
External stimuli-responsive nanoparticles for spatially and temporally controlled delivery of CRISPR–Cas genome editors
Ruosen
Xie
 ab,
Yuyuan
Wang
bc and
Shaoqin
Gong
*abcd
ab,
Yuyuan
Wang
bc and
Shaoqin
Gong
*abcd
aDepartment of Materials Science and Engineering, University of Wisconsin–Madison, Madison, Wisconsin 53706, USA
bWisconsin Institute for Discovery, University of Wisconsin–Madison, Madison, Wisconsin 53715, USA
cDepartment of Biomedical Engineering, University of Wisconsin–Madison, Madison, Wisconsin 53715, USA
dDepartment of Chemistry, University of Wisconsin–Madison, Madison, Wisconsin 53706, USA. E-mail: shaoqingong@wisc.edu
First published on 29th June 2021
Abstract
The CRISPR–Cas9 system is a powerful tool for genome editing, which can potentially lead to new therapies for genetic diseases. To date, various viral and non-viral delivery systems have been developed for the delivery of CRISPR–Cas9 in vivo. However, spatially and temporally controlled genome editing is needed to enhance the specificity in organs/tissues and minimize the off-target effects of editing. In this review, we summarize the state-of-the-art non-viral vectors that exploit external stimuli (i.e., light, magnetic field, and ultrasound) for spatially and temporally controlled genome editing and their in vitro and in vivo applications.
 Ruosen Xie | Mr Ruosen Xie obtained his M.S. degree from the University of Wisconsin–Madison. He is currently a Ph.D. candidate in Prof. Shaoqin Gong's research group. His research interest is in the development of novel non-viral vectors for in vitro and in vivo delivery of CRISPR genome editors. |
 Yuyuan Wang | Dr Yuyuan Wang obtained his Ph.D. degree from the University of Wisconsin–Madison in 2019. He is currently a postdoctoral associate in Prof. Shaoqin Gong's research group. His research interest is in the development of novel non-viral vectors for in vitro and in vivo delivery of drugs, nucleic acids and CRISPR genome editors. |
 Shaoqin Gong | Prof. Shaoqin “Sarah” Gong obtained her Ph.D. degree from the University of Michigan-Ann Arbor in 1999. She is currently a Vilas Distinguished Professor at the University of Wisconsin–Madison. Her current research focuses on the design, synthesis, characterization, and evaluation of innovative biomaterials for various biomedical applications including drug delivery and tissue engineering. |
1. Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)-associated (Cas) nuclease systems are revolutionary genome editing tools developed in recent years.1 CRISPR–Cas systems were first discovered in microbes, which possess adaptive immunity against foreign genes. Such immunity employs RNA-guided nucleases to bind and cut the invading nucleic acid with specific sequences.1 One of these systems is CRISPR–Cas9, a class 2 type II DNA-targeting endonuclease that offers precise and efficient genome editing in mammalian cells, and thus it is of great interest.2–4 A CRISPR–Cas9 nuclease assembles with a CRISPR RNA (crRNA, complementary to the target sequence) and a trans-activating crRNA (tracrRNA, binding scaffold for Cas9), or a single guide RNA (sgRNA, a fusion of crRNA and tracrRNA), to form a ribonucleoprotein (RNP).1,2 The Cas9 RNP can bind with the targeting sequence near the protospacer adjacent motif (PAM) in the genomic DNA. Thereafter, Cas9 generates a double-strand break guided by sequence-specific recognition of the crRNA.1,2 Genome editing occurs during the cellular DNA repair pathways, including (1) non-homologous ending joining (NHEJ), which induces random insertions or deletions and thus causes gene disruption, (2) homology-directed repair (HDR) in the presence of a donor DNA template, which allows precise editing, and (3) microhomology-mediated end joining (MMEJ), which generates large deletions and insertions.Given that CRISPR–Cas9 allows accurate, efficient, and adaptable genome editing in mammalian cells, it has been investigated as a potential therapeutic strategy for genetic diseases, (e.g., sickle cell disease, β-thalassemia, and Duchenne muscular dystrophy)5–8 and non-genetic diseases (e.g., cancers).9,10 However, for in vivo applications, the development of vectors enabling safe, efficient, and targeted delivery of the CRISPR–Cas9 machinery remains challenging. Viral vectors, including adenovirus, adeno-associated virus, retrovirus, and lentivirus, have been utilized for CRISPR–Cas9 delivery. While viral vectors may induce high genome editing efficiencies, they also face several challenges, including low packaging capacity, immunogenicity, potential mutagenesis, and high cost for large-scale manufacturing.11,12 Non-viral vectors, on the other hand, have the potential to overcome these shortcomings. Non-viral vectors can be engineered to deliver DNA, mRNA (Cas9 mRNA + sgRNA), or RNP as the payload for CRISPR–Cas9 genome editing. Among these different forms of CRISPR genome editors, mRNA and RNP may be more desirable because they can provide relatively more transient and more efficient genome editing with limited off-target effects, by avoiding cellular transcription and/or translation. In contrast, DNA tends to express Cas9 more persistently and consequently leads to more off-target DNA cleavages and an immune response to Cas9 nuclease.13,14
Non-viral vectors can be also equipped with stimuli-responsive properties to activate CRISPR–Cas9 payloads by certain physiological and/or external triggers for spatially and temporally controlled genome editing. This is an important aspect for genome editing as spatial control allows organ- or tissue-specific genome editing to limit unwanted editing in non-targeted organs or tissues, whereas the temporal control allows transient and on-demand genome editing, which reduces the off-target effect and immunogenicity generated by long-term expression and prolonged activity of genome editors. Although challenging, judiciously engineered non-viral vectors have the potential to achieve such desirable features.
It is known that the in vivo delivery of genome editing machinery needs to overcome a series of biological barriers before editing target cells (Fig. 1).12 A delivery system must (1) avoid recognition and clearance by immune cells; (2) resist enzymatic degradation by nucleases and proteases; (3) be internalized by target cells; and (4) escape from endosomes/lysosomes and release payloads into the cytoplasm. Moreover, some payloads (e.g., DNA and RNP) must be transported into the nucleus, while some payloads (e.g., DNA and mRNA) must be transcribed and/or translated to express Cas9 RNP. Engineering a stimuli-responsive delivery system to regulate genome editing is similar to adding a “switch” to the pristine vehicle. This “switch” allows turning “on” the delivery process or the activity of genome editors. Indeed, most of the reported stimuli-responsive systems follow the same principle – that is, the delivery system or the payload is intentionally engineered unable to overcome one specific biological barrier. As a result, the function of the payload is turned off. However, in the presence of a stimulus, the function of the payload is turned on as the stimulus serves as a trigger to enable the delivery system or the payload to overcome the specific biological barrier. Nanovectors that harness particular internal stimuli (e.g., pH,15,16 glutathione,17–22 ATP,23 and other substances inside the cell24) in tissues or cells have been developed recently for in vivo stimuli-responsive genome editing (reviewed previously11,12,25–30). In this review, we focus on the recent developments in non-viral vectors that can be triggered by external stimuli (i.e., light, magnetic field, and ultrasound) for CRISPR–Cas genome editor delivery to achieve spatially and temporally controlled genome editing (Table 1).
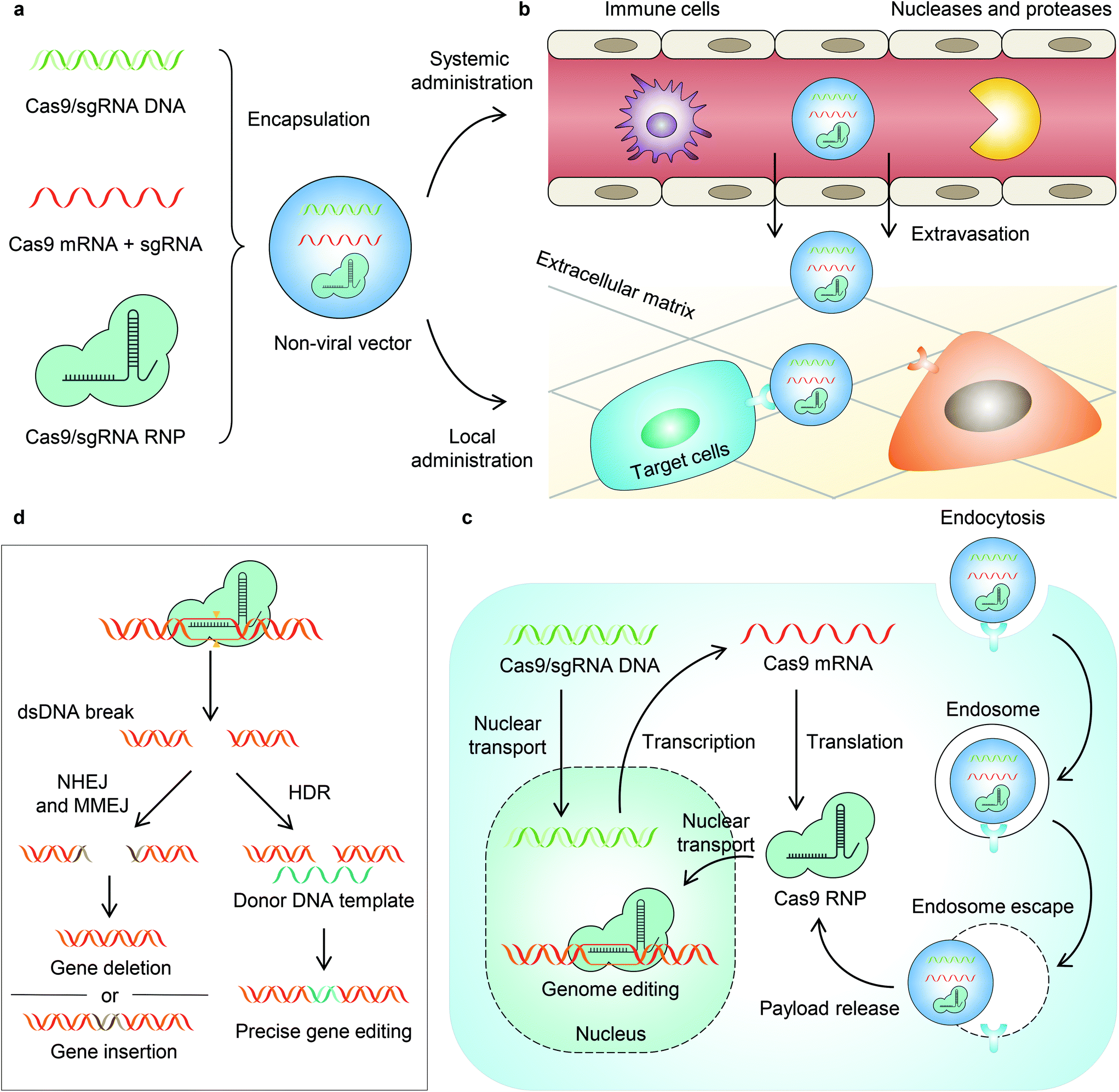 | ||
| Fig. 1 Schematic illustration of nanoparticle-enabled in vivo delivery of genome editors, from nanoparticle fabrication to genome editing in the nucleus. a. Nanoparticle preparation and administration. CRISPR genome editors (e.g., DNA, mRNA, sgRNA, and Cas9 RNP) are first encapsulated in the non-viral nanoparticles, which can then be administered through either systemic or local injections. b. Extracellular barriers for delivery. The nanoparticles encapsulating genome editors must minimize the recognition and clearance by immune cells and protect payloads from enzymatic degradation by nucleases and proteases. After extravasation from the bloodstream or local administration, the nanoparticle must also diffuse through the extracellular matrix and get internalized by target cells via endocytosis. c. Intracellular barriers for delivery. The endocytosed nanoparticles need to escape from endosomes/lysosomes and then release payloads into the cytoplasm. The released DNA and mRNA have to undergo transcription and/or translation to express Cas9 RNPs, which enter the nuclease for genome editing. d. Genome editing mechanisms. Genome editing occurs after Cas9 nuclease binds the target gene sequence and generates a dsDNA break, via NHEJ, MMEJ, or HDR. NHEJ is an error-prone pathway that induces gene deletion or gene insertion. MMEJ generates large deletions and insertions. HDR occurs in the presence of a donor DNA template to allow precise genome editing. | ||
| Major componenta | Payload | Stimuli | Injection routeb | In vivo targets/models | Ref. |
|---|---|---|---|---|---|
| a CPP, cell-penetrating peptide; Au NP, gold nanoparticle; PEI, polyethyleneimine; Au NR, gold nanorod; UCNP, upconverting nanoparticle; MNP, magnetic nanoparticle; and PLGA, poly(lactide-co-glycolide). b i.t., intratumoral; i.m, intramuscular; i.v. intravenous. | |||||
| CPP–Au NP with lipids | Plasmid | Light, 514 nm | i.t. | Melanoma | 31 |
| PEI-coated Au NR | Heat-inducible plasmid | Light, 1064 nm | i.m., i.t., i.v. | Muscle carcinoma, and hepatic failure | 32 |
| PEI-coated Au NR | Heat-inducible plasmid | Light, 1064 nm | i.t. | Metastatic melanoma | 33 |
| Semiconducting polymer | Plasmid | Light, 808 nm | i.t. | Colorectal carcinoma | 34 |
| Semiconducting polymer | Plasmid | Light, 680 nm | Ex vivo | Adenocarcinoma | 37 |
| Self-assembled amphiphilic polymer | Cas9 RNP | Light, 671 nm | i.v. | Nasopharyngeal carcinoma | 38 |
| Liposome | Cas9 RNP | Light, 690 nm | Microinjection in zygotes | N/A | 39 |
| PEI-coated UCNP | Cas9 RNP | Light, 980 nm | i.t. | Carcinoma | 41 |
| UCNP with charge reversal polymer | Plasmid | Light, 980 nm | i.t. | Hepatocellular carcinoma | 43 |
| Baculovirus vector with MNP | Plasmid | Magnetic field | i.t., i.v. | Hepatocellular carcinoma, liver | 48 |
| Au nanowire | Cas9 RNP | Ultrasound | N/A | N/A | 56 |
| Liposome with microbubble | Cas9 RNP | Ultrasound | Tropical | Androgenic alopecia | 57 |
| PLGA NPs with lipid coating and microbubble | Plasmid | Focused ultrasound | i.v. | Glioblastoma | 60 |
2. Photoresponsive non-viral CRISPR–Cas9 delivery nanosystems
Light-controlled delivery of genome editors is desirable due to its distinctive features, including its non-invasiveness, spatial specificity, and temporal control ability.31 Photoactivatable Cas9 RNPs, consisting of photocaged or split Cas9 nucleases, have been developed and reviewed recently.31 However, most of these systems can only be triggered by UV or visible light, which limit their in vivo applications, because tissues can absorb and scatter the light at these wavelengths and result in low penetration depth (<1 mm).31,32 Moreover, these approaches are more focused on the development of a Cas9 RNP payload, which still requires a carrier for intracellular delivery for their functions. In comparison with the sophisticated, bio-engineered Cas9 RNP, photoresponsive non-viral vectors have two advantages: (1) far-red and near-infrared lights can be used as triggers, thereby achieving deeper penetration into tissues; and (2) regular payloads, instead of specially bioengineered payloads, can be used for this approach, thereby ensuring high genome editing efficiency and specificity and scalability for manufacturing.2.1. Photothermal approaches
The photothermal effect is a phenomenon associated with electromagnetic radiation. Nanoparticles have been engineered as photothermal agents for biomedical applications, particularly for photothermal therapy.33,34 In terms of biomolecule delivery, the heat generated upon light irradiation can induce payload release from the delivery system, facilitate endosome/lysosome escape of the payload, and/or trigger the activity of CRISPR genome editors. In this way, genome editing can be spatially and temporally controlled by light.The first delivery system following this concept was reported in 2017 by Wang and Zhang et al.35 In this work, the nanoplatform was fabricated with gold nanoparticles, which generate heat upon light irradiation due to the localized surface plasmon resonance effect. Cell-penetrating peptide-conjugated gold nanoparticles were complexed with Cas9/sgRNA plasmids. The complex was then coated with cationic lipids to construct a 101 nm-sized nanosystem with a zeta potential of +36 mV (Fig. 2a). The photothermal effect generated by laser irradiation (514 nm, 24 mW cm−2, 20 min) was sufficient to induce the disassembly of the nanoparticle to release the payload and facilitate the endosome/lysosome escape of the payload, leading to efficient genome editing. The in vivo genome editing was studied in a xenograft melanoma mouse model, in which the nanoparticle was intratumorally injected and the tumor site was irradiated with light. The tumor growth was then significantly inhibited due to the nanoparticle-mediated disruption of Plk-1, a gene that is overexpressed in melanoma and contributes to tumor progression.
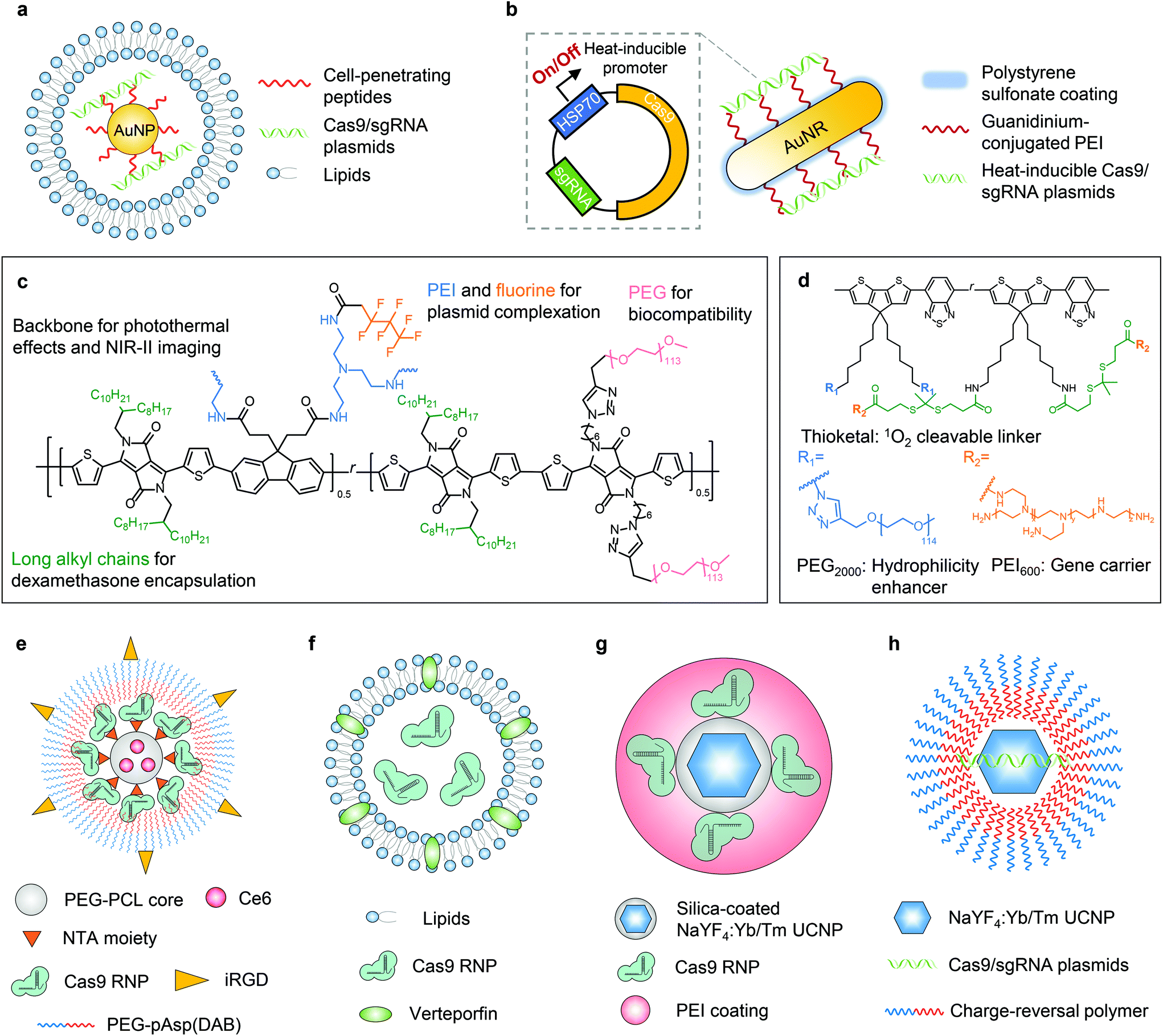 | ||
| Fig. 2 Schematic illustrations of photoresponsive CRISPR–Cas9 delivery nanosystems. a. Cell-penetrating peptide-conjugated gold nanoparticles were complexed with Cas9/sgRNA plasmids, which were then coated with cationic lipids to yield a nanosystem. b. Gold nanorods were coated with PEI and complexed with a RNP-expressing plasmid. The plasmid contained a heat-inducible promoter, HSP70, thus the elevated local temperature induced by external light can switch on the expression of Cas9/sgRNA. c. SPPF was constructed by sequentially conjugating alkyl side chains, polyethylene glycol (PEG) chains, and fluorinated PEI to the backbone of the initial semiconducting polymer. d. pSPN polymer backbone was able to generate singlet oxygen under NIR irradiation, whereas PEI brushes were conjugated through thioketal moieties which can be cleaved by singlet oxygen. e. T-CC-NP had a core–shell structure. The micellar core was formed by NTA-PEG-PCL to encapsulate a photosensitizer Ce6. The shell was constructed by His-tagged Cas9 RNP, which binds to the NTA moiety through nickel coordination, and the Cas9 RNP was then coated with iRGD-PEG-pAsp(DAB). f. The liposome system delivering the Cas9 RNP was constructed by the lipid bilayer incorporated with a clinically used photosensitizer verteporfin. g. The NaYF4:Yb/Tm UCNP was coated with a silica shell, where Cas9 RNPs were covalently conjugated via o-nitrobenzyl ester linkages. The UCNPs–Cas9 conjugates were thereafter complexed with PEI. h. Cas9/sgRNA plasmids were complexed with NaYF4:Yb/Tm UCNPs with a bridging layer of the charge-reversal polymer. | ||
The photothermal effect of gold nanomaterials was also utilized in another system.36 Gold nanorods, serving as thermal transducers, were coated with cationic polymer polyethyleneimine (PEI) and complexed with RNP-expressing plasmids (cationic polymer-coated Au nanorods, termed “APCs”, Fig. 2b). The plasmid contained a heat-inducible promoter, HSP70, and thus the elevated local temperature induced by external light can switch on the expression of Cas9/sgRNA to achieve light-controlled programmable genome editing. APCs can be activated by light at 1064 nm (0.33 W cm−2, 5–30 min) due to the photothermal effect of gold nanorods. NIR within the second near-infrared (NIR-II) optical window (1000–1700 nm) can provide deeper tissue penetration for in vivo applications than NIR-I or visible light.31 The in vivo genome editing was studied in (1) a transplantation model via ex vivo cell transfection; (2) muscles via intramuscular injection; (3) xenograft tumor via peritumoral injection; and (4) liver via intravenous injection, respectively. The in vivo therapeutic effects were further investigated in a xenograft tumor model with the peritumoral injection of APCs, which efficiently interrupted the expression of Plk-1 and led to inhibition of tumor growth. The galactose-modified APCs can also treat fulminant hepatic failure after intravenous injection, while genome editing in diseased livers restored the liver functions. Remarkably, the optogenetic activation of Cas9/sgRNA expression was proved in all the aforementioned in vivo models, although the uniquely engineered plasmid employing the heat-inducible promotor was required for this design.
Based on APCs, Tang et al. have recently expanded the application to cancer immunotherapy.37 In this work, gold nanorods were surface-modified with guanidinium-conjugated PEI to promote tissue penetration and intracellular delivery of CRISPR–Cas9. Given that the payload was a Cas9/sgRNA plasmid with a heat-inducible promoter, the genome editing can be controlled by photothermal effects. After peritumoral injection of nanoparticles in the primary tumor, NIR-II irradiation (1064 nm, 0.33 W cm−2, 30 min) not only triggered PD-L1 gene knockout in tumor cells, but also promoted immunogenic cell death through the photothermal effects. The synergy of PD-L1 gene disruption and photothermal therapy efficiently triggered the adaptive immunity against tumors. Therefore, significant in vivo therapeutic effects were proved in terms of suppression of primary and distant tumors, inhibition of tumor metastasis, and prevention of tumor recurrence.
Other than gold nanomaterials, semiconducting polymers can also be employed in stimuli-responsive nanoplatforms because they have excellent photothermal conversion efficiency and photostability. Li et al. reported a semiconducting polymer brush (SPPF) that can be complexed with Cas9 RNP-expressing plasmids for light-triggered remote control of genome editing.38 The SPPF was constructed by sequentially conjugating alkyl side chains, polyethylene glycol (PEG) chains, and fluorinated PEI to the backbone of the initial semiconducting polymer (Fig. 2c). Under laser exposure (808 nm, 0.45 W cm−2, 10 min), the SPPF can serve as a photothermal transducer to release the payload and facilitate endosome/lysosome escape. The SPPF also has in vivo NIR-II imaging capability following intratumoral injection. The in vivo genome editing was further verified by deep sequencing, but the therapeutic efficacy including tumor suppression and the survival rate of animals were not investigated.
2.2. Photodynamic approaches
Apart from photothermal effects, light irradiation can also be harnessed to generate reactive oxygen species (ROS, e.g., singlet oxygen molecules) in the presence of photosensitizing substances. The photodynamic approach can be used for treating cancers when the ROS level induced by a photosensitizer is sufficient to cause cell death. So far, many nanoplatforms have been developed for photodynamic therapy.39,40 Nonetheless, when the light-induced ROS level is controlled low enough to limit cytotoxicity, such a system can serve as a light-controlled delivery vehicle for biomolecules via ROS-responsive mechanisms, or promote endosome/lysosome escape of the payloads by singlet oxygen, one of the major ROS.Lyu and He et al. reported the first nanosystem applying this concept to control genome editing with a NIR photolabile semiconducting polymer nanotransducer (pSPN) design.41 The polymer backbone was able to generate singlet oxygen under NIR irradiation, whereas PEI brushes were conjugated through thioketal moieties which can be cleaved by singlet oxygen (Fig. 2d). Therefore, the release of the complexed Cas9/sgRNA plasmids was controlled by NIR. To study the in vivo editing efficiency, HeLa cells were transfected with pSPNs and subcutaneously injected into mice. With laser exposure (680 nm, 0.3 W cm−2, 20 min), genome editing was demonstrated by the expression of GFP in cells as the result of genome editing. However, the efficacy of pSPNs after local or systemic injections and the in vivo therapeutic effects need more thorough study.
Deng and Li et al. investigated a spatially controlled tumor-specific gene editing approach with synergistic drug effects.42 The nanoparticle (denoted as “T-CC-NP”) had a core–shell structure (Fig. 2e). The micellar core was formed by nitrilotriacetic acid (NTA)-terminal amphiphilic polymers (i.e., NTA-PEG-PCL) to encapsulate the photosensitizer Ce6. The shell was constructed with His-tagged Cas9 RNP, which binds to the NTA moiety through nickel coordination, and the Cas9 RNP was then coated with iRGD-PEG-pAsp(DAB) to enhance the cellular uptake of T-CC-NP and targeting of cancer cells. The resulting T-CC-NP had neutral surface charges, and the size was ∼110 nm in diameter. In vivo studies revealed that after intravenous injections, T-CC-NP rapidly accumulated in the tumor, and the tumor-specific gene editing can be activated by NIR (671 nm, 0.8 W cm−2, 1 min). Moreover, the photodynamic process also efficaciously inhibited tumor growth and exhibited a synergistic effect with Cas9-mediated genome editing. Instead of subtly controlling the generated ROS level to a critical value that can only induce endosome/lysosome escape of payloads without severe cytotoxicity, the major focus of this work was to eliminate cancer cells, so the photodynamic process was not restrained. The combination of Cas9, on the other hand, was to disrupt Nrf2, a gene that prevents cancer cells from being killed by ROS and promotes tumor progression. The spatially activated genome editing only occurred in the cells at/surrounding the tumor site that were irradiated by laser, and thus editing in normal organs and tissues was prevented. Notwithstanding the relatively elaborate fabrication process of T-CC-NP, this system is promising for spatially controlled genome editing in other organs/tissues, given that this system is applicable for systemic administration.
Aksoy et al. reported a liposome system employing a similar concept.43 A clinically used photosensitizer, verteporfin, was incorporated into the lipid bilayer (Fig. 2f). Under light illumination (690 nm, 0.15 mW cm−2, 5 min), verteporfin generated singlet oxygen which immediately oxidized the unsaturated lipid molecules, thereby leading to the destabilization of liposomes and the subsequent release of the payloads while also enhancing the endosome/lysosome escape of payloads. The in vivo light-responsive genome editing was tested in a reporter zebrafish model, where eGFP was specifically expressed in the slow-muscle fibers. Liposomes encapsulating Cas9 RNP were injected into single-cell zebrafish embryos (newly fertilized eggs through the completion of the first zygotic cell cycle). The embryos irradiated by light exhibited a dramatic decrease in the number of eGFP-positive muscle fibers at 72 hours post-fertilization, compared with those without light irradiation. One advantage of this system is its simple structure and preparation process, which is beneficial for clinical applications, yet the genome editing efficiency has not been evaluated in mammalian animals.
2.3. Photochemical approaches
To date, several photochemical reactions have been developed for biomolecule delivery, including photolysis (e.g., o-nitrobenzyl derivatives), photoisomerization (e.g., azobenzene derivatives, and spiropyran derivatives), and photopolymerization (e.g., acrylates and methacrylates).32,44 However, most photochemical reactions as those mentioned above can only be triggered by UV or visible light, which limit their in vivo applications due to the limited penetration depth of UV/visible light. Lanthanide-doped upconverting nanoparticles (UCNPs) have been developed to convert NIR light to UV or visible light to initiate the photochemical reaction while taking advantage of the NIR penetrability. Pan et al. first developed a UCNP-based nanosystem for in vivo CRISPR–Cas9 delivery.45 The NaYF4:Yb/Tm UCNP was coated with a silica shell, where Cas9 RNPs were covalently conjugated via o-nitrobenzyl ester linkages. UCNPs can covert NIR to UV light to cleave this linkage and release Cas9 RNPs. The UCNPs–Cas9 conjugates were thereafter complexed with PEI to assist endosomal escape (denoted as UCNPs–Cas9@PEI, Fig. 2g). The in vivo genome editing was studied by intratumoral injection of UCNPs–Cas9@PEI in a xenograft tumor model. Controlled by NIR radiation (980 nm, 2.0 W cm−2, 20 min pulse wave), UCNPs–Cas9@PEI efficiently edited the Plk-1 gene in tumor cells and led to inhibition of tumor growth. Although genome editing was demonstrated in a NIR-controlled manner, the site-specificity of Cas9 RNP conjugation is yet to be investigated, as the reported method might generate crosslinking of Cas9 RNPs with UCNPs and lead to a loss of catalytic activity.46Wu et al. developed another NaYF4:Yb/Tm UCNP-based nanosystem for NIR-controlled genome editing.47 Instead of covalently conjugating Cas9 RNPs, Cas9/sgRNA plasmids were complexed to this system with a bridging layer of charge-reversal polymers (Fig. 2h). Under the irradiation of NIR (980 nm, 1.5 W cm−2, 30 min), UV light emitted from UCNPs can break the o-nitrobenzyl ester linker in the polymer to expose the carboxyl groups. Therefore, the original cationic polymer was converted to an anionic polymer, leading to the release of anionic plasmids by electrostatic repulsion, although the subcellular trafficking pathway was not investigated. After the intratumoral injection in a xenograft tumor model, the nanoparticle efficiently inhibited the expression of Plk-1, so the tumor growth was effectively suppressed. This system has the potential to be re-engineered to deliver mRNA or RNP, which can provide more transient genome editing to further reduce latent genotoxicity.
As described above, several promising photoresponsive systems have been developed for spatial and temporal control of genome editing. Currently, the major limitation of these systems is still the restricted light penetration depth in tissues, even with NIR-II, which at most can penetrate a few millimeters below the surface of the skin.31,48 For clinical translation, large animals (e.g., porcine models and non-human primates) demand much deeper penetration of light signals to optically control genome editing in specific regions of interest. Other light sources with excellent penetrability, like X-ray, could be potentially utilized to engineer non-viral vectors for genome editing. In addition, photoresponsive delivery systems are often fabricated with inorganic nanoparticles, including gold nanoparticles and lanthanide-based upconverting nanoparticles. These inorganic nanoparticles may not be excreted from the body by renal clearance and thus potentially induce long-term toxicity.31 Biodegradable inorganic nanoparticles, renal clearable inorganic nanoparticles, or biocompatible small molecule or macromolecule alternatives are thus optimal for building photoresponsive delivery systems. Future studies also need to pay more attention to the biosafety issue, as photothermal and photodynamic approaches may potentially generate excessive heat and singlet oxygen and cause DNA damage.
3. Magnetic responsive non-viral CRISPR–Cas9 delivery nanosystems
The magnetic field can act on nucleic acid delivery vectors associated with magnetic nanoparticles, direct vectors towards target cells, and lead to efficient nucleic acid delivery. This method is termed “magnetofection” and has been investigated and developed for the last two decades for in vitro and in vivo applications.49,50 For in vivo genome editing, utilizing magnetic forces can not only promote the cell entry of genome editor-encapsulated vectors, but more importantly, also provide a spatial control that facilitates the vector accumulation in certain organs or tissues for targeted genome editing. Notably, the magnetic field can provide deep penetration depth in animal tissues, which makes it an attractive stimulus for clinical translation.51A magnetic responsive vector for in vivo genome editing was first reported by Zhu et al. in 2018.52 Recombinant baculovirus vectors (BVs) have a large DNA packaging capacity (>38 kbps) and can transduce many types of mammalian cells efficiently. BVs cannot replicate in mammalian cells or cause genome integration, so the transgene expression is transient. However, in vivo applications of BVs were hindered because BVs can activate the complement system which then inactivates BVs. Employing this property, the serum inactivation of BVs was used as an “off” switch to circumvent BVs from inducing systemic genome editing. To incorporate an “on” switch that can be triggered by magnetic fields, the BVs were complexed with magnetic nanoparticles (i.e., MNP–BVs). The MNPs were covalently conjugated with cell-penetrating peptide-PEG to facilitate complexation with BVs and enhance cellular uptake (Fig. 3). Under a locally administered magnetic field, MNP–BVs were able to overcome the inactivation and led to spatially controlled genome editing in subcutaneous tumors via systemic or intratumoral injection, or in the liver via systemic administration. A hybrid vector with viral and non-viral compositions provides a smart approach enabling spatiotemporal control and ensuring efficient genome editing simultaneously. However, the immunogenicity of the vector needs more investigation.
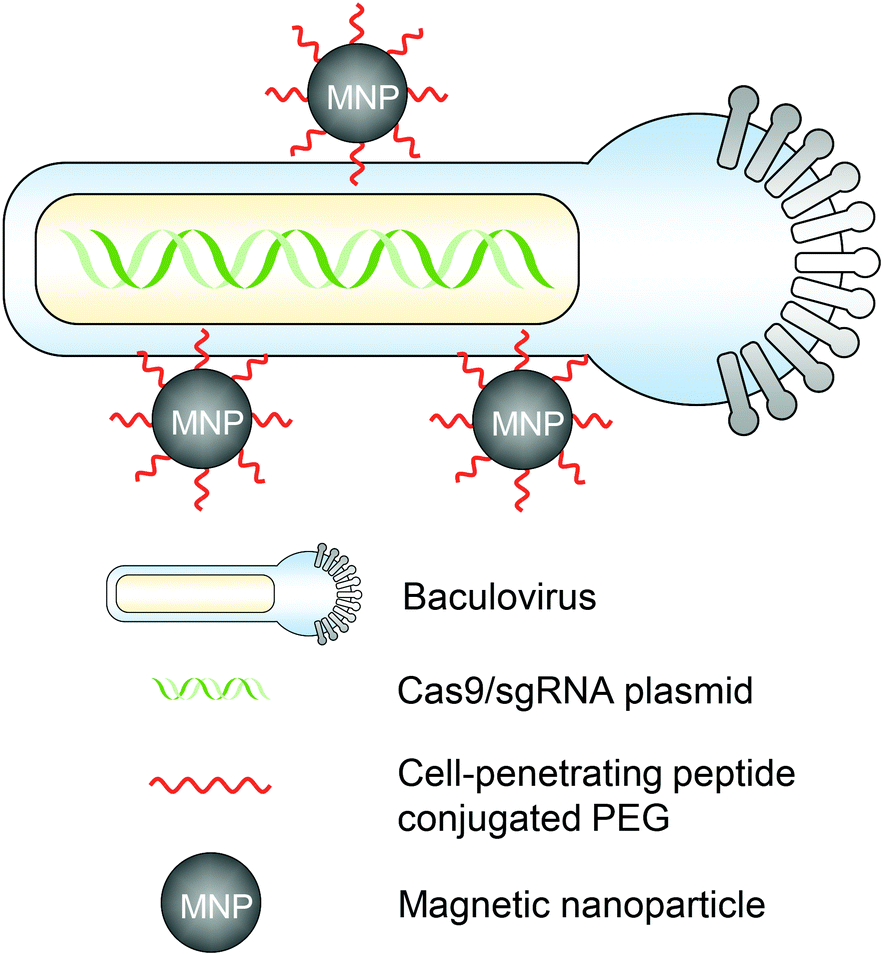 | ||
| Fig. 3 A schematic illustration of the magnetic responsive CRISPR–Cas9 delivery nanosystem. MNP–BVs were formed by the complexation of BVs with MNPs. The MNPs were covalently conjugated with cell-penetrating peptide–PEG to facilitate complexation with BVs via electrostatic interactions. | ||
MNP–BVs is currently the only system for in vivo genome editing controlled by a magnetic field, although so far, many magnetic responsive delivery systems have been reported for chemotherapy or gene therapy.53,54 Therefore, there is still room for the development and optimization of novel platforms following this concept. One major obstacle is that magnetic responsive systems, in order to respond to the magnetic field, are usually composed of superparamagnetic iron oxide nanoparticles, which raise the concern about their long-term accumulation in organs and tissues and potential toxicity.55–57 Future studies may resolve these problems by using magnetic nanoparticles that possess better biocompatibility, faster biodegradation, and/or capability to be removed via renal clearance.
4. Ultrasound-responsive non-viral CRISPR–Cas9 delivery nanosystems
Ultrasound can facilitate the transport of cell membrane-impermeable compounds into living cells by temporally inducing cell membrane openings, and thus has been developed in the last two decades for biomolecule delivery, which is also termed “sonoporation”.50,58,59 Similar to the magnetic field, ultrasound can also provide spatial and temporal control of engineered vectors to enable organ- or tissue-specific genome editing.The first ultrasound-responsive vector for CRISPR–Cas9 intracellular delivery was reported by Hansen-Bruhn et al. in 2018.60 Cas9 RNPs were immobilized onto the gold nanowire (i.e., nanomotor) surface through disulfide bonds (Fig. 4a). Propelled by ultrasound, the nanomotor can move actively and get internalized into the cytoplasm of cells, where the disulfide bond can be cleaved by glutathione, and the Cas9 RNP can be released to induce genome editing. The nanomotor exposed to ultrasound for 5 min induced up to 80% gene knockout efficiency in B16F10 melanoma cells, compared to 30% generated by its static counterpart. The promising features of the nanomotor favor various applications, but its in vivo genome editing efficacy remains unexplored.
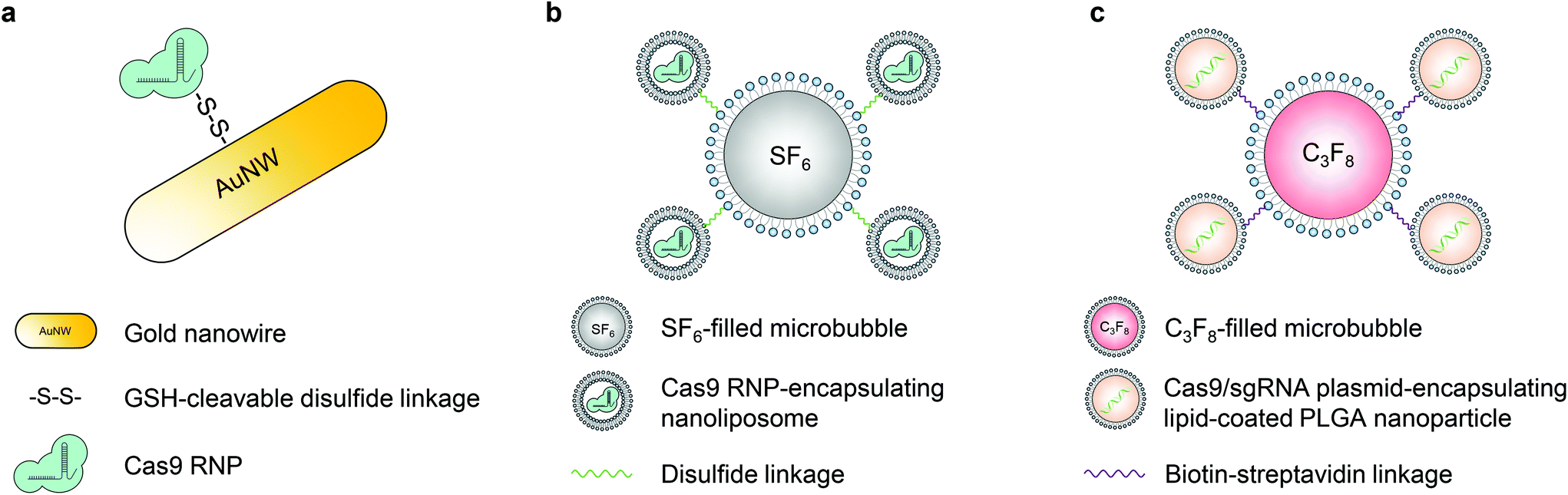 | ||
| Fig. 4 Schematic illustrations of ultrasound-responsive CRISPR–Cas9 delivery nanosystems. a. Cas9 RNPs were immobilized onto the gold nanowire (i.e., nanomotor) surface through disulfide bonds. b. The Cas9 RNP was first encapsulated in ∼100 nm nanoliposomes via a film hydration method. The nanoliposome was then conjugated to a sulfur hexafluoride-filled microbubble via a disulfide linkage, resulting in a microbubble–nanoliposome complex. c. LPHNs with a PLGA core and a lipid coating were used to encapsulate Cas9 RNP-expressing plasmids. LPHNs were then decorated with cRGD and conjugated to octafluoropropane-filled microbubbles through biotin–avidin interactions. | ||
Microbubbles consist of gas-filled cores and stabilized shells.58 This unique structure enables microbubbles to amplify the biophysical effects of ultrasound by cavitation. The oscillation of microbubbles upon ultrasound exposure leads to the formation of transient pores in the cell membrane, so microbubbles have been applied in sonoporation to enhance the cell membrane permeability.58 Multiple types of microbubbles (and nanobubbles) have been developed so far with various features.61,62 Ryu et al. developed an ultrasound-activatable microbubble conjugated nanoliposome system for Cas9 RNP delivery to treat androgenic alopecia.63 The Cas9 RNP was first encapsulated in ∼100 nm nanoliposomes via a film hydration method. The nanoliposome was then conjugated to a sulfur hexafluoride-filled microbubble via a disulfide linkage, resulting in a microbubble–nanoliposome complex (MB–NL) with a size of ∼1.2 μm (Fig. 4b). The MB–NL encapsulating Cas9 RNPs targeting the mouse SRD5A2, a gene that is responsible for the pathogenesis of male pattern baldness, was then tested in a testosterone-induced androgenic alopecia mouse model. It was found that mice topically treated with both SRD5A2-targeting MB–NLs and ultrasound exhibited similar hair generation (up to 90%) to healthy mice at the 7th week, while its counterpart without ultrasound treatment showed a negligible therapeutic effect. Indeed, ultrasound increased the penetration of MB–NLs into dermal papilla cells, where the knockout of SRD5A2 reduced the apoptosis of dermal papilla cells and facilitated the proliferation of hair germ cells. Therefore, hair regeneration was promoted. Notably, this platform can be potentially developed for treating skin diseases, for example, melanoma.
Another important application of ultrasound in the delivery of biomolecules is the use of focused ultrasound (FUS). The blood–brain barrier (BBB) is a major impediment to the delivery of biomolecules into the central nervous system. In the presence of microbubbles and with the use of FUS, the permeability of the BBB can be significantly enhanced. A non-invasive FUS treatment can temporally (several hours), reversibly, and locally disrupt the BBB, providing a window for the substances in the bloodstream to transport into the brain.64,65 Combining FUS with nanoparticles delivering CRISPR can thus potentially enable spatially controlled genome editing in the brain. Recently, a lipid-polymer hybrid nanoparticle (LPHN) was reported for this purpose.66 LPHNs (∼180 nm in diameter) with a PLGA core and a lipid coating were used to encapsulate Cas9 RNP-expressing plasmids. LPHNs were then decorated with cRGD and conjugated to octafluoropropane-filled microbubbles (∼1.8 μm in diameter) through biotin–avidin interactions (Fig. 4c).66,67 The in vivo genome editing was tested in a T98G orthotopic xenograft model. FUS in combination with microbubbles significantly promoted nanoparticle delivery to the tumor in the brain after intravenous injections. Effective knockout of O6-methylguanine-DNA methyltransferase expression suppressed tumor growth and enhanced the mouse survival rate. However, the effect of microbubble-mediated tumor vasculature damage on tumor suppression has not been explored.
An ultrasound-responsive delivery system for genome editing is of great interest, particularly because ultrasound can temporally and non-invasively disrupt the BBB, which is notoriously challenging for delivery to the central nervous system via systemic administration. However, further studies are required to investigate the efficacy and specificity of ultrasound-mediated genome editing in the brain.
5. Conclusion and outlook
Although a number of stimuli-responsive nanoplatforms have emerged for spatially and temporally controlled genome editing, there is still a need to develop more advanced nanovectors for more specific, efficient, and safe genome editing controlled by various modalities. Most of the reviewed systems were locally administered (e.g., via intratumoral injection), and external stimuli were only applied around the injection sites. This may not be sufficient to fully evaluate the efficacy of the spatial control of genome editing, as spatial control is more beneficial for carriers that are systemically administered for organ-/tissue-specific genome editing. Additionally, the temporal control of genome editing has been less studied. The transient activity of genome editors is crucial to limit the off-target effects of genome editing. Furthermore, the spatially and temporally controlled genome editing systems reported thus far are mostly focused on delivering CRISPR–Cas9 for NHEJ in target cells. The controlled delivery of donor DNA templates allowing precise gene correction via HDR has not been explored. Stimuli-responsive delivery systems for other genome editors, such as Cas12 for DNA editing, Cas13 for RNA editing, dCas9 for transcriptional regulation and epigenetic modifications, base editors (e.g., cytosine and adenine base editors), and prime editors, are of interest for broader genome editing applications.68 Besides, the large-scale manufacturing of effective but complicated nanoparticles following Good Manufacturing Practice (GMP) guidelines may be challenging. We envision that, with more development and understanding, the spatiotemporally controlled genome editing approaches will continue to enrich and fulfill the requirements for clinical translation in the near future.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to acknowledge the financial support of the National Institutes of Health (R01HL129785, 1-UG3-NS-111688-01, R01HL143469, 1R24 EY032434).References
- G. J. Knott and J. A. Doudna, Science, 2018, 361, 866–869 CrossRef CAS PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto, N. Habib, P. D. Hsu, X. Wu, W. Jiang, L. A. Marraffini and F. Zhang, Science, 2013, 339, 819–823 CrossRef CAS PubMed.
- P. Mali, L. Yang, K. M. Esvelt, J. Aach, M. Guell, J. E. DiCarlo, J. E. Norville and G. M. Church, Science, 2013, 339, 823–826 CrossRef CAS PubMed.
- C. Long, J. R. McAnally, J. M. Shelton, A. A. Mireault, R. Bassel-Duby and E. N. Olson, Science, 2014, 345, 1184–1188 CrossRef CAS PubMed.
- C. Long, L. Amoasii, A. A. Mireault, J. R. McAnally, H. Li, E. Sanchez-Ortiz, S. Bhattacharyya, J. M. Shelton, R. Bassel-Duby and E. N. Olson, Science, 2016, 351, 400–403 CrossRef CAS PubMed.
- C. E. Nelson, C. H. Hakim, D. G. Ousterout, P. I. Thakore, E. A. Moreb, R. M. C. Rivera, S. Madhavan, X. Pan, F. A. Ran, W. X. Yan, A. Asokan, F. Zhang, D. Duan and C. A. Gersbach, Science, 2016, 351, 403–407 CrossRef CAS PubMed.
- H. Frangoul, D. Altshuler, M. D. Cappellini, Y.-S. Chen, J. Domm, B. K. Eustace, J. Foell, J. de la Fuente, S. Grupp, R. Handgretinger, T. W. Ho, A. Kattamis, A. Kernytsky, J. Lekstrom-Himes, A. M. Li, F. Locatelli, M. Y. Mapara, M. de Montalembert, D. Rondelli, A. Sharma, S. Sheth, S. Soni, M. H. Steinberg, D. Wall, A. Yen and S. Corbacioglu, N. Engl. J. Med., 2020, 384, 252–260 CrossRef PubMed.
- H. Yin, W. Xue and D. G. Anderson, Nat. Rev. Clin. Oncol., 2019, 16, 281–295 CrossRef CAS PubMed.
- F. J. Sánchez-Rivera and T. Jacks, Nat. Rev. Cancer, 2015, 15, 387–393 CrossRef PubMed.
- S. Tong, B. Moyo, C. M. Lee, K. Leong and G. Bao, Nat. Rev. Mater., 2019, 4, 726–737 CrossRef CAS PubMed.
- H. Yin, K. J. Kauffman and D. G. Anderson, Nat. Rev. Drug Discovery, 2017, 16, 387 CrossRef CAS PubMed.
- J. A. Zuris, D. B. Thompson, Y. Shu, J. P. Guilinger, J. L. Bessen, J. H. Hu, M. L. Maeder, J. K. Joung, Z.-Y. Chen and D. R. Liu, Nat. Biotechnol., 2015, 33, 73–80 CrossRef CAS PubMed.
- C. T. Charlesworth, P. S. Deshpande, D. P. Dever, J. Camarena, V. T. Lemgart, M. K. Cromer, C. A. Vakulskas, M. A. Collingwood, L. Zhang, N. M. Bode, M. A. Behlke, B. Dejene, B. Cieniewicz, R. Romano, B. J. Lesch, N. Gomez-Ospina, S. Mantri, M. Pavel-Dinu, K. I. Weinberg and M. H. Porteus, Nat. Med., 2019, 25, 249–254 CrossRef CAS PubMed.
- Y. Wang, P. K. Shahi, R. Xie, H. Zhang, A. A. Abdeen, N. Yodsanit, Z. Ma, K. Saha, B. Pattnaik and S. Gong, J. Controlled Release, 2020, 324, 194–203 CrossRef CAS PubMed.
- S. K. Alsaiari, S. Patil, M. Alyami, K. O. Alamoudi, F. A. Aleisa, J. S. Merzaban, M. Li and N. M. Khashab, J. Am. Chem. Soc., 2017, 140, 143–146 CrossRef PubMed.
- G. Chen, A. A. Abdeen, Y. Wang, P. K. Shahi, S. Robertson, R. Xie, M. Suzuki, B. R. Pattnaik, K. Saha and S. Gong, Nat. Nanotechnol., 2019, 14, 974–980 CrossRef CAS PubMed.
- G. Chen, B. Ma, Y. Wang and S. Gong, ACS Appl. Mater. Interfaces, 2018, 10, 18515–18523 CrossRef CAS PubMed.
- Y. Wang, B. Ma, A. A. Abdeen, G. Chen, R. Xie, K. Saha and S. Gong, ACS Appl. Mater. Interfaces, 2018, 10, 31915–31927 CrossRef CAS PubMed.
- J. Liu, J. Chang, Y. Jiang, X. Meng, T. Sun, L. Mao, Q. Xu and M. Wang, Adv. Mater., 2019, 1902575 CrossRef PubMed.
- M. Wang, J. A. Zuris, F. T. Meng, H. Rees, S. Sun, P. Deng, Y. Han, X. Gao, D. Pouli, Q. Wu, I. Georgakoudi, D. R. Liu and Q. Xu, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2868–2873 CrossRef CAS PubMed.
- R. Rouet, B. A. Thuma, M. D. Roy, N. G. Lintner, D. M. Rubitski, J. E. Finley, H. M. Wisniewska, R. Mendonsa, A. Hirsh, L. de Oñate, J. Compte Barrón, T. J. McLellan, J. Bellenger, X. Feng, A. Varghese, B. A. Chrunyk, K. Borzilleri, K. D. Hesp, K. Zhou, N. Ma, M. Tu, R. Dullea, K. F. McClure, R. C. Wilson, S. Liras, V. Mascitti and J. A. Doudna, J. Am. Chem. Soc., 2018, 140, 6596–6603 CrossRef CAS PubMed.
- X. Yang, Q. Tang, Y. Jiang, M. Zhang, M. Wang and L. Mao, J. Am. Chem. Soc., 2019, 141, 3782–3786 CrossRef CAS PubMed.
- C. Liu, T. Wan, H. Wang, S. Zhang, Y. Ping and Y. Cheng, Sci. Adv., 2019, 5, eaaw8922 CrossRef CAS PubMed.
- T. Wan, D. Niu, C. Wu, F. J. Xu, G. Church and Y. Ping, Mater. Today, 2019, 26, 40–66 CrossRef CAS.
- J. Li, J. J. Røise, M. He, R. Das and N. Murthy, Adv. Drug Delivery Rev., 2021, 168, 99–117 CrossRef CAS PubMed.
- L. Li, S. Hu and X. Chen, Biomaterials, 2018, 171, 207–218 CrossRef CAS PubMed.
- W. Cai, T. Luo, L. Mao and M. Wang, Angew. Chem., Int. Ed., 2021, 60, 8596–8606 CrossRef CAS PubMed.
- Y. Lyu, C. Yang, X. Lyu and K. Pu, Small, 2021, 17, e2005222 CrossRef PubMed.
- S. Zhang, J. Shen, D. Li and Y. Cheng, Theranostics, 2021, 11, 614–648 CrossRef CAS PubMed.
- Y. Chen, X. Yan and Y. Ping, ACS Mater. Lett., 2020, 2, 644–653 CrossRef CAS.
- E. R. Ruskowitz and C. A. DeForest, Nat. Rev. Mater., 2018, 3, 17087 CrossRef CAS.
- D. Jaque, L. Martínez Maestro, B. del Rosal, P. Haro-Gonzalez, A. Benayas, J. L. Plaza, E. Martín Rodríguez and J. García Solé, Nanoscale, 2014, 6, 9494–9530 RSC.
- N. Fernandes, C. F. Rodrigues, A. F. Moreira and I. J. Correia, Biomater. Sci., 2020, 8, 2990–3020 RSC.
- P. Wang, L. Zhang, W. Zheng, L. Cong, Z. Guo, Y. Xie, L. Wang, R. Tang, Q. Feng, Y. Hamada, K. Gonda, Z. Hu, X. Wu and X. Jiang, Angew. Chem., Int. Ed., 2017, 57, 1491–1496 CrossRef PubMed.
- X. Chen, Y. Chen, H. Xin, T. Wan and Y. Ping, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2395–2405 CrossRef CAS PubMed.
- H. Tang, X. Xu, Y. Chen, H. Xin, T. Wan, B. Li, H. Pan, D. Li and Y. Ping, Adv. Mater., 2021, 33, 2006003 CrossRef CAS PubMed.
- L. Li, Z. Yang, S. Zhu, L. He, W. Fan, W. Tang, J. Zou, Z. Shen, M. Zhang, L. Tang, Y. Dai, G. Niu, S. Hu and X. Chen, Adv. Mater., 2019, 31, 1901187 CrossRef PubMed.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 CrossRef CAS PubMed.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef PubMed.
- Y. Lyu, S. He, J. Li, Y. Jiang, H. Sun, Y. Miao and K. Pu, Angew. Chem., Int. Ed., 2019, 58, 18197–18201 CrossRef CAS PubMed.
- S. Deng, X. Li, S. Liu, J. Chen, M. Li, S. Y. Chew, K. W. Leong and D. Cheng, Sci. Adv., 2020, 6, eabb4005 CrossRef CAS PubMed.
- Y. A. Aksoy, B. Yang, W. Chen, T. Hung, R. P. Kuchel, N. W. Zammit, S. T. Grey, E. M. Goldys and W. Deng, ACS Appl. Mater. Interfaces, 2020, 12, 52433–52444 CrossRef CAS PubMed.
- S. Wu and H.-J. Butt, Adv. Mater., 2016, 28, 1208–1226 CrossRef CAS PubMed.
- Y. Pan, J. Yang, X. Luan, X. Liu, X. Li, J. Yang, T. Huang, L. Sun, Y. Wang, Y. Lin and Y. Song, Sci. Adv., 2019, 5, eaav7199 CrossRef CAS PubMed.
- D. N. Nguyen, T. L. Roth, P. J. Li, P. A. Chen, R. Apathy, M. R. Mamedov, L. T. Vo, V. R. Tobin, D. Goodman, E. Shifrut, J. A. Bluestone, J. M. Puck, F. C. Szoka and A. Marson, Nat. Biotechnol., 2020, 38, 44–49 CrossRef CAS PubMed.
- Y. Wu, J. Zheng, Q. Zeng, T. Zhang and D. Xing, Nano Res., 2020, 13, 2399–2406 CrossRef CAS.
- Y. Nihongaki, T. Otabe, Y. Ueda and M. Sato, Nat. Chem. Biol., 2019, 15, 882–888 CrossRef CAS PubMed.
- C. Plank, O. Zelphati and O. Mykhaylyk, Adv. Drug Delivery Rev., 2011, 63, 1300–1331 CrossRef CAS PubMed.
- A. K. Fajrial, Q. Q. He, N. I. Wirusanti, J. E. Slansky and X. Ding, Theranostics, 2020, 10, 5532–5549 CrossRef CAS PubMed.
- M.-N. Hsu and Y.-C. Hu, Nat. Biomed. Eng., 2019, 3, 83–84 CrossRef PubMed.
- H. Zhu, L. Zhang, S. Tong, C. M. Lee, H. Deshmukh and G. Bao, Nat. Biomed. Eng., 2019, 3, 126–136 CrossRef CAS PubMed.
- H. Y. Yang, Y. Li and D. S. Lee, Adv. Ther., 2018, 1, 1800011 CrossRef.
- Y.-L. Liu, D. Chen, P. Shang and D.-C. Yin, J. Controlled Release, 2019, 302, 90–104 CrossRef CAS PubMed.
- M. Mahmoudi, A. Simchi, A. Milani and P. Stroeve, J. Colloid Interface Sci., 2009, 336, 510–518 CrossRef CAS PubMed.
- N. Singh, G. J. Jenkins, R. Asadi and S. H. Doak, Nano Rev., 2010, 1, 5358 CrossRef PubMed.
- M. Mahmoudi, S. Laurent, M. A. Shokrgozar and M. Hosseinkhani, ACS Nano, 2011, 5, 7263–7276 CrossRef CAS PubMed.
- I. Lentacker, I. De Cock, R. Deckers, S. C. De Smedt and C. T. W. Moonen, Adv. Drug Delivery Rev., 2014, 72, 49–64 CrossRef CAS PubMed.
- A. Delalande, S. Kotopoulis, M. Postema, P. Midoux and C. Pichon, Gene, 2013, 525, 191–199 CrossRef CAS PubMed.
- M. Hansen-Bruhn, B. Esteban-Fernandez de Avila, M. Beltrán-Gastélum, J. Zhao, D. E. Ramírez-Herrera, P. Angsantikul, K. Vesterager Gothelf, L. Zhang and J. Wang, Angew. Chem., Int. Ed., 2018, 57, 2657–2661 CrossRef CAS PubMed.
- B. Helfield, Ultrasound Med. Biol., 2019, 45, 282–300 CrossRef PubMed.
- A. Upadhyay and S. V. Dalvi, Ultrasound Med. Biol., 2019, 45, 301–343 CrossRef PubMed.
- J.-Y. Ryu, E.-J. Won, H. A. R. Lee, J. H. Kim, E. Hui, H. P. Kim and T.-J. Yoon, Biomaterials, 2020, 232, 119736 CrossRef CAS PubMed.
- K. Hynynen, N. McDannold, N. Vykhodtseva, S. Raymond, R. Weissleder, F. A. Jolesz and N. Sheikov, J. Neurosurg., 2006, 105, 445–454 CAS.
- H.-L. Liu, M.-Y. Hua, H.-W. Yang, C.-Y. Huang, P.-C. Chu, J.-S. Wu, I.-C. Tseng, J.-J. Wang, T.-C. Yen and P.-Y. Chen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15205–15210 CrossRef CAS PubMed.
- Q. Yang, Y. Zhou, J. Chen, N. Huang, Z. Wang and Y. Cheng, Int. J. Nanomed., 2021, 16, 185 CrossRef PubMed.
- A. Mukherjee, A. K. Waters, P. Kalyan, A. S. Achrol, S. Kesari and V. M. Yenugonda, Int. J. Nanomed., 2019, 14, 1937 CrossRef CAS PubMed.
- A. V. Anzalone, L. W. Koblan and D. R. Liu, Nat. Biotechnol., 2020, 38, 824–844 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |