
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocatalytic access to betazole using a one-pot multienzymatic system in continuous flow†
Maria
Romero-Fernandez
 a and
Francesca
Paradisi
*ab
a and
Francesca
Paradisi
*ab
aSchool of Chemistry, University of Nottingham, University Park, NG7 2RD, Nottingham, UK
bDepartment of Chemistry, Biochemistry and Pharmaceutical Sciences, University of Bern, Freiestrasse 3, Bern, Switzerland. E-mail: francesca.paradisi@dcb.unibe.ch
First published on 24th May 2021
Abstract
As an alternative to classical synthetic approaches for the production of betazole drug, a one-pot biocatalytic system for this pharmaceutical molecule from its alcohol precursor has been developed. An ω-transaminase, an alcohol dehydrogenase and a water-forming NADH oxidase for in situ cofactor recycling have been combined to catalyse this reaction, yielding 75% molar conversion in batch reactions with soluble enzymes. This multienzyme system was then co-immobilised through a newly established protocol for sequential functionalization of a methacrylate-based porous carrier to enable tailored immobilisation chemistries for each enzyme. This pluri-catalytic system has been set up in a continuous flow packed-bed reactor, generating a space–time yield of up to 2.59 g L−1 h−1 with 15 min residence and a constant supply of oxygen for in situ cofactor recycling through a segmented air–liquid flow. The addition of an in-line catch-and-release column afforded >80% product recovery.
Introduction
The use of isolated enzymes as catalysts has been proposed in the fabrication of important pharmaceutical intermediates. In fact, the ACS Green Chemistry Institute's Pharmaceutical Round Table has published a list of 10 key research areas aiming to encourage the integration of green chemistry and engineering into the pharmaceutical industry, and biocatalysis can provide methodologies for many of these research areas.1 Moreover, the use of one-pot biocatalytic cascade systems in synthetic applications provides several economic and environmental benefits: fewer unit operations, minimized solvent and reactor volume, higher throughputs and space–time yields and reduced waste. In addition, it avoids the separation and purification of intermediates and can drive equilibria towards product formation, consequently enhancing overall yields.2–4 One of the challenges currently faced for the use of biocatalysis in synthetic applications stems from enzymes’ limited stability and water solubility, which hinders their recovery from the aqueous solution and imposes a major cost-contribution to the process. However, significant mitigation of this problem can be achieved by enzyme immobilisation, as it can improve enzyme stability and lead to the development of heterogeneous biocatalysts, thus increasing enzyme reusability and simplifying product work-up.5Betazole is an established drug in the pharmaceutical industry and a member of the pyrazole derivative family. This pharmaceutical molecule is an analogue of histamine and a H2-receptor agonist with the ability of stimulating the secretion of gastric acid.6,7 Clinical studies confirmed that betazole stimulation allowed an increase in both serum pepsinogen and gastric acid secretion even in patients with chronic Chagas’ disease subjected to surgical treatment, where reduced gastric acid secretion is relevant, since chagasic achalasia surgical treatment alters the esophagogastric junction anatomy.8
The first synthesis of substituted pyrazoles was carried out in 1883 by Knorr.9 For betazole, a two step synthesis was proposed by Jones, R.G.7, which evolved from a longer synthetic route developed in 1949.10 Hydrazine was first reacted with γ-pyrone in an alcohol solution, affording 3-pyrazoleacetaldehyde hydrazone in practically quantitative yields. A Ni-mediated catalytic hydrogenation of the latter compound afforded betazole in 80% yield.7 Nowadays, several relevant synthetic methods are being used to access the substituted pyrazoles.11
As an alternative to classical synthetic approaches, the design of a biocatalytic synthetic route for betazole has the potential to increase the sustainable production of this drug, but to date it has not been explored. In particular, the biocatalytic synthesis of the primary amine of betazole is relevant, since biocatalysis is a proven powerful tool in the synthesis of (enantiopure) amines, which are key intermediates in the fabrication of numerous active pharmaceutical ingredients (APIs).1 Both ω-transaminases (TA)12–17 and amine dehydrogenases (AmDHs)18,19 catalyse the selective production of primary amines from carbonyl compounds.
Furthermore, the synthesis of amines from the corresponding alcohols offers significant advantages since the alcohol is mostly easily accessible.20 In fact, different metal-catalysed methodologies have been proposed to carry out this reaction.21 No enzyme is however known to directly aminate an alcohol, and multienzyme systems have been proposed to catalyse this reaction in a two-step process: alcohol oxidation to form a carbonyl group, followed by reductive amination of the latter to produce a primary amine. Despite the apparent simplicity of the process, the technical challenges involved in establishing a reliable, sustainable and highly yielding system are significant. To date, only very few such multienzyme systems have been reported: galactose oxidase/TA/alanine dehydrogenase (AlaDH);22 alcohol dehydrogenase (ADH)/TA/AlaDH;23,24 ADH/TA/lactate dehydrogenase;24 ADH/AmDH;25 and alcohol oxidase/TA/AlaDH/catalase.26
The ADH-catalysed oxidation of alcohols requires stoichiometric amounts of oxidised nicotinamide adenine dinucleotide cofactors i.e. NAD(P)+, which represent a major cost contribution to the system. Therefore, an in situ regeneration system for the oxidised cofactor is essential for ensuring the cost-efficiency of the process. The integration of water-forming NAD(P)H oxidase (NOX) enzymes would appear to be ideal,27 as NAD(P)H is oxidized at the expense of the most innocuous and cheapest oxidant, O2, and producing H2O as a by-product.27 Several water-forming NOX have been described in the literature.28–30 However, the application of water-forming NOX to in situ cofactor recycling in continuous flow methodologies, which are increasing in popularity in the pharmaceutical industry, is not a straightforward process. O2 transfer from the gas to the aqueous liquid phase faces several well-known restrictions.31 Although efficient O2–liquid transfer can be achieved in conventional small-scale batch reactors by high levels of aeration and agitation,32 providing O2 to enzymatic reactions is especially challenging in standard biocatalytic flow reactors operating only with a liquid phase, where there is little gas (air)–liquid contact.
The aim of this work is to expand the applicability of synthetic biocatalysis by overcoming existing challenges that limit its implementation, adopting as a case study the sustainable production of betazole, a pyrazole derivative of pharmacological interest. For this purpose, biocatalysis and flow chemistry have been integrated to develop a one-pot multienzyme system in a continuous flow packed-bed reactor (PBR). Moreover, a novel protocol of multienzyme co-immobilisation on a polymethacrylate-based porous bead carrier has been developed. This system has been successfully applied to catalyse the direct synthesis of the primary amine of betazole from the corresponding alcohol.
Results and discussion
One-pot multienzymatic cascade combining HLADH, HEWT and LpNOX
A one-pot enzymatic cascade reaction to catalyse the direct synthesis of the primary amine of betazole from the corresponding alcohol was designed: ADH and TA, together with water-forming NOX as an in situ cofactor regeneration system. The use of the S-selective horse liver alcohol dehydrogenase (HLADH),33,34 the S-selective ω-transaminase from Halomonas elongata (HeWT),35–37 and the water-forming NADH oxidase from Lactobacillus pentosus (LpNOX)29,38 was proposed (Scheme 1).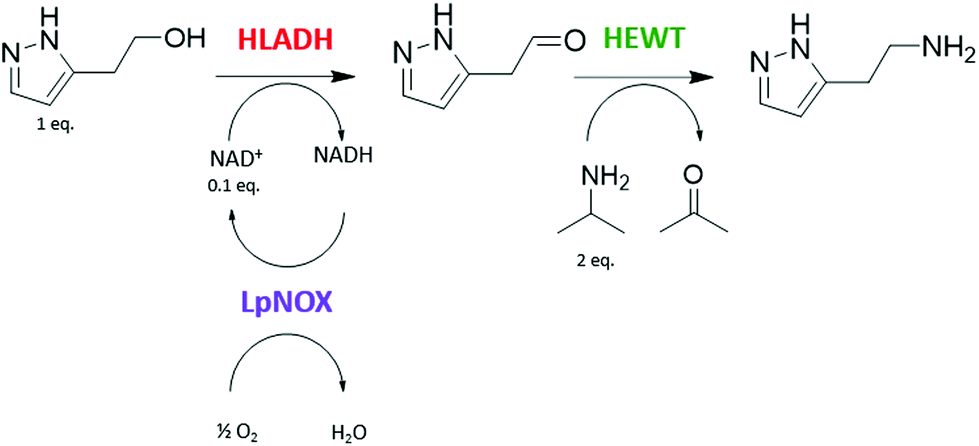 | ||
| Scheme 1 One-pot multienzyme system catalysing the functional group interconversion of the alcohol group of 2-(1H-pyrazol-3-yl)ethanol to the corresponding primary amine to produce a betazole drug. | ||
Initially, the catalytic activity of this multienzyme system on the alcohol group of 2-(1H-pyrazol-3-yl)ethanol was tested in batch reactions at a 10 mM scale with a catalytic amount of NAD+ (0.1 mol equivalents), using isopropylamine (IPA) as the amino-donor (2 mol equivalents) and the required enzymatic cofactors. High molar conversion (m.c.), 75%, to betazole was achieved after 21.5 hours under the specified reaction conditions (Table 1). HLADH and HEWT can therefore catalyse the functional group interconversion of the alcohol of 2-(1H-pyrazol-3-yl)ethanol into the corresponding primary amine to yield betazole in a one-pot system. The presence of LpNOX, despite its known low stability,29 increases the reaction velocity, meaning that in situ NAD+ recycling pushes the reaction equilibrium towards alcohol oxidation (Fig. S1†). This is likely due to the high redox potential of the O2/H2O couple that results in a strong thermodynamic driving force.27
Tailored multienzymatic co-immobilised system
To increase the reusability of this multienzyme system in continuous flow and simplify product work-up,39 the immobilisation of HLADH, HEWT and LpNOX was achieved. LpNOX, in particular, proved to be highly challenging. Among all the immobilisation methods proposed, covalent immobilisation offers significant advantages as it can improve enzyme stability by rigidification throughout their structure,40 and it was successfully attained for HLADH34,41 and HEWT.13 However, despite extensive testing with both epoxy and glyoxyl chemistries, the obtained recovered activity of LpNOX was very low even at low catalyst loading (5 mg g−1) achieving a maximum of 23% with partial immobilisation yield (Table S1†). An alternative immobilisation method for LpNOX based on ionic adsorption was selected, and this increased the recovered activity to 33% while providing the added benefit of enabling the desorption of the catalyst for replacement with fresh enzyme following exhaustion of the activity.In addition to the high conversions obtained by multistep flow biocatalysis in serial reactors for the synthesis of different molecules,37 one-pot flow biocatalysis based on a multistep enzymatic synthesis in a single reactor offers significant advantages.1,4,20 Moreover, immobilising the different enzymes on the same intrapore bead surface improves further the performance of biocatalytic cascade systems due to facilitated intrapore diffusion of substrates and cofactors between different enzyme active sites.42–44 Although co-immobilisation of multienzyme systems is highly challenging since there is no universal immobilisation chemistry or carrier, selective and optimal immobilisation of the different enzymes can be achieved by functionalization of the carrier surface with different reactive groups.42,44,45 However, no examples have been reported on polymethacrylate-based carriers, which are relevant for continuous flow applications.36,46,47 Therefore, a strategy for the co-immobilisation of this multienzyme system was developed on a porous bead carrier based on a polymethacrylate polymer matrix.
HLADH, HEWT and LpNOX enzymes were co-immobilised using tailored immobilisation chemistries for each of them. This was achieved by a novel protocol of sequential functionalization of a methacrylate carrier with different reactive groups (Scheme 2) to pursue optimal immobilisation for each of the enzymes and facilitate the diffusion of substrates between them. The epoxy groups displayed on the carrier surface were partially hydrolysed to diol groups in a controlled manner, and diol groups were then oxidised to glyoxyl groups. The carrier initial epoxy group density was determined to be 94 μmol gcarrier−1. After partial hydrolysis with H2SO4 and oxidation with NaIO4 treatment, the resulting glyoxyl group density was 78.3 μmol gcarrier−1, of which 14% resulted from the transformation of epoxy into glyoxyl groups, and 86% resulted from the transformation of diol groups initially present in the carrier into glyoxyl groups. The initial loading of epoxy plus diol groups of the carrier was determined to be 161 μmol gcarrier−1 (Table S2†). Since this strategy also makes use of the diol groups already present in the carrier, it offers the advantage of a better usage of the carrier surface and a higher efficiency of the resulting catalyst.
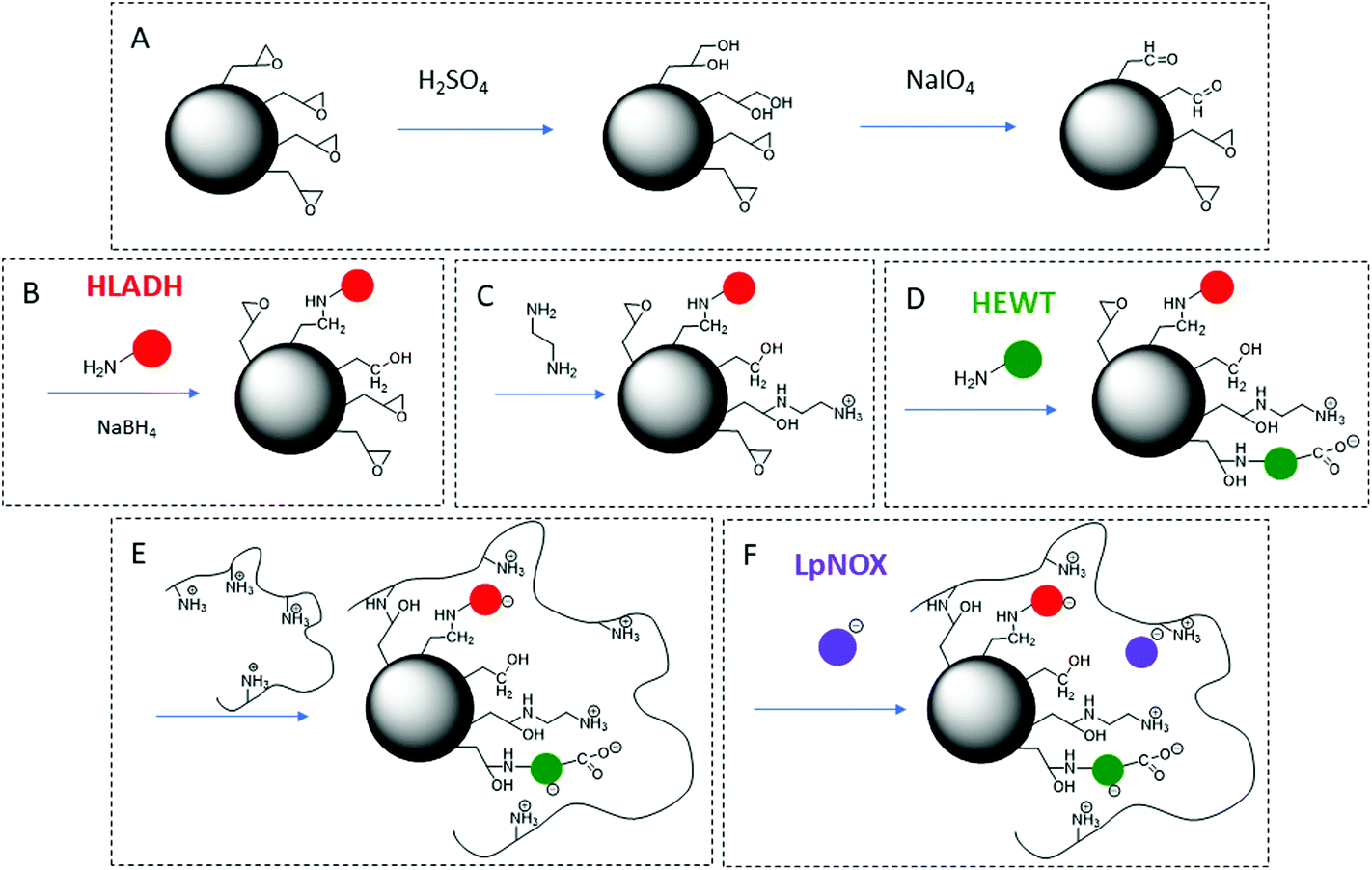 | ||
| Scheme 2 Sequential functionalization of the polymethacrylate-based porous bead carrier and co-immobilisation strategy of HLADH, HEWT and LpNOX. (A) Partial hydrolysis of epoxy groups to diol groups in a controlled manner, and oxidation of diol groups to glyoxyl groups. (B) HLADH covalent immobilisation on glyoxyl groups at pH 10 and 4 °C for 1 h. (C) Modification of some of the epoxy groups to amine groups upon reaction with ethylenediamine at pH 8.5. (D) HEWT covalent immobilisation on amino-epoxy groups at pH 8 and 4 °C for 4 h. (E) Blocking of epoxy groups with polyethyleneimine. (F) Immobilisation of LpNOX by ionic adsorption at pH 7 and 4 °C for 4 h. | ||
The resulting glyoxyl groups were used to immobilise the first enzyme, HLADH, by the multipoint-covalent attachment methodology previously described (Fig. S2†).40,46 This methodology is developed under alkaline conditions, where amine groups from lysine residues of the enzyme react with glyoxyl groups to form a Schiff's base which yields a secondary amine upon reduction with NaBH4.40 A minor proportion of HLADH was expected to immobilise via epoxy groups (Fig. S4†). Some of the remaining epoxy groups (∼12%) were then modified to amine groups upon reaction with ethylenediamine (EDA). The second enzyme, HEWT, was covalently immobilised by the two-step amino-epoxy strategy previously reported.48,49 This two-step approach is based on the first stabilisation of enzyme molecules by ionic interaction between their anionic residues and amine groups of the carrier, prior to the covalent bond formation between their nucleophilic groups and nearby epoxy groups on the carrier.48 The aim of this strategy is to overcome the low reactivity of epoxy groups found in enzyme immobilisation.48 Upon addition of polyethyleneimine (PEI), at least some of the unreacted epoxy groups covalently reacted with the amine groups of PEI, which was also used as a scaffold for the immobilisation by ionic adsorption43,50,51 of the third enzyme, LpNOX. With this strategy, ionic adsorption of enzyme molecules is established by ionic interactions between amine groups of PEI and aspartic and glutamic acid residues of the enzyme.43
Overall good values of immobilisation yield and recovered activity were obtained for the three enzymes (Table 2). Note that the low reported recovered activity for the HLADH could be possibly attributed to a mass transfer limitation at this high catalyst loading52 (Fig. S3†). High stability of HLADH and HEWT was achieved upon covalent immobilisation, and sufficient stability of LPNOX was reached by ionic adsorption on PEI (Fig. S5†). As a proof of concept, LpNOX could be efficiently desorbed by incubation with 0.2 M NaCl and fresh LpNOX could be immobilised on the same catalyst yielding 35% of the initial specific immobilised activity while preserving HLADH and HEWT immobilised activities (Table S3†) demonstrating the feasibility of the approach.
| Immobilisation yielda (%) | Recovered activitya (%) | Specific immobilised activitya (U gcarrier−1) | |
|---|---|---|---|
| a Immobilisation yield (%), recovered activity (%) and specific immobilised activity (U gcarrier−1) were calculated as described in the Experimental section. | |||
| HLADH | 100 | 9 | 2.3 |
| HEWT | 93 | 34 | 6.1 |
| LpNOX | 85 | 33 | 11.2 |
The catalytic activity of the resulting co-immobilised multienzyme system was evaluated on the alcohol group of 2-(1H-pyrazol-3-yl)ethanol in batch reactions under the same conditions as used with soluble enzymes. High m. c., 70%, to the corresponding primary amine was also achieved after 3 hours of reaction under specified conditions (Table 3). To demonstrate the reusability of this co-immobilised HLADH-HEWT-LpNOX biocatalyst, it was used to catalyse 3 batch reaction cycles. Remarkably, the catalytic efficiency of this multienzyme system was practically unaltered for 2 batch reaction cycles of 3 h (Table 3). After the second reaction cycle, the catalytic efficiency of the co-immobilised multienzyme system decreased considerably. This is likely due to the low stability of LpNOX as the activity of immobilised LpNOX on this multienzyme system dropped to 35% after 2 h of incubation under batch reaction conditions of temperature, pH and agitation, and to 1.6% after 24 h (Fig. S5†).
Flow biocatalysis with external oxygen supply
A continuous flow PBR was set-up using this co-immobilised multienzyme system and optimized for the functional group interconversion of the alcohol of 2-(1H-pyrazol-3-yl)ethanol into the corresponding primary amine to synthesize the betazole drug. A liquid phase containing 10 mM substrate, catalytic amounts of NAD+ (0.1 mol equivalents) and IPA (2 mol equivalents), and the required enzymatic cofactors was flowed into the packed-bed reactor with a residence time of 30 min, obtaining 10% m. c. (Scheme 3, black).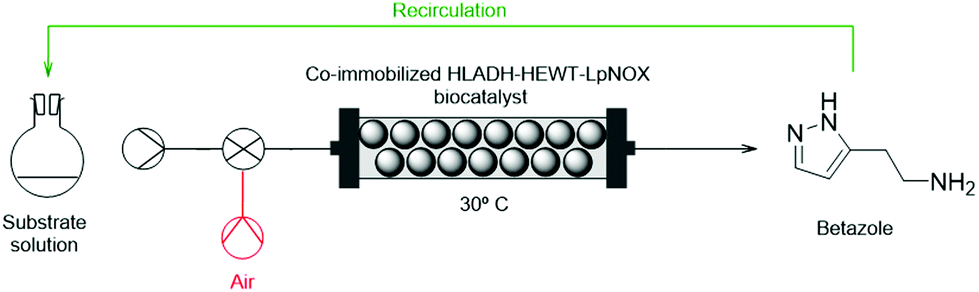 | ||
| Scheme 3 Continuous biocatalytic production of the betazole drug in a PBR. Substrate solution: 10 mM 2-(1H-pyrazol-3-yl)ethanol in phosphate buffer (0.05 M, pH 8), 1 mM NAD+, 20 mM IPA, 1 mM FAD, 0.1 mM PLP. T = 30 °C, P = atmospheric pressure. Reactor volume = 3.66 mL. In red: PBR implementing segmented liquid–gas flow composed of substrate solution/air. In green: PBR implementing recirculation of reaction solution (9.151 mL reaction solution volume). | ||
In systems requiring O2 as a substrate, as is the case for NOX catalysts, standard flow reactors can be highly limiting unless constant supply of O2 to the liquid phase can be guaranteed (e.g. tube-in-tube reactors,53 segmented air–liquid flow reactors32), or a strategy to increase the O2 concentration in the liquid phase (e.g. pressurized flow reactors31) is implemented. A simple solution when a packed-bed reactor is employed can be achieved with a segmented gas–liquid flow strategy composed of substrate solution/air. To date this was only reported with whole cell catalysts.32 Here this approach was trialled with cell-free immobilised catalysts to ensure a constant reservoir of oxygen to supply the liquid phase for the LpNOX-catalysed in situ cofactor regeneration in this PBR (Scheme 3, red).
A segmented 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 substrate solution/air flow was flowed into the PBR with 15 min residence time. This biphasic flow allowed alcohol amination with 50% m. c. To our knowledge, only one example of enzymatic amination of alcohols in continuous flow has been previously reported.54 However, the present work is the first example in the literature of a multienzyme system applied to amination of alcohols in continuous flow that uses TA, which is mostly relevant given the broad range of applications of this type of enzyme in the production of APIs and their intermediates.17
:50 substrate solution/air flow was flowed into the PBR with 15 min residence time. This biphasic flow allowed alcohol amination with 50% m. c. To our knowledge, only one example of enzymatic amination of alcohols in continuous flow has been previously reported.54 However, the present work is the first example in the literature of a multienzyme system applied to amination of alcohols in continuous flow that uses TA, which is mostly relevant given the broad range of applications of this type of enzyme in the production of APIs and their intermediates.17
Recirculation of the reaction mixture through the PBR increased the m. c. to 72% highlighting a possible limitation in the residence time. A recirculation strategy of the reaction mixture (Scheme 3, green) was then further implemented.
A biphasic flow of 50:50 substrate solution/air and a residence time of 4 × 15 min pushed the conversion to 84%. Very low increment of space time yield (STY) with further passes was observed after the first pass, where substrate concentration drops to 5 mM. This suggests a km issue and poor enzymatic efficiency below such a threshold (≈5 mM) (Fig. S6†). When the reaction scale was increased to 50 mM, and the same flow reaction conditions were applied, 23% m. c. to betazole was obtained after 1 pass through this PBR and 66% after 4 passes. STY and catalyst productivity were higher at the 50 mM scale, although conversion was lower (Table 4). At the 30 mM scale, a compromise between conversion and space time yield was found. At this substrate concentration, 16% m. c. to betazole was obtained after 1 pass and 69% after 4 passes. After 20 passes, 85% m. c. was achieved.
| Scale (mM) | Residence time (min) | M. c.a (%) | STYb (gbetazole L−1 h−1) | Catalyst productivityb (μmolbetazole h−1 mgenzyme−1) |
|---|---|---|---|---|
| a Determined by HPLC. b STY (gbetazole L−1 h−1) and catalyst productivity (μmolbetazole h−1 mgenzyme−1) calculated as described in the ESI.† | ||||
| 10 | 1 × 15 min | 50 | 1.11 | 0.40 |
| 2 × 15 min | 72 | 1.61 | 0.58 | |
| 4 × 15 min | 84 | 1.86 | 0.67 | |
| 30 | 1 × 15 min | 16 | 1.07 | 0.39 |
| 2 × 15 min | 35 | 2.28 | 0.84 | |
| 4 × 15 min | 69 | 4.53 | 1.67 | |
| 20 × 15 min | 85 | 5.60 | 2.07 | |
| 50 | 1 × 15 min | 23 | 2.59 | 0.96 |
| 2 × 15 min | 44 | 4.91 | 1.81 | |
| 4 × 15 min | 66 | 7.37 | 2.72 | |
Assuming a volumetric composition of oxygen in air of 21%, the input oxygen flow rate was 0.025 mL min−1. At 30 °C and 1 atm, this would be equivalent to 1.012 μmoloxygen min−1 flowing to the PBR in the gas phase. The molar conversion to betazole at the 10 mM scale and 15 min residence time after 1 pass was 50%, which corresponded to 0.60 μmol product per min. At this reaction scale, the input NAD+ concentration was 1 mM; therefore, 0.12 μmolNAD+ min−1 was flowed into the system. Since the stoichiometry of the ADH-catalysed alcohol oxidation and concomitant NAD+ reduction is 1 to 1, 0.48 μmolNAD+ min−1 must be recycled in 1 pass (at least). This would indicate that 0.24 μmoloxygen min−1 were consumed in the enzymatic reaction under these conditions. In the case of the 50 mM scale reactions, the oxygen intake rate by the enzymatic reaction in the first pass was estimated to be 0.39 μmoloxygen min−1. Moreover, a segmented liquid–gas flow was required to ensure a constant reservoir of oxygen to supply the liquid phase and make the reaction work (only 10% m. c. was obtained without air supply in the 10 mM reaction). Therefore, 0.24 μmoloxygen min−1 were then supplied from the gas to the liquid phase in the 10 mM scale reaction, and 0.39 μmoloxygen min−1 in the 50 mM scale reaction, under the applied flow conditions, and consumed by the enzymatic reaction.
At the 50 mM scale without any recirculation, this one-pot multienzymatic system yields 2.59 gbetazole L−1 h−1 in continuous flow, which is mostly relevant since it represents the first example of a biocatalytic synthetic step in the fabrication of betazole. As opposed to classical synthetic routes to this pharmaceutical molecule,7,11 this metal-free biocatalytic approach allows the synthesis of the primary amine of betazole from the corresponding alcohol by means of a highly yielding system using only water as the solvent at 30 °C and atmospheric pressure, and without the formation of unwanted side-products. Moreover, the co-immobilisation strategy of this multienzyme system increased biocatalyst reusability, thus increasing the catalyst productivity in continuous flow. Considering a window of 5 h of operational stability of the co-immobilised multienzyme system under specified flow conditions (Table 4), the accumulated catalyst productivity is 4.8 μmolbetazole mgenzyme−1.
Finally, a step to trap the primary amine-containing molecules (which include also unreacted IPA as well as betazole) was integrated in the system. A tailor-made aldehyde resin was packed in a column and placed downstream of the recirculating flow reaction. This amine catch-and-release step had an efficiency of 93% with respect to the recovered betazole after desorption with 0.2% HCl solution (Fig. S8†). The IPA could be successfully removed by freeze-drying the eluted fractions, as shown by 1H NMR and 13C NMR. The betazole-HCl salt form of the product was isolated with an apparent 94% yield. The difference with the reaction conversion is due to the presence of residual phosphate salts from the buffer as confirmed by 31P NMR.
Conclusions
This work represents the first example of implementation of a biocatalysed synthetic step in the production of betazole drugs, and one of the few examples of biocatalytic amination of alcohols in a continuous flow PBR. It is also the first cascade which uses TA in continuous flow for the reductive amination of the formed carbonyl group, which is mostly relevant given the broad range of applications of TA in the production of APIs and their intermediates.17 This biocatalytic strategy could also be integrated with other bio/chemo-catalysed steps to design alternative routes of synthesis of this drug molecule. The developed segmented gas–liquid flow strategy ensured a constant reservoir of oxygen which further promotes the application of water-forming NOX as recycling systems in continuous flow PBRs, thus further contributing to the combination of biocatalysis and flow technologies. The immobilisation of three different enzymes with tailored chemistries shows how optimal conditions can be achieved on the same carrier even for catalysts with very different stability profiles. While the stability of LpNOX was not significantly improved, the possibility of replenishing the exhausted catalyst, without affecting the other enzymes, was demonstrated. The method described here has therefore substantial potential in the synthesis of pure primary amines from the corresponding alcohol intermediates within the production of numerous APIs, and in the integration of greener synthetic approaches in the pharmaceutical industry.Experimental section
Materials
All chemicals and reagents used in this work were purchased from Sigma-Aldrich unless specified otherwise. GoTaq® DNA polymerase was purchased from Promega. NheI-HF®, EcoRV-HF® and T4 DNA ligase were acquired from New England Biolabs. A QIAquick PCR Purification Kit and a QIAquick Gel Extraction Kit were purchased from Qiagen. XL10-Gold® Ultracompetent cells were purchased from Agilent. E. coli BL21(DE3) Star competent cells were purchased from Themo Fisher Scientific. Anhydrotetracycline was purchased from Cayman Chemical Company. StrepTrap HP 5 mL and HisTrap HP 1 mL columns were acquired from GE Healthcare. Nicotinamide adenine dinucleotide oxidised (NAD+) and reduced (NADH) forms were purchased from Apollo Scientific. Relisorb® EP400SS was donated by Resindion. Polyethyleneimine (PEI) (M.N. 60000, 50% aq. solution, branched) was purchased from Acros Organics. 2-(1H-Pyrazol-3-yl)ethanol and betazole were acquired from Fluorochem.
Cloning of HLADH
The gene coding the horse alcohol dehydrogenase-E-isoenzyme (GenBank accession number M64864) was synthesized by GeneArt Gene Synthesis service from Thermo Fisher Scientific. The gene was amplified by PCR with GoTaq® DNA polymerase with the oligonucleotide primers FWD-HLADH (5′-AATG![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif)
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif)
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif)
![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) TGGAGCCACCCGCAGTTCGAAAAAGGCGCCATGAGCACCGCAG-3′) and RV-HLADH (5′-TAGTTAATTAAAAGGTCAGAAT-3′). The primers were designed to incorporate NheI and EcoRV restriction sites, respectively (underlined). The manufacturer's protocol for GoTaq® DNA polymerase PCR in 50 μL reaction was used. PCR was carried out with 10 μL 5× GoTaq® buffer; 2.5 μL 10 mM FWD-HLADH primer; 2.5 μL 10 mM RV-HLADH primer; 32.5 μL nuclease-free water; 1 μL 100 ng μL−1 plasmid containing the HLADH gene; and 1 μL GoTaq® DNA polymerase. PCR conditions were 2 min of at 72 °C and 35 cycles of 1 min denaturation at 95 °C, 1 min annealing at 60 °C and 2 min extension 72 °C. The 35 cycles were followed by 7 min of final elongation at 72 °C. The amplified DNA was purified with a QIAquick PCR Purification Kit. The purified PCR product was digested with NheI-HF® and EcoRV-HF® for 2 h at 37 °C. The digested gene was gel-extracted using a QIAquick Gel Extraction Kit and cloned into the pASK-IBA5plus vector harbouring Lactobacillus brevis ADH, kindly donated by Prof. Kroutil, digested with the same restriction enzymes. Ligation was developed with T4 DNA ligase following the manufacturer's guidelines and the resulting product was used to transform E. coli XL10-Gold® Ultracompetent cells. The construct was verified by sequencing and the obtained plasmid was named pASK-IBA5plus-HLADH.
TGGAGCCACCCGCAGTTCGAAAAAGGCGCCATGAGCACCGCAG-3′) and RV-HLADH (5′-TAGTTAATTAAAAGGTCAGAAT-3′). The primers were designed to incorporate NheI and EcoRV restriction sites, respectively (underlined). The manufacturer's protocol for GoTaq® DNA polymerase PCR in 50 μL reaction was used. PCR was carried out with 10 μL 5× GoTaq® buffer; 2.5 μL 10 mM FWD-HLADH primer; 2.5 μL 10 mM RV-HLADH primer; 32.5 μL nuclease-free water; 1 μL 100 ng μL−1 plasmid containing the HLADH gene; and 1 μL GoTaq® DNA polymerase. PCR conditions were 2 min of at 72 °C and 35 cycles of 1 min denaturation at 95 °C, 1 min annealing at 60 °C and 2 min extension 72 °C. The 35 cycles were followed by 7 min of final elongation at 72 °C. The amplified DNA was purified with a QIAquick PCR Purification Kit. The purified PCR product was digested with NheI-HF® and EcoRV-HF® for 2 h at 37 °C. The digested gene was gel-extracted using a QIAquick Gel Extraction Kit and cloned into the pASK-IBA5plus vector harbouring Lactobacillus brevis ADH, kindly donated by Prof. Kroutil, digested with the same restriction enzymes. Ligation was developed with T4 DNA ligase following the manufacturer's guidelines and the resulting product was used to transform E. coli XL10-Gold® Ultracompetent cells. The construct was verified by sequencing and the obtained plasmid was named pASK-IBA5plus-HLADH.
Enzyme expression
The plasmid harbouring HLADH (pASK-IBA5plus-HLADH) was transformed into E. coli BL21(DE3) Star. 1 L flasks containing 300 mL of Terrific Broth media were inoculated with 10 mL of an overnight culture in LB and left to grow at 37 °C and 180 rpm until OD600 reached 0.6–0.7. At that point, the expression was induced with 0.4 μM anhydrotetracycline (final concentration) and the cultures were left to grow for 16 h at 20 °C and 180 rpm. The previously reported expression protocol of HEWT was followed.35 For LpNOX, a single colony of E. coli BL21 (DE3) Star cells previously transformed with the plasmid pET28a-LpNOX, kindly donated by Prof. Sieber,29 was inoculated in autoinduction media ZYP-5052. The cultures were left to grow at 37 °C and 180 rpm for 20 h. The cells were collected by centrifugation at 4500 rpm and stored at −20 °C until further use.Enzyme purification
The cells containing expressed HLADH enzyme were resuspended in 20 mM sodium phosphate buffer, 0.28 M NaCl, and 6 mM KCl pH 7.4. Cells were disrupted by sonication with pulses of 5 seconds on and 5 seconds off at 60% amplitude for 12 min. The insoluble fraction was separated by centrifugation at 14500 rpm for 60 min. The supernatant was filtered (0.45 μm) and loaded into a StrepTrap HP 5 mL column using an ÄKTA™ start FPLC (GE Healthcare). The protein was eluted with 20 mM sodium phosphate buffer, 0.28 M NaCl, 6 mM KCl and 2.5 mM D-desthiobiotin pH 7.4. HLADH was dialysed twice against 50 mM Tris buffer pH 8 at 4 °C. For HEWT, cells were resuspended in 50 mM potassium phosphate buffer, 0.1 M NaCl, 0.1 mM PLP and 30 mM imidazole pH 8. The same sonication and centrifugation conditions as in the case of HADH were used. The supernatant was filtered (0.45 μm) and HEWT was purified using the ÄKTA™ start FPLC as previously described.35 HEWT was dialysed twice against 50 mM potassium phosphate buffer 0.1 mM PLP pH 8 at 4 °C. Cells containing expressed LpNOX enzyme were resuspended in 50 mM potassium phosphate buffer, 0.5 M NaCl, 20 mM imidazole, 10% (v/v) glycerol, and 0.01 mM flavin adenine dinucleotide (FAD) pH 8. After cell disruption and insoluble fraction separation under the same conditions as before, the filtered supernatant was loaded into a HisTrap HP 1 mL column using the ÄKTA™ start FPLC. The protein was eluted with 50 mM potassium phosphate buffer, 0.5 M NaCl, 0.5 M imidazole, 10% (v/v) glycerol, and 0.01 mM FAD pH 8, and dialysed twice against 50 mM Tris buffer 0.01 mM FAD pH 7.5 at 4 °C.
Enzymatic activity assays and determination of protein concentration
All enzyme assays were performed in triplicate in 96-well microplates (unless specified otherwise) using an Epoch 2 Microplate Spectrophotometer (Biotek). For HLADH and HeWT, the activity assays were performed as previously indicated,34,35 at 25 °C and pH 8 (potassium phosphate buffer). LpNOX activity assay was performed following the depletion of NADH at 340 nm using 0.1 mM NADH in 50 mM phosphate buffer at 25 °C and pH 8. One unit of enzymatic activity was defined as the oxidation of 1 μmol of cofactor per minute.For the immobilised enzyme, the activity was measured using the same conditions as for the free enzyme. For HLADH and HEWT, 20 mg of the immobilised biocatalyst were added to a 10 mL reaction mixture. At regular intervals of time during 10 min, a sample was taken to measure the absorbance at 340 nm or 245 nm, for HLADH or HEWT, respectively. For LpNOX, 20 mg of the immobilised biocatalyst were added to 40 mL of reaction mixture, and the same protocol was followed. The specific activity of HLADH, HEWT and LpNOX immobilised biocatalysts (U gcarrier−1) was defined as: the formation of NADH in μmol per minute and g of biocatalyst (HLADH); the formation of acetophenone in μmol per minute and g of biocatalyst (HEWT); and the depletion of NADH in μmol per minute and g of biocatalyst (LpNOX).
HLADH and HEWT protein concentration were determined by UV absorption at 280 nm using an Epoch Take3 Micro-Volume Plate. The extinction coefficients 22460 M−1 cm−1 and 62340 M−1 cm−1, at 280 nm, measured in water, were respectively estimated for HLADH and HEWT using the ExPASy ProtParam tool, accessible from the ExPASy website (http://www.expasy.ch). The LpNOX concentration was determined using a Bradford assay with bovine serum albumin (BSA) as the standard, as previously reported.29
Sequential functionalization of the methacrylate carrier and co-immobilisation of HLADH, HEWT and LpNOX
3 g of Relisorb® EP400SS were resuspended in 30 mL of 0.1 M H2SO4, and the suspension was incubated for 30 min at room temperature under mild agitation. The hydrolysed carrier was filtered, washed with H2O and resuspended in 30 mL of 10 mM NaIO4. After 2 h incubation at room temperature under mild agitation, the resulting carrier was filtered and washed with H2O (Scheme 2A). The resulting glyoxyl groups were quantified as previously described.46 A 30 mL solution containing 60 mg of pure HLADH in 100 mM NaHCO3 pH 10 was added to the resin containing glyoxyl groups. The suspension was incubated for 1 h at 4 °C under mild agitation, and 30 mg of NaBH4 were then added and further incubated for 30 min at 4 °C under mild agitation. HLADH activity in the remaining liquid phase before NaBH4 addition was measured. The immobilised HLADH biocatalyst was filtered and washed with H2O (Scheme 2B). 2 samples of 20 mg of the immobilised HLADH biocatalyst were taken to determine the specific activity of the immobilised HLADH enzyme (in duplicate) as described before. The rest was added to a 30 mL solution of 300 mM ethylenediamine (EDA) in 100 mM NaHCO3 pH 8.5. The suspension was incubated for 2 h at room temperature under mild agitation. The EDA-modified immobilised HLADH biocatalyst was filtered and washed with H2O (Scheme 2C). A 30 mL solution containing 15 mg of pure HEWT in 50 mM potassium phosphate buffer 0.1 mM pyridoxal 5′-phosphate (PLP) pH 8 was added to this biocatalyst. After 4 h incubation at 4 °C under mild agitation, the resulting co-immobilised HEWT-HLADH biocatalyst was filtered, washed with H2O (Scheme 2D), and added to a 60 mL solution of 0.05 g PEI per mL in 25 mM potassium phosphate buffer pH 7.5. The suspension was incubated for 16 h at room temperature under mild agitation, and the resulting co-immobilised HEWT-HLADH biocatalyst was filtered and washed with H2O (Scheme 2E). 4 samples of 20 mg of the co-immobilised HEWT-HLADH biocatalyst were taken to determine the specific activity of the immobilised HEWT and HLADH enzymes (in duplicate). A 30 mL solution containing 15 mg of pure LpNOX in 25 mM potassium phosphate buffer 0.1 mM FAD pH 7 was added to this biocatalyst. After 4 h incubation at 4 °C under mild agitation, the resulting co-immobilised LpNOX-HEWT-HLADH biocatalyst was filtered and washed with H2O (Scheme 2F). 2 samples of 20 mg of the co-immobilised LpNOX-HEWT-HLADH biocatalyst were taken to determine the specific activity of the immobilised LpNOX enzyme (in duplicate).The immobilised activity (U gcarrier−1) was calculated as the difference between initial offered activity per gram of carrier and remaining activity in the liquid phase per gram of carrier at the immobilisation end point. The immobilisation yield (%) was calculated as the ratio between immobilised activity and initial offered activity per gram of carrier. The recovered activity (%) was calculated as the ratio between the specific activity of the immobilised enzyme and the immobilised activity.
Batch reaction of biocatalytic synthesis of the betazole drug from 2-(1H-pyrazol-3-yl)ethanol
Batch reactions with pure soluble enzymes were performed at 30 °C in 5 mL of reaction mixture containing 10 mM 2-(1H-pyrazol-3-yl)ethanol, 1 mM NAD+, 200 mM IPA, 0.1 mM PLP, 0.1 mM FAD, 50 mM potassium phosphate buffer pH 8, HLADH (2 mg mL−1), HEWT (0.2 mg mL−1), and LpNOX (0.1 mg mL−1). Batch reactions with the co-immobilised multienzyme system were performed at 30 °C in 1 mL of reaction mixture containing 10 mM 2-(1H-pyrazol-3-yl)ethanol, 1 mM NAD+, 20 mM IPA, 0.1 mM PLP, 0.1 mM FAD, 50 mM potassium phosphate buffer pH 8, and 0.2 g of the co-immobilised LpNOX-HEWT-HLADH biocatalyst. The reusability test of the co-immobilised LpNOX-HEWT-HLADH biocatalyst was developed in 3 h reaction cycles under the same conditions. After a reaction cycle, the co-immobilised LpNOX-HEWT-HLADH biocatalyst was filtered, washed with H2O and added to a fresh reaction mixture. The reactions were monitored by HPLC, as described in the Analytical methods section.Continuous flow reaction of biocatalytic synthesis of the betazole drug from 2-(1H-pyrazol-3-yl)ethanol
An Omnifit glass column (6.6 mm i.d. × 100 mm length) was filled with 3 g of the co-immobilised LpNOX-HEWT-HLADH biocatalyst to set up a PBR (reactor volume, 3.66 mL). In all the reactions performed, the temperature was set to 30 °C. A 10 mM substrate solution (2-(1H-pyrazol-3-yl)ethanol) containing 1 mM NAD+, 20 mM IPA, 0.1 mM PLP, and 1 mM FAD in 50 mM potassium phosphate buffer pH 8.0 was prepared. For the single-phase flow reaction, this 10 mM substrate solution was directed into the PBR. The flow rate was kept at 122 μL min−1 to allow a residence time of 30 min. For two-phase flow reactions without recirculation, the 10 mM substrate solution was flowed into the PBR at 122 μL min−1 to allow a residence time of 15 min with a peristaltic pump. Air was flowed into the PBR at the same flow rate with a second peristaltic pump and mixed with the substrate solution stream using a T-junction. The resulting 50:50 substrate solution/air segmented flow was directed into the PBR. Samples were taken from the exiting flow streams of single-phase and two-phase flow systems and analysed by HPLC, as described in the Analytical method section. For the two-phase flow reactions with recirculation, a 50 mM substrate solution 5 mM NAD+, 100 mM IPA, 0.1 mM PLP, 1 mM FAD in 50 mM potassium phosphate buffer pH 8.0; and a 30 mM substrate solution 3 mM NAD+, 60 mM IPA, 0.1 mM PLP, 1 mM FAD in 50 mM potassium phosphate buffer pH 8.0 were prepared. 9.151 mL of the 10 mM, the 30 mM or the 50 mM substrate solution were flowed at 122 μL min−1 to allow a residence time of 15 min. The exiting flow stream was placed in the same container as the substrate solution. Air was flowed into the PBR at the same flow rate. After one pass of the 9.151 mL substrate solution (after 75 min), a sample was taken and analysed by HPLC. The other samples were taken after 2 and 4 (after 2.5 h and 5 h, respectively).
Amine catch-and-release in continuous flow
3 g of Relisorb® HG400SS were resuspended in 30 mL of 20 mM NaIO4. After 2 h incubation at room temperature under mild agitation, the resulting carrier was filtered and washed with H2O. The resulting aldehyde groups were quantified as previously described.46 An Omnifit glass column (6.6 mm i.d. × 100 mm length) was filled with 3 g of this tailor-made aldehyde resin (column volume, 3.66 mL). The product mixture resulting from the 30 mM flow reaction (implemented with segmented flow and recirculation) was flowed to the system. The flow rate was set up to 122 μL min−1 to allow a residence time of 30 min (temperature off). 4 column volumes of 50 mM phosphate buffer pH 8 were flowed to the system for washing. The elution of the trapped primary amines by the aldehyde groups was carried out with 4 column volumes of 0.2% HCl.Analytical methods
100 μL of sample with a maximum concentration of substrate (10 mM) were added to 900 μL of MilliQ H2O. The samples were analysed by HPLC (Accucore™ aQ C18 Polar Endcapped HPLC Column, Thermo Fisher Scientific (2.6 μm, 4.6 × 150 mm), measuring at 210 nm, using an isocratic method with H2O 0.1%TFA) over 5 minutes with a flow rate of 0.8 mL min−1. The retention times of the different substances were: 2-(1H-pyrazol-3-yl)ethanol 3.45 min, and betazole 2.38 min. Molar conversions were calculated through a standard curve of the product.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank Prof. Sieber (TU Munich) for kindly providing the plasmid harbouring the LpNOX gene. The authors wish to acknowledge the Biotechnology and Biological Research Council [grant number BB/P002536/1] and H2020 ERACoBioTech programme (project ID: 61 HOMBIOCAT) also funded through the Biotechnology and Biological Sciences Research Council [grant number BB/R021287/1].Notes and references
- R. A. Sheldon and D. Brady, ChemSusChem, 2019, 12, 2859–2881 CrossRef CAS PubMed.
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 4464–4480 CrossRef CAS.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- S. P. France, L. J. Hepworth, N. J. Turner and S. L. Flitsch, ACS Catal., 2017, 7, 710–724 CrossRef CAS.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- G. J. Durant, C. R. Ganellin and M. E. Parsons, J. Med. Chem., 1975, 18, 905–909 CrossRef CAS PubMed.
- R. G. Jones and M. J. Mann, J. Am. Chem. Soc., 1953, 75, 4048–4052 CrossRef CAS.
- J. R. M. Da Rocha, U. Ribeiro, I. Cecconello, R. A. A. Sallum, F. Takeda, A. Nasi and S. Szachnowicz, Dis. Esophagus., 2009, 22, 606–610 CrossRef CAS PubMed.
- L. Knorr, Ber. Dtsch. Chem. Ges., 1883, 16, 2597–2599 CrossRef.
- R. G. Jones, J. Am. Chem. Soc., 1949, 71, 3994–4000 CrossRef CAS.
- K. Karrouchi, S. Radi, Y. Ramli, J. Taoufik, Y. N. Mabkhot, F. A. Al-Aizari and M. Ansar, Molecules, 2018, 23, 134 CrossRef PubMed.
- R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2014, 4, 129–143 CrossRef CAS.
- M. Planchestainer, M. L. Contente, J. Cassidy, F. Molinari, L. Tamborini and F. Paradisi, Green Chem., 2017, 19, 372–375 RSC.
- L. Leipold, D. Dobrijevic, J. W. E. Jeffries, M. Bawn, T. S. Moody, J. M. Ward and H. C. Hailes, Green Chem., 2019, 21, 75–86 RSC.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- I. V. Pavlidis, M. S. Weiß, M. Genz, P. Spurr, S. P. Hanlon, B. Wirz, H. Iding and U. T. Bornscheuer, Nat. Chem., 2016, 8, 1076–1082 CrossRef CAS PubMed.
- S. A. Kelly, S. Pohle, S. Wharry, S. Mix, C. C. R. Allen, T. S. Moody and B. F. Gilmore, Chem. Rev., 2018, 118, 349–367 CrossRef CAS PubMed.
- S. K. Au, B. R. Bommarius and A. S. Bommarius, ACS Catal., 2014, 4, 4021–4026 CrossRef CAS.
- T. Knaus, W. Böhmer and F. G. Mutti, Green Chem., 2017, 19, 453–463 RSC.
- J. H. Schrittwieser, S. Velikogne, M. Hall and W. Kroutil, Chem. Rev., 2018, 118, 270–348 CrossRef CAS PubMed.
- S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853–1864 CrossRef.
- M. Fuchs, K. Tauber, J. Sattler, H. Lechner, J. Pfeffer, W. Kroutil and K. Faber, RSC Adv., 2012, 2, 6262–6265 RSC.
- J. H. Sattler, M. Fuchs, K. Tauber, F. G. Mutti, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Angew. Chem., Int. Ed., 2012, 51, 9156–9159 CrossRef CAS PubMed.
- K. Tauber, M. Fuchs, J. H. Sattler, J. Pitzer, D. Pressnitz, D. Koszelewski, K. Faber, J. Pfeffer, T. Haas and W. Kroutil, Chem. – Eur. J., 2013, 19, 4030–4035 CrossRef CAS PubMed.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- M. Pickl, M. Fuchs, S. M. Glueck and K. Faber, ChemCatChem, 2015, 7, 3121–3124 CrossRef CAS PubMed.
- W. Kroutil, H. Mang, K. Edegger and K. Faber, Curr. Opin. Chem. Biol., 2004, 8, 120–126 CrossRef CAS PubMed.
- B. Geueke, B. Riebel and W. Hummel, Enzyme Microb. Technol., 2003, 32, 205–211 CrossRef CAS.
- C. Nowak, B. Beer, A. Pick, T. Roth, P. Lommes and V. Sieber, Front. Microbiol., 2015, 6, 957 Search PubMed.
- J. T. Park, J. I. Hirano, V. Thangavel, B. R. Riebel and A. S. Bommarius, J. Mol. Catal. B: Enzym., 2011, 71, 159–165 CrossRef CAS.
- J. M. Bolivar, A. Mannsberger, M. S. Thomsen, G. Tekautz and B. Nidetzky, Biotechnol. Bioeng., 2019, 116, 503–514 CrossRef CAS PubMed.
- L. Tamborini, P. Fernandes, F. Paradisi and F. Molinari, Trends Biotechnol., 2018, 36, 73–88 CrossRef CAS PubMed.
- D. Quaglia, J. A. Irwin and F. Paradisi, Mol. Biotechnol., 2012, 52, 244–250 CrossRef CAS PubMed.
- D. Quaglia, M. Pori, P. Galletti, E. Emer, F. Paradisi and D. Giacomini, Process Biochem., 2013, 48, 810–818 CrossRef CAS.
- L. Cerioli, M. Planchestainer, J. Cassidy, D. Tessaro and F. Paradisi, J. Mol. Catal. B: Enzym., 2015, 120, 141–150 CrossRef CAS.
- M. Planchestainer, M. L. Contente, J. Cassidy, F. Molinari, L. Tamborini and F. Paradisi, Green Chem., 2017, 19, 372–375 RSC.
- M. L. Contente and F. Paradisi, Nat. Catal., 2018, 1, 452–459 CrossRef CAS.
- C. Nowak, A. Pick, L.-I. Csepei and V. Sieber, ChemBioChem, 2017, 18, 1944–1949 CrossRef CAS PubMed.
- M. Romero-Fernández and F. Paradisi, Curr. Opin. Chem. Biol., 2020, 55, 1–8 CrossRef PubMed.
- G. Fernández-Lorente, F. Lopez-Gallego, J. Bolivar, J. Rocha-Martin, S. Moreno-Perez and J. Guisan, Curr. Org. Chem., 2015, 19, 1719–1731 CrossRef.
- J. M. Bolivar, L. Wilson, S. A. Ferrarotti, J. M. Guisán, R. Fernández-Lafuente and C. Mateo, J. Biotechnol., 2006, 125, 85–94 CrossRef CAS PubMed.
- J. Rocha-Martin, A. Acosta, J. M. Guisan and F. López-Gallego, ChemCatChem, 2015, 7, 1939–1947 CrossRef CAS.
- S. Velasco-Lozano, E. S. da Silva, J. Llop and F. López-Gallego, ChemBioChem, 2018, 19, 395–403 CrossRef CAS PubMed.
- L. Trobo-Maseda, A. H. Orrego, J. M. Guisan and J. Rocha-Martin, Int. J. Biol. Macromol., 2020, 157, 510–521 CrossRef CAS PubMed.
- J. Rocha-Martín, B. de las Rivas, R. Muñoz, J. M. Guisán and F. López-Gallego, ChemCatChem, 2012, 4, 1279–1288 CrossRef.
- M. Romero-Fernández, S. Moreno-Perez, A. H. Orrego, S. de Oliveira, R. I. Santamaría, M. Díaz, J. M. Guisan and J. Rocha-Martin, Bioresour. Technol., 2018, 266, 249–258 CrossRef PubMed.
- M. L. Contente and F. Paradisi, Nat. Catal., 2018, 1, 452–459 CrossRef CAS.
- C. Mateo, G. Fernández-Lorente, O. Abian, R. Fernández-Lafuente and J. M. Guisán, Biomacromolecules, 2000, 1, 739–745 CrossRef CAS PubMed.
- C. Mateo, R. Torres, G. Fernández-Lorente, C. Ortiz, M. Fuentes, A. Hidalgo, F. López-Gallego, O. Abian, J. M. Palomo, L. Betancor, B. C. C. Pessela, J. M. Guisan and R. Fernández-Lafuente, Biomacromolecules, 2003, 4, 772–777 CrossRef CAS PubMed.
- S. Velasco-Lozano, A. I. Benítez-Mateos and F. López-Gallego, Angew. Chem., Int. Ed., 2017, 56, 771–775 CrossRef CAS PubMed.
- A. I. Benítez-Mateos, M. L. Contente, S. Velasco-Lozano, F. Paradisi and F. López-Gallego, ACS Sustainable Chem. Eng., 2018, 6, 13151–13159 CrossRef.
- L. Illanes, A. González, J. M. Gómez, J. M. Valencia and P. Wilson, Electron. J. Biotechnol., 2010, 13, 1 Search PubMed.
- L. Yang and K. F. Jensen, Org. Process Res. Dev., 2013, 17, 927–933 CrossRef CAS.
- M. P. Thompson, S. R. Derrington, R. S. Heath, J. L. Porter, J. Mangas-Sanchez, P. N. Devine, M. D. Truppo and N. J. Turner, Tetrahedron, 2019, 75, 327–334 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01095f |
| This journal is © The Royal Society of Chemistry 2021 |