
Phenylboronic acid-functionalized polyaminoglycoside as an effective CRISPR/Cas9 delivery system†
Meiyu
Shao
,
Yu
Qi
,
Dandan
Sui
* and
Fu-Jian
Xu
 *
*
Key Laboratory of Biomedical Materials of Natural Macromolecules (Beijing University of Chemical Technology), Ministry of Education, Beijing Laboratory of Biomedical Materials, College of Materials Science and Engineering, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: xufj@mail.buct.edu.cn; suidd@mail.buct.edu.cn
First published on 3rd March 2021
Abstract
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) gene editing technology is a promising approach for cancer therapy, and its application practice urgently requires a safe and effective gene carrier. In this work, we focus on the design of a phenylboronic acid (PBA)-functionalized, disulfide bonded branched polyaminoglycoside (SS-HPT-P) as a robust delivery vector of the CRISPR-Cas9 system. SS-HPT-P showed great tumor-targeting performance, reduction-responsive degradability, and gene transfection ability. The typical pCas9-surv (one CRISPR-Cas9 plasmid that targets and knocks out the survivin gene) delivery mediated by SS-HPT-P exhibited gene editing performance in the A549 cell line, confirming the feasibility of SS-HPT-P to effectively deliver the CRISPR-Cas9 system. SS-HPT-P/pCas9-surv could effectively inhibit the proliferation of tumor cells both in vitro and in vivo, suggesting the potential of PBA-functionalized nanocarriers for cancer gene therapy. The present work provides a promising approach for the treatment of malignant tumors.
1. Introduction
The CRISPR-Cas9 system is a precise, robust genome editing tool that is not only useful for elucidating the function of target genes in biology and disease, but also a powerful tool for gene therapy.1–5 Cancer results from multiple genetic and epigenetic abnormalities, including the abnormal activation of proto-oncogenes.6–9 The knockout of proto-oncogenes can effectively inhibit the growth and survival of tumor cells, providing a suitable application platform for the CRISPR-Cas9 system.10–12 In spite of its potential, the CRISPR-Cas9 technology for cancer gene therapy depends on the development of a delivery system that can target tumor cells efficiently and can be delivered safely.13–15 In recent years, cationic polymers such as polyethyleneimine (PEI),16 branched poly(β-amino ester)s,17,18 aminoglycosides,19 lipids and peptides20,21 have attracted more and more attention as non-viral gene vectors owing to their relatively large gene payloads, non-immunogenicity, safe nature and convenient preparation process.22,23Ligand-mediated targeting of nanocarriers to cancer cells is an attractive strategy to improve the efficiency of gene delivery and avoid side effects on healthy tissues. Phenylboronic acid (PBA) and its derivatives can recognize sialic acid (SA), and can form stable borate ester with SA not only at the physiological pH value, but also in acidic media such as tumor acidic microenvironments.24,25 Recently, the paradigm of using PBA-installed polymeric nanocarriers for advanced cancer therapy has been well established.26,27 Furthermore, disulfide bonds would undergo cleavage by reduced glutathione, the content of which is much higher in tumor cells than that in normal cells.28 Degradable polycations with disulfide bonds can improve the biocompatibility of organisms and facilitate the release of nucleic acids in the reductive environment of tumors. Notably, reduction-responsive branched polyaminoglycoside (SS-HPT) based on hydroxyethyl disulfide diglycidyl ether (HDDE) and tobramycin has already been explored for effective gene delivery.19,28
Herein, PBA-functionalized SS-HPT (SS-HPT-P) was proposed for use in a tumor-targeting CRISPR-Cas9 delivery system (Scheme 1). SS-HPT-P, which contains large amounts of PBA groups and disulfide bonds, exhibits the ability of SA-mediated targeting aggregation and reduction-responsive degradation in tumor cells. The fine transfection properties of SS-HPT-P were investigated in HEK 293 and A549 cell lines through a series of experiments including the cytotoxicity and gene transfection efficiency. Subsequently, to verify the feasibility of SS-HPT-P to deliver the CRISPR-Cas9 genome editing system, we chose an activated oncogene survivin as the targeting gene. As a member of the inhibitor of apoptosis family, survivin is upregulated in multiple cancers including lung cancer, but is rarely detectable in normal tissues.29–31 The transfection of pCas9-surv (one CRISPR-Cas9 plasmid that targets and knocks out the survivin gene) mediated by SS-HPT-P in the A549 cell line demonstrated the high efficiency of gene editing. Further studies in vitro and in vivo demonstrated that the knockout of survivin mediated by SS-HPT-P significantly inhibited the migration and proliferation of tumor cells, especially in combination with temozolomide (TMZ, one widely used cancer-associated drug32).
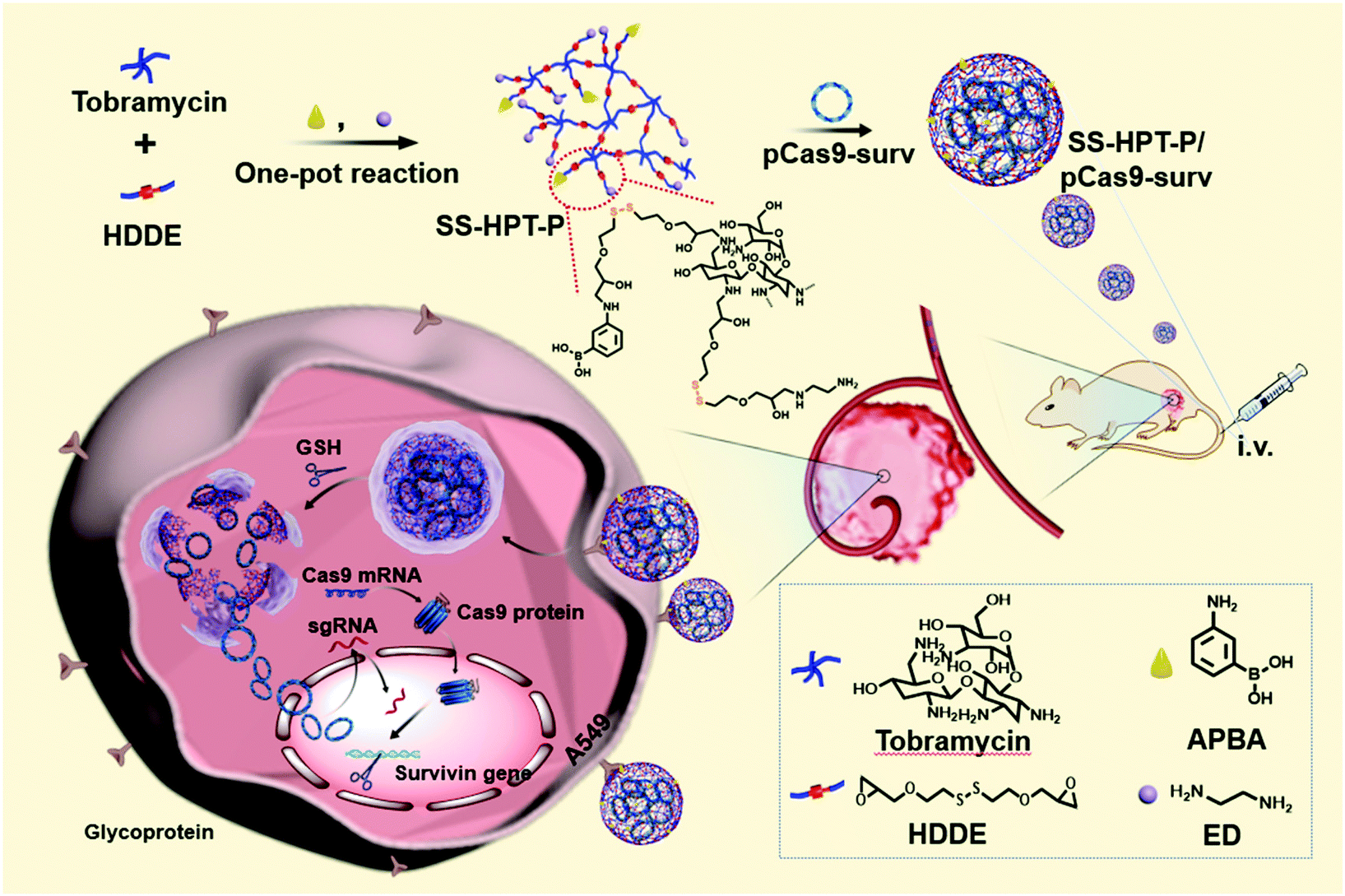 | ||
| Scheme 1 Schematic illustration of the preparation of PBA-functionalized polyaminoglycosides (SS-HPT-P) and its resultant delivery process of pCas9-surv for the treatment of carcinoma. | ||
2. Experimental
2.1 Preparation of SS-HPT and SS-HPT-P
HDDE and SS-HPT were prepared according to our earlier work.23 As a reduction-responsive linker, 2.5 mmol HDDE was added into 5 mL of DMSO containing 1 mmol tobramycin to reach an equal stoichiometric ratio of amine and epoxy groups. 300 μL of TEA was added after the reaction mixture was degassed for 10 min using nitrogen gas. Next, the reaction proceeded under a nitrogen atmosphere with stirring at 40 °C for 24 h. For the ring-opening reaction involving SS-HPT, 2.5 (or 1.25) mmol of APBA was dissolved in DMSO (1 or 0.5 mL) for SS-HPT-P2 (or SS-HPT-P1). After stirring at 40 °C for 8 h, an excess amount of ED (1.0 mL) was added into the mixture to terminate the branching reaction by reacting for another 1 h at 60 °C. Finally, the crude products were dialyzed (MWCO, 3500 Da) against deionized water to remove unreacted small molecules prior to lyophilization.2.2 Transfection assay with reporter genes in vitro
Transfection performances of SS-HPT, SS-HPT-P1 and SS-HPT-P2 polycations were first evaluated in HEK 293 and A549 cell lines using plasmid pRL-CMV (pDNA, encoding Renilla luciferase) as the reporter gene. HEK 293 and A549 cell lines were purchased from American Type Culture Collection (ATCC, Rockville, MD). The cells were seeded at a density of 6 × 104 per well in 500 μL of medium in a 24-well plate and incubated for 24 h. Subsequently, SS-HPT/pDNA and SS-HPT-P/pDNA complexes (containing 1.0 μg of pDNA) at various mass ratios ranging from 10 to 50 were added to each well. After 4 h of cultivation, 500 μL of fresh medium was added to replace the original mixtures and cultured for another 20 h. The luciferase gene expression was evaluated with a commercial kit (Renilla Luciferase Assay Kit) using a luminometer (Berthold Lumat LB 9507, USA).2.3 Cytotoxicity assay
The cytotoxicity of SS-HPT, SS-HPT-P1 and SS-HPT-P2 at a series of concentrations was evaluated through the thiazolyl blue tetrazolium bromide (MTT) assay as described in our previous work.33 Briefly, 2 × 104 cells were seeded into each well of 96-well plates with 100 μL of medium under typical conditions. After culturing for 24 h, the medium was replaced with 100 μL of fresh medium containing SS-HPT/pDNA, SS-HPT-P1/pDNA and SS-HPT-P2/pDNA complexes with different weight ratios ranging from 10 to 50. The gold standard branched polyethylenimine (PEI, ∼25 kDa) at its optimal N/P ratio of 10 was used as a control. After incubating for 4 h, the cells were treated with 100 μL of MTT solution (0.5 mg mL−1). After incubating for another 4 h, the cells were washed with PBS buffer solution three times to remove redundant dyes. Finally, the absorbance of individual wells was measured at a wavelength of 570 nm using a microplate reader (Model 680, Bio-Rad, UK) to detect the relative cell viability.2.4 Determination of SA expression on cellular surfaces
The amount of typical SA on the surfaces of different cell lines was detected according to the literature.34 Cells (106 mL−1) were suspended in Hanks’ balanced salt solution (pH 7.4) and incubated with two types of FITC–lectin conjugates (FITC–MAL and FITC–SNL) at a final concentration of 4 μg mL−1 at 4 °C for 1 h. After incubation, the cells were collected to analyze the mean fluorescence intensity (MFI) using a flow cytometer (FCM, Beckman Coulter, USA), where the data from a total of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events were calculated.
000 events were calculated.
2.5 PBA competition assay
In order to study the PBA-mediated targeting and better transfection performance of SS-HPT-P2, the PBA competition assay was carried out. The cells were cultured with a medium containing free APBA (concentration from 0 to 0.1 mg mL−1) for 4 h, and then SS-HPT/pDNA and SS-HPT-P2/pDNA complexes (containing 1.0 μg of pDNA) at an optimal mass ratio were added to each well. Other transfection procedures were consistent with those described above.2.6 Transfection and gene editing assays with pCas9-surv in vitro
Plasmid pCas9-surv which encodes Cas9 and GFP as well as transcribes survivin sgRNA (5′-TCTTGAATGTAGAGATGCGG-3′) was purchased from Nanjing GenScript Biotechnology Co., Ltd (China). To evaluate the potential transfection performances of plasmid pCas9-surv mediated by SS-HPT and SS-HPT-P2, A549 cell lines were transfected with the complexes of SS-HPT/pCas9-surv (optimal weight ratio, w/w = 40) and SS-HPT-P2/pCas9-surv (optimal weight ratio, w/w = 30) in vitro. The transfection procedures were consistent with the steps described above. A Leica DMIL 3000B fluorescence microscope was used to image the transfected cells, and the percentage of GFP positive cells was detected by flow cytometry.Subsequently, the gene editing ability of SS-HPT-P2/pCas9-surv was investigated. In detail, 2 × 105 cells were seeded into each well of 6-well plates with 2 mL of medium under typical conditions. After culturing for 24 h, the cells were treated with PBS, SS-HPT/pCas9-surv and SS-HPT-P2/pCas9-surv complexes (containing 6 μg of pCas9-surv), respectively. After incubating for 72 h, the transfected cells were collected, and genomic DNA was extracted using a DNA Gel Extraction Kit according to the procedure (Biolabs Inc., New England). DNA sequencing assay and T7 endonuclease I (T7EI) mismatch detection assay were carried out in accordance with the method described in previous literature.31
Down-regulated survivin expression was detected by the western blot assay. 72 h after transfection, the total proteins of A549 cells were extracted with a Protein Extraction Kit, and 100 μg of each protein extract was separated by electrophoresis on 12% SDS-PAGE and transferred to a nitrocellulose membrane. The expression level of survivin was detected by anti-survivin antibody. β-Tubulin was used as load control and the expression of β-Tubulin was detected by a monoclonal anti-β-Tubulin. Anti-survivin antibody and anti-β-Tubulin antibody were purchased from Abcam Co., Ltd (USA).
2.7 Cell migration and proliferation assay
For investigating the migration and proliferation ability of A549 cells treated with different treatments, transwell, cloning formation, wound healing and proliferation assays were performed as described in a previous study.14In the transwell assay, 100 μL of matrigel was evenly spread on the surface of the transwell chamber membrane. After incubation at 37 °C for 1 h, a total of 1 × 105 transfected A549 cells (in 200 μL of medium with 50 g L−1 BSA) were added into the upper layer of transwell chambers. Add 650 μL of medium with 10% FBS to the lower wells. After 48 h of incubation, the cells were fixed in 4% paraformaldehyde for 30 min, and then stained with 0.2% crystal violet solution for 10 min. The nonmigratory cells were scraped from the upper layer of the filter with cotton swabs. The treated cells at the lower layer of the filters through pores were imaged.
In the cloning formation assay, 1 × 103 A549 cells per well were seeded into a 6-well plate and treated with PBS, SS-HPT-P2/pDNA, TMZ (8 μg), SS-HPT-P2/pCas9-surv (containing 4 μg of pCas9-surv), and TMZ+SS-HPT-P2/pCas9-surv (containing 8 μg of TMZ and 4 μg of pCas9-surv), respectively. After ten days, the treated cells were washed with cold PBS, fixed with 4% paraformaldehyde for 30 minutes, and stained with 0.2% crystal violet solution for 10 minutes before imaging.
In the wound healing assay, a total of 7 × 104 A549 cells per well were seeded in a 12-well plate and cultured for 24 h. Then, the cells were treated with PBS, SS-HPT-P2/pDNA, TMZ (4 μg), SS-HPT-P2/pCas9-surv (containing 2 μg of pCas9-surv), and TMZ+SS-HPT-P2/pCas9-surv (containing 4 μg of TMZ and 2 μg of pCas9-surv). After 4 h of incubation, the medium was replaced with fresh DMEM with 10% FBS. The cell layers were scratched using pipette tips when about 90% confluence was reached. Subsequently, the cells were cultured continuously and the wound healing was imaged and quantified at 0, 24, 48, 72 and 96 h.
In the cell proliferation ability assay, A549 cells were seeded in a 6-well plate at a density of 2 × 105 cells per well in 2 mL of medium and incubated for 24 h. PBS, SS-HPT-P2/pDNA, TMZ (12 μg), SS-HPT-P2/pCas9-surv (containing 6 μg of pCas9-surv), and TMZ+SS-HPT-P2/pCas9-surv (containing 12 μg of TMZ and 6 μg of pCas9-surv) were then added to each well, respectively. The treated cells were collected by trypsin digestion at 24 h, 48 h, 72 h and 96 h, and then counted under an optical microscope (Leica DFC425 C, Germany).
2.8 In vivo tumor inhibition assay
Nude BALB/c mice (female, 6-week-old) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd (China). Animal studies were approved by the Ethical Committee of Chinese Academy of Medical Sciences (CAMS) and Peking Union Medical College, and performed under legal protocols. To evaluate in vivo tumor inhibitory activity, A549 cell lines (2 × 106) subcutaneously injected into the backside of BALB/c nude mice (six-week-old, female). Firstly, SS-HPT and SS-HPT-P2 were labeled with Cy7 to observe the bio-distribution (3 mice per group). The SS-HPT/pCas9-surv (containing 25 μg of pCas9-surv, w/w = 40) and SS-HPT-P2/pCas9-surv (containing 25 μg of pCas9-surv, w/w = 30) complexes were injected into the nude mice through the tail vein. After injection, the fluorescence distribution was continuously observed and imaged using an IVIS Spectrum system (PerkinElmer, USA) for 1–6 h.For the tumor inhibitory assay (4 mice per group), 150 μL of PBS, SS-HPT-P2/pDNA (containing 25 μg of pDNA, w/w = 30), TMZ (200 μg), SS-HPT-P2/pCas9-surv (containing 25 μg of pCas9-surv, w/w = 30), and TMZ+SS-HPT-P2/pCas9-surv (containing 200 μg of TMZ and 25 μg of pCas9-surv) were injected through a tail vein in different groups every two days. Mice weights and tumor volumes were measured every two days, too. On Day 12, all the mice were sacrificed, and the tumors were excised, imaged and weighted. The main organs and tumors from different groups were collected in 4% formaldehyde solution for later histological analysis. For immunological analysis, anti-survivin (1:2000, Santa Cruz, CA, USA) was used to detect survivin expression in the tumor tissues.
2.9 Statistical analysis
In this work, P* < 0.05 is represented by one star, P** < 0.01 is represented by two stars. The detailed analysis process is shown in the ESI.† The editing efficiencies of gene editing were analyzed using an online tool called TIDE (Tracking of Indels by Decomposition, https://tide.nki.nl/).3. Results and discussion
3.1 Preparation and characterization of SS-HPT-P
The synthetic route and biological application of SS-HPT-P are illustrated in Scheme 1. SS-HPT-P was prepared via amino-epoxy ring-opening polymerization. In detail, the amino groups of tobramycin with the diepoxide groups of HDDE continue to undergo ring-opening reactions to form a branched polymer. A certain amount of APBA molecules subsequently reacted with redundant epoxy groups on HDDE to produce SS-HPT-P. After a predetermined reaction period, the redundant epoxy groups were treated with an excess of ED. In this work, SS-HPT and two SS-HPT-Ps (SS-HPT: Mn = 8.97 × 103 g mol−1 and PDI = 1.77; SS-HPT-P1: Mn = 9.48 × 103 g mol−1 and PDI = 1.84; and SS-HPT-P2: Mn = 9.56 × 103 g mol−1 and PDI = 1.95) were prepared. Typical 1H NMR spectra of SS-HPT and SS-HPT-P are shown in Fig. 1a. The extra signals from δ = 6.61 to 7.29 ppm referred to the protons on the benzene ring of (–C6H4–) of phenylboronic acid. Based on the 1H NMR analysis, SS-HPT-P2 contained more PBA moieties than SS-HPT-P1.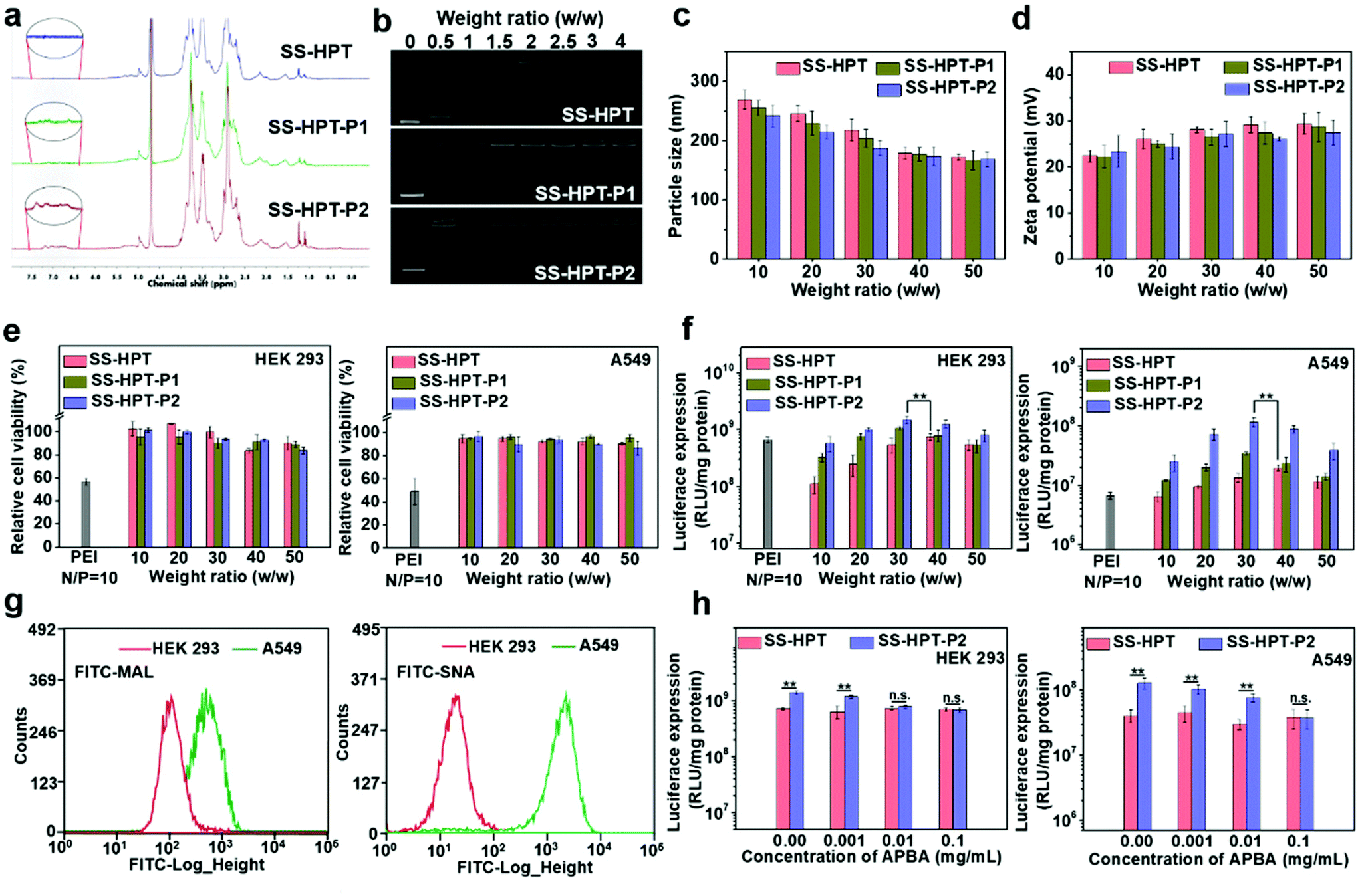 | ||
| Fig. 1 Biophysical properties and in vitro transfection properties of SS-HPT-based materials. (a) 1H NMR (400 MHz) spectra of SS-HPT, SS-HPT-P1 and SS-HPT-P2. (b) Agarose gel electrophoresis images of SS-HPT/pDNA, SS-HPT-P1/pDNA and SS-HPT-P2/pDNA complexes at various w/w ratios. (c) Particle size and (d) zeta potential of SS-HPT/pDNA, SS-HPT-P1/pDNA and SS-HPT-P2/pDNA complexes (at various w/w ratios) in aqueous solution. (e) Cell viability and (f) luciferase expression of HEK 293 and A549 cell lines treated with SS-HPT/pDNA, SS-HPT-P1/pDNA and SS-HPT-P2/pDNA complexes at various weight ratios, in comparison with those of the PEI/pDNA complex. (g) Sialic acid expression on the surface of HEK 293 and A549 cells determined by FITC–MAL and FITC–SNA assay (expressed as FITC fluorescence intensity). (h) Luciferase expression of HEK 293 and A549 cell lines transfected by SS-HPT/pDNA (w/w = 40) and SS-HPT-P2/pDNA (w/w = 30) complexes at various concentrations of APBA. | ||
3.2 Biophysical characterization of SS-HPT-P
Cationic vectors can condense negatively charged pDNA via electrostatic interactions and form nanocomplexes to facilitate endocytosis and gene transfection.35 Agarose gel electrophoresis showed that SS-HPT and SS-HPT-P retard the mobility of pDNA completely at the w/w ratios of 1.5 and above, while SS-HPT-P2 condensed pDNA more strongly at a weight ratio of 0.5 owing to the introduction of the PBA moiety, which improves the ability to condense pDNA, probably due to the facile linkages between boronic acid and the phosphate of pDNA (Fig. 1b).36Since complexes are usually internalized into cells via endocytic pathways, the particle size and the zeta-potential of the complexes are of crucial importance. As depicted in Fig. 1c, all the complexes were around 200 nm in diameter, which can readily undergo endocytosis.37 Meanwhile, the above complexes maintained positive net surface potentials (about 25 mV), which will produce good affinity for negative cell surfaces (Fig. 1d).
Biodegradability is an excellent characteristic of gene carriers, which can reduce the toxicity and improve the biocompatibility of the carriers. As a result of the disulfide bonds, SS-HPT and SS-HPT-P possessed satisfactory reductive degradability. The stable and reductive degradation morphologies of carriers/pDNA complexes were observed via AFM imaging, which showed that the intact nanocomplexes were dispersed in spherical aggregation, while in the presence of the sulfhydryl reducing agent dithiothreitol (DTT), the complex could not maintain stable nanoparticles, and depolymerized into fragments (see the ESI†). This phenomenon indicated that reduction-responsive SS-HPT and SS-HPT-P could become unstable and promote the release of nucleic acid in an intra-cellular reductive environment.
In conclusion, the above biophysical properties demonstrated that SS-HPT-based vectors, especially SS-HPT-P2 possessed better nucleic acid carrying capacity and good reduction-responsive degradation ability, indicating that SS-HPT-based vectors are promising gene carriers.
3.3 In vitro transfection assay with reporter plasmids
Low cytotoxicity is a basic condition for an excellent gene delivery system. The cytotoxicity studies of SS-HPT/pDNA and SS-HPT-P/pDNA complexes at various mass ratios from 10 to 50 in HEK 293 and A549 cell lines were performed using the MTT assay (see the ESI;†Fig. 1e). A dose-dependent tendency was observed for the cytotoxicity of both cell lines mediated by SS-HPT, SS-HPT-P1 and SS-HPT-P2. More importantly, benefitting from the biodegradable disulfide bonds and rich hydroxyl groups, SS-HPT-based polymers showed evidently lower cytotoxicity (cell survival rates above 80%) than the gold standard PEI (cell survival rates about 50%).Furthermore, the favorable endocytosis efficiency of complexes is an important premise for effective transfection. The cellular internalization of SS-HPT/pDNA and SS-HPT-P2/pDNA complexes in A549 cells was imaged and subsequently determined using flow cytometry. As presented in the ESI,† the intuitive images display the cellular uptake of complexes, where the pDNA was labeled by YOYO-1 in green and cell nuclei were stained by DAPI in blue. It is obvious that SS-HPT-P2/pDNA exhibited a stronger signal than SS-HPT/pDNA. The percentages of YOYO-1 positive cells treated with SS-HPT-P2/pDNA (92%) were much more than those treated with SS-HPT/pDNA (81%). These results also indicate that the affinity property induced by the PBA groups can benefit cellular uptake. Such favorable cellular uptake is supposed to benefit the improvement in transfection ability.
The gene transfection performance of SS-HPT-based polymers in vitro was first analyzed with the reporter plasmid pRL-CMV in the selected HEK 293 and A549 cell lines at various mass ratios from 10 to 50, where the gold-standard branched PEI (25 kDa) at its optimal N/P ratio of 10 (see the ESI†) was taken as the control.31 In general, the transfection efficiency first increased and then decreased with an increase of weight ratios. Notably, the SS-HPT-based vectors, especially for SS-HPT-P2, exhibited much higher transfection efficiencies at most weight ratios than those of PEI (25 kDa) at its optimal N/P ratio in both cell lines. As shown in Fig. 1f, it was noticed that the optimal weight ratios of SS-HPT-based complexes were 30–40 for both cells. Compared with SS-HPT, SS-HPT-P exhibited much higher luciferase transfection efficiencies at most weight ratios, especially in A549 cell lines (Fig. 1f). Moreover, SS-HPT-P2 with more PBA groups exhibited better transfection ability than SS-HPT-P1, presumably because of the stronger ability to condense pDNA, which has been shown above. In addition, increased targeting is a possible reason for better transfection ability.
Targeting can increase the probability of complexes binding to tumor cells, which is conducive to endocytosis and further improved the transfection ability. To explore targeting, we focused on the differences between tumor cells and non-tumor cells, and we found that a large amount of sialic acid is present on the surface of tumor cells compared to that of non-tumor cells. To verify this, the contents of sialic acid on the surface of A549 cells (tumor cells) and HEK 293 cells (non-tumor cells) have been checked by two types of FITC–agglutinin conjugators, FITC–MAL and FITC–SNA lectins, which can specifically recognize sialic acid.38 Flow cytometry analysis showed that the content of sialic acid on the surface of A549 cells was indeed significantly higher than that of HEK 293 cells (Fig. 1g). This difference between tumor cells and non-tumor cells provides a good basis for the targeted delivery.
In addition, several research studies have reported that phenylboronic acid can rapidly combine with sialic acid to form boronate ester.39 To take advantage of this specific binding ability, phenylboric acid was modified to SS-HTP, which facilitates the targeted binding of the complex to tumor cells. Transfection efficiencies mediated by SS-HPT-P2 and negative control SS-HPT were measured in both cell lines in the absence or presence of free APBA, which can also bind sialic acid on the cell surface. For negative control SS-HPT, no obvious differences were observed between the transfection efficiencies in the absence and presence of an APBA agent. While SS-HPT-P2 exhibited better transfection performance at the optimal mass ratio in the absence of a free APBA agent in both cells, but with an increase of APBA concentration, transfection efficiency mediated by SS-HPT-P2 gradually decreased until it was equivalent to SS-HPT (0.1 mg mL−1 APBA in A549 cells and 0.01 mg mL−1 APBA in HEK 293 cells) (Fig. 1h). This trend is more pronounced in A549 cells because there is a large amount of sialic acid on the cell surface. In view of the above, the introduction of a PBA group provided SS-HPT-P2 with better affinity for sialic acid on the tumor cell surface to improve the transfection efficiency, which can be competitively antagonized by free APBA.
3.4 In vitro transfection assay with therapeutic pCas9-surv
The selected plasmid pCas9-surv encoding Cas9 and GFP concurrently transcribing single guide RNA (sgRNA)14 was delivered into the lung cancer cell line A549. As reported, pCas9-surv could exactly knock out the survivin gene via the Cas9 protein under the guidance of sgRNA, which would limit the proliferation and enhance the apoptosis of cancer cells.14,31 Based on the above results, SS-HPT-P2 showed low cytotoxicity and good gene transfection performance. Thus, it was supposed to be a potentially splendid pCas9-surv delivery system. GFP expression was performed to visualize the transfection performances of SS-HPT/pCas9-surv and SS-HPT-P2/pCas9-surv in A549 cells at their optimal mass ratios (Fig. 2a; ESI†). After 24 h of transfection, the GFP-positive cells in the SS-HPT-P2/pCas9-surv treatment group were more than those in the SS-HPT/pCas9-surv treatment group. Flow cytometry quantitative analysis of the transfection efficiency suggested that GFP-positive cells mediated by SS-HPT/pCas9-surv and SS-HPT-P2/pCas9-surv were about 12% and 20%, respectively. The better transfection performance of SS-HPT-P2 was consistent with the luciferase gene transfection results (Fig. 1f).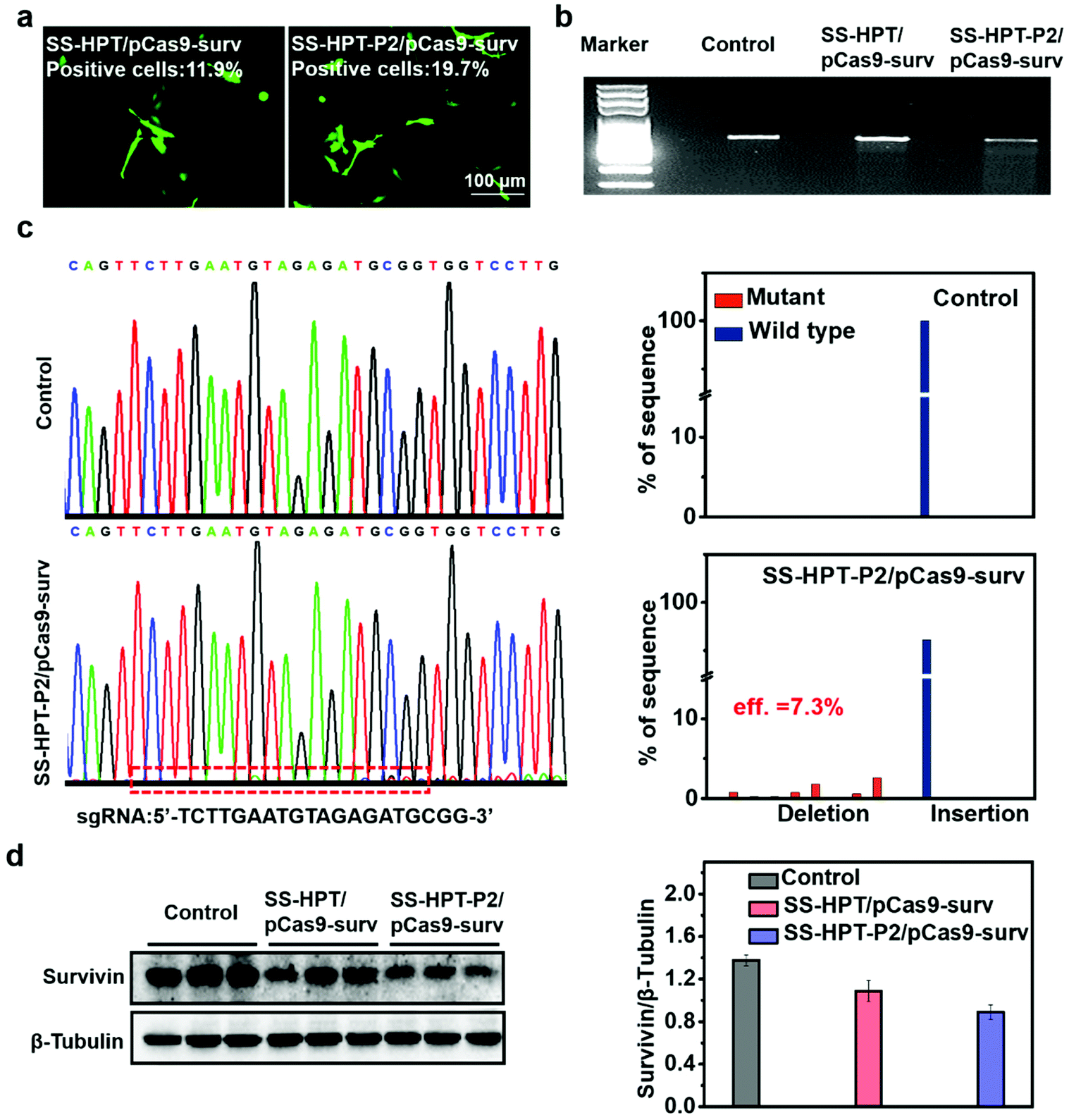 | ||
| Fig. 2 In vitro transfection assay with therapeutic pCas9-surv. (a) Fluorescence microscope images of GFP expression in A549 cells 24 h after transfection of SS-HPT/pCas9-surv or SS-HPT-P2/pCas9-surv complexes. (b) Detection of enzymatic digestion with T7EI enzyme on the survivin locus in the A549 cell line treated with PBS (control), SS-HPT/pCas9-surv or SS-HPT-P2/pCas9-surv complexes. (c) DNA sequencing and result analysis of the targeted loci in control and SS-HPT-P2/pCas9-surv groups. (d) Western blot image and quantitative analysis of survivin expression in Control, SS-HPT/pCas9-surv and SS-HPT-P2/pCas9-surv groups 72 h after transfection. | ||
To further investigate SS-HPT-P2/Cas9-mediated gene disruption at a specific genomic region, the T7EI mismatch detection assay was performed, which can evaluate the activity of the CRISPR-Cas9 system (Fig. 2b). We found that the SS-HPT-P2/pCas9-surv complex successfully triggered cleavage in targeted loci in comparison with the control group (PBS treatment). Furthermore, DNA sequencing was used to confirm the mutations at the targeted loci of PCR fragments. Compared with the control group, the miscellaneous peaks appeared in the middle of the target loci and extended to the 3′ end of the gene sequence in the SS-HPT-P2/pCas9-surv complex group, which confirmed that gene editing had occurred (Fig. 2c). The gene editing efficiency of the SS-HPT-P2/pCas9-surv system was predicted to be 7.3% in the A549 cell line. In accordance with the above results, the western blot assay (Fig. 2d) showed that the gene editing mediated by HPT-P2/pCas9-surv complexes in A549 cells successfully downregulated the survivin protein level. Compared to the SS-HPT/pCas9-surv group, the A549 cells treated with SS-HPT-P2/pCas9-surv down-regulated the expression of survivin more significantly, suggesting that SS-HPT-P2/pCas9-surv is a more effective genome-editing system.
It has been reported that down-regulation of oncogene survivin expression would inhibit the malignant proliferation of cancer cells. In addition, the knockout of survivin can increase cancer cells’ sensitivity to anti-tumor drugs.14,31 Therefore, the combination of pCas9-surv with TMZ was proposed for better performance of cancer inhibition. Matrigel transwell, plate cloning formation, wound scratch and cell proliferation assays were used to evaluate the invasion and migration of A549 cells under different treatments. First, matrigel transwell assays were carried out to examine the invasion ability of A549 cells. Compared with the blank control group and SS-HTP-P2/pDNA group, the invasiveness of A549 cells in the TMZ or pCas9-surv group was obviously reduced, and the TMZ+SS-HPT-P2/pCas9-surv group possessed the best inhibitory effect on cell invasion (Fig. 3a).
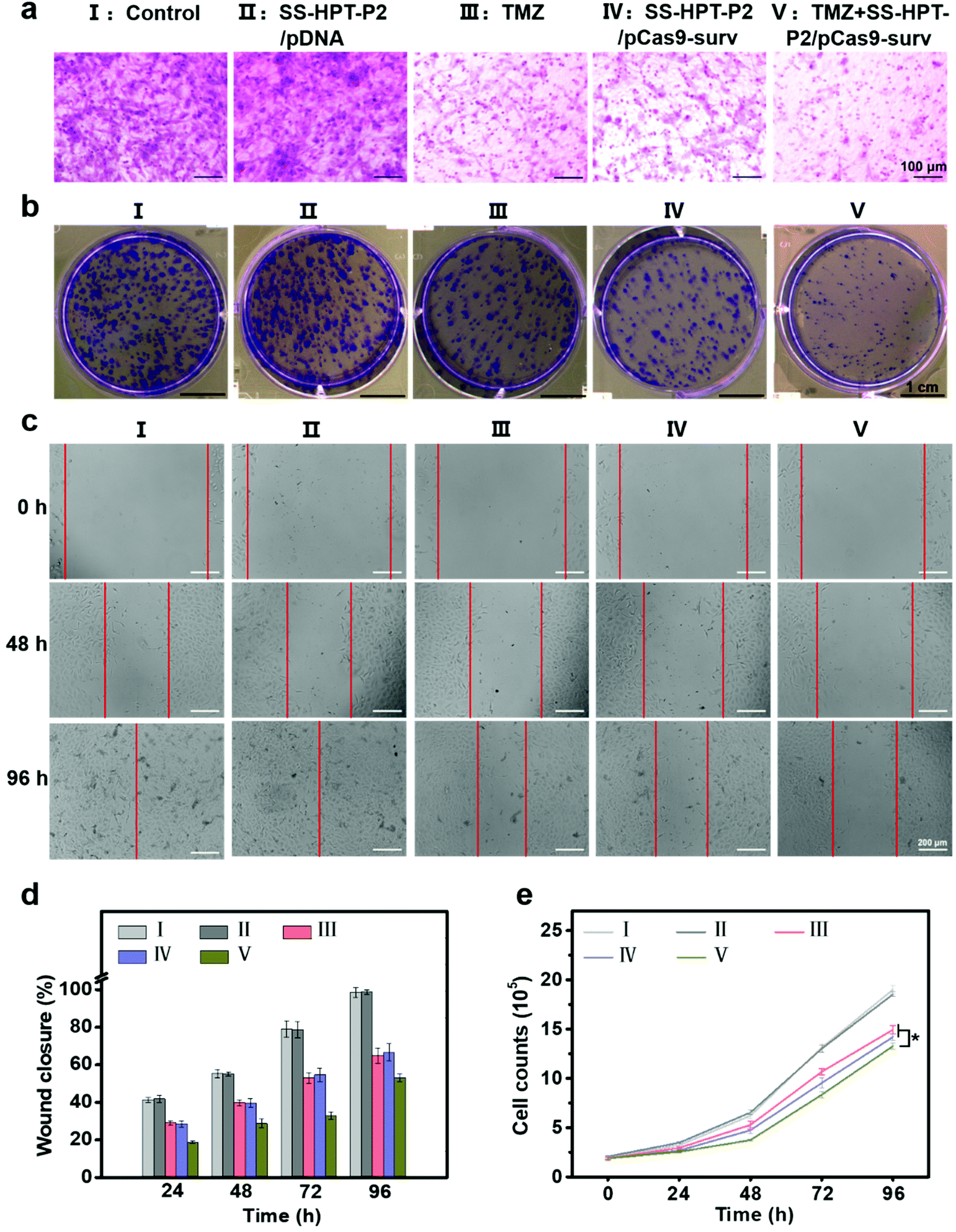 | ||
| Fig. 3 Cell migration and proliferation assay. (a) Representative photographs of A549 cells on the surface of lower chambers of Matrigel transwell after treatment with PBS (control), SS-HPT-P2/pDNA, TMZ, SS-HPT-P2/pCas9-surv or TMZ+SS-HPT-P2/pCas9-surv for 48 h. (b) Representative images of cloning formation after different treatments. (c) Representative images and (d) quantitative analysis of cell migration of A549 cells in different treatment groups from 0 to 96 h. (e) Cell proliferation of A549 cells in different treatment groups from 0 to 96 h. | ||
Next, plate cloning formation assays showed that SS-HPT-P2/pCas9-surv could inhibit cell proliferation and tumorigenic ability more effectively, which had less crystal violet aggregation compared with the control and SS-HPT-P2/pDNA groups. Specifically, in combination with TMZ, SS-HPT-P2/pCas9-surv caused the least crystal violet aggregation, which further inhibited cell proliferation and tumor formation (Fig. 3b).
The ability of pCas9-surv to inhibit cancer cell migration was further demonstrated by wound scratch analysis. As shown in Fig. 3c and the ESI,† it is obvious that the cell wound closure rate treated with SS-HPT-P2/pCas9-surv or TMZ was lower than that of the control group. In particular, cell migration was inhibited to a greater extent by SS-HPT-P2/pCas9-surv in combination with TMZ, which was consistent with previous studies that survivin inhibition can improve the sensitivity of tumor cells to TMZ. The wound closure rate of A549 cells from 0 to 96 h in different treatment groups was calculated (Fig. 3d).
Finally, the cell proliferation experiment was carried out, and the cell proliferation data obtained in 0–96 h from different treatment groups were statistically analyzed. The results showed that compared with the other four groups, the cell growth in the TMZ+SS-HPT-P2/pCas9-surv treatment group was always the slowest (Fig. 3e).
All the above results confirmed that SS-HPT-P2 has prominent nucleic acid delivery performance and can down-regulate survivin to inhibit the biological activities of cancer cell migration and proliferation (especially in combination with anti-neoplasia drugs).
3.5 In vivo tumor inhibition assay
Encouraged by the above good transfection performance of SS-HPT-P2 in vitro, an animal experimental model was established to investigate the tumor inhibition efficacy of pCas9-surv mediated by SS-HPT-P2 (Scheme 1). A549 tumor-bearing BALB/C nude mice were successfully induced and randomly divided into 5 groups, namely the Control group, SS-HPT-P2/pDNA group, TMZ group, SS-HPT-P2/pCas9-surv group and TMZ+SS-HPT-P2/pCas9-surv group.It has been proved that PBA can specifically bind to sialic acid on the surface of tumor cells in vitro, helping SS-HPT-P2 target tumor cells. Fluorescence imaging of tumor model animals would be used to verify the existence of such targeting under the interference of various complex environmental factors in vivo. SS-HPT and SS-HPT-P2 were conjugated with Cy7-NHS, which can track the distribution of the complex, mixed with pCas9-surv and injected into the tail vein of the mice. As shown in Fig. 4a and b, the SS-HPT-P2/pCas9-surv complex was rapidly enriched into the tumor after tail vein injection. Two hours later, the fluorescence intensity reached the peak. Compared to SS-HPT/pCas9-surv, SS-HPT-P2/pCas9-surv showed a higher degree of fluorescence enrichment at the tumor site, which was consistent with the results of cell endocytosis. At 2–4 hours after injection, the fluorescence intensity of the tumor in the SS-HPT-P2/pCas9-surv group was about 10 times higher than that in the SS-HPT/pCas9-surv group. The above data indicated that SS-HPT-P2/pCas9-surv showed obvious advantages in the accumulation rate and amount at the tumor site.
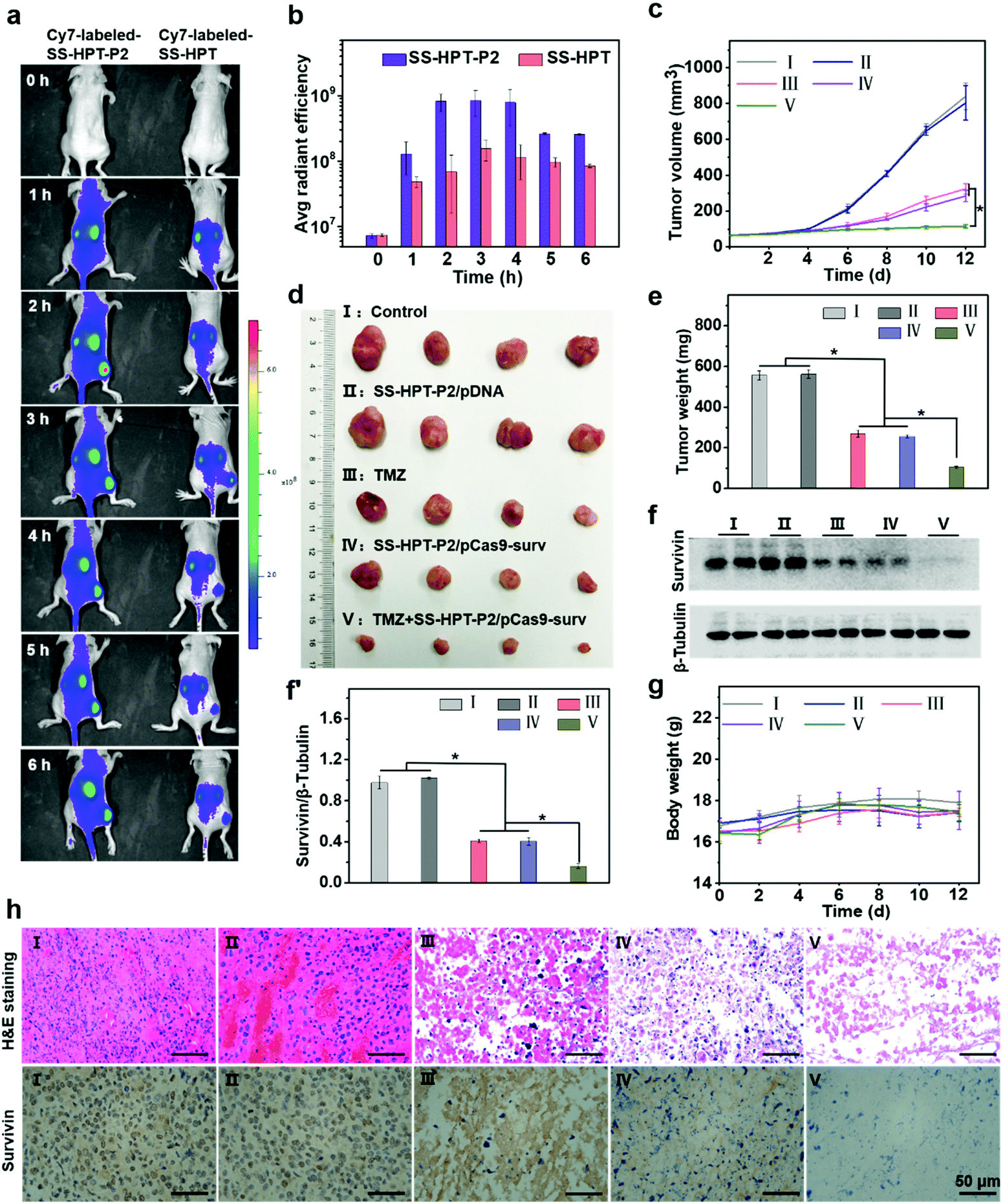 | ||
| Fig. 4 In vivo tumor inhibition performance: (a) in vivo biological distribution and (b) quantitative analysis of SS-HPT/pCas9-surv and SS-HPT-P2/pCas9-surv complexes after intravenous injection 0–6 h were analyzed using an IVIS in vivo imaging system. (c) Tumor volumes in different treatment groups measured using a vernier caliper. (d) Photographs and (e) weights of subcutaneous tumors excised 12 days after different treatments. (f) Western blot image and (f′) quantitative analysis of survivin expression in tumor tissues after various treatments. (g) Body weight of mice during the treatment. (h) H&E staining and immunohistochemical images of tumor tissues in groups I–V. | ||
A tumor-bearing mouse model was established by subcutaneous injection of A549 lung cancer cell lines. Seven days after injection, the tumor-bearing mice were divided into five treatment groups and treated intravenously every two days, and tumor sizes were measured using a vernier caliper throughout the treatment (Fig. 4c). Tumor growth curves showed that tumor size continued to increase in both the PBS (control group) and the SS-HPT-P2/pDNA treatment group, while the tumor growth rate decreased significantly in the TMZ group and the SS-HPT-P2/pCas9-surv group. Furthermore, the TMZ+SS-HPT-P2/pCas9-surv group had the best anti-tumor efficacy, which was consistent with the in vitro enhanced killing effect of tumor cells. After 12 days of treatment, the mice were sacrificed, and the tumor tissues were dissected, imaged and weighed (Fig. 4d and e). The tumor tissue images and weighing results intuitively showed that the tumors in the TMZ+SS-HPT-P2/pCas9-surv treatment group were obviously smaller than that in the other groups, and the growth of tumor was indeed inhibited.
The survivin expression in tumors after different treatments was assessed by the western blot assay (Fig. 4f and f′). The expression of survivin protein in TMZ and SS-HPT-P2/pCas9-surv treated tumors was lower than that in the control group. In particular, the expression of survivin protein was the least in the TMZ+SS-HPT-P2/pCas9-surv group, indicating that the combination of pCas9-surv and TMZ had a better therapeutic effect. In addition, no significant weight loss was observed due to the good biocompatibility of SS-HPT-P2 in vivo (Fig. 4g).
After 12 days of treatment, tumors from mice in different treatment groups were collected for haematoxylin and eosin (H&E) staining and immunohistochemistry analysis (Fig. 4h). H&E staining was used to evaluate the therapeutic efficacy at the histological level. The cell morphologies of the PBS group and SS-HPT-P2/pDNA group were normal and the nuclei were complete. In contrast, the cells in the TMZ group and SS-HPT-P2/pCas9-surv group were fragmented and nuclear pyknosis occurred. In particular, the TMZ+SS-HPT-P2/pCas9-surv group had the most severe cell disintegration and the lowest nuclear integrity, and no intact cell morphology could be observed under a microscope. These results indicated that the TMZ+SS-HPT-P2/pCas9-surv group induced extensive apoptosis in tumor tissues and played an admirable role in tumor suppression and treatment.
Immunohistochemistry (IHC) was performed to detect the expression level of survivin protein in tumor tissues of different treatment groups. The results showed that compared with the PBS group, the SS-HPT-P2/pDNA group and the TMZ group, the expression of survivin in the SS-HPT-P2/pCas9-surv group was significantly decreased, while the expression of survivin in the TMZ+SS-HPT-P2/pCas9-surv group was the least and almost not observed. In conclusion, it can be inferred that the effective inhibition and treatment of tumor is due to survivin knockout by SS-HPT-P2/pCas9-surv, while the addition of TMZ makes the apoptosis of tumor cells more significant.
Finally, in order to evaluate the biosafety of SS-HPT-P2, the heart, liver, spleen, lungs, kidneys and other major organs of each treatment group were collected for H&E staining. The results showed no abnormalities or significant damage to the organs (Fig. 5). The above results in vivo indicate that SS-HPT-P2 has good biosafety and is a potential gene carrier for the treatment of cancer.
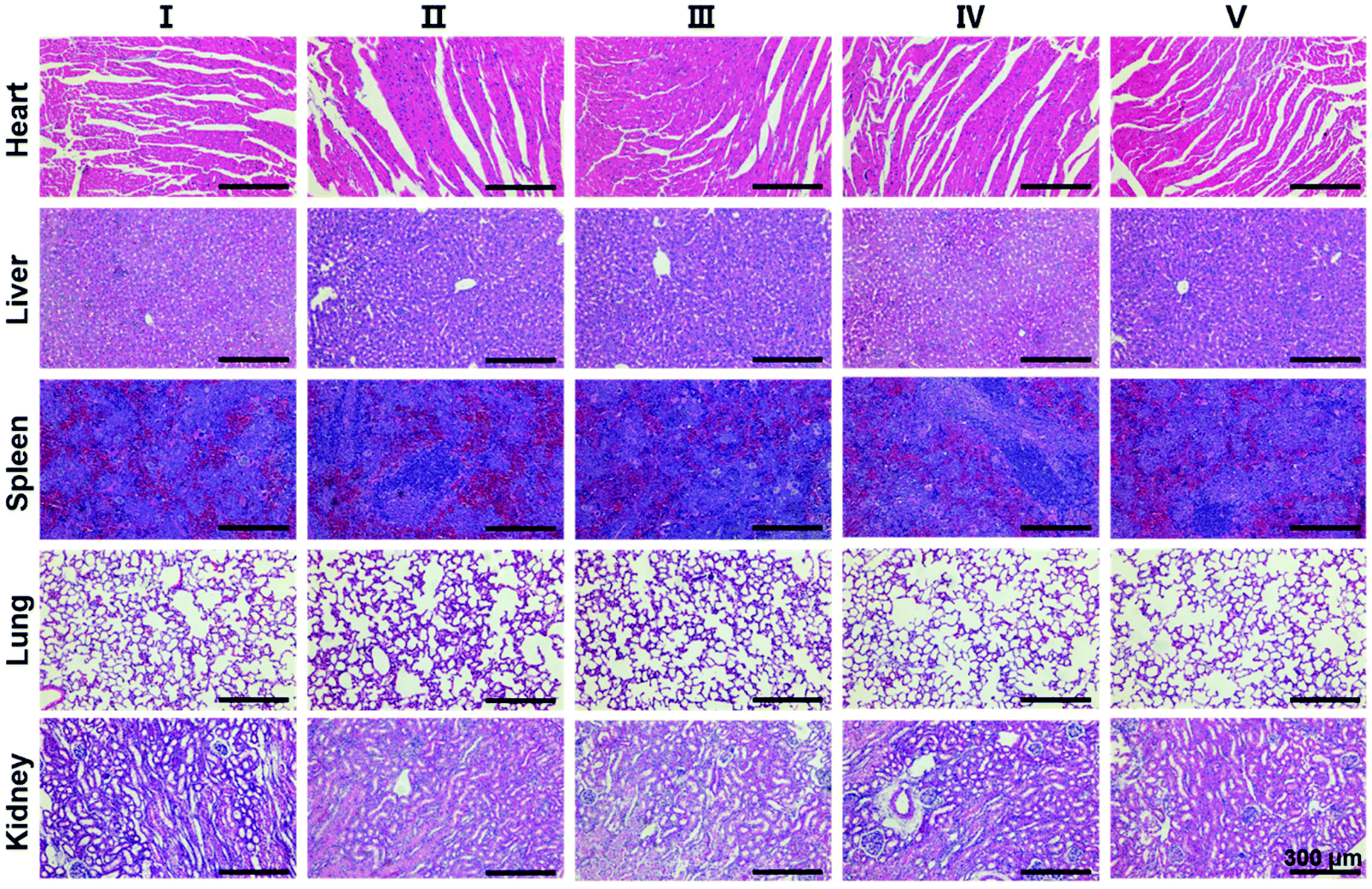 | ||
| Fig. 5 H&E staining images of main organs of mice from different treatment groups. | ||
4. Conclusions
In this study, a PBA-functionalized branched polymer (SS-HPT-P) was successfully designed and synthesized via one-pot ring-opening reactions as a safe and effective CRISPR-Cas9 delivery system for the treatment of carcinoma. SS-HPT-P has good biocompatibility, reductive responsive degradation and low cytotoxicity. Moreover, benefiting from good tumor targeting and gene transfection performances, SS-HPT-P can effectively mediate pCas9-surv to realize gene editing, and inhibit tumor proliferation and migration in vitro and in vivo. In addition, the SS-HPT-P/pCas9-surv complex can enhance the sensitivity of cancer cells to anti-tumor drugs, and the anti-tumor effect can be further enhanced when used in combination with TMZ. This study provides an effective strategy for the application of CRISPR-Cas9 gene editing in cancer therapy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant numbers 51733001 and 51903012) and the Beijing Outstanding Young Scientist Program (BJJWZYJH01201910010024).References
- P. D. Hsu, D. A. Scott, J. A. Weinstein, F. A. Ran, S. Konermann, V. Agarwala, Y. Li, E. J. Fine, X. Wu, O. Shalem, T. J. Cradick, L. A. Marraffini, G. Bao and F. Zhang, Nat. Biotechnol., 2013, 31, 827–832 Search PubMed.
- P. Mali, K. M. Esvelt and G. M. Church, Nat. Methods, 2013, 10, 957–963 Search PubMed.
- F. A. Ran, P. D. Hsu, J. Wright, V. Agarwala, D. A. Scott and F. Zhang, Nat. Protoc., 2013, 8, 2281–2308 Search PubMed.
- O. Shalem, N. E. Sanjana and F. Zhang, Nat. Rev. Genet., 2015, 16, 299–311 Search PubMed.
- X.-J. Lu, H.-Y. Xue, Z.-P. Ke, J.-L. Chen and L.-J. Ji, J. Med. Genet., 2015, 52, 289–296 Search PubMed.
- C. Greenman, P. Stephens and R. Smith, Nature, 2007, 446, 153–158 Search PubMed.
- I. B. Weinstein, Science, 2002, 297, 63–64 Search PubMed.
- I. B. Weinstein and A. K. Joe, Nat. Clin. Pract. Oncol., 2006, 3, 448–457 Search PubMed.
- I. B. Weinstein and A. Joe, Cancer Res., 2008, 68, 3077–3080 Search PubMed.
- T. Zhan, N. Rindtorff, J. Betge, M. P. Ebert and M. Boutros, Semin. Cancer Biol., 2019, 55, 106–119 Search PubMed.
- F. A. Khan, N. S. Pandupuspitasari, C.-J. Huang, A. Zhou, M. Jamal, A. Zohaib, F. A. Khan, M. R. Hakim and S. J. Zhang, Oncotarget, 2016, 7, 52541–52552 Search PubMed.
- M. Martinez-Lage, P. Puig-Serra, P. Menendez, R. Torres-Ruiz and S. Rodriguez-Perales, Biomedicines, 2018, 6, 105 Search PubMed.
- M. A. Zaimy, N. Saffarzadeh, A. Mohammadi, H. Pourghadamyari, P. Izadi, A. Sarli, L. K. Moghaddam, S. R. Paschepari, H. Azizi, S. Torkamandi and J. Tavakkoly-Bazzaz, Cancer Gene Ther., 2017, 24, 233–243 Search PubMed.
- Y. Qi, Y. L. Liu, B. R. Yu, Y. Hu, N. S. Zhang, Y. Zheng, M. Yang and F.-J. Xu, Adv. Sci., 2020, 7, 2001424 Search PubMed.
- C. E. Nelson and C. A. Gersbach, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 637–662 Search PubMed.
- D. Zhong, Y. P. Jiao, Y. Zhang, W. Zhang, N. Li, Q. H. Zuo, Q. Wang, W. Xue and Z. H. Liu, Biomaterials, 2013, 34, 294–305 Search PubMed.
- S. Liu, Y. S. Gao, D. Z. Zhou, M. Zeng, F. Alshehri, B. Newland, J. Lyu, J. O'Keeffe-Ahern, U. Greiser, T. Y. Guo, F. Z. Zhang and W. X. Wang, Nat. Commun., 2019, 10, 3307 Search PubMed.
- D. Z. Zhou, L. Cutlar, Y. S. Gao, W. Wang, J. O'Keeffe-Ahern, S. McMahon, B. Duarte, F. Larcher, B. J. Rodriguez, U. Greiser and W. X. Wang, Sci. Adv., 2016, 2, e1600102 Search PubMed.
- Y. J. Huang, X. K. Ding, Y. Qi, B. R. Yu and F.-J. Xu, Biomaterials, 2016, 106, 134–143 Search PubMed.
- J. P. Gratton, J. J. Yu, R. W. Babbitt, R. S. Scotland, R. Hickey, F. J. Giordano and W. C. Sessa, Nat. Med., 2003, 9, 357–362 Search PubMed.
- M. M. A. Elwakil, I. A. Khalil, Y. H. A. Elewa, K. Kusumoto, Y. Sato, N. Shobaki, Y. Kon and H. Harashima, Adv. Funct. Mater., 2019, 29, 1807677 Search PubMed.
- J. J. Green, R. Langer and D. G. Anderson, Acc. Chem. Res., 2008, 41, 749–759 Search PubMed.
- R. Kanasty, J. R. Dorkin, A. Vegas and D. Anderson, Nat. Mater., 2013, 12, 967–977 Search PubMed.
- K. Djanashvili, L. Frullano and J. A. Peters, Chem. – Eur. J., 2005, 11, 4010–4018 Search PubMed.
- S. Deshayes, H. Cabral, T. Ishii, Y. Miura, S. Kobayashi, T. Yamashita, A. Matsumoto, Y. Miyahara, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2013, 135, 15501–15507 Search PubMed.
- T. Y. Lan and Q. Q. Guo, Nanotechnol. Rev., 2019, 8, 548–561 Search PubMed.
- H. Liu, Y. Li, K. Sun, J. Fan, P. Zhang, J. Meng, S. Wang and L. Jiang, J. Am. Chem. Soc., 2013, 135, 7603–7609 Search PubMed.
- W. H. Fan, M. Y. Shao, J. W. Zhang, G. S. Jin, F.-J. Xu and F. S. Liu, Adv. Funct. Mater., 2019, 29, 1970069 Search PubMed.
- G. N. Zhao, Q. H. Wang, Z. Z. Wu, X. C. Tian, H. Yan, B. J. Wang, P. X. Dong, H. Watari, L. M. Pfeffer, Y. Q. Guo, W. Li and J. M. Yue, Mol. Cancer Ther., 2020, 19, 2233–2245 Search PubMed.
- D. C. Altieri, Oncogene, 2003, 22, 8581–8589 Search PubMed.
- Y. Qi, H. Q. Song, H. H. Xiao, G. Cheng, B. R. Yu and F.-J. Xu, Small, 2018, 14, 1803061 Search PubMed.
- Y. Cui, A. Naz, D. H. Thompson and J. Irudayaraj, Mol. Pharmaceutics, 2015, 12, 1279–1288 Search PubMed.
- S. Duan, B. Yu, C. X. Gao, W. Yuan, J. Ma and F.-J. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 29334–29342 Search PubMed.
- J. Wang, Z. H. Zhang, X. Wang, W. Wu and X. Q. Jiang, J. Controlled Release, 2013, 168, 1–9 Search PubMed.
- C. Xu, Y. Z. Z. Zhang, K. Xu, J.-J. Nie, B. R. Yu, S. J. Li, G. Cheng, Y. L. Li, J. Du and F.-J. Xu, Nat. Commun., 2019, 10, 3184 Search PubMed.
- B. A. Deore and M. S. Freund, Macromolecules, 2009, 42, 164–168 Search PubMed.
- T. Bus, C. Englert, M. Reifarth, P. Borchers, M. Hartlieb, A. Vollrath, S. Hoeppener, A. Traeger and U. S. Schubert, J. Mater. Chem. B, 2017, 5, 1258–1274 Search PubMed.
- M. A. V. Matta, D. S. Alviano, J. N. S. S. Couceiro, M. Nazareth, L. Meirelles, C. S. Alviano and J. Angluster, Parasitol. Res., 1999, 85, 293–299 Search PubMed.
- P. Qi, F. J. Chen, Z. L. Zhong and R. X. Zhuo, Chem. Commun., 2010, 46, 5888–5890 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of materials, agarose gel electrophoresis assay, detection of particle sizes and zeta potentials, AFM characterization, cell culture, intracellular uptake assay and statistical analysis. See DOI: 10.1039/d1bm00185j |
| This journal is © The Royal Society of Chemistry 2021 |