis with or without illumination for 100 min. (D) Bactericidal kinetics of different materials in the presence of light for 100 min. Reprinted from ref. 407. | |
 |
| | Fig. 29 (A) Structures of Ru(II) and Ir(III) polypyridyl complexes used as photosensitizers in photo-catalyst systems. (B) Photoinduced Ru*(bpy)32+ used as electros mediators, receiving or donating electrons. Reprinted from ref. 429. | |
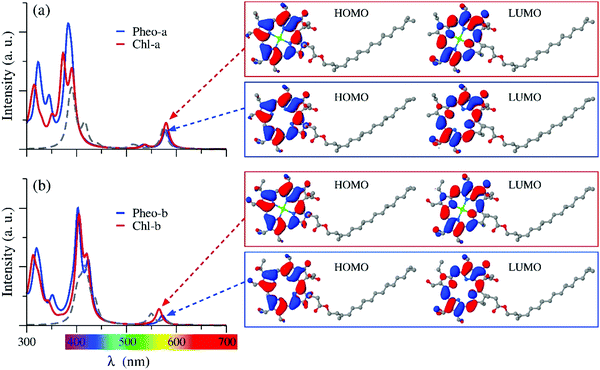 |
| | Fig. 30 Absorption spectra of Chl-a and Pheo-a systems (a) and Chl-b a Pheo-b systems (b). Reprinted from ref. 381. | |
Some polypyridine complexes are promising antibacterial candidates. Polypyridyl ligands have been identified to be lipophilic in nature, which endows polypyridyl good biocompatibility owing to the easy access and interaction between the materials and the cell lipopolysaccharide layer (LPS).430 It was illustrated that the intercalation in biochemistry is a process that can be extensively used to explain the mechanisms of the inactivation of cells for some drugs,391 and the heterocyclic polypyridyl group of molecules has been proven to be typical intercalators.431,432 According to Zhang et al., the electron-acceptor group and large hydrophobic group in the intercalative ligand are beneficial for the combination of ruthenium compounds with DNA.433 The biological application of Ru(II) polypyridyl compounds is increasing due to the awareness of the good bioactive properties of ruthenium for the inactivation of cancer, microorganisms, and even viruses.434,435 For example, a titanium alloy coated with a ruthenium complex exhibited hopeful antibacterial and antifungal activity and low cytotoxicity to mammalian cells, representing that ruthenium complexes are broad-spectrum antibacterial materials.434 Matshwele's team391 synthesized Ru(II)/(III) complexes as antibacterial agents, which exhibited high inactivation capability towards non-drug-resistant bacterial species such as S. aureus and K. pneumoniae, and even their drug-resistant strains such as MRSA and MDR K. pneumoniae. The bacterial DNA was completely denatured, as shown by the characterization of DNA gel electrophoresis, indicating the strong affinity of Ru(II)/(III) complexes towards DNA, and further inducing DNA cleavage by intercalation. According to Nambagari,392 three new cobalt(III) polypyridyl complexes were successfully proven to possess antibacterial properties. Our team proved that a ferrocene-substituted carborane ruthenium(II)–arene complex (FcRuSB) can be utilized as an inducer to effectively reverse the drug resistance of pathogens.436 Yi et al.393 also confirmed that three iridium(III) polypyridyl compounds are promising antibacterial candidates.
4.4.3.3 Natural organic dye hybrids for bacterial inactivation.
(1) Chlorophyll.
Naturally occurring pigments (such as chlorophyll, cytochrome, and vitamin B12) play crucial roles in various life processes and are referred as the “pigments of life”.437 Chlorophyll, belonging to the class of porphyrin macrocycle derivatives, has been extensively employed as a photosensitizer in light-harvesting systems, which is because porphyrin-based compounds with 18π-electron conjugated macrocycles possess distinguished catalytic, photochemical and photophysical properties.438,439 These materials are expected to strongly absorb in the visible and even NIR region as efficient photocatalysts.440 Bevilacqua et al.381 analyzed the absorption spectra of chlorophyll enabled in the near UV and visible light range. Moreover, pheophytin (Pheo-) a and b (with no Mg(II) ion) displayed a slight red-shift compared with the integrated chlorophyll.
Chlorin derivatives, including chlorophylls as Mg-containing chlorins, are presently investigated for antimicrobial PDT to inactivate some important microbial pathogens.441 For example, Ferro's team442 proved the strong inactivation activity of liposome-delivered chlorophyll a towards S. aureus (MRSA), which was more pronounced with selective targeting using liposome as carriers. Kustov et al.443 successfully synthesized water-soluble chlorophyll pigments as novel potential photosensitizers to deal with bacteria and fungi. They indicated that chlorophyll with a positive charge (such as N(CH3)3+ groups) could treat wound infections and bacterial biofilms. Similarly, another positively charged chlorophyll derivative, lysine–chlorin E6(pL–ce6) conjugates, are effective against pathogens in the oral cavity, while oral epithelial cells can be well preserved.441 Particularly, some naturally antibacterial materials such as antimicrobial peptides all have a pronounced polycationic charge, which plays an important role in binding with negatively charged bacteria due to electrostatic attraction.444,445
(2) Cytochrome c.
Cytochrome c (Cyt c) is a hemeprotein intensely combined with the inner mitochondria membrane by electrostatic interactions. It serves as a vital electron transporter in the respiratory chain.446,447 Wang et al.448 firstly reported that Cyt c could be used as a carrier of porphyrin-based photosensitizers to improve their photoinactivation efficiency against the Gram-negative bacteria E. coli. It was shown that the positively charged porphyrin Cyt c was on the surfaces of bacteria via TEM measurement and fluorescence confocal microscopy. Also, the ζ potential analysis clearly demonstrated that porphyrin Cyt c and E. coli were efficiently integrated via electrostatic interactions.
(3) Rubiadin.
Rubiadin, also named alizarin, is a typical medium dye extracted from the root of Rubiaceae.449 Rubiadin has exhibited strong inhibition towards a wide range of bacteria, especially to Staphylococcus aureus in recent studies.450,451 The photogenic superoxide anion (O2˙−) and singlet molecular oxygen (1O2) have been shown to be principle mechanisms of this dye for bacterial inactivation, revealing its potential as a drug for photodynamic antimicrobial chemotherapy.452 It was reported that Alizarin Red S sensitization of TiO2 broadened its light absorption range and improved the photo-quantum efficiency, and therefore enhanced the antimicrobial activity of TiO2.373 Rubiadin also exhibited low toxicity in mice via acute toxicity trials,451 which reflects the good biocompatibility of this dye for further medical applications. For example, rubiadin derivatives have been applied as a biological dressing for improving the protection and preventing bacterial infection of skin.453
(4) Anthocyanin.
Anthocyanins are naturally occurring polyphenols454 that contribute to the high antioxidant activity in some plants.455 They are well known due to their low cost, hydrophilia owing to their hydroxyl groups (˙OH), and biosafety as sensitized dyes.385,456,457 Hilal et al.375 observed that anthocyanin-sensitized ZnO nanoparticles increased the loss of E. coli by 10% compared to naked ZnO under the same light conditions. It was shown that the photoinactivation process resulted in the mineralization of the bacterial membrane and intracellular organic components, which leaked after cell rupture. Moreover, the compounds from the photodegradation of anthocyanin were confirmed to be mineral and innoxious, revealing its potential as a sensitized dye. Tu's team458 invented a smart calcium alginate fiber modified by anthocyanin dyed–hydroxypropyl trimethyl ammonium chloride chitosan (HACC). It was shown that the modification of anthocyanin dyed–HACC had strengthened the dyeing and antibacterial activity of calcium alginate fiber, which is a potential candidate for wound healing.
4.4.4 Application limitation and prospect of dye-sensitized semiconductors.
In the photoinduced process, the dye is always inevitably degraded by oxidation, resulting in a limited number of cycles of the materials. Although porphyrin and polypyridine-based semiconductors have been widely used due to their good photosensitization activity, consideration should be given to the biotoxicity of metal-based dyes, which may degrade to harmful substances in solution under photochemical conditions, and thus each dye should be considered for its form of existence after degradation. In addition, synthetic dyes should be observed for incidental effects and adverse drug interactions.459 Thus, it seems that natural pigments are more attractive. The inherent drawbacks of dyes, for example, poor photon/thermal stability, poor water solubility, and low affinity for bacteria hinder their further applications.460,461 Some claim more specific requirements, including simplified and cheap synthetic procedures, acceptable stability, preferential binding or absorption by target cells or tissues, and low side effects. Thus, the strategy of functional encapsulation or modification of dye-sensitized semiconductors as a nanocarrier and delivery platform has been employed to improve their physicochemical properties, material utilization, and selectivity of target bacteria.
5. Conclusion and prospects
In this review, we comprehensively discussed the research status of the modification strategies of photocatalytic semiconductors for bacterial inactivation, including band structure analysis, synthetic techniques, mechanism of photogenerated charge separation and reactive sites, ROS-induced antibacterial mechanism, and biosafety issues for further medical applications. Although the photo-induced antibacterial strategy has attracted extensive attention, research on the design and application of photoactive hybrid semiconductors for bacterial inactivation remains in its infancy, and thus further developments are evidently required. Some challenges should be considered before further large-scale applications.
5.1 Limitations and challenges
(1) In current research, more semiconductor hybrids depend on visible-light-driven energy with limited light penetration depth, which only suits for bacteria disinfection in the environment or superficial wounds, but not deep tissue infections such as organs inflammations.462
(2) E. coli or S. aureus are used as the model bacteria in most reports. However, different bacteria possess distinct physiological structures and death mechanisms, and thus it is required to explore the distinctions of the interactions between photocatalytic semiconductors and different bacteria. Besides, the existing photocatalytic semiconductors can deal with bacteria due to their broad-spectrum antibacterial properties, but they have no selectivity for bacteria and cannot achieve specific selection and treatment of target bacteria.
(3) The photocatalytic antibacterial activity is affected by various factors such as material morphology, synthesis technique, dosage, irradiation intensity and time, and the reaction environment such as pH and solute species. At present, it is difficult to compare two different modification strategies because an operational evaluation standard has not been developed for photocatalysis. In addition, the actual application environment of photocatalysis is more complicated. For example, it is inclined to hypoxia and low pH in bacterial infections.463 The affinity of photocatalytic materials and bacteria, antibacterial activity and stability of photocatalytic semiconductors in a specific microenvironment remains unclear.
(4) Although photo-induced ROS generation has become a hopeful route to kill antibiotic-resistant bacteria and biofilm rapidly and effectively, the photo-sensitive performance of antibacterial properties occurs only in the presence of light. Furthermore, long-time light exposure may cause phototoxicity and even tissue hypoxia, which largely restrict the clinical application of photoactive semiconductors.
(5) At present, most photocatalytic semiconductors are still in the laboratory stage, and many problems remain for further commercial industrial applications, such as simplified synthesis routes suitable for large-scale production and long-term assessment of product environmental impacts.
5.2 Future prospects
(1) A more comprehensive and in-depth investigation of the photo-induced antibacterial mechanisms of semiconductor hybrids is crucial for their further optimization and enhancement of their photocatalytic performance, which is beneficial for the design and development of photocatalytic antibacterial semiconductors. The mechanism of the interaction between photocatalytic semiconductors and bacteria, including photo-induced ROS antibacterial behaviors and the interface reactions between both need to be further revealed and understood. Advanced in situ characterization technology and theoretical calculations may facilitate the exploration of the relevant mechanisms, which may significantly deepen our understanding on the photocatalytic antibacterial process.
(2) For the further practical use of photocatalytic semiconductors for antibacterial applications, researchers have designed and developed some photocatalytic reactors464 with the aim of the expansion from the laboratory-scale process to commercially/industrially feasible applications and promotion of the recovery and reuse of photocatalysts. These reactors are also beneficial to enhance ROS generation and storage, which may solve the contradiction between high efficiency and long-acting inactivation.
(3) Synergistic antibacterial strategies should be considered in further applications. With the use of two or more inactivation mechanisms, synergistic antibacterial strategies exhibit enhanced antibacterial effects, and meanwhile avoid the cell cytotoxicity and drug resistance caused by the excessive use of a single ingredient. For example, photocatalysis and physical contact effects, in which the sharp edges of semiconductors can stab bacteria.465 Photocatalysis and photothermal effects,466 in which photon energy can be converted into heat to cause photothermal ablation of bacteria. Photocatalysis and the release of metal ions, in which the release behaviors of copper ions is beneficial to kill bacteria.248 Besides, magnetic photocatalytic materials such as Fe3O4 can utilize magnetism to improve the separation and recovery of the material and even achieve magnetic targeting to bacterial infections.467
(4) Functional surface modifications of photocatalytic semiconductors can further improve their effects. For example, photocatalytic semiconductors such as dye-sensitized semiconductors possess relatively poor hydrophilicity, which can be improved by introducing O-containing functional groups, such as hydroxyl (–OH) and carboxyl (–COOH).468 To overcome the unsatisfactory biocompatibility of metal-based semiconductors, biological coatings such as PDA or chitosan can be modified on their surface to improve their biocompatibility and adhesion to bacteria.180 In addition, physical encapsulation or chemical coupling can be used to construct a semiconductor carrier and delivery platform (such as SiO2 microspheres) to improve the delivery efficiency of antibacterial drugs or achieve a sustained release function.371
(5) With the rise of theranostics, the visualization and specific identification of bacterial infections are of great significance. Semiconductors can be combined with imaging molecules (such as fluorescent molecules/radionuclide molecules) to achieve visual imaging of bacteria;469 meanwhile, specific recognition molecules (such as lectin for food-borne pathogens470) of bacteria can be coupled to improve the targeting and selectivity to the target bacteria and realize real-time dynamic monitoring of antibacterial effects. Thus, the theranostic strategy has become an appealing direction for photocatalytic disinfection.
Abbreviation
| ROS | Reactive oxygen species |
| 2D | Two dimensional |
| 0D | One dimensional |
| 1D | One dimensional |
| NIR | Near infrared ray |
| G(+) | Gram-positive |
| G(−) | Gram-negative |
| BPs | Black phosphorus |
| PL intensity | Photoluminescence intensity |
| HRTEM | High resolution transmission electron microscopy |
| Redox | Oxidation–reduction reactions |
| CFM | Chitosan functionalized magnetic MoS2 |
| EPR study | Electron paramagnetic resonance study |
| PCR assay | Polymerase chain reaction assay |
| BCA assay | Bicinchoninic acid assay |
| Ce6 | Chlorin e6 |
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (91753106), Primary Research & Development Plan of Jiangsu Province (BE2019716), and the National Key Research and Development Program of China (2017YFA0205300).
References
- J. Perez, F. Javier Contreras-Moreno, F. Javier Marcos-Torres, A. Moraleda-Munoz and J. Munoz-Dorado, Comput. Struct. Biotechnol. J., 2020, 18, 2547–2555 CrossRef CAS PubMed.
- L. A. Rantz, Calif. Med., 1954, 81, 1–3 CAS.
- P. B. Mead, Semin. Perinatol., 1977, 1, 101–111 CAS.
- J. S. Pitton, Ergeb. Physiol., Biol. Chem. Exp. Pharmakol., 1972, 65, 15–93 CAS.
- Z. Zhou, B. Li, X. Liu, Z. Li, S. Zhu, Y. Liang, Z. Cui and S. Wu, ACS Appl. Bio Mater., 2021, 4, 3909–3936 CrossRef CAS.
- Y. Liu, X. Zeng, X. Hu, Y. Xia and X. Zhang, Sol. RRL, 2021, 5, 2000594 CrossRef CAS.
- N. A. Daub, F. Aziz, M. Aziz, J. Jaafar, W. N. W. Salleh, N. Yusof and A. F. Ismail, Water, Air, Soil Pollut., 2020, 231, 461 CrossRef CAS.
- X. Kong, X. Liu, Y. Zheng, P. K. Chu, Y. Zhang and S. Wu, Mater. Sci. Eng., R, 2021, 145, 100610 CrossRef.
- Y. Hu, Q. Li, H. Liu, B. Kong and Q. Chen, Food Sci., 2020, 41, 232–238 Search PubMed.
- P. Ganguly, C. Byrne, A. Breen and S. C. Pillai, Appl. Catal., B, 2018, 225, 51–75 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- T. Matsunaga, R. Tomoda, T. Nakajima and H. Wake, FEMS Microbiol. Lett., 1985, 29, 211–214 CrossRef CAS.
- M. D. Hernandez-Alonso, F. Fresno, S. Suarez and J. M. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 RSC.
- M. S. Zhu, C. Y. Zhai, L. Q. Qiu, C. Lu, A. S. Paton, Y. K. Du and M. C. Goh, ACS Sustainable Chem. Eng., 2015, 3, 3123–3129 CrossRef CAS.
- H. M. Sung-Suh, J. R. Choi, H. J. Hah, S. M. Koo and Y. C. Bae, J. Photochem. Photobiol., A, 2004, 163, 37–44 CrossRef CAS.
- A. B. Djurisic, Y. H. Leung and A. M. C. Ng, Mater. Horiz., 2014, 1, 400–410 RSC.
- L. Armelao, D. Barreca, G. Bottaro, A. Gasparotto, C. Maccato, C. Maragno, E. Tondello, U. L. Stangar, M. Bergant and D. Mahne, Nanotechnology, 2007, 18(37), 375709 CrossRef.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- F. I. Alzakia, B. Tang, S. J. Pennycook and S. C. Tan, Mater. Horiz., 2020, 7, 3325–3338 RSC.
- M. Zeng, M. Chen, D. Huang, S. Lei, X. Zhang, L. Wang and Z. Cheng, Mater. Horiz., 2021, 8, 758–802 RSC.
- W. Wang, Y. Yu, T. An, G. Li, H.
Y. Yip, J. C. Yu and P. K. Wong, Environ. Sci. Technol., 2012, 46, 4599–4606 CrossRef CAS PubMed.
- Y. Wang, F. Huang, D. Pan, B. Li, D. Chen, W. Lin, X. Chen, R. Li and Z. Lin, Chem. Commun., 2009, 6783–6785, 10.1039/b912137d.
- T. Ashikaga, M. Wada, H. Kobayashi, M. Mori, Y. Katsumura, H. Fukui, S. Kato, M. Yamaguchi and T. Takamatsu, Mutat. Res., 2000, 466, 1–7 CAS.
- P. C. Maness, S. Smolinski, D. M. Blake, Z. Huang, E. J. Wolfrum and W. A. Jacoby, Appl. Environ. Microbiol., 1999, 65, 4094 CrossRef CAS PubMed.
- J. Li, X. Liu, L. Tan, Y. Liang, Z. Cui, X. Yang, S. Zhu, Z. Li, Y. Zheng, K. W. K. Yeung, X. Wang and S. Wu, Small Methods, 2019, 3, 1900048 CrossRef.
- Y. Y. Zhang, X. Y. Bai, X. M. Wang, K. K. Shiu, Y. L. Zhu and H. Jiang, Anal. Chem., 2014, 86, 9459–9465 CrossRef CAS PubMed.
- G. Chen, J. Wang, C. Wu, C.-Z. Li, H. Jiang and X. Wang, Langmuir, 2012, 28, 12393–12399 CrossRef CAS PubMed.
- H. C. Chang, X. M. Wang, K. K. Shiu, Y. L. Zhu, J. L. Wang, Q. W. Li, B. A. Chen and H. Jiang, Biosens. Bioelectron., 2013, 41, 789–794 CrossRef CAS PubMed.
- L. Wang, Y. Y. Zhang, C. S. Cheng, X. L. Liu, H. Jiang and X. M. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 18441–18449 CrossRef CAS PubMed.
- C. H. Li, X. L. Liu, Y. Y. Zhang, Y. Chen, T. Y. Du, H. Jiang and X. M. Wang, Anal. Chim. Acta, 2016, 933, 66–74 CrossRef CAS PubMed.
- Y. K. Zheng, W. W. Liu, Y. Chen, H. Jiang and X. M. Wang, J. Mater. Chem. B, 2018, 6, 3650–3654 RSC.
- R. M. Mazhabi, L. Q. Ge, H. Jiang and X. M. Wang, Biosens. Bioelectron., 2018, 107, 54–61 CrossRef PubMed.
- V. Ivády, I. A. Abrikosov, E. Janzén and A. Gali, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87(20), 205201 CrossRef.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2009, 130, 074103 CrossRef PubMed.
- S. Cavo, J. A. Berger and P. Romaniello, Phys. Rev. B, 2020, 101(11), 115109 CrossRef CAS.
- C.-F. Fu, X. Wu and J. Yang, Adv. Mater., 2018, 30, 1802106 CrossRef PubMed.
- R. G. Parr, S. R. Gadre and L. J. Bartolotti, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 2522 CrossRef CAS PubMed.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef CAS.
- S. Bai, L. L. Wang, Z. Q. Li and Y. J. Xiong, Adv. Sci., 2017, 4(1), 1600216 CrossRef PubMed.
- A. Lipovsky, L. Levitski, Z. Tzitrinovich, A. Gedanken and R. Lubart, Photochem. Photobiol., 2012, 88, 14–20 CrossRef CAS PubMed.
- G. Munuera, A. Navio and V. Rivesarnau, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 2747–2749 RSC.
- Z. Yuan, B. Tao, Y. He, J. Liu, C. Lin, X. Shen, Y. Yu, C. Mu, P. Liu and K. Cai, Biomaterials, 2019, 217, 119290 CrossRef CAS PubMed.
- V. A. Johnson and K. Larkhorovitz, Phys. Rev., 1951, 82, 763 CAS.
- X. X. Sun, X. Q. Wang, P. Wang, B. W. Sheng, M. Li, J. Su, J. Zhang, F. Liu, X. Rong, F. J. Xu, X. L. Yang, Z. X. Qin, W. K. Ge and B. Shen, Opt. Mater. Express, 2017, 7, 904–912 CrossRef CAS.
- Y. F. Yan, W. J. Yin, Y. L. Wu, T. T. Shi, N. R. Paudel, C. Li, J. Poplawsky, Z. W. Wang, J. Moseley, H. Guthrey, H. Moutinho, S. J. Pennycook and M. M. Al-Jassim, J. Appl. Phys., 2015, 117(11), 112807 CrossRef.
- F. X. Ye, A. Ohmori and C. J. Li, Surf. Coat. Technol., 2004, 184, 233–238 CrossRef CAS.
- T. T. Zheng, J. Y. Xu, Z. J. Zhang and H. B. Zeng, RSC Adv., 2015, 5, 99658–99663 RSC.
- Y. Y. Hu, N. Berdunov, C. A. Di, I. Nandhakumar, F. J. Zhang, X. K. Gao, D. B. Zhu and H. Sirringhaus, ACS Nano, 2014, 8, 6778–6787 CrossRef CAS PubMed.
- Y. Miyoshi, K. Takahashi, T. Fujimoto, H. Yoshikawa, M. M. Matsushita, Y. Ouchi, M. Kepenekian, V. Robert, M. P. Donzello, C. Ercolani and K. Awaga, Inorg. Chem., 2012, 51, 456–462 CrossRef CAS PubMed.
- C. L. Guo, Z. Tian, Y. J. Xiao, Q. X. Mi and J. M. Xue, Appl. Phys. Lett., 2016, 109(20), 203104 CrossRef.
- J. Cao, J. D. Querales-Flores, A. R. Murphy, S. Fahy and I. Savic, Phys. Rev. B, 2018, 98(20), 205202 CrossRef.
- Y. Li, W. Zhang, J. Niu and Y. Chen, ACS Nano, 2012, 6, 5164–5173 CrossRef CAS PubMed.
- F. Vatansever, W. C. M. A. de Melo, P. Avci, D. Vecchio, M. Sadasivam, A. Gupta, R. Chandran, M. Karimi, N. A. Parizotto, R. Yin, G. P. Tegos and M. R. Hamblin, FEMS Microbiol. Rev., 2013, 37, 955–989 CrossRef CAS PubMed.
- K. W. Goodman and V. E. Henrich, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 10450–10456 CrossRef CAS PubMed.
- C. M. Yim, C. L. Pang and G. Thornton, Phys. Rev. Lett., 2010, 104(3), 036806 CrossRef CAS PubMed.
- N. Serpone, J. Phys. Chem. B, 2006, 110, 24287–24293 CrossRef CAS PubMed.
- O. Lupan, L. Chow, G. Y. Chai, L. Chernyak, O. Lopatiuk-Tirpak and H. Heinrich, Phys. Status Solidi A, 2008, 205, 2673–2678 CrossRef CAS.
- J. Kiwi and V. Nadtochenko, Langmuir, 2005, 21, 4631–4641 CrossRef CAS PubMed.
- M. Wang, J. Ioccozia, L. Sun, C. Lin and Z. Lin, Energy Environ. Sci., 2014, 7, 2182–2202 RSC.
- R. Konaka, E. Kasahara, W. C. Dunlap, Y. Yamamoto, K. C. Chien and M. Inoue, Redox Rep., 2001, 6, 319–325 CrossRef CAS PubMed.
- R. Shen, D. Ren, Y. Ding, Y. Guan, Y. H. Ng, P. Zhang and X. Li, Sci. China Mater., 2020, 63, 2153–2188 CrossRef CAS.
- A. Jańczyk, E. Krakowska, G. Stochel and W. Macyk, J. Am. Chem. Soc., 2006, 128, 15574–15575 CrossRef PubMed.
- F. P. Koffyberg and F. A. Benko, J. Appl. Phys., 1982, 53, 1173–1177 CrossRef CAS.
- A. O. Musa, T. Akomolafe and M. J. Carter, Sol. Energy Mater. Sol. Cells, 1998, 51, 305–316 CrossRef CAS.
- G. Moretti and H. P. Beck, Surf. Interface Anal., 2019, 51, 1359–1370 CrossRef CAS.
- Q. Zhang, K. Zhang, D. Xu, G. Yang, H. Huang, F. Nie, C. Liu and S. Yang, Prog. Mater. Sci., 2014, 60, 208–337 CrossRef CAS.
- Z. Wang, P. K. Nayak, J. A. Caraveo-Frescas and H. N. Alshareef, Adv. Mater., 2016, 28, 3831–3892 CrossRef CAS PubMed.
- Q. B. Zhang, K. L. Zhang, D. G. Xu, G. C. Yang, H. Huang, F. D. Nie, C. M. Liu and S. H. Yang, Prog. Mater. Sci., 2014, 60, 208–337 CrossRef CAS.
- O. Akhavan, J. Phys. D: Appl. Phys., 2008, 41, 235407 CrossRef.
- E. Apen, B. R. Rogers and J. A. Sellers, J. Vac. Sci. Technol., A, 1998, 16, 1227–1232 CrossRef CAS.
- O. Akhavan and E. Ghaderi, Surf. Coat. Technol., 2010, 205, 219–223 CrossRef CAS.
- A. Torres, C. Ruales, C. Pulgarin, A. Aimable, P. Bowen, V. Sarria and J. Kiwi, ACS Appl. Mater. Interfaces, 2010, 2, 2547–2552 CrossRef CAS PubMed.
- M. Paschoalino, N. C. Guedes, W. Jardim, E. Mielczarski, J. A. Mielczarski, P. Bowen and J. Kiwi, J. Photochem. Photobiol., A, 2008, 199, 105–111 CrossRef CAS.
- T. Kaneto, K. W. Kim and M. A. Littlejohn, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 16257–16266 CrossRef CAS PubMed.
- S. Kobayashi, M. Saitoh, Y. Nakabayashi and K. Uchida, Appl. Phys. Lett., 2007, 91(20), 203506 CrossRef.
- A. D. de Meux, G. Pourtois, J. Genoe and P. Heremans, Phys. Rev. B, 2018, 97(4), 045208 CrossRef.
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603–2636 RSC.
- J. Xing, H. B. Jiang, J. F. Chen, Y. H. Li, L. Wu, S. Yang, L. R. Zheng, H. F. Wang, P. Hu, H. J. Zhao and H. G. Yang, J. Mater. Chem. A, 2013, 1, 15258–15264 RSC.
- Y. F. Sun, S. Gao, F. C. Lei and Y. Xie, Chem. Soc. Rev., 2015, 44, 623–636 RSC.
- B. K. Yoo, Z. X. Su, J. M. Thomas and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 503–508 CrossRef CAS PubMed.
- Y. Wang, H. Sun, S. Tan, H. Feng, Z. Cheng, J. Zhao, A. Zhao, B. Wang, Y. Luo, J. Yang and J. G. Hou, Nat. Commun., 2013, 4, 2214 CrossRef PubMed.
- W. K. Wang, J. J. Chen, Z. Z. Lou, S. Kim, M. Fujitsuka, H. Q. Yu and T. Majima, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18827–18833 CrossRef CAS PubMed.
- H. Pang, F. Gao and Q. Y. Lu, Chem. Commun., 2009, 1076–1078, 10.1039/b816670f.
- V. Lakshmi Prasanna and R. Vijayaraghavan, Langmuir, 2015, 31, 9155–9162 CrossRef CAS PubMed.
- R. Gunder, J. A. Marquez-Prieto, G. Gurieva, T. Unold and S. Schorr, CrystEngComm, 2018, 20, 1491–1498 RSC.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- Y. Li, J. Niu, W. Zhang, L. Zhang and E. Shang, Langmuir, 2014, 30, 2852–2862 CrossRef CAS PubMed.
- T. B. van Driel, K. S. Kjaer, R. W. Hartsock, A. O. Dohn, T. Harlang, M. Chollet, M. Christensen, W. Gawelda, N. E. Henriksen, J. G. Kim, K. Haldrup, K. H. Kim, H. Ihee, J. Kim, H. Lemke, Z. Sun, V. Sundstrom, W. K. Zhang, D. L. Zhu, K. B. Moller, M. M. Nielsen and K. J. Gaffney, Nat. Commun., 2016, 7, 13678 CrossRef CAS PubMed.
- X. A. Shen, Y. A. Small, J. Wang, P. B. Allen, M. V. Fernandez-Serra, M. S. Hybertsen and J. T. Muckerman, J. Phys. Chem. C, 2010, 114, 13695–13704 CrossRef CAS.
- V. Lakshmi Prasanna and R. Vijayaraghavan, Langmuir, 2015, 31, 9155–9162 CrossRef CAS PubMed.
- X. Yang, J. Li, T. Liang, C. Ma, Y. Zhang, H. Chen, N. Hanagata, H. Su and M. Xu, Nanoscale, 2014, 6, 10126–10133 RSC.
- A. C. Ferrari, F. Bonaccorso, V. Fal'ko, K. S. Novoselov, S. Roche, P. Boggild, S. Borini, F. H. Koppens, V. Palermo, N. Pugno, J. A. Garrido, R. Sordan, A. Bianco, L. Ballerini, M. Prato, E. Lidorikis, J. Kivioja, C. Marinelli, T. Ryhanen, A. Morpurgo, J. N. Coleman, V. Nicolosi, L. Colombo, A. Fert, M. Garcia-Hernandez, A. Bachtold, G. F. Schneider, F. Guinea, C. Dekker, M. Barbone, Z. Sun, C. Galiotis, A. N. Grigorenko, G. Konstantatos, A. Kis, M. Katsnelson, L. Vandersypen, A. Loiseau, V. Morandi, D. Neumaier, E. Treossi, V. Pellegrini, M. Polini, A. Tredicucci, G. M. Williams, B. H. Hong, J. H. Ahn, J. M. Kim, H. Zirath, B. J. van Wees, H. van der Zant, L. Occhipinti, A. Di Matteo, I. A. Kinloch, T. Seyller, E. Quesnel, X. Feng, K. Teo, N. Rupesinghe, P. Hakonen, S. R. Neil, Q. Tannock, T. Lofwander and J. Kinaret, Nanoscale, 2015, 7, 4598–4810 RSC.
- F. Schedin, A. K. Geim, S. V. Morozov, E. W. Hill, P. Blake, M. I. Katsnelson and K. S. Novoselov, Nat. Mater., 2007, 6, 652–655 CrossRef CAS PubMed.
- M. Zhou, X. W. Lou and Y. Xie, Nano Today, 2013, 8, 598–618 CrossRef CAS.
- J. Wang, H. Shi, L. Jiang and L. Liu, J. Funct. Mater., 2019, 50(10), 10063–10073 Search PubMed.
- K. Khan, A. K. Tareen, M. Aslam, R. U. R. Sagar, B. Zhang, W. C. Huang, A. Mahmood, N. Mahmood, K. Khan, H. Zhang and Z. Y. Guo, Nano-Micro Lett., 2020, 12(1), 167 CrossRef CAS PubMed.
- X. D. Sun, L. T. Shi, H. W. Huang, X. M. Song and T. Y. Ma, Chem. Commun., 2020, 56, 11000–11013 RSC.
- B. Luo, G. Liu and L. Wang, Nanoscale, 2016, 8, 6904–6920 RSC.
- B. Luo, G. Liu and L. Z. Wang, Nanoscale, 2016, 8, 6904–6920 RSC.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- W. J. Wang, J. C. Yu, D. H. Xia, P. K. Wong and Y. C. Li, Environ. Sci. Technol., 2013, 47, 8724–8732 CrossRef CAS PubMed.
- Z. H. Li, W. L. Wang, Q. Liu, Z. G. Zhang and X. M. Fang, Mater. Res. Bull., 2018, 106, 152–161 CrossRef CAS.
- W. W. Wang, L. Wang, W. B. Li, C. Feng, R. Qiu, L. K. Xu, X. D. Cheng and G. Q. Shao, Mater. Lett., 2019, 234, 183–186 CrossRef CAS.
- C. Lee, H. Yan, L. E. Brus, T. F. Heinz, J. Hone and S. Ryu, ACS Nano, 2010, 4, 2695–2700 CrossRef CAS PubMed.
- Z. Liang, R. Shen, Y. H. Ng, P. Zhang, Q. Xiang and X. Li, J. Mater. Sci. Technol., 2020, 56, 89–121 CrossRef.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 136805 CrossRef PubMed.
- S. M. Shinde, K. P. Dhakal, X. Chen, W. S. Yun, J. Lee, H. Kim and J. H. Ahn, NPG Asia Mater., 2018, 10, e468 CrossRef CAS.
- Z. He and W. Que, Appl. Mater. Today, 2016, 3, 23–56 CrossRef.
- T. B. Wendumu, G. Seifert, T. Lorenz, J. O. Joswig and A. Enyashin, J. Phys. Chem. Lett., 2014, 5, 3636–3640 CrossRef CAS PubMed.
- R. J. Tian, R. X. Fei, S. Q. Hu, T. S. Li, B. J. Zheng, Y. Shi, J. L. Zhao, L. J. Zhang, X. T. Gan and X. M. Wang, Phys. Rev. B, 2020, 101, 7 Search PubMed.
- V. Tran, R. Soklaski, Y. Liang and L. Yang, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 235319 CrossRef.
- T. Sakthivel, X. Huang, Y. Wu and S. Rtimi, Chem. Eng. J., 2020, 379, 122297 CrossRef CAS.
- S. Banerjee and S. K. Pati, Phys. Chem. Chem. Phys., 2016, 18, 16345–16352 RSC.
- R. Xu, J. Yang, Y. Zhu, H. Yan, J. Pei, Y. W. Myint, S. Zhang and Y. Lu, Nanoscale, 2016, 8, 129–135 RSC.
- K. F. Mak, C. Lee, J. Hone, J. Shan and T. F. Heinz, Phys. Rev. Lett., 2010, 105, 4 CrossRef PubMed.
- H. S. Lee, S.-W. Min, Y.-G. Chang, M. K. Park, T. Nam, H. Kim, J. H. Kim, S. Ryu and S. Im, Nano Lett., 2012, 12, 3695–3700 CrossRef CAS PubMed.
- Y. Li, X. Li, H. Zhang, J. Fan and Q. Xiang, J. Mater. Sci. Technol., 2020, 56, 69–88 CrossRef.
- Y. F. Sun, Q. H. Liu, S. Gao, H. Cheng, F. C. Lei, Z. H. Sun, Y. Jiang, H. B. Su, S. Q. Wei and Y. Xie, Nat. Commun., 2013, 4, 2899 CrossRef PubMed.
- F. Yao, Y. Cai, Z. Xiao, G. Zhang, R.-J. Xie and C. Jin, 2D Mater., 2020, 8, 025004 CrossRef.
- X. Tian, Y. Sun, S. Fan, M. D. Boudreau, C. Chen, C. Ge and J.-J. Yin, ACS Appl. Mater. Interfaces, 2019, 11, 4858–4866 CrossRef CAS PubMed.
- C. Liu, D. S. Kong, P. C. Hsu, H. T. Yuan, H. W. Lee, Y. Y. Liu, H. T. Wang, S. Wang, K. Yan, D. C. Lin, P. A. Maraccini, K. M. Parker, A. B. Boehm and Y. Cui, Nat. Nanotechnol., 2016, 11, 1098–1104 CrossRef CAS PubMed.
- C. Liu, D. Kong, P. C. Hsu, H. Yuan, H. W. Lee, Y. Liu, H. Wang, S. Wang, K. Yan, D. Lin, P. A. Maraccini, K. M. Parker, A. B. Boehm and Y. Cui, Nat. Nanotechnol., 2016, 11, 1098–1104 CrossRef CAS PubMed.
- W. T. Zhang, S. Shi, Y. R. Wang, S. X. Yu, W. X. Zhu, X. Zhang, D. H. Zhang, B. W. Yang, X. Wang and J. L. Wang, Nanoscale, 2016, 8, 11642–11648 RSC.
- J. A. Lemire, J. J. Harrison and R. J. Turner, Nat. Rev. Microbiol., 2013, 11, 371–384 CrossRef CAS PubMed.
- M. Tatullo, F. Genovese, E. Aiello, M. Amantea, I. Makeeva, B. Zavan, S. Rengo and L. Fortunato, Materials, 2019, 12, 11 Search PubMed.
- W. Liu, Y. Zhang, Y. Zhang and A. Dong, Chem. – Eur. J., 2020, 26, 2478–2485 CrossRef CAS PubMed.
- J. Shao, C. Ruan, H. Xie, Z. Li, H. Wang, P. Chu and X.-F. Yu, Adv. Sci., 2018, 5, 1700848 CrossRef PubMed.
- S. M. Shinde, K. P. Dhakal, X. Chen, W. S. Yun, J. Lee, H. Kim and J.-H. Ahn, NPG Asia Mater., 2018, 10, e468 CrossRef CAS.
- C. Y. Lan, Z. Shi, R. Cao, C. Li and H. Zhang, Nanoscale, 2020, 12, 11784–11807 RSC.
- B. Liu, H. H. Tai, H. Y. Liang, E. Y. Zheng, M. Sahoo, C. H. Hsu, T. C. Chen, C. A. Huang, J. C. Wang, T. H. Hou and C. S. Lai, Mater. Chem. Front., 2020, 4, 1756–1763 RSC.
- B. W. H. Baugher, H. O. H. Churchill, Y. F. Yang and P. Jarillo-Herrero, Nano Lett., 2013, 13, 4212–4216 CrossRef CAS PubMed.
- K. Kaasbjerg, T. Low and A. P. Jauho, Phys. Rev. B, 2019, 100(11), 115409 CrossRef CAS.
- G. Z. He, T. Dong, Z. C. Yang and P. Ohlckers, Chem. Mater., 2019, 31, 9917–9938 CrossRef CAS.
- S. Thurakkal and X. Y. Zhang, Adv. Sci., 2020, 7(2), 1902359 CrossRef CAS PubMed.
- L. Tan, J. Li, X. Liu, Z. Cui, X. Yang, K. Yeung, H. Pan, Y. Zheng, X. Wang and S. Wu, Small, 2017, 14, 1703197 CrossRef PubMed.
- C. Mao, Y. Xiang, X. Liu, Z. Cui, X. Yang, Z. Li, S. Zhu, Y. Zheng, K. Yeung and S. Wu, ACS Nano, 2018, 12(2), 1747–1759 CrossRef CAS PubMed.
- J. M. Pirie, Chem. Ind., 1951, 65 Search PubMed.
- M. Zhu, X. M. Liu, L. Tan, Z. D. Cui, Y. Q. Liang, Z. Y. Li, K. Wai, K. Yeung and S. L. Wu, J. Hazard. Mater., 2020, 383, 121122 CrossRef CAS PubMed.
- F. Raza, D. Yim, J. H. Park, H. I. Kim, S. J. Jeon and J. H. Kim, J. Am. Chem. Soc., 2017, 139, 14767–14774 CrossRef CAS PubMed.
- X. Xin, S. H. Li, N. Zhang, Z. R. Tang and Y. J. Xu, Appl. Catal., B, 2019, 245, 343–350 CrossRef CAS.
- U. Baig, M. A. Gondal, M. A. Dastageer, A. B. Khalil and S. M. Zubair, J. Photochem. Photobiol., B, 2018, 187, 113–119 CrossRef CAS PubMed.
- Y. Peng, B. Z. Lu and S. W. Chen, Adv. Mater., 2018, 30, 25 Search PubMed.
- Z. Huang, X. Gu, Q. Cao, P. Hu, J. Hao, J. Li and X. Tang, Angew. Chem., Int. Ed., 2012, 51, 4198–4203 CrossRef CAS PubMed.
- S. C. Warren and E. Thimsen, Energy Environ. Sci., 2012, 5, 5133–5146 RSC.
- S. K. Cushing and N. Wu, J. Phys. Chem. Lett., 2016, 7, 666–675 CrossRef CAS PubMed.
- W. Fan and M. K. Leung, Molecules, 2016, 21(2), 180 CrossRef PubMed.
- S. Yu, Y. H. Kim, S. Y. Lee, H. D. Song and J. Yi, Angew. Chem., Int. Ed., 2014, 53, 11203–11207 CrossRef CAS PubMed.
- L. M. Lai, X. R. Jiang, S. Y. Han, C. Q. Zhao, T. Y. Du, F. U. Rehman, Y. K. Zheng, X. Q. Li, X. L. Liu, H. Jiang and X. M. Wang, Langmuir, 2017, 33, 9018–9024 CrossRef CAS PubMed.
- T. Y. Du, C. Q. Zhao, F. U. Rehman, L. M. Lai, X. Q. Li, Y. Sun, S. H. Luo, H. Jiang, M. Selke and X. M. Wang, Nano Res., 2017, 10, 2626–2632 CrossRef CAS.
- Z. He, R. Liu, C. Xu, Y. Lai, W. Shan and J. Liu, Appl. Catal., B, 2021, 285, 119775 CrossRef CAS.
- Y. Lei, F. Mehmood, S. Lee, J. Greeley, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, R. J. Meyer, P. C. Redfern, D. Teschner, R. Schlogl, M. J. Pellin, L. A. Curtiss and S. Vajda, Science, 2010, 328, 224–228 CrossRef CAS PubMed.
- C. S. Wang, J. Y. Li, C. Amatore, Y. Chen, H. Jiang and X. M. Wang, Angew. Chem., Int. Ed., 2011, 50, 11644–11648 CrossRef CAS PubMed.
- X. Y. Lv, W. Ge, Q. W. Li, Y. L. Wu, H. Jiang and X. M. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 11025–11031 CrossRef CAS PubMed.
- C. Q. Zhao, T. Y. Du, F. U. Rehman, L. M. Lai, X. L. Liu, X. R. Jiang, X. Q. Li, Y. Chen, H. Zhang, Y. Sun, S. H. Luo, H. Jiang, M. Selke and X. M. Wang, Small, 2016, 12, 6255–6265 CrossRef CAS PubMed.
- J. Ye, X. W. Dong, H. Jiang and X. M. Wang, J. Mater. Chem. B, 2017, 5, 691–696 RSC.
- S. Shaikh, F. U. Rehman, T. Y. Du, H. Jiang, L. H. Yin, X. M. Wang and R. J. Chai, ACS Appl. Mater. Interfaces, 2018, 10, 26056–26063 CrossRef CAS PubMed.
- Y. Zhao, X. Li, H. Li and L. He, New J. Chem., 2021, 45, 15378–15385 RSC.
- X. Weng, B. Shi, A. Liu, J. Sun, Y. Xiong, H. Wan, S. Zheng, L. Dong and Y.-W. Chen, Appl. Surf. Sci., 2019, 497, 143747 CrossRef CAS.
- G. D. Wu, G. Q. Zhao, J. H. Sun, X. Z. Cao, Y. F. He, J. T. Feng and D. Q. Li, J. Catal., 2019, 377, 271–282 CrossRef CAS.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS.
- R. Kavitha, P. M. Nithya and S. Girish Kumar, Appl. Surf. Sci., 2020, 508, 145142 CrossRef CAS.
- L. Zhou, H. Y. Zhang, H. Q. Sun, S. M. Liu, M. O. Tade, S. B. Wang and W. Q. Jin, Catal. Sci. Technol., 2016, 6, 7002–7023 RSC.
-
C. Moslah, G. A. Mousdis, M. Kandyla, G. Petropoulou and M. Ksibi, in Nanostructured Materials for the Detection of Cbrn, ed. J. Bonca and S. Kruchinin, 2018, pp. 71–89 DOI:10.1007/978-94-024-1304-5_6.
- M. A. Barakat, R. I. Al-Hutailah, E. Qayyum, J. Rashid and J. N. Kuhn, Environ. Technol., 2014, 35, 137–144 CrossRef CAS PubMed.
- S. F. Su, L. M. Ye, Q. M. Tian, W. B. Situ, X. L. Song and S. Y. Ye, Appl. Surf. Sci., 2020, 515, 145964 CrossRef CAS.
- L. Shang, B. Li, W. Dong, B. Chen, C. Li, W. Tang, G. Wang, J. Wu and Y. Ying, J. Hazard. Mater., 2010, 178, 1109–1114 CrossRef CAS PubMed.
- R. Gupta, N. K. Eswar, J. M. Modak and G. Madras, Catal. Today, 2018, 300, 71–80 CrossRef CAS.
- L. Q. Mei, S. Zhu, W. Y. Yin, C. Y. Chen, G. J. Nie, Z. J. Gu and Y. L. Zhao, Theranostics, 2020, 10, 757–781 CrossRef CAS PubMed.
- G. Pedroza-Herrera, I. E. Medina-Ramirez, J. A. Lozano-Alvarez and S. E. Rodil, Catal. Today, 2020, 341, 37–48 CrossRef CAS.
- Q. Zhang, X. M. Liu, L. Tan, Z. D. Cui, Z. Y. Li, Y. Q. Liang, S. L. Zhu, K. W. K. Yeung, Y. F. Zheng and S. L. Wu, Chem. Eng. J., 2020, 383, 123088 CrossRef CAS.
- V. Amenta, K. Aschberger, M. Arena, H. Bouwmeester, F. B. Moniz, P. Brandhoff, S. Gottardo, H. J. P. Marvin, A. Mech, L. Q. Pesudo, H. Rauscher, R. Schoonjans, M. V. Vettori, S. Weigel and R. J. Peters, Regul. Toxicol. Pharmacol., 2015, 73, 463–476 CrossRef PubMed.
- A. Zane, R. Zuo, F. A. Villamena, A. Rockenbauer, A. M. D. Foushee, K. Flores, P. K. Dutta and A. Nagy, Int. J. Nanomed., 2016, 11, 6459–6470 CrossRef CAS PubMed.
- R. Masa, A. Deak, G. Braunitzer, Z. Toth, J. Kopniczky, I. Pelsoczi-Kovacs, K. Ungvari, I. Dekany and K. Turzo, J. Nanosci. Nanotechnol., 2018, 18, 3916–3924 CrossRef CAS PubMed.
- V. Rodríguez-González, C. Terashima and A. Fujishima, J. Photochem. Photobiol., C, 2019, 40, 49–67 CrossRef.
- A. J. Misra, S. Das, A. P. H. Rahman, B. Das, R. Jayabalan, S. K. Behera, M. Suar, A. J. Tamhankar, A. Mishra, C. S. Lundborg and S. K. Tripathy, J. Colloid Interface Sci., 2018, 530, 610–623 CrossRef CAS PubMed.
- R. Wang, B. Zhang, Z. Liang, Y. He, Z. Wang, X. Ma, X. Yao, J. Sun and J. Wang, Appl. Catal., B, 2019, 241, 167–177 CrossRef CAS.
- Y. Li, W. Zhang, J. F. Niu and Y. S. Chen, ACS Nano, 2012, 6, 5164–5173 CrossRef CAS PubMed.
- P. C. Maness, S. Smolinski, D. M. Blake, Z. Huang, E. J. Wolfrum and W. A. Jacoby, Appl. Environ. Microbiol., 1999, 65, 4094–4098 CrossRef CAS PubMed.
- Y. Y. Wu, Y. Z. Zhou, H. Xu, Q. G. Liu, Y. Li, L. L. Zhang, H. G. Liu, Z. G. Tu, X. N. Cheng and J. Yang, ACS Sustainable Chem. Eng., 2018, 6, 14082–14094 CrossRef CAS.
- Y. Wu, L. Zhang, Y. Zhou, L. Zhang, Y. Li, Q. Liu, J. Hu and J. Yang, Chin. J. Catal., 2019, 40, 691–702 CrossRef CAS.
- I. Teixeira, J. Quiroz, M. Homsi and P. Camargo, J. Braz. Chem. Soc., 2020, 31, 211–229 CAS.
- A. Kasahara, K. Nukumizu, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Phys. Chem. B, 2003, 107, 791–797 CrossRef CAS.
- H. Kato and A. Kudo, J. Phys. Chem. B, 2001, 105, 4285–4292 CrossRef CAS.
- Y. H. Xu, H. R. Chen, Z. X. Zeng and B. Lei, Appl. Surf. Sci., 2006, 252, 8565–8570 CrossRef CAS.
- X. L. Liu, H. Jiang, J. Ye, C. Q. Zhao, S. P. Gao, C. Y. Wu, C. H. Li, J. C. Li and X. M. Wang, Adv. Funct. Mater., 2016, 26, 8694–8706 CrossRef CAS.
- A. Mittal, B. Mari, S. Sharma, V. Kumari, S. Maken, K. Kumari and N. Kumar, J. Mater. Sci.: Mater. Electron., 2019, 30, 3186–3207 CrossRef CAS.
- W. Y. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669–13679 CrossRef.
- H. Yu, H. Irie, Y. Shimodaira, Y. Hosogi, Y. Kuroda, M. Miyauchi and K. Hashimoto, J. Phys. Chem. C, 2010, 114, 16481–16487 CrossRef CAS.
- S. K. Loeb, P. J. J. Alvarez, J. A. Brame, E. L. Cates, W. Choi, J. Crittenden, D. D. Dionysiou, Q. Li, G. Li-Puma, X. Quan, D. L. Sedlak, T. David Waite, P. Westerhoff and J.-H. Kim, Environ. Sci. Technol., 2019, 53, 2937–2947 CrossRef CAS PubMed.
- L. Brus, J. Phys. Chem., 1986, 90, 2555–2560 CrossRef CAS.
- M. I. Litter, Appl. Catal., B, 1999, 23, 89–114 CrossRef CAS.
- Z. F. Shen, F. F. Li, J. R. Lu, Z. D. Wang, R. Li, X. C. Zhang, C. M. Zhang, Y. W. Wang, Y. F. Wang, Z. P. Lv, J. X. Liu and C. M. Fan, J. Colloid Interface Sci., 2021, 584, 174–181 CrossRef CAS PubMed.
- K. Z. Qi, X. H. Xing, A. Zada, M. Y. Li, Q. Wang, S. Y. Liu, H. X. Lin and G. Z. Wang, Ceram. Int., 2020, 46, 1494–1502 CrossRef CAS.
- S. Yadav and G. Jaiswar, J. Chin. Chem. Soc., 2017, 64, 103–116 CrossRef CAS.
- P. Sanitnon, S. Chiarakorn, C. Chawengkijwanich, S. Chuangchote and T. Pongprayoon, J. Aust. Ceram. Soc., 2020, 56, 579–590 CrossRef CAS.
- Z. Zha, S. Zhang, Z. Deng, Y. Li, C. Li and Z. Dai, Chem. Commun., 2013, 49, 3455–3457 RSC.
- D. L. Han, Y. J. Han, J. Li, X. M. Liu, K. W. K. Yeung, Y. F. Zheng, Z. D. Cui, X. J. Yang, Y. Q. Liang, Z. Y. Li, S. L. Zhu, X. B. Yuan, X. B. Feng, C. Yang and S. L. Wu, Appl. Catal., B, 2020, 261, 12 CrossRef.
- H. Irie, K. Kamiya, T. Shibanuma, S. Miura, D. A. Tryk, T. Yokoyama and K. Hashimoto, J. Phys. Chem. C, 2009, 113, 10761–10766 CrossRef CAS.
- M. Vautier, C. Guillard and J. M. Herrmann, J. Catal., 2001, 201, 46–59 CrossRef CAS.
- C. L. Yu, H. B. He, Q. Z. Fan, W. Y. Xie, Z. Liu and H. B. Ji, Sci. Total Environ, 2019, 694, 133727 CrossRef CAS PubMed.
- M. A. Mohamed, N. A. Rahman, M. F. M. Zain, L. J. Minggu, M. B. Kassim, J. Jaafar, S. Samad, M. S. Mastuli and R. J. Wong, J. Alloys Compd., 2020, 820, 153143 CrossRef CAS.
- J. C. Yu, J. G. Yu, W. K. Ho, Z. T. Jiang and L. Z. Zhang, Chem. Mater., 2002, 14, 3808–3816 CrossRef CAS.
- W. Wang, M. O. Tade and Z. P. Shao, Prog. Mater. Sci., 2018, 92, 33–63 CrossRef CAS.
- X. Bai, J. Jia, Y. Y. Du, X. Y. Hu, J. L. Li, E. Z. Liu and J. Fan, Appl. Surf. Sci., 2020, 503, 144298 CrossRef CAS.
- M. Rahbar, M. Mehrzad, M. Behpour, S. Mohammadi-Aghdam and M. Ashrafi, Nanotechnology, 2019, 30(50), 505702 CrossRef CAS PubMed.
- V. Etacheri, C. Di Valentin, J. Schneider, D. Bahnemann and S. C. Pillai, J. Photochem. Photobiol., C, 2015, 25, 1–29 CrossRef CAS.
- Q. Q. Yin, R. Qiao and G. X. Tong, Prog. Chem., 2014, 26, 1619–1632 CAS.
- Y. J. Jang, M. D. Bhatt, J. Lee, S. H. Choi, B. J. Lee and J. S. Lee, Adv. Energy Mater., 2018, 8, 1702636 CrossRef.
- H. Zhang, Y. Zhang, J. Yin, Z. Li, Q. Zhu and Z. Xing, Chem. Phys., 2018, 513, 86–93 CrossRef CAS.
- C. Zhu, M. Zhu, Y. Sun, Y. Zhou, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong, Y. Liu and Z. Kang, Appl. Catal., B, 2018, 237, 166–174 CrossRef CAS.
- I. Milošević, S. Rtimi, A. Jayaprakash, B. van Driel, B. Greenwood, A. Aimable, M. Senna and P. Bowen, Colloids Surf., B, 2018, 171, 445–450 CrossRef PubMed.
- M. Zheng and J. Wu, Appl. Surf. Sci., 2009, 255, 5656–5661 CrossRef CAS.
- K. Qi, B. Cheng, J. Yu and W. Ho, J. Alloys Compd., 2017, 727, 792–820 CrossRef CAS.
- Y. Xu, K. Lv, Z. Xiong, W. Leng, W. Du, D. Liu and X. Xue, J. Phys. Chem. C, 2007, 111, 19024–19032 CrossRef CAS.
- C. Di Valentin, G. Pacchioni and A. Selloni, J. Phys. Chem. C, 2009, 113, 20543–20552 CrossRef CAS.
- N. U. M. Nor and N. A. S. Amin, J. CO2 Util., 2019, 33, 372–383 CrossRef CAS.
- F. Amano, M. Nakata, J. J. M. Vequizo and A. Yamakata, ACS Appl. Energy Mater., 2019, 2, 3274–3282 CrossRef CAS.
- N. Najibi Ilkhechi, M. R. Akbarpour, R. Yavari and Z. Azar, J. Mater. Sci.: Mater. Electron., 2017, 28, 16658–16664 CrossRef CAS.
- H. Liu and L. Gao, Chem. Lett., 2004, 33, 730–731 CrossRef CAS.
- K. Gupta, J. Kumar, R. P. Sing, A. Pandey, P. Rai and A. Pandey, Mater. Technol., 2019, 34, 765–775 CrossRef CAS.
- S. Gao, W. Yang, J. Xiao, B. Li and Q. Li, J. Mater. Sci. Technol., 2019, 35, 610–614 CrossRef.
- X. Niu, W. Yan, C. Shao, H. Zhao and J. Yang, Appl. Surf. Sci., 2019, 466, 882–892 CrossRef CAS.
- S. Na Phattalung, S. Limpijumnong and J. Yu, Appl. Catal., B, 2017, 200, 1–9 CrossRef CAS.
- N. Serpone, E. Borgarello and M. Gratzel, J. Chem. Soc., Chem. Commun., 1984, 342–344, 10.1039/c39840000342.
- R. Shen, K. He, A. Zhang, N. Li, Y. H. Ng, P. Zhang, J. Hu and X. Li, Appl. Catal., B, 2021, 291, 120104 CrossRef CAS.
- R. Shen, Y. Ding, S. Li, P. Zhang, Q. Xiang, Y. H. Ng and X. Li, Chin. J. Catal., 2021, 42, 25–36 CrossRef CAS.
- P. Zhang and X. W. Lou, Adv. Mater., 2019, 31, 1900281 CrossRef PubMed.
- L. Liu, T. Hu, K. Dai, J. Zhang and C. Liang, Chin. J. Catal., 2021, 42, 46–55 CrossRef CAS.
- J. X. Low, J. G. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 20 CrossRef PubMed.
- Y. N. Jia, S. H. Zhan, S. L. Ma and Q. X. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 6841–6851 CrossRef CAS PubMed.
- T. W. Ng, L. S. Zhang, J. S. Liu, G. C. Huang, W. Wang and P. K. Wong, Water Res., 2016, 90, 111–118 CrossRef CAS PubMed.
- Y. Jia, S. Zhan, S. Ma and Q. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 6841–6851 CrossRef CAS PubMed.
- T. W. Ng, L. Zhang, J. Liu, G. Huang, W. Wang and P. K. Wong, Water Res., 2016, 90, 111–118 CrossRef CAS PubMed.
- M. Cho, H. Chung, W. Choi and J. Yoon, Appl. Environ. Microbiol., 2005, 71, 270 CrossRef CAS PubMed.
- L. Armelao, D. Barreca, G. Bottaro, A. Gasparotto, C. Maccato, C. Maragno, E. Tondello, U. L. Stangar, M. Bergant and D. Mahne, Nanotechnology, 2007, 18, 7 Search PubMed.
- K. Hayashi, K. Nozaki, Z. Tan, K. Fujita, R. Nemoto, K. Yamashita, H. Miura, K. Itaka and S. Ohara, Materials, 2020, 13(1), 78 CrossRef CAS PubMed.
- K. R. Raghupathi, R. T. Koodali and A. C. Manna, Langmuir, 2011, 27, 4020–4028 CrossRef CAS PubMed.
- S. K. Ray, D. Dhakal, J. Hur and S. W. Lee, Nanotechnology, 2020, 31, 16 CrossRef PubMed.
- Y. Li and Y. F. Cheng, J. Mater. Chem. B, 2018, 6, 1458–1469 RSC.
- X. Tian, Y. R. Sun, S. H. Fan, M. D. Boudreau, C. Y. Chen, C. C. Ge and J. J. Yin, ACS Appl. Mater. Interfaces, 2019, 11, 4858–4866 CrossRef CAS PubMed.
- L. Xiong, H. Yu, C. Nie, Y. Xiao, Q. Zeng, G. Wang, B. Wang, H. Lv, Q. Li and S. Chen, RSC Adv., 2017, 7, 51822–51830 RSC.
- L. Xiong, T. W. Ng, Y. Yu, D. Xia, H. Y. Yip, G. Li, T. An, H. Zhao and P. K. Wong, Electrochim. Acta, 2015, 153, 583–593 CrossRef CAS.
- J. Huang, W. Ho and X. Wang, Chem. Commun., 2014, 50, 4338–4340 RSC.
- J. H. Thurston, N. M. Hunter and K. A. Cornell, RSC Adv., 2016, 6, 42240–42248 RSC.
- D. Xia, Z. Shen, G. Huang, W. Wang, J. C. Yu and P. K. Wong, Environ. Sci. Technol., 2015, 49, 6264–6273 CrossRef CAS PubMed.
- Y. Zhong, C. Peng, Z. He, D. Chen, H. Jia, J. Zhang, H. Ding and X. Wu, Catal. Sci. Technol., 2021, 11(1) 10.1039/d0cy01847c.
- S. Adhikari, A. Banerjee, N. K. Eswar, D. Sarkar and G. Madras, RSC Adv., 2015, 5, 51067–51077 RSC.
- M. M. Li, D. G. Li, Z. R. Zhou, P. F. Wang, X. Y. Mi, Y. G. Xia, H. T. Wang, S. H. Zhan, Y. Li and L. M. Li, Chem. Eng. J., 2020, 382, 122762 CrossRef CAS.
- Y. Li, J. Zhao, G. S. Zhang, L. L. Zhang, S. Y. Ding, E. X. Shang and X. H. Xia, Water Res., 2019, 161, 251–261 CrossRef CAS PubMed.
- A. Senthilraja, B. Krishnakumar, R. Hariharan, A. Sobral, C. Surya, N. A. A. John and M. Shanthi, Sep. Purif. Technol., 2018, 202, 373–384 CrossRef CAS.
- M. C. Wu, T. H. Lin, K. H. Hsu and J. F. Hsu, Appl. Surf. Sci., 2019, 484, 326–334 CrossRef CAS.
- C. Liang, H. Guo, L. Zhang, M. Ruan, C.-G. Niu, H.-P. Feng, X.-J. Wen, N. Tang, H.-Y. Liu and G.-M. Zeng, Chem. Eng. J., 2019, 372, 12–25 CrossRef CAS.
- L. Jing, Y. Xu, S. Huang, M. Xie, M. He, H. Xu, H. Li and Q. Zhang, Appl. Catal., B, 2016, 199, 11–22 CrossRef CAS.
- L. Jing, Y. Xu, C. Qin, J. Liu, S. Huang, M. He, H. Xu and H. Li, Mater. Res. Bull., 2017, 95, 607–615 CrossRef CAS.
- D. Han, Y. Han, J. Li, X. Liu, K. W. K. Yeung, Y. Zheng, Z. Cui, X. Yang, Y. Liang, Z. Li, S. Zhu, X. Yuan, X. Feng, C. Yang and S. Wu, Appl. Catal., B, 2020, 261, 118248 CrossRef CAS.
- N. F. A. Neto, K. N. Matsui, C. A. Paskocimas, M. R. D. Bomio and F. V. Motta, Mater. Sci. Semicond. Process., 2019, 93, 123–133 CrossRef.
- D. Neena, M. Humayun, D. Z. Lu, V. B. Mohan, D. J. Fu and W. Gao, Ceram. Int., 2020, 46, 5278–5288 CrossRef.
- Q. F. Zhao, M. Wang, H. Yang, D. Shi and Y. Z. Wang, Ceram. Int., 2018, 44, 5145–5154 CrossRef CAS.
- Y. Wang, X. Xue and H. Yang, Ceram. Int., 2014, 40, 12533–12537 CrossRef CAS.
- V. Etacheri, G. Michlits, M. K. Seery, S. J. Hinder and S. C. Pillai, ACS Appl. Mater. Interfaces, 2013, 5, 1663–1672 CrossRef CAS PubMed.
- R. S. Pessoa, V. P. dos Santos, S. B. Cardoso, A. C. O. C. Doria, F. R. Figueira, B. V. M. Rodrigues, G. E. Testoni, M. A. Fraga, F. R. Marciano, A. O. Lobo and H. S. Maciel, Appl. Surf. Sci., 2017, 422, 73–84 CrossRef CAS.
- N. Tahmasebizad, M. T. Hamedani, M. Shaban Ghazani and Y. Pazhuhanfar, J. Sol-Gel Sci. Technol., 2020, 93, 570–578 CrossRef CAS PubMed.
- K. V. Divya Lakshmi, T. Siva Rao, J. Swathi Padmaja, I. Manga Raju and M. Ravi Kumar, Chin. J. Chem. Eng., 2019, 27, 1630–1641 CrossRef.
- Y. Z. Wang, Y. S. Wu, H. Yang, M. Wang, X. G. Shi, C. Wang and S. W. Zhang, Mater. Technol., 2018, 33, 48–56 CrossRef CAS.
- N. S. Leyland, J. Podporska-Carroll, J. Browne, S. J. Hinder, B. Quilty and S. C. Pillai, Sci. Rep., 2016, 6, 24770 CrossRef CAS PubMed.
- K. Ravichandran, R. Uma, B. Sakthivel, S. Gobalakrishnan and P. K. Praseetha, J. Mater. Sci.: Mater. Electron., 2016, 27, 1609–1615 CrossRef CAS.
- R. Michal, E. Dworniczek, M. Caplovicova, M. Gregor, L. U. Caplovic, A. Seniuk, P. Kus and G. Plesch, Ceram. Int., 2014, 40, 5745–5756 CrossRef CAS.
- P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS PubMed.
- D. Xia, W. Wang, R. Yin, Z. Jiang, T. An, G. Li, H. Zhao and P. K. Wong, Appl. Catal., B, 2017, 214, 23–33 CrossRef CAS.
- X. Zeng, Z. Wang, G. Wang, T. R. Gengenbach, D. T. McCarthy, A. Deletic, J. Yu and X. Zhang, Appl. Catal., B, 2017, 218, 163–173 CrossRef CAS.
- Y. Xiang, Q. Zhou, Z. Li, Z. Cui, X. Liu, Y. Liang, S. Zhu, Y. Zheng, K. W. K. Yeung and S. Wu, J. Mater. Sci. Technol., 2020, 57, 1–11 CrossRef.
- H. Yan, R. Wang, R. Liu, T. Xu, J. Sun, L. Liu and J. Wang, Appl. Catal., B, 2021, 291, 120096 CrossRef CAS.
- T. W. Ng, L. Zhang, J. Liu, G. Huang, W. Wang and P. K. Wong, Water Res., 2016, 90, 111–118 CrossRef CAS PubMed.
- S. Zhang, C. Liu, X. Liu, H. Zhang, P. Liu, S. Zhang, F. Peng and H. Zhao, Appl. Microbiol. Biotechnol., 2012, 96, 1201–1207 CrossRef CAS PubMed.
- P. Ju, Y. Wang, Y. Sun and D. Zhang, Dalton Trans., 2016, 45, 4588–4602 RSC.
- V. Siyahi, A. Habibi-Yangjeh, S. Latifi-Navid and A. Asadi, Mater. Express, 2015, 5, 201–210 CrossRef CAS.
- G. Tong, F. Du, W. Wu, R. Wu, F. Liu and Y. Liang, J. Mater. Chem. B, 2013, 1, 2647 RSC.
- O. Akhavan, R. Azimirad, S. Safa and M. M. Larijani, J. Mater. Chem., 2010, 20, 7386 RSC.
- O. Akhavan, R. Azimirad and S. Safa, Mater. Chem. Phys., 2011, 130, 598–602 CrossRef CAS.
- M. J. Muñoz-Batista, M. Ferrer, M. Fernández-García and A. Kubacka, Appl. Catal., B, 2014, 154–155, 350–359 CrossRef.
- Y. Jin, Z. Dai, F. Liu, H. Kim, M. Tong and Y. Hou, Water Res., 2013, 47, 1837–1847 CrossRef CAS PubMed.
- W. Wang, J. C. Yu, D. Xia, P. K. Wong and Y. Li, Environ. Sci. Technol., 2013, 47, 8724–8732 CrossRef CAS PubMed.
- D. L. Guan, C. G. Niu, X. J. Wen, H. Guo, C. H. Deng and G. M. Zeng, J. Colloid Interface Sci., 2018, 512, 272–281 CrossRef CAS PubMed.
- S. J. Gu, D. Zhang, S. R. Luo and H. Yang, Int. J. Photoenergy, 2019, 2019, 6307858 CrossRef.
- M. Padervand, Appl. Nanosci., 2016, 6, 1119–1126 CrossRef CAS.
- J. Liu, L. Huang, Y. Li, L. Yang, C. Wang, J. Liu, Y. Song, M. Yang and H. Li, J. Colloid Interface Sci., 2021, 600, 344–357 CrossRef CAS PubMed.
- C. Chen, D. Wang, Y. Li, H. Huang and Y. Ke, Appl. Surf. Sci., 2021, 565, 150534 CrossRef CAS.
- Y. Sun, X. Qi, R. Li, Y. Xie, Q. Tang and B. Ren, Opt. Mater., 2020, 108, 110170 CrossRef CAS.
- A. Enesca and L. Andronic, Nanomaterials, 2021, 11(4), 871 CrossRef CAS PubMed.
- J. Low, J. Yu, M. Jaroniec, S. Wageh and A. A. Al-Ghamdi, Adv. Mater., 2017, 29, 1601694 CrossRef PubMed.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS.
- R. He, H. Liu, H. Liu, D. Xu and L. Zhang, J. Mater. Sci. Technol., 2020, 52, 145–151 CrossRef.
- Q. Xu, D. Ma, S. Yang, Z. Tian, B. Cheng and J. Fan, Appl. Surf. Sci., 2019, 495, 143555 CrossRef CAS.
- S. Wageh, A. A. Al-Ghamdi, R. Jafer, X. Li and P. Zhang, Chin. J. Catal., 2021, 42, 667–669 CrossRef CAS.
- R. C. Shen, X. Y. Lu, Q. Q. Zheng, Q. Chen, Y. H. Ng, P. Zhang and X. Li, Sol. RRL, 2021, 5(7), 2100177 CrossRef CAS.
- S. Li, C. Wang, M. Cai, F. Yang, Y. Liu, J. Chen, P. Zhang, X. Li and X. Chen, Chem. Eng. J., 2022, 428, 131158 CrossRef CAS.
- P. Xia, S. Cao, B. Zhu, M. Liu, M. Shi, J. Yu and Y. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5218–5225 CrossRef CAS PubMed.
- J. Wang, G. Wang, B. Cheng, J. Yu and J. Fan, Chin. J. Catal., 2021, 42, 56–68 CrossRef CAS.
- S. P. Adhikari, H. R. Pant, J. H. Kim, H. J. Kim, C. H. Park and C. S. Kim, Colloids Surf., A, 2015, 482, 477–484 CrossRef CAS.
- V. Shanmugam, A. L. Muppudathi, S. Jayavel and K. S. Jeyaperumal, Arabian J. Chem., 2020, 13, 2439–2455 CrossRef CAS.
- S. Vignesh, S. Suganthi, J. K. Sundar and V. Raj, J. Ind. Eng. Chem., 2019, 76, 318–332 CrossRef.
- J. Xu, Q. Z. Gao, X. J. Bai, Z. P. Wang and Y. F. Zhu, Catal. Today, 2019, 332, 227–235 CrossRef CAS.
- M. J. Munoz-Batista, O. Fontelles-Carceller, M. Ferrer, M. Fernandez-Garcia and A. Kubacka, Appl. Catal., B, 2016, 183, 86–95 CrossRef CAS.
- W. Zhang, C. Yu, Z. Sun and S. Zheng, J. Microbiol. Biotechnol., 2018, 28, 957–967 CrossRef CAS PubMed.
- Y. Wu, Y. Zhou, H. Xu, Q. Liu, Y. Li, L. Zhang, H. Liu, Z. Tu, X. Cheng and J. Yang, ACS Sustainable Chem. Eng., 2018, 6, 14082–14094 CrossRef CAS.
- Q. Zhang, X. Quan, H. Wang, S. Chen, Y. Su and Z. L. Li, Sci. Rep., 2017, 7, 3128 CrossRef PubMed.
- Y. Y. Qin, Y. C. Guo, Z. Q. Liang, Y. J. Xue, X. L. Zhang, L. Yang and J. Tian, Chin. Chem. Lett., 2021, 32, 1523–1526 CrossRef CAS.
- P. Dhandapani, M. S. AlSalhi, R. Karthick, F. M. Chen, S. Devanesan, W. Kim, A. Rajasekar, M. Ahmed, M. J. Aljaafreh and A. Muhammad, J. Hazard. Mater., 2021, 409, 124661 CrossRef CAS PubMed.
- J. W. Xu, Y. Li, X. M. Zhou, Y. Z. Li, Z. D. Gao, Y. Y. Song and P. Schmuki, Chem. – Eur. J., 2016, 22, 3947–3951 CrossRef CAS PubMed.
- H. Y. Sun, Z. Q. Yang, Y. N. Pu, W. W. Dou, C. Y. Wang, W. H. Wang, X. P. Hao, S. G. Chen, Q. Shao, M. Y. Dong, S. D. Wu, T. Ding and Z. H. Guo, J. Colloid Interface Sci., 2019, 547, 40–49 CrossRef CAS PubMed.
- W. J. Wang, G. Y. Li, T. C. An, D. K. L. Chan, J. C. Yu and P. K. Wong, Appl. Catal., B, 2018, 238, 126–135 CrossRef CAS.
- H. Li, H. Yu, X. Quan, S. Chen and Y. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 2111–2119 CrossRef CAS PubMed.
- J. Li, Y. C. Yin, E. Z. Liu, Y. N. Ma, J. Wan, J. Fan and X. Y. Hu, J. Hazard. Mater., 2017, 321, 183–192 CrossRef CAS PubMed.
- X. Y. Zhao, M. M. Chen, H. L. Wang, L. Xia, M. Guo, S. W. Jiang, Q. Wang, X. J. Li and X. F. Yang, Mater. Sci. Eng., C, 2020, 116, 111221 CrossRef CAS PubMed.
- B. K. Liu, X. L. Han, Y. Wang, X. Fan, Z. Y. Wang, J. T. Zhang and H. Z. Shi, J. Mater. Sci.: Mater. Electron., 2018, 29, 14300–14310 CrossRef CAS.
- Y. Jia, S. Zhan, S. Ma and Q. Zhou, ACS Appl. Mater. Interfaces, 2016, 8, 6841–6851 CrossRef CAS PubMed.
- T. S. Natarajan, K. R. Thampi and R. J. Tayade, Appl. Catal., B, 2018, 227, 296–311 CrossRef CAS.
- J. Li, S. Zhang, W. Guo, H. Qi, H. Liang and L. Mao, Chin. J. Semicond., 2005, 26, 2416–2421 CAS.
- D. Zhang, Z. Yang, J. Hao, T. Zhang, Q. Sun and Y. Wang, Chemosphere, 2021, 276, 130226 CrossRef CAS PubMed.
- J. J. Zhao, J.-S. Zhang, F. Zhang, W. Wang, H. R. He, W. Y. Cai and J. Wang, Chin. Phys. B, 2019, 28(12), 126801 CrossRef CAS.
- C. F. Yu, K. Wang, P. Y. Yang, S. N. Yang, C. Lu, Y. Z. Song, S. Y. Dong, J. Y. Sun and J. H. Sun, Appl. Surf. Sci., 2017, 420, 233–242 CrossRef CAS.
- A. Kumar, G. Sharma, A. Kumari, C. Guo, M. Naushad, D. V. N. Vo, J. Iqbal and F. J. Stadler, Appl. Catal., B, 2021, 284, 119808 CrossRef CAS.
- Y. Zhou, M. Yu, H. Liang, J. Chen, L. Xu and J. Niu, Appl. Catal., B, 2021, 291, 120105 CrossRef CAS.
- M. Guo, Z. Xing, T. Zhao, Z. Li, S. Yang and W. Zhou, Appl. Catal., B, 2019, 257, 117913 CrossRef CAS.
- J. Yu, J. Low, W. Xiao, P. Zhou and M. Jaroniec, J. Am. Chem. Soc., 2014, 136, 8839–8842 CrossRef CAS PubMed.
- J. Zhang, L. Zhang, W. Yu, F. Jiang, E. Zhang, H. Wang, Z. Kong, J. Xi and Z. Ji, J. Am. Ceram. Soc., 2017, 100, 5274–5285 CrossRef CAS.
- J. Zhang, L. Zhang, Y. Shi, G. Xu, E. Zhang, H. Wang, Z. Kong, J. Xi and Z. Ji, Appl. Surf. Sci., 2017, 420, 839–848 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- J. Li, E. Z. Liu, Y. N. Ma, X. Y. Hu, J. Wan, L. Sun and J. Fan, Appl. Surf. Sci., 2016, 364, 694–702 CrossRef CAS.
- Q. Zhang, X. Liu, L. Tan, Z. Cui, Z. Li, Y. Liang, S. Zhu, K. W. K. Yeung, Y. Zheng and S. Wu, Chem. Eng. J., 2020, 383, 123088 CrossRef CAS.
- B. Han, S. Liu, Y.-J. Xu and Z.-R. Tang, RSC Adv., 2015, 5, 16476–16483 RSC.
- G. P. Neupane, K. Zhou, S. Chen, T. Yildirim, P. Zhang and Y. Lu, Small, 2019, 15, 1804733 CrossRef PubMed.
- Y. Li, P. Zhang, D. Wan, C. Xue, J. Zhao and G. Shao, Appl. Surf. Sci., 2020, 504, 144361 CrossRef CAS.
- Z. Jiang, Q. Chen, Q. Zheng, R. Shen, P. Zhang and X. Li, Acta Phys.-Chim. Sin., 2020, 0, 2010059 Search PubMed.
- Z. Xu, J. Jiang, Q. Zhang, G. Chen, L. Zhou and L. Li, J. Colloid Interface Sci., 2020, 563, 131–138 CrossRef CAS PubMed.
- W. Zhao, J. Liu, Z. Ding, J. Zhang and X. Wang, J. Alloys Compd., 2020, 813, 152234 CrossRef CAS.
- Z. Lou and C. Xue, CrystEngComm, 2016, 18, 8406–8410 RSC.
- R. Zhang, L. Huang, Z. Yu, R. Jiang, Y. Hou, L. Sun, B. Zhang, Y. Huang, B. Ye and Y. Zhang, Electrochim. Acta, 2019, 323, 134845 CrossRef CAS.
- D. Ren, Z. Liang, Y. H. Ng, P. Zhang, Q. Xiang and X. Li, Chem. Eng. J., 2020, 390, 124496 CrossRef CAS.
- H. J. Cheng, J. G. Hou, O. Takeda, X. M. Guo and H. M. Zhu, J. Mater. Chem. A, 2015, 3, 11006–11013 RSC.
- D. He, Z. Zhang, Y. Xing, Y. Zhou, H. Yang, H. Liu, J. Qu, X. Yuan, J. Guan and Y.-N. Zhang, Chem. Eng. J., 2020, 384, 123258 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
-
Y. X. Xing, H. Zhu, G. F. Chang, M. Yu, M. Zhao, F. L. Yue and Iop, 2018 4th International Conference on Environmental Science and Material Application, 2019, vol. 252.
- G. A. da Collina, F. Freire, V. D. Barbosa, C. B. Correa, H. R. Nascimento, A. C. R. T. Horliana, D. D. T. da Silva, R. A. Prates and C. Pavani, Photodiagn. Photodyn. Ther., 2020, 29, 101612 CrossRef PubMed.
- N. Maldonado-Carmona, T. S. Ouk, M. J. F. Calvete, M. M. Pereira, N. Villandier and S. Leroy-Lhez, Photochem. Photobiol. Sci., 2020, 19, 445–461 CrossRef CAS PubMed.
- R. Santus, Pathol. Biol., 1991, 39, 54–58 CAS.
- G. K. Couto, B. S. Pacheco, V. M. Borba, J. C. Rodrigues, T. L. Oliveira, N. V. Segatto, F. K. Seixas, T. V. Acunha, B. A. Iglesias and T. Collares, J. Photochem. Photobiol., B, 2020, 202, 111725 CrossRef CAS PubMed.
- M. Q. Yang, J. R. Deng, D. Guo, J. Zhang, L. X. Yang and F. S. Wu, Org. Biomol. Chem., 2020, 18, 569 RSC.
- W. Zhu, Y. H. Gao, P. Y. Liao, D. Y. Chen, N. N. Sun, P. A. N. Thi, Y. J. Yan, X. F. Wu and Z. L. Chen, Eur. J. Med. Chem., 2018, 160, 146–156 CrossRef CAS PubMed.
- C. Xu, W. Hu, N. Zhang, Y. Qi, J.-J. Nie, N. Zhao, B. Yu and F.-J. Xu, Biomaterials, 2020, 248, 120031 CrossRef CAS PubMed.
- A. Sadiki, E. M. Kercher, H. B. Lu, R. T. Lang, B. Q. Spring and Z. S. Zhou, Photochem. Photobiol., 2020, 96, 596–603 CrossRef CAS PubMed.
- Y. L. He, S. J. Wang, L. W. Zhang, J. Xin, J. Wang, C. P. Yao, Z. X. Zhang and C. C. Yang, J. Biomed. Opt., 2016, 21, 9 CrossRef PubMed.
- N. Zhang, L. Wen, J. Yan and Y. Liu, Chem. Pap., 2020, 74, 389–406 CrossRef CAS.
- O. Bettucci, T. Skaltsas, M. Calamante, A. Dessì, M. Bartolini, A. Sinicropi, J. Filippi, G. Reginato, A. Mordini, P. Fornasiero and L. Zani, ACS Appl. Energy Mater., 2019, 2, 5600–5612 CrossRef CAS.
- X. H. Zhang, T. Y. Peng and S. S. Song, J. Mater. Chem. A, 2016, 4, 2365–2402 RSC.
- Y. Li, C. Xie, S. Peng, G. Lu and S. Li, J. Mol. Catal. A: Chem., 2008, 282, 117–123 CrossRef CAS.
- Q. Li and G. Lu, J. Mol. Catal. A: Chem., 2007, 266, 75–79 CrossRef CAS.
- Y. Li, M. Guo, S. Peng, G. Lu and S. Li, Int. J. Hydrogen Energy, 2009, 34, 5629–5636 CrossRef CAS.
- T. Shimidzu, T. Iyoda and Y. Koide, J. Am. Chem. Soc., 1985, 107, 35–41 CrossRef CAS.
- Z. Jin, X. Zhang, G. Lu and S. Li, J. Mol. Catal. A: Chem., 2006, 259, 275–280 CrossRef CAS.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS PubMed.
- J. Xu, Y. Li and S. Peng, Int. J. Hydrogen Energy, 2015, 40, 353–362 CrossRef CAS.
- C. Kong, S. Min and G. Lu, Int. J. Hydrogen Energy, 2014, 39, 4836–4844 CrossRef CAS.
- C. Kong, S. Min and G. Lu, ACS Catal., 2014, 4, 2763–2769 CrossRef CAS.
- X. Zhang, L. Yu, C. Zhuang, T. Peng, R. Li and X. Li, ACS Catal., 2014, 4, 162–170 CrossRef CAS.
- X. Zhang, T. Peng, L. Yu, R. Li, Q. Li and Z. Li, ACS Catal., 2015, 5, 504–510 CrossRef CAS.
- K. Hirano, E. Suzuki, A. Ishikawa, T. Moroi, H. Shiroishi and M. Kaneko, J. Photochem. Photobiol., A, 2000, 136, 157–161 CrossRef CAS.
- K. Hirano, E. Suzuki, A. Ishikawa, T. Moroi, H. Shiroishi and M. Kaneko, J. Photochem. Photobiol., A, 2000, 136, 157–161 CrossRef CAS.
- D. N. Furlong, D. Wells and W. H. F. Sasse, J. Phys. Chem., 1986, 90, 1107–1115 CrossRef CAS.
- P. Chanhom, N. Charoenlap, C. Manipuntee and N. Insin, J. Magn. Magn. Mater., 2019, 475, 602–610 CrossRef CAS.
- M. Giridhar, H. S. BhojyaNaik, R. Vishwanath, C. N. Sudhamani, M. C. Prabakar and R. Kenchappa, Mater. Today: Proc., 2017, 4, 11671–11678 Search PubMed.
- L. Zan, S. Wang, W. Fa, Y. Hu, L. Tian and K. Deng, Polymer, 2006, 47, 8155–8162 CrossRef CAS.
-
Y. Xing, H. Zhu, G. Chang, M. Yu, M. Zhao, F. Yue and Iop, in 2018 4th International Conference on Environmental Science and Material Application, 2019, vol. 252.
- A. Zyoud, M. Dwikat, S. Al-Shakhshir, S. Ateeq, J. Shteiwi, A. Zu'bi, M. H. S. Helal, G. Campet, D. Park, H. Kwon, T. W. Kim, M. Kharoof, R. Shawahna and H. S. Hilal, J. Photochem. Photobiol., A, 2016, 328, 207–216 CrossRef CAS.
- M. A. Khan, S. Mutahir, F. Wang, J. W. Zhou, W. Lei and M. Xia, Sol. Energy, 2019, 186, 204–214 CrossRef CAS.
- X. Yang, D. Wang, Y. Shi, J. Zou, Q. Zhao, Q. Zhang, W. Huang, J. Shao, X. Xie and X. Dong, ACS Appl. Mater. Interfaces, 2018, 10, 12431–12440 CrossRef CAS PubMed.
- M. Giridhar, H. S. BhojyaNaik, R. Vishwanath, C. N. Sudhamani, M. C. Prabakar and R. Kenchappa, Mater. Today: Proc., 2017, 4, 11671–11678 Search PubMed.
- A. Galstyan, Chem. – Eur. J., 2020, 27, 1903–1920 CrossRef PubMed.
- C. Yan, X. Shao, Q. Shu, Y. Teng, Y. Qiao, P. Guan, X. Hu and C. Wang, Int. J. Biol. Macromol., 2021, 186, 839–848 CrossRef CAS PubMed.
- A. C. Bevilacqua, M. H. Kohler, B. A. Iglesia and P. C. Piquini, Comput. Mater. Sci., 2019, 158, 228–234 CrossRef CAS.
- Y. Sun, Y. L. Sun, C. Dall'Agnese, X. F. Wang, G. Chen, O. Kitao, H. Tamiaki, K. Sakai, T. Ikeuchi and S. Sasaki, ACS Appl. Energy Mater., 2018, 1, 2813–2820 CrossRef CAS.
- Y. Jiao, J.-F. Xu, Z. Wang and X. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 22635–22640 CrossRef CAS PubMed.
- J. H. van Wonderen, D. Li, S. E. H. Piper, C. Y. Lau, L. P. Jenner, C. R. Hall, T. A. Clarke, N. J. Watmough and J. N. Butt, ChemBioChem, 2018, 19, 2206–2215 CrossRef CAS PubMed.
- A. H. Zyoud, F. Saleh, M. H. Helal, R. Shawahna and H. S. Hilal, J. Nanomater., 2018, 2018, 2789616 Search PubMed.
-
Y. X. Xing, H. Zhu, G. F. Chang, M. Yu, M. Zhao and F. L. Yue, 2018 4th International Conference on Environmental Science and Material Application, 2019, vol. 252.
- M. Hosseini-Sarvari, M. Koohgard, S. Firoozi, A. Mohajeri and H. Tavakolian, New J. Chem., 2018, 42, 6880–6888 RSC.
- K. S. Yao, D. Y. Wang, C. Y. Chang, W. Y. Ho and L. Y. Yang, J. Nanosci. Nanotechnol., 2008, 8, 2699–2702 CrossRef CAS PubMed.
- Y. W. Zhao, Q. W. Shang, J. C. Yu, Y. J. Zhang and S. Q. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 11783–11791 CrossRef CAS PubMed.
- H. H. Mohamed, I. Hammami, S. Akhtar and T. E. Youssef, Composites, Part B, 2019, 176, 107314 CrossRef CAS.
- J. T. P. Matshwele, F. Nareetsile, D. Mapolelo, P. Matshameko, M. Leteane, D. O. Nkwe and S. Odisitse, J. Chem., 2020, 2020, 2150419 Search PubMed.
- N. Nambagari, S. Perka, R. K. Vuradi and S. Satyanarayana, Nucleosides, Nucleotides Nucleic Acids, 2019, 38, 400–417 CrossRef CAS PubMed.
- Q. Y. Yi, W. Y. Zhang, M. He, F. Du, X. Z. Wang, Y. J. Wang, Y. Y. Gu, L. Bai and Y. J. Liu, J. Biol. Inorg. Chem., 2019, 24, 151–169 CrossRef CAS PubMed.
- J. Lu, Z. Li, W. An, L. Liu and W. Cui, Nanomaterials, 2019, 9(9), 1321 CrossRef CAS PubMed.
- Y. Tian, K. E. Martin, J. Y. T. Shelnutt, L. Evans, T. Busani, J. E. Miller, C. J. Medforth and J. A. Shelnutt, Chem. Commun., 2011, 47, 6069–6071 RSC.
- N. N. Zhang, L. Wen, J. Y. Yan and Y. Liu, Chem. Pap., 2020, 74, 389–406 CrossRef CAS.
- Y. Zhu, Y. Huang, Q. Li, D. Zang, J. Gu, Y. Tang and Y. Wei, Inorg. Chem., 2020, 59, 2575–2583 CrossRef CAS PubMed.
- T. Kim, S. Ham, S. H. Lee, Y. Hong and D. Kim, Nanoscale, 2018, 10, 16438–16446 RSC.
- S. Saraswat, A. Desireddy, D. Zheng, L. Guo, H. P. Lu, T. P. Bigioni and D. Isailovic, J. Phys. Chem. C, 2011, 115, 17587–17593 CrossRef CAS.
- H. V. R. Maraka, R. M. Al-Shammari, N. Al-Attar, S. G. Lopez, T. E. Keyes and J. H. Rice, Mater. Res. Express, 2014, 1, 045038 CrossRef CAS.
- S. P. Wang, W. Lin, X. Wang, T. Y. Cen, H. Xie, J. Huang, B. Y. Zhu, Z. Zhang, A. Song, J. Hao, J. Wu and S. Li, Nat. Commun., 2019, 10, 1399 CrossRef PubMed.
- Z. Wang, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126, 15954–15955 CrossRef CAS PubMed.
- S. Verma, A. Ghosh, A. Ghosh and H. N. Ghosh, Chem. – Eur. J., 2011, 17, 3458–3464 CrossRef CAS PubMed.
- C. Costa-Coquelard, S. Sorgues and L. Ruhlmann, J. Phys. Chem. A, 2010, 114, 6394–6400 CrossRef CAS PubMed.
- Y. L. He, S. Wang, L. Zhang, J. Xin, J. Wang, C. Yao, Z. Zhang and C. C. Yang, J. Biomed. Opt., 2016, 21, 128001 CrossRef PubMed.
- Y. He, J. Xiong, C. Yao, W. Jing and Z. Zhang, Proc. SPIE, 2015, 9449, 944910 CrossRef.
- Y. Zhao, Q. Shang, J. Yu, Y. Zhang and S. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 11783–11791 CrossRef CAS PubMed.
- C. Q. Zhao, F. U. Rehman, Y. L. Yang, X. Q. Li, D. Zhang, H. Jiang, M. Selke, X. M. Wang and C. Y. Liu, Sci. Rep., 2015, 5, 11518 CrossRef PubMed.
- C. Q. Zhao, F. U. Rehman, H. Jiang, M. Selke, X. M. Wang and C. Y. Liu, Sci. China: Chem., 2016, 59, 637–642 CrossRef CAS.
- F. U. Rehman, C. Q. Zhao, C. Y. Wu, H. Jiang, M. Selke and X. M. Wang, RSC Adv., 2015, 5, 107285 RSC.
- F. U. Rehman, C. Q. Zhao, C. Y. Wu, X. Q. Li, H. Jiang, M. Selke and X. M. Wang, Nano Res., 2016, 9, 3305–3321 CrossRef CAS.
- F. U. Rehman, C. Q. Zhao, H. Jiang, M. Selke and X. M. Wang, Photodiagn. Photodyn. Ther., 2016, 13, 267–275 CrossRef CAS PubMed.
- F. U. Rehman, C. Q. Zhao, H. Jiang, M. Selke and X. M. Wang, J. Nanosci. Nanotechnol., 2016, 16, 12691–12694 CrossRef CAS.
- J. Schindler, Y. Zhang, P. Traber, J. F. Lefebvre, S. Kupfer, M. Demeunynck, S. Grafe, M. Chavarot-Kerlidou and B. Dietzek, J. Phys. Chem. C, 2018, 122, 83–95 CrossRef CAS.
- B. Tambosco, K. Segura, C. Seyrig, D. Cabrera, M. Port, C. Ferroud and Z. Amara, ACS Catal., 2018, 8, 4383–4389 CrossRef CAS.
- D. van der Westhuizen, K. G. von Eschwege and J. Conradie, Data Brief, 2019, 27, 104759 CrossRef PubMed.
- T. P. Yoon, Acc. Chem. Res., 2016, 49, 2307–2315 CrossRef CAS PubMed.
- J. D. Knoll, S. M. Arachchige and K. J. Brewer, ChemSusChem, 2011, 4, 252–261 CAS.
- K. Murata, M. Araki, A. Inagaki and M. Akita, Dalton Trans., 2013, 42, 6989–7001 RSC.
- Y. Sasaki, H. Kato and A. Kudo, J. Am. Chem. Soc., 2013, 135, 5441–5449 CrossRef CAS PubMed.
- J. E. Martin, E. J. Hart, A. W. Adamson, H. Gafney and J. Halpern, J. Am. Chem. Soc., 1972, 94, 9238–9240 CrossRef CAS.
-
S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, in Photochemistry and Photophysics of Coordination Compounds I, ed. V. Balzani and S. Campagna, Springer Berlin Heidelberg, Berlin, Heidelberg, 2007, pp. 117–214 DOI:10.1007/128_2007_133.
- J. K. McCusker, Acc. Chem. Res., 2003, 36, 876–887 CrossRef CAS PubMed.
- M. Kuss-Petermann and O. S. Wenger, Chem. – Eur. J., 2017, 23, 10808–10814 CrossRef CAS PubMed.
- R. Konduri, H. Ye, F. M. MacDonnell, S. Serroni, S. Campagna and K. Rajeshwar, Angew. Chem., Int. Ed., 2002, 41, 3185–3187 CrossRef CAS PubMed.
- J. I. Goldsmith, W. R. Hudson, M. S. Lowry, T. H. Anderson and S. Bernhard, J. Am. Chem. Soc., 2005, 127, 7502–7510 CrossRef CAS PubMed.
- H. P. Liang, A. Acharjya, D. A. Anito, S. Vogl, T. X. Wang, A. Thomas and B. H. Han, ACS Catal., 2019, 9, 3959–3968 CrossRef CAS.
- W. F. Tian, Y. Q. He, X. R. Song, H. X. Ding, J. Ye, W. J. Guo and Q. Xiao, Adv. Synth. Catal., 2020, 362, 1032–1038 CrossRef CAS.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- B. G. Tweedy, Phytopathology, 1964, 54, 910–914 Search PubMed.
-
B. Norden, P. Lincoln, B. Akerman and E. Tuite, in Metal Ions in Biological Systems, Vol 33: Probing of Nucleic Acids by Metal Ion Complexes of Small Molecules, ed. A. Sigel and H. Sigel, 1996, vol. 33, pp. 177–252 Search PubMed.
- A. D. Richards and A. Rodger, Chem. Soc. Rev., 2007, 36, 471–483 RSC.
- H. Zhang, Med. Chem., 2016, 6 DOI:10.4172/2161-0444.1000338.
- N. E. A. El-Gamel and A. M. Fekry, Bioelectrochemistry, 2015, 104, 35–43 CrossRef CAS PubMed.
- F. F. Li, J. G. Collins and F. R. Keene, Chem. Soc. Rev., 2015, 44, 2529–2542 RSC.
- S. Li, C. Wu, X. Tang, S. Gao, X. Zhao, H. Yan and X. Wang, Sci. China: Chem., 2013, 56, 595–603 CrossRef CAS.
- A. R. Battersby, C. J. R. Fookes, G. W. J. Matcham and E. McDonald, Nature, 1980, 285, 17–21 CrossRef CAS PubMed.
- L. Cheng, R. S. Assary, X. H. Qu, A. Jain, S. P. Ong, N. N. Rajput, K. Persson and L. A. Curtiss, J. Phys. Chem. Lett., 2015, 6, 283–291 CrossRef CAS PubMed.
- L. Castro and M. Buhl, J. Chem. Theory Comput., 2014, 10, 243–251 CrossRef CAS PubMed.
- G. Hautier, A. Jain and S. P. Ong, J. Mater. Sci., 2012, 47, 7317–7340 CrossRef CAS.
- N. S. Soukos, L. A. Ximenez-Fyvie, M. R. Hamblin, S. S. Socransky and T. Hasan, Antimicrob. Agents Chemother., 1998, 42, 2595 CrossRef CAS PubMed.
- S. Ferro, F. Ricchelli, G. Mancini, G. Tognon and G. Jori, J. Photochem. Photobiol., B, 2006, 83, 98–104 CrossRef CAS PubMed.
- A. V. Kustov, D. V. Belykh, N. L. Smirnova, E. A. Venediktov, T. V. Kudayarova, S. O. Kruchin, I. S. Khudyaeva and D. B. Berezin, Dyes Pigm., 2018, 149, 553–559 CrossRef CAS.
- E. A. Groisman, Trends Microbiol., 1994, 2, 444–449 CrossRef CAS PubMed.
- M. Vaara and T. Vaara, Antimicrob. Agents Chemother., 1983, 24, 107–113 CrossRef CAS PubMed.
- P. Giege, J. M. Grienenberger and G. Bonnard, Mitochondrion, 2008, 8, 61–73 CrossRef CAS PubMed.
- N. C. Robinson, J. Bioenerg. Biomembr., 1993, 25, 153–163 CrossRef CAS PubMed.
- G. Y. Jiang, W. H. Lei, Y. J. Hou and X. S. Wang, New J. Chem., 2012, 36, 2180–2183 RSC.
- M. F. B. Vázquez, L. R. Comini, J. M. Milanesio, S. C. N. Montoya, J. L. Cabrera, S. Bottini and R. E. Martini, J. Supercrit. Fluids, 2015, 101, 170–175 CrossRef.
- Y. Wang, W. H. Chen, G. Y. Chen, X. P. Song, D. S. Zhang, B. Fu and Y. Chen, Chin. J. Org. Chem., 2014, 34, 522–525 CrossRef CAS.
- S. C. N. Montoya, A. M. Agnese, C. Perez, I. N. Tiraboschi and J. L. Cabrera, Phytomedicine, 2003, 10, 569–574 CrossRef PubMed.
- L. R. Comini, S. C. Núñez Montoya, P. L. Páez, G. A. Argüello, I. Albesa and J. L. Cabrera, J. Photochem. Photobiol., B, 2011, 102, 108–114 CrossRef CAS PubMed.
-
J. Y. Shin, Y. S. Kim, H. S. Cho and S. H. Lee, US Pat., KR2017114738-A, 2017 Search PubMed.
- B. A. S. Reyes, E. C. Dufourt, J. Ross, M. J. Warner, N. C. Tanquilut and A. B. Leung, Stud. Nat. Prod. Chem., 2018, 55, 111–143 Search PubMed.
-
E. Lima, M. J. Martínez-Ortiz, E. Fregoso and J. Méndez-Vivar, in Studies in Surface Science and Catalysis, ed. R. Xu, Z. Gao, J. Chen and W. Yan, Elsevier, 2007, vol. 170, pp. 2110–2115 Search PubMed.
- W. A. Dhafina, M. Z. Daud and H. Salleh, Optik, 2020, 207, 163808 CrossRef CAS.
- C. Diaz-Uribe, W. Vallejo, G. Camargo, A. Munoz-Acevedo, C. Quinones, E. Schott and X. Zarate, J. Photochem. Photobiol., A, 2019, 384, 112050 CrossRef CAS.
- L. Cui, J. J. Hu, W. Wang, C. Yan, Y. Guo and C. Tu, Cellulose, 2020, 27, 6367–6381 CrossRef CAS.
- Q. Q. Yang, A. K. Farha, G. Kim, K. Gul, R. Y. Gan and H. Corke, Trends Food Sci. Technol., 2020, 97, 341–354 CrossRef CAS.
- L. Sobotta, P. Skupin-Mrugalska, J. Piskorz and J. Mielcarek, Eur. J. Med. Chem., 2019, 175, 72–106 CrossRef CAS PubMed.
- J. Oyim, C. A. Omolo and E. K. Amuhaya, Front. Chem., 2021, 384, 112050 Search PubMed.
- X. Lu, R. Chen, J. Lv, W. Xu, H. Chen, Z. Ma, S. Huang, S. Li, H. Liu, J. Hu and L. Nie, Acta Biomater., 2019, 99, 363–372 CrossRef CAS PubMed.
- R. Lood, K. W. Waldetoft and P. Nordenfelt, Future Microbiol., 2015, 10, 1659–1668 CrossRef CAS PubMed.
- C. Zhang, Y. Li, D. Shuai, Y. Shen and D. Wang, Chem. Eng. J., 2019, 355, 399–415 CrossRef CAS.
- S. Archana, K. Y. Kumar, B. K. Jayanna, S. Olivera, A. Anand, M. K. Prashanth and H. B. Muralidhara, J. Sci.: Adv. Mater. Devices, 2018, 3, 167–174 Search PubMed.
- J.-D. Xiao and H.-L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS PubMed.
- S. Kumar, T. Surendar, B. Kumar, A. Baruah and V. Shanker, J. Phys. Chem. C, 2013, 117, 26135–26143 CrossRef CAS.
- Y. F. Cheng, Y. H. Mei, G. Sathishkumar, Z. S. Lu, C. M. Li, F. Wang, Q. Y. Xia and L. Q. Xu, Colloids Interface Sci. Commun., 2020, 35, 100241 CrossRef CAS.
- F. Aldeek, C. Mustin, L. Balan, T. Roques-Carmes, M.-P. Fontaine-Aupart and R. Schneider, Biomaterials, 2011, 32, 5459–5470 CrossRef CAS PubMed.
- G. Yang, P. Li, L. Hu, X. Meng, Y. Wang and H. Xu, Microbiology, 2019, 46, 1136–1145 Search PubMed.
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2021 |

 a and
Xuemei
Wang
a and
Xuemei
Wang