
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating the role of non-covalent interactions in unexpectedly high and selective CO2 uptake and catalytic conversion of porphyrin-based ionic organic polymers†
Sinem T.
Kostakoğlu
a,
Yurii
Chumakov
bc,
Yunus
Zorlu
 a,
Ali E.
Sadak
d,
Serpil
Denizaltı
e,
Ayşe G.
Gürek
a and
Mehmet M.
Ayhan
*a
a,
Ali E.
Sadak
d,
Serpil
Denizaltı
e,
Ayşe G.
Gürek
a and
Mehmet M.
Ayhan
*a
aGebze Technical University, Department of Chemistry, Kocaeli, 41400, Turkey. E-mail: menafayhan@gtu.edu.tr
bGebze Technical University, Department of Physics, Kocaeli, 41400, Turkey
cInstitute of Applied Physics, MD-2028, Chisinau, Moldova
dTUBITAK UME, Chemistry Group Laboratories, TR-41470, Gebze-Kocaeli, Turkey
eEge University, Department of Chemistry, Bornova, İzmir, Turkey
First published on 12th April 2021
Abstract
Here, we present viologen-porphyrin based ionic covalent organic polymers (H2-ICOP and Zn-ICOP) with multiple CO2-philic sites. The specific surface areas of H2-ICOP and Zn-ICOP were found to be 9 m2 g−1 and 20 m2 g−1, respectively. CO2 uptake analyses reveal that H2-ICOP exhibits very high CO2 capture uptake (62.9 mg g−1), which is one of the highest values among previously reported ICOPs. The results indicate very efficient non-covalent interactions between H2-ICOP and CO2. The possible non-covalent interactions of hydrogen (OCO2⋯H–N), tetrel (CCO2⋯N, CCO2⋯Cl−), pnicogen (OCO2⋯N+), and spodium bonds (OCO2⋯Zn) between CO2 and H2-ICOP and Zn-ICOP are investigated via symmetry adapted perturbation theory (SAPT) analysis and electrostatic potential maps (MEP). The strength of non-covalent interactions in H2-ICOP and Zn-ICOP is decreasing in the following order ΔEC⋯N > ΔEC⋯Cl− > ΔEO⋯N+ and ΔEZn⋯O > ΔEC⋯Cl− > ΔEC⋯N > ΔEO⋯N+, respectively. The major CO2 uptake contribution comes from CCO2⋯N tetrel bonding (−22.02 kJ mol−1) interactions for H2-ICOP, whereas OCO2⋯Zn spodium bonding (−21.065 kJ mol−1) interactions for Zn-ICOP. H2-ICOP has more CO2-philic moieties with powerful non-covalent interactions compared to Zn-ICOP, which is in good agreement with the experimental results. Furthermore, the CO2 catalytic conversion performances of Zn-ICOP and H2-ICOP gave good yields of 83% and 54%, respectively. Surprisingly, Zn-ICOP, despite having significantly lower CO2 uptake capacity, displayed better catalytic activity than H2-ICOP, owing to a higher number of counter anions (Cl−) on its surface, which shows the crucial role of the counter anion (Cl−) in the mechanism of this catalytic reaction.
1. Introduction
The rise of the atmospheric carbon dioxide (CO2) concentration has become a widespread concern due to its link to the greenhouse effect and threat to the environment.1 Therefore, viable solutions for efficient capture and utilization of CO2 (CCU) have received much attention in the past decades.2,3 Among various CO2 removal technologies, physical adsorbents such as zeolites, metal–organic frameworks (MOFs), covalent organic frameworks (COFs) and porous covalent organic polymers (COPs) came into focus owing to their high CO2 capture capacities, catalytic transformation, and low energy requirements for regeneration.4–8 Especially COPs have evolved for CCU due to their high specific surface areas, easily tunable pore structures, good chemical and thermal stabilities, tailored surface manipulation with polar groups, and variation in the synthesis.9–13A high selective uptake of CO2 is known to arise from large surface areas and non-covalent interactions between CO2 and surfaces of materials. In the past decade, significant effort has been mainly focused on increasing the surface areas of materials and the non-covalent interactions between CO2 and surfaces of materials have not attracted enough attention.14 One should note that a large surface area is one of the parameters, but not the only one, that allows for high CO2 capture. However, materials with a low surface area may still exhibit large CO2 capture capacity with efficient non-covalent interactions with CO2. Therefore, it is vitally important to identify and estimate the strength of these non-covalent interactions in terms of the contribution to the CO2 capture capacity.15,16 CO2 is known to be able to form simultaneous non-covalent interactions via its electron-rich terminal oxygen atoms and its electron-deficient central carbon atom.16,17 The electron-deficient central carbon atom acts as a Lewis acid (LA) and can form tetrel bonding interactions with anions or lone-pair-possessing atoms such as N, O, S, and F.18–21 On the other hand, the electron-rich terminal oxygen atom acts as a Lewis base (LB) and can form hydrogen bonding with hydrogen atoms and, as recently reported, halogen bonding with halogen atom (I, Br, Cl) containing moieties.22–29
In particular, electron-rich nitrogen-based functional groups, such as amines, triazoles, triazine cores, imines, tetrazoles, benzimidazoles, azo linkages, and viologens, continue to be a dominant polar functional group to enhance the CO2 adsorption capacity and CO2/N2 selectivity thanks to their multiple non-covalent interactions with CO2.20,27,30–38 Among nitrogen based adsorbents, viologen linked ionic covalent organic polymers (ICOPs) are of particular importance in CCU technologies because they not only have all the advantages of COPs but also possess charged cationic skeletons paired with counter-ions which provide additional CO2 affinity owing to their electrostatic interactions with CO2 molecules.35,39–42 Consequently, the CO2 uptake capacity of viologen linked ICOPs is found to be significantly higher compared to their neutral counterparts.34,43,44 Moreover, unlike other porous materials, selective recognition of viologen linked ICOPs can be manipulated by simply controlling the redox state of viologen or the nature of the counter-ions for the desired application.39,41
In recent years, several cationic viologen linked ICOPs with good surface areas have been reported for CCU.34,39,43–49 The general understanding of these studies is that the high CO2 capture capacity of viologen linked ICOPs is credited mostly to tetrel bonding interactions between the counter anions and CO2.50 Commonly, nitrogen-based groups in CCU applications are preferred for their Lewis basic character to form non-covalent interactions with the electron-deficient central carbon (Lewis acid) of CO2. However, in the case of viologen, the quaternary nitrogen (N+) atom lacks lone electron pairs, and hence acts as a Lewis acid; consequently, the formation of efficient interactions between cation (N+) nitrogen and the electron-rich terminal oxygen atoms of CO2, commonly referred to as pnicogen bonds, is highly possible.19,51 Yet, the pnicogen (OCO2⋯N+) interaction is often disregarded, and its contribution to CO2 capture has not been reported.
However, despite their attractive features for CCU applications, only a limited number of viologen linked ICOPs have been reported in the literature, mainly due to their low physicochemical stability. This drawback can be overcome by the incorporation of viologens into polymeric systems with suitable rigid and symmetric macrocycles such as porphyrin.52 The incorporation of porphyrin brings three advantages. First, unstable cationic species get stabilized by extended delocalized polymeric structures. Second, porphyrin is another excellent N containing molecular building block, which can increase the CO2 uptake and selectivity through the CO2 interaction with pyrroles (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, –NH). Third, the metalation of the porphyrin core with Lewis acid metals such as zinc(II) can promote additional non-covalent interactions with the electron-rich terminal oxygen atoms of CO2, known as spodium bonding.53 In addition to their multiple CO2-philic functional sides, both viologen and porphyrin are also known to exhibit effective CO2 catalytic conversion, which makes them very promising bifunctional materials in CCU applications.46–48,54–56
N, –NH). Third, the metalation of the porphyrin core with Lewis acid metals such as zinc(II) can promote additional non-covalent interactions with the electron-rich terminal oxygen atoms of CO2, known as spodium bonding.53 In addition to their multiple CO2-philic functional sides, both viologen and porphyrin are also known to exhibit effective CO2 catalytic conversion, which makes them very promising bifunctional materials in CCU applications.46–48,54–56
Herein, based on the above considerations, we report viologen-linked porphyrin ICOPs (H2-ICOP and Zn-ICOP) synthesized through the Zincke reaction of H2 and Zn(II) tetrakis(4-aminophenyl) porphyrin with viologen Zincke salt, as presented in Scheme 1.
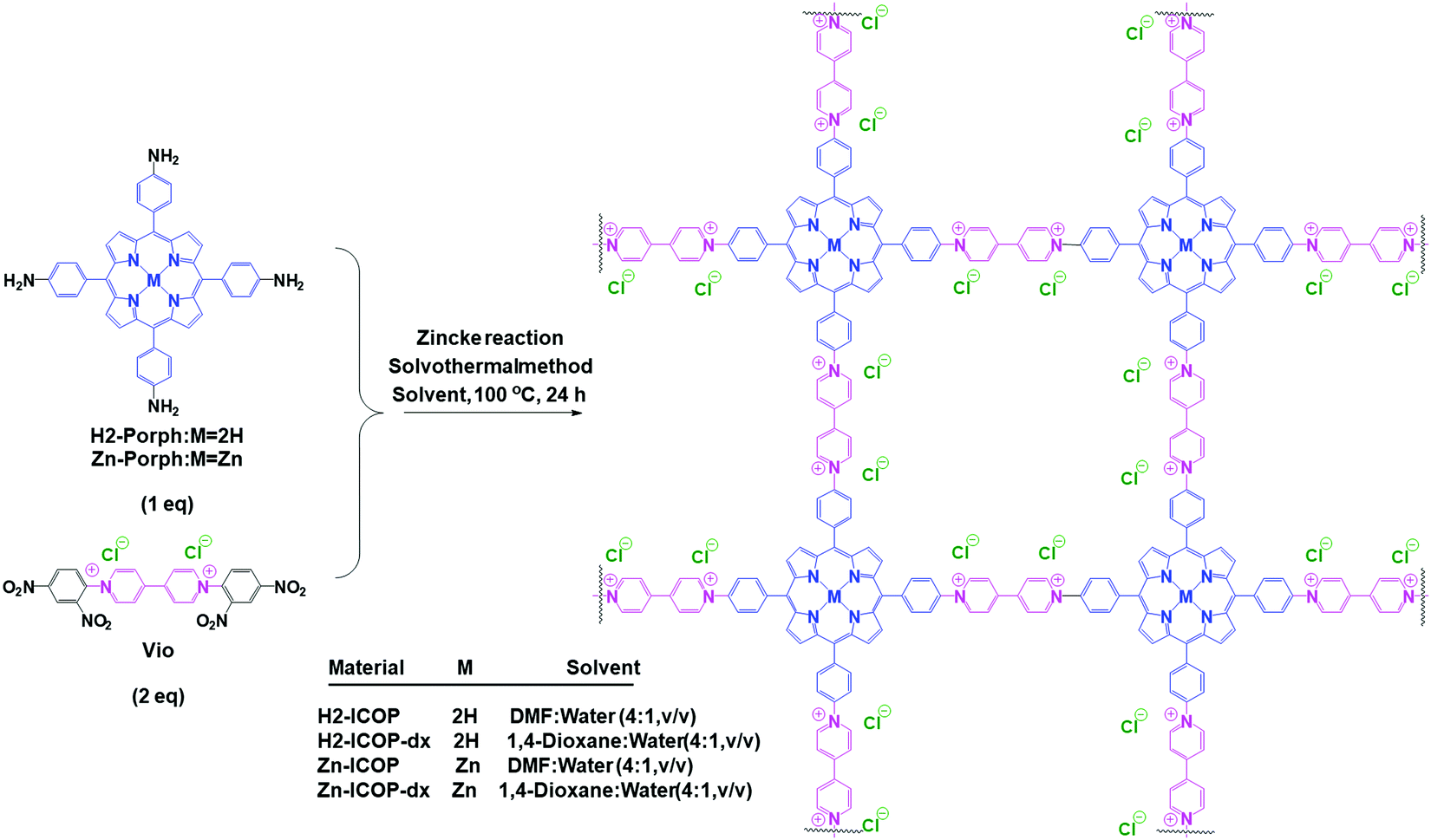 | ||
| Scheme 1 Schematic representation of the synthesis of the viologen-linked porphyrin ICOPs via a Zincke reaction under solvothermal conditions. | ||
The resultant ICOPs were characterized with various experimental analyses including FTIR spectroscopy, solid-state 13C NMR spectroscopy, energy-dispersive X-ray spectroscopy (EDX), diffuse reflectance spectroscopy (DRS), thermogravimetric analysis (TGA), powder X-ray diffraction (PXRD), field emission scanning electron microscopy (SEM), transmission electron microscopy (TEM) and dynamic light scattering (DLS). Furthermore, their CO2 capture capacity, CO2/N2 selectivity, and CO2 catalytic conversion performances were explored. Brunauer–Emmett–Teller (BET) surface area analyses reveal that both H2-ICOP (9 m2 g−1) and Zn-ICOP (20 m2 g−1) possess very low surface areas. Yet, despite their nearly non-porous surfaces, especially H2-ICOP exhibits a very large CO2 capture capacity (62.9 mg g−1) and CO2/N2 selectivity, which indicates powerful non-covalent interactions with CO2. Detailed theoretical analyses have been conducted to decode insights into these unexpected large CO2 capture capacities and CO2/N2 selectivity. The strengths of possible non-covalent interactions, such as hydrogen, tetrel, pnicogen, and spodium bonding, between CO2 molecules and the H2-ICOP and Zn-ICOP surfaces were calculated, in terms of the contribution to the CO2 capture capacity. Additionally, the CO2 catalytic conversion performances of Zn-ICOP and H2-ICOP gave good yields of 83% and 54% in mild conditions, respectively.
2. Results and discussion
2.1. Synthesis and characterization
The synthesis of the ionic covalent organic polymers (H2-ICOP and Zn-ICOP) was carried out under solvothermal conditions by the Zincke reaction of 1,1′-bis(2,4-dinitrophenyl)-[4,4′-bipyridine]-1,1′-diium dichloride (Vio) with either 5,10,15,20-tetrakis(4-aminophenyl)porphyrin (H2-Porph) or 5,10,15,20-tetrakis(4-aminophenyl)porphyrinato zinc(II) (Zn-Porph) in a mixture of 1,4-dioxane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (4:1, v/v) or DMF:water (4:1, v/v) (Scheme 1, for details see the ESI†). A gel-like material was obtained in the DMF:water mixture, whereas a powder was obtained in the 1,4-dioxane:water mixture (Fig. S1, ESI†). Washing the solids with the appropriate solvents is found to be crucial to remove unreacted starting materials and any smaller oligomers that may have formed. Therefore, the solids were washed with ethanol and DMSO several times until a colourless solution was obtained. The purification procedure was completed by washing with water and acetone. Complementary methods were used for the detailed structural characterization of the ICOPs. The formation of the ICOPs is confirmed by Fourier-transform infrared (FTIR) spectroscopy and solid-state 13C NMR. In FTIR (Fig. 1), the decline of the characteristic –NH2 peak at 3345 cm−1 and –NO2 peak at 1343 cm−1 and the appearance of the characteristic pyridinium CN peak at 1628 cm−1 for H2-ICOP and 1656 cm−1 for Zn-ICOP indicate the loss of the 2,4-dinitroaniline group and the formation of viologen-linked ICOPs. Besides, the peaks belonging to the porphyrin ring at 964 and 790 cm−1 for H2-ICOP and at 993 and 794 cm−1 for Zn-ICOP confirm the formation of the expected materials. The same structures are obtained for H2-ICOP-dx and Zn-ICOP-dx with identical FTIR as shown in Fig. S2 (ESI†). The solid-state 13C NMR spectra (Fig. S3, ESI†) of the materials show broad multi-peaks between δ = 100 and 170 ppm. The peaks at 151 ppm for H2-ICOP and 152 ppm for Zn-ICOP can be attributed to the carbon signals in the bipyridinium moieties, whereas the peaks at 122 ppm for H2-ICOP and Zn-ICOP correspond to phenyl carbons of the porphyrin subunit.
:water (4:1, v/v) or DMF:water (4:1, v/v) (Scheme 1, for details see the ESI†). A gel-like material was obtained in the DMF:water mixture, whereas a powder was obtained in the 1,4-dioxane:water mixture (Fig. S1, ESI†). Washing the solids with the appropriate solvents is found to be crucial to remove unreacted starting materials and any smaller oligomers that may have formed. Therefore, the solids were washed with ethanol and DMSO several times until a colourless solution was obtained. The purification procedure was completed by washing with water and acetone. Complementary methods were used for the detailed structural characterization of the ICOPs. The formation of the ICOPs is confirmed by Fourier-transform infrared (FTIR) spectroscopy and solid-state 13C NMR. In FTIR (Fig. 1), the decline of the characteristic –NH2 peak at 3345 cm−1 and –NO2 peak at 1343 cm−1 and the appearance of the characteristic pyridinium CN peak at 1628 cm−1 for H2-ICOP and 1656 cm−1 for Zn-ICOP indicate the loss of the 2,4-dinitroaniline group and the formation of viologen-linked ICOPs. Besides, the peaks belonging to the porphyrin ring at 964 and 790 cm−1 for H2-ICOP and at 993 and 794 cm−1 for Zn-ICOP confirm the formation of the expected materials. The same structures are obtained for H2-ICOP-dx and Zn-ICOP-dx with identical FTIR as shown in Fig. S2 (ESI†). The solid-state 13C NMR spectra (Fig. S3, ESI†) of the materials show broad multi-peaks between δ = 100 and 170 ppm. The peaks at 151 ppm for H2-ICOP and 152 ppm for Zn-ICOP can be attributed to the carbon signals in the bipyridinium moieties, whereas the peaks at 122 ppm for H2-ICOP and Zn-ICOP correspond to phenyl carbons of the porphyrin subunit.
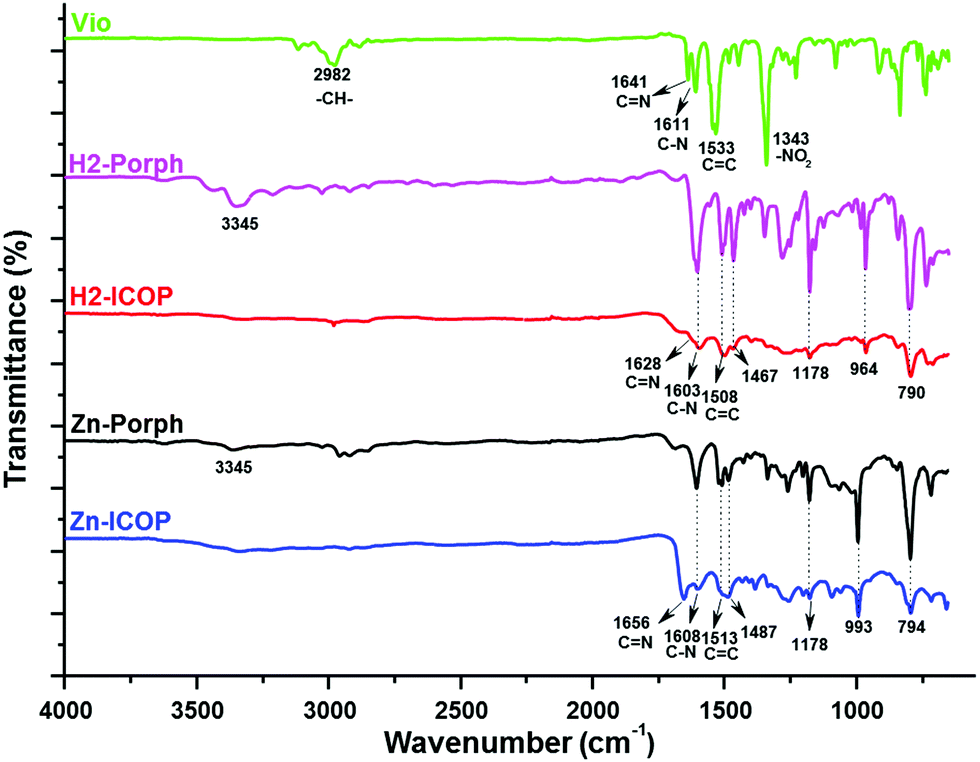 | ||
| Fig. 1 FTIR spectra of H2-ICOP and Zn-ICOP in comparison with the corresponding monomers (H2-Porph, Zn-Porph and Vio). | ||
The diffuse reflectance spectroscopy (DRS) of H2-ICOP and Zn-ICOP displays the characteristic Soret and Q bands of the porphyrin at 436 nm and 535 nm for H2-ICOP, and 447 nm and 538 nm for Zn-ICOP (Fig. 2a). Moreover, characteristics of viologen radical bands were observed at 627 nm for H2-ICOP and 586 nm for Zn-ICOP. H2-ICOP and Zn-ICOP exhibit an electron paramagnetic resonance (EPR) signal (Fig. 2b). The radical character of these ICOPs is the result of charge transfer from the aromatic porphyrin core to the viologen unit and is an indication of the highly conjugated structures.57
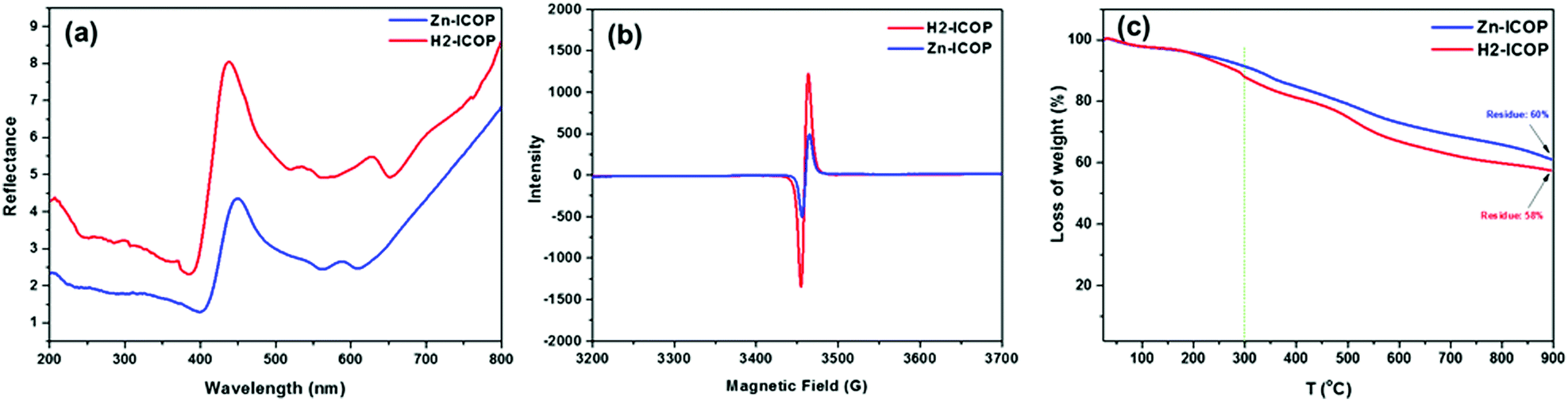 | ||
| Fig. 2 DR spectra (a), EPR signals (b) and TGA curves (c) of H2-ICOP and Zn-ICOP. | ||
Thermogravimetric analysis (TGA) shows that H2-ICOP and Zn-ICOP exhibit thermal stability up to 300 °C with slight weight losses (Fig. 2c). However, it is noteworthy that both materials retain about 60% of their initial mass even at 900 °C, indicating good thermal stability. H2-ICOP and Zn-ICOP have similar experimental PXRD profiles with low angle diffraction peaks in the vicinity of 2θ values of 5°, which is characteristic for expected large unit-cell parameters (Fig. S4, ESI†). However, the broadening of peaks in both profiles in combination with a large peak at ca. 22.5° indicates a semi-crystalline nature of the studied materials. To determine the structures of H2-ICOP and Zn-ICOP, the geometry optimization of constructed models having the eclipsed (AA) and staggered (AB) topology was performed using the ABINIT code using the following unit cell parameters: a = b = 35.563, c = 4.09 (7.8 for the staggered structure) Å, α = β = γ = 90°, space group P1. The comparisons of both the experimental and calculated PXRD profiles based on the relaxed models are presented in Fig. S4a for H2-ICOP and in Fig. S4b (ESI†) for Zn-ICOP. The experimental PXRD profiles of both ICOPs are in good agreement with the simulated staggered AB-stacking model (Fig. S4d, ESI†). Since the PXRD patterns of H2-ICOP-dx and Zn-ICOP-dx showed an amorphous nature rather than a semi-crystalline structure, H2-ICOP and Zn-ICOP were used for further investigations. Elemental mapping by energy dispersive spectroscopy (EDS) exhibits the elements C, N, and Cl for H2-ICOP and C, N, Cl, and Zn for Zn-ICOP (Fig. S5, ESI†), which is consistent with the compositions of our ICOPs. Dynamic light scattering (DLS) analyses showed two particle populations with an average colloidal particle size in the range between 1 and 10 μm for both ICOPs, suggesting a polydisperse nature (Fig. S6, ESI†). The stability of colloidal systems of H2-ICOP and Zn-ICOP was evaluated with zeta potential measurements in water (pH = 7.4) (Fig. S7, ESI†). Interestingly, the zeta potential of H2-ICOP shows a positive value (+25), whereas Zn-ICOP shows a negative zeta potential value (−21.6). These high absolute values of the zeta potential of H2-ICOP and Zn-ICOP indicate a stable state of their colloidal systems, which is also confirmed by observation of the Tyndall effect (Fig. S8, ESI†). Furthermore, the zeta potential data not only reflect the stability of the colloidal systems of H2-ICOP and Zn-ICOP dispersions in water but also can be used to explain the adsorption and conversion mechanisms of CO2, which we will discuss in related sections.
Depending on the solvent mixture used, structures with different morphologies were obtained. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) studies reveal that the ICOPs obtained by using the DMF:water mixture (H2-ICOP and Zn-ICOP) exhibit a sheet-like morphology, whereas the ICOPs obtained by using the 1,4-dioxane:water mixture (H2-ICOP-dx and Zn-ICOP-dx) exhibit a spherical morphology (Fig. 3). Uniform spheres with an average diameter of 1.4 μm for H2-ICOP-dx and 0.65 μm for Zn-ICOP-dx were observed by both SEM (Fig. 3b and d and Fig. S9, ESI†) and TEM analysis (Fig. 3f and h).
 | ||
| Fig. 3 Microscopic characterization [SEM (top row) and TEM (bottom row)] of H2-ICOP (a and e) and Zn-ICOP (c and g) prepared in DMF:water (4:1, v/v) and obtained as plates; and H2-ICOP-dx (b and f) and Zn-ICOP-dx (d and h) prepared in 1,4-dioxane:water (4:1, v/v) and obtained as spheres. | ||
2.2. CO2 uptake and CO2/N2 selectivity performance
To investigate the permanent porosity of H2-ICOP and Zn-ICOP, krypton (Kr) adsorption–desorption isotherms were measured via the Brunauer–Emmett–Teller (BET) model, at 77 K (for details see the ESI†). As shown in Fig. 4b, both H2-ICOP and Zn-ICOP show typical type-II reversible adsorption isotherms and their porosities are found to be mesoporous in the range of 3–20 nm, whereas the nitrogen (N2) adsorption–desorption isotherms show non-porous surfaces. The specific surface areas of H2-ICOP and Zn-ICOP were found to be 9 m2 g−1 and 20 m2 g−1, respectively (Fig. 4a). This difference in the BET surface areas can be explained by the zeta potential values of H2-ICOP (+25) and Zn-ICOP (−21.6) (Fig. S7, ESI†). The positive zeta potential value suggests that the pores are occupied by nearly all of the chloride counter ions and they do not remain on the surface of H2-ICOP, whereas the negative zeta potential value indicates that some chloride counter-ions remain on the surface of Zn-ICOP. Notably, the surface areas of both ICOPs are one of the lowest values reported to date for any ICOP. These relatively low values are likely due to the pores of the materials being occupied by chloride counterions and their semi-crystalline state and staggered morphology with an AB stacking sequence (Fig. S4d, ESI†).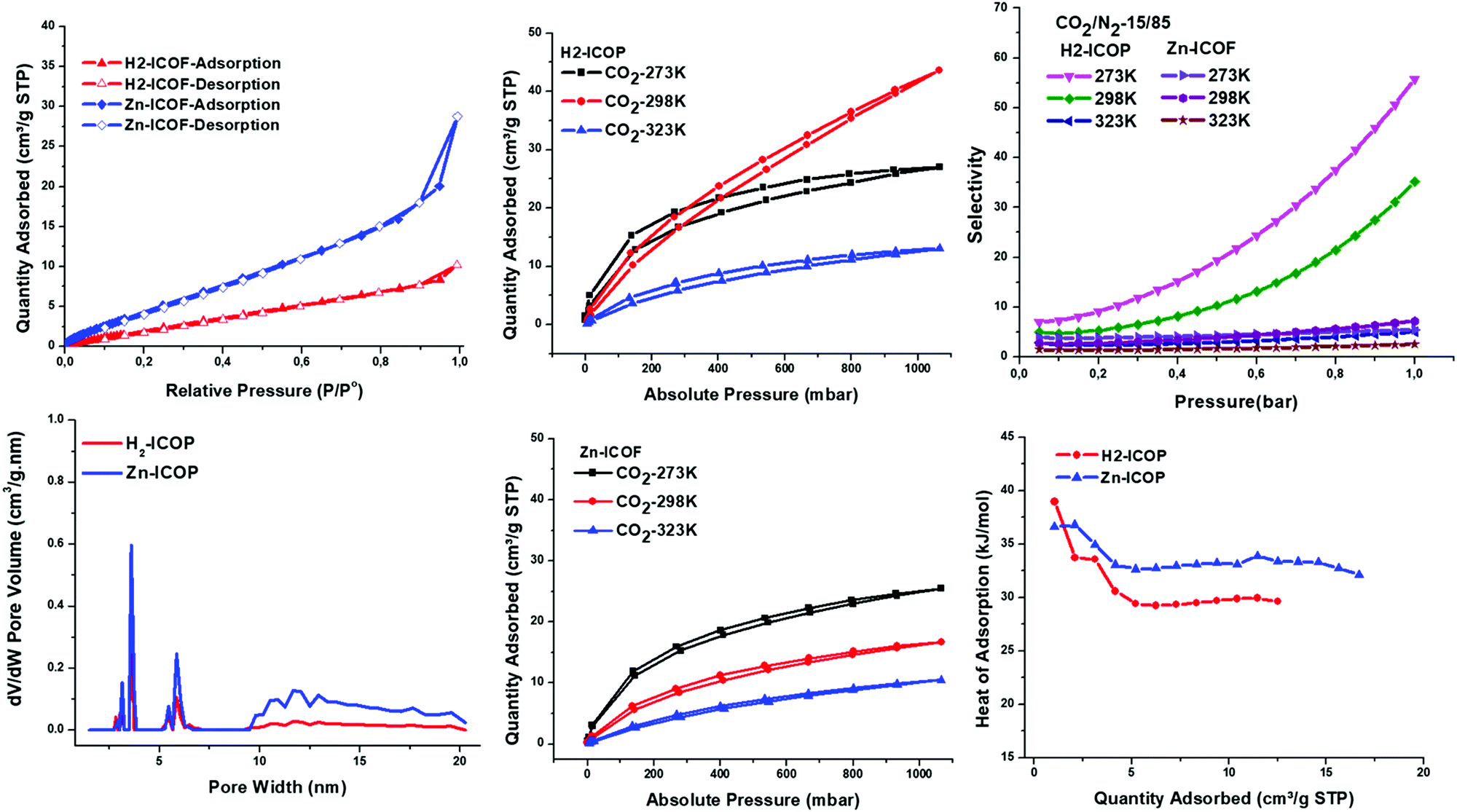 | ||
| Fig. 4 (a) Kr adsoption/desorption isotherms of H2-ICOP and Zn-ICOP measured at 77 K, filled and empty symbols represent adsorption and desorption, respectively. (b) Pore size distribution of H2-ICOP and Zn-ICOP calculated from Kr isotherms calculated using NLDFT. CO2 adsorption isotherms of H2-ICOP (c) and Zn-ICOP (d) collected up to 1 bar at 273, 298, and 323 K. (e) CO2/N2 (IAST method) selectivity of H2-ICOP and Zn-ICOP at 273 K, 298 K, and 323 K. (f) The isosteric heat of adsorption (Qst) plots of CO2 for H2-ICOP and Zn-ICOP. | ||
Owing to their multiple CO2-philic sides, despite their low surface areas, we explored the affinity of H2-ICOP and Zn-ICOP toward CO2 and N2. To evaluate their performance for CO2 capture, we carried out temperature-dependent CO2 uptake measurements at 273, 298, and 323 K up to 1 bar (Fig. 4c and d). The CO2 capture capacity of H2-ICOP, despite the lower BET surface area, is found to be up to 26 (42.4 mg g−1, 273 K), 44 (62.9 mg g−1, 298 K), and 13 cm3 g−1 (17.3 mg g−1, 323 K) at 1 bar, which is much higher than that of Zn-ICOP, 25 (39.3 mg g−1, 273 K), 16.7 (24 mg g−1, 298 K) and 10.2 cm3 g−1 (13.9 mg g−1, 323 K) at 1 bar, respectively. Notably, the CO2 uptake capacities are not saturated for H2-ICOP within the pressure range, suggesting that the CO2 uptake capacities can be further improved with increased pressure. Remarkably, the CO2 capture capacities of H2-ICOP and Zn-ICOP are one of the highest values reported among previously reported viologen based ICOFs and ICOPs with much larger surface areas (Table 1). The CO2 capture capacity of these viologen linked ICOFs and ICOPs is attributed mainly to tetrel bonding interactions between the counter anions and CO2. However, as the positive zeta potential value of H2-ICOP shows that the pores are occupied by nearly all of the chloride counter ions and they do not remain on the surface, yet H2-ICOP has a very high CO2 capture capacity, the results indicate that H2-ICOP, due to our molecular design, possesses much stronger non-covalent interactions with CO2 than the counter anions.
| ICOP | BET surface area (m2 g−1) | CO2 adsorption (mg g−1) | CO2/N2 selectivity (15/85) (IAST) 1 bar | Q st (kJ mol−1) | Pressure (bar) | Ref. | |||
|---|---|---|---|---|---|---|---|---|---|
| 273 K | 298 K | 323 K | 273 K | 298 K | |||||
| H2-ICOP | 9 | 42.4 | 62.9 | 17.3 | 56.5 | 35 | 39 | 1 | This work |
| Zn-ICOP | 20 | 39.3 | 24 | 13.9 | 5.6 | 6.9 | 36 | 1 | This work |
| PCP-Cl | 755 | 101 | 61.4 | — | 34 | — | 28.5 | 1 | 39 |
| V-PCIF-Cl | 174 | 86.7 | 62.4 | — | — | — | 56.6 | 1 | 43 |
| POV-V1 | 812 | — | 40.5 | — | — | — | 25 | 1.1 | 49 |
| POV-V2 | 960 | — | 55.9 | — | — | — | 24 | 1.1 | 49 |
| c-CTF-400 | 744 | 126 | 83 | 52 | — | — | 49 | 1 | 34 |
| c-CTF-450 | 861 | 99 | 62 | 38 | — | — | 46 | 1 | 34 |
| c-CTF-500 | 1247 | 133 | 80 | 47 | — | — | 43 | 1 | 34 |
| SYSU-Zn@IL1 | 38 | 68 | 40 | — | — | — | 27.2 | 1 | 47 |
To gain further insights from the non-covalent interactions between the materials and CO2 molecules, the CO2 isosteric heat of adsorption (Qst) of H2-ICOP and Zn-ICOP was obtained with the Clausius–Clapeyron equation from the CO2 uptake isotherms recorded (Fig. 4f). The Qst values for CO2 were found to be 39 and 36 kJ mol−1 for H2-ICOP and Zn-ICOP, respectively, at zero coverage. This difference of the Qst value for the two polymers shows that the non-covalent interactions of the CO2 molecules with the skeleton of H2-ICOP are more efficient than with Zn-ICOP, which may explain the higher CO2 capture capacity of H2-ICOP compared to Zn-ICOP. It is also worth mentioning that the Qst values of the polymers are moderate, not exceeding 50 kJ mol−1, which implies that CO2 is physically adsorbed by both ICOPs and the recyclability of the adsorbent at a low energy penalty.
Additionally, to assess the potential in separation of CO2 from flue gas (>70% N2), the adsorption selectivity of CO2 over N2 (15/85) of H2-ICOP and Zn-ICOP was calculated with Myers and Prausnitz's ideal adsorbed solution theory (IAST) at three different temperatures (273, 298, and 320 K) up to 1 bar (Fig. 4e and Table 1).58 The selectivity of CO2 from CO2/N2 (15/85) of H2-ICOP and Zn-ICOP is 55.7 and 5.5 at 273 K, 35.1 and 7.2 at 298 K, and 5.1 and 2.55 at 323 K, respectively. As expected, H2-ICOP exhibits much higher selectivity compared to Zn-ICOP, which also indicates powerful CO2 affinity of H2-ICOP.
2.3. Computational analysis of non-covalent bonds
The high CO2 capture and selectivity capacities of H2-ICOP and Zn-ICOP, despite their low surface areas, clearly show the powerful non-covalent interactions between CO2 molecules and multiple CO2-philic moieties of H2-ICOP and Zn-ICOP. Theoretical analyses (for details of the computational methodology see the ESI†) have been performed to get insights into the possible non-covalent interactions between CO2 molecules and the surfaces of H2-ICOP and Zn-ICOP, and their contribution to the selective CO2 uptake of these compounds.To model the mentioned non-covalent interactions, the fragments H2-ICOP and Zn-ICOP were ‘cut off’ from the polymeric chains and the linkers were replaced by hydrogen atoms. H2-ICOP and Zn-ICOP contain two porphyrin cores, a chlorine counterion, and a positively charged viologen linker. The σ-hole formation on the surface of H2-ICOP, Zn-ICOP, and CO2 has been visualized using electrostatic potential maps (MEP) and the results are displayed in Fig. 5 along with the corresponding electrostatic potential values. The calculated MEP surfaces show that a maximum of positive potential (σ-hole) involves a region with the cationic viologen subunits and Zn-ICOP porphyrin core (Zn) as well as the carbon atom CCO2, which shows the Lewis acid character of these regions. The negative electrostatic potential is located in the vicinity of oxygen OCO2, chlorine atoms, and nitrogen atoms in the H2-ICOP porphyrin core (Fig. 6). Thus, the positive and negative MEP distributions of H2-ICOP, Zn-ICOP, and CO2 indicate that attractive interactions are favourable for the formation of the above mentioned non-covalent bonds.
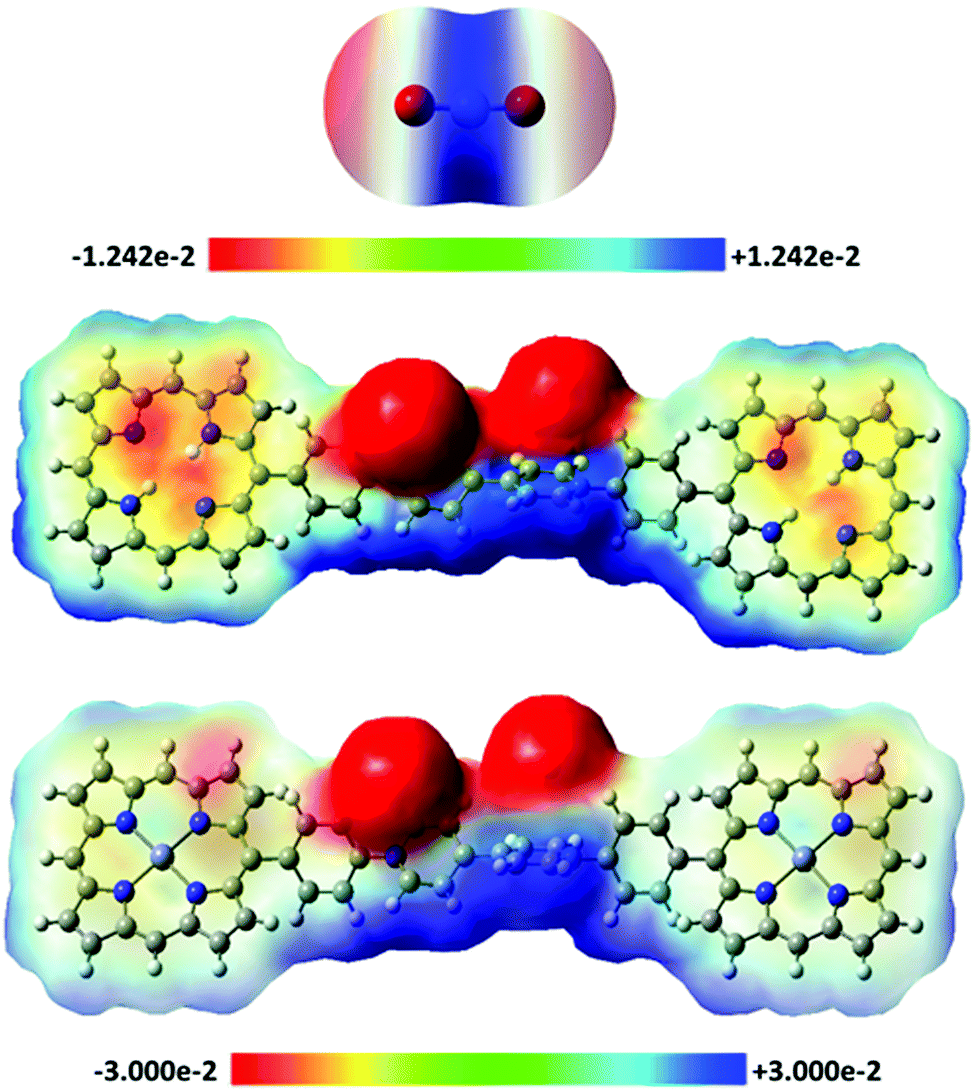 | ||
| Fig. 5 Electrostatic potentials mapped on the molecular surfaces of CO2, H2-ICOP and Zn-ICOP. The values of the MEPs lie in the intervals −7.794, +7.794 kcal mol−1 for CO2 and −18.825, +18.825 kcal mol−1 for H2-ICOP and Zn-ICOP. | ||
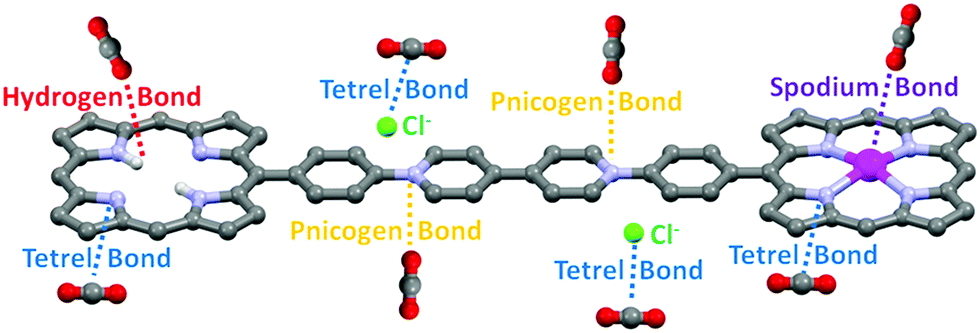 | ||
| Fig. 6 Scheme of possible non-covalent interactions between CO2 and H2-ICOP and Zn-ICOP. | ||
The MEP analyses have revealed possible non-covalent interactions, such as hydrogen, tetrel, pnicogen, and spodium bonds formed between CO2 molecules and the surfaces of H2-ICOP and Zn-ICOP, and their contribution to the selective CO2 uptake of these compounds (Fig. 6).
These noncovalent interactions can be uniformly defined based on the positive electrostatic region present on an atom due to the anisotropic distribution of the electron density. A positive electrostatic region present on an atom along a σ-polymer is called a σ-hole. The size and magnitude of a σ-hole are dependent on both the nature of its atom and the electron-withdrawing ability of groups attached to this atom. These σ-holes can have an attractive interaction with electron donors such as Lewis bases, anions or radicals. Depending on the σ-hole origins, these interactions are called tetrel bonding (group IV), pnicogen bonding (group V), chalcogen bonding (group VI), halogen bonding (group VII) or spodium bonding (group XII).59
Symmetry adapted perturbation theory (SAPT) analysis, which partitions the attractive forces into electrostatic (Eelst), exchange-repulsion (Eexch), induction (Eind), and dispersion (Edisp) terms, has been carried out to calculate the interaction energy of hydrogen (OCO2⋯H), tetrel (CCO2⋯N, CCO2⋯Cl−), pnicogen (OCO2⋯N+), and spodium bonds (OCO2⋯Zn) in H2-ICOP and Zn-ICOP and estimate the preferable binding sites to capture CO2 molecules on their surfaces (Table 3). For all studied non-covalent interactions, the calculated attractive components Eelst, Eind, and Edisp provide sufficient stabilization to overcome the repulsive exchange component Eexch, and the resultant interaction energy (Eint) values are negative, except hydrogen bonding. The value of Eexch for hydrogen bonds in H2-ICOP is very large and suppresses the other attractive components, and therefore the resulting interaction energy is positive, which indicates that the formation of H-bonds between CO2 and –NH (porp) is not favourable (Table 2 and Fig. 7d). Dispersion forces (Edisp) are found to be dominant in the attractive binding energies of pnicogen (OCO2⋯N+) and CCO2⋯N tetrel bonds and spodium bonds (OCO2⋯Zn), while electrostatic interactions (Eelst) dominate in CCO2⋯Cl− tetrel bonds in both compounds. The percentage contribution of the dispersion term in the overall attractive forces for pnicogen bonds consists of 73% in CO2-H2-ICOP and CO2-Zn-ICOP, while for CCO2⋯N tetrel bonds in these systems it is equal to 58% and 66%, respectively. For spodium bonds the dispersion term constitutes 53%. For CCO2⋯Cl− tetrel bonds in both compounds the percentage contribution of electrostatic interactions in the overall attractive forces consists of 58%.
| Compound | LB⋯LA | Bond type | R | E elst | E exch | E ind | E disp | E int |
|---|---|---|---|---|---|---|---|---|
| a The D–H⋯A angles for both CO2 are 125.8 and 118.7°. | ||||||||
| H2-ICOP | OCO2⋯N+ | Pnicogen | 3.115 | −6.1 | 16.8 | −2.0 | −22.2 | −13.6 |
| Cl−⋯CCO2 | Tetrel | 3.42 | −16.3 | 11.0 | −3.8 | −7.9 | −17.0 | |
| N⋯CCO2 | Tetrel | 2.976 | −21.7 | 42.3 | −5.5 | −37.1 | −22.0 | |
| OCO2a⋯H | Hydrogen | 1.961 | −177.6 | 398.9 | −21.2 | −105.9 | +94.3 | |
| Zn-ICOP | OCO2⋯N+ | Pnicogen | 3.115 | −6.2 | 16.8 | −2.0 | −22.3 | −13.7 |
| Cl−⋯CCO2 | Tetrel | 3.42 | −16.4 | 11.0 | −3.8 | −7.9 | −17.1 | |
| N⋯CCO2 | Tetrel | 2.976 | −14.2 | 42.7 | −5.2 | −38.4 | −15.2 | |
| OCO2⋯Zn | Spodium | 2.6 | −19.8 | 30.2 | −5.8 | −28.8 | −21.1 | |
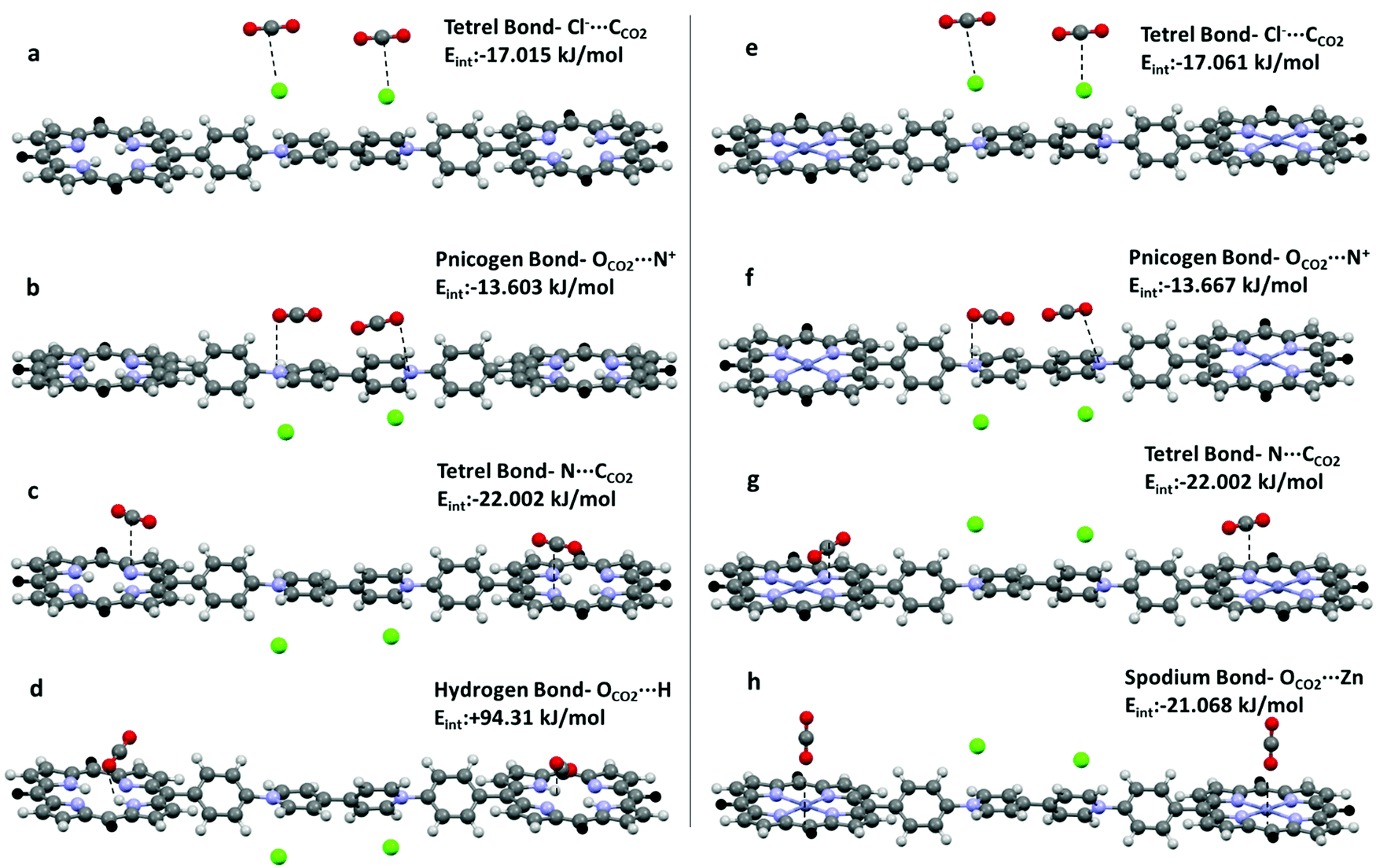 | ||
| Fig. 7 Formation of (a, e, c and g) tetrel, (b and f) pnicogen, (d) hydrogen and (h) spodium bonds in H2-ICOP and Zn-ICOP. | ||
The strength of the non-covalent interactions in CO2-H2-ICOP and CO2-Zn-ICOP is decreasing in the following order ΔEC⋯N > ΔEC⋯Cl−> ΔEO⋯N+ and ΔEZn⋯O > ΔEC⋯Cl− > ΔEC⋯N > ΔEO⋯N+, respectively (Table 2 and Fig. 7a–h). The binding energy of CCO2⋯N tetrel bonds in CO2-H2-ICOP is −22.02 kJ mol−1 and is the strongest among the studied non-covalent interactions. Interestingly, upon Zn metalation in the porphyrin core, the binding energy of CCO2⋯N tetrel bonds in CO2-Zn-ICOP is decreased to −15.163 kJ mol−1, probably due to an increase of the positive electrostatic potential around nitrogen atoms (Fig. 6) in the porphyrin core, which resulted in the formation of weaker tetrel bonds (CCO2⋯N) with CO2. In addition, further analyses were performed to calculate the binding energy of the simultaneous presence, on the same porphyrin core, of two CCO2⋯N tetrel bonds in H2-ICOP and spodium and CCO2⋯N tetrel bonds in Zn-ICOP. The analyses reveal that the simultaneous presence of spodium and CCO2⋯N tetrel bonds is impossible on the same porphyrin core in Zn-ICOP because of the strong repulsion between two CO2 molecules (Table S1 and Fig. S10, ESI†), while two tetrel bonds may form in the H2-ICOP porphyrin core because of negligible repulsion between CO2 molecules (Fig. S11 and Table S1, ESI†). Overall, in our fragment model, H2-ICOP can potentially form two pnicogen, two CCO2⋯Cl− and four CCO2⋯N tetrel bonds with CO2, while Zn-ICOP can form two pnicogen, two CCO2⋯Cl− tetrel, and two spodium bonds with CO2. The major CO2 uptake contribution comes from the four CCO2⋯N tetrel bonding interactions for H2-ICOP, whereas the two spodium bonding interactions for Zn-ICOP. In conclusion, H2-ICOP has more binding sites with more powerful non-covalent interactions that lead to an enhancement of the CO2 uptake compared to Zn-ICOP, which is in good agreement with the experimental results.
2.4. The catalytic performance in CO2 conversion into cyclic carbonates
To evaluate the catalytic activities of viologen-linked porphyrin H2-ICOP and Zn-ICOP, the cycloaddition of CO2 to propylene oxide was chosen as a model reaction and the results are listed in Table 3. The reactions were conducted under 1.0 MPa CO2 pressure and solvent and co-catalyst free conditions at 80 °C (for details see the ESI†).|
|
||||||
|---|---|---|---|---|---|---|
| Catalyst | Loading | T (°C) | P CO2 (bar) | t (h) | Yield (%) | Ref. |
| a This work (yields are determined by 1H NMR). | ||||||
| CTF-P-HSA | 0.1 g | 130 | 6.9 | 4 | 83 | 60 |
| cCTF | 4 wt% | 90 | 10 | 12 | 99 | 34 |
| PCP-Cl | 5 wt% | 100 | 30 | 12 | 99 | 39 |
| Zn-CIF2-C2H5 | 0.18 mol% | 120 | 25 | 5 | 98 | 61 |
| TBB-Bpy-a | 0.08 g | 120 | 10 | 4 | 99 | 48 |
| COF-salen-Zn | 0.1 mol% | 100 | 20 | 3 | 90 | 62 |
| SYSU-Zn@IL1 | 0.16 mol% | 80 | 10 | 12 | 62 | 47 |
| H2-ICOP | 0.16 mol% | 80 | 10 | 12 | 54 | |
| Zn-ICOP | 0.16 mol% | 80 | 10 | 12 | 83 | |

The compared catalysts Zn-ICOP and H2-ICOP gave yields of 83% and 54%, respectively. Surprisingly, Zn-ICOP, despite having a significantly lower CO2 uptake capacity, displayed better catalytic activity than H2-ICOP. The high activity of Zn-ICOP can be attributed to the synergistic effect of Lewis acid Zn sites and a nucleophile counter anion (Cl−). The zeta potential data revealed that Zn-ICOP possesses a counter anion (Cl−) on its surface, whereas H2-ICOP does not, which indicates the crucial role of the counter anion (Cl−) in the mechanism of this catalytic reaction.46,47 Notably, the catalytic activity of Zn-ICOP was achieved with lowered temperature and CO2 pressure, in a solvent-free reaction system, and was comparable to that of most of the reported examples (Table 3).
The catalytic performance of Zn-ICOP was compared with that of the corresponding polymer SYSU-Zn@IL1(Br−) since both structures are based on Zn–porphyrin and viologen. SYSU-Zn@IL1, with a BET surface area of 38 m2 g−1, has a nearly identical chemical composition to Zn-ICOP. Under similar conditions, Zn-ICOP had 83% conversion of PO, whereas SYSU-Zn@IL1(Br−) could only afford a PO conversion of 62%. The higher activity of Zn-ICOPz(Cl−) is mainly attributed to the higher nucleophilicity of its counter anions (Cl− > Br−).
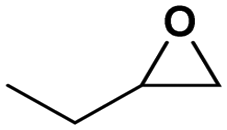
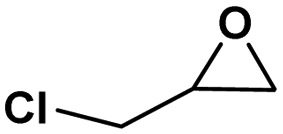
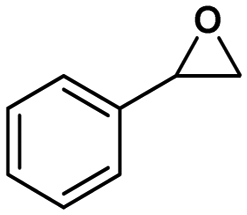
Under the optimized reaction conditions, the catalytic activity of Zn-ICOP was further studied over a variety of epoxides. As shown in Table 4, various epoxides are efficiently converted into the desired products with moderate yields under mild conditions by Zn-ICOP. With the increase of the size of epoxides, a steady decrease in the yield of cyclic carbonates was observed from PO (83%) and 1,2-epoxybutane (42%) to styrene oxide (4%), likely owing to their increasing steric hindrance, except for epichlorohydrin (77%) (Table 4). The deviation of epichlorohydrin's yield from the trend, despite its larger size, is probably due to additional non-covalent interactions between the chlorine functional group and CO2.
| Epoxide |
|
|
|
|
| Product |
|
|
|
|
| Conversion (%) | 83 | 42 | 77 | 4 |
3. Conclusion
Two viologen-porphyrin based ionic covalent organic polymers were constructed by a one-pot facile Zincke reaction. The BET surface area analyses reveal that both H2-ICOP (9 m2 g−1) and Zn-ICOP (20 m2 g−1) possess a very low surface area. Yet, despite their nearly non-porous surfaces, especially H2-ICOP exhibits a very large CO2 capture capacity and CO2/N2 selectivity, which indicates efficient non-covalent interactions with CO2. The theoretical analyses show that H2-ICOP can potentially form two pnicogen, two CCO2⋯Cl− and four CCO2⋯N tetrel bonds with CO2, while Zn-ICOP can form two pnicogen, two CCO2⋯Cl− tetrel, and two spodium bonds with CO2. The strength of the non-covalent interactions in H2-ICOP and Zn-ICOP is decreasing in the following order ΔEC⋯N > ΔEC⋯Cl− > ΔEO⋯N+ and ΔEZn⋯O > ΔEC⋯Cl− > ΔEC⋯N > ΔEO⋯N+, respectively. The binding energy of CCO2⋯N tetrel bonds in CO2-H2-ICOP is −22.02 kJ mol−1 and is the strongest among the studied non-covalent interactions. The major CO2 uptake contribution comes from the four CCO2⋯N tetrel bonding interactions for H2-ICOP, whereas the two spodium bonding interaction for Zn-ICOP. Furthermore, the CO2 catalytic conversion performances of Zn-ICOP and H2-ICOP gave encouraging yields of 83% and 54%, respectively. Surprisingly, Zn-ICOP, even with a significantly lower CO2 uptake capacity, displayed better catalytic activity than H2-ICOP, probably due to the crucial role of the counter anion (Cl−) in the mechanism of this catalytic reaction.Overall, we have demonstrated here that ICOPs with multiple CO2-philic sites can indeed exhibit very large CO2 capture capacity, CO2/N2 selectivity, and highly efficient catalytic activity of CO2 cycloaddition of epoxides, owing to efficient non-covalent interactions with CO2, in spite of low surface areas. These results set a useful example for the importance of non-covalent interactions with CO2 for the capture and conversion of CO2 into value-added products. Therefore, we believe that these findings will prove to be of key importance to the design of bifunctional charged materials for CCU applications.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the TUBITAK ULAKBIM High Performance and Grid Computing Center (TRUBA resources), Turkey, for computational facilities. Y. Zorlu acknowledges financial support from The Science Academy, Young Scientist Award (BAGEP).References
- V. Scott, S. Gilfillan, N. Markusson, H. Chalmers and R. S. Haszeldine, Nat. Clim. Change, 2013, 3, 105–111 CrossRef CAS.
- R. M. Cuéllar-Franca and A. Azapagic, J. CO2 Util., 2015, 9, 82–102 CrossRef.
- A. Modak, P. Bhanja, S. Dutta, B. Chowdhury and A. Bhaumik, Green Chem., 2020, 22, 4002–4033 RSC.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- G. Singh, J. Lee, A. Karakoti, R. Bahadur, J. Yi, D. Zhao, K. Albahily and A. Vinu, Chem. Soc. Rev., 2020, 49, 4360–4404 RSC.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac, Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- G. Sneddon, A. Greenaway and H. H. P. Yiu, The Potential Applications of Nanoporous Materials for the Adsorption, Separation, and Catalytic Conversion of Carbon Dioxide, Adv. Energy Mater., 2014, 4, 1301873–1301892 CrossRef.
- A. A. Olajire, J. CO2 Util., 2017, 17, 137–161 CrossRef CAS.
- R. Dawson, E. Stöckel, J. R. Holst, D. J. Adams and A. I. Cooper, Energy Environ. Sci., 2011, 4, 4239–4245 RSC.
- P. Bhanja, A. Modak and A. Bhaumik, ChemCatChem, 2019, 11, 244–257 CrossRef CAS.
- W. Wang, M. Zhou and D. Yuan, J. Mater. Chem. A, 2017, 5, 1334–1347 RSC.
- H. A. Patel, J. Byun and C. T. Yavuz, ChemSusChem, 2017, 10, 1303–1317 CrossRef CAS PubMed.
- R. Bera, M. Ansari, A. Alam and N. Das, J. CO2 Util., 2018, 28, 385–392 CrossRef CAS.
- M. Cox and R. Mokaya, Sustainable Energy Fuels, 2017, 1, 1414–1424 RSC.
- S. K. Shukla and J. P. Mikkola, Phys. Chem. Chem. Phys., 2018, 20, 24591–24601 RSC.
- K. M. De Lange and J. R. Lane, J. Chem. Phys., 2011, 135, 0–8 CrossRef CAS PubMed.
- G. Chang, Z. Shang, T. Yu and L. Yang, J. Mater. Chem. A, 2016, 4, 2517–2523 RSC.
- M. Li, J. Lei, G. Feng, J. U. Grabow and Q. Gou, Spectrochim. Acta, Part A, 2020, 238, 118424 CrossRef CAS PubMed.
- A. C. Legon, Phys. Chem. Chem. Phys., 2017, 19, 14884–14896 RSC.
- X. Tan, L. Kou, H. A. Tahini and S. C. Smith, Sci. Rep., 2015, 5, 1–8 Search PubMed.
- D. Nam, P. De Luna, A. Rosas-hernández, A. Thevenon, F. Li, T. Agapie, J. C. Peters, O. Shekhah, M. Eddaoudi and E. H. Sargent, Molecular enhancement of heterogeneous CO2 reduction, Nat. Mater., 2020, 19, 266–276 CrossRef CAS PubMed.
- B. Dash, J. Mol. Model., 2018, 24, 1–8 CrossRef CAS PubMed.
- P. Kanoo, R. Matsuda, H. Sato, L. Li, N. Hosono and S. Kitagawa, Chem. – Eur. J., 2020, 26, 2148–2153 CrossRef CAS PubMed.
- S. S. Rao and S. P. Gejji, J. Phys. Chem. A, 2016, 120, 1243–1260 CrossRef CAS PubMed.
- L. M. Azofra and S. Scheiner, Tetrel, chalcogen, and CH⋅⋅⋅O hydrogen bonds in complexes pairing carbonylcontaining molecules with 1, 2, and 3 molecules of CO2, J. Chem. Phys., 2015, 142, 034307-1–034307-9 CrossRef PubMed.
- L. Lodeiro, R. Contreras and R. Ormazábal-Toledo, J. Phys. Chem. B, 2018, 122, 7907–7914 CrossRef CAS PubMed.
- X. Y. Luo, X. Fan, G. L. Shi, H. R. Li and C. M. Wang, J. Phys. Chem. B, 2016, 120, 2807–2813 CrossRef CAS PubMed.
- H. Y. Li, Y. X. Lu, X. Zhu, C. J. Peng, J. Hu, H. L. Liu and Y. Hu, Sci. China: Chem., 2012, 55, 1566–1572 CrossRef CAS.
- X. Zhu, Y. Lu, C. Peng, J. Hu, H. Liu and Y. Hu, J. Phys. Chem. B, 2011, 115, 3949–3958 CrossRef CAS PubMed.
- V. Chernikova, O. Shekhah, Y. Belmabkhout and M. Eddaoudi DOI:10.1021/acsanm.0c00909.
- S. K. Das, P. Bhanja, S. K. Kundu, S. Mondal and A. Bhaumik, ACS Appl. Mater. Interfaces, 2018, 10, 23813–23824 CrossRef CAS PubMed.
- C. Liao, Z. Liang, B. Liu, H. Chen, X. Wang and H. Li, ACS Appl. Nano Mater., 2020, 3, 2889–2898 CrossRef CAS.
- Y. Fu, Z. Wang, S. Li, X. He, C. Pan, J. Yan and G. Yu, ACS Appl. Mater. Interfaces, 2018, 10, 36002–36009 CrossRef CAS PubMed.
- O. Buyukcakir, S. H. Je, S. N. Talapaneni, D. Kim and A. Coskun, ACS Appl. Mater. Interfaces, 2017, 9, 7209–7216 CrossRef CAS PubMed.
- Y. P. Zhao, Y. Li, C. Y. Cui, Y. Xiao, R. Li, S. H. Wang, F. K. Zheng and G. C. Guo, Inorg. Chem., 2016, 55, 7335–7340 CrossRef CAS PubMed.
- J. Wang, W. Sng, G. Yi and Y. Zhang, Chem. Commun., 2015, 51, 12076–12079 RSC.
- A. E. Sadak, Microporous Mesoporous Mater., 2021, 311, 110727 CrossRef CAS.
- V. A. Online, M. M. Unterlass, F. Emmerling, M. Antonietti and J. Weber, From dense monomer salt crystals to CO2 selective microporous polyimides via solid-state polymerization, Chem. Commun., 2014, 50, 430–432 RSC.
- O. Buyukcakir, S. H. Je, D. S. Choi, S. N. Talapaneni, Y. Seo, Y. Jung, K. Polychronopoulou and A. Coskun, Chem. Commun., 2016, 52, 934–937 RSC.
- J. Ding, C. Zheng, L. Wang, C. Lu, B. Zhang, Y. Chen, M. Li, G. Zhai and X. Zhuang, J. Mater. Chem. A, 2019, 7, 23337–23360 RSC.
- C. Hua, B. Chan, A. Rawal, F. Tuna, D. Collison, J. M. Hook and D. M. D’Alessandro, J. Mater. Chem. C, 2016, 4, 2535–2544 RSC.
- W. Hui, X. M. He, X. Y. Xu, Y. M. Chen, Y. Zhou, Z. M. Li, L. Zhang and D. J. Tao, J. CO2 Util., 2020, 36, 169–176 CrossRef CAS.
- G. Chen, X. Huang, Y. Zhang, M. Sun, J. Shen, R. Huang, M. Tong, Z. Long and X. Wang, Chem. Commun., 2018, 54, 12174–12177 RSC.
- Y. Zhang, K. Zhang, L. Wu, K. Liu, R. Huang, Z. Long, M. Tong and G. Chen, RSC Adv., 2020, 10, 3606–3614 RSC.
- T. Škorjanc, D. Shetty, M. A. Olson and A. Trabolsi, ACS Appl. Mater. Interfaces, 2019, 11, 6705–6716 CrossRef PubMed.
- Y. Zhang, K. Liu, L. Wu, H. Zhong, N. Luo, Y. Zhu, M. Tong, Z. Long and G. Chen, ACS Sustainable Chem. Eng., 2019, 7, 16907–16916 CrossRef CAS.
- Y. Chen, R. Luo, Q. Xu, J. Jiang, X. Zhou and H. Ji, ACS Sustainable Chem. Eng., 2018, 6, 1074–1082 CrossRef CAS.
- Y. Leng, D. Lu, P. Jiang, C. Zhang, J. Zhao and W. Zhang, Catal. Commun., 2016, 74, 99–103 CrossRef CAS.
- C. Hua, B. Chan, A. Rawal, F. Tuna, D. Collison, J. M. Hook and D. M. D’Alessandro, J. Mater. Chem. C, 2016, 4, 2535–2544 RSC.
- T. Lu, J. Zhang, Q. Gou and G. Feng, Phys. Chem. Chem. Phys., 2020, 22, 8467–8475 RSC.
- S. J. Grabowski, Struct. Chem., 2019, 30, 1141–1152 CrossRef CAS.
- T. Skorjanc, D. Shetty, F. Gándara, L. Ali, J. Raya, G. Das, M. A. Olson and A. Trabolsi, Chem. Sci., 2020, 11, 845–850 RSC.
- A. Bauzá, I. Alkorta, J. Elguero, T. J. Mooibroek and A. Frontera, Angew. Chem., 2020, 132, 17635–17640 CrossRef.
- N. Manoranjan, D. H. Won, J. Kim and S. I. Woo, J. CO2 Util., 2016, 16, 486–491 CrossRef CAS.
- S. Zahedi and E. Safaei, J. CO2 Util., 2020, 42, 101308 CrossRef CAS.
- X. Jiang, F. Gou, X. Fu and H. Jing, J. CO2 Util., 2016, 16, 264–271 CrossRef CAS.
- G. Das, T. Skorjanc, S. K. Sharma, F. Gándara, M. Lusi, D. S. Shankar Rao, S. Vimala, S. Krishna Prasad, J. Raya, D. S. Han, R. Jagannathan, J. C. Olsen and A. Trabolsi, J. Am. Chem. Soc., 2017, 139, 9558–9565 CrossRef CAS PubMed.
- A. L. Myers and J. M. Prausnitz, AlChE J., 1965, 11, 121–127 CrossRef CAS.
- R. Parajuli, Curr. Sci., 2016, 110, 495–498 Search PubMed.
- J. Roeser, K. Kailasam and A. Thomas, ChemSusChem, 2012, 5, 1793–1799 CrossRef CAS PubMed.
- J. Liu, G. Zhao, O. Cheung, L. Jia, Z. Sun and S. Zhang, Chem. – Eur. J., 2019, 25, 9052–9059 CrossRef CAS PubMed.
- H. Li, X. Feng, P. Shao, J. Chen, C. Li, S. Jayakumar and Q. Yang, J. Mater. Chem. A, 2019, 7, 5482–5492 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and analysis details; photographs of gel-like or powder ICOPs; FTIR of H2-ICOP-dx and Zn-ICOP-dx; CP/MAS 13C-NMR; PXRD with simulated patterns, EDS, DLS and zeta potentials for H2-ICOP and Zn-ICOP; SEM images of H2-ICOP-dx and Zn-ICOP-dx; formation of simultaneous tetrel bonds in H2-ICOP and Zn-ICOP; SAPT results for the interaction between two CO2 molecules placed at the same porphyrin fragments in H2-ICOP and Zn-ICOP. See DOI: 10.1039/d1ma00217a |
| This journal is © The Royal Society of Chemistry 2021 |