
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments and perspectives in CdS-based photocatalysts for water splitting
Jamal Abdul
Nasir
 ac,
Zia ur
Rehman
*a,
Syed Niaz Ali
Shah
a,
Azam
Khan
a,
Ian S.
Butler
b and
C. Richard A.
Catlow
*cd
ac,
Zia ur
Rehman
*a,
Syed Niaz Ali
Shah
a,
Azam
Khan
a,
Ian S.
Butler
b and
C. Richard A.
Catlow
*cd
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad, 45320, Pakistan. E-mail: zrehman@qau.edu.pk; hafizqau@yahoo.com
bDepartment of Chemistry, McGill University, 801 Sherbrooke St. West, Montreal, Quebec H3A 0B8, Canada
cDepartment of Chemistry, Kathleen Lonsdale Materials Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: c.r.a.catlow@ucl.ac.uk
dSchool of Chemistry, Cardiff University, Park Place, Cardiff, CF10 3AT, UK
First published on 12th August 2020
Abstract
Over the past few years, many approaches have been developed progressively to produce hydrogen (H2) from water under solar light irradiation. This process of fuel production is clean, potentially cost-effective, and environment-friendly. At present, however, current technologies are unable to meet the industrial requirements because of high cost, low photoresponse, and insufficient catalytic performance. Among water splitting photocatalysts, CdS is considered to be an interesting and important material owing to its low cost, prominent catalytic activity, high absorption in the visible spectrum, and the suitable positions of its conduction (CB) and valence (VB) bands. There are, however, some associated problems such as the rapid recombination of photogenerated electron–hole pairs and photocorrosion that have severely hampered its practical usage. The efficient conversion of water to H2 depends on the extent to which the charge carriers, especially the electrons, are first generated and then have sufficient life-time for their effective utilization. This review highlights work over the past several years to improve the photocatalytic efficiency and stability of CdS for H2 production from water.
 Jamal Abdul Nasir | Jamal Abdul Nasir received his MSc and MPhil degree in Inorganic/Analytical Chemistry from Quaid-i-Azam University Islamabad Pakistan in 2014 and 2016 respectively. He joined the same institute as a PhD student in September 2016 and completed his coursework and research under the supervision of Dr Zia ur Rehman. He has published more than 8 research articles in various journals of international repute. Currently, he is a part of Prof. Richard's group at University College London. He has a decent research background in material science, especially in photocatalysis and holds a keen interest in the area of QM/MM calculation for zeolite-based materials to investigate NOx decomposition, reaction mechanism, and designing photoactive materials for H2 production and CO2 reduction. |
 Zia ur Rehman | Dr Zia ur Rehman is working as a Tenured Associate Professor at Quaid-i-Azam University Islamabad, Pakistan. His group is involved in the synthesis of new materials for biomedical, clean energy, and environmental applications. He has published 110 research articles in various journals of international repute, and co-author of a book entitled “DNA Binding and DNA Extraction: Methods, Applications, and Limitations”. In his short academic carrier, 6 PhDs and 38 MPhil students obtained their degrees in his direction. His teaching and research efforts have been recognized by the Dr Abus Salam Award from TWAS and Pakistan Academy of Sciences (PAS), Dr Atta-ur-Rahman Gold Medal (PAS), and CSP Gold Medal (The Chemical Society of Pakistan). He is a young member of the PAS and International Committee Member of ACCC. |
 Syed Niaz Ali Shah | Dr Syed Niaz Ali Shah did his postdoc (2017/10 to 2019/10) from Tsinghua University, Beijing, China. He got his PhD degree in analytical chemistry from the Department of Chemistry, Tsinghua University, Beijing, China, in July 2017. He got his MPhil degree in inorganic/analytical chemistry from the Department of Chemistry, Quaid-i-Azam University, Islamabad, Pakistan, in 2014, and Master's degree in analytical chemistry from the Institute of Chemical Sciences, University of Peshawar, Pakistan, in 2011. His research interests mainly focus on chemiluminescent nanomaterials and sensors. He is the author of more than 18 peer-reviewed articles. |
 Azam Khan | Dr Azam Khan received his MPhil degree in Inorganic Chemistry from Hazara University Mansehra Pakistan in 2010. He completed a PhD in Inorganic/Analytical Chemistry under the supervision of Dr Zia ur Rehman at the Department of Chemistry Quaid-i-azam University Islamabad, Pakistan. His current research is focused on the design and synthesis of metal sulfide nanoparticles for energy and environmental applications. He has published more than 15 articles in various scientific areas. |
 Ian S. Butler | Professor Ian S. Butler was educated in U.K. at the University of Bristol and, following two years as a Fulbright postdoctoral scholar in the U.S.A, he joined the faculty in the Department of Chemistry at McGill University in 1966. He has served as Department Chair and Associate Vice-Principal (Research). He is an Honorary Member of the Spectroscopy Society of Canada, a Fellow of both the Chemical Institute of Canada and the Royal Society of Chemistry (U.K.). He has supervised over 200 researchers, resulting in the co-authorship of about 560 publications. He has also published several text books on General Chemistry, Inorganic Chemistry and Erbium. His honours include the Gerhard Herzberg Award for Excellence in Spectroscopy and the David Thomson Award for Excellence in Graduate Teaching and Supervision. His current research focuses on structural changes induced by high pressures and variable temperatures, bioinorganic chemistry, biomass conversion, mechanochemistry and art forensics. He has been married to his wife Pamela, a former dancer with American Ballet Theatre and a Professor of Political Science, for 55 years and they have four children and 15 grandchildren. |
 Richard A. Catlow | Professor C. Richard A. Catlow obtained his doctorate from the University of Oxford and subsequently held academic appointments at University College London, Keele University, the Daresbury Laboratory, and the Royal Institution (RI). He was made director of the Davy-Faraday Research Laboratory (DFRL) in 1998, a position that he held until 2007. In 2002, he was appointed Head of the chemistry department at UCL, becoming the first person to lead the UCL chemistry department and the DFRL at the same time. Five years later, he left the RI to become full-time at UCL, where he was named Dean of Science. He is also Vice-President and Foreign Secretary of the Royal Society – the Academy of Science of the UK and the Commonwealth. His research concentrates on computational chemistry and materials sciences and has published over 1000 research articles in this field. He currently has a joint professorial position with UCL and Cardiff University. His research interests are in the field of the chemistry of complex materials, including those for applications in catalytic and energy applications, investigated by a combination of computational and experimental techniques. |
Introduction
Energy is an essential part of economic activity. Today, most energy is produced from fossil fuel combustion, which as is well-known results in the emission of carbon dioxide, one of the major greenhouse gases responsible for climate change.1–4 Moreover, the amount of available fossil fuel is finite and is continuing to be depleted. The total worldwide power demand, however, which was 17 TW in 2010, is expected to rise to 27 TW by 2040.5 It is widely accepted that it is essential to solve future energy crises and current environmental threats by identifying a clean, safe, and renewable energy source. Solar energy technologies are attractive alternatives to fossil fuels as the total solar energy received by the Earth's surface in an hour is enough to meet the annual global energy demand.6–8 The challenge of energy storage and integration into the current power grid can, however, be a hindrance to the implementation of these technologies.9 An alternative promising approach is provided by photocatalytic water splitting for the production of H2.10,11 However, a suitable photocatalyst needs to be efficient, stable, and to consist of Earth-abundant elements.12 Many attempts have been made to develop such a catalyst from non-precious, Earth-abundant elements13,14 and considerable progress has been made, but the search is still underway.Amongst the earliest research on photoelectrochemistry was the work of Brattain,15 who studied germanium electrolyte junctions. This work was quickly followed by that of other groups.16,17 Later, Bard et al.18 demonstrated the photocatalytic behaviour of suspended semiconductor (SC) nanoparticles (NPs) and Nozik et al.19 explored the idea of photochemical diodes. The concept of water splitting through photoelectrochemistry was first established in 1972 by Honda and Fujishima using titania electrodes.20,21 Since then, various SC photoelectrodes and photocatalysts have been examined.22–25 Photochemical water splitting reactions require chemical energy from the direct sunlight source that involves a large positive change in Gibbs free energy (an endoergic reaction, ≈237 kJ mol−1). This process is similar to photosynthetic reactions in plants26 and is thus often regarded as artificial photosynthesis (eqn (1)). The absorption of light by a photocatalyst is required to produce a large enough potential to perform water splitting; moreover, the electron–hole life-time in this context is very important and should be sufficient for the redox reactions to proceed.27
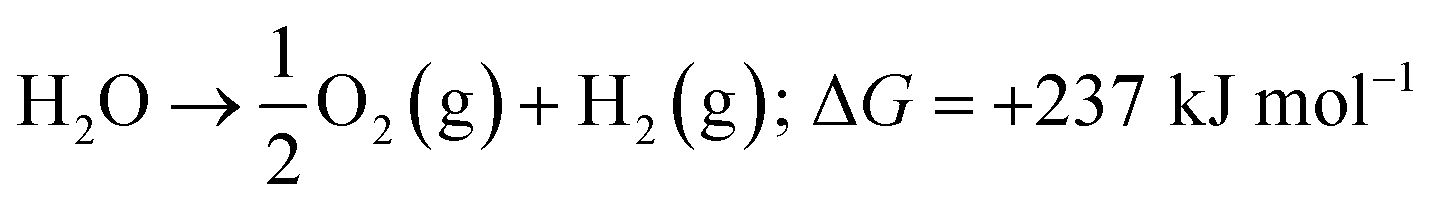 | (1) |
The reaction needs a potential of 1.23 eV and the band gap of the SC in this context should, therefore, be greater than 1.23 eV (<1000 nm) and less than 3.0 eV (>400 nm) so that it can be operated in the UV/visible region.
Recently, two primary approaches have been applied to establish an effective photocatalytic water splitting system28 (Fig. 1). In one approach, a single photocatalyst is used to drive the overall process. In this case, the SC must have a suitable band gap to harvest visible light and a sufficient thermodynamic potential for the water-splitting redox reactions. As a consequence of these restrictions, the number of efficient and stable photocatalysts for one-step water splitting is limited.29,30 In an alternative approach, two different photocatalysts are used and a two-step excitation mechanism is followed.18 This idea was taken from heterojunction31 and Z-scheme32–35 processes (like natural photosynthesis). The formation of a heterojunction between two photocatalysts allows charges to be transferred from one photocatalyst to another, thereby enhancing the life-time of the photogenerated charges. Similarly, the Z-scheme has many advantages over a one-step mechanism as two different SCs are combined with the introduction of a redox mediator species [electron donor/acceptor (A/D) pair], which acts as a shuttle between the two SCs.36 The separation of the gases produced (H2 and O2) is also easier in this case. An advantage of this approach is that an SC with either an oxidation potential or a reduction potential sufficient to effect water splitting can be used as a half-reaction in a two-step process.37,38
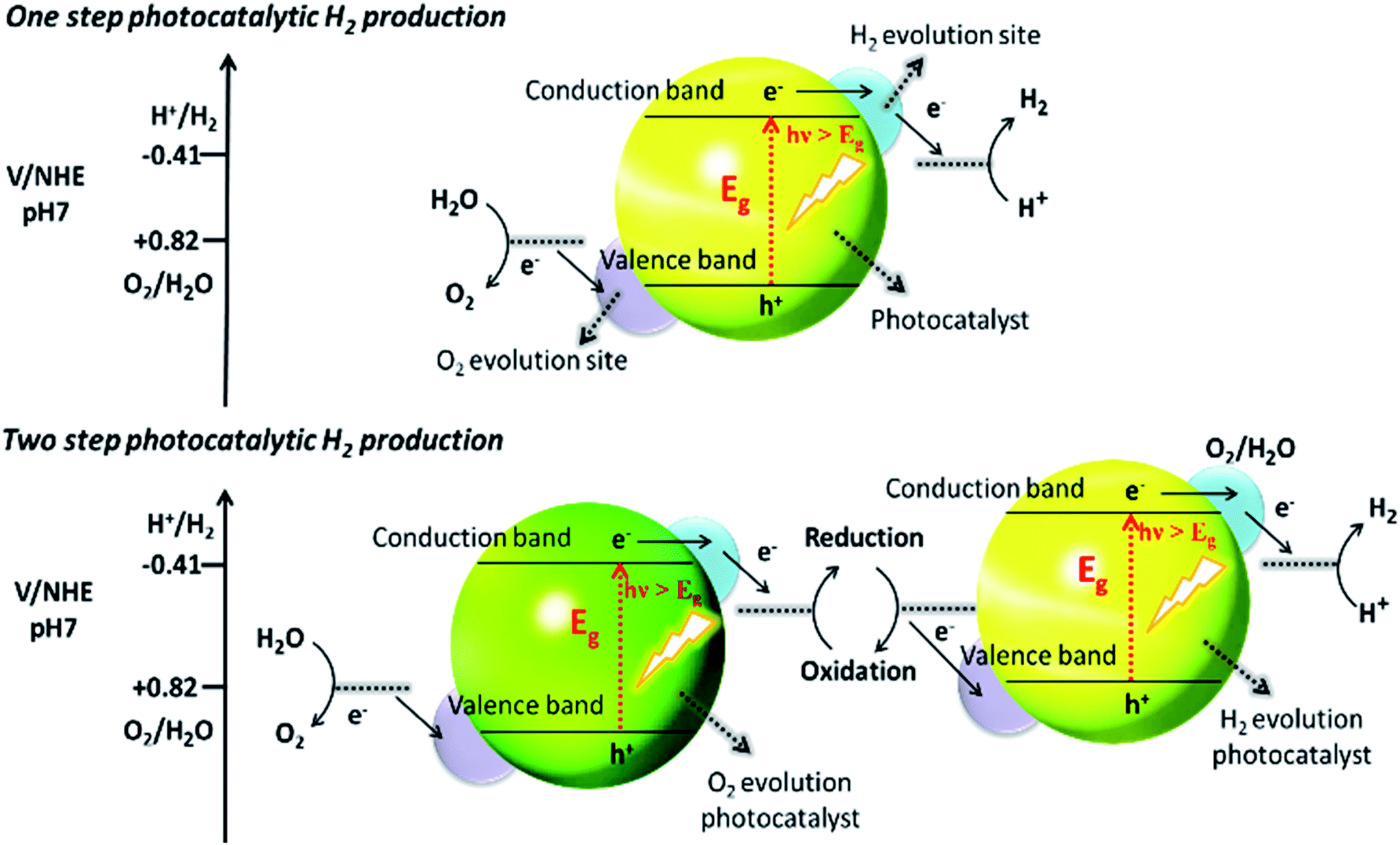 | ||
| Fig. 1 Schematic illustration of photocatalytic water splitting by one-step and two-step photoexcitation system. Reproduced with permission from ref. 28. | ||
Since the initial breakthrough in the splitting of water on n-TiO2 electrodes (photoelectrochemically),20 extensive work has been conducted on TiO2-based catalysts.39–41 Unfortunately, TiO2 has a band gap energy of about 3.0–3.2 eV and absorbs UV light, <400 nm,42 which hampers its use as a photocatalyst, because the solar spectrum contains only 3–4% of UV light. Consequently, there is an urgent need to develop a SC material that can harvest the copious amount of visible light (43%) in the solar spectrum. Additionally, photocatalysts for water oxidation should have a suitable redox potential for the CB and VB levels at neutral pH, together with a narrower band gap (<3.1 eV) capable of absorbing visible light irradiation.28 In this context, the location of the VB of the SC should be sufficiently positive to oxidize the water molecule O2/H2O (1.23 V vs. NHE) and the CB should be negative enough for the reduction of a proton (H+/H2 0 V vs. NHE).43 Among the visible-responsive photocatalysts,44 much work has focused on CdS, because of its low cost, appropriate band gap, and electron affinity that best fit the visible light absorption spectrum.45–49 The photocatalytic properties of CdS are somewhat limited though, because of several issues such as (i) photocorrosion and (ii) aggregation of CdS NPs during photocatalysis, which causes a large decrease in surface area and loss of active-sites. To mitigate the photocorrosion and to increase the overall efficiency of CdS, common sacrificial reagents such as Na2S/Na2SO3,50 ethanol,51 formic acid,52 and other organic and inorganic reagents are used.10,53–55 The benefits and detriments of these sacrificial agents will be discussed later. Other approaches for enhancing the activity of CdS include the control of the particle sizes56 (i.e., preparation of quantum-sized particles), tuning the morphology of the NPs,57 loading of co-catalysts58 (i.e., doping with noble and non-noble metals) and the combination of CdS with other SC materials to form mixed photocatalysts.59 Among these, the combination of different band gap SCs and the loading of suitable co-catalysts onto CdS are effective approaches for enhancing photocatalytic activity.60–62 More generally, the optimization of any material for photocatalytic activity requires that the following three fundamental factors be considered: (i) the absorption of light for generation of charges, (ii) the separation of the charges especially electrons and their effective transportation and (iii) the utilization of electrons for overall water splitting.
To improve the light absorption of wide-band gap SC materials, band gap engineering,63–67 surface plasmonic enhancement, doping with narrow-band gap SC materials,68–70 dye sensitization,71,72 and disorder engineering73,74 have been employed. To avoid charge recombination and assist charge transport, various morphologies of SC materials have been tuned on a nanoscale by the formation of nanospheres, nanorods, quantum dots, nanotubes and nanosheets.49,70,75,76 In addition to morphology tuning, co-catalyst incorporation, heterojunction creation, and Z-scheme developments have also proved highly effective for improving this second step. The third factor is encouraged by surface functionalization and by tailoring the reaction pathway through the use of sacrificial reagents. Co-catalysts play a dual role: (i) preventing recombination and (ii) acting as active sites either for activation of water molecules or for the reduction of protons. In the co-catalyst strategy, holes or electrons are scavenged by the co-catalysts once they are generated within the photocatalyst. Oxidative co-catalysts77 scavenge holes, while reductive co-catalysts78 trap electrons. By introducing an oxidative co-catalyst, such as CoII, not only is it possible to scavenge holes and hinder the recombination process but also it can provide an opportunity for the possible utilization of electrons.26,28,59,79–83
In the context of these challenges and approaches, the remainder of this review summarises recent developments in the study of CdS-based photocatalysts used for water splitting and considers the likely future development of the field; and we will consider CdS-based systems in the broader context of the field of water splitting by photocatalysts.
CdS as a photocatalyst: fundamentals and challenges
Visible-light-driven photocatalysts that are capable of the efficient production of H2 have good light absorption capacity and sufficiently negative flat band potentials. Widely investigated materials include TiO2Fe2O3, WO3,84 and the other materials referred to in Fig. 2, but especially CdS, which is one of the most widely investigated photocatalysts used in water splitting and in several other processes because of its suitable band gap of around 2.4 eV. This band gap is sufficiently negative for proton reduction (H2O/H2), as shown in Fig. 2.85 The band gap, of course, satisfies the thermodynamic requirement.86 Various other aspects, as noted above, affect the photocatalytic process, such as charge separation, and transportation, and life-time, over-potentials, and the pH of the system. Associated issues, such as photocorrosion and particle aggregation, also limit the photocatalytic activity of CdS. | ||
| Fig. 2 The band gap positions for CdS and other SCs relative to the redox potential of water. | ||
Photocorrosion is a common phenomenon and one of the greatest challenges with CdS or CdS-based photocatalysts, which hinders their practical use on an industrial scale. This phenomenon arises when (i) the sulfur ions are attacked by photogenerated holes and are oxidized into elemental sulphur; (ii) the photogenerated holes lead to the anodic corrosion under irradiation; and (iii) the defects created on the CdS surface during photocatalysis facilitate redox reactions on the surface under aerobic conditions.87 Photocorrosion has greatly limited the use of CdS,88 but fortunately, it can be inhibited and the stability of CdS can be improved by incorporation of an oxide layer,89 combining it with other materials (microporous and mesoporous),51,90 coupling with metallic/non-metallic catalysts,91,92 coating its surface,93 heterojunction formation,94 morphology tuning,95 cocatalysts incorporation,96 and the addition of sacrificial reagents.97 Meissner et al. have incorporated various catalysts such as Ru2O and Rh2O3 into CdS-mono-grain membranes to obtain more insight into water cleavage by suppressing the corrosion of the CdS nanocatalyst.98
According to previous reports, corrosion of CdS has been observed mostly in an acidic medium.99 The reaction media have a profound effect on the photocatalytic activity of CdS. To tailor the reaction pathway, increasing the pH of the media results in holes transferring from the photocatalyst to the scavenger (i.e. ethanol) as reported by Simon et al.78 In basic media, the release and transfer of holes operates as a shuttle and the redox couple of ˙OH/−OH affords electrons for proton reduction, while the hydroxyl anions are regenerated through water dissociation. In general, the process of hole scavenging from the VB of photocatalyst to the sacrificial electron donor is mainly favorable at high pH. On the other hand, operating the photoreaction in acid media not only makes hole transportation difficult but also causes the corrosion of photocatalyst.
As the pH of the reaction media affects the cleavage of a water molecule as well the stability of photocatalyst, such studies should be carried out in basic media to avoid corrosion as well as to assist water splitting. Similarly, Torimoto et al. reported that metal chalcogenides such as CdS are photocorroded.100 Also, Hu et al. showed that the photocorrosion in CdS based photocatalysts of sulphide ions by photo generated holes makes CdS unstable for photocatalysis.101 Similarly, other reports showed that photogenerated holes attack sulfide ions accompanied by leaching of the toxic Cd2+ ions.26,102 The concentration effect of Cd2+ ions103 and the kinetics during photocorrosion of CdS have been also studied in this regard using in situ Raman spectroscopy.104 A significant effect on photocorrosion of different environments (air and argon) was noted and the catalytic performance was changed in the two atmospheres, but the structural characteristics remained the same. Furthermore, the rate of H2 formation in an argon environment was much higher than in air. The decrease in activity was attributed to the “lattice stress “of CdS in air (oxygen environment). This term is related to some surface substitution of S2− ions by O2− ions in the CdS lattice. The increase in the lattice stress during photoreaction could be due to the rapid interaction between CdS and the adsorbed O2 leading to a decrease in activity. Furthermore, it is anticipated that the increase in strain and lattice stress is the initial sign of photocorrosion in CdS based photocatalyst, which, however, can be difficult to investigate through traditional techniques such as XRD. It is, therefore, necessary to develop a new CdS-based photocatalyst to overcome these difficulties, as will now be discussed.
Pure SC NPs like those of CdS are prone to surface damage as discussed above. Some surface modification of the CdS nanocatalyst is therefore needed,105 which may, however, appreciably change the photocatalytic, chemical, and optical properties.106 Numerous methods of modification have, moreover, been reported to tune the catalytic properties of CdS, such as noble metal loading,107–109 ion doping,110 SC composite formation,111 and metal ion-implantation.112
Although, CdS has an excellent ability to absorb visible light, however, the extension of its absorption into the entire visible range and in some cases to the IR region can be achieved by some modification such as surface plasmonic enhancement. Moreover, modification has advantages such as suppressing electron–hole recombination, enhancing photostability, and creating new sites on the surface, which may lead to the creation of a new layer, thus improving the selectivity and overall efficiency of the photocatalyst, as has been reported for composites such as PbS/CdS113 colloidal nanocrystals, ZnSe/CdS core–shell,114,115 CdS/HgS/CdS quantum dot116 and co-sensitized CdS/CdSe quantum dots.117,118 In addition, the surface modification of a nano-chalcogenide is often achieved by the incorporation of metal ions onto the surface of NPs, as reported for CdS, e.g., Mn,119,120 Ag,121,122 Zn,123 Hg,124 Ni,78etc. Electron utilization can be encouraged by surface functionalization and by tailoring the reaction pathway through the use of sacrificial reagents to enhance the water splitting reaction.
These and other challenges will be reviewed further in later sections. Meanwhile, in the next section, we address in greater detail the fundamental issues of charge generation and lifetimes.
Charge generation in CdS
Electron generation is the initial step in a photocatalytic H2 production process and the extent and ability to reduce the desired species by the generated electrons are crucial factors in the H2 evolution reaction (HER). The range of energy (300–2500 nm) supplied by the sun, comprises ∼52% near infrared (>760 nm), ∼43% visible (400 to 760 nm), and 3–4% ultraviolet (<400 nm) light. A photocatalyst with a narrow band gap has a good spectral response capturing photons with an energy corresponding to its band gap. As stated earlier, CdS, a narrow band gap SC, harvests nearly ∼38% of the visible spectrum. As noted, efforts have been made to extend the absorption of the CdS up to the full visible range and the IR region. Two different approaches can be used to widen the spectral response of photocatalysts: self-modification and extra-modification. Self-modification refers to modifying a photocatalysts itself through structural defects and surface modification, while extra-modification means adding some additional material. Enhancing the spectral response through self-modification might increase the chances of recombination and hence, would not be a suitable approach for CdS-like photocatalysts, as it already has a narrow band gap. Moreover, narrowing the band gap through self-modification causes a reduction of the CB, which weakens the reduction capacity of the photogenerated electrons. Therefore, many attempts have been made to widen the spectral response without decreasing the band gap of CdS by extra-modification. For example, the introduction of foreign light-harvesting materials is an effective method of modifying a CdS photocatalyst. However, the light blockage may occur on the surface of the catalyst due to foreign light-harvesting materials, and this effect should be taken into consideration. A more detailed discussion follows in the next section.Modification of a narrow-band gap SC
The combination of photocatalysts for the purpose of the enhanced spectral response is an obvious way of extending the working spectrum. To acquire a broad spectral response, the wide band SCs are primarily co-sensitized by narrow band SCs such as CdS, CdTe, CdSe, PbS, which mainly serve as sensitizers in place of dye molecules. Among these, CdS is considered one of the most promising contenders owing to its broadly tuneable band gap and is, therefore, typically used to sensitize the wide band SCs in order to harvest the visible light irradiation, Wang et al. have used CdS with a double-sized incorporated CdSe nanohybrid co-sensitized with ZnO nanowires for photochemical H2 production.125 To develop this double-sided design, the thick ZnO nanowires were doped onto the indium tin oxide (ITO) substrate to which CdS and CdSe QDs were sensitized on each side (Fig. 3a). As a result, the photoanode displayed good absorption over almost the entire spectrum of the visible region, with an excellent incident-photon-to-current-conversion efficiency (IPCE) of ∼45% (at 0 V vs. Ag/AgCl). Besides, the direct interaction between the quantum dot and the nanowire in the double-sided architecture makes it a promising system for effective charge collection as compared to a co-sensitized single-sided layer. Commensurate with this study, a different strategy was adopted by Vuong et al., who prepared ZnO nanorods, sensitized with CdS QDs and carbon QDs (CQDs).126 The ZnO nanorods on ITO glass were hydrothermally synthesized followed by calcination (500 °C) in air and pure ambient H2 atmospheric pressure at 400 °C. The extension of absorbance within the visible and IR region was made possible following the sensitization of the H:ZnO surface with CdS and CQDs. The optimized H:ZnO/CdS/CQDs displayed a better photocurrent of ∼12.82 mA cm−2 at 0 V under solar light irradiation. In addition, a novel CdS-intercalated zirconium titanium phosphate (CdS–ZTP) has been prepared by Parida et al., to extend the light absorption toward the visible spectrum while meanwhile inhibiting the fast recombination of charges.127 Besides, they also found that the surface area of CdS–ZTP material gradually increased with an increased amount of CdS (up to 15 wt%), which is essentially an indirect pillaring effect created by CdS NPs. Pillaring is an important phenomenon that is employed to cross-link the inorganic zirconium-layers and includes materials such as PO3Zr and ZTP, etc. This concept was first described by Dines et al.128, who used rigid aryl groups to cross-link the PO3Zr layers. Intercalation of pillared materials leads to increases in the porosity and surface area of the layered material, which ultimately have an effect on the catalytic activity.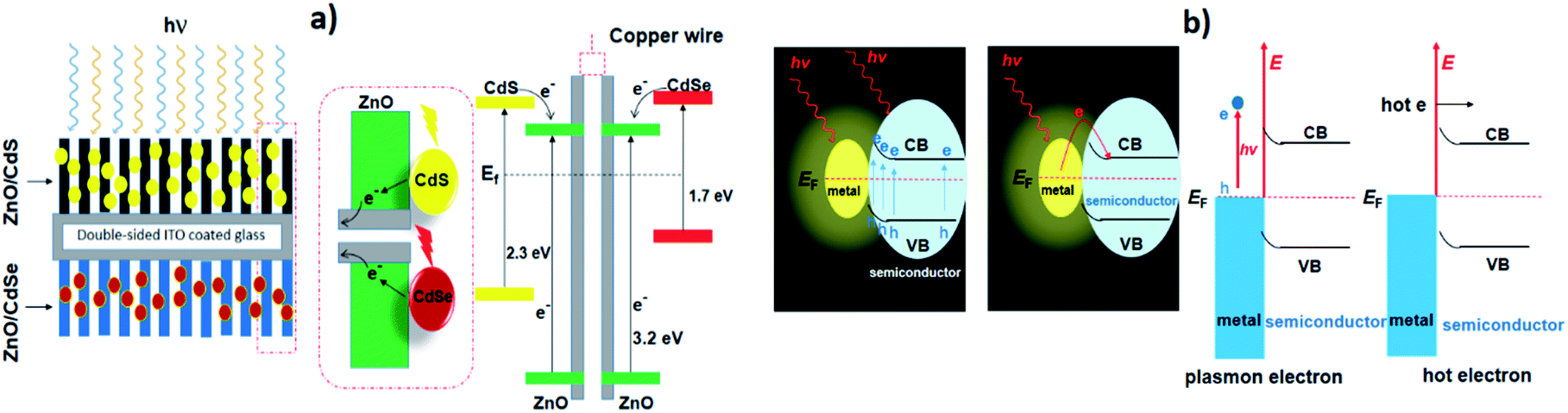 | ||
| Fig. 3 (a) The architecture and the energy diagram of the double-sided CdS–ZnO–ZnO–CdSe nanohybrids. The mid dashed box represents the CdS–ZnO and CdSe–ZnO interfaces. Reproduced with permission from ref. 125, (b) schematic representation of plasmon-enhanced chemical reactions enhancement of light absorption and hot-electron effect. Reproduced with permission from ref. 129. | ||
Plasmonic electron generation
In conductive nanocrystals, localized surface plasmon resonance (LSPR) is often used to boost the absorption capacities of the photocatalysts. Plasmonic materials are employed to enhance the photoresponse of materials in the visible or NIR region. Importantly, the LSPR generally takes place at a higher wavelengths and where there is a coupling between the plasmonic material and the photocatalyst. Jiang et al. reported two important effects of the LSPR: (i) the absorption of light and (ii) the hot electron phenomenon (Fig. 3b).129There is a strong electromagnetic field localized around a plasmonic material when excited. The SC absorption overlaps with the created plasmon resonance of the plasmonic material and is subjected to a strong field, so the generation of electrons is greatly increased. This feature is termed the “plasmonic enhancement of light absorption effect”. Another effect of (LSPR) is the “hot electron effect” (plasmonic sensitization effect), which refers to the relocation of electrons at the interface between the SC and plasmonic materials.
Duan et al. have introduced the concept of multilayer nano-shell plasmonic photocatalysis for photocatalytic production of H2 by water splitting.130 The optical parameters of the Ag/SiO2/CdS material were investigated and the researchers confirmed that the light absorption ability of this hybrid was markedly enhanced via LSPR. By amending the intermediate layers, the resonance wavelength was tuned so that one of the thinner intermediate layers led to a redshift. In another study, hot-electron instillation onto the CdS rod employing optically excited plasmons in the Au tip led to ∼2.75% quantum yield. The possibility of further optimization by directing the sizes and morphology of the plasmonic and excitonic domains may lead to a well-designed photocatalyst that can absorb in the broader range of the solar spectrum.131 Several mechanisms have been proposed to explain the overall chemistry involved and among the many plasmonic materials, the noble metals Ag and Au are the most popular. To prevent the direct transfer of electrons from CdS to the plasmonic material, Torimoto et al.132 introduced SiO2 around the Au metal. Shell CdS NPs immobilized on Au metal coated with SiO2 particles were designed; the latter served as an insulator to avoid the direct injection of electrons to Au particles from the CB of CdS (Fig. 4). The activity in this regard is dependent on the relocation of electrons from CdS to Au, because of the locally generated electric field, created by the photoexcitation of the LSPR peak of the Au particles.
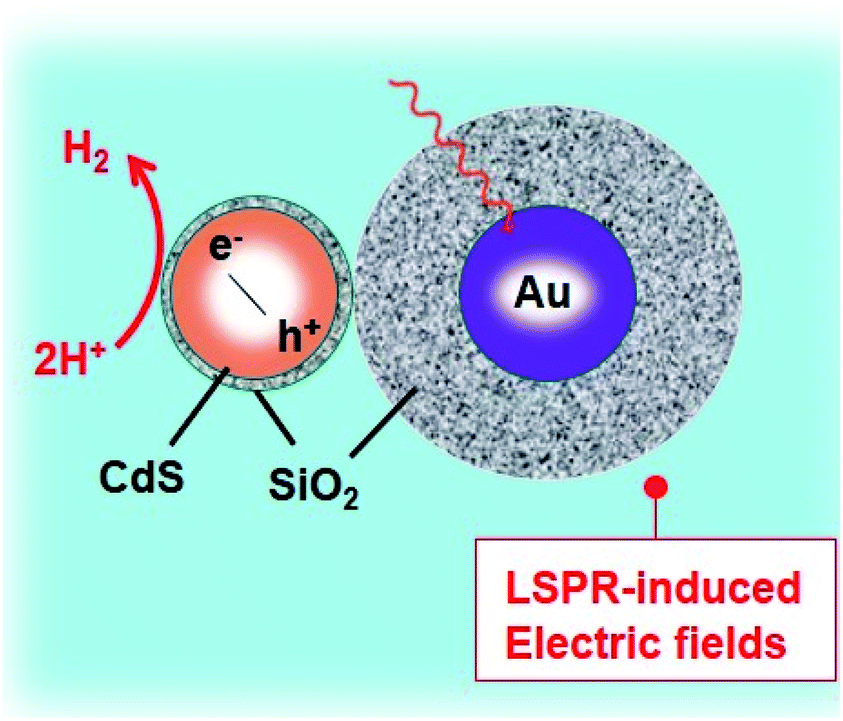 | ||
| Fig. 4 LSPR-induced electric field immobilized on Au core-SiO2. Reproduced with permission from ref. 132. | ||
The distance between Au and CdS materials greatly depends on the size of the Au particle, i.e. larger particle sizes lead to improved photocatalytic activity.
Based on the LSPR mechanism, Au has been used to modify CdS to enhance its photocatalytic performance.133 Li et al. have developed a sandwich CdS–Au–TiO2 heterostructure as a photoanode for H2 generation from water.134 The Au NPs which fit in between TiO2 and CdS-QD play two roles: (i) as an electron transmitter, which assists the electron transportation between TiO2 and CdS; and (ii) as a plasmonic photosensitizer to extend the absorption spectrum to longer wavelengths. A photocurrent of 4.07 mA cm−2 at 0 V (vs. Ag/AgCl) in the presence of Au was achieved with a solar-to-chemical energy conversion efficiency of 2.8%. The incorporation of Au NPs reduces the trap-state, promotes the charge-transfer, hampers the charge recombination, and compensates the undesirable effect of surface trap states. The plasmonic technique is found to provide a swift transfer of the plasmonic hot carriers, a phenomenon that encourages the relocation of electrons at the interface between the SC and the plasmonic material. The interfacial Schottky barrier height (an energy barrier for an electron at SC–metal junction) controls not only the back-transfer of electrons but also controls the plasmonic hot-electron transfer. To gain insight into mechanistic aspects of Au-doped CdS for H2 generation, the CdS–Au plasmon exciton interaction (as explained above) has been studied by transient absorption spectroscopy. The excitation of the optical plasmons in the Au tip jumps to a hot-electron containing CdS. Impressively, by employing these methodologies, an excellent QE of ∼2.75% has been achieved.131
Charge life times in CdS
The transportation of photogenerated charges to the target site is essential in photocatalysis. Most of these pairs are lost through recombination.135 To address this problem, the concentration of defects in a catalyst material can be reduced by shortening the distance between the charges (electron/hole) and the target site on the surface of the photocatalyst. For this purpose, morphology engineering, structural engineering, cocatalyst strategies, the use of heterojunctions and Z-schemes for CdS as a model photocatalyst, have been investigated.Morphology and structural engineering for induced charge separation
The process of introducing anisotropy into a photocatalytic system is referred to as morphology engineering, which can help in the survival of electrons. Various SC nanostructures have been produced with different shapes, sizes, crystallinities, and morphologies. In addition, several methods have been reported for the nanoscale formation of CdS, such as solvothermal, chemical vapor deposition (CVD), and thermal decomposition processes.136–139 All these approaches aim at controlling the morphology, crystallinity, and size of the CdS nanocrystals in the form of nanorods, cubes, tetrapods, hexagons, and pyramids. For example, CdS nanoribbons have been prepared via a vapour transport procedure,140 while CdS nanowires were obtained using a silica template141 and dendritic CdS nanostructures were hydrothermally synthesized.142 It is crucial to control the size and morphology of the materials, as the resulting physiochemical properties are strongly affected. However, it is difficult to obtain a nanoscale CdS single crystal that possesses a high dispersibility, even by using surfactants to decrease agglomeration. Consequently, CdS single crystals have been obtained via several processes involving reverse micelles, organic solvents, or under harsh conditions.143,144 The preparation of such crystalline materials with good dispersibility in aqueous phases leads to the possibility of hydrophobic–hydrophilic interactions. This approach promotes charge separation and enhances the rate of photocatalytic reactions.As mentioned earlier, there are two types of recombination that limit the photocatalytic activity of CdS. One occurs in the bulk, while the other happens on the surface of the photocatalyst. Recombination in the bulk has been overcome by fabricating various CdS nanostructures to facilitate the channelling of electrons. Recently, Kundu et al. have prepared different morphologies of CdS and employed them as photoanodes for photoelectrochemical H2 generation.3 They envisaged that the enhanced activity of the CdS microspheres mostly depends on the shape rather than the effective surface area. Further analysis of these morphologies revealed that the microspheres were mainly composed of one-dimensional (1-D) structures, such as nanoneedles and nanorods with small surface areas of 19.0 m2 g−1 and 7.5 m2 g−1, respectively. Moreover, it was claimed that nanorods and nanoneedles exhibit a better activity than do nanobelts and nanowires despite their large surface areas (32.8 m2 g−1 and 83.9 m2 g−1, respectively). The significantly enhanced activity of all these materials has been attributed to superior charge transport and reduced charge recombination. Besides, they also demonstrated that both surface area and shape contribute equally to the catalytic activity of CdS nanostructures. They fabricated different CdS crystal morphologies by a simple hydrothermal method in which they controlled the volume ratio between water and ethylenediamine (en)3 (Fig. 5A and B). Other factors, such as temperature, precursor ratio, and precursor combinations were also examined. The variation in just the CdS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water ratio (1:1, 1:3, 3:1) led to the formation of different CdS nanostructures, such as nanobelts, nanorods, and nanoplates.
:water ratio (1:1, 1:3, 3:1) led to the formation of different CdS nanostructures, such as nanobelts, nanorods, and nanoplates.
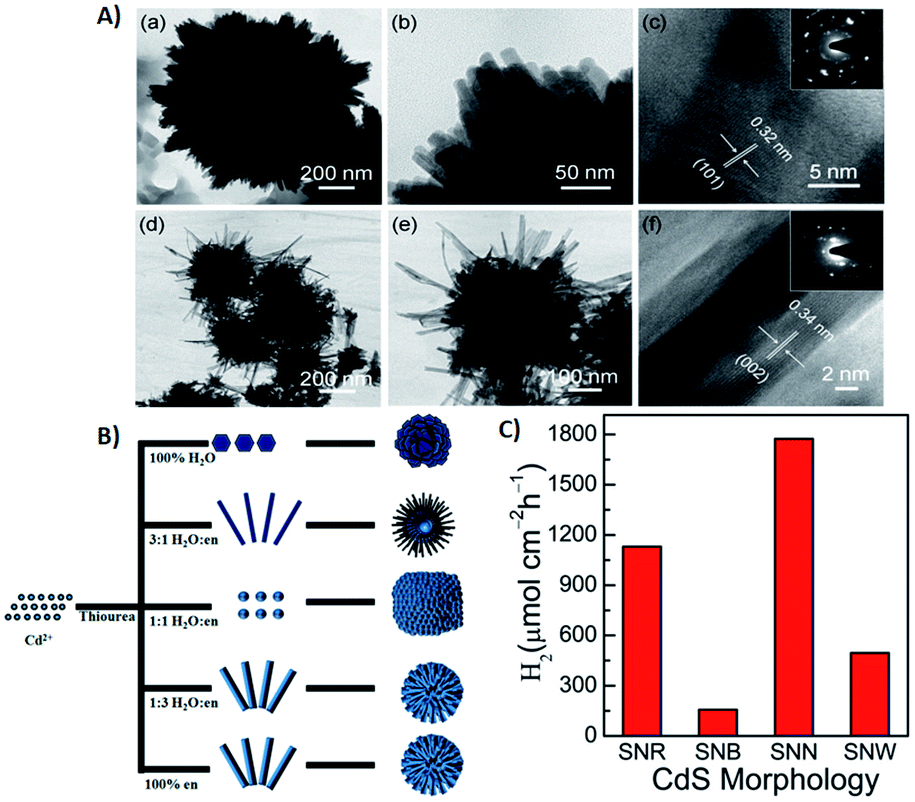 | ||
| Fig. 5 (A) (a and b) TEM images of CdS nanorods obtained with 3:1 water:en solvent; (c) HRTEM and inset SAED pattern; (d and e) TEM images of CdS nanobelts obtained with 1:3 water:en solvent; (f) HRTEM and inset SAED pattern, (B) schematic diagram of synthesized CdS morphologies in different solvent media. Reprinted with permission from ref. 3, (C) photoelectrochemical H2 generation rate of different CdS morphologies under visible light irradiation. Reprinted with permission from ref. 145. | ||
The efficacy of the particle in facilitating charge transport in the crystal may be important. To examine this effect, Pham et al. developed a convenient method to produce highly single-crystalline CdS nanocrystals using cetyltrimethylammonium bromide (CTAB, a capping agent) and demonstrated the impact of CTAB on the crystallinity of CdS single crystals to improve the photocatalytic activity of CdS.145 The interactions between the crystalline facets (001) and the tailored surfactants (CTAB) are the cause of this uniform single-crystalline growth of CdS nanocrystals along the (100) direction. CTAB has been used in the preparation of other metal sulfides,146,147 but there have only been a few reports regarding CdS. The crystal growth of CdS can be well controlled in the presence of a passive agent such as a surfactant. Moreover, by changing the precursor (sulfur source) and solvent ratio, CdS microspheres with different blocks, such as nano-wires (SNW), nanorods (SNR), nanobelts (SNB) and nano-needles (SNN), were obtained.145 These morphologies were further tested quantitatively as photocatalysts for photocatalytic water splitting (Fig. 5C).
Among the nanostructures obtained, the catalytic activity of CdS SNR and SNN were found to be better than those of SNW and SNB since the recombination rate in SNW/SNB was more than SNR and SNN due to a large number of defect centres in the former. The main advantage of this work over other studies was the lower temperature (150 °C) and a shorter time (8 h). Similar studies were conducted to fabricate spherical CdS nanostructures using a water/en mixed solvent at ≥180 °C for 24 h via hydrothermal route.148,149
Guo et al. have demonstrated that the efficiency in the photocatalytic behavior of nanocrystalline materials depends not only on size, surface chemistry, and composition but also on the morphology.150 They successfully controlled the morphology of 3-D assemblies of CdS nanocrystals using an additive-free solvothermal method. Further work revealed that the pH of the reaction media and the cadmium sources also have a marked effect on the morphology of the resulting CdS crystals. All these approaches demonstrate that the catalyst morphology is pivotal in obtaining effective charge separation. Bao et al. have produced hollow nanorods and porous nanosheets via a two-step aqueous route involving the precipitation of a Cd(OH)2 intermediate followed by ion exchange (S2−/OH−).151 A very high H2 evolution rate of 4.1 mmol−1 was observed under 420 nm irradiation, corresponding to the highest QE (60.34%) so far reported for CdS when loaded with 3–5 nm Pt nanocrystals. Another morphology-controlled synthesis of CdS photocatalysts has been reported by Wang et al. who fabricated novel 3-dimensional (3-D) flower-like materials by a template-free solvothermal technique using Cd(NO3)2·4H2O and thiourea and an L-histidine (chelating agent).152 The comparative study of both pure and 3-D “flower-like” CdS was investigated for photocatalytic water splitting (Fig. 6a). The flower-like CdS exhibited better photocatalytic activity (13 times of CdS). For the CdS with L-histidine, the rate of H2 was increased up to 376.7 μmol h−1 using an aqueous medium comprising 0.5 M of Na2S and Na2SO3 while for the CdS without L-histidine, the rate was only 29.2 μmol h−1 (Fig. 6b). Similarly, the photocurrent response of the flower-like CdS material was better (i.e. double) than that of pure CdS. This enhancement in performance was ascribed to the effective charge separation and to the high surface area of CdS/L-histidine. A growth mechanism was proposed for flower-like CdS. The Cd2+ ions in the solution were captured by the imidazole ring of L-histidine, thus avoiding the agglomeration of CdS NPs.
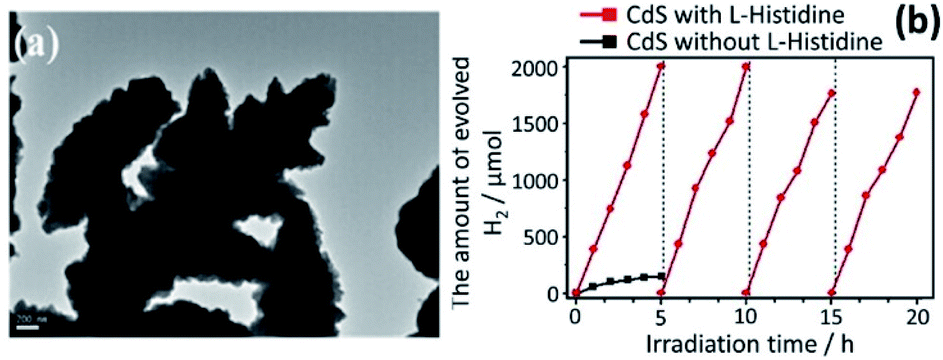 | ||
| Fig. 6 (a) Morphological analysis of flower-like CdS through TEM analysis, and (b) photogeneration of H2 on flower-like CdS with and without L-histidine. Reprinted with permission from ref. 152. | ||
Novel multi-armed CdS nanorods were synthesized by Yu et al.153 by using a solvothermal method employing dodecylamine as a solvent. The nanorods fabricated by this method displayed prolonged activity in the presence of Pt and lactic acid. The rate of H2 production, 1.21 mmol−1 over 12 h, was about 3.2 times greater than with simple CdS NPs. Moreover, with a substantially lower content of Pt (0.23 wt%), a QE of 51% was achieved. The high activity was ascribed to the positive synergistic effect created by several aspects, such as an adaptation of the hexagonal phase, nanorod morphology, good crystallization, and greater pore volume. Additionally, the hexagonal CdS nanorods revealed good photostability and no photocorrosion after recycling. Lamellar and granular CdS nanostructures of different shapes, e.g. nanorods, branches, and cauliflowers, have been synthesized via a hydrothermal route using deionized water (as a coordination agent) and en as a template.154 Interestingly, the morphology of the CdS NPs was dictated by the concentration of en and both Cd(NO3)2·4H2O and thiourea. The nucleation of CdS during the decomposition of Cd–en complex is the key step for determining the shape, which in turn depends on the en concentration. A 30% en content leads to the formation of branched CdS NPs, while a 70% en content yields the cauliflower shaped particles. In the presence of pure en, however, hexagonal CdS nanorods were obtained. The morphologies were tested for photocatalytic H2 production (2577 μmol produced by 0.05 g CdS nanorods over a 4 h period). The rate of H2 production for the CdS nanorods was approximately 43 times faster than agglomerated lamellar and granular CdS which is attributed to the controlled and effective charge separation in the nanorods, due to channelling of electrons.
Apart from morphology tuning for inducing charge survival, the strategy of directional charge separation has also been applied at a molecular level between structurally engineered CdS and photoelectron conducting species. This approach affords another type of self-modification for electron survival. The electric field generated between CdS and the conducting species is the basis of this separation. Liao et al. have described how a conducting head-to-tail (HT) poly-(3-hexylthiophene-2,5-diyl) (P3HT) polymer can be used to control the morphological aspect of CdS nanocrystals as well as the photoelectron conduction for effectual charge separation.155 All these reports reveal that directional charge separation can be obtained by structural tuning.
Cocatalysts loaded CdS for charge separation
The separation and collection of photogenerated charges and their transportation to the reaction sites greatly reduce the chances of recombination and so charge carriers are protected. The function of a cocatalyst is to catalyse the photocatalytic reaction rather than triggering the HER under light irradiation. Moreover, the cocatalyst acts as a sink for collecting the photogenerated charges as well as serving as the source of active sites for HER.77 It is generally believed that H2 activity in the presence of cocatalysts can be increased by at least one order of magnitude. Noble metal-loaded-CdS, such as Pt/CdS,156 Pd/CdS,157 Au/CdS,158 Rh/CdS159 and Ag/CdS,121 are promising photocatalysts for water splitting. And if we take Pt as an example, the mechanism of H2 generation from water splitting using CdS as a photocatalyst is illustrated in Fig. 7.160 Under solar light irradiation, the separation of photogenerated charges occurs at the interphase between Pt and CdS. The lower Fermi level of Pt compared to CdS and the higher work function is the key driver behind the effective charge separation. Electrons can be relayed easily from the CB level of CdS to the Fermi level of Pt.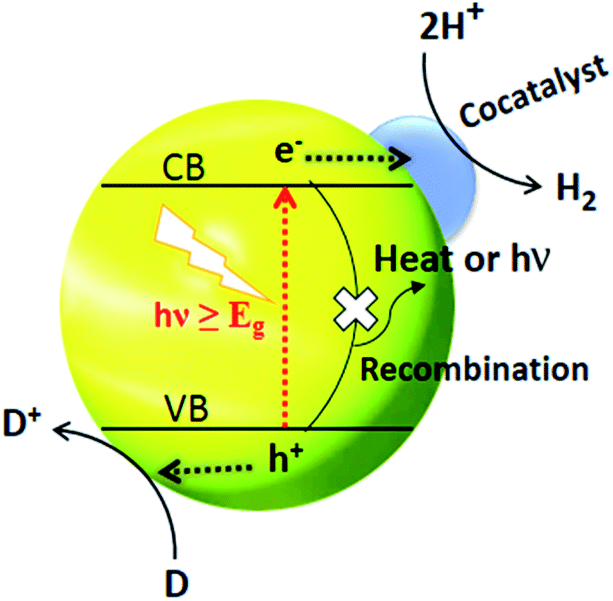 | ||
| Fig. 7 Cocatalyst triggered photocatalytic process where D and D+ represent an electron donor and its oxidized form respectively. Reproduced with permission from ref. 160. | ||
This transfer of electrons creates an electron enrichment (−ve) at Pt and an electron deficiency (+ve) at CdS. The relay of electron and holes away from each other occurs due to the drift current, the force opposing the transfer of electrons.
Ultimately, a dynamic balance is reached, whereby the current resulting from the different electronic levels (e− flow from CdS to Pt) is the same as the drift current (e− flow from Pt to CdS).
Yan et al. have prepared Pt–PdS/CdS, a photocatalyst that achieved a QE of up to 93% and produced 8.77 mmol h−1 H2 with the extremely low loading of Pt (0.30%) and PdS (0.13%) as cocatalysts to CdS.163 Besides, the Pt–PdS/CdS material showed high stability for more than 100 h. Also, Xin and coworkers deposited Pt as a cocatalyst onto CdS via coprecipitation and subsequent annealing.164 The H2 evolution activity reached a maximum at 450 °C (∼300 μmol h−1 g−1) and decreased beyond this temperature, which may be due to the incomplete oxidation of H2PtCl6 and poor crystallinity of the catalyst. In addition, Yao et al. demonstrated the rates of H2 evolution from photochemically treated Pt/CdS, which were compared with those from untreated Pt/CdS; the H2 production rates were 4.8 μmol h−1 and 644.1 μmol h−1, respectively.156 Jin et al. investigated the effect of reduction (NaBH4 reduction and photo-reduction deposition) for Pt deposited onto a CdS photocatalyst.165 Pt deposition via reduction using NaBH4 resulted in a higher photocatalytic activity than when Pt was deposited through the photoreduction process. A significant H2 evolution rate of 1.49 mmol h−1 (QE of 61.7%) was obtained with a minor amount of Pt (0.3%) loaded onto CdS. The reduction method strongly influences the size and distribution of the Pt NPs, which in turn alters the activity of the photocatalyst.
The effects of the morphology of the Pt particles on the activity of the Pt/CdS material has been investigated. In this study, nanocubic and nanospherical Pt particles were prepared and deposited on CdS separately.166 The H2 production rate was 52% higher in the case of the Pt nanocubes. Besides, Pt nanocubes (5.7 and 4.0 nm), loaded onto CdS, exhibited 52% and 31% more activity compared to the same size of nano-spherical Pt particles. This feature arises from several factors: first, spherical particles have smaller volumes than the cubic ones, and, therefore, the possibility of aggregation in the case of spherical Pt NPs is higher than with the Pt nanocubes. Second, the atom density of crystal planes of Pt nanocubes for proton adsorption is higher than that of the nanosphere; and third, the contact of CdS crystals with Pt nanocubes is better than that of the Pt nanospheres, which reduces the impedance between CdS and Pt particles and, therefore, prevents energy loss during charge transportation.
Sahu et al. have studied the kinetics of H2 production using alumina as support under visible irradiation; the reaction rate initially increased and then decreased.167 They observed a negligible induction period upon illumination of the catalyst, which is due to the readsorption of sulfide ions on the Pt/CdS catalyst surface, while the decrease in the rate was attributed to the deactivation of the catalyst by H2. Zhou et al. anchored CdS and Pt NPs in a highly dispersed form in the mesoporous channels of zeolite beta by applying a two-step pore modification strategy.168 A rate of 3.09 mmol h−1 g−1 was achieved which could be ascribed to the synergetic catalytic chemistry between Pt and CdS, the interaction between CdS and the zeolite matrix, and the highly dispersive form of the CdS and Pt particles.
Sakamoto et al. successfully deposited 1 nm Pt NPs on a CdS nanostructure using a wet-chemical method and reported that Pt deposition shows a higher catalytic activity.169 To prepare this system, first, Pt ions are accumulated on the S 21 inch surface of CdS, followed by a reduction of oleylamine to form the Pt core. These cores grow gradually as more Pt ions are reduced and accumulate on the Pt NPs. Depending on the amount of oleylamine used, the diameter of the Pt NPs remains approximately 1 nm. Shen et al. dispersed Ag2S in the CdS nanostructure to investigate its activity toward H2 production in water splitting with the sacrificial reagent Na2S/Na2SO3 being present as a hole trapper.170 The Ag2S/CdS material (with Ag2S 5% by weight) exhibited better photoactivity with an external quantum yield (EQY) of 0.7%. Also, Li et al. have studied the hydrothermal transformation of the cubic-phase CdS nanocrystal into its hexagonal form by treatment with Na3PO4 at 180 °C for 12 h.171 They also studied the effects of hydrothermal time, Pt loading, and phosphate concentration on H2 evolution from aqueous solutions (containing formic acid as a hole scavenger). The phase alteration from the cubic to the hexagonal form was markedly promoted by the phosphate ions. The hexagonal CdS photocatalyst exhibited a higher rate of H2 activity compared to the cubic phase. Moreover, Pt (0.025 wt%) loaded CdS showed excellent activity with an apparent quantum yield of 21.4%.
Large-scale CdS nanorods, nanostrips, and plates have been synthesized via solvothermal techniques using a cadmium oxalate precursor and (NH4)2S as a sulfiding agent.172 The aspect ratio of the nanorod lengths varied from 3 to 30 nm. Comparative photocatalytic activity studies were conducted on a CdS, calcined CdS, and noble-metal loaded CdS nanorods. The nanorod loaded with Pt proved to be highly effective compared to bulk CdS. The 0.2% Pt loaded sample evolved 26 mL of H2 in 6 h, while the activity of bare CdS was less efficient. It was further noted that beyond the optimum amount of Pt deposition, the partial blockage of the CdS surface by metallic Pt NPs occurs, which shields it from the adsorbing photons.
Wang et al. have prepared wool-supported Pd co-catalysts, which were then loaded at an optimal weight percentage (3.0 wt%) onto CdS.173 A high photocatalytic H2 production activity rate of 1555 μmol h−1 was observed. The better catalytic performance was credited to the recycled co-catalyst, wool–Pd that introduced an oxidised (PdS) and reduced (Pd) cocatalyst. For direct conversion of solar into H2 energy, Yan et al. have developed a photocatalyst composed of Pt–PdS doped CdS with a QE of 93%.108 The better QE was achieved with very minimal loadings of Pt and PdS onto CdS (0.13 wt% of PdS and 0.30 wt% of Pt). In combination with a noble metal, transition metal oxides proved to be highly effective in HER. Yao et al. have used Pd and Cr2O3 nanocomposites as cocatalysts and loaded them onto a CdS photocatalyst.174 The HER was higher for Pd–Cr2O3/CdS (2.27 mL min−1) than for the plain Pd metal (1.64 mL min−1) loaded catalyst at room temperature. The shape-dependent effect of Pd nanostructures on the photoactivity of CdS has been investigated by Luo et al.175 Nano-cubes (NCs) and nano-octahedra (NOTs) of Pd nanostructures were prepared by a morphology-controlled method and were deposited onto commercial CdS, and the photocatalytic activity of the Pd/CdS was determined in the presence of an ammonium sulfite (sacrificial reagent). The NCs were enclosed by six {100} facets, while the NOTs were enclosed by eight {111} planes. The Pd-NCs doped CdS (Pd-NCs/CdS) exhibited higher activity (1.38 times than that of Pd-NOTs/CdS) due to the Pd {100} crystal facets of the Pd NCs and the higher electrochemical active surface area (ECSA).
Many studies have explored the synthesis of PdS and its use as a cocatalyst on CdS, e.g., Chen et al. have developed efficient supports for overall water splitting (in the presence of PdS and Na2S/Na2SO3 as a hole scavenger).176 The 1 wt% loading of PdS onto CdS, prepared by an in situ coprecipitation method, exhibited better photoactivity and stability than prepared by hydrothermal methods. Bi-metal cocatalyst incorporation on CdS, e.g., Pt–Pd NPs, at an appropriate loading has been studied by Li and coworkers.177 A notable H2 evolution rate of 25.28 mmol h−1 g−1 (Pt–Pd/CdS) was recorded under visible irradiation (300 W Xe light, λ > 420 nm), which was 2.4 and 3.8 times higher than that obtained for Pt/CdS and Pd/CdS respectively. The improved catalytic activity of Pt–Pd/CdS was ascribed to the effective suppression of charge recombination.
Owing to the unique optical, magnetic, electronic and catalytic properties, Au NPs have appeared as a new class of material.178 Recently, Zhao et al. found that combining CdS QDs and Au NPs under certain conditions results in the transfer of electrons and energy between particles (Au NPs and CdS QDs).179 The CdS-QDs/Au-NPs nanohybrids apparently promote charge separation and enhanced catalytic activity. Similarly, Xing et al. have prepared the novel nanohybrid, CdS-QDs/Au-NPs@POM, by a self-assembly scheme.180 The POM (H3PW12O40) material was employed as an encapsulating, reducing, and bridging molecule in the hybrid system. Interestingly, the resulting nanohybrid exhibited very high photocatalytic H2 evolution in water splitting.
Also, Shemesh et al. have prepared two novel nanohybrid materials containing CdS as the photocatalyst and Pd compounds (PdO and Pd4S) as cocatalysts.181 The CdS–PdO material was prepared by an aqueous phase hydrolysis method and CdS–Pd4S was designed via a high-temperature treatment (using a reduction method) followed by cation-substitution. The desired CdS nanorods of varying diameter were prepared and injected into a solution of Pd(II) acetylacetonate. The new nanohybrids were tested for the photoreduction of water. The nanohybrid maintained the CdS morphology, but some sites clearly showed more contrast at the tips of the CdS nanorods (Fig. 8a).
 | ||
| Fig. 8 (a) (a) Pd4S growth on CdS nanorods; (b) size distribution of CdS nanorods decorated by Pd4S; (c) close-up of the sample shown in (a); (d) HRTEM of a single CdS–Pd4S nanohybrid of the sample shown in (b); (e) PdO growth on CdS nanorods; (f) CdS nanorods decorated with large dots of PdO. Reprinted with permission from ref. 181. (b) TEM (A and B) and HAADF (C and D) images of CdSe@CdS nanorod with Au–Pt bimetallic. Reprinted with permission from ref. 182 | ||
Palladium oxide (PdO), was more catalytically active than other small NPs, such as gold. Nano-SCs may elevate the CB level due to quantum confinement effects to assist in the reduction process and make it more thermodynamically favorable (0.03% yields). Moreover, the small NPs may exhibit a greater number of reactive step-edge sites and therefore, boost the photocatalytic activity.
Further improvement in the productivity of photocatalytic water splitting was accomplished by Kalisman et al., who used the bimetal Au–Pt as a cocatalyst182 (Fig. 8b). They prepared CdSe@CdS nanorods tipped with Au decorated Pt. Simply, by varying and adjusting the composition and shape-engineering of Au–Pt, they achieved an impressive H2 production rate compared to pure Pt. The strategy of coupling Au with Pt, grown on CdSe@CdS nanorods leads to the formation of a promising single catalytic reduction site.
The synthesis of colloidal tetrashaped CdS nanocrystals via a low-temperature phosphine-free method and in the presence of sodium diethylcarbamodithioate or thioacetamide has been undertaken.183 Using different sulfur sources, the growth of CdS particles was controlled to tune the arm length and diameter of a CdS tetrapod. Then, Au incorporation onto CdS to form the Au/CdS nanohybrid was demonstrated towards HER. In the presence of Au, the tetrapod CdS nanohybrid displayed excellent photoactivity.
To study the role of the oxidation catalyst, Tseng et al. have studied the charge-transfer relations between the photoexcited CdS nanorods and the water oxidation catalyst resulting from [Ru(bpy) (tpy)Cl]+.184 The photogenerated hole in the VB of CdS oxidized Ru2+ to Ru3+ (100 ps) followed by a reduction of Ru3+ back to Ru2+ (10–100 ns). The advantage of this slow dynamic electron-transfer is that it may provide an opportunity for holes to oxidize water molecules.
Non-noble metals doped CdS photocatalysts, such as Ni–CdS QDs, have been reported to be one of the most robust materials for the artificial photocatalytic release of H2 from water. CdS QDs, capped with 3-mercaptopropionic acid, can be reacted in situ with Ni salts for the synthesis of Ni hybrid CdS QDs. As compared to CdTe and CdSe QDs, surface-modified CdS QDs have a greater affinity for Ni2+ and very high efficiency for H2 production.
Liu and coworkers have managed to dope CdS with Mn2+ and found that the crystal phase, band gap, and morphology of CdS remain intact.195 A doping content of 0.5 wt% doubled the H2 production rate (300 μmol h−1 gcat−1) compared to that for bare CdS. The Mn2+ ions serve as shallow trapping sites for electron–hole pair separation. Moreover, the apparent quantum yield (AQY) value reached 5% using 0.5 wt% doping indicating that the photoactivity of CdS can be enhanced by Mn2+ doping. As discussed in the previous section, the noble metals (Pt, Pd, Rh) are very effective active sites for proton reduction (as they act as electrons traps).196,197 Similarly, a non-noble metal, such as Ni and its compounds such as NiS, NiO, Ni(OH)2, and Ni@C, can play the same role as a cocatalyst in the decomposition of water.
To use Ni and Ni compounds as cocatalysts, researchers managed to synthesize successfully various combinations of photocatalysts, such as Ni/CdS, NiS/CdS, Ni(OH)2/CdS, Ni@C/CdS, in efforts to improve the efficiency of the CdS SC.185–189 These modifications have several advantages with respect to release H2 from water, as well as providing an economically cheap material for the cocatalyst. In general, the high activity of a NiS/CdS photocatalyst was attributable to the construction of a p–n junction between the NiS and CdS interface, which hampered the recombination of the charge carriers thereby enhancing the H2 production rate. The activity of Ni(OH)2/CdS was improved significantly, up to a factor of 136 compared to bare CdS (using Na2SO3 and Na2S aqueous solutions).185 The Ni(OH)2 loaded CdS-nanorods composite was synthesized using a precipitation method. A special focus was given to the influence of the Ni(OH)2 species on the photoactivity of CdS using a triethanolamine aqueous solution. The 23 mol% optimized Ni(OH)2 loaded CdS generated an H2 production rate of 5085 μmol h−1 g−1 with 28% QE at 420 nm, exceeding by a factor of 145 and 1.3 compared to pure CdS and 1 wt% loaded Pt CdS nanorods respectively. The improved activity was credited to the suitable reduction potential of Ni2+/Ni0 (Ni2+ + 2e− = Ni0, E0 = −0.23 V), which was higher than the CB potential of CdS (−0.7 V) and is less than the reduction potential of H+/H2 (2H+ + 2e− = H2, E0 = 0.0 V). In addition, the role of Ni0 is to suppress charge recombination when acting as a reduction co-catalyst in water splitting; Ni0 can easily channel the transfer of electrons and accelerate the rate of H2 generation.
Simon et al. have employed hydroxyl anion/radicals as redox mediators to transport holes from Ni/CdS to the scavenger.78 A rate of 63 mmol h−1 g−1 and an apparent QE of 53% were achieved under 447 nm laser irradiation. The high activity was ascribed to the fast-hole channelling effect that confers long-term photostability on CdS and thus improves overall water splitting. Peng et al. have prepared Ni@C/CdS and tested it as a photocatalyst (using Na2SO3 and Na2S as a hole scavenger) in aqueous solution for the production of H2 from water.198 A rate of 3.328 mmol h−1 g−1 was observed using a 300 W Xe-lamp (I > 420 nm). The improved performance was attributed to the deposition of metallic Ni on the CdS surface, which was supported by graphite-like carbon acting as an electron acceptor.
Li et al. have synthesized graphene nanosheets decked with CdS clusters for use as a visible responsive candidate for highly efficient H2 production.199 The graphene/CdS nanocomposite had an excellent H2 release rate of 1.12 mmol h−1, i.e., 4.87 times greater than that for pure CdS NPs. Moreover, for a 1.0 wt% graphene and 0.5% Pt content, the QE obtained at 420 nm was 22.5%. The improved activity was ascribed to the electron collector ability of graphene and the channelling of these electrons, thus extending the lifespan of the charge carriers. The electrons were promoted from the VB of CdS to the CB and then were channelled in three ways. First, the electrons were trapped by the deposited Pt, followed by the carbon atoms in the graphene nanosheets and then by the metallic Pt on the graphene nanosheets. The adsorbed H+ ions were reduced by these electrons to generate H2 gas. As previously mentioned, the CB of CdS is more negative than the reduction potential of H+/H2; however, using bare CdS, the H2 activity was negligible due to the rapid recombination of the photogenerated electron–hole pairs. In the presence of graphene, the electron collection and transfer are enhanced, which suppresses the charge recombination process. Importantly, in the presence of graphene, the proton reduction reaction not only occurs on the surface of the catalyst, but also over the graphene nanosheets themselves199 (Fig. 9). In the case of N-graphene/CdS, the electrons are transferred preferentially from CdS to the N-doped graphene.192 The recombination of the photogenerated charges, in this case, was effectively suppressed, which enhanced the photocatalytic activity. The activity order (N-graphene/CdS > graphene/CdS > GO/CdS > CdS) in this regard showed that N-graphene/CdS has better stability for H2 production over a period of more than 30 h, revealing that N-graphene, acting as a cocatalyst, can inhibit CdS from photocorrosion. Thus, N-graphene is a promising cocatalyst for use in developing a high-performance CdS photocatalyst for H2 generation.
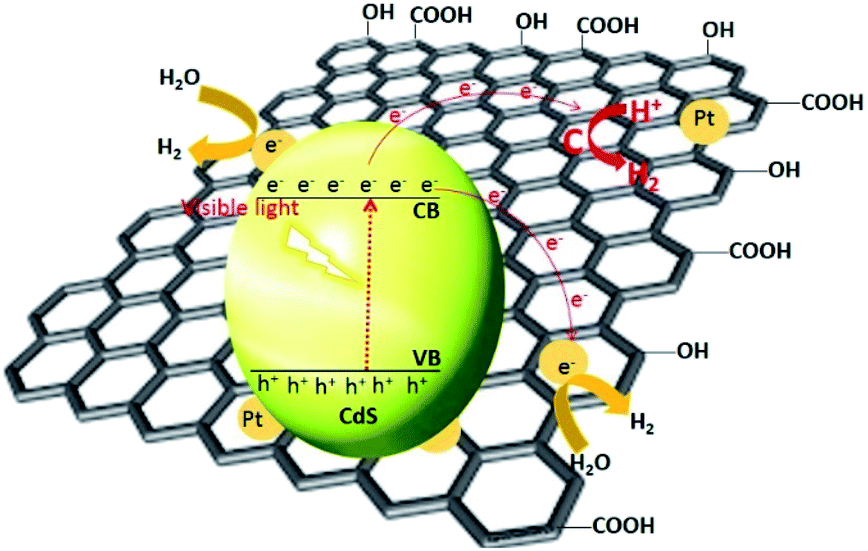 | ||
| Fig. 9 Proposed mechanism for the charge separation and transfer in the graphene–CdS under visible light irradiations. Reproduced with permission from ref. 199. | ||
CdS-based heterojunctions
A heterojunction is simply a hybrid photocatalytic system constructed from two different SCs. The built-in electric field is created at the interface of two SCs owing to the difference in Fermi levels. There are three types of heterojunctions that are classified as the band alignment straddling gap (type-I), the staggering gap (type-II), and the broken gap (type-III)200 (Fig. 10).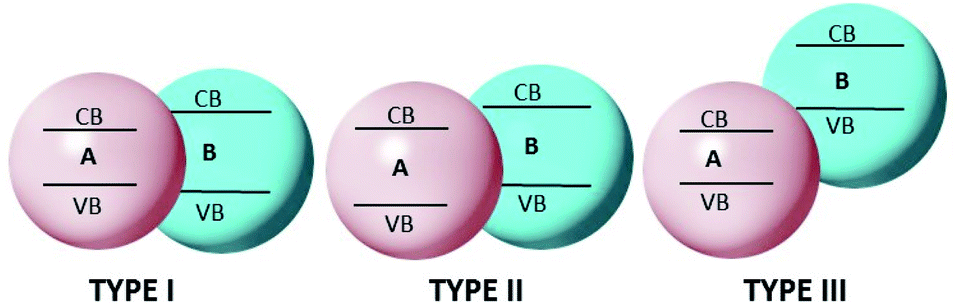 | ||
| Fig. 10 Band alignment in type I, II, and III heterojunctions. | ||
In type-I heterojunctions, the VB and CB levels of the narrower band gap are enclosed in those of the larger one. Also, in a type-I heterojunction, charges are pushed towards a narrower band-gap SC, where they easily recombine. In type-II heterojunctions, CB and VB levels are staggered between two SCs, thus creating an accumulation of opposite charges at the respective sites and so preventing recombination of the charges. Type-II heterojunctions, which are considered to be more effective, comprise two classes. One class is formed by a single SC having two phases, i.e., a heterophase, such as the heterojunction formed with the TiO2(B)–anatase heterophase. The second is formed by two different SCs, and has been demonstrated to be highly efficient, e.g., the CdS/CdSe201 and CdS/g-C3N4,202 CdS/TiO2 (ref. 203) heterojunctions. In type-III heterojunctions, charge suppression is hard to achieve as the CB level of one SC lies just below that of the other SC, where charge recombination is dominant. These types of heterojunctions are rarely applied in photocatalysis.
The SCs BiVO4,204 Ta3N5 (ref. 205) and Ag3PO4 (ref. 206) are known for their role in O2 evolution, while the CdS photocatalyst is especially efficient for H2 evolution;47 however, as discussed above, some associated issues hamper its activity. Hence, CdS-based heterojunctions have been developed to overcome the recombination phenomenon, poor stability, and particles agglomeration. Notable examples include the formation of a Pt–PdS/CdS heterojunction with a very stable QE of 93% for H2 generation when it contains Ag2S/CdS.170 The holes in Ag2S oxidize sulfite ions, which are the hole scavengers. Besides, the junction formed between CdS and TiO2 nanotubes results in improved photocatalytic performance because of the ease of electron transportation from the CB of CdS to the CB of TiO2.207 Moreover, in the presence of a reduction metal cocatalyst, such as Pt, the heterojunction system displays the same electron-transfer phenomenon in a more controlled way.208
A very high current density (12 mA cm−2 at 0 V vs. Ag/AgCl) has been recorded for CdS/ZnO/ZnO/CdSe nanowire arrays in PEC water splitting.125 In this heterojunction system, the position of the Fermi levels of all species (CdS, CdSe and ZnO) are so aligned that the CB of CdSe and CdS are close enough to permit electrons (created in CdSe) to delocalize and redirect to ZnO via the CdS layer. In a report published by Moriya et al., a thin coating of CdS on Pt/CuGaSe2 – an active p–n heterojunction – achieved not only a 10 days long term performance for photoelectrochemical (PEC) water splitting, but also had 6 times greater photocurrent response than did Pt/CuGaSe2 (ref. 209) (Fig. 11). The markedly enhanced activity was ascribed to the facile charge suppression between these two materials because of the p–n junction created and the optimum increase in the thickness of the depleted region at the interphase of solid-electrolyte. Moreover, the different position of the VB offset between the two materials (0.98 V) was the reason behind the electron diffusion into CdS from CuGaSe2 (CB –0.8 V and VB +0.9 V vs. NHE). More details on different CdS based heterojunctions are discussed in the following sub-sections.
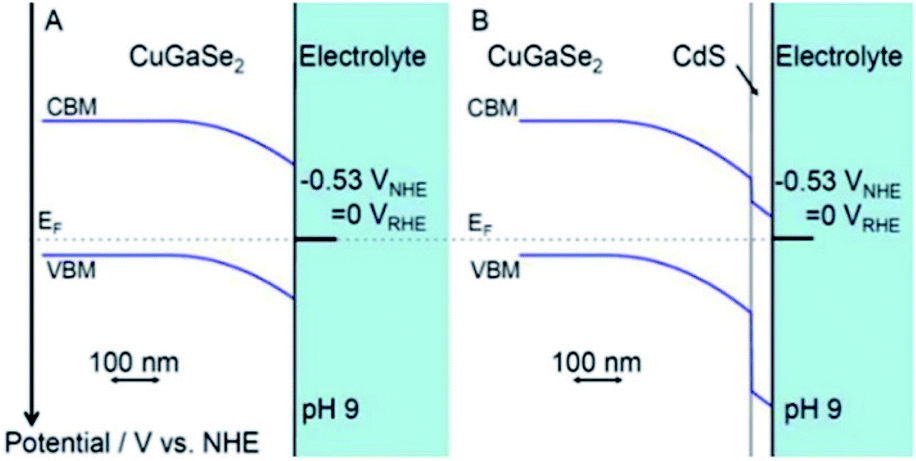 | ||
| Fig. 11 Band alignment at interfaces for (A) CuGSe2 and (B) CdS/CuGSe2 electrodes. Reprinted with permission from ref. 209. | ||
A 2-D hybrid system, consisting of CdS nanosheets and an MoS2 layer, has been developed via a two-step hydrothermal and ultrasonic treatment. The loading of an appreciable amount of MoS2 onto CdS nanosheets enhanced their photocatalytic activity. Furthermore, the optimized 2-D CdS/MoS2 (1.0 wt%) achieved a 1.75 mmol h−1 g−1 rate of H2 from water containing sulfide and sulfite ions. This rate was greater by a factor of ∼2 compared with pure CdS nanosheets. The intimate 2D–2D contact coupling interface in the material promoted and transported charges to the surface sites. To suppress further the recombination process, formic and lactic acids were introduced as sacrificial reagents.213 Other electron donor species can also be used to enhance the photocatalytic activity of such junctions, such as lactic acid as reported by Zong et al.190
Recently, Xie et al. have reported a unique charge-transfer phenomenon for CdS–ZnS core–shell particles in photocatalytic H2 generation (with 792 μmol h−1 g−1) via water splitting.218 They found that electrons and holes cannot transfer to the ZnS shell because the CB energy is higher compared to CdS. Also, the VB of ZnS is more positive than is that in CdS, with the acceptor states lying within the band gap of ZnS, thus allowing hole transfer from the VB of ZnS shell to CdS and promoting efficient charge separation. To overcome the problem associated with type-I heterojunctions, Zhuang et al. have prepared a multi-node sheath to accomplish adaptation from a type-I to type-II heterojunction.219 The [ZnS–CdS]–ZnS–[ZnS–CdS]–ZnS-hetero-nanorods alone displayed type-I behaviour. At the ZnS/CdS interface, the charges are close enough to recombine. After introducing Au, the position of CdS bands alters, which facilitates electron transfer to CdS from Au. The electrons are shifted from CdS to ZnS, creating holes in the VB of CdS, thus separating the charges, therefore converting into a type-II example.
Inorganic-organic CdS@g-C3N4 core–shell nanorods have been prepared by a hydrothermal method.220 These materials displayed excellent stability against light illumination as compared to pure CdS NRs. Besides, a photocurrent density of 1.16 mA cm−2 was obtained (2.5 times of pure CdS NRs). Interestingly, more than 85% of the initial photocurrent was maintained for CdS@g-C3N4, while for pure CdS NRs it dropped to 20% after 3600 s of continuous illumination. Some studies are reported on the g-C3N4/CdS heterojunction, including a systematic calculation of both the energy of the band gap and charge transfer.221 The contact between CdS (110) and g-C3N4 is known as a van der Waals heterojunction. The formation of a standard type-II heterostructure was the result of changed positions of the band edges of g-C3N4 and CdS. Moreover, the calculated Bader charge and the charge density difference show that the separation of the charges at the g-C3N4/CdS interface was due to the internal generated electric field.221 These observations provide evidence for the band structures of the hybrid g-C3N4/CdS and its photocatalytic efficiency.
CdS/g-C3N4 heterojunction nano-cone arrays, constructed on Si (100) substrates, have also been investigated via the plasma sputtering reaction deposition (PSRD) and pulsed laser deposition (PLD) techniques.222 The g-C3N4 nano-cone arrays prepared by PSRD were single crystalline in nature with a [001] growth direction, while deposition of CdS NPs shells by the PLD method had a hexagonal wurtzite structure. The bare g-C3N4 nano-cones showed an intense distinctive PL peak at 454 nm. While preparing the hybrid with CdS, peak quenching occurred, showing that the chances of charge recombination had been extensively suppressed because of the spatial charge separation in the CdS/g-C3N4 heterojunctions. The absorption edges of the CdS/g-C3N4 heterojunctions displayed slight red shifts when CdS was deposited onto g-C3N4.
Some CdS/g-C3N4 nanowires have been produced by changing the contents of g-C3N4 using a combined solvothermal and chemisorption synthetic route.223 These nanowires have been investigated for H2 production using Na2S and Na2SO3 (λ ≥ 420 nm). Preferentially, g-C3N4 was coated on CdS (after successful spontaneous adsorption), which resulted in an enhanced photocatalytic activity (H2: 4152 μmol h−1, g-C3N4: 2 wt%). Also, the g-C3N4 coating improved the photostability of the catalyst for long term photocatalytic activity. This high activity was ascribed to the synergistic effect created between CdS and g-C3N4, which suppressed the charge recombination and redirected the holes from CdS towards g-C3N4.
Similarly, CdS QDs coupled with g-C3N4, were prepared through a chemical impregnation process. The effect of CdS loading (varying dosage of CdS) on the g-C3N4 catalytic activity was investigated in methanol/aqueous solution.224 This positive synergistic coordination between CdS and g-C3N4 leads to effective charge separation and enhanced catalytic activity. A 30 wt% CdS content was shown to be the optimum amount, which evolved 17.27 μmol h−1 (∼9 times of g-C3N4). The CdS QDs/g-C3N4 nanohybrid had a bathochromic shift and exhibited a robust absorption capability for the visible light.
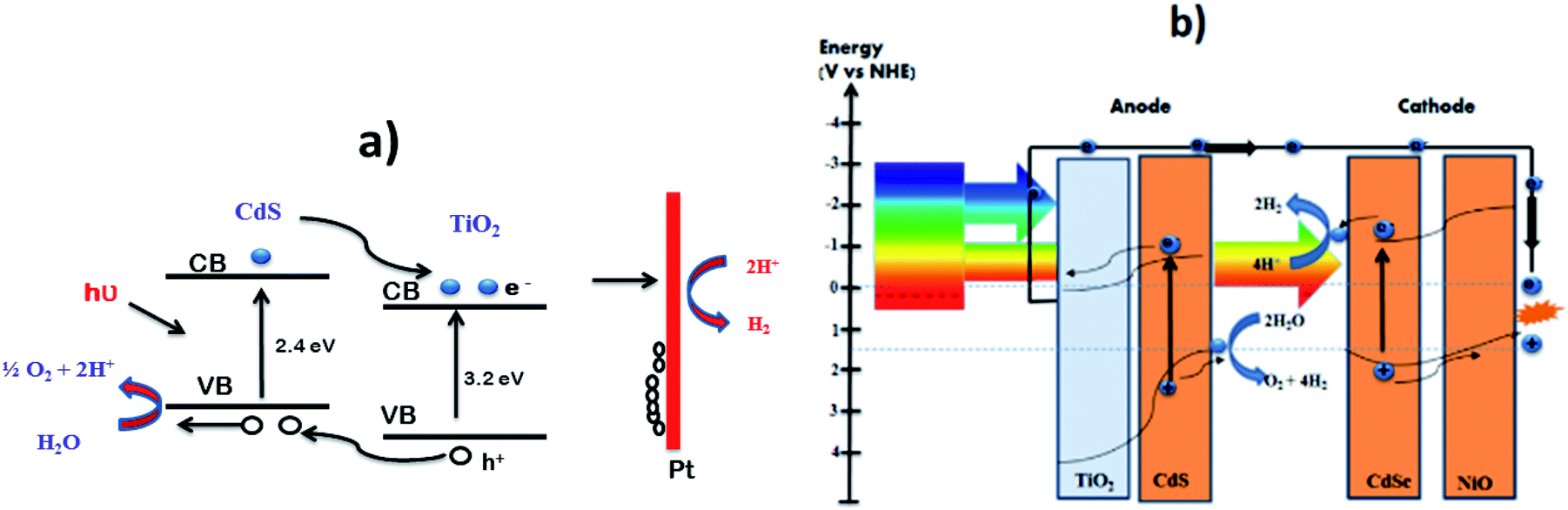 | ||
| Fig. 12 Photoelectrochemical water splitting. (a) Mechanism of PEC water splitting by CdS–TiO2 nanocomposite. Reproduced with permission from ref. 225 (b) photoelectrolysis cell consisting of CdS coupled TiO2 photoanode and CdSe coupled NiO photocathode. Reproduced with permission from ref. 227. | ||
Corrosion-resistant photoelectrodes for efficient water splitting consist of surface-modified CdS QDs coupled with TiO2 as the photoanode and CdSe QD coupled with NiO as the photocathode227 (Fig. 12b).
The QDs in these photoelectrodes were surface passivated with ZnS shells. These photoelectrodes have an efficiency comparable to that of natural photosynthesis (0.17%) and exhibit excellent photostability in visible light. The surface-modified CdS QDs impregnated with TiO2 represent an efficient photoanode and CdSe modified by NiO acts as a photocathode where the Cd chalcogenide is protected by a ZnS layer. The optimized system exhibited a photo efficiency of 0.17% comparable to that of natural photosynthesis.
Because of their high molar absorptivity, CdS QDs have been used as potential candidates in photo-electrochemistry and photovoltaics. CdS QDs are deposited on TiO2-NTs for efficient water splitting. However, CdS NPs tend to aggregate on the surface of TiO2-NTs, which causes a considerable decrease of contact between these two SCs. The aggregation of CdS QDs results in a low absorption of light and a decrease in charge separation and the charge-transfer rate. Surface-modified CdS QDs have a notable degree of dispersion and impregnation with TiO2-NTs extends exposure to longer wavelengths and promotes the efficient shifting of electrons between CdS QDs and TiO2-NTs.228
Although CdS QDs are potential contenders for water splitting, the rapid rate of electron–hole pair recombination limits their application in water splitting. However, CdS QDs capped with different surface modifying agents show remarkable properties. Stabilizing agents decrease the size of the CdS NP and result in broadening of the band-gap energy, which preserves the potential barrier for free electrons and holes, and lowers their rate of recombination. In surface-modified CdS QDs, free electrons are readily available in the CB, which is used for photoelectrochemical reduction of H+ to H2. CdS QDs have size-tunable band gaps (3.5 to 2.2 eV). These surface-modified CdS QDs can transfer electrons from the CB level of wide-band gap SCs, thus improving the efficiency of energy conversion and providing new opportunities for harvesting energy.
CdS-sensitized TiO2 NPs have been proposed as efficient materials for photo-electrochemical water splitting under visible light irradiation229 and have attracted substantial attention worldwide. However, surface-modified CdS QDs also have important properties that make them versatile for photoelectrochemical water splitting.230 The CB level of surface-modified CdS is higher than that of TiO2, while the CB of CdSe is lower than that of TiO2. Therefore, CdS NPs can be efficiently used for the transfer of electrons to TiO2. But, because of its band gap energy of 2.25 eV in the bulk, the absorption range is limited to below 550 nm. Conversely, CdSe NPs have an extended absorption range (below 730 nm), but their CB level lies below the CB level of TiO2. Advantageously, these materials have been used as co-sensitizers for TiO2 films. However, poor performance was reported for the reversed structure, i.e., TiO2/CdSe/CdS. From knowledge of the Fermi levels of CdS and CdSe, a stepwise method for the construction of TiO2/CdS/CdSe was described, which is an efficient electrode in photoelectrochemical cells for water splitting118 (Fig. 13). The TiO2–CdS nanohybrids displayed improved performance for water splitting compared to bare TiO2. The cascade structure of TiO2, sensitized with surface modified CdS and CdSe NPs, achieves the equilibrium of the band edge via alignment of the Fermi levels. Interfacial transfer of an electron between the QDs and TiO2 is promoted, thus increasing the photocurrent.231 ZnO nanowires and TiO2 sensitized with surface-modified CdS and CdSe QDs represent electrodes with a high photocurrent density of 12 and 14.9 mA cm−2, respectively, resulting in high efficiency for water splitting.
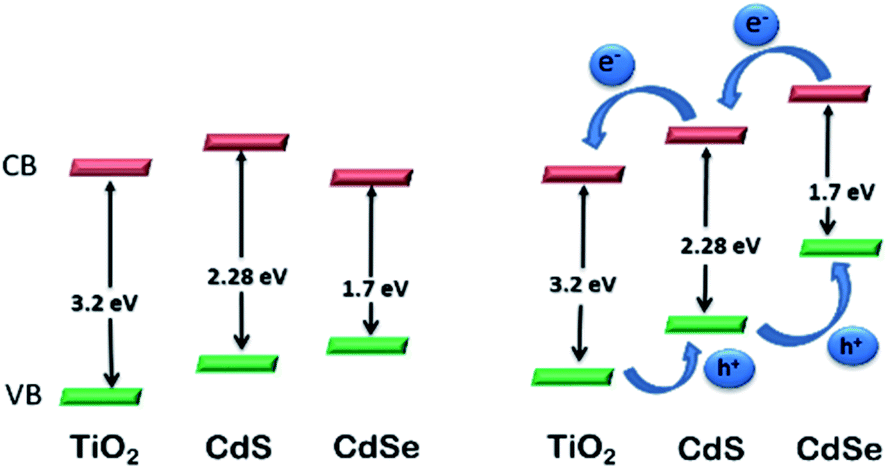 | ||
| Fig. 13 Relative energy levels in TiO2 photoelectrode co-sensitized with CdS and CdSe NPs. Reproduced with permission from ref. 118. | ||
Recently, a core–shell nanowire of ZnO/ZnS/CdS/CuInS2 was prepared which achieved a better photocurrent response (10.5 mA cm−2) in the presence of Na2S/Na2SO3 as an electrolyte with an IPCE of 57.7% at 480 nm at 0 V vs. Ag/AgCl.241 The p–n junction designed between CdS and CulnS2 maximizes the absorption of visible light and avoids the possibility of recombination. In Au-based junctions such as the CdS–Au–TiO2 heterojunction (a detailed discussion of which follows in the next section), Au acts as a plasmonic sensitizer and charge facilitator, which results in a photocurrent response of 4.07 mA cm−2 at 0 V (vs. Ag/AgCl) with an STH efficiency of 2.8% under full arc irradiation.134 The heterojunction allows the transfer of electrons and prolongs the lifetime of charges and so increases the rate of reaction. These photoelectrons were transferred from CdS via Au to TiO2 at a lower incident wavelength (I < 525 nm). At longer wavelengths (525 nm–725 nm), however, CdS and TiO2 were not excited and an extra plasmonic occurrence to transfer energy from the excited Au to TiO2 takes place via a hot electron phenomenon as discussed earlier.
The above approach is somewhat different, but it has a very important and unique mechanism, which certainly offers an enhanced solar absorption capacity for photocatalytic water splitting. A heterojunction between CdS and Au (CdS/Au) exhibited excellent photocatalytic activity (ca. 4 mA cm−2 at 0 V vs. Ag/AgCl, Na2S/Na2SO3 electrolyte) owing to the facile relocation of an electron from CdS to Au.242 On the other hand, it has been reported that Au incorporation has an effect on H2 production compared to Pt.243 Tongying et al. fabricated some double-heterojunction nanowire (CdSe and CdSe/CdS NWs) photocatalysts decorated with Pt and achieved an H2 generation rate of 435 μmol h−1 g−1.244
To develop a single junction from a core–shell structure, the coating of the SC is a good approach as it prolongs the lifetime of the charge carriers, which was achieved by hindering the influence of surface defects, which promote recombination, while in the double junction, the electrons are easily mobilized from the shell to the metal. Besides, the NiS/CdS p–n junction revealed notable H2 photocatalytic activity because of the enhanced charge-transfer phenomenon at the junction of two SCs.186 Similarly, a junction constructed between WS2 and CdS displayed an excellent photocatalytic activity with an H2 generation rate of 420 μmol h−1 g−1, i.e., a higher response than CdS but lower than that for a Pt/CdS electrode.245
CdS-based Z-scheme photocatalysis
As we have noted, heterojunctions play an important role in efficiently separating the charges. Unfortunately, the reduction capacity of the electron decreases as they move to a lower CB. As discussed earlier, Z-type natural photosynthesis is considered an effective mechanism for solar energy conversion. It consists of a two-step photoexcitation and comprises three types.32–35 The first is “natural” photosynthesis for which the two SCs do not necessarily have to be in contact. The electron acceptor/donor (A/D) pairs need to transfer charges to each other, as in I−/IO3− and Fe2+/Fe3+. The second and third types are A/D-free systems (known as solid-state Z-schemes). The second type contains the A/D pair which replaces the conductor as an electron mediator, while the third type has no need of a conductor.246Zhang et al. have prepared a CdS/WO3 heterojunction for the clean generation of H2via photolysis of water.247 Interestingly, even though WO3 cannot be used alone in water splitting, it enhances the photoactivity of CdS through a Z-scheme in the presence of lactate as an electron donor. An activity for H2 production of 369 μmol h−1 g−1 using CdS/WO3 (20 wt% CdS) was achieved (5 times greater than that of CdS) with lactic acid.32 More recently, a RGO based ternary CdS/RGO/g-C3N4 hybrid system for H2 generation by Z-scheme electron transportation has been designed.
By using a hydrothermal process, the two SCs exfoliated g-C3N4 nanosheets, and a CdS-coupled RGO can be coupled to construct a Z-scheme pathway. Compared to the bare CdS photocatalyst, the CdS/RGO/g-C3N4 hybrid showed more efficient H2 generation owing to the better charge separation and photostability of the system. The RGO had a crucial role in channelling electrons between the interfaces of the two SCs through the Z-scheme route. An H2 generation rate of 676.5 μmol h−1 g−1 with an AQE of 36.5% and 1980.2 μmol g−1 was achieved using the ternary hybrid material. Only an optimal amount of g-C3N4 (50 wt%) was loaded onto the hybrid system, as an excess amount beyond this limit (say, 70 wt%) would block the incoming light from the surface of CdS. The proposed Z-scheme charge transfer in the CdS/RGO/g-C3N4 composite was explained based on the band potentials of the two SCs and the electron mediation ability of the RGO. Under light irradiation, the electrons are readily promoted from the VBs to CBs of the two SCs (g-C3N4 and CdS). Consequently, the strong oxidation potential possessed by the VB of CdS (∼1.88 eV) resulted predominantly in the oxidation of the sacrificial reagent. Meanwhile, the electrons from CdS flow into the RGO as CdS was grown in situ on the RGO nanosheets via a hydrothermal process. The Fermi level of the RGO and the CB of CdS were close in energy and so a very short distance for charge transfer was created. Subsequently, the VB holes of g-C3N4 were filled with electrons from the RGO (electron shift from CdS to g-C3N4via RGO). As a consequence, the excess electrons residing in the CB of g-C3N4 reduced the H+ ions of water to evolve H2 gas.
Solid–solid contacts create a number of defects, to reduce which, the third type of Z-scheme has been developed. The electron mediator is removed to reduce the solid–solid interface contact area between the two SCs. Then, to augment electron transfer, a CdS/WO3 nanohybrid has been prepared by Huang et al.248 In this case, the function of the RGO is to facilitate the transfer of electrons and not to act as an electron mediator. Under light irradiation, both SCs generated electron–hole pairs where the CB electrons of CdS can readily move to the VB of WO3 in the case where the electron mediator is absent. This transfer occurs due to the electrostatic interaction between CdS and WO3 and operates as a Z-scheme.249 The direct coupling of the components may not only avoid any back reactions and meanwhile can improve the reaction efficiency. Similarly, another simple technique was established for the preparation of CdS–Au–TiO2 (an anisotropic nanojunction) on which Au was spatially fixed as an electron-transfer agent. This ternary system displays much higher activity than did either a single- and a two-component system. The higher activity achieved for the three-component system may be due to anisotropic electron movement between TiO2 and CdS. As a consequence of the energy band of the three-component system (λ > 400 nm) (Fig. 14), the CB electrons reduce the methyl viologen (MV2+) rather than being transferred to Au owing to the CdS size quantization.250 The holes in the VB of CdS are subsequently filled by electrons from Au, as indicated by the red shift of λmax (electron transfer I, Au → CdS). Meanwhile, at λex > 300 nm, the holes from the VB of TiO2 have a strong ability to oxidize the solvent, while the electrons from the CB of TiO2 flow into the Au (confirmed by the blue shift of λmax for electron transfer II, TiO2 → Au). The instantaneous electron transfers following I and II path (vectorial electron transfers) occur due to the excitation of both CdS and TiO2 under the MV2+ reduction condition.
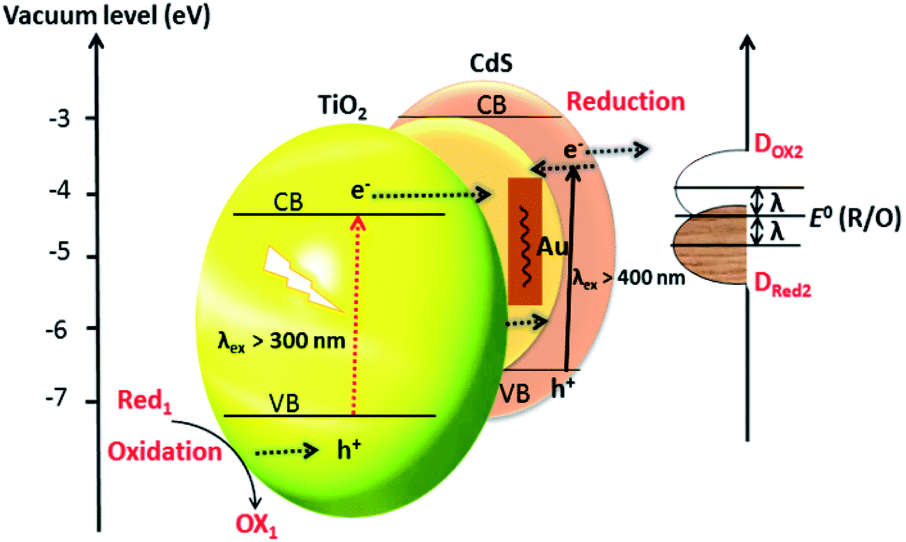 | ||
| Fig. 14 Schematic energy diagram for the Z-scheme electron transfer mechanism. Energy band illustration of CdS–Au–TiO2 system where E0 (R/O) is the standard electrode potential of MV+/MV2+ and DRed2 and DOx2 illustrate the occupied and unoccupied states, respectively. I represent the reorganization energy. Reproduced with permission from ref. 250. | ||
In view of the above discussion, Z-scheme photocatalysis has significant advantages because of the excellent reduction ability compared to heterojunctions. The available electrons reside in a higher CB rather than transferring to the lower one, which hinders the charge recombination very efficiently. It should be recognized that both heterojunction and Z-schemes have their own roles and specialties.
Given the above-mentioned approaches, the properties of some important CdS based photocatalysts are listed in Table 1 to provide an overall view of the modifications that have been made to CdS for the purpose of photocatalytic water splitting.
| Photocatalyst | Mass (g)/volume (mL) | Light source (W/nm) | Sacrificial reagent | Cocatalysts | Rate (μmol h−1 g−1) | QE (%) 420 nm | Reference |
|---|---|---|---|---|---|---|---|
| CdS nanowire | 0.1/100 | 500 Hg/<400 | Na2S/Na2SO3 | Pt | ≈60 | — | 251 |
| Hexagonal CdS | 0.1/200 | 300 Xe/≥420 | Lactic acid | Pt | 740.9 | — | 160 |
| CdS | 0.1/100 | 500 Hg/>420 | Na2S/Na2SO3 | WC | ≈1360 | — | 252 |
| Hexagonal CdS | 0.3/200 | 300 Xe/≥420 | Na2S/Na2SO3 | Pt | 1600 | — | 253 |
| Nanoporous CdS | 0.15/200 | 300 Xe/≥420 | Na2S/Na2SO3 | Pt | 600.34 | — | 254 |
| Mesoporous CdS | 0.1/50 | 400 Hg | Na2S/Na2SO3 | Pt | 14150 |
— | 255 |
| CdS microcrystal | 0.03/40 | 500 Xe/≥400 | Na2S/Na2SO3 | Pt | 4600 | — | 256 |
| CdS/ZnS | 0.1/10 | 00 Ha UV | Na2S/Na2SO3 | — | ≈40.5 | — | 257 |
| CdS–Zn1−xCdxS | 0.05/80 | 350 Xe/>400 | Na2S/Na2SO3 | — | ≈2300 | 6.3 | 258 |
| RGO–Zn1−xCdxS | 0.05/80 | Solar simulator | Na2S/Na2SO3 | — | 1820.4 | 23.4 | 259 |
| CdS/ZnS/In2S3 | 0.015/320 | 300 Xe/>400 | Na2S/Na2SO3 | — | 8100 | 40.9 | 260 |
| Pt–PdS–CdS | 0.3/200 | 300 Xe/>420 | Na2S/Na2SO3 | Pt | 29230 |
93 | 261 |
| CdS–AgGaS2 | 0.1 | 450 Hg/≥420 | Na2S/Na2SO3 | Pt | 2960 | 19.7 | 260 |
| Ag2S/CdS | 0.1/120 | Hg–Xe/>400 | Na2S/Na2SO3 | Ag2S & Pt | 870.4 | — | 262 |
| N-GR/CdS | 0.2/300 | 300 Xe/>420 | Na2S/Na2SO3 | — | 1050 | 192 | |
| CdS/GR | 0.02/80 | 350 Xe/≥420 | Lactic acid | Pt | 56000 |
22.5 | 263 |
| RGO–ZnCdS | 0.05/80 | Solar simulator | Na2S/Na2SO3 | — | 1820.4 | 23.4 | 264 |
| CdS/TiO2 nanotube | 0.15/300 | 300 Xe/≥420 | Na2S/Na2SO3 | Pt | 2080 | 43.4 | 76 |
| CdS/TiO2 | 0.1/50 | 350 Xe/≥400 | Na2S/Na2SO3 | — | — | 8.9 | 265 |
| CdS/TiO2 | 0.0125/25 | 450 Xe/>420 | Na2S/Na2SO3 | Pt | 6720 | 4.5 | 266 |
| CdS/TiO2 | 0.1/100 | 450 Hg/≥420 | Na2S/Na2SO3 | Pt | ≈110 | — | 267 |
| PANI–PbS–CdS | 0.2/200 | 300 Xe/>430 | Na2S/Na2SO3 | — | 16600 |
— | 268 |
| CdS/TaON | 0.2/200 | 300 Xe/=420 | Na2S/Na2SO3 | Pt | 3160.5 | 31 | 269 |
| CdS/TNT | 0.2/190 | 500 Xe/≥430 | Na2S/Na2SO3 | Pt | 1760.7 | 25.5 | 270 |
| MWCNTS/CdS | 0.035/100 | 300 Xe/≥420 | Na2S/Na2SO3 | — | 4980 | — | 271 |
| LaMnO3/CdS | 0.1/60 | 300 Xe/≥420 | Na2S/Na2SO3 | — | 370.5 | — | 272 |
| CdS/Re cellulose | 0.05/100 | 250 Xe/≥420 | Na2S/Na2SO3 | Pt | 1320.3 | — | 273 |
| CdS/CdWO4 | 0.05/200 | 500 Xe | Na2S/Na2SO3 | — | 1800.5 | — | 274 |
| MoS2/CdS | 0.1/200 | 300 Xe | Lactic acid | MoS2 | ≈5300 | — | 190 |
| CdS/zeolite | 0.1/50 | 400 Hg UV | Na2S/Na2SO3 | Pt | 6000 | — | 75 |
| Z–CdS | 0.2/200 | 300 Xe | Na2S/Na2SO3 | RuO2 | ≈6300 | — | 275 |
| CdS/Ta2O5 | 0.05/50 | Xe/400–800 | Lactic acid | — | ≈9000 | — | 276 |
| CeO2/CdS | 0.05/100 | 300 Xe | Na2S/Na2SO3 | — | 15640 |
— | 277 |
| Z–CdS–Cd | 0.1/300 | 300 Xe | Na2S/Na2SO3 | Pt | 19200 |
— | 278 |
| SrS/CdS | 0.2/200 | 350 Xe/≥430 | Na2S/Na2SO3 | — | — | 2.85 | 69 |
| NiO–CdS | 0.2/50 | 500 halogen | Na2S/Na2SO3 | — | 740.5 | 6 | 279 |
| Sr/CdS | 0.2/50 | 350 Xe | Na2S/Na2SO3 | — | 1230 | 10 | 280 |
| Ni(OH)2/CdS | 0.05/80 | 300 Xe/≥420 | Triethalamine | Pt | 5080.4 | 28 | 281 |
| CdS/TaON | 0.2/200 | 300 Xe/=420 | Na2S/Na2SO3 | Pt | 3160.5 | 31 | 282 |
| CdS/Au/g-C3N4 | 0.5/60 | 300 Xe/>420 | Methanol | Pt | 19.02 | 283 | |
| CdS/g-C3N4 | 0.5/60 | 300 Xe/>420 | Methanol | Pt | 4152 | 4.3 | 283 |
| CdS-QD's/g-C3N4 | 0.1/120 | 300 Xe/>400 | Methanol | Pt | 17.27 | — | 224 |
| WS2/CdS | 0.1/200 | 300 Xe/≥400 | Lactic acid | Pt | 4200 | — | 245 |
| WS2–CdS | 0.02/10 | 300 Xe/≥400 | Lactic acid | — | 2000 | — | 284 |
| MoS2/CdS | 0.1/10 | 300 Xe/>420 | Lactic acid | — | 5400 | — | 190 |
| NiOx/CdS | 0.1/100 | 300 Xe/>400 | Na2S/Na2SO3 | — | 590.8 | 8.6 | 285 |
| CuS/CdS | 0.2/200 | 500 Xe/>420 | Na2S/Na2SO3 | — | 3300 | — | 286 |
| PdS/CdS | 0.2/200 | 300 Xe/>430 | Na2S/Na2SO3 | — | 4450 | — | 176 |
| NiS/CdS | 0.05/80 | 300 Xe/>420 | Na2S/Na2SO3 | NiS | 1131 | 6.1 | 287 |
| CdS/K2Ti3.9Nb0.1O9 | 0.1/20 | 300 Xe/>420 | Na2S/Na2SO3 | — | 4700 | — | 45 |
| CdS–zeolite | 0.1/50 | 400 Hg/>420 | Na2S/Na2SO3 | — | 102 (0.1 gcat) | — | 75 |
| CdS | 0.2/100 | Solar driven | — | — | 340 | — | 288 |
| Co(OH)2/CdS | 0.1/100 | 500 Xe/>420 | — | Co | 61 | — | 289 |
| Ni/CdS | 0.1/50 | 300 Xe/>420 | (NH4)2SO3 | Ni | 25848 |
26.8 | 290 |
| CdS/CNTs | 0.1/180 | 350 Xe/>420 | Na2S/Na2SO3 | — | 794.6 | — | 291 |
| CdS/CNTs | 0.1/180 | 350 Xe/>420 | Na2S/Na2SO3 | NiS | 12130 |
— | 291 |
| CdS/ZnS | 0.1/10 | 500 halogen/>420 | Na2S/Na2SO3 | — | 45 | — | 69 |
| CdSeZnS/ZTP | 0.02/20 | 125 Hg/>420 | Na2S/Na2SO3 | — | 2142.7 | 9.6 | 292 |
| ZnS–CuS–CdS | 0.1/150 | 150 Xe/>420 | — | — | 837.6 | — | 293 |
| GO/CdS | 0.02/80 | 350 Xe/>420 | — | Pt | 1120 | 22.5 | 294 |
| Ni(OH)2–CdS/g-C3N4 | 0.001/20 | 300 Xe/>420 | Na2S/Na2SO3 | Ni | 115.18 | 16.7 | 295 |
| Nih–CdS QDs | 0.0008/10 | 500 Hg/>400 | — | — | 74.6 | 12.2 | 296 |
| CdS/Pt/Ga2O3 | 0.001/1 | 300 Xe/>420 | — | — | 995.8 | 43.6 | 297 |
| CdS/Pt/In2O3 | 0.001/1 | 300 Xe/>420 | — | — | 1032.2 | 45.3 | 297 |
| Au–Pt–CdS/SBA-15 | 0.1/20 | 500 halogen/>420 | — | — | 133.3 | 298 |
Charge utilization in CdS
After the generation and survival of photogenerated charges, the use of electrons for the HER is the overall objective of photocatalytic water splitting. The generated electrons reach the sites that generally reside on the surface of photocatalyst where they are in contact at the solid/liquid interface. The electrons on these sites transfer from the solid into the liquid phase to carry on the subsequent HER. In this section, we, therefore, discuss how to improve the use of photogenerated electrons.Surface functionalization of CdS
Photocatalysis is a surface phenomenon. The photogenerated electrons reside on active sites that are on the photocatalyst or cocatalyst surface. A common approach is to load an optimized amount of cocatalyst (reduction cocatalyst) onto the photocatalyst. As mentioned before, co-catalysts play a dual role: (i) preventing recombination and (ii) acting as active sites either for activation of water molecules or for the reduction of protons. In the co-catalyst strategy, holes or electrons are scavenged by the co-catalysts once they are generated within the photocatalyst. For example, by introducing an oxidative co-catalyst, such as CoII, not only is it possible to scavenge holes and hinder the recombination process but also provide an opportunity for the possible utilization of electrons.26,28,59,79–83Another strategy is to extend the specific surface area of an SC (CdS) by decreasing its particle size. Unfortunately, very small-sized particles create additional problems. In this section, the functionalization of the CdS surface without the addition of additional materials will be discussed.
Lunawat et al. immobilized a CdS nanocatalyst on porous polymeric support (polystyrene).299 The higher activity and longer life of coated CdS nanocatalyst in water splitting is the result of its hydrophobic nature. The number of active sites on the CdS surface can be increased by the immobilized polystyrene support. Thote et al. have prepared CdS NPs stabilized by the 2-D covalent organic framework (COF) to enhance photocatalytic H2 production via water splitting.300 The enhancement in activity has been ascribed to the presence of a p-conjugated polymer backbone and the abundant 2-D hetero-interface and high surface-active sites, which act as efficient supports for stabilizing the photocatalyst and prolonging the lifetime of the photogenerated electrons. The effect of the coupling interface between CdS and the COF has been investigated. A comparison of the calculated flat-band potentials of CdS (0.64 V vs. NHE) and the COF (0.54 V, vs. NHE) suggests that CdS has a higher Fermi level than does the COF. The hybrid CdS–COF (90:10) has a flat-band potential of 0.59 V, which indicates that a new Fermi level has been designed between CdS and COF. In addition, the number of active sites formed by the preparation of this hybrid has increased markedly.
Wilker et al. have functionalized CdS NRs with mercaptocarboxylate surface-capping ligands to examine the role of ligands that passivate the sites of CdS with respect to transferring electrons to [FeFe] hydrogenase (from Clostridium acetobutylicum) for H2 generation.301
The HER obtained matched the QE of the electron transfer. The authors suggested that H2 production could be improved by introducing suitable surface-capping ligands that better-directed electrons to the target sites. Furthermore, Lin et al. demonstrated that poly(methyl methacrylate)–poly(methacrylic) (PMMA/PMAA) CdS nanocrystals have a shielded hybrid microsphere structure due to the formation of colloidal arrays.302 They managed to prepare photonic crystals, which possessed both angular and spectral electromagnetic resonance structures. Through quantization effects, optical functionalization together with spectral control was achieved, via the in situ CdS nanocrystal growth on the surface of the microsphere.
Tailoring the pathway of reaction media
Once the electrons have crossed the solid/liquid interface, the ultimate goal is then to fix the pathway. It is understood that holes are utilized by either oxidation cocatalysts or by the scavengers thus allowing the free electrons to reduce the protons (H+). This process is complex, leading to considerable conjecture regarding the mechanism. In almost all photocatalytic systems, a reduction cocatalyst, for example, is first loaded onto the photocatalyst (usually residing on the surface of the catalyst) forming a steric obstacle for the scavenger that suppressed electron–hole recombination. By tailoring the pathway, Simon et al. have reported that increasing the pH of the reaction media results in holes transferring from the photocatalyst to the scavenger (ethanol)78 (Fig. 15a). The release and transfer of holes operate as a shuttle and the redox couple of ˙OH/−OH affords electrons for proton reduction, while the hydroxyl anions are regenerated through water dissociation. More importantly, through this shuttle mechanism, the slow hole scavenging pathway is substituted by two faster ones that involve redox mediator species. This redox shuttle technique greatly enhances the photocatalytic H2 production compared to that for Ni/CdS (without ethanol). Hong et al. have prepared the ternary heterostructural system, ZnS–CuS–CdS, to use electrons more effectively303 (Fig. 15b). They aimed to achieve synergy with their system and, therefore, decrease the CdS content and increase the CuS content resulting in an appropriate improvement in the efficacy of the transfer of electrons to CuS. Optimizing the composition of this multi-component system will make possible the improved utilization of electrons for photolysis of water. Under illumination, the transfer of electrons from the VB of ZnS to CuS takes place, resulting in the partial reduction of CuS to Cu2S. Meanwhile, because of the favourable VB position of CdS, electrons transfer first to ZnS and then to CuS via a cascade pathway.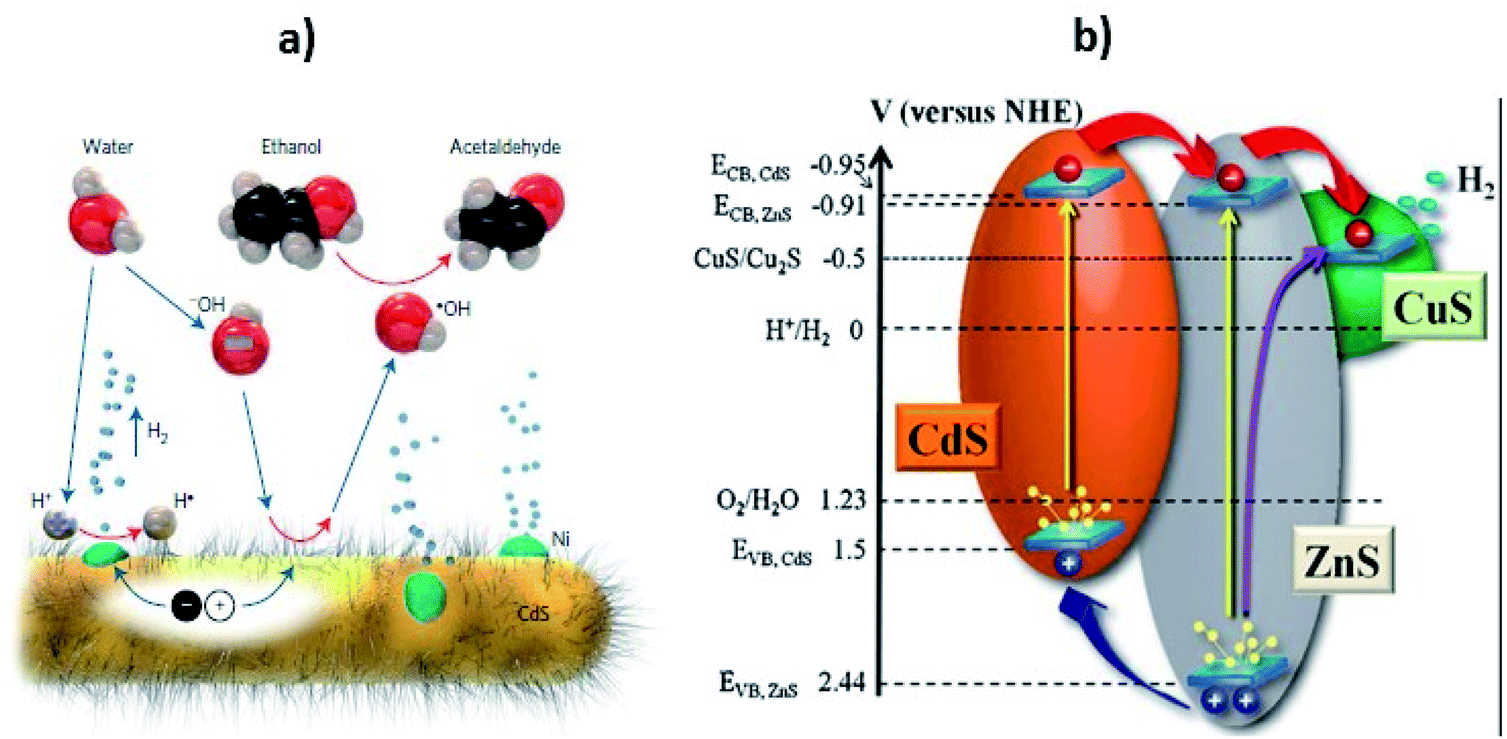 | ||
| Fig. 15 (a) The proposed hole shuttle mechanism for H2 generation over the cysteine-stabilized CdS nanorods. The hydroxyl anions scavenge the holes and carry away the positive charges which in turn oxidize the ethanol to acetaldehyde. The blue arrow represents the movement of species and red arrows denote a redox reaction. Reprinted with permission from ref. 78. (b) Charge transfer illustration in ZnS–CuS–CdS photocatalyst. Reprinted with permission from ref. 303. | ||
Effect of sacrificial reagents
As has been emphasized above, it is difficult to achieve efficient photochemical H2 production via water-splitting using TiO2 and CdS without the addition of a sacrificial agent (hole scavenger/electron trapper). The anodic decomposition of CdS in the absence of a hole scavenger occurs at +0.32 V at pH 7 (eqn (2)).10,52,304| CdS + 2h+ → Cd2+ + S; Edecomp = +0.32 V (NHE, pH 7) | (2) |
The complex redox chemistry related to complete water splitting to afford gaseous H2 evolution involves four electrons. There are two factors related to the use of sacrificial reagents.
First, in the case of using a hole scavenger, O2 is not generated. Therefore, the reversible reaction for a generated water molecule is suppressed, which in turn, increases the H2 production and avoids a gas separation stage. Secondly, the H2 release rate can be reduced by the formation of an oxidized sacrificial reagent layer upon the surface of the photocatalyst.305 In the presence of an electron-donor species like alcohol, S2−, or SO23−, however, the holes in the VB of the SC will oxidize the sacrificial reagent instead of corroding the photocatalyst, thus facilitating the reduction of water by the CB electrons.
Conversely, an electron-acceptor sacrificial reagent is irreversibly reduced by an electron (in the CB of the SC), which facilitates water oxidation by VB holes10,78,185,306–308 (Fig. 16a). This phenomenon is related to the photodegradation of an organic contaminant oxidized by photogenerated holes. The most common sacrificial agents used are lactic acid,165 formic acid,52 S2−/SO32−,50 ethanol,51 and some other organic and inorganic reagents.10,53–55 Several organic compounds, such as organic acids, alcohol, and hydrocarbons are used as electron donors with CdS for photocatalytic H2 generation.
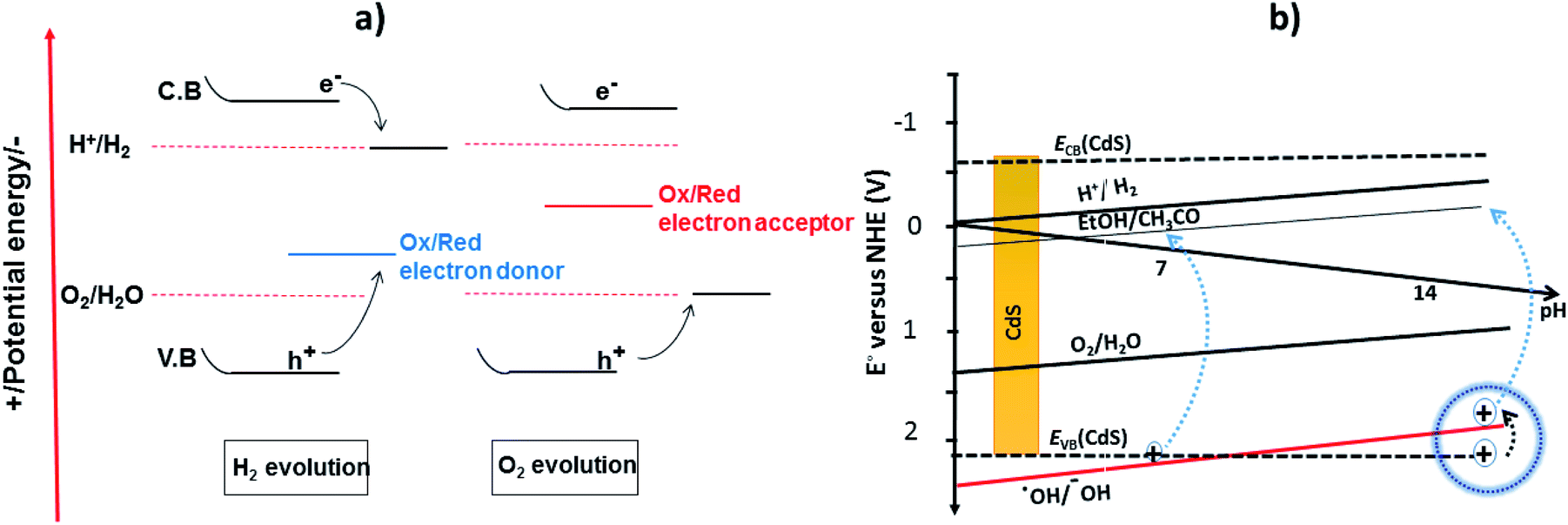 | ||
| Fig. 16 Sacrificial reagents assisted photocatalytic water splitting. (a) Redox mediated reaction mechanism in the presence of sacrificial reagents. Reproduced with permission from ref. 307 and (b) the band edge position of CdS (CB and VB) and the relative redox potential of water, hydroxyl anion, and ethanol oxidation. Reproduced with permission from ref. 78. | ||
Alcohols, such as ethanol and methanol, react irreversibly with the photogenerated holes, thus preventing photo-corrosion of the photocatalyst as well as enhancing the probability of electron utilization, which in turn, increases the stability and QE of the photocatalyst. The continuous addition of electron donors is required, however, for sustained H2 generation, because of its continuous consumption.78,309,310 This approach is somewhat limited at lower pH due to the reduction potential of protons vs. NHE. As mentioned earlier regarding the report of Simon et al., the scavenging of holes from CdS (in the VB) is based on the ˙OH/OH− redox couple, functioning as a shuttle.78 This redox-mediated mechanism significantly boosts the H2 production over Ni/CdS (in the presence of ethanol as a scavenger). Moreover, hole scavenging from the VB of CdS by OH− ions are only favorable at high pH (Fig. 16b).
Harada et al. have tested the oxidation ability of CdS using several sacrificial reagents and discovered that not all the sacrificial reagent is prone to oxidation by the photogenerated holes.311 The main point is the compatibility of the oxidation potentials of the photocatalyst (VB potential) and sacrificial reagent. The higher the oxidation potential of the VB, the easier the species can be oxidized by the photogenerated holes. Zong et al. developed a MoS2/CdS photocatalytic system for H2 generation using different sacrificial agents with their activity order being lactic acid > glycerol > glycol > ethanol > methanol.312 Inorganic electron donors such as sulfide (S2−) and sulfite (SO32−) ions can also act as hole scavengers. These two sulfur-containing anions allow electrons to be utilized in the HER and are the most commonly used sacrificial agents. As discussed earlier, in the presence of electron donor species, CdS is considered to be an efficient photocatalyst for the generation of H2 from aqueous systems.174,223,313–316 The VB level of CdS is positive enough (+2.2 V vs. NHE) for the oxidation of sulfur-containing compounds.317 As mentioned before, there are two advantages of using S2− and/or SO32− as hole scavengers for CdS over alcohols: (i) they are easily oxidized by VB holes, which reduces the undesired anodic photo corrosion and (ii) the dissolved Cd2+ in a solution can reform CdS upon reaction with S2−. In aqueous methanol solution, however, it is not practical to reform CdS. The very high QE (93%) was achieved upon employing Pt–PdS/CdS as the photocatalyst in the presence of S2−/SO32−.318 Berr et al. applied transient absorption spectroscopy on colloidal Pt-decorated CdS nanorods in the presence and absence of Na2SO3 as the hole scavenger.319 Notably, the migration of photoelectrons from the CB of CdS to the active site of Pt was slow without the use of a hole scavenger and resulted in no significant H2 generation. The possible mechanistic steps for the H2 production in the presence of S2−/SO32− as electron-donor sacrificial agents are given in eqn (3)–(8).
| 2H2O + 2e− → H2 + 2OH− | (3) |
| SO32− + 2OH− + 2h+ → SO42− + H2O | (4) |
| S2O32− + 2h+ → S2O62− | (5) |
| 2S2− + 2h+ → S22− | (6) |
| SO32− + S2− + 2h+ → S2O32− | (7) |
| S22− + SO32− → S2O32− + S2− | (8) |
Some inorganic ions also act as sacrificial agents for a CdS photocatalyst, such as Ce3+, Fe2+, Ni2+, and I−.78,320 Unfortunately, it is necessary constantly to add these sacrificial agents to keep the reaction viable, which is both inconvenient and not cost-effective. Therefore, the replacement of inorganic sacrificial agents with organic ones is the best approach to generate H2 from water using a CdS photocatalyst.
The choice of electron donors as sacrificial agents for water splitting is quite extensive when compared to the range of possible electron acceptors. Nevertheless, electron acceptor agents have their own advantages, but they must be used where the photocatalyst is stable enough to oxidation.
Moreover, there is some concern with using only an oxidation process. As an example, Ag+ has been employed an electron acceptor (eqn (9)–(11)).
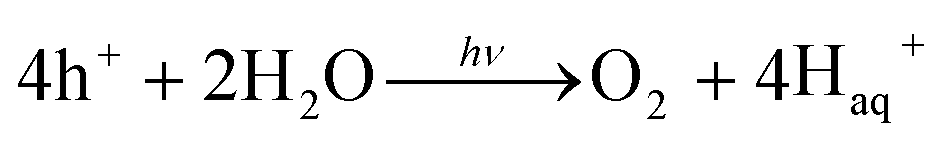 | (9) |
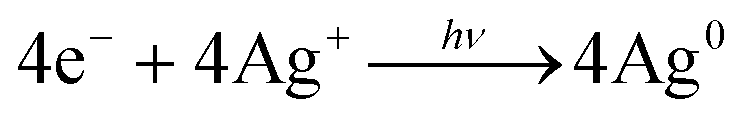 | (10) |
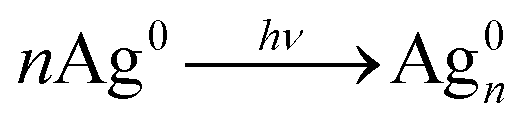 | (11) |
Conclusions and future perspective
Photocatalysis is a highly important multifunctional technology in the areas of energy and environmental remediation, especially photocatalytic H2 production and CO2 reduction.28,322 Solar H2 generation from water is a key strategy in addressing the world's energy shortage by utilising the ample solar energy directly in the form of molecular hydrogen, a green fuel.323,324 Although photocatalytic technology has grown and been applied to using solar energy to generate H2 and other fuels, there are still challenges with its efficiency. As we have shown in this review, to enhance the efficiency of this technology, the three critical steps i.e., charge generation, charge survival, and its proper utilization need significant development, both for solar light conversion to electricity and photocatalytic H2 production. Among these, the first cornerstone is to improve the light absorption ability of the material for effective charge generation. CdS is basically a visible light-responsive photocatalyst; its absorption properties can be modified up to the full visible range or the IR region. As is evident from many literature reports, the strategies of combining two photocatalysts or plasmonic electron generation have been found to be very successful in enhancing the light absorption capacities of photocatalyst. Moreover, deliberately introducing structural defects and chemical impurities also widen the spectral response of CdS.Suppression of electron–hole recombination is the key to all photocatalytic processes and is crucial for charge survival. The strategies recommended for reducing the rate of charge recombination in CdS include: (i) anisotropic induction and morphology engineering, which effectively suppresses charge recombination; (ii) co-catalyst incorporation, which extracts electrons from the CB level of the photocatalyst; (iii) induced heterojunction formation, which pushes the charges in opposite directions; and (iv) Z-scheme photocatalysis which, like natural photosynthesis, involves a two-step photoexcitation and also hinders the recombination process and promotes charge separation as do heterojunctions. However, the reduction ability of an electron and the prevention of the backward reaction of water splitting is often better in Z-scheme photocatalysis than in the heterojunctions. Moreover, elevating the position of the CB level through crystal engineering has also proved a highly feasible approach to improve the ability (for reduction of a proton) of the photogenerated electrons. Furthermore, to increase the efficacy of the photocatalyst, the search for cost-effective and efficient cocatalysts should be continued to replace Pt-based cocatalysts. After these two processes, presumably the charges especially electrons reach the sites and are utilized for the HER. The main protagonists are electrons, which are emitted from the photocatalyst and enter smoothly into the solid/liquid interface. Surface functionalization for charge transportation can be achieved by surface modification (an increased number of active sites) and by introducing foreign materials having high conductivities. Currently, the available efficiency of CdS-based photocatalysts is relatively low due to the fast recombination of the electron–hole pair, the backward reaction, and the photo-corrosion effect. To make these processes more feasible, the continual addition of electron-donor species into the reaction medium is needed. These electron-donor agents irreversibly consume photogenerated holes and allow electrons to be used for proton reduction as well as hindering the undesired charge recombination phenomenon. Interestingly, the use of biomass (low-cost and renewable source) from animals and plants may well provide sacrificial reagents for water-splitting, at almost no cost; and both H2 production and waste-water treatment could be performed in parallel.
Finally, all the above strategies play key roles in improving the performance of CdS-based photocatalysts. There are several CdS-based photocatalysts that have not yet been tested for H2 generation from water; more information on them is still needed. In addition, the reaction kinetics and molecular mechanisms must be considered carefully when further developing CdS-based photocatalytic systems. Nevertheless, it is vitally important to pursue such an ambitious goal for meeting future energy needs. The industrial application of photocatalytic H2 production will also require comprehensive studies to scale-up laboratory studies and improve the low photocatalytic efficiency of the photocatalysts. The design of a robust and efficient photocatalytic system remains a major future challenge. This review has indicated where some of the fundamental developments will be needed.
List of abbreviations
| CB | Conduction band |
| VB | Valence band |
| H2 | Hydrogen |
| CO2 | Carbon dioxide |
| SC | Semiconductor |
| NHE | Normal hydrogen electrode |
| NMs | Nanomaterials |
| NPs | Nanoparticles |
| NCs | Nanocrystals |
| NTs | Nanotubes |
| HER | Hydrogen evolution reaction |
| RHE | Reversible hydrogen electrode |
| DFT | Density functional theory |
| RGO | Reduced graphene oxide |
| GO | Graphene oxide |
| XPS | X-ray photoelectron spectroscopy |
| SEM | Scanning electron microscopy |
| FESEM | Field emission scanning electron microscopy |
| STEM | Scanning tunnelling electron microscopy |
| TEM | Transmission electron microscopy |
| HRTEM | High resolution transmission electron microscopy |
| TDA | Transient differential absorption |
| A/D pair | Electron acceptor/donor |
| ZTP | Zirconium titanium phosphate |
| QD | Quantum dot |
| IPCE | Incident-photon-to-current-conversion efficiency |
| LSPR | Localized surface plasmon resonance |
| CTAB | Cetyltrimethylammonium bromide |
| AQY | Apparent quantum yield |
| AQE | Apparent quantum efficiency |
| EQY | External quantum yield |
| QE | Quantum efficiency |
| HT | Head-to-tail |
| P3HT | Poly-(3-hexylthiophene-2,5-diyl) |
| NCs | Nanocubes |
| NOTs | Nanooctahedrons |
| ECSA | Electrochemical active surface area |
| POM | Polyoxometalate |
| PSRD | Plasma sputtering reaction |
| PLD | Pulsed laser deposition |
| IPCE | Incident photon-to-current conversion efficiency |
| COF | Covalent organic frame work |
| PMMA/PMAA | Poly-methyl methacrylate–poly-methacrylic |
| TMBs | Transition metal borides |
| NIR | Near infrared |
| UV | Ultraviolet |
| ITO | Indium tin oxide |
| CQDs | Carbon quantum dots |
| 2D | Two dimensional |
| 1D | One dimensional |
| PL | Photoluminescence |
| STH | Solar-to-hydrogen efficiency |
| PEC | Photoelectrochemical |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Higher Education Commission of Pakistan.Notes and references
- N. Panwar, S. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 CrossRef.
- F. E. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- J. Kundu, S. Khilari and D. Pradhan, ACS Appl. Mater. Interfaces, 2017, 9, 9669–9680 CrossRef CAS.
- C.-H. Liao, C.-W. Huang and J. Wu, Catalysts, 2012, 2, 490–516 CrossRef CAS.
- U. EIA, US Energy Information Administration, Washington, DC, 2013, pp. 60–62.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- N. S. Lewis, G. Crabtree, A. J. Nozik, M. R. Wasielewski, P. Alivisatos, H. Kung and R. Ellingson, Basic Research Needs for Solar Energy Utilization, Report of the Basic Energy Sciences Workshop on Solar Energy Utilization, DOESC (USDOE Office of Science (SC)), 2005 Search PubMed.
- N. Panwar, S. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 CrossRef.
- Z. Yang, J. Zhang, M. C. Kintner-Meyer, X. Lu, D. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- A. Züttel, A. Remhof, A. Borgschulte and O. Friedrichs, Philos. Trans. R. Soc., A, 2010, 368, 3329–3342 CrossRef.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- W. Brattain and C. Garrett, Bell Labs Tech. J., 1955, 34, 129–176 CrossRef.
- H. Gerischer, J. Electrochem. Soc., 1966, 113, 1174–1182 CrossRef CAS.
- Y. V. Pleskov and Y. Y. Gurevich, Semiconductor photoelectrochemistry, 1986 Search PubMed.
- A. J. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- A. J. Nozik, Annu. Rev. Phys. Chem., 1978, 29, 189–222 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- A. Fujishima, Nature, 1972, 238, 37–38 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- M. Matsuoka, M. Kitano, M. Takeuchi, K. Tsujimaru, M. Anpo and J. M. Thomas, Catal. Today, 2007, 122, 51–61 CrossRef CAS.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- M. B. Wilker, K. J. Schnitzenbaumer and G. Dukovic, Isr. J. Chem., 2012, 52, 1002–1015 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS.
- Y. Lee, H. Terashima, Y. Shimodaira, K. Teramura, M. Hara, H. Kobayashi, K. Domen and M. Yashima, J. Phys. Chem. C, 2007, 111, 1042–1048 CrossRef CAS.
- S. Ma, J. Xie, J. Wen, K. He, X. Li, W. Liu and X. Zhang, Appl. Surf. Sci., 2017, 391, 580–591 CrossRef CAS.
- L. J. Zhang, S. Li, B. K. Liu, D. J. Wang and T. F. Xie, ACS Catal., 2014, 4, 3724–3729 CrossRef CAS.
- P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS.
- W. Li, C. Feng, S. Dai, J. Yue, F. Hua and H. Hou, Appl. Catal., B, 2015, 168, 465–471 CrossRef.
- B. Qiu, Q. Zhu, M. Du, L. Fan, M. Xing and J. Zhang, Angew. Chem., 2017, 129, 2728–2732 CrossRef.
- J. Low, C. Jiang, B. Cheng, S. Wageh, A. A. Al-Ghamdi and J. Yu, Small Methods, 2017, 1, 1700080 CrossRef.
- K. Sayama, K. Mukasa, R. Abe, Y. Abe and H. Arakawa, Chem. Commun., 2001, 2416–2417 RSC.
- H. Kato, M. Hori, R. Konta, Y. Shimodaira and A. Kudo, Chem. Lett., 2004, 33, 1348–1349 CrossRef CAS.
- M. Ni, M. K. Leung, D. Y. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- S. Kaur and V. Singh, J. Hazard. Mater., 2007, 141, 230–236 CrossRef CAS.
- R. M. Navarro Yerga, M. C. Álvarez Galván, F. Del Valle, J. A. Villoria de la Mano and J. L. Fierro, ChemSusChem, 2009, 2, 471–485 CrossRef CAS.
- R. Abe, J. Photochem. Photobiol., C, 2010, 11, 179–209 CrossRef CAS.
- W. Shangguan and A. Yoshida, J. Phys. Chem. B, 2002, 106, 12227–12230 CrossRef CAS.
- H. Yin, Y. Wada, T. Kitamura and S. Yanagida, Environ. Sci. Technol., 2001, 35, 227–231 CrossRef CAS.
- M. Matsumura, S. Furukawa, Y. Saho and H. Tsubomura, J. Phys. Chem., 1985, 89, 1327–1329 CrossRef CAS.
- J. F. Reber and M. Rusek, J. Phys. Chem., 1986, 90, 824–834 CrossRef CAS.
- L. Amirav and A. P. Alivisatos, J. Phys. Chem. Lett., 2010, 1, 1051–1054 CrossRef CAS.
- J. S. Jang, S. M. Ji, S. W. Bae, H. C. Son and J. S. Lee, J. Photochem. Photobiol., A, 2007, 188, 112–119 CrossRef CAS.
- S. Y. Ryu, W. Balcerski, T. Lee and M. R. Hoffmann, J. Phys. Chem. C, 2007, 111, 18195–18203 CrossRef CAS.
- Y. J. Zhang, L. Zhang and S. Li, Int. J. Hydrogen Energy, 2010, 35, 438–444 CrossRef CAS.
- X. Zong, G. Wu, H. Yan, G. Ma, J. Shi, F. Wen, L. Wang and C. Li, J. Phys. Chem. C, 2010, 114, 1963–1968 CrossRef CAS.
- J. Jin, J. Yu, G. Liu and P. K. Wong, J. Mater. Chem. A, 2013, 1, 10927–10934 RSC.
- Y. Li, Y. Hu, S. Peng, G. Lu and S. Li, J. Phys. Chem. C, 2009, 113, 9352–9358 CrossRef CAS.
- M. F. Kuehnel, D. W. Wakerley, K. L. Orchard and E. Reisner, Angew. Chem., Int. Ed., 2015, 54, 9627–9631 CrossRef CAS.
- W.-T. Yao, S.-H. Yu, S.-J. Liu, J.-P. Chen, X.-M. Liu and F.-Q. Li, J. Phys. Chem. B, 2006, 110, 11704–11710 CrossRef CAS.
- X. Song, W. Yao, B. Zhang and Y. Wu, Int. J. Photoenergy., 2012, 1–5 CrossRef.
- S. J. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- J. R. Darwent and G. Porter, J. Chem. Soc., Chem. Commun., 1981, 145–146 RSC.
- M. Matsumura, Y. Saho and H. Tsubomura, J. Phys. Chem., 1983, 87, 3807–3808 CrossRef CAS.
- F. Del Valle, A. Ishikawa, K. Domen, J. V. de La Mano, M. Sánchez-Sánchez, I. González, S. Herreras, N. Mota, M. Rivas and M. Á. Galván, Catal. Today, 2009, 143, 51–56 CrossRef CAS.
- E. Borgarello, J. Kiwi, M. Graetzel, E. Pelizzetti and M. Visca, J. Am. Chem. Soc., 1982, 104, 2996–3002 CrossRef CAS.
- F. Zuo, L. Wang, T. Wu, Z. Zhang, D. Borchardt and P. Feng, J. Am. Chem. Soc., 2010, 132, 11856–11857 CrossRef CAS.
- R. Niishiro, R. Konta, H. Kato, W.-J. Chun, K. Asakura and A. Kudo, J. Phys. Chem. C, 2007, 111, 17420–17426 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS.
- X. Li, N. Kikugawa and J. Ye, Adv. Mater., 2008, 20, 3816–3819 CrossRef CAS.
- A. Kongkanand, K. Tvrdy, K. Takechi, M. Kuno and P. V. Kamat, J. Am. Chem. Soc., 2008, 130, 4007–4015 CrossRef CAS.
- A. Deshpande, P. Shah, R. Gholap and N. M. Gupta, J. Colloid Interface Sci., 2009, 333, 263–268 CrossRef CAS.
- C. Xing, Y. Zhang, W. Yan and L. Guo, Int. J. Hydrogen Energy, 2006, 31, 2018–2024 CrossRef CAS.
- S. Pelet, J.-E. Moser and M. Grätzel, J. Phys. Chem. B, 2000, 104, 1791–1795 CrossRef CAS.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2006, 6, 215–218 CrossRef CAS.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS.
- M. Salari, S. H. Aboutalebi, A. Aghassi, P. Wagner, A. J. Mozer and G. G. Wallace, Phys. Chem. Chem. Phys., 2015, 17, 5642–5649 RSC.
- M. Sathish, B. Viswanathan and R. Viswanath, Int. J. Hydrogen Energy, 2006, 31, 891–898 CrossRef CAS.
- C. Li, J. Yuan, B. Han, L. Jiang and W. Shangguan, Int. J. Hydrogen Energy, 2010, 35, 7073–7079 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS.
- T. Simon, N. Bouchonville, M. J. Berr, A. Vaneski, A. Adrović, D. Volbers, R. Wyrwich, M. Döblinger, A. S. Susha and A. L. Rogach, Nat. Mater., 2014, 13, 1013–1018 CrossRef CAS.
- M. A. Khan, Z.-u. Rehman, J. A. Nasir, M. Hafeez, M. Arshad, N. Z. Ali, I. F. Teixieira and I. McPherson, ChemSusChem, 2018, 11, 2587–2592 CrossRef.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS.
- M. Ni, M. K. Leung, D. Y. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 RSC.
- G. Wang, X. Yang, F. Qian, J. Z. Zhang and Y. Li, Nano Lett., 2010, 10, 1088–1092 CrossRef CAS.
- X. Yue, S. Yi, R. Wang, Z. Zhang and S. Qiu, Sci. Rep., 2016, 6, 22268 CrossRef CAS.
- Y. Xu and M. A. Schoonen, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- L. Cheng, Q. Xiang, Y. Liao and H. Zhang, Energy Environ. Sci., 2018, 11, 1362 RSC.
- D. Meissner, R. Memming and B. Kastening, J. Phys. Chem., 1988, 92, 3476–3483 CrossRef CAS.
- W. Shangguan and A. Yoshida, J. Phys. Chem. B, 2002, 106, 12227–12230 CrossRef CAS.
- T. Hirai and Y. Bando, J. Colloid Interface Sci., 2005, 288, 513–516 CrossRef CAS.
- Y. Yang, Y. Zhang, Z. Fang, L. Zhang, Z. Zheng, Z. Wang, W. Feng, S. Weng, S. Zhang and P. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 6950–6958 CrossRef CAS.
- Y. Zhong, G. Zhao, F. Ma, Y. Wu and X. Hao, Appl. Catal., B, 2016, 199, 466–472 CrossRef CAS.
- A. Wu, C. Tian, Y. Jiao, Q. Yan, G. Yang and H. Fu, Appl. Catal., B, 2017, 203, 955–963 CrossRef CAS.
- J. Hou, C. Yang, Z. Wang, S. Jiao and H. Zhu, RSC Adv., 2012, 2, 10330–10336 RSC.
- J. Yu, Y. Yu, P. Zhou, W. Xiao and B. Cheng, Appl. Catal., B, 2014, 156, 184–191 CrossRef.
- H. Yu, X. Huang, P. Wang and J. Yu, J. Phys. Chem. C, 2016, 120, 3722–3730 CrossRef CAS.
- W. Zhang, Y. Wang, Z. Wang, Z. Zhong and R. Xu, Chem. Commun., 2010, 46, 7631–7633 RSC.
- D. Meissner, R. Memming and B. Kastening, Chem. Phys. Lett., 1983, 96, 34–37 CrossRef CAS.
- D. Meissner, R. Memming, L. Shuben, S. Yesodharan and M. Grätzel, Ber. Bunsen-Ges. Phys. Chem., 1985, 89, 121–124 CrossRef CAS.
- T. Torimoto, M. Hashitani, T. Konishi, K.-i. Okazaki, T. Shibayama and B. Ohtani, J. Nanosci. Nanotechnol., 2009, 9, 506–513 CrossRef CAS.
- Y. Hu, X. Gao, L. Yu, Y. Wang, J. Ning, S. Xu and X. W. D. Lou, Angew. Chem., 2013, 125, 5746–5749 CrossRef.
- M.-Q. Yang, C. Han and Y.-J. Xu, J. Phys. Chem. C, 2015, 119, 27234–27246 CrossRef CAS.
- Y. Bessekhouad, N. Chaoui, M. Trzpit, N. Ghazzal, D. Robert and J. Weber, J. Photochem. Photobiol., B, 2006, 183, 218–224 CrossRef CAS.
- L. Ma, M. Liu, D. Jing and L. Guo, J. Mater. Chem. A, 2015, 3, 5701–5707 RSC.
- A. V. Isarov and J. Chrysochoos, Langmuir, 1997, 13, 3142–3149 CrossRef CAS.
- L. Spanhel, M. Haase, H. Weller and A. Henglein, J. Am. Chem. Soc., 1987, 109, 5649–5655 CrossRef CAS.
- Y. Zhu, Z. Chen, T. Gao, Q. Huang, F. Niu, L. Qin, P. Tang, Y. Huang, Z. Sha and Y. Wang, Appl. Catal., B, 2015, 163, 16–22 CrossRef CAS.
- H. Yan, J. Yang, G. Ma, G. Wu, X. Zong, Z. Lei, J. Shi and C. Li, J. Catal., 2009, 266, 165–168 CrossRef CAS.
- V. M. Daskalaki, M. Antoniadou, G. Li Puma, D. I. Kondarides and P. Lianos, Environ. Sci. Technol., 2010, 44, 7200–7205 CrossRef CAS.
- P. Srivastava, P. Kumar and K. Singh, J. Nanopart. Res., 2011, 13, 5077 CrossRef CAS.
- Z. Gao, N. Liu, D. Wu, W. Tao, F. Xu and K. Jiang, Appl. Surf. Sci., 2012, 258, 2473–2478 CrossRef CAS.
- S. Chandramohan, A. Kanjilal, J. K. Tripathi, S. N. Sarangi, R. Sathyamoorthy and T. Som, J. Appl. Phys., 2009, 105, 123507 CrossRef.
- M. V. Kovalenko, R. D. Schaller, D. Jarzab, M. A. Loi and D. V. Talapin, J. Am. Chem. Soc., 2012, 134, 2457–2460 CrossRef CAS.
- Z. Ning, H. Tian, C. Yuan, Y. Fu, H. Qin, L. Sun and H. Ågren, Chem. Commun., 2011, 47, 1536–1538 RSC.
- A. Nemchinov, M. Kirsanova, N. N. Hewa-Kasakarage and M. Zamkov, J. Phys. Chem. C, 2008, 112, 9301–9307 CrossRef CAS.
- M. Braun, C. Burda and M. A. El-Sayed, J. Phys. Chem. A, 2001, 105, 5548–5551 CrossRef CAS.
- Y. L. Lee and Y. S. Lo, Adv. Funct. Mater., 2009, 19, 604–609 CrossRef.
- Y.-L. Lee, C.-F. Chi and S.-Y. Liau, Chem. Mater., 2010, 22, 922–927 CrossRef CAS.
- A. K. Gupta and R. Kripal, Spectrochim. Acta, Part A, 2012, 96, 626–631 CrossRef CAS.
- Z. K. Heiba, M. B. Mohamed and N. Imam, J. Alloys Compd., 2015, 618, 280–286 CrossRef CAS.
- S. Huang, Y. Lin, J. Yang, X. Li, J. Zhang, J. Yu, H. Shi, W. Wang and Y. Yu, RSC Adv., 2013, 3, 20782–20792 RSC.
- H. Duan and Y. Xuan, Sol. Energy Mater. Sol. Cells, 2014, 121, 8–13 CrossRef CAS.
- L. A. González, I. Carreón-Moncada and M. A. Quevedo-López, Mater. Lett., 2017, 192, 161–164 CrossRef.
- B. A. Korgel and H. G. Monbouquette, Langmuir, 2000, 16, 3588–3594 CrossRef CAS.
- G. Wang, X. Yang, F. Qian, J. Z. Zhang and Y. Li, Nano Lett., 2010, 10, 1088–1092 CrossRef CAS.
- N. M. Vuong, J. L. Reynolds, E. Conte and Y.-I. Lee, J. Phys. Chem. C, 2015, 119, 24323–24331 CrossRef CAS.
- K. Parida, N. Biswal, D. Das and S. Martha, Int. J. Hydrogen Energy, 2010, 35, 5262–5269 CrossRef CAS.
- M. B. Dines, P. M. DiGiacomo, K. P. Callahan, P. C. Griffith, R. H. Lane and R. E. Cooksey, in Chemically Modified Surfaces in Catalysis and Electrocatalysis, ed. J. S. Miller, ACS Symposium Series 192, ACS, Washington, DC, 1982, p. 223 Search PubMed.
- R. Jiang, B. Li, C. Fang and J. Wang, Adv. Mater., 2014, 26, 5274–5309 CrossRef CAS.
- H. Duan and Y. Xuan, Phys. E, 2011, 43, 1475–1480 CrossRef CAS.
- K. Wu, W. E. Rodríguez-Córdoba, Y. Yang and T. Lian, Nano Lett., 2013, 13, 5255–5263 CrossRef CAS.
- T. Torimoto, H. Horibe, T. Kameyama, K.-i. Okazaki, S. Ikeda, M. Matsumura, A. Ishikawa and H. Ishihara, J. Phys. Chem. Lett., 2011, 2, 2057–2062 CrossRef CAS.
- G. Yu, X. Wang, J. Cao, S. Wu, W. Yan and G. Liu, Chem. Commun., 2016, 52, 2394–2397 RSC.
- J. Li, S. K. Cushing, P. Zheng, T. Senty, F. Meng, A. D. Bristow, A. Manivannan and N. Wu, J. Am. Chem. Soc., 2014, 136, 8438–8449 CrossRef CAS.
- T. Peng, K. Li, P. Zeng, Q. Zhang and X. Zhang, J. Phys. Chem. C, 2012, 116, 22720–22726 CrossRef CAS.
- J. Ge and Y. Li, Adv. Funct. Mater., 2004, 14, 157–162 CrossRef CAS.
- M. Muruganandham, Y. Kusumoto, C. Okamoto, A. Muruganandham, M. Abdulla-Al-Mamun and B. Ahmmad, J. Phys. Chem. C, 2009, 113, 19506–19517 CrossRef CAS.
- J. Yang, J.-H. Zeng, S.-H. Yu, L. Yang, G.-e. Zhou and Y.-t. Qian, Chem. Mater., 2000, 12, 3259–3263 CrossRef CAS.
- H. Chu, X. Li, G. Chen, W. Zhou, Y. Zhang, Z. Jin, J. Xu and Y. Li, Cryst. Growth Des., 2005, 5, 1801–1806 CrossRef CAS.
- S. Kar, B. Satpati, P. Satyam and S. Chaudhuri, J. Phys. Chem. B, 2005, 109, 19134–19138 CrossRef CAS.
- R. Thiruvengadathan and O. Regev, Chem. Mater., 2005, 17, 3281–3287 CrossRef CAS.
- W. Qingqing, X. Gang and H. Gaorong, Cryst. Growth Des., 2006, 6, 1776–1780 CrossRef.
- N. Pinna, K. Weiss, J. Urban and M.-P. Pileni, Adv. Mater., 2001, 13, 261–264 CrossRef CAS.
- Y. Cheng, Y. Wang, F. Bao and D. Chen, J. Phys. Chem. B, 2006, 110, 9448–9451 CrossRef CAS.
- L. Q. Pham, T.-K. Van, H. G. Cha and Y. S. Kang, CrystEngComm, 2012, 14, 7888–7890 RSC.
- L. Zhang, J. C. Yu, M. Mo, L. Wu, Q. Li and K. W. Kwong, J. Am. Chem. Soc., 2004, 126, 8116–8117 CrossRef CAS.
- P. Zhang and L. Gao, J. Mater. Chem. A, 2003, 13, 2007–2010 RSC.
- S. Xiong, B. Xi, C. Wang, G. Zou, L. Fei, W. Wang and Y. Qian, Chem.–Eur. J., 2007, 13, 3076–3081 CrossRef CAS.
- A. Phuruangrat, T. Thongtem and S. Thongtem, Mater. Lett., 2009, 63, 1538–1541 CrossRef CAS.
- Y. Guo, J. Wang, L. Yang, J. Zhang, K. Jiang, W. Li, L. Wang and L. Jiang, CrystEngComm, 2011, 13, 5045–5048 RSC.
- N. Bao, L. Shen, T. Takata and K. Domen, Mater. Lett., 2007, 20, 110–117 Search PubMed.
- Q. Wang, J. Lian, J. Li, R. Wang, H. Huang, B. Su and Z. Lei, Sci. Rep., 2015, 5, 1–9 CrossRef.
- J. Yu, Y. Yu and B. Cheng, RSC Adv., 2012, 2, 11829–11835 RSC.
- B. Zhang, W. Yao, C. Huang, Q. Xu and Q. Wu, Int. J. Hydrogen Energy, 2013, 38, 7224–7231 CrossRef CAS.
- H.-C. Liao, S.-Y. Chen and D.-M. Liu, Macromolecules, 2009, 42, 6558–6563 CrossRef CAS.
- W. Yao, X. Song, C. Huang, Q. Xu and Q. Wu, Catal. Today, 2013, 199, 42–47 CrossRef CAS.
- Q. Wang, J. Li, N. An, Y. Bai, X. Lu, J. Li, H. Ma, R. Wang, F. Wang and Z. Lei, Int. J. Hydrogen Energy, 2013, 38, 10761–10767 CrossRef CAS.
- I. Majeed, M. A. Nadeem, M. Al-Oufi, M. A. Nadeem, G. Waterhouse, A. Badshah, J. Metson and H. Idriss, Appl. Catal., B, 2016, 182, 266–276 CrossRef CAS.
- X. Chen and W. Shangguan, Front. Energy Res., 2013, 7, 111 CrossRef.
- S. Huang, Y. Lin, J.-H. Yang and Y. Yu, in Nanotechnology for Sustainable Energy, ACS Publications, 2013, pp. 219–241 Search PubMed.
- Y. Wang, Y. Wang and R. Xu, J. Phys. Chem. C, 2013, 117, 783–790 CrossRef CAS.
- L. A. Silva, S. Y. Ryu, J. Choi, W. Choi and M. R. Hoffmann, J. Phys. Chem. C, 2008, 112, 12069–12073 CrossRef CAS.
- H. Yan, J. Yang, G. Ma, G. Wu, X. Zong, Z. Lei, J. Shi and C. Li, J. Catal., 2009, 266, 165–168 CrossRef CAS.
- G. Xin, B. Yu, Y. Xia, T. Hu, L. Liu and C. Li, J. Phys. Chem. C, 2014, 118, 21928–21934 CrossRef CAS.
- J. Jin, J. Yu, G. Liu and P. K. Wong, J. Mater. Chem. A, 2013, 1, 10927–10934 RSC.
- M. Luo, W. Yao, C. Huang, Q. Wu and Q. Xu, J. Mater. Chem. A, 2015, 3, 13884–13891 RSC.
- N. Sahu, S. Upadhyay and A. Sinha, Int. J. Hydrogen Energy, 2009, 34, 130–137 CrossRef CAS.
- X. Zhou, H. Chen, Y. Sun, K. Zhang, X. Fan, Y. Zhu, Y. Chen, G. Tao and J. Shi, Appl. Catal., B, 2014, 152, 271–279 CrossRef.
- M. Sakamoto, A. Xiong, R. Kanakubo, T. Ikeda, T. Yoshinaga, K. Maeda, K. Domen and T. Teranishi, Chem. Lett., 2012, 41, 1325–1327 CrossRef CAS.
- S. Shen, L. Guo, X. Chen, F. Ren and S. S. Mao, Int. J. Hydrogen Energy, 2010, 35, 7110–7115 CrossRef CAS.
- Y. Li, L. Tang, S. Peng, Z. Li and G. Lu, CrystEngComm, 2012, 14, 6974–6982 RSC.
- C. Janet and R. Viswanath, Nanotechnology, 2006, 17, 5271 CrossRef CAS.
- Q. Wang, J. Li, N. An, Y. Bai, X. Lu, J. Li, H. Ma, R. Wang, F. Wang, Z. Lei and W. Shangguan, Int. J. Hydrogen Energy, 2013, 38, 10761–10767 CrossRef CAS.
- W. Yao, C. Huang, N. Muradov and T. Ali, Int. J. Hydrogen Energy, 2011, 36, 4710–4715 CrossRef CAS.
- M. Luo, W. Yao, C. Huang, Q. Wu and Q. Xu, RSC Adv., 2015, 5, 40892–40898 RSC.
- Q. Chen, C. Suo, S. Zhang and Y. Wang, Int. J. Photoenergy, 2013, 1 DOI:10.1155/2013/149586.
- X. Li, H. Liu, S. Liu, J. Zhang, W. Chen, C. Huang and L. Mao, Int. J. Hydrogen Energy, 2016, 41, 23015–23021 CrossRef CAS.
- P. D. Cozzoli, T. Pellegrino and L. Manna, Chem. Soc. Rev., 2006, 35, 1195–1208 RSC.
- W.-W. Zhao, J. Wang, J.-J. Xu and H.-Y. Chen, Chem. Commun., 2011, 47, 10990–10992 RSC.
- X. Xing, R. Liu, X. Yu, G. Zhang, H. Cao, J. Yao, B. Ren, Z. Jiang and H. Zhao, J. Mater. Chem. A, 2013, 1, 1488–1494 RSC.
- Y. Shemesh, J. E. Macdonald, G. Menagen and U. Banin, Angew. Chem., 2011, 123, 1217–1221 CrossRef.
- P. Kalisman, L. Houben, E. Aronovitch, Y. Kauffmann, M. Bar-Sadan and L. Amirav, J. Mater. Chem. A, 2015, 3, 19679–19682 RSC.
- Y. Du, B. Chen, Z. Yin, Z. Liu and H. Zhang, Small, 2014, 10, 4727–4734 CrossRef CAS.
- H.-W. Tseng, M. B. Wilker, N. H. Damrauer and G. Dukovic, J. Am. Chem. Soc., 2013, 135, 3383–3386 CrossRef CAS.
- J. Ran, J. Yu and M. Jaroniec, Green Chem., 2011, 13, 2708–2713 RSC.
- J. Zhang, S. Z. Qiao, L. Qi and J. Yu, Phys. Chem. Chem. Phys., 2013, 15, 12088–12094 RSC.
- J. Meng, F. Li, Y. Hu, L. Xu, Z. Sun and J. Liu, Mater. Res. Bull., 2013, 48, 2111–2116 CrossRef CAS.
- W. Zhang, Y. Wang, Z. Wang, Z. Zhong and R. Xu, Chem. Commun., 2010, 46, 7631–7633 RSC.
- T. Peng, X. Zhang, P. Zeng, K. Li, X. Zhang and X. Li, J. Catal., 2013, 303, 156–163 CrossRef CAS.
- X. Zong, H. Yan, G. Wu, G. Ma, F. Wen, L. Wang and C. Li, J. Am. Chem. Soc., 2008, 130, 7176–7177 CrossRef CAS.
- J. S. Jang, D. J. Ham, N. Lakshminarasimhan, W. y. Choi and J. S. Lee, Appl. Catal., A, 2008, 346, 149–154 CrossRef CAS.
- L. Jia, D.-H. Wang, Y.-X. Huang, A.-W. Xu and H.-Q. Yu, J. Phys. Chem. C, 2011, 115, 11466–11473 CrossRef CAS.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS.
- L. Jia, D.-H. Wang, Y.-X. Huang, A.-W. Xu and H.-Q. Yu, J. Phys. Chem. C, 2011, 115, 11466–11473 CrossRef CAS.
- M. Liu, Y. Du, L. Ma, D. Jing and L. Guo, Int. J. Hydrogen Energy, 2012, 37, 730–736 CrossRef CAS.
- J. Yang, H. Yan, X. Wang, F. Wen, Z. Wang, D. Fan, J. Shi and C. Li, J. Catal., 2012, 290, 151–157 CrossRef CAS.
- A. J. Frank, Z. Goren and I. Willner, J. Chem. Soc., Chem. Commun., 1985, 1029–1030 RSC.
- T. Peng, X. Zhang, P. Zeng, K. Li, X. Zhang and X. Li, J. Catal., 2013, 303, 156–163 CrossRef CAS.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS.
- Y.-P. Yuan, L.-W. Ruan, J. Barber, S. C. J. Loo and C. Xue, Energy Environ. Sci., 2014, 7, 3934–3951 RSC.
- H. Chauhan, Y. Kumar, J. Dana, B. Satpati, H. N. Ghosh and S. Deka, Nanoscale, 2016, 8, 15802–15812 RSC.
- L. Chen, Y. Xu and B. Chen, Appl. Catal., B, 2019, 256, 117848 CrossRef CAS.
- Y. Huang, J. Chen, W. Zou, L. Zhang, L. Hu, M. He, L. Gu, J. Deng and X. Xing, Dalton Trans., 2016, 45, 1160–1165 RSC.
- Y. K. Kho, W. Y. Teoh, A. Iwase, L. Mädler, A. Kudo and R. Amal, ACS Appl. Mater. Interfaces, 2011, 3, 1997–2004 CrossRef CAS.
- Y. Hou, F. Zuo, Q. Ma, C. Wang, L. Bartels and P. Feng, J. Phys. Chem. C, 2012, 116, 20132–20139 CrossRef CAS.
- S. S. K. Ma, K. Maeda, T. Hisatomi, M. Tabata, A. Kudo and K. Domen, Chem.–Eur. J., 2013, 19, 7480–7486 CrossRef CAS.
- W.-T. Sun, Y. Yu, H.-Y. Pan, X.-F. Gao, Q. Chen and L.-M. Peng, J. Am. Chem. Soc., 2008, 130, 1124–1125 CrossRef CAS.
- H. Park, W. Choi and M. R. Hoffmann, J. Mater. Chem., 2008, 18, 2379–2385 RSC.
- M. Moriya, T. Minegishi, H. Kumagai, M. Katayama, J. Kubota and K. Domen, J. Am. Chem. Soc., 2013, 135, 3733–3735 CrossRef CAS.
- J. Xu and X. Cao, Chem. Eng. J., 2015, 260, 642–648 CrossRef CAS.
- Y. Liu, Y.-X. Yu and W.-D. Zhang, J. Phys. Chem. C, 2013, 117, 12949–12957 CrossRef CAS.
- S. Liang, Z. Zhou, X. Wu, S. Zhu, J. Bi, L. Zhou, M. Liu and L. Wu, Molecules, 2016, 21, 213 CrossRef.
- S. Ma, J. Xie, J. Wen, K. He, X. Li, W. Liu and X. Zhang, Appl. Surf. Sci., 2017, 391, 580–591 CrossRef CAS.
- D. Jiang, Z. Sun, H. Jia, D. Lu and P. Du, J. Mater. Chem. A, 2016, 4, 675–683 RSC.
- J. Zhang, L. Wang, X. Liu, X. a. Li and W. Huang, J. Mater. Chem. A, 2015, 3, 535–541 RSC.
- J. Zhang, Y. Wang, J. Zhang, Z. Lin, F. Huang and J. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 1031–1037 CrossRef CAS.
- C. W. Raubach, Y. V. de Santana, M. M. Ferrer, V. M. Longo, J. A. Varela, W. Avansi, P. G. Buzolin, J. R. Sambrano and E. Longo, Chem. Phys. Lett., 2012, 536, 96–99 CrossRef CAS.
- Y. P. Xie, Z. B. Yu, G. Liu, X. L. Ma and H.-M. Cheng, Energy Environ. Sci., 2014, 7, 1895–1901 RSC.
- T. T. Zhuang, Y. Liu, M. Sun, S. L. Jiang, M. W. Zhang, X. C. Wang, Q. Zhang, J. Jiang and S. H. Yu, Angew. Chem., 2015, 127, 11657–11662 CrossRef.
- Y. Li, X. Wei, H. Li, R. Wang, J. Feng, H. Yun and A. Zhou, RSC Adv., 2015, 5, 14074–14080 RSC.
- J. Liu, J. Phys. Chem. C, 2015, 119, 28417–28423 CrossRef CAS.
- Z. Xu, H. Li, Z. Wu, J. Sun, Z. Ying, J. Wu and N. Xu, J. Phys. Chem. C, 2016, 4, 7501–7507 CAS.
- J. Zhang, Y. Wang, J. Jin, J. Zhang, Z. Lin, F. Huang and J. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 10317–10324 CrossRef CAS.
- L. Ge, F. Zuo, J. Liu, Q. Ma, C. Wang, D. Sun, L. Bartels and P. Feng, J. Phys. Chem. C, 2012, 116, 13708–13714 CrossRef CAS.
- S. Chaguetmi, F. Mammeri, M. Pasut, S. Nowak, H. Lecoq, P. Decorse, C. Costentin, S. Achour and S. Ammar, J. Nanopart. Res., 2013, 15, 2140 CrossRef.
- Y. Yin, Z. Jin and F. Hou, Nanotechnology, 2007, 18, 495608 CrossRef.
- H. B. Yang, J. Miao, S.-F. Hung, F. Huo, H. M. Chen and B. Liu, ACS Nano, 2014, 8, 10403–10413 CrossRef CAS.
- T. Jafari, E. Moharreri, A. S. Amin, R. Miao, W. Song and S. L. Suib, Molecules, 2016, 21, 900 CrossRef.
- Y. Li, L. Zhang, W. Wu, P. Dai, X. Yu, M. Wu and G. Li, Nanoscale Res. Lett., 2014, 9, 270 CrossRef.
- P. Lv, W. Fu, H. Yang, H. Sun, Y. Chen, J. Ma, X. Zhou, L. Tian, W. Zhang and M. Li, CrystEngComm, 2013, 15, 7548–7555 RSC.
- S. T. Kochuveedu, J. Nanomater., 2016, 2016, 12 CrossRef.
- Y.-Y. Hsu, N.-T. Suen, C.-C. Chang, S.-F. Hung, C.-L. Chen, T.-S. Chan, C.-L. Dong, C.-C. Chan, S.-Y. Chen and H. M. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 22558–22569 CrossRef CAS.
- K. Li, M. Han, R. Chen, S. L. Li, S. L. Xie, C. Mao, X. Bu, X. L. Cao, L. Z. Dong and P. Feng, Adv. Mater., 2016, 28, 8906–8911 CrossRef CAS.
- Y. Huang, J. Chen, W. Zou, L. Zhang, L. Hu, M. He, L. Gu, J. Deng and X. Xing, Dalton Trans., 2016, 45, 1160–1165 RSC.
- K. Li, M. Han, R. Chen, S. L. Li, S. L. Xie, C. Mao, X. Bu, X. L. Cao, L. Z. Dong and P. Feng, Adv. Mater., 2016, 28, 8906–8911 CrossRef CAS.
- S. Cao, Y. Chen, L. Kang, Z. Lin and W.-F. Fu, J. Mater. Chem., 2015, 3, 18711–18717 RSC.
- J. Song, H. Zhao, R. Sun, X. Li and D. Sun, Energy Environ. Sci., 2017, 10, 225–235 RSC.
- Y. Zhong, Y. Shao, F. Ma, Y. Wu, B. Huang and X. Hao, Nano Energy, 2017, 31, 84–89 CrossRef CAS.
- Z. Lin, L. Li, L. Yu, W. Li and G. Yang, J. Mater. Chem. A, 2017, 5, 5235–5259 RSC.
- W. Jiang, Y. Liu, R. Zong, Z. Li, W. Yao and Y. Zhu, J. Mater. Chem. A, 2015, 3, 18406–18412 RSC.
- Y.-X. Yu, W.-X. Ouyang, Z.-T. Liao, B.-B. Du and W.-D. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 8467–8474 CrossRef CAS.
- X. Wang, C. Liow, D. Qi, B. Zhu, W. R. Leow, H. Wang, C. Xue, X. Chen and S. Li, Adv. Mater., 2014, 26, 3506–3512 CrossRef CAS.
- J. U. Bang, S. J. Lee, J. S. Jang, W. Choi and H. Song, J. Phys. Chem. Lett., 2012, 3, 3781–3785 CrossRef CAS.
- P. Tongying, F. Vietmeyer, D. Aleksiuk, G. Ferraudi, G. Krylova and M. Kuno, Nanoscale, 2014, 6, 4117–4124 RSC.
- X. Zong, J. Han, G. Ma, H. Yan, G. Wu and C. Li, J. Phys. Chem. C, 2011, 115, 12202–12208 CrossRef CAS.
- K. Iwashina, A. Iwase, Y. H. Ng, R. Amal and A. Kudo, J. Am. Chem. Soc., 2015, 137, 604–607 CrossRef CAS.
- W.-K. Jo and N. C. S. Selvam, Chem. Eng. J., 2017, 317, 913–924 CrossRef CAS.
- Y. Huang, Y. Liu, D. Zhu, Y. Xin and B. Zhang, J. Mater. Chem. A, 2016, 4, 13626–13635 RSC.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782–786 CrossRef CAS.
- Y. Nosaka, J. Phys. Chem., 1991, 95, 5054–5058 CrossRef CAS.
- J. S. Jang, U. A. Joshi and J. S. Lee, J. Phys. Chem. C, 2007, 111, 13280–13287 CrossRef CAS.
- J. S. Jang, D. J. Ham, N. Lakshminarasimhan, W. y. Choi and J. S. Lee, Appl. Catal., A, 2008, 346, 149–154 CrossRef CAS.
- N. Bao, L. Shen, T. Takata, K. Domen, A. Gupta, K. Yanagisawa and C. A. Grimes, J. Phys. Chem. C, 2007, 111, 17527–17534 CrossRef CAS.
- N. Bao, L. Shen, T. Takata and K. Domen, Chem. Mater., 2007, 20, 110–117 CrossRef.
- M. Sathish and R. Viswanath, Catal. Today, 2007, 129, 421–427 CrossRef CAS.
- M. Muruganandham, Y. Kusumoto, C. Okamoto, A. Muruganandham, M. Abdulla-Al-Mamun and B. Ahmmad, J. Phys. Chem. C, 2009, 113, 19506–19517 CrossRef CAS.
- A. Deshpande, P. Shah, R. Gholap and N. M. Gupta, J. Colloid Interface Sci., 2009, 333, 263–268 CrossRef CAS.
- G. De, A. Roy and S. Bhattacharya, Int. J. Hydrogen Energy, 1995, 20, 127–131 CrossRef CAS.
- J. Zhang, J. Yu, M. Jaroniec and J. R. Gong, Nano Lett., 2012, 12, 4584–4589 CrossRef CAS.
- Z. Shen, G. Chen, Q. Wang, Y. Yu, C. Zhou and Y. Wang, Nanoscale, 2012, 4, 2010–2017 RSC.
- J. Yang, H. Yan, X. Wang, F. Wen, Z. Wang, D. Fan, J. Shi and C. Li, J. Catal., 2012, 290, 151–157 CrossRef CAS.
- S. Shen, L. Guo, X. Chen, F. Ren and S. S. Mao, Int. J. Hydrogen Energy, 2010, 35, 7110–7115 CrossRef CAS.
- A. Ye, W. Fan, Q. Zhang, W. Deng and Y. Wang, Catal. Sci. Technol., 2012, 2, 969–978 RSC.
- J. Zhang, J. Yu, M. Jaroniec and J. R. Gong, Nano Lett., 2012, 12, 4584–4589 CrossRef CAS.
- D. He, M. Chen, F. Teng, G. Li, H. Shi, J. Wang, M. Xu, T. Lu, X. Ji and Y. Lv, Superlattices Microstruct., 2012, 51, 799–808 CrossRef CAS.
- H. Park, W. Choi and M. R. Hoffmann, J. Mater. Chem. A, 2008, 18, 2379–2385 RSC.
- J. S. Jang, H. G. Kim, U. A. Joshi, J. W. Jang and J. S. Lee, Int. J. Hydrogen Energy, 2008, 33, 5975–5980 CrossRef CAS.
- S. Zhang, Q. Chen, D. Jing, Y. Wang and L. Guo, Int. J. Hydrogen Energy, 2012, 37, 791–796 CrossRef CAS.
- J. Hou, Z. Wang, W. Kan, S. Jiao, H. Zhu and R. Kumar, J. Mater. Chem. A, 2012, 22, 7291–7299 RSC.
- Y. Chen, L. Wang, G. M. Lu, X. Yao and L. Guo, J. Mater. Chem., 2011, 21, 5134–5141 RSC.
- T. Peng, P. Zeng, D. Ke, X. Liu and X. Zhang, Energy Fuels, 2011, 25, 2203–2210 CrossRef CAS.
- T. Kida, G. Guan and A. Yoshida, Chem. Phys. Lett., 2003, 371, 563–567 CrossRef CAS.
- D. Ke, S. Liu, K. Dai, J. Zhou, L. Zhang and T. Peng, J. Phys. Chem. C, 2009, 113, 16021–16026 CrossRef CAS.
- L. Wang and W. Wang, CrystEngComm, 2012, 14, 3315–3320 RSC.
- X. Wang, G. Liu, G. Q. Lu and H.-M. Cheng, Int. J. Hydrogen Energy, 2010, 35, 8199–8205 CrossRef CAS.
- L. Xu, W. Shi and J. Guan, Catal. Commun., 2012, 25, 54–58 CrossRef CAS.
- X.-H. Lu, S.-L. Xie, T. Zhai, Y.-F. Zhao, P. Zhang, Y.-L. Zhang and Y.-X. Tong, RSC Adv., 2011, 1, 1207–1210 RSC.
- X. Wang, G. Liu, L. Wang, Z. G. Chen, G. Q. M. Lu and H. M. Cheng, Adv. Energy Mater., 2012, 2, 42–46 CrossRef.
- Z. Khan, M. Khannam, N. Vinothkumar, M. De and M. Qureshi, J. Mater. Chem., 2012, 22, 12090–12095 RSC.
- X. Wang, G. Liu, G. Q. Lu and H.-M. Cheng, Int. J. Hydrogen Energy, 2010, 35, 8199–8205 CrossRef CAS.
- J. Ran, J. Yu and M. Jaroniec, Green Chem., 2011, 13, 2708–2713 RSC.
- J. Hou, Z. Wang, W. Kan, S. Jiao, H. Zhu and R. Kumar, J. Mater. Chem., 2012, 22, 7291–7299 RSC.
- X. Ding, Y. Li, J. Zhao, Y. Zhu, Y. Li, W. Deng and C. Wang, APL Mater., 2015, 3, 104410 CrossRef.
- J. Chen, X. J. Wu, L. Yin, B. Li, X. Hong, Z. Fan, B. Chen, C. Xue and H. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1210–1214 CrossRef CAS.
- X. Chen, W. Chen, H. Gao, Y. Yang and W. Shangguan, Appl. Catal., B, 2014, 152, 68–72 CrossRef.
- L. J. Zhang, T. F. Xie, D. J. Wang, S. Li, L. L. Wang, L. P. Chen and Y. C. Lu, Int. J. Hydrogen Energy, 2013, 38, 11811–11817 CrossRef CAS.
- J. Zhang, S. Z. Qiao, L. Qi and J. Yu, Phys. Chem. Chem. Phys., 2013, 15, 12088–12094 RSC.
- E. Baniasadi, I. Dincer and G. Naterer, Appl. Catal., A, 2013, 455, 25–31 CrossRef CAS.
- L. J. Zhang, R. Zheng, S. Li, B. K. Liu, D. J. Wang, L. L. Wang and T. F. Xie, ACS Appl. Mater. Interfaces, 2014, 6, 13406–13412 CrossRef CAS.
- H. Wang, W. Chen, J. Zhang, C. Huang and L. Mao, Int. J. Hydrogen Energy, 2015, 40, 340–345 CrossRef CAS.
- X. Wang, M. Liu, Q. Chen, K. Zhang, J. Chen, M. Wang, P. Guo and L. Guo, Int. J. Hydrogen Energy, 2013, 38, 13091–13096 CrossRef CAS.
- N. Biswal, D. Das, S. Martha and K. Parida, Int. J. Hydrogen Energy, 2011, 36, 13452–13460 CrossRef CAS.
- E. Hong, D. Kim and J. H. Kim, Ind. Eng. Chem. Res., 2014, 20, 3869–3874 CrossRef CAS.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS.
- Z. Yan, Z. Sun, X. Liu, H. Jia and P. Du, Nanoscale, 2016, 8, 4748–4756 RSC.
- J. J. Wang, Z. J. Li, X. B. Li, X. B. Fan, Q. Y. Meng, S. Yu, C. B. Li, J. X. Li, C. H. Tung and L. Z. Wu, ChemSusChem, 2014, 7, 1468–1475 CrossRef CAS.
- Y. X. Pan, H. Zhuang, J. Hong, Z. Fang, H. Liu, B. Liu, Y. Huang and R. Xu, ChemSusChem, 2014, 7, 2537–2544 CrossRef CAS.
- P. S. Lunawat, R. Kumar and N. M. Gupta, Catal. Lett., 2008, 121, 226–233 CrossRef CAS.
- P. S. Lunawat, S. Senapati, R. Kumar and N. M. Gupta, Int. J. Hydrogen Energy, 2007, 32, 2784–2790 CrossRef CAS.
- J. Thote, H. B. Aiyappa, A. Deshpande, D. Díaz Díaz, S. Kurungot and R. Banerjee, Chem.–Eur. J., 2014, 20, 15961–15965 CrossRef CAS.
- M. B. Wilker, J. K. Utterback, S. Greene, K. A. Brown, D. W. Mulder, P. W. King and G. Dukovic, J. Phys. Chem. C, 2018, 122, 741–750 CrossRef CAS.
- Y. Lin, J. Zhang, E. Sargent and E. Kumacheva, Appl. Phys. Lett., 2002, 81, 3134–3136 CrossRef CAS.
- E. Hong, D. Kim and J. H. Kim, J. Ind. Eng. Chem., 2014, 20, 3869–3874 CrossRef CAS.
- N. Buehler, K. Meier and J. F. Reber, J. Phys. Chem., 1984, 88, 3261–3268 CrossRef CAS.
- J. Schneider and D. W. Bahnemann, J. Phys. Chem. Lett., 2013, 4, 3479–3483 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CrossRef CAS.
- A. Kudo, Int. J. Hydrogen Energy, 2006, 31, 197–202 CrossRef CAS.
- T. Kawai and T. Sakata, J. Chem. Soc., Chem. Commun., 1980, 694–695 RSC.
- M. J. Berr, P. Wagner, S. Fischbach, A. Vaneski, J. Schneider, A. S. Susha, A. L. Rogach, F. Jäckel and J. Feldmann, Appl. Phys. Lett., 2012, 100, 223903 CrossRef.
- H. Harada, T. Sakata and T. Ueda, J. Am. Chem. Soc., 1985, 107, 1773–1774 CrossRef CAS.
- X. Zong, G. Wu, H. Yan, G. Ma, J. Shi, F. Wen, L. Wang and C. Li, J. Phys. Chem. C, 2010, 114, 1963–1968 CrossRef CAS.
- T. Peng, K. Li, P. Zeng, Q. Zhang and X. Zhang, J. Phys. Chem. C, 2012, 116, 22720–22726 CrossRef CAS.
- S. Shen and L. Guo, Mater. Res. Bull., 2008, 43, 437–446 CrossRef CAS.
- W. Yao, X. Song, C. Huang, Q. Xu and Q. Wu, Catal. Today, 2013, 199, 42–47 CrossRef CAS.
- D. Ke, S. Liu, K. Dai, J. Zhou, L. Zhang and T. Peng, J. Phys. Chem. C, 2009, 113, 16021–16026 CrossRef CAS.
- S. Chen and L.-W. Wang, Chem. Mater., 2012, 24, 3659–3666 CrossRef CAS.
- H. Yan, J. Yang, G. Ma, G. Wu, X. Zong, Z. Lei, J. Shi and C. Li, J. Catal., 2009, 266, 165–168 CrossRef CAS.
- M. J. Berr, A. Vaneski, C. Mauser, S. Fischbach, A. S. Susha, A. L. Rogach, F. Jäckel and J. Feldmann, Small, 2012, 8, 291–297 CrossRef CAS.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- A. B. Ellis, S. W. Kaiser, J. M. Bolts and M. S. Wrighton, J. Am. Chem. Soc., 1977, 99, 2839–2848 CrossRef CAS.
- H. Fujiwara, H. Hosokawa, K. Murakoshi, Y. Wada, S. Yanagida, T. Okada and H. Kobayashi, J. Phys. Chem. B, 1997, 101, 8270–8278 CrossRef CAS.
- N. S. Lewis, Nature, 2001, 414, 589 CrossRef CAS.
- K. Sanderson, Nature, 2008, 452, 400–402 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |