
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceActive faceted nanoporous ruthenium for electrocatalytic hydrogen evolution†
Abdillah Sani Bin
Mohd Najib
 abc,
Muhammad
Iqbal
b,
Mohamed Barakat
Zakaria
d,
Shusaku
Shoji
e,
Yohei
Cho
e,
Xiaobo
Peng
b,
Shigenori
Ueda
fg,
Ayako
Hashimoto
g,
Takeshi
Fujita
h,
Masahiro
Miyauchi
e,
Yusuke
Yamauchi
*di and
Hideki
Abe
*ab
abc,
Muhammad
Iqbal
b,
Mohamed Barakat
Zakaria
d,
Shusaku
Shoji
e,
Yohei
Cho
e,
Xiaobo
Peng
b,
Shigenori
Ueda
fg,
Ayako
Hashimoto
g,
Takeshi
Fujita
h,
Masahiro
Miyauchi
e,
Yusuke
Yamauchi
*di and
Hideki
Abe
*ab
aGraduate School of Science and Technology, Saitama University, Shimo-Okubo 255, Saitama 338-8570, Japan. E-mail: BE.Hideki@Anims.go.jp
bNational Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
cSchool of Mechanical engineering, Faculty of engineering, Universiti Teknologi Malaysia, Skudai, Johor Bahru, 81310, Malaysia
dAustralian Institute for Bioengineering, Nanotechnology (AIBN) and School of Chemical Engineering, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: y.yamauchi@uq.edu.au
eDepartment of Materials Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, Ookayama 2-12-1, Meguro-ku, Tokyo 152-8552, Japan
fSynchrotron X-ray Station at SPring-8, National Institute for Materials Science, 1-1-1 Kouto, Sayo, Hyogo 679-5148, Japan
gNational Institute for Materials Science, 1-2-1 Sengen, Tsukuba, Ibaraki 305-0047, Japan
hSchool of Environmental Science and Engineering, Kochi University of Technology, 185 Miyanokuchi, Tosayamada, Kami City, Kochi 782-8502, Japan
iDepartment of Plant & Environmental New Resources, Kyung Hee University, 1732 Deogyeong-daero, Giheung-gu, Yongin-si, Gyeonggi-do 446-701, South Korea
First published on 22nd September 2020
Abstract
Nanoporous ruthenium (np-Ru) comprising reaction active facets is synthesized from a ruthenium–cerium (Ru2Ce) alloy precursor. Spontaneous nanophase separation of Ru2Ce in an oxidative atmosphere results in a lamellar nanocomposite consisting of Ru metal and cerium oxide (CeO2). Selective leaching of the Ru–CeO2 nanocomposite in sulfuric acid yields np-Ru with a high surface area of 48 m2 g−1, predominantly surrounded by reaction active {10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0} facets. Active-faceted np-Ru efficiently catalyzed the hydrogen evolution reaction (HER) in acidic media at an overpotential of +74 mV vs. the reversible hydrogen electrode, which is 20 mV lower than that for carbon-supported Ru (Ru/C) and rather close to +58 mV for state-of-the-art Pt/C. Np–Ru has great potential in broad applications such as hydrogen battery electrodes because of its low HER overpotential and scalable synthetic protocol.
0} facets. Active-faceted np-Ru efficiently catalyzed the hydrogen evolution reaction (HER) in acidic media at an overpotential of +74 mV vs. the reversible hydrogen electrode, which is 20 mV lower than that for carbon-supported Ru (Ru/C) and rather close to +58 mV for state-of-the-art Pt/C. Np–Ru has great potential in broad applications such as hydrogen battery electrodes because of its low HER overpotential and scalable synthetic protocol.
Nanoporous materials are one of the confocal subjects of broad research on catalysts, absorbates and sensors due to their large surface area as a stage for different molecular events.1,2 Metallic nanoporous materials, i.e., nanoporous metals, are of particular and growing interest because of their high thermal/electric conductivity, magnetic , and plasmonic properties.3–5 There are a number of successful attempts to tailor the nanopores in size, dispersity and connectivity, using templates such as block copolymers, liquid crystals and/or mesoporous silica.6–11 The nanoporous metals synthesized in such bottom-up approaches are inherently an aggregate of individual clusters deposited to the template surface. Different approaches such as dealloying of prepared alloy precursors may yield more favorable nanoporous metals for electrocatalytic applications because of their continuously connected network structure.12–17 The so far developed dealloying methods can, however, hardly tailor the surface atomic arrangement of nanoporous metals: dealloyed metals are usually surrounded by randomly developed facets, some of which can inhibit targeted surface reactions.18
Herein, we propose another versatile method to synthesize facet-tailored nanoporous metals, utilizing a spontaneous nanophase-separation phenomenon of alloy precursors. An alloy precursor consisting of the target metal and an oxyphilic metal counterpart is subjected to an oxidative atmosphere to promote internal oxygen diffusion. Selective oxidation of the counterpart metal results in a spatially ordered metal-oxide nanocomposite. As the result of a strong metal–oxygen interaction at the epitaxial interface, the metal phase is allowed to develop energetically unfavorable crystal planes. The oxide counterpart is finally leached out in solvent to yield the targeted nanoporous metal that is surrounded by reaction active facets.
Precious-Group Metals (PGMs) especially platinum (Pt) serve as prominent catalysts for different chemical reactions.19–22 Electrolytic hydrogen (H2) evolution from aqueous solution (Hydrogen Evolution Reaction: HER) can potentially produce CO2-free H2, yet is precluded from broad use due to the lack of reaction active and economically sound electrocatalysts.23 It is highly desirable to materialize nanoporous ruthenium (np-Ru) as an efficient alternative to the state-of-the-art, expensive Pt electrocatalysts for the HER (Pt: 30 $ g−1; Ru: 8 $ g−1 – Johnson Matthey). However, to date, most of the attempts to synthesize np-Ru by conventional dealloying24,25 and organometallic decomposition26 methods resulted in the emergence of random facets which diminished the catalytic performance: the {100} facets of hexagonal Ru are theoretically demonstrated as most HER active, but less readily developed than any of the other energetically favorable facets.27
To demonstrate the priority of the proposed approach for nanoporous metals, we have successfully synthesized np-Ru from an precursor alloy consisting of Ru and a highly oxyphilic metal counterpart, cerium (Ce) (Fig. 1).28 A Ru2Ce alloy was first subjected to an oxidative environment to promote nanophase separation into a Ru–CeO2 nanocomposite. The Ru and CeO2 phases were ordered in a nanometer-thick (∼5 nm) lamellar structure because oxygen atoms diffuse throughout the precursor so as to extend an epitaxial interface of the CeO2{110} and Ru{100} planes. Selective leaching of the CeO2 phase in sulfuric acid (H2SO4) finally yielded phase-pure, highly crystalline np-Ru that was predominantly surrounded by {100} facets. The active faceted np-Ru efficiently promoted the HER in acidic media at a lower overpotential than carbon-supported Ru (Ru/C) with a higher turn-over frequency (TOF) at +100 mV vs. the reversible hydrogen electrode (RHE) compared to previously reported metallic np-Ru (Fig. S1 and S2†).26
 | ||
| Fig. 1 Synthetic protocol for active faceted np-Ru. Heating the Ru2Ce alloy precursor in an oxidative atmosphere results in a nanophase-separated metal-oxide composite having an extended Ru{100}/CeO2{110} epitaxial interface. The nanocomposite is finally leached in sulfuric acid to selectively dissolve CeO2, yielding active faceted np-Ru. | ||
The Ru2Ce alloy precursor was prepared by melting Ru and Ce metal ingots using an arc torch in a pure Ar atmosphere. The precursor alloy was then powdered with a mortar, sieved to 50–60 μm in size and heated in a mixture of carbon monoxide (CO), oxygen and argon gas (CO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2:Ar = 1:1:98 in volumetric ratio; flow rate = 60 cm3 s−1) at 600 °C for 12 hours. Powder X-ray diffractometry (pXRD) demonstrated that the reflections from the Ru2Ce precursor were fully substituted by those from Ru metal and CeO2 after the atmosphere treatment (Fig. S3†). Cross-sectional scanning transmission electron microscopy (STEM) observation further showed that the atmosphere-treated Ru2Ce alloy was converted into a Ru–CeO2 nanocomposite consisting of a highly ordered lamellar structure with a spatial period of 5 nm (Fig. 2a, b; see Fig. S4†). The bright and dark contrasted areas in Fig. 2a correspond to the metal Ru and CeO2 phases, respectively, as supported by compositional mapping (Fig. 2b; see Fig. S5† for details).
:O2:Ar = 1:1:98 in volumetric ratio; flow rate = 60 cm3 s−1) at 600 °C for 12 hours. Powder X-ray diffractometry (pXRD) demonstrated that the reflections from the Ru2Ce precursor were fully substituted by those from Ru metal and CeO2 after the atmosphere treatment (Fig. S3†). Cross-sectional scanning transmission electron microscopy (STEM) observation further showed that the atmosphere-treated Ru2Ce alloy was converted into a Ru–CeO2 nanocomposite consisting of a highly ordered lamellar structure with a spatial period of 5 nm (Fig. 2a, b; see Fig. S4†). The bright and dark contrasted areas in Fig. 2a correspond to the metal Ru and CeO2 phases, respectively, as supported by compositional mapping (Fig. 2b; see Fig. S5† for details).
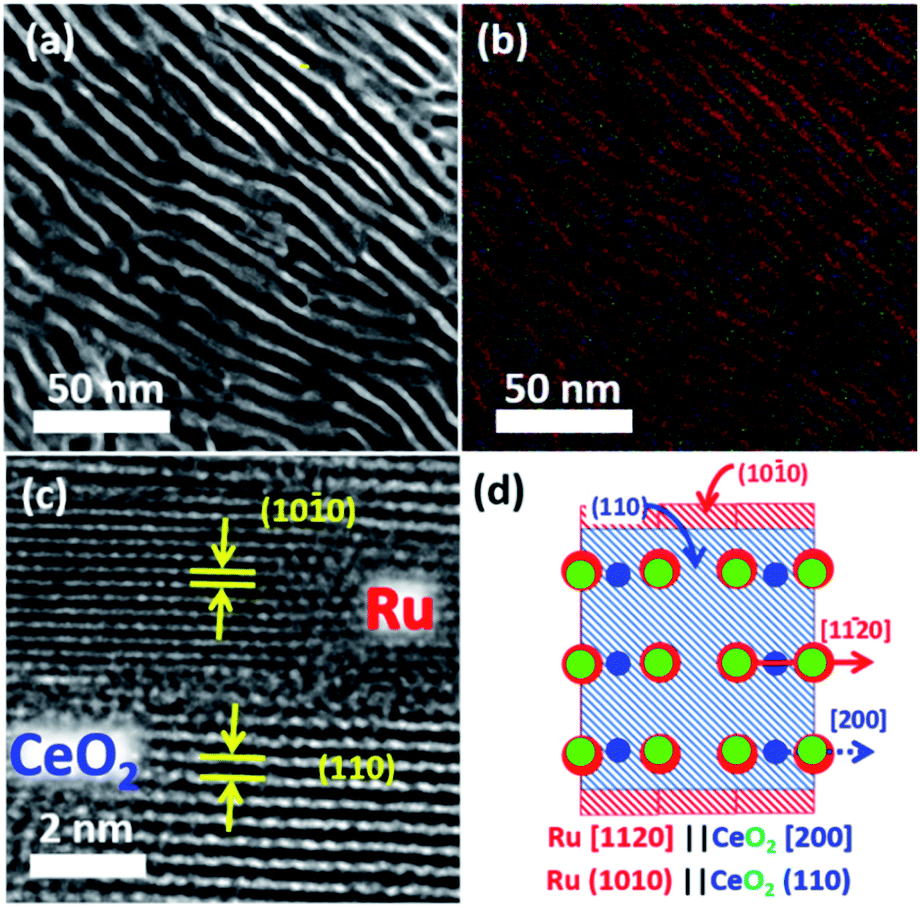 | ||
| Fig. 2 (a) Cross-sectional annular-dark field (ADF) scanning transmission electron microscopy (STEM) image and (b) compositional mapping image of a nanophase-separated Ru–CeO2 composite obtained from precursor Ru2Ce via atmosphere treatment. The red and dark green stripes correspond to the Ru and CeO2 phases, respectively. (c) High-resolution transmission electron microscopy (TEM) image of the Ru–CeO2 interface. (d) A schematic model for the epitaxial relationship between the Ru{100} and CeO2{110} planes. The red, blue and green circles correspond to the Ru, Ce and O atoms, respectively. | ||
The Ru phase was developed such that the Ru{100} and CeO2{110} planes were oriented with an epitaxial relationship (Fig. 2c; S3 and S7†). The interatomic distance of oxygen on the CeO2 (110) planes along the [100] axis, 2.7 Å, matches with the interatomic distance of Ru on the Ru (100) planes along the [11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0] axis, 2.7 Å (Fig. S8†).29 This lattice matching may minimize the interfacial energy between the metal and oxide phases to favor the epitaxial Ru{100}/CeO2{110} interface. Note that in the pXRD spectra, the {100} reflections were clearly visible for np-Ru due to the preferred growth of the {100} facets, while much weak reflections were recognized for directly dealloyed Ru2Ce having polycrystalline nature (Fig. S3†).
0] axis, 2.7 Å (Fig. S8†).29 This lattice matching may minimize the interfacial energy between the metal and oxide phases to favor the epitaxial Ru{100}/CeO2{110} interface. Note that in the pXRD spectra, the {100} reflections were clearly visible for np-Ru due to the preferred growth of the {100} facets, while much weak reflections were recognized for directly dealloyed Ru2Ce having polycrystalline nature (Fig. S3†).
Subsequent selective leaching of CeO2 from the Ru–CeO2 nanocomposite (Fig. 3a) in H2SO4 solution resulted in the targeted, faceted np-Ru (Fig. 3b). The CeO2 phase was completely removed as evidenced by pXRD and hard X-ray photoemission spectroscopy (HAXPES) (Fig. S3 and S9†). The Ru{100} facets, which were extended via the formation of the Ru{1010}/Ce{110} interface in the nanophase separation of the precursor alloy, became fully exposed to the environment after the elimination of the CeO2 matrix (Fig. 3c and d, see Fig. S10† for FFT analysis). The specific surface area of np-Ru was quantified by BET (Brunauer–Emmett–Teller) surface area measurements as 48 m2 g−1. The nitrogen (N2) adsorption–desorption isotherm demonstrated a hysteresis trend, showing that np-Ru consisted of slit-shaped and/or lamellar pores, as expected from the transmission electron microscopy (TEM) observations (Fig. S11†).
 | ||
| Fig. 3 (a) Cross-sectional ADF STEM image of a nanophase-separated Ru–CeO2 nanocomposite (bright: Ru; dark: CeO2) and (b) a bright-field transmission electron microscopy (TEM) image of active faceted np-Ru obtained via a selective leaching of the Ru–CeO2 nanocomposite (dark: Ru; bright: vacuum). (c) Bright-field TEM images of active faceted np-Ru. The atomic fringes in (d) correspond to the Ru{100} planes. | ||
Cyclic Voltammetry (CV) was conducted in 0.5 M H2SO4 solution using a rotating glassy-carbon disk electrode to identify the preferential facets of np-Ru. Fig. 4a presents the CV profiles for np-Ru and carbon-supported Ru (Ru/C; loading weight: 5%). Ru/C showed a typical CV profile for polycrystalline Ru with no preferential facets. By contrast, np-Ru showed a CV profile having distinct peaks at +0.30 V and +0.52 V vs. RHE, corresponding to the electrochemical oxidation of chemisorbed hydrogen atoms over the {100} and {0001} facets, respectively.30 As expected from the TEM characterization (Fig. 2d–f and S7†), np-Ru was predominantly surrounded by the {100} and {0001} facets.
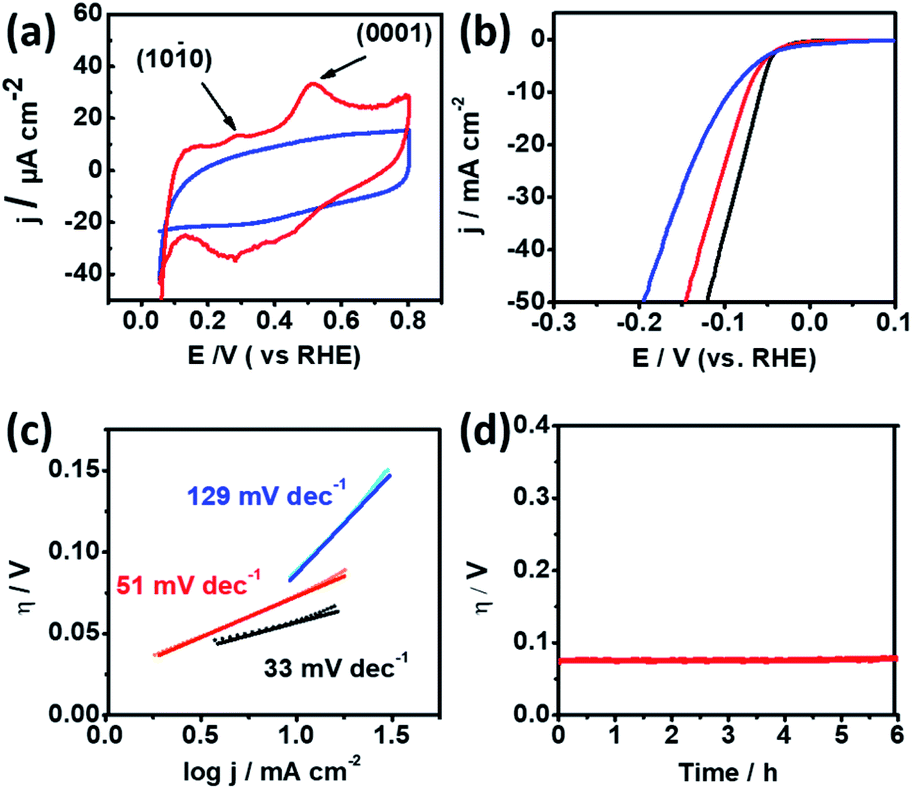 | ||
| Fig. 4 (a) Cyclic Voltammetry (CV) profiles for np-Ru (red) and Ru/C (blue), and (b) Linear Sweep Voltammetry (LSV) profiles and the corresponding (c) Tafel plots for np-Ru (red), Ru/C (blue) and Pt black (black). The corresponding Tafel slopes are shown in the inset. (d) Chronogalvanometry profile for np-Ru (red) acquired at a HER current density of 10 mV s−1. All the experiments were conducted in 0.5 M H2SO4 solution using platinum wire as the counter electrode. | ||
Linear Sweep Voltammetry (LSV) was performed in 0.5 M H2SO4 in a potential range from +100 mV to −400 mV vs. RHE to evaluate the HER performance of the faceted np-Ru catalyst (by considering overpotential at current density, j of 10 mA cm−2).26,31,32 As a comparison, Ru/C and Pt black were also tested under the same conditions. The polarization curves resulting from LSV demonstrated that np-Ru had an overpotential of −74 mV. This value was significantly low compared to the overpotential of −94 mV for Ru/C, and rather close to that for the state-of-the-art Pt/C, −58 mV (Fig. 4b).
We performed LSV using a graphite rod as the counter electrode to avoid possible contamination by redeposition of Pt (Fig. S12†).33 Another control LSV experiment was conducted in a perchloric acid (HClO4) electrolyte to avoid possible contamination by adsorption of sulfuric anions (SO42−) (Fig. S13†). The np-Ru catalyst was superior to Ru/C in the both cases in terms of higher onset potentials. np-Ru also showed higher HER mass activity than a non-supported metal Ru catalyst (Fig. S14, see the ESI† for the synthetic details of metal Ru). The electrochemical surface areas (ECSAs) for np-Ru and Ru/C were determined to be 39.60 and 73.87 m2 g−1, respectively, by measuring the double-layer capacitance in the electrolyte solution (Fig. S15, see the ESI† for experimental details).34–38 The current density normalized to the ECSA finally supported that np-Ru was superior to Ru/C in HER activity (Fig. S16†).
Furthermore, the Tafel slopes derived from LSV for np-Ru, +51 mV dec−1, was much lower than that for Ru/C, +129 mV dec−1, and again closer to that of Pt black, +33 mV dec−1. The np-Ru catalyst promoted the HER not only at a lower overpotential but also kinetically fast compared to Ru/C (Fig. 4c). In addition, the exchange current density at zero overpotential (J0) from the Tafel extrapolation method for np-Ru, PtB, and Ru/C was estimated to be 22.67 mA cm−2, 17.25 mA cm−2, and 6.638 mA cm−2, respectively (Fig. S17†). The np-Ru catalyst exhibited a higher exchange current density than Ru/C, indicating that the np-Ru surface had a so low energy barrier that charge is readily transported between the electrolyte and the catalyst surface.
It is acknowledged that there are three rate-determining steps associated with the HER in acidic media, resulting in different Tafel slopes: reduction of hydronium ions, H3O+ to H adatoms, Hads (Volmer reaction: H3O+ + e− + M → Hads + H2O; A = 120 mV dec−1), electrochemical desorption (Heyrovsky reaction: Hads + H3O+ + e− → H2 + H2O + M; A = 40 mV dec−1) and pairing of Hads to form molecular H2 (Tafel reaction: 2Hads → H2 + 2M; A = 30 mV dec−1) where M corresponds to the surface empty site.39,40
Based on the calculated Tafel slopes, the rate-determining step for Ru/C is assigned to the Volmer reaction in which H3O+ reduction to Hads was relatively slow. By contrast, the np-Ru catalyst promotes the HER predominantly via the Volmer–Heyrovsky reaction, where Hads was readily available to be desorbed via electrochemical pathways. As theoretically demonstrated by Li et al., the Ru {100} and {0001} facets moderately adsorb Hads compared to the other facets including {110}.27 The moderately adsorbed Hads onto the {100} facets can serve as an active intermediate for the other reactions, i.e., hydronium ions to molecular H2 (Heyrovsky) as for np-Ru, whereas too strongly adsorbed Hads depletes the surface empty site, M, slowing down further generation of active Hads (Volmer) as for Ru/C.41 Electrochemical impedance spectroscopy demonstrated a small arc diameter for np-Ru compared to that for Ru/C, showing that np-Ru had a lower resistance at the electrode/electrolyte interface (Fig. S18†). The low electrode/electrolyte resistance is associated with better diffusion of hydrogen, electrolyte, and intermediate species.34 The improved HER catalytic activity of np-Ru is thus partly attributed to the lowered charge-transfer resistance at the catalyst/electrode interface.
The catalyst stability of np-Ru was evaluated by chronogalvanometry in 0.5 M H2SO4 solution at +10 mA (Fig. 4d). The active faceted np-Ru exhibited a stable overpotential over 6 hours, supporting the reaction stability of the developed Ru{100} facets (see also Fig. S19† for chronoamperometric tests). TEM characterization has also confirmed that the np-Ru catalyst after the stability test retained not only the nanoporous structure but also the catalytically active {100} facets (Fig. S20 and S21†).
Conclusions
A nanoporous ruthenium (np-Ru) catalyst surrounded by reaction-active {100} facets has been successfully materialized via leaching of a lamellar Ru–CeO2 nanocomposite that spontaneously emerges from a Ru2Ce precursor alloy in an oxidative atmosphere. Np-Ru can be a rational alternative to Pt catalysts because of its low hydrogen evolution reaction (HER) overpotential, high H2 generation rate, long-term reaction stability, and low material cost. The active faceted np-Ru can be further improved in catalytic performance through hybridization with promoter nanomaterials such as carbon nanotubes and/or graphene materials, contributing to future social implementation of CO2-free hydrogen resources.42,43 Finally, the proposed material design strategy for np-Ru, i.e., the utilization of nanophase separation of precursor alloys, will open up unexplored synthetic routes toward a wide variety of nanoporous metals.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was preliminarily supported by the JST CREST program (grant no. JPMJCR15P1) and the JST PRESTO program towards “Innovative Catalysts” (grant no. JPMJPR17S7). This work was carried out by utilizing the facility of the NIMS TEM Station and was supported by the Global Research Center for Environment and Energy based on Nanomaterials Science. The HAXPES measurements were performed with the approval of the NIMS Synchrotron X-ray Station (Proposal No. 2016A4607, 2016B4600, 2017A4602, 2017B4605, 2018A4600, 2018B4601, 2019A4600 and 2019B4601). This work was performed in part at the Queensland node of the Australian National Fabrication Facility (ANFF), a company established under the National Collaborative Research Infrastructure Strategy to provide nano and microfabrication facilities for Australia’s researchers. This work was also partly funded by KAKENHI (JP16H02293).Notes and references
- S. Bhattacharyya, Y. Mastai, R. Narayan Panda, S.-H. Yeon and M. Z. Hu, J. Nanomater., 2014, 2014, 275796 CrossRef
.
-
S. Ameen, M. S. Akhtar, R. Godbole and H.-S. Shin, An Introduction to Nanoporous Materials, IntechOpen, 2019 Search PubMed
- R. Ron, E. Haleva and A. Salomon, Adv. Mater., 2018, 30, 1706755 CrossRef
- Y. Tang, B. Tang, Q. Li, J. Qing, L. Lu and K. Chen, Exp. Therm. Fluid Sci., 2013, 44, 194–198 CrossRef CAS
-
Q. Xu, H. Wang, X. Jiang and Y. Yamauchi, in Nanoporous Materials, CRC Press, 2013, pp. 183–200 Search PubMed
- B. Jiang, C. Li, Ö. Dag, H. Abe, T. Takei, T. Imai, M. S. A. Hossain, M. T. Islam, K. Wood, J. Henzie and Y. Yamauchi, Nat. Commun., 2017, 8, 1–8 CrossRef
- B. R. Wiesenauer and D. L. Gin, Polym. J., 2012, 44, 461–468 CrossRef CAS
- X. Zhang, W. Lu, J. Dai, L. Bourgeois, J. Yao, H. Wang, J. R. Friend, D. Zhao and D. R. MacFarlane, Sci. Rep., 2014, 4, 1–5 Search PubMed
- H. Luo, L. Sun, Y. Lu and Y. Yan, Langmuir, 2004, 20, 10218–10222 CrossRef CAS
- J. H. Nam, Y. Y. Jang, Y. U. Kwon and J. Do Nam, Electrochem. Commun., 2004, 6, 737–741 CrossRef CAS
- V. Malgras, Q. Ji, Y. Kamachi, T. Mori, F.-K. Shieh, K. C.-W. Wu, K. Ariga and Y. Yamauchi, Bull. Chem. Soc. Jpn., 2015, 88, 1171–1200 CrossRef CAS
- L. Zhang, H. Chang, A. Hirata, H. Wu, Q. K. Xue and M. Chen, ACS Nano, 2013, 7, 4595–4600 CrossRef CAS
- P. Haldar and A. Chatterjee, Acta Mater., 2017, 127, 379–388 CrossRef CAS
- L. Zeng, C. You, X. Cai, C. Wang, X. Zhang and T. Liang, J. Mater. Res. Technol., 2020, 9, 6909–6915 CrossRef CAS
- C. Qin, M. Zhang, B. Li, Y. Li and Z. Wang, Mater. Lett., 2020, 258, 126823 CrossRef CAS
- Q. Lu, G. S. Hutchings, W. Yu, Y. Zhou, R. V. Forest, R. Tao, J. Rosen, B. T. Yonemoto, Z. Cao, H. Zheng, J. Q. Xiao, F. Jiao and J. G. Chen, Nat. Commun., 2015, 6, 1–8 CrossRef
- Y. T. Kim, P. P. Lopes, S. A. Park, A. Y. Lee, J. Lim, H. Lee, S. Back, Y. Jung, N. Danilovic, V. Stamenkovic, J. Erlebacher, J. Snyder and N. M. Markovic, Nat. Commun., 2017, 8, 1–8 CrossRef CAS
- J. Erlebacher and I. McCue, Acta Mater., 2012, 60, 6164–6174 CrossRef CAS
- S. Yang, Y. J. Tak, J. Kim, A. Soon and H. Lee, ACS Catal., 2017, 7, 1301–1307 CrossRef CAS
- P. Xie, T. Pu, A. Nie, S. Hwang, S. C. Purdy, W. Yu, D. Su, J. T. Miller and C. Wang, ACS Catal., 2018, 8, 4044–4048 CrossRef CAS
- X. Chen, X. Chen, S. Cai, J. Chen, W. Xu, H. Jia and J. Chen, Chem. Eng. J., 2018, 334, 768–779 CrossRef CAS
- G. Arteaga, L. M. Rivera-Gavidia, S. J. Martínez, R. Rizo, E. Pastor and G. García, Surfaces, 2019, 2, 16–31 CrossRef
- G. Zhao, K. Rui, S. X. Dou and W. Sun, Adv. Funct. Mater., 2018, 28, 1803291 CrossRef
- M. Hakamada, J. Motomura, F. Hirashima and M. Mabuchi, Mater. Trans., 2012, 53, 524–530 CrossRef CAS
- Q. Zhou, H. Yang and C. Xu, Int. J. Hydrogen Energy, 2016, 41, 12714–12721 CrossRef CAS
- S. Drouet, J. Creus, V. Collière, C. Amiens, J. García-Antón, X. Sala and K. Philippot, Chem. Commun., 2017, 53, 11713–11716 RSC
- Y. Li, L. A. Zhang, Y. Qin, F. Chu, Y. Kong, Y. Tao, Y. Li, Y. Bu, D. Ding and M. Liu, ACS Catal., 2018, 8, 5714–5720 CrossRef CAS
- A. S. B. M. Najib, X. Peng, A. Hashimoto, S. Shoji, T. Iida, Y. Bai and H. Abe, Chem.–Asian J., 2019, 14, 201900542 CrossRef
- S. Gates-Rector and T. Blanton, Powder Diffr., 2019, 34, 352–360 CrossRef CAS
-
N. S. Marinkovic, M. B. Vukmirovic and R. R. Adzic, in Modern Aspects of Electrochemistry, Springer, New York, 2008, pp. 1–52 Search PubMed
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS
- Y. Gorlin and T. F. Jaramillo, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS
- M. B. Zakaria, RSC Adv., 2016, 6, 10341–10351 RSC
- M. B. Zakaria, M. Hu, M. Pramanik, C. Li, J. Tang, A. Aldalbahi, S. M. Alshehri, V. Malgras and Y. Yamauchi, Chem.–Asian J., 2015, 10, 1541–1545 CrossRef CAS
- A. A. Alireza, N. Hamnabard, S. M. H. Meshkati, M. Pakan and Y. H. Ahn, Dalton Trans., 2019, 48, 5429–5443 RSC
- Z. Zhou, L. Wei, Y. Wang, H. E. Karahan, Z. Chen, Y. Lei, X. Chen, S. Zhai, X. Liao and Y. Chen, J. Mater. Chem. A, 2017, 5, 20390–20397 RSC
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS
- B. Seo, D. S. Baek, Y. J. Sa and S. H. Joo, CrystEngComm, 2016, 18, 6083–6089 RSC
- L. X. Chen, Z. W. Chen, Y. Wang, C. C. Yang and Q. Jiang, ACS Catal., 2018, 8, 8107–8114 CrossRef CAS
- A. Kahyarian, B. Brown and S. Nesic, J. Electrochem. Soc., 2017, 164, H365–H374 CrossRef CAS
- T. Shinagawa, A. T. Garcia-Esparza and K. Takanabe, Sci. Rep., 2015, 5, 1–21 Search PubMed
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2016, 138, 16174–16181 CrossRef CAS
- D. H. Kweon, M. S. Okyay, S.-J. Kim, J.-P. Jeon, H.-J. Noh, N. Park, J. Mahmood and J.-B. Baek, Nat. Commun., 2020, 11, 1278 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta04223d |
| This journal is © The Royal Society of Chemistry 2020 |