
Semiconductor-based photocatalysts for photocatalytic and photoelectrochemical water splitting: will we stop with photocorrosion?
Sha
Chen
ab,
Danlian
Huang
 *ab,
Piao
Xu
ab,
Wenjing
Xue
ab,
Lei
Lei
ab,
Min
Cheng
ab,
Rongzhong
Wang
ab,
Xigui
Liu
ab and
Rui
Deng
ab
*ab,
Piao
Xu
ab,
Wenjing
Xue
ab,
Lei
Lei
ab,
Min
Cheng
ab,
Rongzhong
Wang
ab,
Xigui
Liu
ab and
Rui
Deng
ab
aCollege of Environmental Science and Engineering, Hunan University, Changsha, 410082, PR China. E-mail: huangdanlian@hnu.edu.cn; Fax: +86-731-88822829; Tel: +86-731-88822829
bKey Laboratory of Environmental Biology and Pollution Control, Hunan University, Ministry of Education, Changsha 410082, PR China
First published on 4th January 2020
Abstract
The status of photocatalytic (PC)/photoelectrochemical (PEC) water splitting as a promising approach to solar-to-chemical energy conversion has increased significantly over the past several decades for addressing the energy shortage. However, the overall energy conversion efficiency is still relatively poor due to the severe photocorrosion in photosensitive semiconductors. Herein, the review begins with the discussion of the photocorrosion mechanism with several typical semiconductors as examples. Then the feasible characterization methods used to evaluate the stability of semiconductors are summarized. Notably, most studies regarding water splitting focus on achieving high efficiency by improving the charge separation and transfer efficiency within the semiconductors. This review focuses on the recent advances in effective strategies for photocorrosion inhibition of semiconductor-based composites with respect to their intrinsic properties and interface charge transfer kinetics, including morphology/size control, heteroatom doping, heterojunction construction, surface modification, and reaction environment regulation. Furthermore, an in-depth investigation of photocorrosion pathways and mechanisms is critical to accurately and effectively address the photocorrosion of semiconductor-based composites to improve PC/PEC water splitting performance in the future.
 Sha Chen | Sha Chen received her B.S. from Jianghan University in 2017. Currently, she is a PhD candidate under the supervision of Prof. Danlian Huang at the College of Environmental Science and Engineering, Hunan University, China. Her current research interests focus on the synthesis and application of functional photocatalysts. |
 Danlian Huang | Danlian Huang received her Ph.D. degree from Hunan University, China, in 2011. She is currently a professor at the College of Environmental Science and Engineering, Hunan University. Her research interests focus on the synthesis and application of functional microbes and functional materials in environmental remediation. |
 Top (from left to right): Piao Xu; Wenjing Xue; Lei LeiBottom (from left to right): Min Cheng; Rongzhong Wang; Xigui Liu; Rui Deng |
1. Introduction
Providing a steady stream of clean and renewable energy for society is a daunting task, especially in an era of increasing fossil energy consumption and carbon dioxide emissions.1–4 Overall water splitting based on photocatalytic (PC) or photoelectrochemical (PEC) technology can convert or store solar energy in chemical bonds, which has attracted growing and intense interest since the landmark work of photocatalytic water splitting reported by Fujishima and Honda in 1972.5–7 Thermodynamically, overall water splitting should straddle the potentials of the hydrogen evolution reaction (−0.41 V vs. NHE) and oxygen evolution reaction (+0.82 V vs. NHE) (eqn (1)–(3)), which is an energetically uphill process.| 2H+ + 2e− → H2(g), ΔE0 = −0.41 V | (1) |
| 2H2O(l) + 4h+ → O2(g) + 4H+, ΔE0 = 0.82 V | (2) |
| H2O(l) → H2(g) + 1/2O2(g), ΔE0 = 1.23 V | (3) |
In terms of practical application, high efficiency is a prerequisite for solar-chemical energy conversion, which depends on the following factors: (i) shortening the carrier transport distance to accelerate the transfer of excited photogenerated carriers to the catalyst surface;8,9 (ii) increasing the specific surface area to enhance the adsorption of water molecules;10 (iii) reducing the feature size over the critical size to facilitate charge carrier separation;11,12 (iv) increasing the band gap accompanying the conduction band (CB) or/and valence band (VB) edge shift to improve the thermodynamic driving force for surface reactions.13 Therefore, the band structure of the semiconductor for overall water splitting under light irradiation should satisfy the following conditions: (i) the band gap should be larger than 1.23 eV to straddle the oxygen evolution reaction potentials; (ii) the CB potential should be more negative than that for water reduction; (iii) the VB potential should be more positive than that for water oxidation.2,14 However, it is difficult to satisfy all the requirements with a single semiconductor photocatalyst; therefore, the existing research is dedicated to solving the above problems and has made breakthrough progress to obtain improved PC/PEC activity. Nevertheless, these improvements are sometimes limited by photostability issues, which results in an initial PC/PEC water splitting performance that decreases over time.
Up to now, TiO2 has been the most widely investigated photocatalytic material for PC/PEC water splitting due to its excellent stability, affordability, and environmental friendliness. However, a severe limitation is that it can only be excited by ultraviolet light to show photocatalytic activity. Alternatively, various emerging semiconductors with excellent visible light response have been developed for PC/PEC water splitting, such as WO3, α-Fe2O3, BiVO4, CdS, Cu2O and so on.15–18 Of these, α-Fe2O3 can effectively absorb solar light with an energy distribution of 40% due to its narrow band gap of 2.1 eV, and its high solar-to-hydrogen (STH) efficiency of 16.8% and long-term stability (1000 h) render it a good candidate for PC/PEC water splitting. However, little success has been obtained on iron oxide due to its fast bulk recombination and short carrier diffusion length of no more than 10 nm.19 Although various strategies have been developed to improve the photocurrent density of α-Fe2O3, the obtained maximum photocurrent density is still far below its estimated theoretical value due to its inherent photoelectrochemical instability. Similar limitations also exist in other narrow band gap semiconductors, as displayed in Table 1.20–34 This photoelectrochemical corrosion refers to the photogenerated charge carriers participating in the self-oxidation and/or reduction of semiconductors rather than water splitting.35,36 As a typical example, metal sulfides are easily oxidized by photoexcited holes, causing the formation of sulfate (SO42−) in the presence of molecular oxygen and the formation of sulfur (S0) in the absence of molecular oxygen under light illumination, thereby resulting in a decrease in photocatalytic performance.37 In view of the long-term photochemical stability of photocatalysts as a prerequisite for practical applications, research interests and efforts should be focused on how to overcome photocorrosion of semiconductor-based composites to achieve a long-term stable performance with respect to PC/PEC water splitting.
| Photocatalysts | Bandgap (Eg, eV) | Band edge positions VRHE (V) | Theoretical photocurrent density STH efficiency | Best reported PEC performance | Ref. |
|---|---|---|---|---|---|
| CuO | 1.7 | VB at 2.16, CB at 0.46 | 35 mA cm−2 | 3.7 mA cm−2 at 1.23 VRHE | 20 |
| BiVO4 | 2.4 | VB at 2.5, CB at 0.1 | 9.2%, 7.5 mA cm−2 | 6.72 mA cm−2 at 1.23 VRHE | 21 |
| Cu2O | 2.2 | VB at 1.92, CB at −0.28 | 18.1%, −14.7 mA cm−2 | −10 mA cm−2 at 0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) VRHE VRHE |
22 |
| WO3 | 2.7 | VB at 2.16, CB at 0.46 | 4.8%, 2.4 mA cm−2 | 2.3 mA cm−2 at 1.23 VRHE | 23 |
| TaON | 2.5 | VB at 2.1, CB at −0.4 | 7.8%, 6 mA cm−2 | 4.57 mA cm−2 at 1.23 VRHE | 24 |
| Ta3N5 | 2.1 | VB at 1.5, CB at −0.6 | 12.6 mA cm−2, 15.9% | 12.1 mA cm−2 at 1.23 VRHE | 25 |
| CuWO4 | 2.2 | VB at 2.6, CB at 0.4 | 13%, 10.7 mA cm−2 | 1.5 mA cm−2 at 1.23 VRHE | 26 |
| CaFe2O4 | 1.9 | VB at 1.3, CB at −0.6 | 21%, 17.5 mA cm−2 | 1.2 mA cm−2 at −0.6 V Ag/AgCl |
27 |
| ZnFe2O4 | ∼2 eV | VB at 2.5, CB at 0.5 | 18%, 14.3 mA cm−2 | 0.24 mA cm−2 at 1.23 VRHE | 28 |
| CuBi2O4 | 1.8 eV | VB at 1.17, CB at −0.63 | 24.1%, 19.7 mA cm−2 | 2.5 mA cm−2 at 0.6 VRHE |
29 |
| CuFeO2 | 1.5 eV | VB at 1.1, CB at −0.4 | 35%, 29 mA cm−2 | 1.51 mA cm−2 at 0.35 VRHE |
30 |
| LaTiO2N | 2.1 eV | VB at 1.9, CB at −0.2 | 15.8%, 12.5 mA cm−2 | 6.5 mA cm−2 at 1.23 VRHE | 31 |
| BaTaO2N | 1.8 eV | VB at 1.31, CB at −0.49 | 24%, 20 mA cm−2 | 4.2 mA cm−2 at 1.2 VRHE | 32 |
| CuInS2 | 1.53 eV | VB at 0.64, CB at −0.89 | 35%, 28 mA cm−2 | 13.0 mA cm−2 at 0 VRHE | 33 |
| CuGaSe2 | 1.7 eV | VB at 1.43, CB at −0.27 | 27.6%, 22.5 mA cm−2 | 20 mA cm−2 at 0 VRHE | 34 |
To date, the amount of research on the photocorrosion inhibition of semiconductors for PEC or/and PC water splitting has been increasing, and many feasible methods have been explored to enhance the photoreaction stability of PC/PEC water splitting systems, such as designing photocatalyst structures, constructing multicomponent composites, and optimizing reaction system conditions. In this regard, a comprehensive and timely overview regarding the latest developments and future directions of effective strategies toward photocorrosion inhibition of semiconductor-based composites in PC/PEC water splitting systems is highly desirable to promote the latest developments in this field of research, which have not yet been reported. Therefore, this review outlines recent advances in strategies to improve the photoreaction stability of PC/PEC water splitting systems, followed by discussing the tactics involved in improving the stability of PC/PEC water splitting systems with different photocorrosion mechanisms.
The review begins with the discussion of photocorrosion mechanisms of semiconductors under different circumstances. Taking typical semiconductors (CdS, ZnO and Cu2O) as examples, the photocorrosion pathway in the presence of photogenerated electrons and/or holes is thoroughly clarified to provide a fundamental understanding of semiconductor instability under light irradiation. Then, a series of feasible characterization methods used to evaluate or confirm the stability of semiconductors are summarized. Most importantly, unlike most studies regarding water splitting aiming to achieve high efficiency by promoting the charge separation/transfer within the semiconductors, the focus of this review is on the recent advances in effective strategies for suppressing photocorrosion of semiconductor-based composites with respect to their intrinsic properties and interface charge transfer kinetics, including morphology/size control, heteroatom doping, heterojunction construction, surface modification, and reaction environment regulation. Finally, the daunting tasks that future research efforts should be focused on are discussed, aiming to accurately and effectively solve the photocorrosion of semiconductors to further improve the PC/PEC water splitting activity and realize the long-term performance of PC/PEC water splitting in practical applications.
2. Understanding the photocorrosion mechanism toward PC/PEC water splitting
A precondition for introducing PC/PEC water splitting into practical applications is to improve the stability of semiconductor materials without sacrificing catalytic activity. However, many semiconductors suffer from severe photocorrosion under light irradiation, which results in a gradual decrease in activity as irradiation time increases.38 An in-depth investigation of the mechanism of semiconductor photocorrosion is conducive to the development of a series of feasible strategies to improve the photoreaction stability of semiconductors. In general, whether semiconductors undergo photochemical corrosion depends on the alignment of the reduction potential (ϕre) relative to the conduction band minimum (CBM) or the oxidation potential (ϕox) relative to the valence band maximum (VBM). As shown in Fig. 1, a semiconductor with a VBM lower than ϕox (O2/H2O) will suffer from oxidation by unconsumed holes, such as CdS, ZnO, ZnS, TiO2 and so on. Similarly, a semiconductor with a CBM higher than ϕre (H+/H2) will suffer from reduction by unconsumed electrons, such as Cu2O, BiVO4, WO3 and so on.39 In this section, the photocorrosion pathway in the presence of photogenerated electrons and/or holes is thoroughly discussed with three typical semiconductors (CdS, ZnO and Cu2O) as examples, which can also be compared to other semiconductors.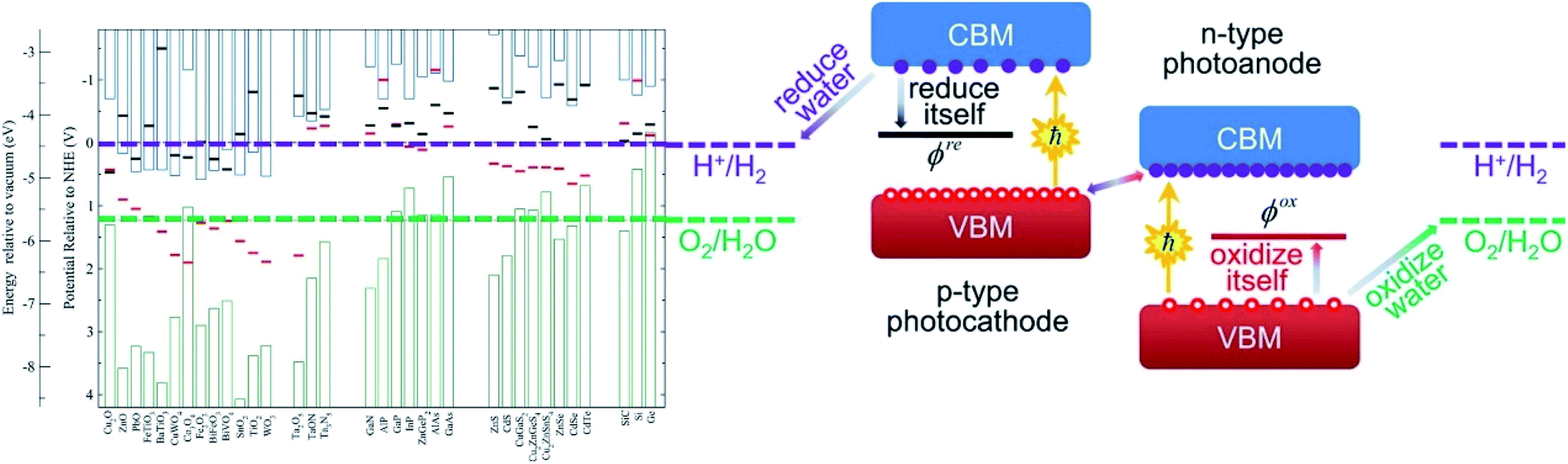 | ||
| Fig. 1 The relationship between band edge position and self-reduced and/or oxidized potential for various semiconductors. Reprinted with permission from ref. 39. Copyright 2012, American Chemical Society. | ||
Sulfide semiconductors are regarded as promising candidates for photocatalytic hydrogen evolution due to their strong visible light absorption capacity and thermodynamically favorable band structure.40 However, photogenerated hole-induced instability leads to a decrease in photocatalytic performance as the irradiation time increases, which greatly limits the application of sulfide semiconductors in photocatalytic hydrogen evolution.41 Taking CdS as an example, under light irradiation, CdS is excited to produce photo-generated electron–hole pairs after absorbing more energy than its band gap. The surface sulfur ions of CdS would be oxidized to S0 in the absence of oxygen (CdS + 2h+ → Cd2+ + S)42 and to SO42− in the presence of oxygen (CdS + 4h+ + H2O + O2 → Cd2+ + SO42− + 4H+)43 if the VB-holes of CdS cannot be consumed or withdrawn in a timely manner, thereby resulting in CdS inactivation.
Similar hole-induced photocorrosion also exists in PEC systems; taking ZnO as an example, the lattice oxygen of ZnO, rather than the oxygen atoms in water molecules, is oxidized by photogenerated holes to release zinc ions (2ZnO + 4h+ → 2Zn2+ + O2), thereby resulting in the dissolution of the ZnO photoelectrode. Generally, the photo-induced dissolution of ZnO involves the following steps. The excited holes are first transferred from the VB to the catalyst surface (Osurface2− + h+ → Osurface− and Osurface− + 3O2− + 3h+ → 2(O − O2−)), followed by the oxidation of the lattice oxygen ((O − O2−) + 2h+ → O2) accompanied by the dissolution of Zn2+(2Zn2+ → 2Zn2+).44
Unlike CdS and ZnO semiconductors, Cu2O, a semiconductor widely used for photocatalytic hydrogen evolution, has a more complex photocorrosion pathway, involving hole-induced oxidation of Cu2O (Cu2O + 2OH− + 2h+ → 2CuO + H2O) and electron-induced reduction of Cu2O (Cu2O + H2O + 2e− → 2Cu + 2OH−).45,46 More importantly, the redox potential of Cu2O is between its CB and VB potential, which causes one or both of the electron and/or hole driven photocorrosion to occur.47 Notably, the effects of environmental media such as O2 and pH on the stability of semiconductors under light irradiation also should be taken into consideration. For instance, whether the photocorrosion product of CdS is SO42− or S0 depends on the presence or absence of oxygen molecules. Besides, CdS suffers from serious photooxidative dissolution under acidic conditions even without light irradiation, while photocorrosion and leaching can be effectively inhibited in a high pH solution.
Based on the above discussion, a decrease in photostability would have a significant adverse effect on PC and/or PEC water splitting performances. Therefore, it is necessary to control the undesired reaction between excited holes or/and electrons and the semiconductor, while promoting the separation and migration of electron–hole pairs to accelerate the photocatalytic redox reaction, thereby resulting in improved PC/PEC water splitting activity and stability. To this end, tremendous strategies have been developed to improve the photoreaction stability of PC/PEC water splitting systems such as morphology/size control, reaction environment regulation, heterojunction construction and so on, which will be discussed in the subsequent section. Notably, a comprehensive understanding of the characterization methods used to evaluate semiconductor stability is essential and will be the focus of the next section.
3. Advanced characterization methods of semiconductor-based catalysts for PC/PEC water splitting
The stability of the photocatalyst is an important indicator to determine whether it can be reused in practical applications, which is usually evaluated by successive cycle tests. That is, the used catalyst is collected and reused for several cycles under the same experimental conditions as the fresh sample.48–51 However, it is not convincing to use the catalyst cycle performance as an indicator to evaluate the photostability of the catalyst, as other factors may also result in a decrease in photocatalytic performance after multiple recycle tests, such as active site blocking, loss of sample during recycling, adsorption of intermediates, or surface changes. Therefore, it is necessary to characterize the surface morphology/structure of the sample after multiple cycles to further determine the stability of the photocatalyst.In general, electron microscopy such as scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS) is applied to analyze the morphological information, crystal structure, and surface properties of the reused catalysts to investigate the structural changes of the photocatalysts before and after the reaction, thereby evaluating the photostability of the sample.52 For instance, Chen et al. reported that the prepared MoS2/CdS catalyst showed a long-term stability toward photocatalytic water splitting, and the hydrogen production was almost the same every 5 h (Fig. 2a).53 The excellent stability can be confirmed by SEM (Fig. 2b) and XRD (Fig. 2c) characterization, in which no significant difference in the XRD pattern and surface morphology was observed between the used and fresh catalysts, suggesting that the photocorrosion of CdS caused by holes can be effectively inhibited. In another example, Zhou et al. found that the photostability of CdS nanorods was greatly improved after being modified with amorphous turbostratic carbon nitride layers (CNx) (Fig. 2d and e).54 Comparing the TEM images of the used sample, no obvious etching was found for the used CdS-Pt/CNx and CdS/CNx-Pt, while severe photocorrosion was detected in the used CdS-Pt sample, indicating that the modification with the CNx layer can effectively inhibit the instability of CdS under light irradiation (Fig. 2f and g).
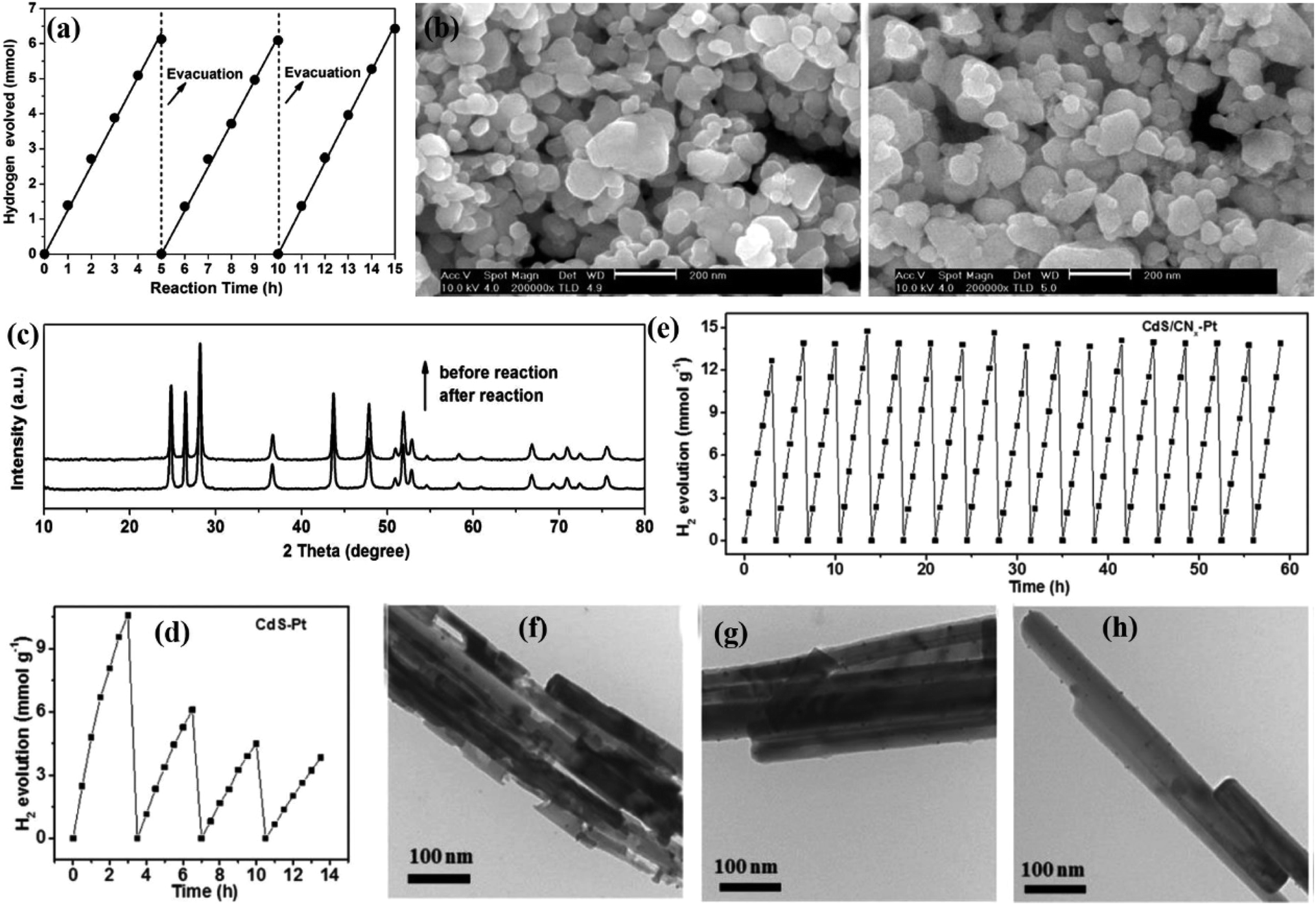 | ||
| Fig. 2 (a) Time course of H2 evolution on 0.9 mol% MoS2/CdS photocatalyst under visible light irradiation; (b) SEM images of 0.9 mol% MoS2/CdS and collected 0.9 mol% MoS2/CdS prepared at 673 K and (c) their XRD patterns. Reprinted with permission from ref. 53. Copyright 2012, Elsevier B.V. The catalytic stability of various photocatalysts for hydrogen production: (d) CdS–Pt and (e) CdS/CNx-Pt, respectively. TEM images of (f) CdS/Pt, (g) CdS–Pt/CNx and (h) CdS/CNx-Pt after cycled H2 production. Reprinted with permission from ref. 54. Copyright 2017, Royal Society of Chemistry. | ||
The photocorrosion of metal sulfide-based composite catalysts is caused by the hole-induced oxidation of surface lattice S2− to S0 and/or SO42−, which results in a change in the surface structures of the metal sulfide in the composite.41 Therefore, investigating the valence state of the element S in the metal sulfide composite before and after multiple cycle reactions is conclusive evidence for evaluating the photoreaction stability of the composite, which can be achieved by XPS characterization. For instance, ternary ZnO–CdS–MoS2 (ZCM) exhibited an improved photocatalytic performance and stability for hydron evolution after light tuning from visible light to UV-vis light, which was attributed to interfacial charge transfer following a traditional type-II heterojunction under visible light while a Z-scheme under UV-vis light (Fig. 3a).55 As proven from the XPS spectra of the fresh and used sample, only little S0 generated by oxidation of the surface lattice S2− by excited holes can be found in the used sample (Fig. 3b); the Cd 3d spectra displayed in Fig. 3c showed that no obvious decrease in intensity could be found under UV-vis irradiation while a significant decrease occurred under visible light irradiation, suggesting higher photocatalytic stability under UV-vis light.
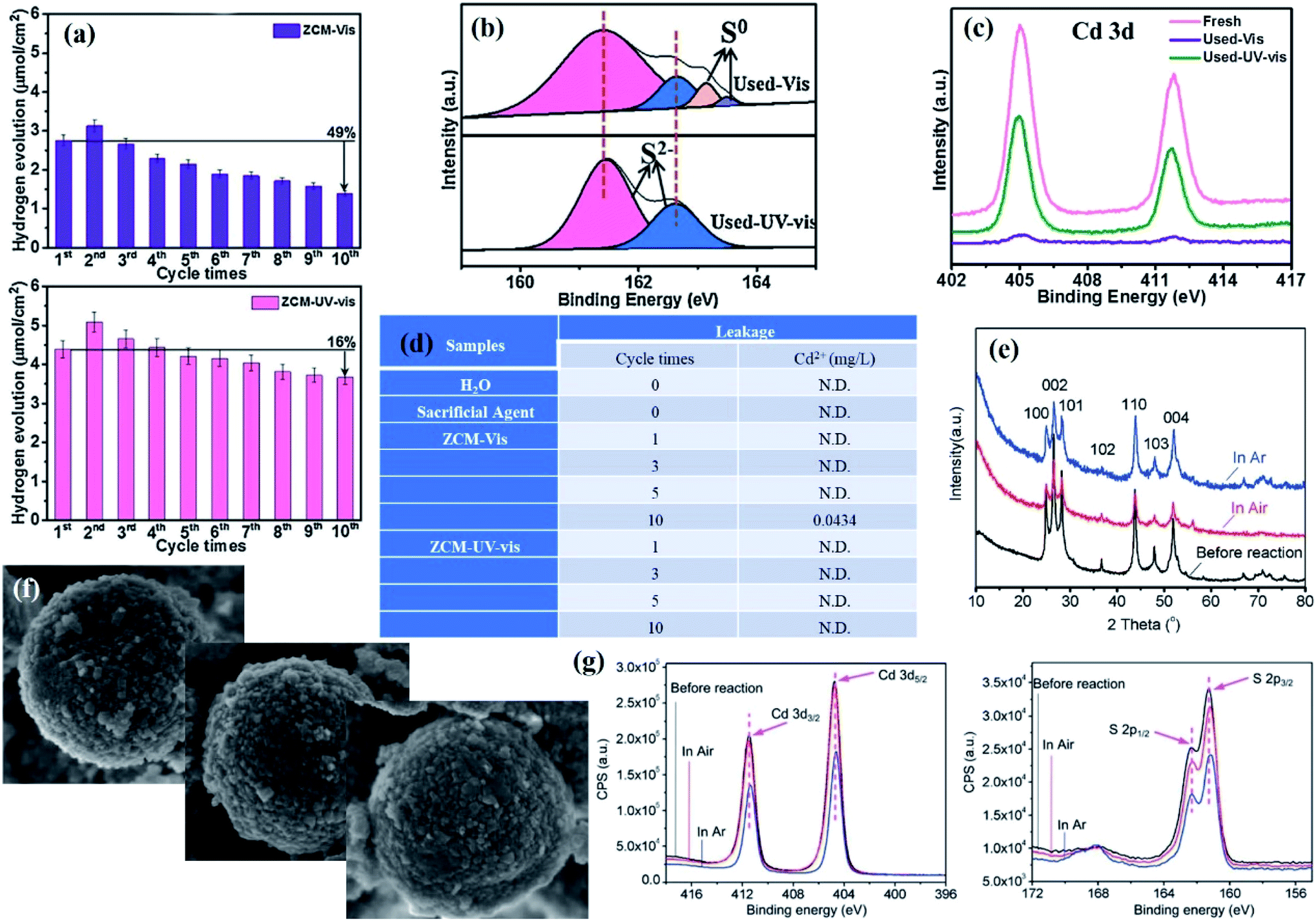 | ||
| Fig. 3 (a) Cycling test of photocatalytic H2 evolution for ZCM under visible light and UV-vis irradiation for 20 h; (b) comparison of S 2p XPS spectra of fresh and used ZCM; (c) XPS survey spectra of Cd 3d for ZCM before and after ten cycles. (d) ICP analysis of Cd2+ leakage in the different samples after different cycles. Reprinted with permission from ref. 55. Copyright 2018, Elsevier B.V. (e) XRD patterns and (f) SEM images of CdS before and after the photocatalytic reaction under air and argon (Ar). (g) XPS spectra of Cd 3d and S 2p for CdS before and after the photocatalytic reaction under air and Ar. Reprinted with permission from ref. 61. Copyright 2015, Royal Society of Chemistry. | ||
In addition to characterizing the changes in morphology, structure, and element valence state of the catalyst before and after the reaction, the use of inductively coupled plasma optical emission spectroscopy (ICP) to determine the concentration and type of ions in the solution after the reaction to evaluate whether the catalyst is dissolved during the photoreaction is also strong evidence to prove the stability of the catalyst.56–58 For instance, since CdS deactivation is caused by the oxidation of S2− by photogenerated holes, monitoring the leakage content of Cd2+ in the reaction process can effectively evaluate the photoreaction stability of CdS-based photocatalysts.59,60 As reported by Xie and co-workers, no detectable Cd2+ leaching was observed after ten cycles in the presence of ZnO–CdS–MoS2 catalysts under UV-vis irradiation, while there was 0.0434 mg L−1 leakage of Cd2+ after visible light irradiation (Fig. 3d), suggesting that the photocorrosion resistance of CdS was greatly enhanced with UV-vis irradiation.55 This difference in stability resulting from the irradiation light source was attributed to the charge transfer following the Z-scheme mechanism under UV-vis irradiation while the type-II charge transfer pathway in the case of visible light irradiation: the former reduced the accumulation of VB holes in CdS, thereby effectively preventing CdS from being oxidized by photogenerated holes.
Although the above characterization techniques can demonstrate photocorrosion of semiconductors to some extent, there is always some photoreaction instability that cannot be demonstrated by conventional characterization due to the uncertainty of the photocatalytic reaction process. For instance, Ma et al. investigated the microstructural changes of CdS before and after the photocatalytic hydrogen evolution reaction with Na2S–Na2SO3 as the electron donor through various techniques.61 No obvious difference was detected between fresh and used CdS by conventional structural characterization such as XRD (Fig. 3e), SEM (Fig. 3f) and XPS (Fig. 3g). However, photocatalytic hydrogen production under argon was much higher than that under air, which indicated that partial photocorrosion of CdS occurred during photocatalytic hydrogen evolution in air, while this change cannot be proven by conventional structural characterization. Since optical phonons can significantly affect the carrier relaxation process and emission efficiency in semiconductor nanostructures, which can be probed by Raman spectroscopy, they further designed a simple photocatalytic system equipped with in situ Raman analysis to monitor the microscopic changes in CdS during the photocatalytic reaction, revealing the relationship between the interfacial crystal lattice and photocorrosion at the spectral level. It was observed that the peak intensity at 500 cm−1 belonging to CdO increased as the reaction proceeded, and the peak intensity at 500 cm−1 was significantly lower under argon than in air during the first 100 min of the reaction, both of which indicated that the surface S atoms were replaced by O (Fig. 4a and b). The increase in lattice stress during the photoreaction in air was attributed to a physically and/or chemically enhanced interaction between absorbed O2 and the photocatalyst surface, which in turn resulted in a lower photocatalytic performance.
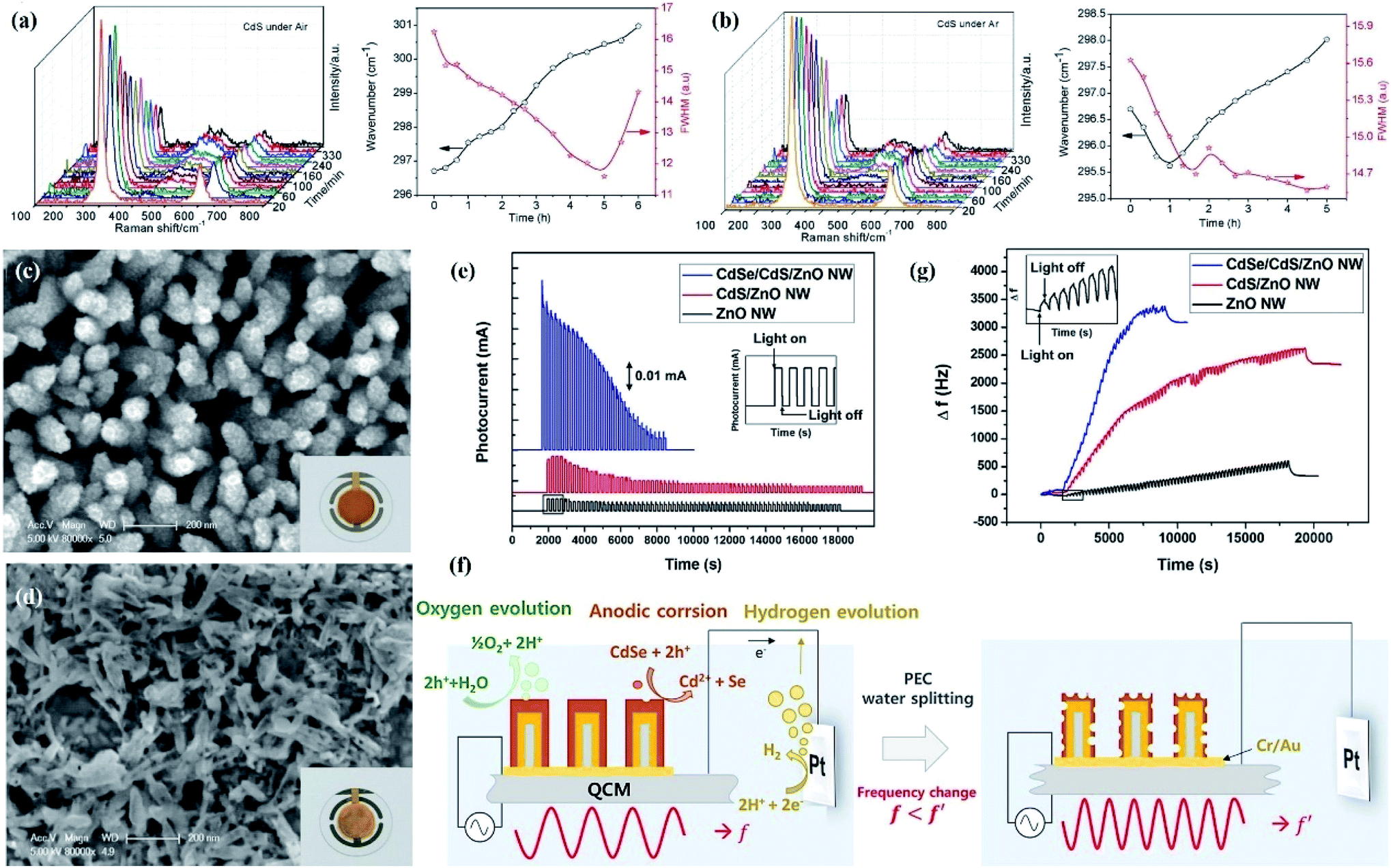 | ||
| Fig. 4 In situ Raman spectra of CdS reacting (a) under air and the corresponding Raman shift and FWHM of the CdS 1LO peak and (b) under Ar and the corresponding Raman shift and FWHM for the 1LO peak of CdS. Reprinted with permission from ref. 61. Copyright 2015, Royal Society of Chemistry. The SEM and schematic images of the fabrication process of CdSe/CdS/ZnO (c) before and (d) after PEC water splitting. Inset images show photographs of QCM grown CdSe/CdS/ZnO; (e) chronoamperometry for ZnO, CdS/ZnO, and CdSe/CdS/ZnO photoanode at an applied potential of 0.0 V vs. RHE under 1 h of sun simulated AM1.5G illumination; (f) schematic diagram illustrating the PEC water splitting process using CdSe/CdS/ZnO grown on QCM; (g) variation in the resonance frequency of ZnO, CdS/ZnO, and CdSe/CdS/ZnO photoanode during PEC water splitting. Reprinted with permission from ref. 67. Copyright 2015, Elsevier B.V. | ||
For the PEC system, the use of narrow bandgap semiconductors, especially quantum dots (QDs) (PbS,62 CdS,63,64 and CdSe QDs65,66) to sensitize photoelectrodes has been widely developed in PEC hydrogen evolution reactions due to the wider light absorption range from UV to visible light and adjustable band gap energy of QDs. Similar to photocatalytic systems, currently developed QDs for sensitized photoelectrodes still have significant limitations, owing to poor photostability and long-term durability. However, considerable studies have proven that the instability of QDs is caused by the anodic photocorrosion since the rate of interfacial hole injection in the electrolyte is slower than the rate at which electrons are transferred to the metal oxide. However, a technique for direct and quantitative analysis of photocorrosion of QDs is still needed. At present, an in situ analysis method is developed to investigate the photocorrosion mechanism of QDs and propose a possible solution to photocorrosion. In a typical example, Choi et al. prepared CdSe/CdS/ZnO nanowire arrays as PEC photoanodes using a quartz crystal microbalance (QCM) as a platform, with simultaneous mass analysis.67 The SEM image showed that the structure of the CdSe/CdS/ZnO photoanode was destroyed after the water splitting reaction, which was due to the anodic photocorrosion of QDs (Fig. 4c and d). The current density–time curve showed that the photocurrent density of CdSe/CdS/ZnO and CdS/ZnO photoanodes decreased sharply with PEC water decomposition until it was consistent with the photocurrent level of the pure ZnO anode (Fig. 4e), which was attributed to anode oxidation caused by photogenerated holes. The photogenerated electrons of QDs can be effectively migrated to ZnO and transfer to counter electrons by electrical connection, while the holes cannot be consumed in a timely manner since the oxygen evolution reaction involving four electrons is more difficult, thereby resulting in accumulated photogenerated holes participating in the anodic corrosion reaction (Fig. 4f). The anodic corrosion in turn caused the decrease in the mass of the quartz crystal microbalance, which can be reflected by the highest resonance frequency variation of CdSe/CdS/ZnO compared to ZnO and CdS/ZnO (Fig. 4g).
Generally, traditional characterization can determine whether the catalyst suffers from photocorrosion during photocatalysis from the changes in morphology, crystal structure, elemental valence of the catalyst before and after the reaction, and composition of the solution after the reaction. However, considering the complexity and uncertainty of the reaction process, further research should focus on the development of in situ monitoring techniques to investigate changes in the morphology and structure of semiconductors in the photoreaction process, which provides more convincing evidence for assessing the photoreaction stability of materials.
4. Strategies to improve the stability of semiconductor-based composites for PC/PEC water splitting
In order to obtain a PC/PEC water splitting system with high activity and long-term stability for practical application, one of the difficulties is how to overcome the photocorrosion of the semiconductor under light irradiation. To date, a considerable number of strategies for this topic have been developed to obtain long-term stable and efficient catalysts. Therefore, the focus of this section is on systematically elaborating various strategies developed to improve the stability of semiconductor composites, such as optimizing the physical/chemical properties of the semiconductor, foreign atom doping, constructing heterojunctions, loading with cocatalysts, and adjusting reaction conditions.4.1. Crystal structure, morphology and size control
Due to the structure-determining function, optimizing the physical properties of the semiconductor can result in an improved stability under light irradiation.68 In most cases, increasing the crystallinity of the semiconductor is beneficial to enhance the photocatalytic activity and stability because the increase in crystallinity can promote the charge transfer and reduce the number of recombination centers of photogenerated electrons and holes. In addition, the morphology and size of the semiconductor depend on its physical and/or chemical properties,69,70 which in turn determines the photocatalytic activity and stability of the catalyst. Accordingly, considerable research studies have focused on designing materials with the desired structural morphology to prevent photo-induced instability in various photocatalytic applications.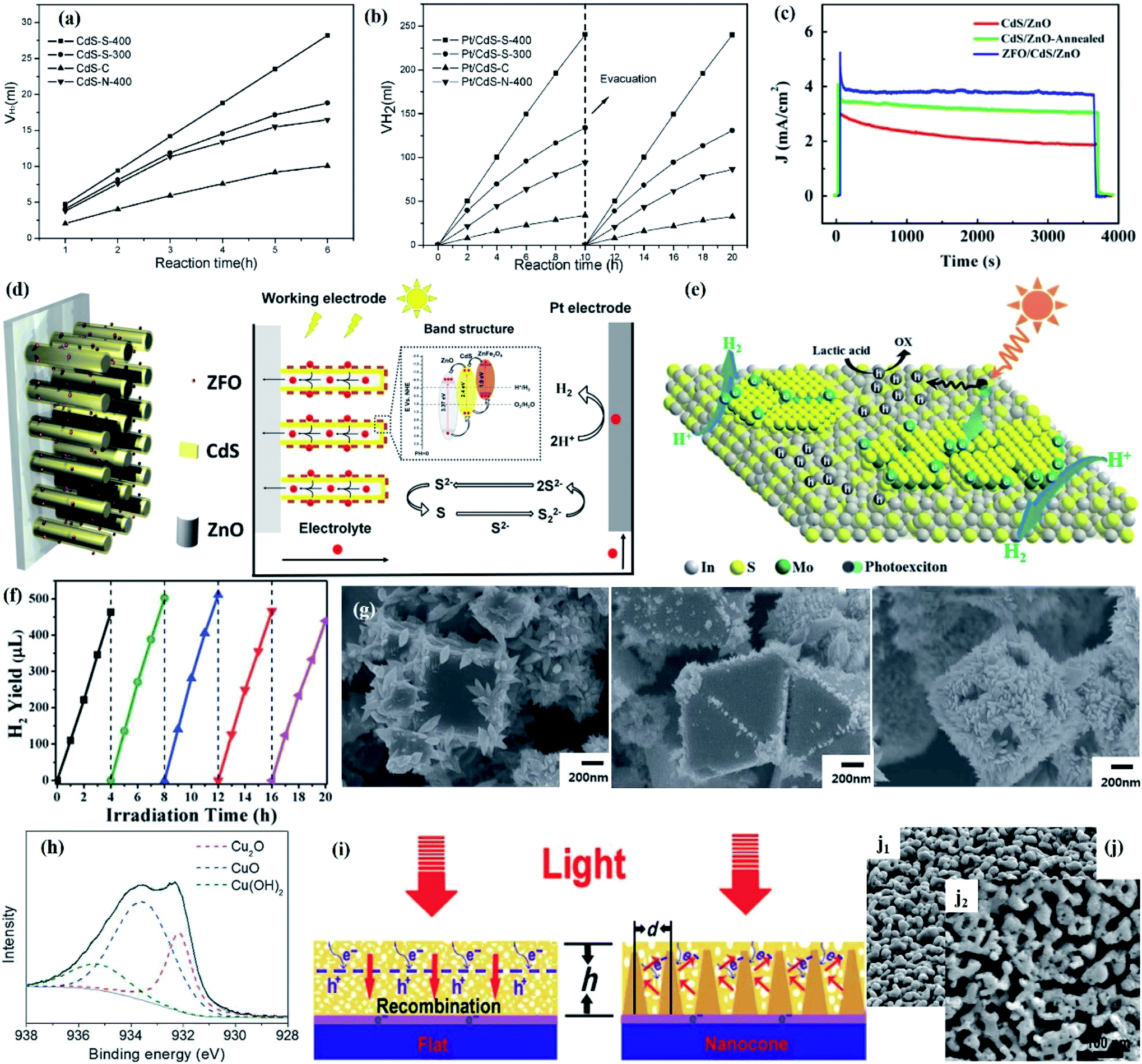 | ||
| Fig. 5 Amount of H2 evolved vs. irradiation time on various CdS samples (a) without Pt loading and (b) with 2 wt% Pt loading. Reprinted with permission from ref. 71. Copyright 2006, American Chemical Society. (c) Stability of CdS/ZnO, annealed CdS/ZnO and ZFO/CdS/ZnO photoanode under illumination; (d) 2D schematic of the ZFO/CdS/ZnO photoanode and the energy band structure of the ZFO/CdS/ZnO heterojunction. Reprinted with permission from ref. 72. Copyright 2016, Elsevier B.V. (e) Photocatalytic mechanism of H2 evolution in the dual-defective MoS2/In2S3 photocatalyst system; (f) cycling test for the IM-1% sample. Reprinted with permission from ref. 73. Copyright 2016, Royal Society of Chemistry. (g) SEM images of cubic, octahedral, and rhombic dodecahedral Cu2O dispersed in deionized water under light irradiation for 9 h; (h) XPS Cu 2p3/2 peak for rhombic dodecahedral Cu2O. Reprinted with permission from ref. 86. Copyright 2015, Royal Society of Chemistry. (i) Schematic illustration of the optical absorption mechanism and electron transport of BiVO4 on the flat substrate and the conductive nanocone substrate; (j) SEM image of Mo:BiVO4 Fe(Ni)OOH before (j1) and after (j2) 10 h of PEC test. Reprinted with permission from ref. 88. Copyright 2016, Science. | ||
However, excellent crystallinity is not always beneficial for improving photocatalytic performance and stability. In a typical example, Fang et al. proposed that dual-defective construction in which defective few-layered MoS2 was grown in situ on defective layered In2S3 could also obtain remarkable performance and impressive photostability.73 The existing abundant disordered crystal structures broke the strict crystal anisotropy of In2S3 and formed homogenous bond situations in all orientations. Therefore, the vast majority of In2S3 molecules remained at similar energy levels, with the exception of those relatively unstable positions occupied by the MoS2 cocatalyst. Under illumination, the photo-excitons generated in the crystallite region extend throughout the In2S3 layer. When concentrated in intimate binding sites, photogenerated electrons expediently flew through the interface channel to defective MoS2 layers that possessed less negative CB potential than In2S3 and readily reduced H+ to H2, while the holes cannot easily find energy-unbalanced bonds to attack to cause photocorrosion due to the isotropy of the disordered structures. In contrast, the holes continued to travel in In2S3 until being consumed by the sacrificial reagents to induce oxidized products (Fig. 5e). Therefore, a remarkable performance and impressive photostability for photocatalytic hydrogen production can be obtained through such dual-defective construction (Fig. 5f).
Besides, in view of the non-directionality in the semiconductor growth process, it is difficult to accurately control the size of a semiconductor simply by optimizing the reaction parameters in the synthesis process. Therefore, the use of the structural characteristics of the host material to orientate the target semiconductor is a feasible strategy to achieve size controllability, with mesoporous materials being the preferred host material.78–80 Taking CdS clusters as an example, theoretically, confining CdS clusters in a porous support can enhance its photostability by reducing the exposed area of CdS and control the size of CdS due to quantum confinement which resulted in a negative shift of the CB edge. As a result, semiconductors with small dimensions allow charge carriers to diffuse rapidly to surface active sites, thereby minimizing the bulk recombination of electron–hole pairs in CdS clusters. In addition, the host porous material provides additional electron transfer channels to promote the separation of charge carriers and further improve the photocatalytic performance. In this regard, Peng et al. prepared CdS incorporated Ti-MCM-48 mesoporous materials for overall water splitting with ethanol as sacrificial electron donor under 300 W Xe lamp irradiation. XPS analysis showed that no obvious difference in the intensity of Cd and S was detected between fresh and used catalysts, suggesting that the stability of CdS was effectively enhanced after encapsulation in the MCM-48 matrix, thereby achieving high water splitting efficiency after three cycles.81
In the case of PEC systems, coating the photoelectrode with a protective overlayer to avoid direct contact of the electrode with the electrolyte is a viable strategy to obtain enhanced stability. The thickness of the protective overlayer depends on the carrier transport lifetime, and the thickness of currently reported photoanode is typically less than 200 nm in order to compensate for the short carrier diffusion length. However, thin protective overlayers usually have high transparency, which allows most of the visible light to pass through, resulting in poor light absorption. In order to improve the light absorption efficiency, depositing a thick protective overlayer on the photoanode by engineered architectures might be a feasible strategy. In a typical example, Qiu et al. constructed an engineered cone-shaped nanostructure to achieve the deposition of an approximately 700 nm-thick nanoporous Mo-doped BiVO4 layer, which effectively overcame the barrier of incompatibility between light absorption capability and carrier transfer distance (Fig. 5i).88 The total thickness of the BiVO4 film was increased after constructing the nanocone arrays due to the shortened charge transfer distance which allowed for efficient collection of charge around the conductive nanocones. Moreover, the unique structure allowed light to be scattered multiple times inside to increase the optical path, thereby further improving the light absorption capacity. This cone-shaped nanostructure resulted in an excellent stability, which could be demonstrated by the stable perovskite solar cell after 10 h and unchanged morphology between the fresh and used sample (Fig. 5j). Moreover, embedded/hierarchical catalysts with a sandwich structure have recently been developed to prevent electrode photocorrosion. For example, Li et al. reported that the stability of Si nanowires could be greatly improved after successively depositing Pt NPs and a TiO2 passivated layer to construct a sandwich structure.89 A more complex sandwich structure was also employed by Yang et al. to improve the stability of black Si (bSi) photoelectrodes, that is, a 20 nm-thick TiO2 layer and Pt NPs were deposited on top of nano-porous Si, followed by the deposition of a 2 nm-thin second TiO2 layer. The prepared photoelectrodes with such a dual-TiO2-layer sandwich structure exhibited more excellent stability than a single (TiO2/Pt/bSi, Pt/TiO2/bSi) or non-protective layer (Pt/bSi).90 Other photoelectrodes with a similar sandwich structure were also reported by the Chidsey group.91 Furthermore, Gu and co-worker designed a GaInP2 photocathode with a graded catalytic-protective layer by annealing an amorphous-MoSx/TiOx-GaInP2 stack at 450 °C.92 Annealing treatment resulted in a large degree of mutual penetration between MoSx, TiO2 and GaInP2, while there was a relatively clear boundary between the components before annealing. As a result, the annealed MoSx/TiOx-GaInP2 photocathode showed an enhanced stability compared to the sample without annealing.
4.2. Doping effects
Heteroatom doping is regarded as an efficient strategy to enhance the performance and stability of semiconductors since the doping of foreign elements can alter the band gap of the semiconductor by introducing an impurity level in the forbidden band or forming a solid solution, which extends its light absorption range and promoted the separation efficiency of electron–hole pairs, thereby reducing the possibility of photocorrosion caused by excited electrons or/and holes.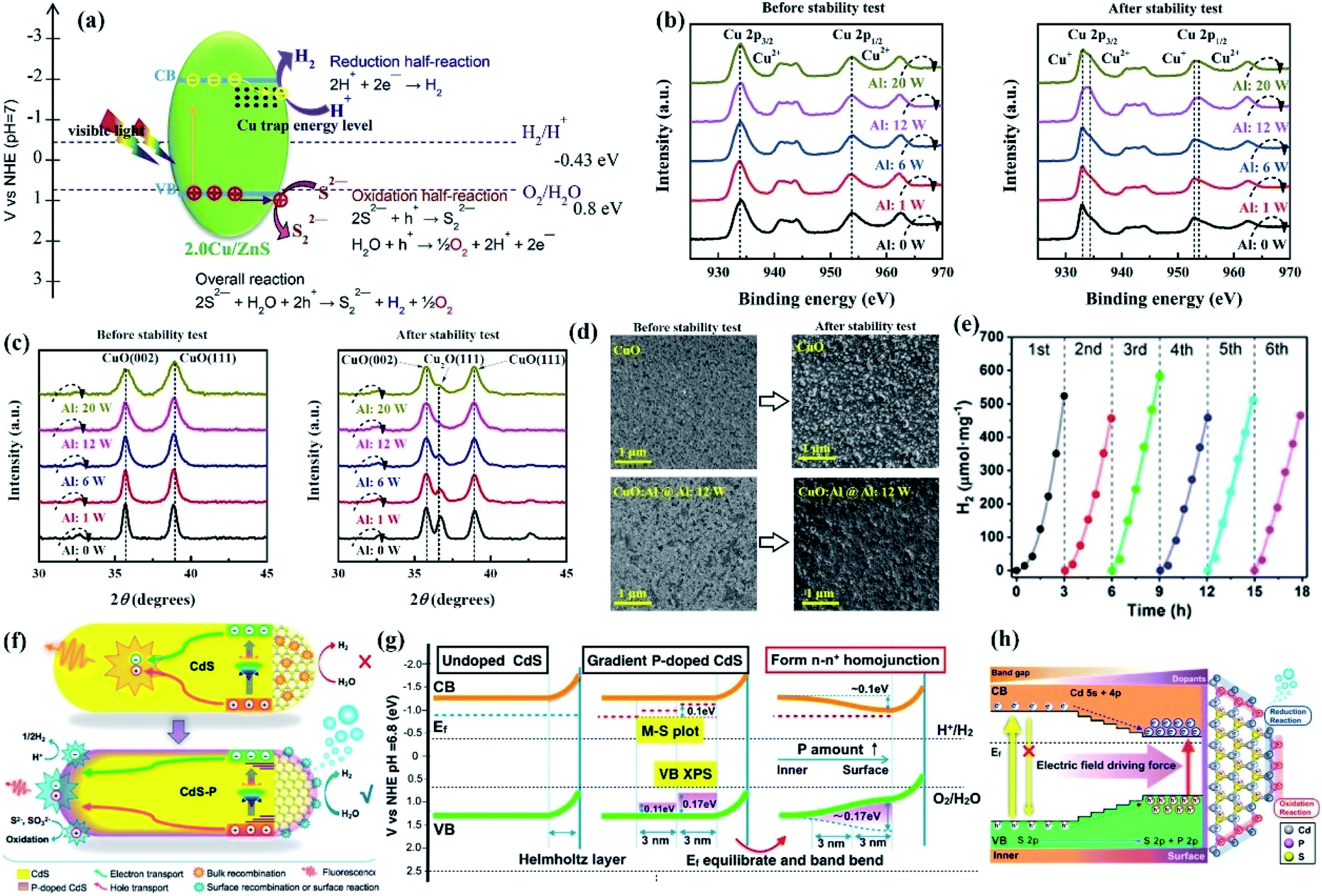 | ||
| Fig. 6 (a) Illustration of the proposed mechanism of Cu-doped ZnS microspheres for photocatalytic hydrogen generation under visible light irradiation. Reprinted with permission from ref. 93. Copyright 2016, Elsevier B.V. (b) Cu 2p XPS and (c) XRD spectra of p-(CuO/CuO:Al) photocathodes before and after the photocorrosion stability test; (d) top-view SEM images of the CuO and CuO:Al (@Al:12 W) photocathodes before and after the photocorrosion stability test. Reprinted with permission from ref. 20. Copyright 2018, Royal Society of Chemistry. (e) Cycling runs of CdS–P1 for photocatalytic H2 evolution; (f) possible carriers transfer in P-doped CdS NRs leading to efficient photocatalytic H2 evolution; (g) the band structure evolution for the formation of homojunctions. (h) Schematic energy level and exciton transfer pathways in the CdS–P homojunction with a gradient distribution of P element from the interior to surface of the catalyst. Reprinted with permission from ref. 95. Copyright 2017, American Chemical Society. | ||
For PEC systems, it has been demonstrated that coating a protective layer on the surface of the photocathode can improve its durability, while it is also limited by a duration of less than one hour. Masudy-Panah reported that long-term stability was achieved in Al-incorporated CuO photocathodes because the photogenerated electrons of CuO could be effectively transferred from the photocathode to the electrolyte, thereby preventing CuO from being reduced by photogenerated electrons.20 The XPS spectra (Fig. 6b) and the XRD peaks (Fig. 6c) of the photocathodes after the stability test showed that the peak of CuO at binding energies of 932.7 and 952.8 eV and the Cu2O XRD peak intensity at a 2θ of 36.45° of the CuO:Al photocathode were much lower than those of the CuO photocathode, suggesting that the incorporation of Al into CuO curbed the reduction of CuO and resulted in improved photoreaction stability. The SEM image of the CuO and CuO:Al photocathodes after the stability test shows the same conclusion (Fig. 6d).
Moreover, since the photocorrosion of ZnS results from the oxidation of lattice S2− by excited holes, lowering the oxidation ability of VB holes is a thermodynamically feasible method to improve the photocorrosion resistance of ZnS. In addition, the energy of the N 2p orbital is higher than that of the O 2p orbital; substituting S with N sites of metal sulfides can reduce the band gap by increasing the VB potential, thereby lowering the oxidizing capability of holes. In this regard, Zhou et al. prepared N-doped ZnS by nitriding ZnS powder in an NH3 atmosphere.96 DFT calculations revealed the VBM of ZnS was increased after nitrogen heteroatoms occupied the S sites, but the CBM still basically consisted of the Zn 4s states and small S 3p states with a small position change. That is, the CB electrons still maintained high reduction ability while the oxidative capacity of holes was weakened, which effectively inhibited the hole-induced photocorrosion of ZnS, resulting in less dissociation in aqueous solution for the N-doped ZnS sample. As a result, N-doped ZnS showed excellent stability for photocatalytic hydrogen evolution under simulated sunlight irradiation for 12 h.
4.3. Hybridization with cocatalysts
It is generally believed that the cocatalyst can act as a reaction site and catalyze the surface redox reaction, promoting charge separation and transfer driven by the interface generated between the cocatalyst and semiconductor. Loading suitable cocatalysts with a larger work function for electron sinks or lowering the overpotential for hole transfer on the semiconductor can effectively alleviate the photocorrosion of the composites, which is due to the fact that the photogenerated charge carriers and accumulated active sites can be consumed in a timely manner or transferred.99–103 In this section, how the cocatalyst enhances the stability of semiconductor photocatalysts in various photocatalytic reactions will be systematically discussed.However, metal cocatalyst loading also has some disadvantages, that is, the metal NPs are generally deposited on the surface of the semiconductor, which causes the metal NPs to be directly exposed to the surrounding reaction environment, resulting in the possibility of metal dissolution during the reaction. As a result, metal NPs may be dissolved during the reaction, resulting in a decrease in photocatalytic performance and stability.115,116 In addition, the metal cocatalysts are detached from the semiconductor domains and are severely aggregated due to the oxidation of the interface by photogenerated holes in the reaction process. In order to overcome the above problems, Choi proposed an effective physical separation between the reduction and oxidation sites by constructing a double shell structure (Fig. 7a).117 The reaction activity of Pt–CdS double shell hollow nanocubes remained at its initial value for up to 20 h without deactivation (Fig. 7b), while the original morphology of the hollow nanocubes was retained (Fig. 7c). In contrast, the performance of Pt–CdS spheres, in which Pt was photo-deposited on commercial CdS spheres, dropped by 50% over 20 h (Fig. 7b), which was due to the detachment and aggregation of Pt from the CdS surface. The improved stability of the Pt–CdS hollow nanocubes indicated that such a double shell structure could effectively consume photogenerated holes from the sacrificial reagents through the external CdS surface. The concept of physical separation was also reported by Jiang et al., where the activity and stability of Cu2ZnSnSe4 regarding PEC water splitting were greatly improved after coating with an In2S3/CdS double layer and the deposition of Pt particles.118 The surface coating of the In2S3 layer acted as a mediator for efficient electron transfer to Pt deposits and a protecting layer to avoid contact between the CdS layer and outer electrolyte solution, resulting in excellent durability. As shown in Fig. 7d–g, an obvious dissolution was detected for Pt/CdS/Cu2ZnSnSe4 after the durability test, while the morphology and structure were well maintained before and after reaction in the case of Pt/In2S3/CdS/Cu2ZnSnSe4, indicating that the photostability of CdS can be effectively improved by coverage with an In2S3 layer to avoid direct contact of the CdS layer with an electrolyte.
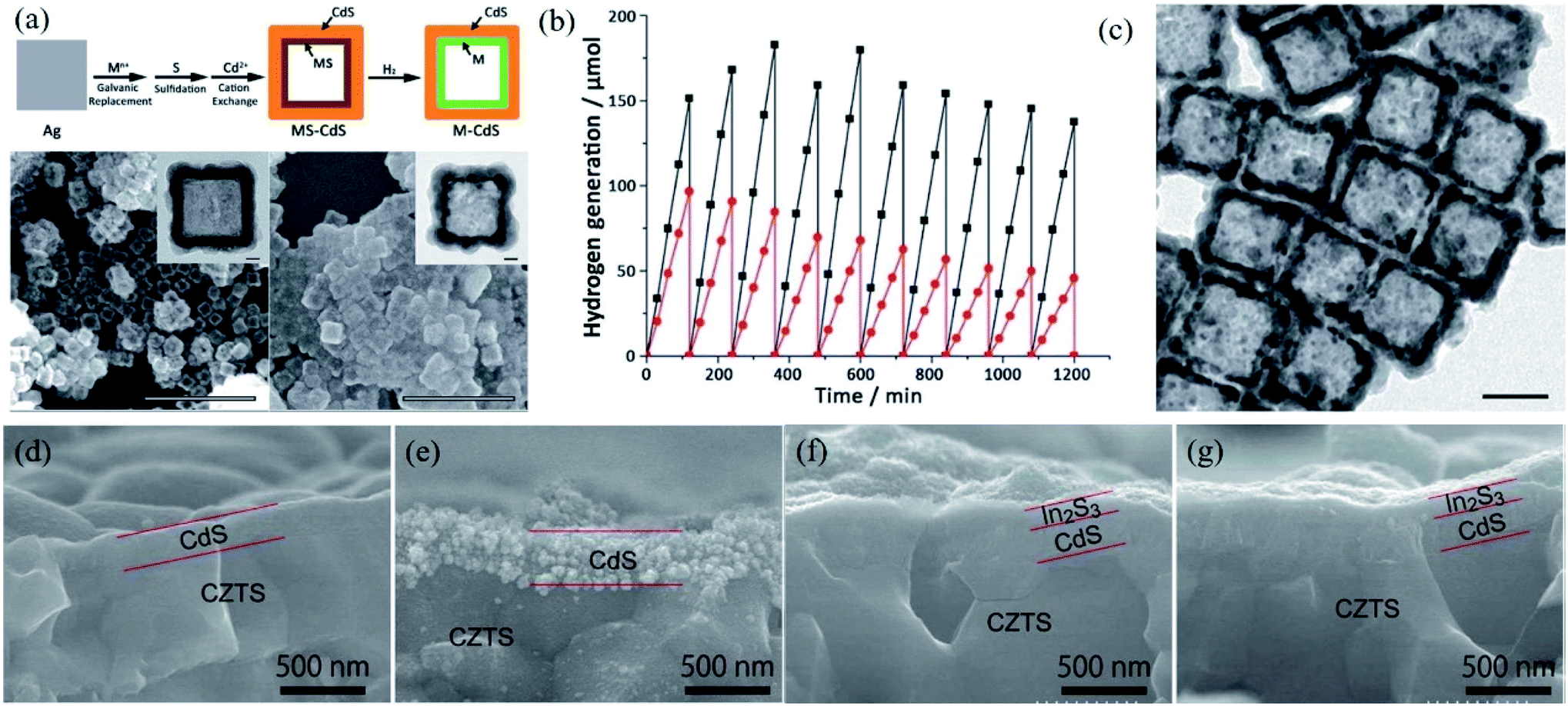 | ||
| Fig. 7 (a) General synthetic approach of M–CdS double shell hollow nanocubes (M ¼Pt, Au, and Pd) and SEM and TEM images of PtS–CdS and Pt–CdS hollow nanocubes; (b) time course of H2 generation using Pt–CdS double shell hollow nanocubes (black) and commercial CdS platinized by photo-deposition (red); (c) TEM image of Pt–CdS hollow nanocubes after 10 cycles of H2 generation. Reprinted with permission from ref. 116. Copyright 2016, Royal Society of Chemistry. (d–g) Cross-sectional SEM images of (d, e) Pt/CdS/CZTS and (f, g) Pt/In2S3/CdS/CZTS photocathodes before (d, f) and after (e, g) a 3 h durability test. Reprinted with permission from ref. 117. Copyright 2015, American Chemical Society. | ||
However, for hole-sensitive semiconductors such as ZnO, CdS, and Cu2O, not only can metal modification accelerate the evacuation of electrons from the semiconductor, but it is also easy to overlook the negative effect of hole accumulation on the stability of these semiconductors. However, the accumulated holes can be consumed in time by the sacrificial agent and/or the reactants in the reaction system, which can suppress the hole-induced dissolution process. However, if the photocorrosion rate caused by accumulated holes is faster than the rate at which it is consumed by the sacrificial agent and/or reactant, severe photocorrosion of the hole-sensitive semiconductor will occur. In view of this, the photoreaction stability of the hole-sensitized semiconductor can be improved as follows: (i) providing sufficient sacrificial agents or increasing the adsorption capacity of the reactants to improve the rate and efficiency of hole consumption; (ii) introducing a hole-cocatalyst for timely evacuation of holes accumulated in the VB of hole-sensitive semiconductors to prevent hole-induced photocorrosion.
Nevertheless, considering that noble metals are expensive and not readily available, the focus of research has gradually shifted from noble metals to the exploration of low-cost cocatalysts,119 in which earth-abundant transition metals such as Co(II),120 Cu(II),121 and Fe(III)122 are considered to be ideal cocatalysts for improving the photoreaction stability of semiconductors. This improved photoreaction stability is attributed to surface-loaded transition metal ions that can act as cocatalysts to accept excited electrons from the semiconductor, thereby inhibiting the recombination of electrons and holes, which in turn improves photocatalytic performance and stability.
Recently, Yuan and co-workers reported that the photocatalytic activity and stability of CdS nanorods could be improved by loading with Co3O4 cocatalysts, where Co3O4 accelerated the consumption of VB holes in CdS, resulting in an increase in the number of available photogenerated electrons. As a result, no significant decrease in hydrogen production was found even after three cycles under visible light irradiation with Na2S–Na2SO3 as the sacrificial reagent.129 In addition to Co3O4, cobalt oxide species CoOx containing both Co(II) and Co(III) is an emerging cocatalyst for rapidly trapping holes, thereby prolonging the lifetime of photogenerated electrons and holes.120 For example, Liu et al. prepared CdS/cobalt oxide core–shell nanorods (CdS/CoOx NRs) by an impregnation-calcination process for photocatalytic hydrogen evolution under a N2 atmosphere using Na2S–Na2SO3 as the sacrificial agent.130 The intimate contact of CdS NRs with the CoOx shell reduced the interfacial resistance of the VB holes of CdS NRs to the CoOx shells, and CoOx as a hole cocatalyst can accept VB holes of CdS NRs, both of which alleviated the hole-induced photocorrosion of CdS semiconductors. As a result, a relatively less decrease in hydrogen production was obtained in the CdS/CoOx system (38%) (Fig. 8a) compared with single CdS (55%) (Fig. 8b). Unlike PC systems, the separation of the photogenerated charge carriers of the PEC system can be improved by inserting a multifunctional layer underneath the electrochemical catalyst. For instance, Chen et al. prepared hierarchical TiO2@CoNi-layered double hydroxide nanotube arrays (TiO2@CoNi-LDHs NTAs) for PEC water splitting (Fig. 8c), which exhibited an excellent long-term photocorrosion resistance, with no more than 1.0% loss of the initial activity even after 10 h of irradiation in a Na2SO4 electrolyte solution (Fig. 8d and e).131 This excellent stability was attributed to the fact that the hierarchical structure facilitated the transfer of excited holes toward the LDHs via CoII–CoIII, CoIII–O2, and H2O oxidation reactions, which in turn protected the surface from photocorrosion and resulted in improved stability. Vijselaar et al. applied a spatioselective method to prepare Si microwire photocathodes with a nickel silicide (NiSi) interlayer between the catalyst and Si microwires.132 Such a design facilitated electron transfer to the NiMo catalysts and simultaneously minimized light intervention. As a result, the Si photocathodes achieved a stable performance in strongly alkaline solutions (1 M KOH). Besides, Guo et al. reported that stable PEC activity was obtained by the introduction of a Ni/NiOx bridging layer between NiFe-LDH and Si nanowires, wherein NiOx with a high ability for hole accumulation and strong binding with NiFe-LDH facilitated the transfer of holes to the catalysts.133 As a result, the NiOx-containing catalysts achieved long-term stability for 60 h in 1.0 M KOH.
 | ||
| Fig. 8 (a) Stability test of CC-20 and (b) pristine CdS. The amount of hydrogen produced on CC-20 dropped to 520.6 μmol after 4 cycles of cycling tests, while that of pristine CdS dropped to 9.0 μmol. Reprinted with permission from ref. 130. Copyright 2018, Elsevier B.V. (c) Schematic illustration of the preparation of the hierarchical TiO2@CoNi-LDHs NTAs via an anodization and subsequent electrodeposition process for highly efficient and long-term stable PEC water splitting; (d) H2 and O2 evolution curves from the hierarchical TiO2@CoNi-LDH NTA electrodes at 1.23 V versus RHE under AM 1.5G for different cycles. (e) The steady-state photocurrent curve of the typical hierarchical TiO2@CoNi-LDH NTA electrode under AM 1.5G illumination at 1.23 V versus RHE for different cycles. Reprinted with permission from ref. 131. Copyright 2016, Wiley-VCH. (f) Schematic theories and the corresponding verification for the photocorrosion-recrystallization strategy of CdS in the 0D/2D CdS/MoS2 nanocomposite; (g) morphology change of the CdS/MoS2 nanocomposite at different visible light irradiation times; (h) H2 evolution during the photocatalytic stability study for over 400 h. Reprinted with permission from ref. 139. Copyright 2016, Elsevier B.V. | ||
Other transition metal compounds, transition metal sulfides such as MoS2 (ref. 134 and 135) and WS2,136,137 are also regarded as an inexpensive alternatives to noble metals to improve the activity and stability of semiconductors for solar energy conversion. For instance, Hao's group reported that the photocorrosion of CdS could be effectively inhibited through a photocorrosion-recrystallization process.138 A comprehensive analysis of crystallization thermodynamics and kinetics indicated that the establishment of a pathway for the reduction of elemental S to S2− or the design of a new system with a large recrystallization reaction constant (k) can inhibit the photo-induced instability of CdS. Since heterogeneous nucleation, especially that of an irregular concave surface, can provide active sites with high nucleation energy compared to homogeneous nucleation, Ma et al. designed and prepared a novel CdS/MoS2 nanocomposite using efficient electron mobility and S vacancies in MoS2 nanosheets.139 The dissociated S elements adsorbed on the surface of the MoS2 nanosheets can be reduced to S2− using lactic acid as a sacrificial agent. Since MoS2 nanosheets, especially the edges and defective regions of these sheets, can provide active sites with high nucleation energy and a large k value for the crystallization of CdS according to heterogeneous nucleation theory, Cd2+ and S2− can recrystallize and CdS grows as solid droplets on the defects and edges of large-scale MoS2 nanosheets (Fig. 8f). The formation of new CdS NPs in the as-designed CdS/MoS2 nanocomposites, followed by the photocorrosion-recrystallization pathway, can effectively prevent the photocorrosion process of CdS (Fig. 8g). When the photocorrosion-recrystallization process reached an equilibrium state, a final CdS/MoS2 nanocomposite with excellent activity and stability was obtained, and the hydrogen evolution rate had no obvious decrease for over 400 h of irradiation (Fig. 8h). Similarly, a CdS/WS2 photocatalyst was also prepared through the photocorrosion-recrystallization strategy since WS2 and MoS2 have extremely similar crystal structures and chemical properties, and as a result, the hydrogen production was maintained at a high level even after irradiation for 200 h.138 Similar in situ catalyst regeneration is also applied to improve the stability of the PEC system. As reported by the Domen group, the photocurrent remained at a high level up to 400 h for a NiFe oxygen evolution catalyst (OEC)-decorated BiVO4 photoanode (NiFe-OEC/Mo:BiVO4/Ni/Sn) with Ni as the contact layer.140 In contrast, an obvious decrease in the photocurrent was detected after 20 h in the NiFe-OEC/Mo:BiVO4/Ti/Sn photoanode with Ti as the contact layer. This difference in stability was attributed to the self-regeneration of NiFe-OEC by the dissolution of Ni2+ in the Ni contact layer, while the Ti contact layer cannot.
As previously described, the oxidative corrosion of hole-sensitive semiconductors can be effectively inhibited by loading with electron cocatalysts; in view of this, MoS2 might be a good choice. For instance, Yuan and his co-workers prepared core–shell structure MoS2–ZnO photocatalysts for hydrogen evolution with Na2S–Na2SO3 as the sacrificial agent.141 Only ca. 6% decrease in hydrogen production compared to that of the fresh sample was detected after three cycles, and no difference in the XRD pattern was found between fresh and used MoS2–ZnO catalysts, indicating an excellent photocatalytic stability for photocatalytic hydrogen production. A similar core–shell structure was also applied in a ternary photocatalytic system, as reported by Guan et al.; the ternary MoS2-RGO/ZnO composite prepared through a two-step solvothermal pathway exhibited long-term stability for photocatalytic hydrogen evolution with Na2S–Na2SO3 as the sacrificial agent.142 The stability experiments showed that ca. 93% hydrogen production compared to that of the first cycle was retained after 12 h of reaction, which was much higher than that of single ZnO, indicating improved stability under light irradiation. In order to investigate the difference in structure between fresh and used samples, XRD characterization was carried out. However, no obvious change in the XRD pattern was found before and after reaction for both single ZnO and MoS2-RGO/ZnO composites, which confirmed the shortcomings of some of the characterization studies described in Section 3. Further XPS analysis of single ZnO can demonstrate the presence of S2− ions, speculating that some of the ZnO was oxidized by holes to ZnS in the presence of a sulfide sacrificial agent, resulting in a relatively poor photocatalytic stability.
Similar to cobalt oxide species, molybdenum sulfide species MoSx, a robustly bound co-catalyst, is applied as a cocatalyst to improve the stability of the PEC water splitting system. For instance, Prabhakar et al. prepared Sb2Se3–MoSx catalysts through a low temperature sulfurization process, which had high photocurrent densities and stability in strongly acidic media (1 M H2SO4). The improved stability was attributed to the fact that the ionization potential of Sb2S3 was higher than that of Se, which resulted in a VB offset at the interface of Sb2S3/Sb2Se3. This offset acted as a hole-blocking layer to avoid interface recombination, endowing the Sb2Se3 thin film with long-term stability in strong acid for 20 h without any photocorrosion.143 Morales-Guio reported that a Cu2O photocathode decorated with MoS2+x exhibited a higher stability compared with the Pt-decorated Cu2O photocathode in a wide range of pH.144
Some other transition metal compounds are also regarded as efficient co-catalysts to improve the stability of photocatalytic reactions in semiconductor-based composites. For instance, core–shell Co(OH)2/CdS with Co(OH)2 decorated with CdS nanowires was prepared by the impregnation synthetic method and exhibited long-term stability for hydrogen evolution, and the hydrogen production was up to 7.52 mmol h−1 g−1 after irradiation with visible light for 12 h.145 This stability was attributed to the fact that Co(OH)2 could effectively consume the CB electrons of CdS to generate Co+ and Co atoms, resulting in an enhanced charge separation efficiency. Transition metal phosphides are similar to zero-valent metals in properties, which exhibit high activity and stability in PC/PEC water reduction regardless of acid or alkali conditions. In this regard, Cao et al. prepared CdS with TMP NP (CoP, Ni2P and Cu3P) cocatalysts loaded for photocatalytic hydrogen evolution with L(+)-lactic acid as the electron donor, of which CoP/CdS composites exhibited the highest and most stable performance.146 The durability tests showed that only a slight decrease in hydrogen production was detected after 50 h of irradiation, suggesting that CoP cocatalysts could greatly improve the photoreaction stability of CdS. In addition, as reported by Cheng et al., FeP/CdS nanorods prepared by grinding showed an excellent stability for photocatalytic hydrogen evolution using lactic acid as a sacrificial reagent.147 FeP/CdS catalysts with 5% FeP loading showed impressive photochemical stability even after irradiation for up to 100 h. The improved stability was attributed to the band bending between CdS and FeP, which promoted the separation of electron–hole pairs.
Furthermore, recent studies have indicated that metallic Ti3C2 can be used as a cocatalyst to improve the mobility of charge carriers within the semiconductors due to the formation of Schottky junctions. In a typical example, Xie et al. prepared a two-dimensional (2D) in-plane CdS/Ti3C2Tx sheet-onto-sheet heterojunction, which not only maximized the CdS–Ti3C2Tx interlayer interaction, but also ensured open surface reaction sites for photocatalysis.148 The stability experiments showed that no significant change in conversion efficiency was found for CdS/Ti3C2Tx, while a significant decrease was detected for single CdS (Fig. 9a). The S 2p XPS peak showed that the polysulfide ratio of CdS (26.0%) was higher than that of CdS/Ti3C2Tx (17.1%) (Fig. 9b), suggesting the retarded light-induced instability of CdS by the modification of Ti3C2Tx, which can also be proved by Cd2+ leakage (Fig. 9c). This enhanced photostability was attributed to the strong interaction between Cd2+ and Ti3C2Tx. The Cd2+ released by CdS photocorrosion were in situ confined on the interlayer interacted Ti3C2Tx, which was demonstrated by the Cd2+ adsorption results (Fig. 9d). As a result, not only could Cd2+ leaching be effectively prevented, but the surface reaction sites were also not damaged. Furthermore, given that one-dimensional (1D) nanostructures with high length-to-diameter ratios are in favor of light adsorption and charge transfer, Xiao and co-workers prepared 1D CdS nanorod/2D Ti3C2 heterojunctions with interfacial coupling for photocatalytic hydrogen evolution.149 It was found that the photocatalytic activity and stability of CdS with respect to hydrogen evolution were obviously improved after the introduction of Ti3C2, with negligible decrease in hydrogen production after five cycles. This enhanced activity and stability was attributed to the fact that the Schottky junction at the interface of CdS and Ti3C2 can accelerate charge separation and transfer within CdS nanorods.
 | ||
| Fig. 9 (a) Photostability of the pure CdS nanosheets and CdS-0.5% MXene for the photoreduction of 4-NA; (b) high resolution XPS spectra of S 2p for the used pure CdS nanosheets and CdS-0.5% MXene after photocatalytic reduction of 4-NA; (c) Cd2+ leakage profiles under visible light irradiation; (d) adsorption of Cd2+ over CdS nanosheets and Ti3C2Tx MXene in the dark. Reprinted with permission from ref. 148. Copyright 2018, Elsevier B.V. (e) Schematic illustration for the chemical anchoring of Co/Ni species on the CdS surface through the controlled decomposition of an M-EDTA precursor and the corresponding HRTEM, EDS elemental mapping and STEM images of the resulting CdS-CoE-300 and CdS-NiE-350; cycling test of photocatalytic H2 evolution over (f) CdS-CoE-300 with 0.7% mole proportion of Co-EDTA to CdS and (g) CdS-NiE-350 with 0.7% mole proportion of Ni-EDTA to CdS. Reprinted with permission from ref. 151. Copyright 2017, Wiley-VCH. (h) XRD patterns and (i) TEM images of Pt–PdS/CdS samples after reaction for 1 h and 25 h; (j) photocatalytic activity of H2 evolution over several other metal sulfide (MS) photocatalysts with the loading of Pt, PdS and Pt–PdS. Reprinted with permission from ref. 152. Copyright 2012, Elsevier B.V. | ||
From an economic point of view, the replacement of precious metals with alternative rich non-precious metal promoters is in line with the concept of sustainable development. For instance, Yu' group prepared dual amorphous TiO2 and Ni(II) co-modified CdS catalysts (Ti(IV)–Ni(II)/CdS) for hydrogen evolution using Na2SO3–Na2S as the sacrificial reagent under visible light irradiation.154 Cycling tests showed that as the cycles increased, the hydrogen production of a single CdS decreased gradually, while Ti(IV)–Ni(II)/CdS maintained a high stable hydrogen evolution under identical conditions, suggesting that the stability of CdS was improved with the co-modification with amorphous Ti(IV) and Ni(II). This excellent stability was due to the fact that the holes could be consumed in a timely manner by the hole-cocatalyst Ti(IV), resulting in more available photogenerated electrons participating in the hydrogen evolution reaction.
4.4. Hybridization with carbon-based materials
Carbon-based materials, such as graphene, reduced graphene oxide (rGO), carbon nitride or carbon quantum dots, are considered to be ideal electron acceptors and transporters due to their high chemical stability and excellent electrical conductivity.155 Hence, constructing carbon-based-semiconductor photocatalysts is a feasible method to promote charge separation, thereby preventing the photocorrosion caused by charge carriers under light irradiation. | ||
| Fig. 10 (a) Potentiostatic curves of CdS, CdS/1LG, CdS/2LG, and CdS/3LG, (b) digital images of the as-prepared CdS, CdS/1LG, CdS/2LG, and CdS/3LG taken at different visible-light irradiation times. The yellow and black areas are the electrodeposited CdS on FTO glass and nail polish, respectively. Reprinted with permission from ref. 156. Copyright 2017, American Chemical Society. (c) Synthesis approach of the graphene/Cu2O/Cu mesh and digital photograph underneath; (d) FESEM images and digital photograph (inset underneath) of the Cu2O/Cu mesh and G-1.0/Cu2O/Cu mesh before and after 20 min photo-stability measurement under illumination of AM 1.5G. Reprinted with permission from ref. 161. Copyright 2014, Royal Society of Chemistry. (e) Schematic of the photoanode composed of graphene and C60 nanoshells on the ZnO inner core surface and the magnified image of chemical bonding between the functional groups and Zn2+ (Zn–O–C bonding); (f) schematic of water oxidation of ZnO-nanocarbon core–shell QDs. Reprinted with permission from ref. 163. Copyright 2015, Elsevier B.V. (g) Photocathode design and synthesis strategy of the C/Cu2O NWA/Cu mesh; (h) photocurrent density decay at 0 V vs. RHE under chopped light illumination (light on/off cycle: 10 s) for stability evaluation of the Cu2O NWA/Cu mesh and 20 nm coated carbon/Cu2O NWA/Cu mesh, respectively. Reprinted with permission from ref. 165. Copyright 2013, American Chemical Society. | ||
Graphene derivatives with properties similar to those of graphene have also been shown to improve the photostability of metal sulfides. For example, Jia et al. bridged CdS nanocrystals with N-graphene nanosheets to prepare N-graphene/CdS photocatalysts, in which N-doped graphene (N-graphene) acted as an electron transfer channel to reduce the recombination of the photogenerated electron–hole pairs.157 Therefore, the photocatalytic performance for water splitting did not show any significant decrease after the reaction for longer than 30 h, indicating improved stability in the N-graphene/CdS composite. What is more, Tran et al. reported that rGO could consume the photogenerated electrons of Cu2O in a timely manner, thereby preventing Cu2O from being reduced to Cu.158 As a result, a high and long-term stability of hydrogen evolution was obtained in the Cu2O-rGO composite. Similarly, Zhang et al. demonstrated that the photo instability of Cu2O concave cubes with oxygen vacancies could be effectively inhibited by the modification of RGO.159 The prepared Cu2O-RGO catalysts still exhibited a stable photocatalytic performance for hydrogen evolution after three recycles with methanol (20%) as the sacrificial agent under 300 W Hg lamp irradiation, which was due to the fact that RGO and oxygen vacancies can effectively transfer photogenerated electrons and prevent the recombination of electron–hole pairs. In another example, Zhang et al. prepared the RGO-ZnCdS composite through a coprecipitation hydrothermal reduction process for photocatalytic hydrogen evolution with Na2S–Na2SO3 as the sacrificial agent under simulated solar irradiation.160 Cycling tests showed that no detectable decrease in hydrogen production was found in RGO-ZnCdS composites after four cycles, However, in the case of single ZnCdS, hydrogen production gradually decreased as the cycles increase. Such an enhanced stability was due to the fact that the photogenerated electrons of ZnCdS were transferred to RGO, thereby preventing ZnCdS from being reduced by photogenerated electrons.
To date, most investigations regarding graphene-based materials focus on the immobilization of nanoparticles on graphene sheets. However, in the case of a PEC system, the one-dimensional (1D) nanostructures such as nanowire arrays (NWAs) or nanotubes are more favored since their unique morphology can offer a direct path for charge carrier transfer, with reduced grain boundaries and high crystallinity, resulting in superior charge transfer ability. For instance, Dubale et al. prepared a 1D-nanocomposite electrode of graphene/Cu2O NWA/Cu mesh combining electrochemical anodization, dip coating and annealing process (Fig. 10c).161 The photocurrent density of the G-1.0/Cu2O/Cu mesh at the end of 20 min exhibited more than five-fold enhancement in photostability (83.3%) compared to the bare Cu2O/Cu mesh (14.5%), demonstrating that graphene exhibited an excellent behavior in suppressing photocorrosion of Cu2O and could be used to accelerate the transfer of photogenerated carriers and lower the recombination of electrons and holes. Furthermore, SEM (Fig. 10d) and XRD analysis showed that Cu NPs over the surface of the Cu2O/Cu mesh sample could be observed, while it was absent for graphene modified Cu2O, suggesting that the incorporation of graphene exhibited interesting protection towards inhibiting photocorrosion of Cu2O, thereby resulting in an enhanced photocurrent density and stability under light irradiation.
For PEC systems, coating the semiconductor with an inert protective layer to avoid direct contact of the electrode and the electrolyte is an effective method to relieve photocorrosion. However, the inert protective layer has poor conductivity, which limits the transfer of charge carriers. Therefore, carbon-based materials with high electrical conductivity are a good substitute for the inert protective layer to enhance the stability and activity of PEC systems. Since both photoreduction in the presence of photogenerated electrons or photooxidation by the consumption of holes will lead to the photocorrosion of Cu2O, the high conductivity of the carbon-based material makes it possible to effectively withdraw the excited electrons away from Cu2O to suppress the photoreduction reaction of Cu2O. In addition, the carbon sheath can also indirectly protect Cu2O from being oxidized to CuO. Therefore, coating a carbon-based protecting layer on Cu2O thin films is a theoretically feasible strategy to inhibit the photocorrosion of Cu2O. For example, Zhang et al. used glucose as the carbon precursor to form a protective carbon coating onto Cu2O NWAs through a simple solution-based process (Fig. 10g).165 The carbon layer with a thickness of 20 nm exhibited optimal performance and stability for water splitting, with more than 6-fold photostability improvement for C-3-/Cu2O NWA/Cu compared to the non-carbon protected Cu2O NWA/Cu mesh photocathode, as illustrated in the stability evaluation (Fig. 10h). In addition, the long-term stability test showed that only 25% decrease of the original photoactivity was detected in the C-3-/Cu2O NWA/Cu mesh while 100% loss was found for the Cu2O NWA/Cu mesh after 1 h of irradiation, and no copper particles can be identified in the SEM image of used C-3-/Cu2O NWA/Cu while some copper particles were found from the SEM image of used Cu2O NWA/Cu, suggesting that the stability of Cu2O can be effectively improved by coating with a carbon protective layer.
Moreover, since the self-photooxidation of Cu2O occurs in the presence of hydroxyl ions (OH−), preventing the Cu2O particles from directly coming into contact with the solvent or the electrolyte can inhibit the photocorrosion of Cu2O. In this case, the hydrophobicity of the carbon material plays an important role in protecting the particles. For instance, Yu et al. prepared carbon layer coated Cu2O mesoporous nanorods on Cu foil through a facile chemical oxidation and subsequent carbonization process.166 Only 7% loss compared to the first run was found after six cycles, while 46% decrease was detected for single Cu2O, indicating the good photostability of Cu2O after modification with the carbon layer. The SEM image of Cu2O and C-2/Cu2O after three cycles and six cycles showed that Cu2O tended to agglomerate and fuse after six cycles; in the case of C-2/Cu2O, only a slight increase in size was found and the 1-D nanorod structure remained well, suggesting superior structure stability, which can also be demonstrated from the XPS spectrum of Cu 2p and surface X-ray diffraction (SXRD) results. As a result, the gradually passivated thin carbon layer acted as a protective layer of Cu2O to greatly improve its photoreaction stability.
4.5. Construction of heterojunctions
One of the factors limiting the photocatalytic performance and stability of single semiconductors is the rapid recombination of photogenerated electrons and holes. Numerous studies have shown that the construction of a heterojunction structure can greatly accelerate the separation of photogenerated electrons and holes. At present, research toward heterojunction construction is mainly concentrated on type-I, type-II, Z-scheme, and p–n heterojunctions, which will be elaborated in detail with convincing examples in the following section.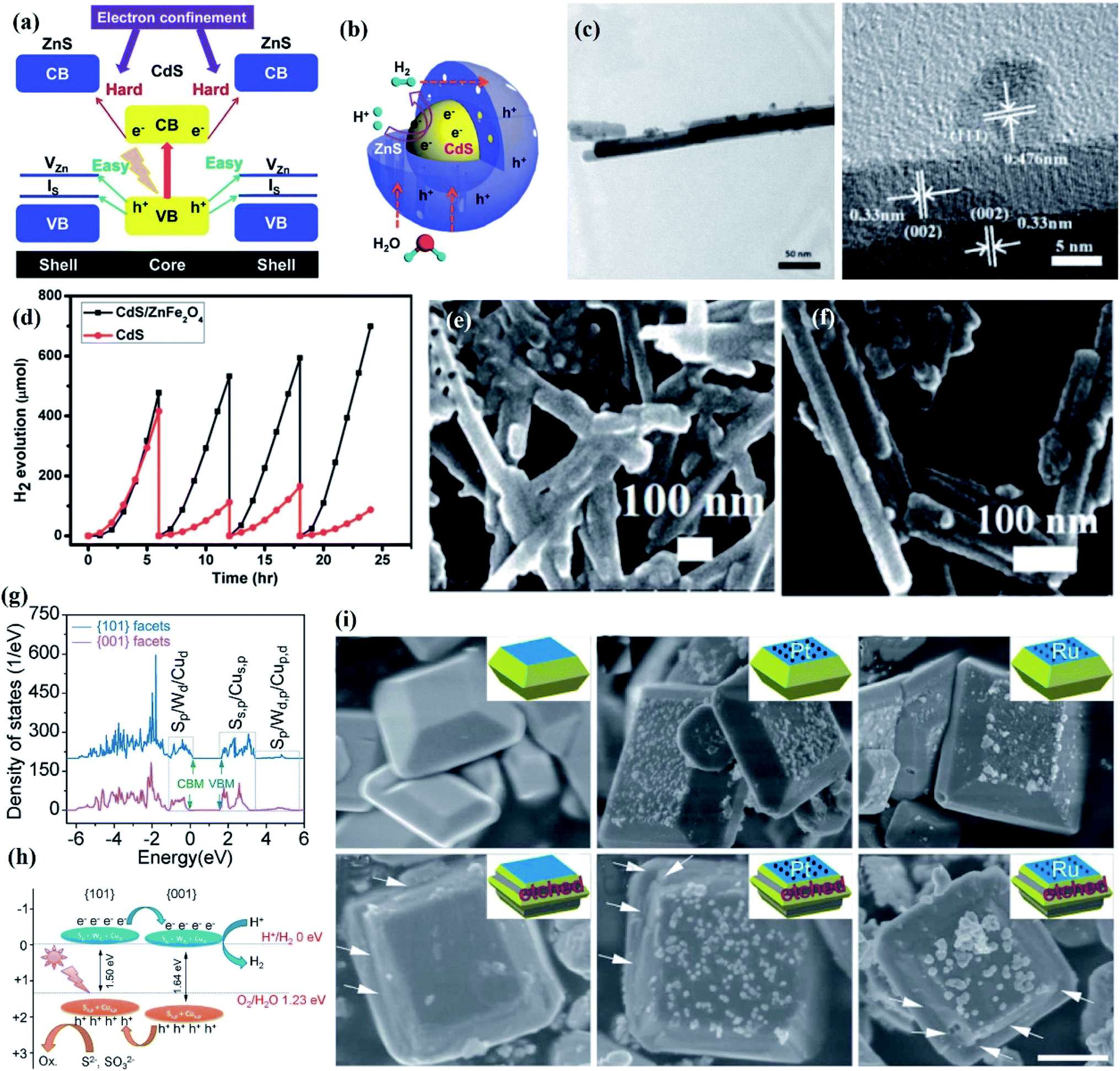 | ||
| Fig. 11 (a) Band structure alignments of the CdS–ZnS core–shell structure; (b) schematic of the photoexcited charge carrier distribution and related photocatalytic reactions in the CdS–ZnS core–shell structure. Reprinted with permission from ref. 168. Copyright 2014, Royal Society of Chemistry. (c) TEM images of ZnFe2O4 decorated CdS nanorods at low and high magnifications; (d) hydrogen evolution of plain and ZnFe2O4 decorated CdS nanorods. SEM images of (e) plain and (f) ZnFe2O4 decorated CdS nanorods after cycling tests; calculated band structure of the Cu2WS4 decahedron. Reprinted with permission from ref. 170. Copyright 2013, Royal Society of Chemistry. (g) Total DOS of the (100) and (101) facets and (h) the corresponding energy levels; (i) TEM images of the recovered Cu2WS4, 5 wt% Pt/Cu2WS4, and 5 wt% Ru/Cu2WS4 photocatalysts after the hydrogen evolution reaction for 5 h with Na2S/Na2SO3 as the sacrificial agent and with only Na2SO3 as the sacrificial agent. Arrows indicate the etched (101) facets. Reprinted with permission from ref. 173. Copyright 2014, Royal Society of Chemistry. | ||
However, the structural features of type I heterojunctions cause the photogenerated electrons and holes of the wide band gap semiconductor to be transferred to the corresponding CB and VB of the narrow band gap semiconductor, which results in a sharp decrease in the number of photogenerated carriers available for surface redox reactions. In view of this, how to promote the transfer of charge carrier from the core to the outer surface is the key to realize the application of type I core/shell nanocrystals in PC/PEC water splitting systems. To this end, type II heterojunctions come into being.
Nowadays, (oxygen) nitrides are reported as a promising material for solar energy conversion owing to their wide visible light absorption, suitable CB/VB position, and excellent photostability. As reported by Dong et al., a 0D/1D Ta3N5/BaTaO2N heterostructure was prepared by an ammonia thermal synthesis process with KBa2Ta5O15 as the precursor, in which BaTaO2N NPs were in situ grown on the surface of Ta3N5 nanorods.171 Due to a type-II heterostructure formed between Ta3N5 and BaTaO2N, the CB electrons of BaTaO2N with a relatively high CB potential were transferred to the CB of Ta3N5, while the VB holes of Ta3N5 were transferred to the VB of BaTaO2N, resulting in the accumulation of photogenerated electrons and holes in two individual semiconductors. As a result, the prepared Ta3N5/BaTaO2N composites exhibited a high and stable photocatalytic performance for water splitting, and no significant drop in H2/O2 production was found after five cycles (150 h).
Moreover, since the stability of semiconductor is also dependent on its structural design, the combination of morphology engineering and a type II heterojunction can achieve composite catalysts with improved stability. As a typical example, a lollipop-shaped uniform Cu@Cu2O/ZnO composite with ultra-small Cu@Cu2O NPs partially coated with ZnO nanorods was designed through a hot-injection synthesis process for photocatalytic hydrogen evolution with Na2S–Na2SO3 as the sacrificial agent under UV-vis irradiation.172 Cycling experiments showed that Cu@Cu2O/ZnO composites maintained a stable hydrogen production after more than 8 h of irradiation. The excellent stability was attributed to the formation of type-II heterojunction between Cu@Cu2O and ZnO, in which ZnO nanorods were selectively epitaxially grown on the (111) facets of Cu nanocrystals, resulting in partial coating of the Cu@Cu2O NPs with ZnO, thereby preventing the surface oxidation of Cu NPs. Moreover, facet-dependent properties also play important roles in photostability improvement due to the influences towards charge transfer efficiency. Based on the partial density of states (DOS) (Fig. 11g), Li et al. found that there were 80 meV and 60 meV band offsets of the CB and VB for the (001) facets relative to the (101) facets of Cu2WS4, which indicated that a type-II staggered band alignment formed between these two facets, resulting in the photogenerated electrons and holes being accumulated in the (001) and (101) facets (Fig. 11h).173 Photocatalytic hydrogen evolution performed in the presence of S2− or S2−/SO32− can maintain the smooth (101) facets; however, an obvious corrosion on the (101) facets can be detected with only Na2SO3 as the sacrificial agent. This difference was determined by whether the holes accumulated in the (101) facets can be consumed in a timely manner by S2− (Fig. 11i). Therefore, the photooxidative etching caused by the holes can be avoided by decreasing the ratio of the (101)/(001) crystal facets.
An all-solid-state Z-scheme photocatalytic system consists of two isolated photochemical systems and a mediator, in which the mediator promotes charge carrier transfer at the contact interface. A typical all-solid-state Z-scheme photocatalytic system, CdS–Ag–TiO2, was constructed by Zhao et al. through a solvothermal process for hydrogen evolution under xenon lamp irradiation, in which Ag NPs and TiO2 quantum dots were successively deposited on the surface of CdS nanowires.176 The recycling test showed that CdS–Ag–TiO2 exhibited the highest stability with no obvious decrease in hydrogen production after four cycles, while nearly 43% loss in hydrogen production was detected for the CdS–TiO2 composite. The poor stability of CdS–TiO2 was attributed to the fact that the excited holes of CdS still remained in the VB of CdS, thereby resulting in hole-induced photocorrosion in the CdS–TiO2 system. Conversely, the VB holes of CdS can be consumed in a timely manner by the CB electrons of TiO2 through the Ag mediator, which not only prevented the hole-induced photocorrosion of CdS, but also left more available photogenerated electrons to participate in the hydrogen evolution reaction. Besides noble metals, readily available and inexpensive media are more environmentally friendly. For example, Wang et al. prepared a ZnO–CdS@Cd heterojunction with the Cd core coated with a ZnO–CdS shell for photocatalytic hydrogen evolution using Na2S–Na2SO3 as the sacrificial agent.177 The prepared ZnO–CdS@Cd exhibited long-term stability without any decrease in hydrogen production after four cycles (20 h). In addition, no change in binding energies of Zn 2p, Cd 3d, and O 1s XPS spectra was found in the used ZnO–CdS@Cd catalyst, and ZnO–CdS@Cd retained a stable morphology after photocatalytic reactions for 20 h. This stable hydrogen evolution performance was due to the following: (i) the CB electrons of ZnO can effectively recombine with the VB holes of CdS through the Cd core mediator, resulting in a prolonged lifetime; (ii) the thin ZnO–CdS shell (15 nm) was able to shorten the diffusion length of charge carrier transport in ZnO–CdS@Cd to hinder the bulk recombination of charge carriers in ZnO or CdS.
More recently, the direct Z-scheme photocatalytic system that omits the process of carriers passing through the electron mediator has attracted widespread concern since Wang et al. constructed the first mediator-free all-solid-state Z-scheme photocatalytic system in 2009.178 Subsequently, more research has been focused on the design and construction of direct Z-scheme photocatalytic systems for widespread application. In a typical example, a CdS/BiVO4 composite with CdS NPs decorated on 1D BiVO4 nanowires was prepared by Zhou and co-worker, which exhibited a high and stable performance for photocatalytic hydrogen evolution with lactic acid as the hole scavenger and Pt as the cocatalyst under visible light irradiation.179 The photocatalytic hydrogen production experiments of Pt/CdS and CdS/BiVO4 in Na2SO3 electrolyte showed that a significant decrease (53%) in hydrogen production was found in the presence of Pt/CdS after five cycles (300 h), while only 17% decrease of hydrogen evolution in the first cycle was detected over CdS/BiVO4, indicating that the oxidation sites were located at BiVO4 instead of CdS. Due to the formation of the Z-scheme heterojunction, the VB holes of CdS can recombine with the CB electrons of BiVO4, which not only retained the strong redox ability of photogenerated charge but also effectively prevented CdS from being oxidized by its holes, thereby resulting in an excellent photocatalytic performance and stability.
In addition, Ma et al. applied atomic layer deposition (ALD) technology to prepare a CdS@ZnO composite with controlled morphology and distribution of the ZnO shell for photocatalytic hydrogen evolution with Na2SO3–Na2S as the sacrificial agent under 225 W Xe arc lamp irradiation.180 Long-term recycling investigation showed that the hydrogen production of Pt-deposited CdS@ZnO catalysts did not change after five cycles, and a stable crystal structure was obtained after multiple cycles, as demonstrated by the XRD pattern. Moreover, ICP was applied to investigate the leaching of Cd2+ ions; the results showed that only 0.01 mg L−1 Cd2+ was detected for Pt-deposited CdS@ZnO after 8 h of irradiation, while 1.39 mg L−1 Cd2+ was detected for Pt/CdS under the same conditions, indicating that the modification with the ZnO shell can effectively prevent CdS from being photo-corroded. The stable photocatalytic performance was due to the Z-scheme charge transfer mechanism between CdS and ZnO, resulting in photogenerated electrons accumulating in the CB of CdS while holes accumulated in the VB of ZnO. In this case, ZnO also might be oxidized by accumulated holes, as demonstrated by ICP results. Therefore, the hole cocatalyst PdS was in situ deposited on the surface of ZnO for timely capture of the VB holes of ZnO, which exhibited an improved stability and activity toward photocatalytic hydrogen evolution. In another work, a ZnO–CdS core–shell heterojunction with 0D CdS NPs coated on the surface of 1D rod-like ZnO was fabricated for photocatalytic hydrogen evolution with Na2S–Na2SO3 as the sacrificial agent under 300 W Xe lamp irradiation (Fig. 12a).181 Cycling experiments showed that no obvious decrease in hydrogen production was found after three cycles (30 h) regardless of ZnO@CdS or RuO2-deposited ZnO@CdS, indicating a high and long-term stable performance for photocatalytic hydrogen evolution. The stability was attributed to the charge transfer between ZnO and CdS following the Z-scheme mechanism, in which the photogenerated electrons in the CB of ZnO recombined with the holes of CdS, leaving electrons in the CB of CdS involved in the hydrogen evolution reaction.
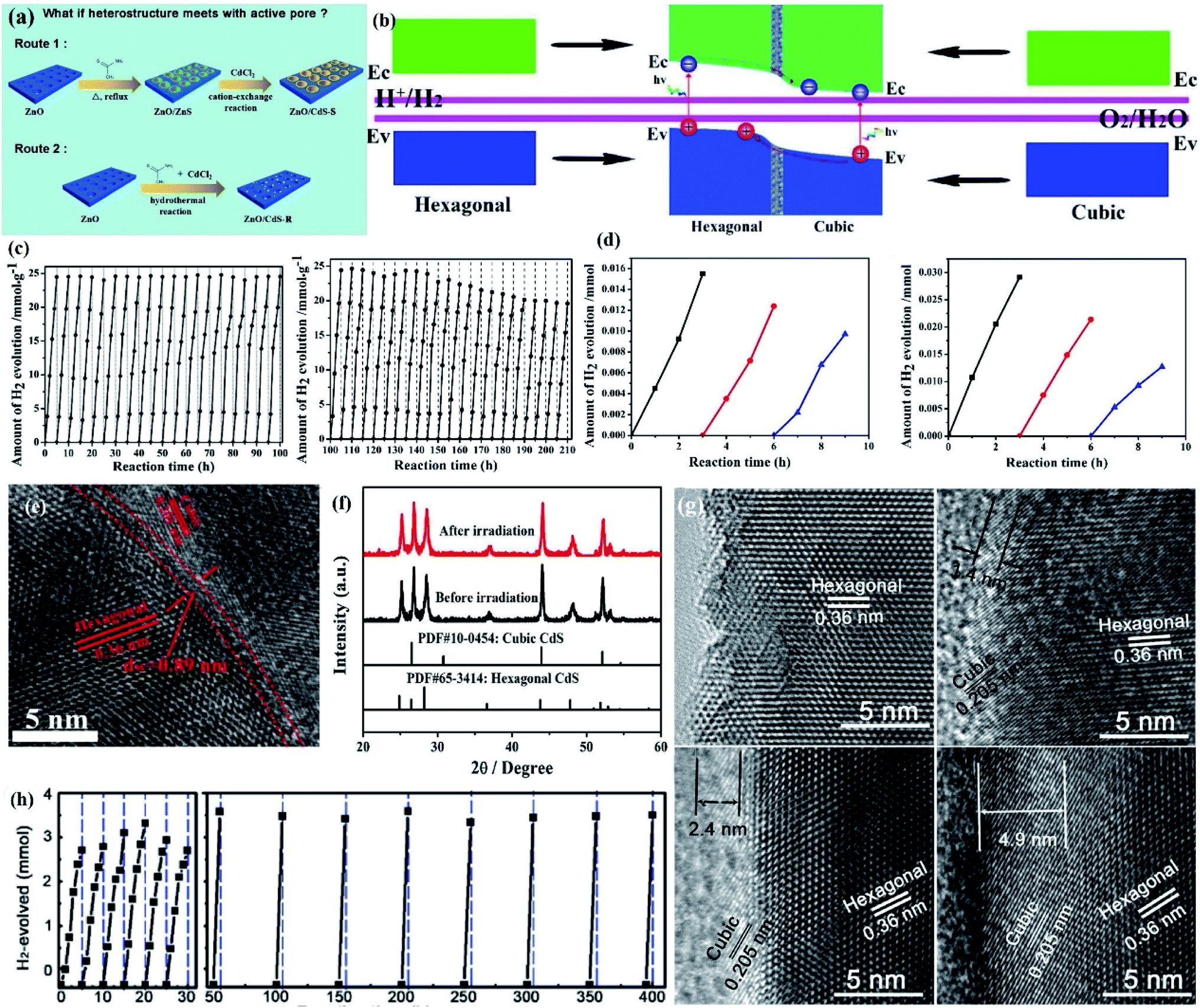 | ||
| Fig. 12 (a) Schematic of the synthesis of selectively and randomly distributed ZnO/CdS heterostructures. Reprinted with permission from ref. 182. Copyright 2018, Royal Society of Chemistry. (b) Schematic theories illustrating the regulation of the band structure through a phase junction with the bonding region; (c) long-term cycle stability of photocatalytic H2 evolution for over 100/200 h; (d) cycle stability of photocatalytic H2 evolution: cubic phase CdS NPs and hexagonal CdS NPs; (e) the HRTEM images and (f) XRD patterns of the CdS NPs after irradiation for 100 h. Reprinted with permission from ref. 185. Copyright 2018, Elsevier B.V. (g) The HRTEM images of CS1, CS12, CS18, and CS25; (h) the photocatalytic stability of CS18 in a vacuum under visible light irradiation. Reprinted with permission from ref. 186. Copyright 2016, Wiley-VCH. | ||
Considering that creating a specific and favorable interface can promote the directional transfer of charge carriers, if the polar (002) facets of ZnO serve as active sites, the heterojunction bonded on these active sites might facilitate the separation of charge carriers. In this regard, Ma et al. investigated the synthesis and properties of both selectively and randomly distributed ZnO/CdS heterostructures for photocatalytic hydrogen evolution with Na2S–Na2SO3 as the sacrificial agent under irradiation with a 300 W Xe lamp.182 The results indicated that ZnO/CdS with selective distribution of heterostructures around the pores exhibited a higher photocurrent response and lower electrochemical impedance than those of ZnO/CdS with randomly distributed heterostructures. Due to the Z-scheme, the ZnO/CdS heterostructure was selectively fabricated at the active pores, resulting in more efficient charge separation, higher hydrogen evolution and stability for the ZnO/CdS heterostructure.
To further improve the long-term stability of CdS-based phase junctions, efforts and interest have further focused on the application of structural engineering in the phase-junction design. For example, Li et al. prepared concentric CdS nanorod phase junction (NRPJ) integrated hexagonal-cubic core–shell architecture with a nanorod morphology through the hydrothermal process for photocatalytic hydrogen evolution with Na2S–Na2SO3 as the sacrificial agent and a 300 W Xe arc lamp as the light source.186 The formation mechanism of the hexagonal/cubic CdS phase junction was investigated by optimizing the ratio of Cd to S and the reaction times (denoted as CSx, x = 1, 6, 12, 18, 25, and 45 h). The high-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) image exhibited a resolved phase junction between the shell and core region, and the stacking mode of the CdS lattice changed from the cubic phase (ABCABC) to the hexagonal phase (ABABAB) (Fig. 12g). Further stability experiments showed that the hydrogen production of the optimal CdS NRPJs was maintained at a high level after 400 h of irradiation, which was much higher than that of hexagonal CdS (only about 10 h) (Fig. 12h). The stable performance of CdS NRPJs was due to the following: (i) the unique core–shell concentric nanorod geometric structure promoted charge transfer through the passivation of surface states and the enhanced tunneling of charge carriers; (ii) the presence of the ultrathin cubic shell not only passivated the surface state of the hexagonal core but also reduced the recombination of charge carriers via enhanced tunneling.
Moreover, although the photogenerated electrons of ZnS with more negative potential is beneficial for the photocatalytic hydrogen evolution reaction, the wide band gap of 3.6 eV greatly limits its light response performance. To solve this problem, it might be a promising strategy to combine wide and narrow bandgap semiconductors to form a solid-solution nanocomposite to obtain a controlled bandgap width through generating new orbital hybridization of atoms. For instance, Hsu and co-workers prepared a Zn1−xCdxS solid solution with a zinc blende-wurtzite phase homojunction in between for photocatalytic hydrogen evolution using Na2S–K2SO3 as the electron donor and a 300 W Xe lamp as the light source.187 In the TEM (HR-TEM) image of Zn0.52Cd0.48S, an interplanar spacing of 3.3 Å was close to the value of the zinc blende phase for the Zn0.52Cd0.48S (111) facet and wurtzite for the (002) facet and d-spacings of 3.2 and 2.1 Å were respectively matched with the values of the (101) and (220) facets in the wurtzite phase, indicating the possible construction of the heterojunction between these zinc blende and wurtzite phases (Fig. 13a). In zinc blende and wurtzite phases, Zn/Cd atoms attached to four S atoms ([Zn/Cd]S4) were connected to each other by sharing the corners to construct a three-dimensional structure to form heterojunctions. After visible light irradiation, the CB electrons of the zinc blende phase would transfer to the CB of the wurtzite phase and the VB holes of the wurtzite phase would diffuse into the VB of the zinc blende phase due to Fermi level equilibration. Therefore, the excited CB electrons in the wurtzite phase would participate in the hydrogen evolution reaction and the VB holes in the zinc blende phase would be consumed by the scavenger, resulting in an improved photocatalytic performance toward hydrogen evolution. More importantly, compared with single CdS, the prepared Zn0.52Cd0.48S has fewer Cd–S bonds, which reduced the exposed area of the sample in the electrolyte, resulting in an enhanced photocatalytic stability. More recently, in order to solve the aggregation of Pt cocatalysts, Ng and co-workers prepared twinned Zn0.5Cd0.5S nanocrystals decorated with highly dispersed sub-2 nm Pt for photocatalytic hydrogen evolution with Na2S–Na2SO3 or ascorbic acid as the sacrificial reagent under 500 W Xe arc lamp irradiation (Fig. 13b).188 Cycling tests showed that the optimal sample (8Pt–Zn0.5Cd0.5S) maintained a hydrogen production of 85.0 under alkaline conditions and 80.8% under acidic conditions after five cycles (30 h) (Fig. 13c and d). Also, no obvious difference in morphology and crystallinity of 8Pt-Zn0.5Cd0.5S was found after the reaction, as demonstrated by the TEM and XRD image. The above results all demonstrated that the prepared sample with zinc blende-wurtzite phase heterojunctions in between could achieve improved activity and stability for photocatalytic hydrogen evolution.
 | ||
| Fig. 13 (a) HR-TEM images of the Zn0.52Cd0.48S sample. Reprinted with permission from ref. 187. Copyright 2015, American Chemical Society. (b) Schematic illustration of the synthesis process for twinned Zn0.5Cd0.5S nanocrystals decorated with highly dispersed ultra-small Pt NPs and aggregated Pt nanoclusters; photostability tests of 8Pt-Zn0.5Cd0.5S for recycling runs of H2 evolution under visible light: time courses of cycling H2 evolution and the corresponding H2 yield (c) in 0.1 M Na2S/0.1 M Na2SO3 and (d) 0.15 M ascorbic acid. Reprinted with permission from ref. 188. Copyright 2018, Elsevier B.V. (e) Schematic illustration of the formation of MoS2/CdS/N-RGO and the corresponding TEM (HR-TEM) image of CdS/N-RGO and MoS2/CdS/N-RGO; (f) schematic illustration of MoS2/CdS/N-RGO; (g) repeated runs of hydrogen generation for the MoS2/CdS/N-RGO hybrid in 7 vol% lactic acid solution under solar light irradiation. Reprinted with permission from ref. 194. Copyright 2015, Royal Society of Chemistry. | ||
Moreover, embedding CdS in a 2D p–n junction composed of MoS2 and N-RGO was proposed to promote the carrier transfer for a better PEC performance and stability (Fig. 13e).194 Since MoS2 and CdS shared the same hexagonal crystalline structure, a high-quality and intimate heterojunction grew together on N-RGO sheets. The space charge region created by the p–n junction of MoS2/N-RGO resulted in effective charge defective layers in MoS2 and N-RGO. Therefore, the excited electrons and holes generated in CdS nanorods would transfer to p-type MoS2 and n-type N-RGO, respectively (Fig. 13f), which improved the efficiency of photocatalytic water splitting and inhibited the hole-induced photocorrosion of CdS (Fig. 13g).
For PEC systems, Zhang et al. reported that the CuSbS2/CdS/Pt p–n heterojunction composite formed by the combination of a solution-processed CuSbS2 thin film and thin CdS layer can achieve a stable performance for PEC hydrogen evolution,195 as demonstrated by a stable cathodic photocurrent under continuous illumination at an applied potential of 0 V vs. RHE during the initial 30 min. However, the photocurrent decayed around 15% of the initial value after 1 h, which was probably attributed to the partial photocorrosion of CdS and the subsequent detachment of the Pt catalyst, but the poor stability can be improved by protective coating using TiO2.
As mentioned above, the formation of heterojunctions can effectively inhibit the photocorrosion of semiconductor-based catalysts, and further research should focus on improving and optimizing some heterojunction systems to achieve long-term photostability. Taking the type-I CdS–ZnS heterojunction as an example, both ZnS and CdS can be excited by visible light to generate excited electrons and holes. The VB holes of CdS would transfer to the corresponding VB of ZnS due to the more positive VB potential of CdS. As a result, the photogenerated holes would accumulate in the VB of ZnS, which was to prevent the photocorrosion of CbS by sacrificing the stability of ZnS since the surface sulfur ions of ZnS might be oxidized after long-term illumination.
4.6. Hybridization with conducting polymers
Conducting polymers with high conductivity, stability, good environmental compatibility, flexibility and controllability are emerging as novel functional materials in artificial photo-redox reactions.196 In particular, due to the excellent conductivity and flexibility of polyaniline (PANI), coating of PANI on the surface of the semiconductor to construct the core–shell structure can greatly improve the stability of the semiconductor.197 This is attributed to the fact that PANI acts as a physical barrier to prevent direct contact between the semiconductor and the external environment, thereby preventing the dissolution of the semiconductor. In addition, the good conductivity of PANI can promote the transfer of charge carriers at the interface and suppress the photocorrosion caused by the accumulation of photogenerated carriers.198 For example, PANI@CdS core–shell nanospheres with a PANI thin layer on porous CdS nanospheres were constructed through a proton doped in situ polymerization method.199 XPS analysis of S 2p3/2 peaks showed a slight increase in binding energy from 151.9 eV in CdS to 152.7 eV in PANI@CdS, indicating that PANI@CdS exhibited better photocorrosion resistance under light irradiation compared to CdS nanospheres. The cycle experiments showed a stable hydrogen production on PANI@CdS core–shell nanospheres after six cycles, and this excellent stability was attributed to the fact that newly formed C–S or N–Cd bonds greatly prevented the reduction of S2− to S0. In another work, as the cycle experiment progressed, the hydrogen evolution rate of single Ta3N5 gradually decreased.200 However, no significant decrease in hydrogen evolution of Ta3N5 coated with thickness-tunable PANI was detected even after six cycles, which was due to the fact that the PANI shell can effectively prevent the contact between the holes and nitrogen anions in Ta3N5, thereby improving the stability of Ta3N5.In addition to PANI, the stability of CdS can also be improved by coupling it with the conjugated polymer polyimide (PI); in situ XPS analysis suggested that the photogenerated electrons in PI would transfer to CdS and combine with the holes therein under light irradiation, which could effectively suppress hole-induced photocorrosion within CdS.201 A further comparative study showed that both PANI and polypyrrole (PPy) can prevent Ta3N5 from self-photocorrosion caused by the oxidation of the nitride of Ta3N5 by self-generated holes, while a relative higher photocatalytic water splitting performance was obtained for Ta3N5/PANI due to the higher electric conductivity of PANI than PPy.202 A similar investigation was also reported by Wang et al., where three types of conducting polymers, PANI, PPy and poly(3,4-ethylenedioxythiophene) (PEDOT) were coated on the surface of CdS nanorods to investigate their photocorrosion resistance and photocatalytic hydrogen evolution.203 The result showed that PANI and PPY shells with abundant N and C atoms can increase the binding energy of Cd and S atoms on the surface of the CdS nanorods, resulting in a relatively stronger driving force for hole transfer from CdS to the polymer. Therefore, the PANI modified PANI@CdS core–shell nanorods exhibited the highest improvement in the hydrogen production rate and photocorrosion resistance in 30 h without deactivation under visible-light irradiation.
4.7. Control of reaction conditions
In general, the photocatalytic performance and stability of semiconductors are also related to the reaction conditions. For instance, a typical pH-dependent semiconductor, CdS, suffered from serious photooxidative dissolution under acidic conditions even without light irradiation, while the photocorrosion and leaching can be effectively inhibited in a high pH solution.204 However, in the case of ZnO, photocorrosion and dissolution occurred regardless of an acidic or alkaline environment.205 Therefore, the optimization of pH should be taken into consideration in various photocatalytic applications to obtain high performance and stability. Similar pH-determined activity also existed in the PEC system, for example, the surface passivation of GaAs by coating with Ga2O3 thin films was a viable method to improve its stability due to the fact that Ga2O3 can reduce the density of surface states and decrease the surface recombination velocity of minority carriers (holes in n-type GaAs). However, the stability of an amorphous or polycrystalline Ga2O3 layer on GaAs largely depended on the pH values of the surrounding environment, which influenced the rate of oxide formation and photoetching. In addition, Sharma investigated the sensitivity of the photocorrosion of GaAs/AlGaAs microstructures with a stack of 30 GaAs quantum well (QW) layers to pH of aqueous solutions ranging between 2.2 and 11.2.206 The photocorrosion rates of microstructures immersed in strong acid (pH 2.2) or alkali (pH 11.2) reached the highest (0.83 nm min−1) due to the solubility of Ga and As oxides (Ga2O3 and As2O3), while that in near neutral or moderate pH environments were significantly reduced (0.33 nm min−1) due to the accumulation of some photocorrosion products on the surface. Notably, regulating the electrolyte composition and controlling the dissolution during the PEC reaction can also improve the stability of the PEC system, which is becoming an increasingly attractive strategy. For instance, the stability of BiVO4 photoanodes was greatly improved by simply adding V2O5 to the electrolyte, resulting in negligible decrease in the photocurrent for 60 h.207 Conversely, both Bi and V were dissolved in the electrolyte in the absence of V2O5, as demonstrated by changes in the XRD pattern after the stability test.At present, the photocorrosion of semiconductor-based catalysts is mainly caused by photogenerated electron/hole-induced redox reactions. However, the effect of newly formed oxygen on the stability of the photocatalytic oxygen evolution reaction also should be considered.208 Moreover, since the dissolved oxygen is about five times higher than that of hydrogen in water, the recombination of hydrogen and oxygen occurs very rapidly on the surface of photocatalysts, which further results in a decrease in oxygen production.209 Generally, there are two strategies to reduce the effects of newly formed oxygen on photocatalytic performance: one is to cover the semiconductor surface with a protective layer to prevent direct contact between the semiconductor and nascent oxygen. For instance, Ning et al. decorated the surface of CdS with chemically inert Al2O3 to form a CdS@Al2O3 core@shell structure for photocatalytic hydrogen production under 300 W xenon lamp irradiation.210 A cycling hydrogen evolution test showed that CdS or CdS–Pt exhibited poor photocatalytic stability due to the rapid recombination of photogenerated electrons and holes, while Pt/CdS@Al2O3 maintained a high and stable hydrogen production after four cycles (Fig. 14a), which was attributed to the use of the Al2O3 shell as a stable oxide matrix to protect the embedded CdS particles from photocorrosion. No obvious changes in phase structure and surface microstructures of Pt/CdS@Al2O3 after multi-cycle were found according to XRD and XPS characterization. High-resolution XPS spectra of S 2p showed that the ratio of S0/S (total) was 2.02% for used CdS while only 1.00% for the used Pt/CdS@Al2O3 catalyst (Fig. 14b), and ICP analysis showed that the concentration of Cd2+ increased gradually with the increase of irradiation time for single CdS, while only negligible Cd2+ was detected in the Pt/CdS@Al2O3 catalyst system (Fig. 14c), indicating good photocorrosion resistance performance of Pt/CdS@Al2O3. However, the hydrogen evolution rate of Pt/CdS@Al2O3 exhibited a slight decrease in the fourth cycle, which may be due to the back recombination of hydrogen and oxygen into water or the presence of nascent oxygen. Inspired by the respiratory system of fish, applying artificial gill to photocatalytic overall water splitting can remove neonatally formed O2 from water, which not only prevented the oxygen-induced photocorrosion but also retarded the reverse reaction of hydrogen and oxygen recombination back to water (Fig. 14d). As demonstrated by the cycling experiments, Pt/CdS@Al2O3 equipped with the artificial gill exhibited a higher performance and enhanced stability for photocatalytic hydrogen evolution after 10 cycles (30 h) (Fig. 14a). Similarly, artificial gill was also applied in Zhen's work, in which a core–shell structured Ni2P@CdS photocatalyst was prepared through a solvothermal process for photocatalytic water splitting under visible light irradiation without the addition of any sacrificial reagent and noble metal loading.211 Cycle experiment, XPS analysis of the used sample and ICP results of the Cd2+ concentration in the reaction solutions all demonstrated that the photocorrosion of CdS was efficiently inhibited. More importantly, O2 evolution close to half of H2 production was achieved in the 10Ni2P@CdS photocatalytic system equipped with artificial gill, demonstrating that the removal of newly formed O2 from water can further improve the photocatalytic performance for water splitting (Fig. 14e). Additionally, since the generated peroxide species (H2O2) in the photocatalytic system is also not conducive to the stability of CdS, Zhen et al. fabricated peroxide decomposing active MnO2 thin-layer over CdS nanowires and assembled oxygen separation artificial gill to remove nascent formed oxygen in the dispersion (Fig. 14f).212 Considering that CdS might be oxidized to CdSO4 by peroxide species, the oxygen generation performance of 20NiSe/MnO2–CdS was much higher than that of 20NiSe/CdS in the dark (Fig. 14g), suggesting that the role of the MnO2 layer over CdS was to decompose hydrogen peroxide in the NiSe/MnO2–CdS composite, which can also be proved by the UV-Vis absorption spectrum (Fig. 14h). As a result, a high and stable performance in hydrogen evolution was achieved for 20NiSe/MnO2–CdS but a low and gradually decreasing performance for CdS was observed during four cycles (Fig. 14i).
 | ||
| Fig. 14 (a) Cycling runs for photocatalytic H2 evolution under visible light over CdS, Pt/CdS, Pt/CdS@Al2O3 with artificial gill and Pt/CdS@Al2O3 without artificial gill; (b) high-resolution XPS spectra of S 2p after photocatalytic reactions for CdS and Pt/CdS@Al2O3; (c) the Cd2+ concentration change in solution of CdS and Pt/CdS@Al2O3 with light irradiation time; (d) the structure of fish gill and the schematic diagram of the photocatalytic reaction system. Reprinted with permission from ref. 210. Copyright 2018, Elsevier B.V. (e) Photocatalytic H2/O2-production over 10Ni2P@CdS in 150 mL pure H2O with artificial gill. Reprinted with permission from ref. 211. Copyright 2018, Elsevier B.V. (f) TEM and HRTEM images of CdS, MnO2–CdS and 20NiSe/MnO2–CdS; (g) time courses of O2 evolution catalyzed by 20NiSe/CdS and 20NiSe/MnO2–CdS in 5% (v/v) H2O2 under dark conditions; (h) the two-electron process for water oxidation to form peroxides; (i) the hydrogen production stability over the 20NiSe/MnO2–CdS photocatalyst with artificial gill under visible light irradiation. Reprinted with permission from ref. 212. Copyright 2018, Elsevier B.V. | ||
In general, photocatalytic water splitting is considered a promising method for developing clean energy alternatives to fossil fuels. Over the past several decades, a great deal of effort and research have focused on obtaining high photocatalytic performance by promoting the charge separation and transfer within semiconductors and have achieved substantial breakthrough. Nevertheless, the overall photocatalytic performance is still relatively poor due to their intrinsic stability under light irradiation. To overcome this bottleneck, the focus is gradually projecting on how to improve the photoreaction stability of semiconductors. In line with the focus of this section, various feasible strategies implemented to suppress photocorrosion, dissolution and improve the stability of semiconductor-based composites have been comprehensively summarized, which includes nanostructure engineering (crystal structure, morphology and size control), heteroatom doping, heterojunction construction, surface modification (hybridization with cocatalysts, carbon-based materials or conducting polymers), and reaction environment regulation. As shown in Table 2, a series of rational modification strategies developed for promising photocatalysts can achieve improved performance with respect to PC/PEC water splitting.
| Electrode configuration | Electrolyte and illumination conditions | AQY STH efficiency | Stability (retained performance) | Note | Ref. |
|---|---|---|---|---|---|
| CdS/ZnO/ZnFe2O4 | 0.5 M Na2S; AM 1.5G | 4.4% at 0.2V | 85% (1 h) | Improved crystallinity of CdS | 57 |
| MoS2/In2S3 | 10% lactic acid; 300 W Xe lamp | — | 100% (20 h) | Dual-defective construction | 58 |
| Cu2O porous foam | 0.5 M Na2SO4; 200 W Xe lamp | −1.7 mA cm−2 at −0.4 V | 7200 s at −0.55 V | Improved electron transfer efficiency by the porous structure | 62 |
| Cu2O with different facet exposures | 100 mL pure water; 300 W Xe lamp | — | The ratio of H2/O2 from 2.13 to 2.26 (72 h) | TiIrOx protective layer | 71 |
| nanocone/Mo:BiVO4/Fe(Ni)OOH | 0.5 M Na2SO3; AM 1.5G | 5.82 ± 0.36 mA cm−2 at 1.23 V; STH: 6.2% | 94.2% (10 h) | Nanoporous photoabsorption layer | 73 |
| Si/TiO2/PtTiO2 | 0.5 M H2SO4; 150 W Xe lamp | — | 85% (24 h) 70% (110 h) | Dual TiO2 protection layer | 75 |
| MoSx/TiOx-GaInP2 | 0.5 M H2SO4; AM 1.5G | 11 mA cm−2 at 0V | 80% (20 h) | Graded catalytic-protective layer | 77 |
| Cu-doped ZnS | 0.05–1.0 M Na2S; 350 W Xe lamp | — | 100% (48 h) | Cation doping | 78 |
| CuO/CuO:Al/ZnO:Al/Au–Pd | 0.1 M Na2SO4; AM 1.5G | 3.7 mA cm−2 at 0V | 92% (1 h) | Cation doping | 79 |
| CdS–P | 0.5 M Na2SO4; 280 W Xe lamp | AQY: 8.2% at 420 nm | 100% (18 h) | Anion doping | 81 |
| N-Doped ZnS | 0.5 M Na2SO4; AM 1.5G | — | 100% (12 h) | Anion doping | 82 |
| Cd0.1SnxZn0.9−2xS | 0.35 M Na2SO3 and 0.25 M Na2S; 500 W halogen lamp | — | 100% (10 h) | Solid solution | 83 |
| (Zn0.95Cu0.05)1−xCdxS | 0.35 M Na2SO3 and 0.25 M Na2S; 300 W Xe lamp | — | 100% (12 h) | Solid solution | 84 |
| CdS/(Au-ReS2) | 0.25 M Na2SO3 and 0.35 M Na2S; 300 W Xe lamp | — | 133% (20 h) | Metal cocatalysts | 100 |
| Pt–CdS double shell hollow nanocubes | 0.25 M Na2SO3 and 0.35 M Na2S; 300 W Xe lamp | 2.84% at 420 nm | 100% (20 h) | Pt–CdS hollow nanocube structure inhibits oxidation on the surface and interface of CdS and Pt detachment | 103 |
| Cu2O/RuOx | 0.5 M Na2SO4; AM 1.5G | STH: 6% | 94% (8 h) | RuOx cocatalyst with a porous nature | 114 |
| Co3O4/CdS | 0.5 M Na2S and 0.5 M Na2SO3; 300 W Xe lamp | — | 100% (9 h) | The accelerated VB-hole transfer of CdS to aqueous solution by Co3O4 | 115 |
| CdS/CoOx | 0.35 M Na2S and 0.25 M Na2SO3; 350 W Xe lamp | — | 62% (24 h) | Superior hole capture capability of CoOx cocatalysts | 116 |
| TiO2@CoNi-layered double hydroxide nanotube arrays | 0.1 M Na2SO4; AM 1.5G | 4.4 mA cm−2 at 1.23 V | ≥99% (10 h) | Reduced accumulation of holes on the surface of the TiO2 by CoNi-LDHs | 117 |
| n+p-Si/SiOx/Ni/NiOx/NiFe-LDH | 1.0 M KOH; AM 1.5G | ∼37 mA cm−2 at 1.23 V | 100% (68 h) | Activated Ni(Ni/NiOx) bridge layer acts as interlayer protection and hole transfer | 119 |
| CdS/WS2 | 0.5 M Na2SO4; 300 W Xe lamp | AQE: 25.03% at 420 nm | 100% (200 h) | Photocorrosion-recrystallization process | 125 |
| MoS2-RGO/ZnO | 0.25 M NaS2 and 0.25 M NaSO3; 300 W Xe lamp | AQE: 0.13% at 365 nm; 0.12% at 380 nm | 93% (12 h) | Improved electron transfer efficiency by RGO | 128 |
| FeP/CdS | 10% v/v lactic acid; 16 mW cm−2 | AQE: >35% at 520 nm | 100% (100 h) | Efficient charge separation within CdS by FeP | 133 |
| Pt–PdS/CdS | 0.5 M Na2S and 0.5 M Na2SO3; 300W Xe lamp | QE: 93% at 420 nm | 100% (25 h) | Pt–PdS dual cocatalysts | 137 |
| Ti(IV)–Ni(II)/CdS | 0.35 M Na2S and 0.25 M Na2SO3; 350 W Xe lamp | — | 86.5% (5 cycles) | Holes can be consumed in a timely manner by the hole-cocatalyst Ti(IV) | 138 |
| Zn1−xCdxS/CdS | 0.35 M Na2S and 0.25 M Na2SO3; 300 W Xe lamp | AQE: 50.1% at 420 nm | 100% (100 h) | The localized acceptor states in ZnS promote the VB holes of CdS to ZnS | 145 |
| Graphene/Cu2O NWA/Cu mesh | 1.0 M Na2SO4; AM 1.5G | −4.8 mA cm−2 at 0 V; ABPE: 3.3% | 83% (20 min) | Enhanced charge separation and transfer by graphene | 146 |
| Carbon nanofiber@ZnIn2S4 | 0.35 M Na2S and 0.25 M Na2SO3; AM 1.5G | AQE: 25.35% at 420 nm; 18.15% at 450 nm; 2.04% at 500 nm | 100% (12 h) | Vector transfer of CB electrons from ZnIn2S4 to carbon nanofibers driven by the hierarchical core–shell structure | 149 |
| C/Cu2ONWAs/Cu mesh | 1.0 M Na2SO4; AM 1.5G | −3.95 mA cm−2; STH: 0.56% | 80.7% (20 min) | The protective role of the carbon layer | 150 |
| CdS@MoS2 | 0.2 M Na2SO4; 300 W Xe lamp | — | 80.2% (12 h) | MoS2 shell promotes the transfer of VB holes from CdS to MoS2 | 152 |
| CdS@ZnS | 0.1 M Na2SO3 and 0.1 M Na2S; 300 W Xe lamp | — | 87% (60 h) | Improved VB-hole transfer from CdS to ZnS by the acceptor states of VZn | 153 |
| Cu@Cu2O/ZnO | 0.75 M Na2S and 1.05 M Na2SO3; 300 W Xe lamp | — | 100% (8 h) | Cu core was passivated by ZnO nanorods | 157 |
| CdS–Ag–TiO2 | 10 mL TEOA and 90 mL DI water; 300 W Xe lamp | — | 100% (20 h) | Improve charge separation and transfer by the Schottky junction | 161 |
| CdS/BiVO4 NWs | 20% lactic acid or 1 M Na2SO3; 300 W Xe lamp | — | 83% (5 h) | Z-Scheme charge separation mechanism | 164 |
| Cubic or hexagonal CdS phase junction | 0.5 M Na2SO4 with 20% lactic acid; 300 W Xe lamp | QE: 41.5% at 420 nm | 80% (150 h) | The suitable bonding region width can be the shield for the phase junction | 170 |
| Hexagonal@cubic CdS phase junction | 0.35 M Na2S and 0.25 M Na2SO3; 300 W Xe lamp | AQE: 43% at 420 nm | 100% (400 h) | Cubic CdS shell can passivate the surface state of hexagonal CdS and reduce the recombination of charge carriers via enhanced tunneling | 171 |
| Pt–Zn0.5Cd0.5S | 0.1 M Na2S and 0.1 M Na2SO3 or 0.15 M ascorbic; 500 W Xe lamp | AQY: 7.15% (0.1M Na2S/Na2SO3) 8.56% (0.15M ascorbic) | 85% (30 h, 0.1 M Na2S/Na2SO3); 81% (30 h, 0.15 M ascorbic) | Enhanced spatial charge separation by the synergistic effects of homojunctions and heterojunctions | 173 |
| CuSbS2/CdS/Pt | 0.5 M Na2SO4; AM 1.5G | −4.2 mA cm−2 at 0 V | 85% (1 h) | The formation of a p–n heterojunction between CuSbS2 and CdS | 180 |
| BiVO4/FeOOH/NiOOH | V2O5 and 1 M borate buffer; AM 1.5G | 5.0 mA cm−2 at 1.9 V | 100% (60 h) | Controlling the dissolution | 190 |
| Pt/CdS@Al2O3 | 0.2 M Na2SO4; 300 W Xe lamp | AQE: 0.11% at 430 nm | 100% (30 h) | Reduced oxygen-induced photocorrosion by the artificial gill | 193 |
5. Conclusions and outlooks
5.1. Concluding remarks
Artificial solar energy-driven technology for hydrogen and/or oxygen generation from water splitting has aroused increasing attention in the scientific community, since this process can generate clean and renewable energy that can replace fossil fuels. Currently, various systems for solar-chemical conversion have been developed, among which water splitting using PC and/or PEC technology is the most widely investigated. Considerable previous research is focused on developing various strategies to improve the photocurrent and efficiency of the PC/PEC system, and significant breakthroughs have been made. However, the long-term efficiency of semiconductor-based catalysts is generally low due to the existence of instability under light irradiation, thereby constraining the overall performance. Therefore, how to improve the stability or durability of semiconductors under light irradiation is a daunting challenge, aimed at obtaining PC/PEC water splitting systems with enhanced activity and stability. In this regard, this is the right and appropriate time to give a comprehensive and state-of-the-art overview with respect to discussing the photocorrosion of photocatalysts and the corresponding measures to improve stability.In line with the focus of this review, the latest advances in strategies that have been performed to inhibit photocorrosion of semiconductors in PC/PEC water splitting systems are comprehensively outlined. Specifically, this review begins with the discussion of photocorrosion mechanisms of semiconductors under different circumstances. In general, semiconductor photocorrosion is due to the photogenerated electrons and/or holes participating in the self-oxidation and/or reduction of semiconductors rather than water splitting. Taking typical semiconductors (CdS, ZnO and Cu2O) as examples, the photocorrosion pathway in the presence of photogenerated electrons and/or holes is thoroughly discussed, which provides a fundamental understanding of semiconductor instability under light irradiation. Subsequently, it is well known that the stability of the photocatalyst is an important indicator to determine whether it can be reused in practical applications, which is usually evaluated by successive cycle tests. However, it is not convincing to use the cycle activity as an indicator to evaluate the stability of the catalyst, as other factors may also result in a decrease in photocatalytic performance after multiple recycle tests, such as active site blocking, loss of sample during recycling, adsorption of intermediates, or surface changes. Therefore, the surface morphological/structural characterization of the sample after multiple cycles is critical to further determine the stability of the photocatalyst. In view of this, various feasible characterization methods used to evaluate or confirm the stability of semiconductors are summarized in the following section.
More importantly, a series of strategies applied to overcome the photocorrosion of the semiconductor are systematically elaborated, with the aim to obtain a PC/PEC water splitting system with high activity and long-term stability for practical application. These modifications include (1) increasing the crystallinity of the semiconductor to reduce the number of recombination centers of photogenerated electron–hole pairs; (2) tailoring the particle size to accelerate the separation and transfer of charge carriers; (3) optimizing the structure/morphology of the semiconductor to obtain improved photocorrosion resistance since the surface atom coordination and surface electronic structure of different morphologies are different; (4) doping with heteroatoms to introduce an impurity level into the forbidden band or form a solid solution to improve charge separation efficiency; (5) loading suitable cocatalysts with a larger work function for electron sinks or lowering the overpotential for hole transfer for timely consumption or transfer of undesired charge carriers; (6) coupling with carbon-based materials and/or conducting polymers used as electron acceptors and transporters to promote charge separation; (7) constructing heterojunctions to accelerate the separation of photogenerated electrons and holes; (8) controlling the reaction conditions to inhibit the photocorrosion caused by the reaction system environment rather than photogenerated carriers. Nevertheless, there is still a long way to go to further optimize the catalytic activity of photocatalysts to achieve large-scale practical applications.
5.2. Fundamental scientific challenges and future outlook
In general, numerous studies have confirmed that rational modifications for semiconductors can be applied to obtain photocatalysts with enhanced activity and stability for PC/PEC water splitting. However, considerable efforts are still needed to further narrow the gap between theoretical efficiency and current achievements, with the aim of addressing many technical challenges in designing the optimal photocatalyst.First, since the photocorrosion of semiconductors is caused by the undesired redox reactions between photogenerated electrons and/or holes and the semiconductor, the photocorrosion rate is largely dependent on the withdrawal efficiency of electrons/holes, that is, the improvement in the charge transfer rate can result in enhanced stability. In this regard, in-depth investigation of the photocorrosion pathway and mechanism is of great significance for accurately and effectively tackling photoreaction instability issues of semiconductor-based materials. Although considerable studies with regard to investigating the photocorrosion mechanism of Cu2O in PC/PEC systems have been made, the photocorrosion pathway is still inconsistent, especially for PEC systems. Besides, the currently widely reported hole-induced photooxidation corrosion mechanism cannot explain the decomposition process of CdS-based photocatalysts under non-aqueous conditions. Notably, the influence of reaction conditions on the performance and stability of semiconductor-based materials cannot be neglected. Further investigations on photocatalytic stability should consider by adjusting the pH of the reaction solution and controlling the reaction atmosphere to alleviate the photoinduced dissolution to some extent.
Second, the photoreaction stability of semiconductor-based materials is generally demonstrated by successive recycling experiments combined with morphology, crystal structure, and surface property characterization of used catalysts. However, some photocorrosion that results in a decrease in activity cannot be convincingly reflected by conventional structural characterization such as XRD, SEM and XPS. Therefore, the development of in situ monitoring technologies to directly reflect the change of semiconductor materials in the photoreaction process is a promising development direction, but there is still much room for exploration in this direction.
Next, as the photoreaction stability is also determined by the crystal structure, morphology, crystal facet and so on, further efforts and attention should be focused on optimizing individual semiconductors. For instance, the photooxidative etching of Cu2WS4 caused by photogenerated holes can be alleviated by decreasing the ratio of the (101)/(001) crystal facets due to the fact that photogenerated electrons and holes tend to accumulate in the (001) and (101) facets, respectively. Therefore, tailoring the crystal structure to explore the relationship between photocatalytic stability and facet-dependent performance should be taken into consideration since surface adsorption ability, electronic structures and intrinsic carrier dynamics are determined by the crystal structures of the semiconductor. It is worth noting that unlike PC water splitting using powder photocatalysts in a suspension system, PEC water splitting is achieved by loading photocatalysts on a conductive substrate. Therefore, how to prepare a crystal facet oriented photoelectrode for PEC water splitting remains a technical obstacle to be overcome, which requires the facet-oriented photocatalyst to form a good contact interface with the conductive substrate. Although the direct growth of the facet-oriented photocatalyst on the surface of conductive substrates is the most commonly used method, the effects of the components in the growth solution on the durability of the substrates should be taken into consideration.
Also, in terms of practical application, chemical impurities present in the actual water environment may chemically react with the photocatalyst, resulting in a gradual decrease in stability over time. A typical example is the splitting of seawater in metal sulfide systems. The presence of chemical salts and impurities in seawater will contaminate metal sulfide systems and increase the required bias voltage, thereby reducing the efficiency of seawater splitting and limiting long-term stability.
Last but not least, most photocathodes show good stability in acidic electrolytes while photoanodes can stably perform in alkaline electrolytes. Hence, achieving high stability of the photocathode in a strong alkali electrolyte and that of the photoanode in strong acid electrolytes is still an arduous challenge. Besides, how to improve the stability of film-based materials with a rough surface still needs to be investigated in depth. Also, seeking promising materials with strong light absorption and excellent photocorrosion resistance in aqueous media to omit the protective layer is a common goal.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This study was financially supported by the Program for the National Natural Science Foundation of China (51879101, 51579098, 51779090, 51709101, 51521006, 51809090 and 51909084), the National Program for Support of Top-Notch Young Professionals of China (2014), the Program for Changjiang Scholars and Innovative Research Team in University (IRT-13R17), and Hunan Provincial Science and Technology Plan Project (2018SK20410, 2017SK2243, and 2016RS3026), and the Fundamental Research Funds for the Central Universities (531119200086, 531118010114, 531107050978 and 541109060031).References
- M. Walter, E. Warren, J. McKone, S. Boettcher, Q. Mi, E. Santori and N. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- Z. Li, W. Luo, M. Zhang, J. Feng and Z. Zou, Energy Environ. Sci., 2013, 6, 347–370 RSC.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- S. Moniz, S. Shevlin, D. Martin, Z. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- T. Wang, Z. Luo, C. Li and J. Gong, Chem. Soc. Rev., 2014, 43, 7469–7484 RSC.
- R. Liu, Z. Zheng, J. Spurgeon and X. Yang, Energy Environ. Sci., 2014, 7, 2504–2517 RSC.
- J. Baxter, C. Richter and C. Schmuttenmaer, Annu. Rev. Phys. Chem., 2014, 65, 423–447 CrossRef CAS PubMed.
- C. Zhen, L. Wang, G. Liu, G. Lu and H. Cheng, Chem. Commun., 2013, 49, 3019–3021 RSC.
- S. Yan, L. Wan, Z. Li and Z. Zou, Chem. Commun., 2011, 47, 5632–5634 RSC.
- F. Osterloh, Chem. Soc. Rev., 2013, 42, 2294–2320 RSC.
- A. Hagfeldt, H. indstrom, S. Sodergren and S. Lindquist, J. Electroanal. Chem., 1995, 381, 39–46 CrossRef.
- S. Sun, W. Wang, D. Li, L. Zhang and D. Jiang, ACS Catal., 2014, 4, 3498–3503 CrossRef CAS.
- J. Ager, M. Shaner, K. Walczak, I. Sharp and S. Ardo, Energy Environ. Sci., 2015, 8, 2811 RSC.
- Q. Li, B. Guo, J. Yu, J. Ran, B. Zhang, H. Yan and J. R. Gong, J. Am. Chem. Soc., 2011, 133, 10878–10884 CrossRef CAS PubMed.
- J. Zhang, J. Yu, Y. Zhang, Q. Li and J. R. Gong, Nano Lett., 2011, 11, 4774–4779 CrossRef CAS PubMed.
- J. Kondo, Chem. Commun., 1998, 3, 357–358 Search PubMed.
- T. Xu, L. Zhang, H. Cheng and Y. Zhu, Appl. Catal., B, 2011, 101, 382–387 CrossRef CAS.
- H. J. Ahn, M. J. Kwak, J. S. Lee, K. Y. Yoon and J. H. Jang, J. Mater. Chem. A, 2014, 2, 19999–20003 RSC.
- S. Masudy-Panah, E. Kong, N. Khiavi, R. Katal and X. Gong, J. Mater. Chem. A, 2018, 6, 11951–11965 RSC.
- Y. Pihosh, I. Turkevych, K. Mawatari, J. Uemura, Y. Kazoe, S. Kosar, K. Makita, T. Sugaya, T. Matsui, D. Fujita, M. Tosa, M. Kondo and T. Kitamori, Sci. Rep., 2015, 5, 11141 CrossRef PubMed.
- L. Pan, J. H. Kim, M. T. Mayer, M. Son, A. Ummadisingu, J. S. Lee, A. Hagfeldt, J. Luo and M. Grätzel, Nat. Catal., 2018, 1, 412–420 CrossRef CAS.
- S. J. Hong, H. Jun and J. S. Lee, Scr. Mater., 2010, 63, 757–760 CrossRef CAS.
- J. Hou, H. Cheng, O. Takeda and H. Zhu, Energy Environ. Sci., 2014, 7, 3758–3768 RSC.
- G. Hitoki, A. Ishikawa, T. Takata, J. N. Kondo, M. Hara and K. Domen, Chem. Lett., 2002, 736 CrossRef CAS.
- J. E. Yourey and B. M. Bartlett, J. Mater. Chem., 2011, 21, 7651–7660 RSC.
- K. Sekizawa, T. Nonaka, T. Arai and T. Morikawa, ACS Appl. Mater. Interfaces, 2014, 6(14), 10969–10973 CrossRef CAS PubMed.
- J. H. Kim, J. H. Kim, J. W. Jang, J. Y. Kim, S. H. Choi, G. Magesh, J. Lee and J. S. Lee, Adv. Energy Mater., 2015, 5, 1401933 CrossRef.
- F. Wang, W. Septina, A. Chemseddine, F. F. Abdi, D. Friedrich, P. Bogdanoff, R. van de Krol, S. D. Tilley and S. P. Berglund, J. Am. Chem. Soc., 2017, 139, 15094–15103 CrossRef CAS PubMed.
- M. S. Prévot, N. Guijarro and K. Sivula, ChemSusChem, 2015, 8, 1359–1367 CrossRef PubMed.
- J. Feng, W. Luo, T. Fang, H. Lv, Z. Wang, J. Gao, W. Liu, T. Yu, Z. Li and Z. Zou, Adv. Funct. Mater., 2014, 24, 3535–3542 CrossRef CAS.
- K. Ueda, T. Minegishi, J. Clune, M. Nakabayashi, T. Hisatomi, H. Nishiyama, M. Katayama, N. Shibata, J. Kubota and T. Yamada, J. Am. Chem. Soc., 2015, 137, 2227–2230 CrossRef CAS PubMed.
- J. Zhao, T. Minegishi, L. Zhang, M. Zhong, G. Wan, M. Nakabayashi, G. Ma, T. Hisatomi, M. Katayama, S. Ikeda, N. Shibata, T. Yamada and K. Domen, Angew. Chem., Int. Ed., 2014, 53, 11808–11812 CrossRef CAS PubMed.
- M. Moriya, T. Minegishi, H. Kumagai, M. Katayama, J. Kubota and K. Domen, J. Am. Chem. Soc., 2013, 135, 3733–3735 CrossRef CAS PubMed.
- A. Bard, J. Photochem., 1979, 10, 59–75 CrossRef CAS.
- S. Chen, D. Huang, G. Zeng, W. Xue, L. Lei, P. Xu, R. Deng, J. Li and M. Cheng, Chem. Eng. J., 2020, 382, 122840 CrossRef CAS; S. Chen and L. Wang, Chem. Mater., 2012, 24, 3659–3666 CrossRef.
- J. Ran, J. Zhang, J. Yu, M. Jaroniec and S. Qiao, Chem. Soc. Rev., 2014, 43, 7787–7812 RSC.
- J. Zheng, W. Sheng, Z. Zhuang, B. Xu and Y. Yan, Sci. Adv., 2016, 2, e1501602 CrossRef PubMed.
- S. Chen and L. W. Wang, Chem. Mater., 2012, 24, 3659–3666 CrossRef CAS.
- D. Huang, Z. Li, G. Zeng, C. Zhou, W. Xue, X. Gong, X. Yan, S. Chen, W. Wang and M. Cheng, Appl. Catal., B, 2019, 240, 153–173 CrossRef CAS.
- A. Davis and C. Huang, Water Res., 1991, 25, 1273–1278 CrossRef CAS.
- D. Meissner, R. Memming, B. Kastening and D. Bahnemann, Chem. Phys. Lett., 1986, 127, 419–423 CrossRef CAS.
- D. Meissner, R. Memming, L. Shuben, S. Yesodharan and M. Grätzel, Berichte der Bunsengesellschaft für physikalische Chemie, 1985, 89, 121–124 CrossRef CAS.
- A. Rudd and C. Breslin, Acta, 2000, 45, 1571–1579 CAS.
- C. Y. Toe, J. Scott, R. Amal and Y. H. Ng, J. Photochem. Photobiol., C, 2019, 40, 191–211 CrossRef CAS.
- C. Morales-Guio, L. Liardet, M. Mayer, S. Tilley, M. Grätzel and X. Hu, Angew. Chem., Int. Ed., 2015, 54, 664–667 CAS.
- L. Huang, F. Peng, H. Yu and H. Wang, Solid State Sci., 2009, 11, 129–138 CrossRef CAS.
- S. Chen, D. Huang, G. Zeng, X. Gong, W. Xue, J. Li, Y. Yang, C. Zhou, Z. Li, X. Yan, T. Li and Q. Zhang, Chem. Eng. J., 2019, 370, 1087–1100 CrossRef CAS.
- S. Guo, X. Li, J. Zhu, T. Tong and B. Wei, Small, 2016, 12, 5692–5701 CrossRef CAS PubMed.
- X. Cui, Y. Wang, G. Jiang, Z. Zhao, C. Xu, A. Duan, J. Liu, Y. Wei and W. Bai, J. Mater. Chem. A, 2014, 2, 20939–20946 RSC.
- X. Chen, Y. He, Q. Zhang, L. Li, D. Hu and T. Yin, J. Mater. Sci., 2010, 45, 953–960 CrossRef CAS.
- D. Huang, H. Luo, C. Zhang, G. Zeng, C. Lai, M. Cheng, R. Wang, R. Deng, W. Xue, X. Gong, X. Guo and T. Li, Chem. Eng. J., 2019, 361, 353–363 CrossRef CAS.
- G. Chen, D. Li, F. Li, Y. Fan, H. Zhao, Y. Luo, R. Yu and Q. Meng, Appl. Catal., A, 2012, 443–444, 138–144 CrossRef CAS.
- X. Zhou, T. Shelton, Z. Xia and Y. Ma, Inorg. Chem. Front., 2017, 4, 1923–1929 RSC.
- Y. Xie, L. Yuan, N. Zhang and Y. Xu, Appl. Catal., B, 2018, 238, 19–26 CrossRef CAS.
- D. Huang, W. Xue, G. Zeng, J. Wan, G. Chen, C. Huang, C. Zhang, M. Cheng and P. Xu, Water Res., 2016, 106, 15–25 CrossRef CAS PubMed.
- X. Guo, Z. Peng, D. Huang, P. Xu, G. Zeng, S. Zhou, X. Gong, M. Cheng, R. Deng, H. Yi, H. Luo, X. Yan and T. Li, Chem. Eng. J., 2018, 347, 74–83 CrossRef CAS.
- D. Huang, X. Qin, Z. Peng, Y. Liu, X. Gong, G. Zeng, C. Huang, M. Cheng, W. Xue, X. Wang and Z. Hu, Ecotoxicol. Environ. Saf., 2018, 153, 229–237 CrossRef CAS PubMed.
- W. Xue, D. Huang, G. Zeng, J. Wan, C. Zhang, R. Xu, M. Cheng and R. Deng, J. Hazard. Mater., 2018, 341, 381–389 CrossRef PubMed.
- R. Wang, D. Huang, Y. Liu, C. Zhang, C. Lai, G. Zeng, M. Cheng, X. Gong, J. Wan and H. Luo, Bioresour. Technol., 2018, 261, 265–271 CrossRef CAS PubMed.
- L. Ma, M. Liu, D. Jing and L. Guo, J. Mater. Chem. A, 2015, 3, 5701–5707 RSC.
- C. Chuang, P. Brown, V. Bulovic and M. Bawendi, Nat. Mater., 2014, 13, 796–801 CrossRef CAS PubMed.
- G. Ai, H. Li, S. Liu, R. Mo and J. Zhong, Adv. Funct. Mater., 2015, 25, 5706–5713 CrossRef CAS.
- C. Guo, J. Xie, H. Yang and C. Li, Adv. Sci., 2015, 2, 1500135–1500141 CrossRef PubMed.
- Y. Lee, C. Chi and S. Liau, Chem. Mater., 2010, 22, 922–927 CrossRef CAS.
- J. Hensel, G. Wang, Y. Li and J. Zhang, Nano Lett., 2010, 10, 478–483 CrossRef CAS PubMed.
- Y. Choi, C. Yim, S. Baek, M. Choi, S. Jeon and K. Yong, Sens. Actuators, B, 2015, 221, 113–119 CrossRef CAS.
- W. Cui, W. Niu, R. Wick-Joliat, T. Moehl and S. D. Tilley, Chem. Sci., 2017, 38, 518–525 Search PubMed.
- T. Grewe and H. Tüysüz, ACS Appl. Mater. Interfaces, 2015, 7, 23153–23162 CrossRef CAS PubMed.
- W. Peng, S. Qu, G. Cong and Z. Wang, Cryst. Growth Des., 2006, 6, 1518–1522 CrossRef CAS.
- D. Jing and L. Guo, J. Phys. Chem. B, 2006, 110, 11139–11145 CrossRef CAS PubMed.
- S. Cao, X. Yan, Z. Kang, Q. Liang, X. Liao and Y. Zhang, Nano Energy, 2016, 24, 25–31 CrossRef CAS.
- Z. Fang, X. Huang, Y. Wang, W. Feng, Y. Zhang, S. Weng, X. Fu and P. Liu, J. Mater. Chem. A, 2016, 4, 13980–13988 RSC.
- H. Tan, R. Amal and Y. Ng, ACS Appl. Mater. Interfaces, 2016, 8, 28607–28614 CrossRef CAS PubMed.
- S. Hong, H. Jun, P. Borse and J. Lee, Int. J. Hydrogen Energy, 2009, 34, 3234–3242 CrossRef CAS.
- M. Singh, D. Jampaiah, A. Kandjani, Y. Sabri, E. Della Gaspera, P. Reineck, M. Judd, J. Langley, N. Cox and J. van Embden, Nanoscale, 2018, 10, 6039–6050 RSC.
- P. Shahbazi and A. Kiani, Int. J. Hydrogen Energy, 2016, 41, 17247–17256 CrossRef CAS.
- M. Hernandez-Alonso, F. Fresno, S. Suarez and J. Coronado, Energy Environ. Sci., 2009, 2, 1231–1257 RSC.
- X. Liu, D. Huang, C. Lai, L. Qin, G. Zeng, P. Xu, B. Li, H. Yi and M. Zhang, Small, 2019, 15, 1–27 CAS.
- S. Ryu, W. Balcerski, T. Lee and M. Hoffmann, J. Phys. Chem. C, 2007, 111, 18195–18203 CrossRef CAS.
- R. Peng, C. Wu, J. Baltrusaitis, N. Dimitrijevic, T. Rajh and R. Koodali, Chem. Commun., 2013, 49, 3221–3223 RSC.
- M. Maisano, M. Dozzi and E. Selli, J. Photochem. Photobiol., C, 2016, 28, 29–43 CrossRef CAS.
- Y. Xie, G. Liu, L. Yin and H. Cheng, J. Mater. Chem., 2012, 22, 6746–6751 RSC.
- A. Pawar, C. Kim, M. Kang and Y. Kang, Nano Energy, 2016, 20, 156–167 CrossRef CAS.
- Z. Zheng, B. Huang, Z. Wang, M. Guo, X. Qin, X. Zhang, P. Wang and Y. Dai, J. Phys. Chem. C, 2009, 113, 14448–14453 CrossRef CAS.
- Y. Kwon, A. Soon, H. Han and H. Lee, J. Mater. Chem. A, 2015, 3, 156–162 RSC.
- P. Bhavani, D. Kumar, S. Jeong, E. Kim, H. Park, S. Hong, M. Gopannagari and D. Reddy, Catal. Sci. Technol., 2018, 8, 1880–1891 RSC.
- Y. Qiu, W. Liu, W. Chen, W. Chen, G. Zhou, P. Hsu, R. Zhang, Z. Liang, S. Fan, Y. Zhang and Y. Cui, S. Adv., 2016, 2, 1501764–1501772.
- S. Li, P. Zhang, X. Song and L. Gao, ACS Appl. Mater. Interfaces, 2015, 7, 18560–18565 CrossRef CAS PubMed.
- F. Yang, J. Aguiar, M. Fairchild, W. Vakki, S. Younan, Y. Zhou, L. Zhuo and J. Gu, Adv. Mater. Interfaces, 2019, 1802085 CrossRef.
- O. Hendricks, R. Tang-Kong, A. Babadi, P. McIntyre and C. Chidsey, Chem. Mater., 2018, 31, 90–100 CrossRef.
- J. Gu, J. Aguiar, S. Ferrere, K. Steirer, Y. Yan, C. Xiao, J. Young, M. Al-Jassim, N. Neale and J. Turner, Nat. Energy, 2017, 2, 16192 CrossRef CAS.
- G. Lee, S. Anandan, S. Masten and J. Wu, Renewable Energy, 2016, 89, 18–26 CrossRef CAS.
- S. Kouser, S. Lingampalli, P. Chithaiah, A. Roy, S. Saha, U. Waghmare and C. Rao, Angew. Chem., 2015, 127, 8267–8271 CrossRef.
- H. Huang, B. Dai, W. Wang, C. Lu, J. Kou, Y. Ni, L. Wang and Z. Xu, Nano Lett., 2017, 17, 3803–3808 CrossRef CAS PubMed.
- Y. Zhou, G. Chen, Y. Yu, Y. Feng, Y. Zheng, F. He and Z. Han, Phys. Chem. Chem. Phys., 2014, 17, 1870–1876 RSC.
- M. Kimi, L. Yuliati and M. Shamsuddin, Int. J. Hydrogen Energy, 2011, 36, 9453–9461 CrossRef CAS.
- W. Zhang, Z. Zhong, Y. Wang and R. Xu, J. Phys. Chem. C, 2008, 112, 17635–17642 CrossRef CAS.
- H. Stein, R. Gutkowski, A. Siegel, W. Schuhmann and A. Ludwig, J. Mater. Chem. A, 2016, 4, 3148–3152 RSC.
- J. Yang, D. Wang, H. Han and C. Li, ACS. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- D. Wang, T. Hisatomi, T. Takata, C. Pan, M. Katayama, J. Kubota and K. Domen, Angew. Chem., Int. Ed., 2013, 52, 11252–11256 CrossRef CAS PubMed.
- J. Zhang, Z. Yu, Z. Gao, H. Ge, S. Zhao, C. Chen, S. Chen, X. Tong, M. Wang, Z. Zheng and Y. Qin, Angew. Chem., Int. Ed., 2017, 56, 816–820 CrossRef CAS PubMed.
- Y. Chen, S. Zhao, X. Wang, Q. Peng, R. Lin, Y. Wang, R. Shen, X. Gao, L. Zhang, G. Zhou, J. Li, A. Xia and Y. Li, J. Am. Chem. Soc., 2016, 138, 4286–4289 CrossRef CAS PubMed.
- X. Chen, Y. Li, X. Pan, D. Cortie, X. Huang and Z. Yi, Nat. Commun., 2016, 7, 12273 CrossRef CAS PubMed.
- A. Sathish and R. Viswanath, Catal. Today, 2007, 129, 421–427 CrossRef.
- J. Zhang, Y. Tang, K. Lee and O. Min, Science, 2010, 327, 1634–1638 CrossRef CAS PubMed.
- U. Banin, Y. Ben-Shahar and K. Vinokurov, Chem. Mater., 2014, 26, 97–110 CrossRef CAS.
- D. Seo, G. Park and H. Song, J. Am. Chem. Soc., 2012, 134, 1221–1227 CrossRef CAS PubMed.
- A. Mau, C. Huang, N. Kakuta, A. Bard, A. Campion, M. Fox, J. White and S. Webber, J. Am. Chem. Soc., 1984, 106, 6537–6542 CrossRef CAS.
- L. Yang, D. Zhong, J. Zhang, Z. Yan, S. Ge, P. Du, J. Jiang, D. Sun, X. Wu, Z. Fan, S. Dayeh and B. Xiang, ACS Nano, 2014, 8, 6979–6985 CrossRef CAS PubMed.
- N. Bao, L. Shen, T. Takata, D. Lu and K. Domen, Chem. Lett., 2014, 8, 6979–6985 Search PubMed.
- Y. Kim and H. Park, Energy Environ. Sci., 2011, 4, 685–694 RSC.
- G. Yuan, C. Hsia, Z. Lin, C. Chiang, Y. Chiang and M. Huang, Chem.–Eur. J., 2016, 22, 12548–12556 CrossRef CAS PubMed.
- J. Liu, K. Chen, G. Pan, Z. Luo, Y. Xie, Y. Li, Y. Lin, Z. Hao, L. Zhou, S. Ding and Q. Wang, Nanoscale, 2018, 10, 19586–19594 RSC.
- H. Li, Z. Bian, J. Zhu, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 4538–4539 CrossRef CAS PubMed.
- C. Chang, K. Orchard, B. Martindale and E. Reisner, J. Mater. Chem. A, 2016, 4, 2856–2862 RSC.
- W. Choi, G. Park, K. Bae, J. Choi, K. Nam and H. Song, J. Mater. Chem. A, 2016, 4, 13414–13418 RSC.
- F. Jiang, T. Harada, Y. Kuang, T. Minegishi, K. Domen and S. Ikeda, J. Am. Chem. Soc., 2015, 137, 13691–13697 CrossRef CAS PubMed.
- Y. Yuan, H. Lu, Z. Yu and Z. Zou, ChemSusChem, 2015, 8, 4113–4127 CrossRef CAS PubMed.
- S. Fang, S. Li, L. Ge, C. Han, P. Qiu and Y. Gao, Dalton Trans., 2017, 46, 10578–10585 RSC.
- P. Wang, Y. Xia, P. Wu, X. Wang, H. Yu and J. Yu, J. Phys. Chem. C, 2014, 118, 8891–8898 CrossRef CAS.
- M. Liu, X. Qiu, M. Miyauchi and K. Hashimoto, J. Am. Chem. Soc., 2013, 135, 10064–10072 CrossRef CAS PubMed.
- H. Kato, K. Asakura and A. Kudo, J. Am. Chem. Soc., 2003, 125, 3082–3089 CrossRef CAS PubMed.
- A. Ishikawa, T. Takata, J. Kondo, M. Hara, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2002, 124, 13547–13553 CrossRef CAS PubMed.
- H. Maghrabi, H. Ali and S. Younis, RSC Adv., 2017, 7, 4409–4421 RSC.
- K. Kalyanasundaram, E. Borgarello, D. Duonghong and M. Grätzel, Angew. Chem., Int. Ed., 1981, 20, 987–988 CrossRef.
- K. Maeda, T. Takata, M. Hara, N. Saito, Y. Inoue, H. Kobayashi and K. Domen, J. Am. Chem. Soc., 2005, 127, 8286–8287 CrossRef CAS PubMed.
- S. D. Tilley, M. Schreier, J. Azevedo, M. Stefik and M. Graetzel, Adv. Funct. Mater., 2014, 24, 303–311 CrossRef CAS.
- J. Yuan, J. Wen, Q. Gao, S. Chen, J. Li, X. Li and Y. Fang, Dalton Trans., 2015, 44, 1680–1689 RSC.
- Y. Liu, S. Ding, X. Liu, Z. Wu, Q. Jiang, T. Zhou, N. Liu and J. Hu, Appl. Catal., B, 2018, 234, 109–116 CrossRef CAS.
- W. Chen, T. Wang, J. Xue, S. Li, Z. Wang and S. Sun, Small, 2017, 13, 1602420 CrossRef PubMed.
- W. Vijselaar, R. Tiggelaar, H. Gardeniers and J. Huskens, ACS Energy Lett., 2018, 3, 1086–1092 CrossRef CAS PubMed.
- B. Guo, A. Batool, G. Xie, R. Boddula, L. Tian, S. Jan and J. Gong, Nano Lett., 2018, 18, 1516–1521 CrossRef CAS PubMed.
- D. Kumar, S. Hong, D. Reddy and T. Kim, J. Mater. Chem. A, 2016, 4, 18551–18558 RSC.
- K. Zhang, S. Qian, W. Kim, J. Kim, X. Sheng, J. Lee and J. Park, Nano Energy, 2017, 34, 481–490 CrossRef CAS.
- W. Zhao, Z. Ghorannevis, L. Chu, M. Toh, C. Kloc, P. Tan and G. Eda, ACS Nano, 2012, 7, 791–797 CrossRef PubMed.
- J. Chen, X. Wu, L. Yin, B. Li, X. Hong, Z. Fan, B. Chen, C. Xue and H. Zhang, Angew. Chem., Int. Ed., 2015, 54, 1210–1214 CrossRef CAS PubMed.
- Y. Zhong, G. Zhao, F. Ma, Y. Wu and X. Hao, Appl. Catal., B, 2016, 27, 466–472 CrossRef.
- F. Ma, Y. Wu, Y. Shao, Y. Zhong, J. Lv and X. Hao, Nano Energy, 2016, 27, 466–474 CrossRef CAS.
- Y. Kuang, Q. Jia, G. Ma, T. Hisatomi, T. Minegishi, H. Nishiyama, M. Nakabayashi, N. Shibata, T. Yamada, A. Kudo and K. Domen, Nat. Energy, 2016, 2, 16191 CrossRef.
- Y. Yuan, F. Wang, B. Hu, H. Lu, Z. Yu and Z. Zou, Dalton Trans., 2015, 44, 10997–11003 RSC.
- Z. Guan, P. Wang, Q. Li, Y. Li, X. Fu and J. Yang, Chem. Eng. J., 2017, 327, 397–405 CrossRef CAS.
- R. Prabhakar, W. Septina, S. Siol, T. Moehl, R. Wick-Joliat and S. Tilley, J. Mater. Chem. A, 2017, 5, 23139–23145 RSC.
- C. Morales-Guio, S. Tilley, H. Vrubel, M. Grätzel and X. Hu, Nat. Commun., 2014, 5, 3059 CrossRef PubMed.
- X. Zhou, J. Jin, X. Zhu, J. Huang, J. Yu, W. Wong and W. Wong, J. Mater. Chem. A, 2016, 4, 5282–5287 RSC.
- S. Cao, Y. Chen, C. Wang, X. Lv and W. Fu, Chem. Commun., 2015, 51, 8708–8711 RSC.
- H. Cheng, X. Lv, S. Cao, Z. Zhao, Y. Chen and W. Fu, Sci. Rep., 2016, 6, 19846 CrossRef CAS PubMed.
- X. Xie, N. Zhang, Z. Tang, M. Anpo and Y. Xu, Appl. Catal., B, 2018, 237, 43–49 CrossRef CAS.
- R. Xiao, C. Zhao, Z. Zou, Z. Chen, L. Tian, H. Xu, H. Tang, Q. Liu, Z. Lin and X. Yang, Appl. Catal., B DOI:10.1016/j.apcatb.2019.118382.
- Y. Huang and B. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14804–14806 CrossRef CAS PubMed.
- G. Zhao, Y. Sun, W. Zhou, X. Wang, K. Chang, G. Liu, H. Liu, T. Kako and J. Ye, Adv. Mater., 2017, 29, 1703258 CrossRef PubMed.
- J. Yang, H. Yan, X. Wang, F. Wen, Z. Wang, D. Fan, J. Shi and C. Li, J. Catal., 2012, 290, 151–157 CrossRef CAS.
- R. Li, H. Han, F. Zhang, D. Wang and C. Li, Energy Environ. Sci., 2014, 7, 1369 RSC.
- H. Yu, X. Huang, P. Wang and J. Yu, J. Phys. Chem. C, 2016, 120, 3722–3730 CrossRef CAS.
- D. Huang, X. Yan, M. Yan, G. Zeng, C. Zhou, J. Wan, M. Cheng, W. Xue and ACS Appl, Mater. Interfaces, 2018, 10, 21035–21055 CrossRef CAS PubMed.
- M. Wang, L. Cai, Y. Wang, F. Zhou, K. Xu, X. Tao and Y. Chai, J. Am. Chem. Soc., 2017, 139, 4144–4151 CrossRef CAS PubMed.
- L. Jia, D. H. Wang, Y. X. Huang, A. W. Xu and H. Q. Yu, J. Phys. Chem. C, 2011, 115, 11466–11473 CrossRef CAS.
- P. Tran, S. Batabyal, S. Pramana, J. Barber, L. Wong and S. Loo, Nanoscale, 2012, 4, 3875–3878 RSC.
- Y. Zhang, X. Cai, D. Guo, H. Zhang, N. Zhou, S. Fang, J. Chen and H. Zhang, J. Mater. Sci.: Mater. Electron., 2019, 30, 7182–7193 CrossRef CAS.
- J. Zhang, J. Yu, M. Jaroniec and J. Gong, Nano Lett., 2012, 12, 4584–4589 CrossRef CAS PubMed.
- A. Dubale, W. Su, A. Tamirat, C. Pan, B. Aragaw, H. Chen, C. Chen and B. Hwang, J. Mater. Chem. A, 2014, 2, 18383–18397 RSC.
- J. Zhang, Y. Wang, J. Jin, J. Zhang, Z. Lin, F. Huang and J. Yu, ACS Appl. Mater. Interfaces, 2013, 5, 10317–10324 CrossRef CAS PubMed.
- J. Kim, S. Bae, W. Kim, M. Jeong, S. Lee, C. Lee, W. Choi, J. Hwang, J. Park and D. Son, Nano Energy, 2015, 13, 258–266 CrossRef CAS.
- Y. Chen, G. Tian, Z. Ren, K. Pan, Y. Shi, J. Wang and H. Fu, ACS Appl. Mater. Interfaces, 2014, 6, 13841–13849 CrossRef CAS PubMed.
- Z. Zhang, R. Dua, L. Zhang, H. Zhu, H. Zhang and P. Wang, ACS Nano, 2013, 7, 1709–1717 CrossRef CAS PubMed.
- L. Yu, G. Li, X. Zhang, X. Ba, G. Shi, Y. Li, P. Wong, J. Yu and Y. Yu, ACS Catal., 2016, 6, 6444–6454, DOI:10.1021/nn3057092.
- Y. Yang, Y. Zhang, Z. Fang, L. Zhang, Z. Zheng, Z. Wang, W. Feng, S. Weng, S. Zhang and P. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 6950–6958 CrossRef CAS PubMed.
- Y. Xie, Z. Yu, G. Liu, X. Ma and H. Cheng, Energy Environ. Sci., 2014, 7, 1895–1901 RSC.
- J. Bandara, C. Udawatta and C. Rajapakse, Science, 2005, 4, 857–861 CAS.
- T. Yu, W. Cheng, K. Chao and S. Lu, Nanoscale, 2013, 5, 7356–7360 RSC.
- B. Dong, J. Cui, Y. Gao, Y. Qi, F. Zhang and C. Li, Adv. Mater., 2019, 31, 1808185 CrossRef PubMed.
- Y. Lou, Y. Zhang, L. Cheng, J. Chen and Y. Zhao, ChemSusChem, 2018, 11, 1505–1511 CrossRef CAS PubMed.
- N. Li, M. Liu, Z. Zhou, J. Zhou, Y. Sun and L. Guo, Nanoscale, 2014, 6, 9695–9702 RSC.
- D. Huang, S. Chen, G. Zeng, X. Gong, C. Zhou, M. Cheng, W. Xue, X. Yan and J. Li, Coord. Chem. Rev., 2019, 385, 44–80 CrossRef.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- W. Zhao, J. Liu, Z. Deng, J. Zhang, Z. Ding and Y. Fang, Int. J. Hydrogen Energy, 2018, 43, 18232–18241 CrossRef CAS.
- X. Wang, G. Liu, L. Wang, Z. Chen, G. Lu and H. Cheng, Adv. Energy Mater., 2012, 2, 42–46 CrossRef.
- X. Wang, G. Liu, Z. Chen, F. Li, L. Wang, G. Lu and H. Cheng, Chem. Commun., 2009, 23, 3452–3454 RSC.
- F. Zhou, J. Fan, Q. Xu and Y. Min, Appl. Catal., B, 2017, 201, 77–83 CrossRef CAS.
- D. Ma, J. Shi, Y. Zou, Z. Fan, X. Ji, C. Niu and L. Wang, Nano Energy, 2017, 39, 183–191 CrossRef CAS.
- X. Wang, G. Liu, G. Lu and H. Cheng, Int. J. Hydrogen Energy, 2010, 35, 8199–8205 CrossRef CAS.
- X. Ma, F. Zhao, Q. Qiang, T. Liu and Y. Wang, Dalton Trans., 2018, 47, 12162–12171 RSC.
- M. Liu, D. Jing, Z. Zhou and L. Guo, Nat. Commun., 2013, 4, 2278 CrossRef PubMed.
- J. Song, H. Zhao, R. Sun, X. Li and D. Sun, Energy Environ. Sci., 2017, 10, 225–235 RSC.
- Z. Ai, G. Zhao, Y. Zhong, Y. Shao, B. Huang, Y. Wu and X. Hao, Appl. Catal., B, 2018, 221, 179–186 CrossRef CAS.
- K. Li, M. Han, R. Chen, S. Li, S. Xie, C. Mao, X. Bu, X. Cao, L. Dong, P. Feng and Y. Lan, Adv. Mater., 2016, 28, 8906–8911 CrossRef CAS PubMed.
- Y. Hsu, N. Suen, C. Chang, S. Hung, C. Chen, T. Chan, C. Dong, C. Chan, S. Chen and H. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 22558–22569 CrossRef CAS PubMed.
- B. Ng, L. Putri, X. Kong, K. Shak, P. Pasbakhsh, S. Chai and A. Mohamed, Appl. Catal., B, 2018, 224, 360–367 CrossRef CAS.
- W. Fan, Q. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2013, 15, 2632–2649 RSC.
- L. Li, P. Salvador and G. Rohrer, Nanoscale Res. Lett., 2014, 6, 24–42 RSC.
- J. Yu, S. Zhuang, X. Xu, W. Zhu, B. Feng and J. Hu, J. Mater. Chem. A, 2015, 3, 1199–1207 RSC.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- Z. Sun, Q. Yue, J. Li, J. Xu, H. Zheng and P. Du, J. Mater. Chem. A, 2015, 3, 10243–10247 RSC.
- K. Zhang, W. Kim, M. Ma, X. Shi and J. Park, J. Mater. Chem. A, 2015, 3, 4803–4810 RSC.
- L. Zhang, Y. Li, X. Li, C. Li, R. Zhang, J. Delaunay and H. Zhu, Nano Energy, 2016, 28, 135–142 CrossRef CAS.
- X. Lu, W. Zhang, C. Wang, T. Wen and Y. Wei, Prog. Polym. Sci., 2011, 36, 671–712 CrossRef CAS.
- K. Lee, S. Cho, S. Park, A. Heeger, C. Lee and S. Lee, Nature, 2006, 441, 65–68 CrossRef CAS PubMed.
- H. Tran, D. Li and R. Kaner, Adv. Mater., 2009, 21, 1487–1499 CrossRef CAS.
- C. Wang, L. Wang, J. Jin, J. Liu and Y. Li, Appl. Catal., B, 2016, 188, 351–359 CrossRef CAS.
- B. Niu and Z. Xu, J. Catal., 2019, 371, 175–184 CrossRef CAS.
- Y. Hu, X. Hao, Z. Cui, J. Zhou, S. Chu, Y. Wang and Z. Zou, Appl. Catal., B, 2020, 260, 118131 CrossRef CAS.
- V. D. Dao, N. Chi, D. Thuan, T. D. Pham, D. T. Tran, M. Nguyen, P. Thao, M. Nguyen, N. Cam, N. Tuong, N. Dang and H. Choi, J. Alloys Compd., 2019, 775, 942–949 CrossRef CAS.
- C. Wang, Z. Hu, H. Zhao, W. Yu, S. Wu, J. Liu, L. Chen, Y. Li and B. Su, J. Colloid Interface Sci., 2018, 521, 1–10 CrossRef CAS PubMed.
- K. Daskalakis and G. Helz, Environ. Sci. Technol., 1992, 26, 2462–2468 CrossRef CAS.
- L. Zhang, H. Cheng, R. Zong and Y. Zhu, J. Phys. Chem. C, 2009, 113, 2368–2374 CrossRef.
- H. Sharma, K. Moumanis and J. Dubowski, J. Phys. Chem. C, 2016, 120, 26129–26137 CrossRef CAS.
- D. Lee and K. Choi, Nat. Energy, 2018, 3, 53–60 CrossRef CAS.
- D. Meissner, R. Memming and B. Kastening, J. Phys. Chem., 1988, 92, 3476–3483 CrossRef CAS.
- W. Gao, W. Zhang and G. Lu, Appl. Catal., B, 2017, 212, 23–31 CrossRef CAS.
- X. Ning, W. Zhen, Y. Wu and G. Lu, Appl. Catal., B, 2018, 226, 373–383 CrossRef CAS.
- W. Zhen, X. Ning, B. Yang, Y. Wu, Z. Li and G. Lu, Appl. Catal., B, 2018, 221, 243–257 CrossRef CAS.
- W. Zhen, X. Ning, M. Wang, Y. Wu and G. Lu, J. Catal., 2018, 367, 269–282 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |