
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeparating bulk and surface processes in NiOx electrocatalysts for water oxidation†
Sacha
Corby‡
 a,
Miguel-García
Tecedor‡
b,
Sven
Tengeler
c,
Céline
Steinert
c,
Benjamin
Moss
a,
Camilo A.
Mesa
a,
Hany F.
Heiba
d,
Anna A.
Wilson
a,
Bernhard
Kaiser
c,
Wolfram
Jaegermann
*c,
Laia
Francàs
*a,
Sixto
Gimenez
*b and
James R.
Durrant
*a
a,
Miguel-García
Tecedor‡
b,
Sven
Tengeler
c,
Céline
Steinert
c,
Benjamin
Moss
a,
Camilo A.
Mesa
a,
Hany F.
Heiba
d,
Anna A.
Wilson
a,
Bernhard
Kaiser
c,
Wolfram
Jaegermann
*c,
Laia
Francàs
*a,
Sixto
Gimenez
*b and
James R.
Durrant
*a
aDepartment of Chemistry and the Centre for Plastic Electronics, Imperial College London, South Kensington, London, SW7 2AZ, UK. E-mail: j.durrant@imperial.ac.uk; laia.francas@uab.cat
bInsitute of Advanced Materials (INAM), University Jaume I, 12071 Castello de la Plana, Spain. E-mail: sjulia@fca.uji.es
cInstitute of Material Science, Technical University Darmstadt, 64287 Darmstadt,, Germany. E-mail: jaegermann@surface.tu-darmstadt.de
dEarth Science and Engineering, Imperial College London, South Kensington, London, SW7 2AZ, UK
First published on 10th August 2020
Abstract
Nickel oxide-based catalysts currently represent the state of the art in electrochemical water oxidation in alkaline pH. However, much of their functionality remains poorly understood, particularly regarding catalytically active sites and mechanism. Herein, we conduct a thickness dependent study of sputter deposited NiOx films by electrochemical impedance spectroscopy and spectroelectrochemistry in order to differentiate bulk oxidation from catalytic activation. We find that while catalytic activation occurs throughout the film bulk, only the upper ≤5 nm of these films are able to participate in the water oxidation reaction, a result that may be critical in the design of next generation co-catalysts to maximise performance and minimise light absorption losses.
Introduction
Stable catalysts comprising Earth-abundant materials are required to generate sustainable yields of solar H2 from water. In particular, the fundamental but kinetically slow 4-hole oxidation of water to molecular oxygen benefits greatly from efficient catalysts, which may be employed either as co-catalysts in conjunction with light absorbing materials (as seen in PEC cells), or solely as electrocatalysts for dark catalysis (as in electrolysers).1–6 NiOOH is a highly promising material for water oxidation, currently challenging the more traditional, precious metal-based catalysts for smallest overpotentials and highest attainable current densities.7–10 With catalytic onsets as low as 1.50 V vs. RHE reported (η = 0.27 V at pH 13), nickel- and iron-based oxides and oxyhydroxides provide a benchmark for the development of future catalysts, and yet much of their current mechanistic function is still under debate.7,10,11 Furthermore, the relationship between the primary oxidation of Ni centres observed before the catalytic onset and further oxidations during catalysis remains unclear.12 Evidence of water penetration within the layered oxyhydroxide structure aligns with reports of catalytic activity beyond the atomic surface,13,14 though other works suggest that the active species are surface-generated oxo and superoxo species.12,15,16 Recent works on the model OER catalyst CoPi by the groups of Boettcher and Dau have indicated that bulk cobalt atoms are active for water oxidation, resulting in a proportionality between catalyst thickness and activity,17,18 but no clear consensus has been reached regarding bulk activity in the family of Ni/Fe oxyhydroxides. Alongside hydration, it is widely reported that the high affinity of nickel oxyhydroxide for iron allows for the latter to be incorporated into the NiOOH structure from trace impurities in alkaline electrolyte, adding complication to mechanistic understanding.19,20 With the electrochemically active area often ill-defined for these materials,21 determining where catalysis occurs remains a challenge, which when solved may greatly aid our understanding of these remarkably high performing materials and determine where future improvements can be targeted.In this work, we shine light on this uncertainty in the field by differentiating ‘bulk’ and ‘surface’ oxidation processes using different thicknesses of highly defective NiOx films sputtered on a conductive substrate. In coupling electrical and optical spectroscopic techniques, we elucidate that the initial oxidation of NiOx to NiOOH is a bulk oxidation process, penetrating deep into the film and thereby allowing incorporation of water and iron impurities into the layered structure. However, the intermediate species accumulated during water oxidation catalysis are limited to the near surface, ≤5 nm, despite the prior activation of the entire film. As such, much of the bulk material only contributes to resistance losses and fine nanostructuring of these electrocatalysts is predicted to greatly enhance performance.
Results and discussion
NiOx thin films were deposited on FTO coated glass by magnetron sputtering under inert atmosphere, the details of which may be found in the ESI†. By controlling the deposition time, films of 5, 10, 20 and 100 nm were synthesised, their thicknesses confirmed by XPS as detailed in previous work.22 The NiOx structure was most readily apparent in the thickest film but the presence of Ni was confirmed for each thickness using EDS, and gave the expected proportionality (Fig. S2†). The as-deposited films were examined by XPS before and after exposure to electrolyte, and the thickest films were also examined by XRD, allowing confirmation of the bunsenite NiOx structure, which is further oxidised with use, as discussed below (Fig. S3–S5†).The films were interrogated electrochemically in 0.1 M KOH electrolyte, reported to afford the highest performances for Ni-based catalysts.23 The initial cyclic voltammogram (CV) for a 10 nm film is shown in Fig. 1 (red trace), with similar CVs for the other thicknesses given in Fig. S6.† A shallow, broad redox wave can be observed ∼150 mV before the catalytic onset at 1.64 VRHE, which we assign to the oxidation of Ni2+ to Ni3+ and the concurrent formation of NiOOH. The catalytic onset or onset potential is defined herein as the potential where the extrapolated faradaic current density reaches 50 μA cm−2, as detailed in the ESI Fig. S7, Section F.† This oxidation and hydration process associated with the Ni2+/Ni3+ transformation has been widely reported in the literature and known to be a necessary step in the activation of these films for water oxidation catalysis.16,24–27 In order to activate as much of the deposited material as possible, 50 CV scans were performed over potentials corresponding to this redox wave, resulting in a clear enhancement in the wave amplitude from the oxidation of more Ni centres, as seen in Fig. 1 (dark trace). An improvement in the catalytic current can also be observed following this activation (see Fig. S6† for electrochemical activation of the other thicknesses), stabilising at the best performance after 50 CVs for the 5, 10 and 20 nm films. While the redox wave growth correlates with greater film activation, the catalytic enhancement also agrees with several reports in literature of Fe incorporation from impurities in standard grade alkaline electrolytes, with Fe traces as low as 1 ppm able to significantly improve the OER activity of similar electrocatalysts.20,28,29
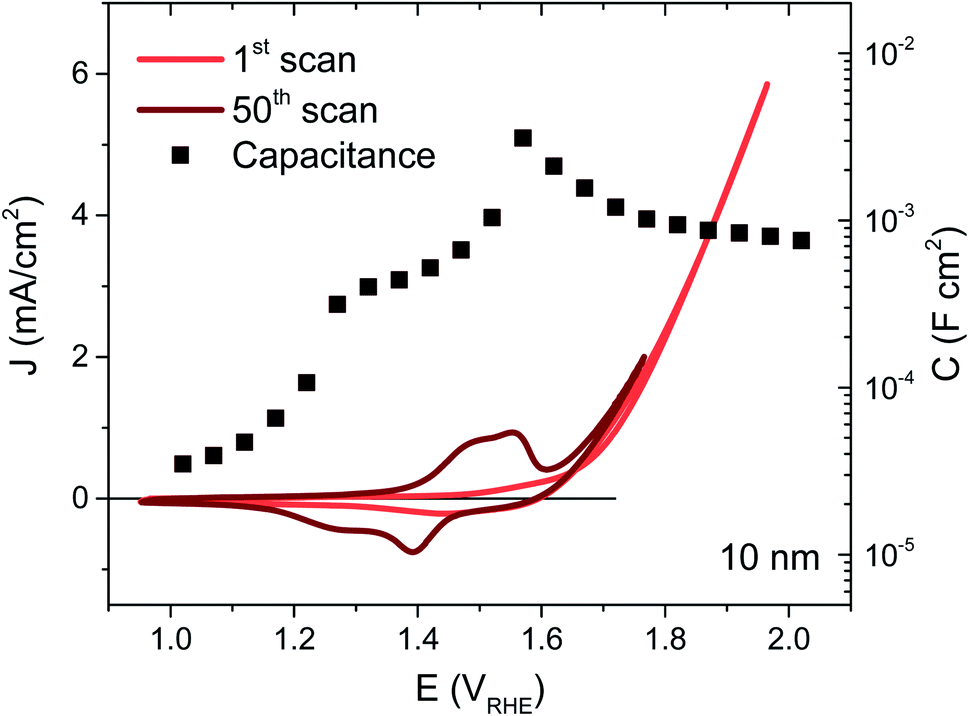 | ||
| Fig. 1 Cyclic voltammetry of 10 nm thick NiOx after 1 scan (red line) and 50 scans (dark red line), and the corresponding capacitance data (black squares) over the same potentials. The CV scan rate was 50 mV s−1 and 0.1 M KOH was used for both impedance and electrochemistry. | ||
The capacitance over the same applied potentials was determined from impedance spectroscopy using the equivalent circuit models given in Fig. S8† and resultant experimental and fitted Nyquist plots (see Fig. S9† for example at 1.6 VRHE). Exemplified in Fig. 1 for the 10 nm sample (squares), the capacitance increases with applied potential with a well-defined peak over the redox wave and a gradual decrease over the catalytic region (evident at each thickness as shown in Fig. S6†). This peak in capacitance is thereby associated with the bulk transformation of Ni(OH)2 to NiOOH, before the onset of catalysis. Furthermore, the capacitance increases with film thickness and the amount of material available. This behaviour is typical of a chemical capacitance, from which an approximate density of states (DOS) may be calculated by dividing by the charge of an electron, as shown in Fig. S10.†30 The agreement in energetic maxima of the DOS between samples and the increase in density of states available with thickness strengthens the argument that the same bulk oxidation is required for each film, as expected. A semi-molecular approach in the analysis of these materials is pertinent given the local nature of the oxidation process (requiring direct water/hydroxide contact),16,27 and while treatment of these films as semiconductors may not be constructive for understanding their mechanistic activity in the catalytic region, Mott–Schottky analysis in the pre-redox region (<1.2 VRHE) reveals a doping density around 1021 cm−3 (Fig. S11†), thereby confirming the highly defective nature of these materials.31 Higher catalytic activities have previously been associated with highly defective and disordered phases of NiOOH.32 Furthermore, the flat band potential obtained from this analysis at 1.2 VRHE agrees well with previously literature reports.33–35 In the case of the 100 nm sample, the relatively large amount of material gives rise to a large and broad redox wave, which proceeds to grow past 50 CV scans. This therefore indicates that the entire bulk of this film is not oxidised to NiOOH by our activation process, as supported by the similarity in before and after XPS data of Ni 2p and O 1s for this sample (Fig. S4 and S5†) and the SEC results shown in Fig. S18.† This has been observed in other metal oxide systems and attributed to increasing inhibition of water and hydroxide ion transfer to the inner depths of the oxide layer.36 As such, the DOS is likely to be underestimated for this thickness and this sample was not considered further.
After immersion in alkaline electrolyte, the oxidation of Ni(OH)2 proceeds via the following reaction: Ni(OH)2 + OH− → NiOOH + H2O + 1e−, with NiOOH, or rather Fe-doped NiOOH (Ni(Fe)OOH), the basis for OER catalysis.32 By comparing the 50th CV scan for 5, 10 and 20 nm thick films, a relationship between film thickness and the redox capacitance (size of the redox wave) can be observed (Fig. 2a). This confirms that this oxidation pre-requisite to catalysis is very much a bulk process, with the electrolyte penetrating deep into the film to form NiOOH throughout. This result correlates with the layered nature of these films, which can swell to incorporate water (hydration process).28,36,37 The 5 nm and 10 nm films appear to have a similar catalytic onset (1.63 and 1.64 VRHE, respectively) after the film activation, whereas the 20 nm film has an earlier onset, at 1.55 VRHE, addressed below.
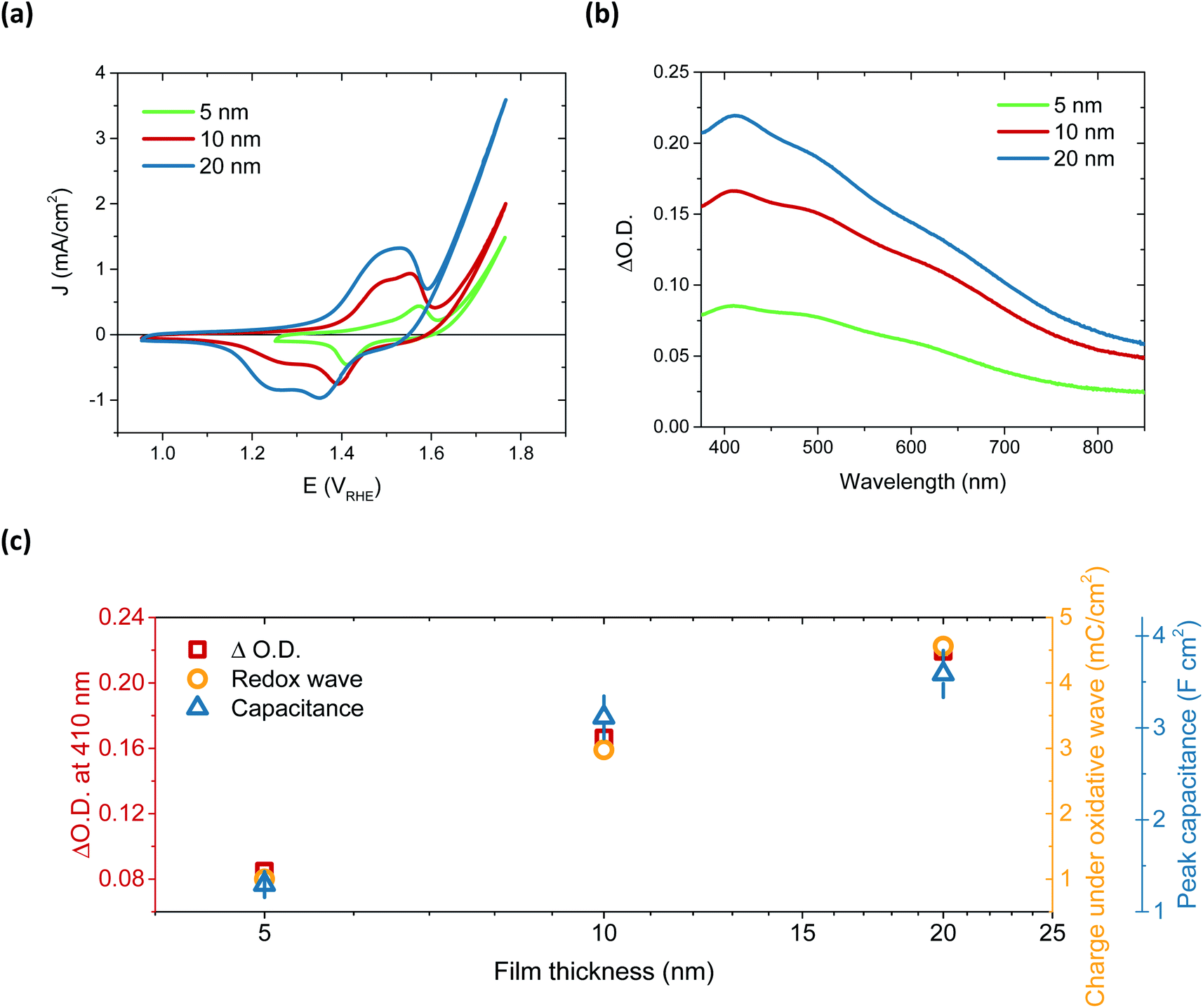 | ||
| Fig. 2 (a) The 50th CV scan for 5 (green), 10 (red) and 20 nm (blue) films, 50 mV s−1 in 0.1 M KOH electrolyte. (b) Spectroelectrochemistry showing the change in absorbance after activation for each sample with respect to the absorbance before activation; 5 nm (green), 10 nm (red), 20 nm (blue). (c) Comparison of the area of the redox wave, peak capacitance value from impedance and absorbance maximum from spectroelectrochemistry of this first oxidation for each thickness. | ||
The activation of Ni(OH)2 to NiOOH also induces a reversible colour change, as previously reported (Fig. S13†),27,38,39 allowing for this transition to be probed by spectroelectrochemistry. By monitoring the change in optical absorption of the films after activation, i.e. subtracting the initial absorption spectra from those obtained after 50 CV scans, the residual spectra of the oxidised species formed were obtained. These are given in Fig. 2b and clearly demonstrate that the same species, NiOOH, is formed in each case, as the spectra are very similar (normalisation shown in Fig. S14†) and agree with other literature reports.24,40 It is equally evident that increasing film thickness results in greater absorption, thereby supporting the electrochemical assignment of this initial Ni(OH)2/NiOOH transition to a bulk oxidation process. This thickness dependent absorption, alongside the increase in chemical capacitance with thickness, is displayed in Fig. 2c. While doubling of the film thickness from 5 nm to 10 nm results in the anticipated doubling of both the optical signal and chemical capacitance, this proportionality is not maintained in doubling again to give 20 nm. This thereby suggests that, as with the 100 nm sample, there remains some small portion of the film which is not adequately hydrated to allow NiOOH to be synthesised.
Performing spectroelectrochemistry across potentials that give rise to catalytic current produces a different result, with the appearance of a new feature. As with the first oxidation, similar difference spectra are produced for each thickness, now with central peaks at 510 nm and 625 nm, as demonstrated in Fig. 3a for the 5, 10 and 20 nm films at 0.1 V after catalytic onset. (See Fig. S15† for independent incremental increases in optical data for each thickness and normalised data in Fig. S16†). With increasing anodic potential, these peaks grow steadily due to the increased accumulation of the oxidised species from which they arise. As these data correspond to steady-state catalysis, the resulting spectra represent the absorbance profile of the population of reactive intermediates accumulated under steady-state conditions to drive water oxidation catalysis. Notably, when comparing the onset of accumulation of these intermediates to the current onset (Fig. 3b), it is clear that a degree of accumulation is necessary for catalytic current to be produced, in line with the multi-step nature of water oxidation.27 It is also evident that this accumulation begins ∼50 mV earlier for the 20 nm film, in agreement with the earlier catalytic onset of this thicker film, and thereby indicating that current onset is directly correlated with the relative ease of oxidised species accumulation in such materials. To understand this behaviour, we carried out Raman spectroscopy on both 5 nm and 20 nm films (Fig. S17†) and observed that the broad emission centred at 550 cm−1 in the 5 nm sample is shifted to higher wavenumbers in the 20 nm sample, which may be indicative of disproportionately higher Fe incorporation in the 20 nm films.32 Increased concentrations of Fe atoms in NiOOH structures have previously been shown to lower the onset for OER catalysis, in agreement with the observations herein.19,20,27,41 Alternatively, the improved onset of the 20 nm films may stem from different proportions of β-NiOOH and γ-NiOOH, which are reported to have difference OER efficiencies and different affinities for Fe.42–44 Appearance of different phases would also be expected to induce changes to the Raman spectrum as seen for the 20 nm film. Unfortunately, the small thicknesses of the samples inhibited adequate determination of the phases present by XRD and thus further elucidation on this point could not be made.
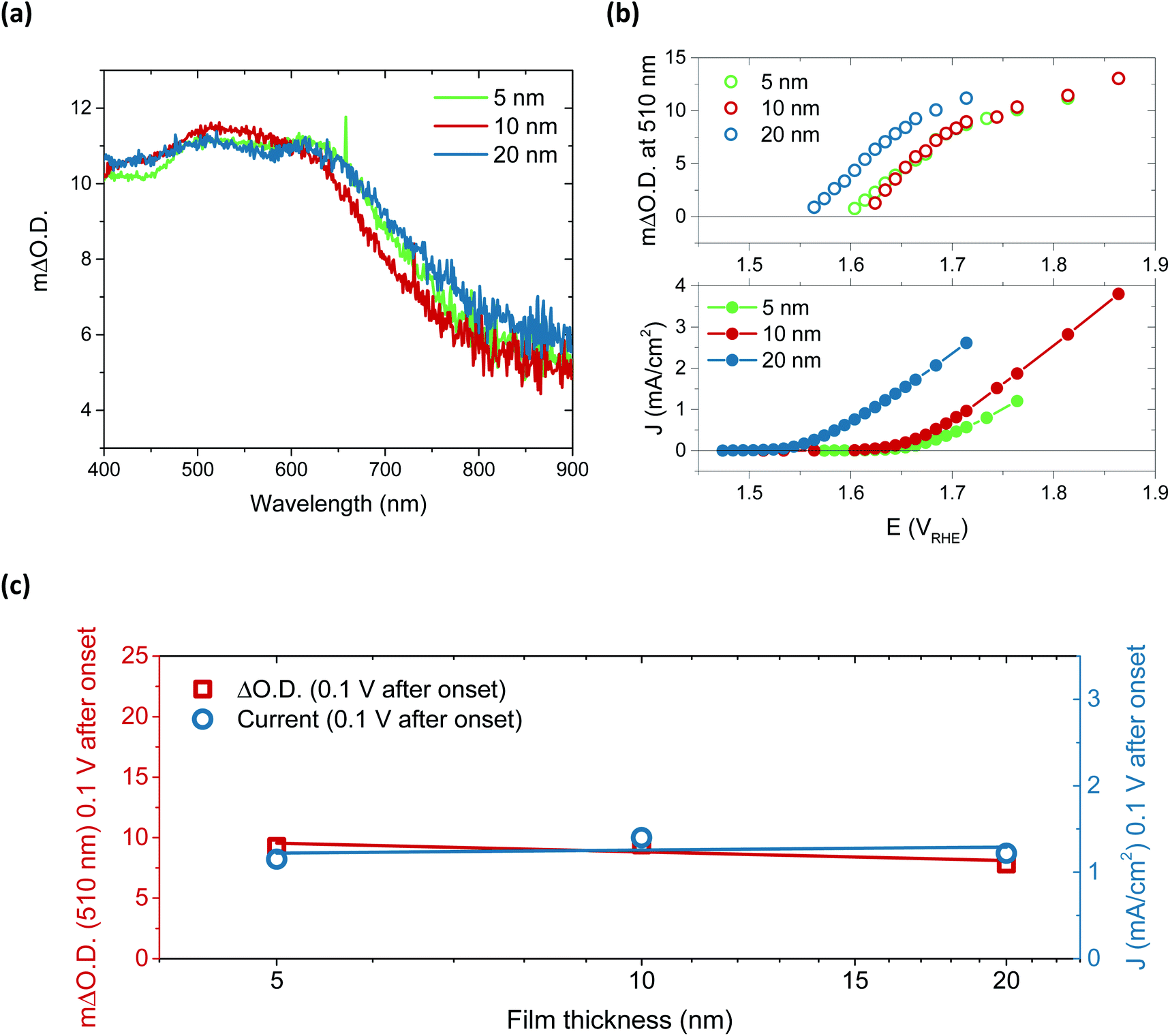 | ||
| Fig. 3 (a) Spectroelectrochemistry data applying 0.1 V beyond the catalytic onset to ensure each sample is examined under the same catalytic regime (this equates to 1.74 VRHE for 5 and 10 nm, and 1.65 VRHE for the 20 nm sample). (b) The change in absorption at 510 nm for each film thickness as a function of applied potential over catalytic range (top), and the corresponding steady-state current obtained simultaneously from the spectroelectrochemistry measurement (bottom) in 0.1 M KOH. In both (a) and (b), 5 nm (green), 10 nm (red), and 20 nm (blue) are shown. (c) The catalytic current and optical density at 510 nm and 0.1 V after current onset against film thickness, revealing no increase in the population of accumulated species with thickness. | ||
Arguably the most striking trend observed in these data is the similarity in optical data at the same current density. These data account for the difference in onset, the ΔO.D. in each case presented at 0.1 V after the respective catalytic onset. Indeed, if we were to shift the data in Fig. 3b by the difference in onset between the samples, we would find that both the optical data in the upper panel and current data in the lower panel would overlap at each thickness across the entire potential range measured. The degree of similarity in the species and currents we observe between samples over catalytic potentials is highlighted in the comparison in Fig. 3c. Both the steady-state current and peak optical amplitude remain approximately constant across each of the samples examined, independent of thickness for the same potential past onset. This result stands in direct contrast to the first oxidation–hydration process (Fig. 2), which demonstrated increasing absorption amplitudes with increasing thickness. The similarity in optical amplitude implies that a similar number of species are accumulating in each sample. The formation of these accumulated species must therefore be limited to the thickness of the thinnest film, i.e. the upper 5 nm or less of NiOOH, despite the initial oxidation of the entirety of the film (Fig. 2).
Furthermore, this result indicates that the accumulating oxidised species are not formed in the ‘bulk’ of the film, presumably due to instability or restricted access to water. With this, we can conclude that the hydration of the NiOx structure observed in Fig. 2 is essential for the greater oxidative conversion of Ni2+ to Ni3+ deep into the structure, but that the quantity of NiOOH generated does not directly correlate to the number of catalytically active sites in the films, with only those centres ≤5 nm of the upper electrode surface able to accumulate species driving water oxidation catalysis, as summarised in Scheme 1. This result is in agreement with pH dependent data reported by Koper and co-workers which show that the catalytic activity of NiOOH in alkaline media is dependent on surface MO− and MOO− species formation,16 as has been reported for other similar materials.45,46 Likewise a recent investigation using isotope labelling of NiFeOxHy particles reports that oxygen evolution is limited to the near-surface region, in line with our conclusions herein.47 By contrast, the DFT work of Doyle et al. reports that, given the hydrated layered structure of these oxyhydroxides, ‘bulk’ sites could contribute to water oxidation catalysis,14 whereas our results indicate that this is not the case in these sputtered films.
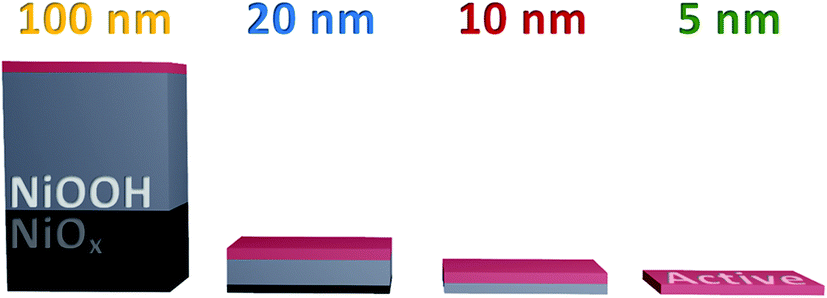 | ||
| Scheme 1 Graphic representation of each thickness of NiOx sample, showing the relative thicknesses of active material to bulk oxidised NiOOH and unaltered NiOx. Only the upper 5 nm or less are active for water oxidation. | ||
In summary, we demonstrate that the activation of NiOx to NiOOH is a bulk oxidation process, concomitant to hydration and Fe incorporation, whereas the intermediates observed during steady-state catalysis in these sputtered films only accumulate at the near surface, ≤5 nm from the atomic interface of NiOOH with the electrolyte. It is not clear from this study alone whether this trend is likely to hold for all layered oxyhydroxide materials with different degrees of layering and porosity. However, this study highlights that the potential redundancy of thicker catalyst layers, which may incur resistance losses, should be considered in the analysis of any new materials for solar fuels applications.
Author contributions
Conceptualisation: S. C., M. G. T., S. T., L. F., S. G., B. K., J. R. D.; sample synthesis: S. T., C. S.; data collection: S. C., M. G. T., B. M., H. F. H., A. W.; data analysis: S. C., M. G. T., B. M.; experimental design/discussion: S. C., M. G. T., C. A. M., L. F., S. G., J. R. D.; visualization: S. C., M. G. T.; writing: S. C.; funding acquisition: W. J., S. G., J. R. D.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the EU FET programme (A-LEAF 732840). S. C. thanks Imperial College London for a Schrödinger Scholarship, L. F. thanks the EU for a Marie Curie fellowship (658270), C. A. M also thanks COLCIENCIAS (call 568) for funding and B. M. thanks the EPRSC for a DTP scholarship. S. G. acknowledges financial support from Ministerio de Ciencia, Innovación y Universidades of Spain (project ENE2017-85087-C3-1-R). W. J. and B. K. are members of the Excellency Graduate School of the German Science Foundation (DFG GSC 1070) “Energy Science and Engineering".References
- T. W. Kim and K.-S. Choi, Nanoporous BiVO4 Photoanodes with Dual-Layer Oxygen Evolution Catalysts for Solar Water Splitting, Science, 2014, 343(6174), 990–994 CrossRef CAS PubMed.
- D. K. Zhong, M. Cornuz, K. Sivula, M. Grätzel and D. R. Gamelin, Photo-assisted electrodeposition of cobalt–phosphate (Co–Pi) catalyst on hematite photoanodes for solar water oxidation, Energy Environ. Sci., 2011, 4(5), 1759–1764 RSC.
- D. K. Zhong, J. Sun, H. Inumaru and D. R. Gamelin, Solar Water Oxidation by Composite Catalyst/α-Fe2O3 Photoanodes, J. Am. Chem. Soc., 2009, 131(17), 6086–6087 CrossRef CAS PubMed.
- J. K. Hurst, Pursuit of Water Oxidation Catalysts for Solar Fuel Production, Science, 2010, 328(5976), 315–316 CrossRef CAS PubMed.
- P. Du and R. Eisenberg, Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: Recent progress and future challenges, Energy Environ. Sci., 2012, 5(3), 6012–6021 RSC.
- F. Malara, A. Minguzzi, M. Marelli, S. Morandi, R. Psaro, V. Dal Santo and A. Naldoni, α-Fe2O3/NiOOH: An Effective Heterostructure for Photoelectrochemical Water Oxidation, ACS Catal., 2015, 5(9), 5292–5300 CrossRef CAS.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, Efficient Water Oxidation Using Nanostructured α-Nickel-Hydroxide as an Electrocatalyst, J. Am. Chem. Soc., 2014, 136(19), 7077–7084 CrossRef CAS PubMed.
- K. Fominykh, P. Chernev, I. Zaharieva, J. Sicklinger, G. Stefanic, M. Döblinger, A. Müller, A. Pokharel, S. Böcklein, C. Scheu, T. Bein and D. Fattakhova-Rohlfing, Iron-Doped Nickel Oxide Nanocrystals as Highly Efficient Electrocatalysts for Alkaline Water Splitting, ACS Nano, 2015, 9(5), 5180–5188 CrossRef CAS PubMed.
- K. Fominykh, J. M. Feckl, J. Sicklinger, M. Döblinger, S. Böcklein, J. Ziegler, L. Peter, J. Rathousky, E.-W. Scheidt, T. Bein and D. Fattakhova-Rohlfing, Ultrasmall Dispersible Crystalline Nickel Oxide Nanoparticles as High-Performance Catalysts for Electrochemical Water Splitting, Adv. Funct. Mater., 2014, 24(21), 3123–3129 CrossRef CAS.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, An Advanced Ni–Fe Layered Double Hydroxide Electrocatalyst for Water Oxidation, J. Am. Chem. Soc., 2013, 135(23), 8452–8455 CrossRef CAS PubMed.
- Z. Zeng, K.-C. Chang, J. Kubal, N. M. Markovic and J. Greeley, Stabilization of ultrathin (hydroxy)oxide films on transition metal substrates for electrochemical energy conversion, Nat. Energy, 2017, 2, 17070 CrossRef CAS.
- F. Lin and S. W. Boettcher, Adaptive semiconductor/electrocatalyst junctions in water-splitting photoanodes, Nat. Mater., 2013, 13, 81 CrossRef PubMed.
- H. S. Ahn and A. J. Bard, Surface Interrogation Scanning Electrochemical Microscopy of Ni1–xFexOOH (0 < x < 0.27) Oxygen Evolving Catalyst: Kinetics of the “fast” Iron Sites, J. Am. Chem. Soc., 2016, 138(1), 313–318 CrossRef CAS PubMed.
- A. D. Doyle, M. Bajdich and A. Vojvodic, Theoretical Insights to Bulk Activity Towards Oxygen Evolution in Oxyhydroxides, Catal. Lett., 2017, 147(6), 1533–1539 CrossRef.
- M. Wehrens-Dijksma and P. H. L. Notten, Electrochemical quartz microbalance characterization of Ni(OH)2-based thin film electrodes, Electrochim. Acta, 2006, 51(18), 3609–3621 CrossRef CAS.
- O. Diaz-Morales, D. Ferrus-Suspedra and M. T. M. Koper, The importance of nickel oxyhydroxide deprotonation on its activity towards electrochemical water oxidation, Chem. Sci., 2016, 7(4), 2639–2645 RSC.
- K. Klingan, F. Ringleb, I. Zaharieva, J. Heidkamp, P. Chernev, D. Gonzalez-Flores, M. Risch, A. Fischer and H. Dau, Water Oxidation by Amorphous Cobalt-Based Oxides: Volume Activity and Proton Transfer to Electrolyte Bases, ChemSusChem, 2014, 7(5), 1301–1310 CrossRef CAS PubMed.
- M. R. Nellist, F. A. L. Laskowski, J. Qiu, H. Hajibabaei, K. Sivula, T. W. Hamann and S. W. Boettcher, Potential-sensing electrochemical atomic force microscopy for in operando analysis of water-splitting catalysts and interfaces, Nat. Energy, 2018, 3(1), 46–52 CrossRef CAS.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M.-J. Cheng, D. Sokaras, T.-C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, Identification of Highly Active Fe Sites in (Ni,Fe)OOH for Electrocatalytic Water Splitting, J. Am. Chem. Soc., 2015, 137(3), 1305–1313 CrossRef CAS PubMed.
- D. A. Corrigan, The Catalysis of the Oxygen Evolution Reaction by Iron Impurities in Thin Film Nickel Oxide Electrodes, J. Electrochem. Soc., 1987, 134(2), 377–384 CrossRef CAS.
- M. Gong and H. Dai, A mini review of NiFe-based materials as highly active oxygen evolution reaction electrocatalysts, Nano Res., 2015, 8(1), 23–39 CrossRef CAS.
- S. Tengeler, B. Kaiser, D. Chaussende and W. Jaegermann, (001) 3C SiC/Ni contact interface: in situ XPS observation of annealing induced Ni2Si formation and the resulting barrier height changes, Appl. Surf. Sci., 2017, 400(Suppl. C), 6–13 CrossRef CAS.
- J. Zaffran, M. B. Stevens, C. D. M. Trang, M. Nagli, M. Shehadeh, S. W. Boettcher and M. Caspary Toroker, Influence of Electrolyte Cations on Ni(Fe)OOH Catalyzed Oxygen Evolution Reaction, Chem. Mater., 2017, 29(11), 4761–4767 CrossRef CAS.
- M. Görlin, J. Ferreira de Araújo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, Tracking Catalyst Redox States and Reaction Dynamics in Ni–Fe Oxyhydroxide Oxygen Evolution Reaction Electrocatalysts: The Role of Catalyst Support and Electrolyte pH, J. Am. Chem. Soc., 2017, 139(5), 2070–2082 CrossRef PubMed.
- R. D. L. Smith, C. Pasquini, S. Loos, P. Chernev, K. Klingan, P. Kubella, M. R. Mohammadi, D. González-Flores and H. Dau, Geometric distortions in nickel (oxy)hydroxide electrocatalysts by redox inactive iron ions, Energy Environ. Sci., 2018, 11(9), 2476–2485 RSC.
- J. Su, L. Guo, N. Bao and C. A. Grimes, Nanostructured WO3/BiVO4 Heterojunction Films for Efficient Photoelectrochemical Water Splitting, Nano Lett., 2011, 11(5), 1928–1933 CrossRef CAS PubMed.
- L. Francàs, S. Corby, S. Selim, D. Lee, C. A. Mesa, R. Godin, E. Pastor, I. E. L. Stephens, K.-S. Choi and J. R. Durrant, Spectroelectrochemical study of water oxidation on nickel and iron oxyhydroxide electrocatalysts, Nat. Commun., 2019, 10(1), 5208 CrossRef PubMed.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, Effects of Fe Electrolyte Impurities on Ni(OH)2/NiOOH Structure and Oxygen Evolution Activity, J. Phys. Chem. C, 2015, 119(13), 7243–7254 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, Nickel–Iron Oxyhydroxide Oxygen-Evolution Electrocatalysts: The Role of Intentional and Incidental Iron Incorporation, J. Am. Chem. Soc., 2014, 136(18), 6744–6753 CrossRef CAS PubMed.
- J. Bisquert, Chemical capacitance of nanostructured semiconductors: its origin and significance for nanocomposite solar cells, Phys. Chem. Chem. Phys., 2003, 5(24), 5360–5364 RSC.
- S. Tengeler, M. Fingerle, W. Calvet, C. Steinert, B. Kaiser, T. Mayer and W. Jaegermann, The Impact of Different Si Surface Terminations in the (001) n-Si/NiOx Heterojunction on the Oxygen Evolution Reaction (OER) by XPS and Electrochemical Methods, J. Electrochem. Soc., 2018, 165(4), H3122–H3130 CrossRef CAS.
- M. W. Louie and A. T. Bell, An Investigation of Thin-Film Ni–Fe Oxide Catalysts for the Electrochemical Evolution of Oxygen, J. Am. Chem. Soc., 2013, 135(33), 12329–12337 CrossRef CAS PubMed.
- K. Darowicki, S. Krakowiak and P. Ślepski, Selection of measurement frequency in Mott–Schottky analysis of passive layer on nickel, Electrochim. Acta, 2006, 51(11), 2204–2208 CrossRef CAS.
- G. Barral, S. Maximovitch and F. Njanjo-Eyoke, Study of electrochemically formed Ni(OH)2 layers by EIS, Electrochim. Acta, 1996, 41(7), 1305–1311 CrossRef CAS.
- B. R. Cruz-Ortiz, M. A. Garcia-Lobato, E. R. Larios-Durán, E. M. Múzquiz-Ramos and J. C. Ballesteros-Pacheco, Potentiostatic electrodeposition of nanostructured NiO thin films for their application as electrocatalyst, J. Electroanal. Chem., 2016, 772, 38–45 CrossRef CAS.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. G. Lyons, Redox and electrochemical water splitting catalytic properties of hydrated metal oxide modified electrodes, Phys. Chem. Chem. Phys., 2013, 15(33), 13737–13783 RSC.
- H. Bode, K. Dehmelt and J. Witte, Zur kenntnis der nickelhydroxidelektrode—I. Über das nickel (II)-hydroxidhydrat, Electrochim. Acta, 1966, 11(8), 1079 CrossRef CAS.
- B. J. Trześniewski, O. Diaz-Morales, D. A. Vermaas, A. Longo, W. Bras, M. T. M. Koper and W. A. Smith, In Situ Observation of Active Oxygen Species in Fe-Containing Ni-Based Oxygen Evolution Catalysts: The Effect of pH on Electrochemical Activity, J. Am. Chem. Soc., 2015, 137(48), 15112–15121 CrossRef PubMed.
- R. M. Bendert and D. A. Corrigan, Effect of Coprecipitated Metal Ions on the Electrochromic Properties of Nickel Hydroxide, J. Electrochem. Soc., 1989, 136(5), 1369–1374 CrossRef CAS.
- Z. K. Goldsmith, A. K. Harshan, J. B. Gerken, M. Vörös, G. Galli, S. S. Stahl and S. Hammes-Schiffer, Characterization of NiFe oxyhydroxide electrocatalysts by integrated electronic structure calculations and spectroelectrochemistry, Proc. Natl. Acad. Sci. U.S.A., 2017, 114(12), 3050–3055 CrossRef CAS PubMed.
- L. Demourgues-Guerlou, L. Fournès and C. Delmas, On the Iron Oxidation State in the Iron-Substituted γ Nickel Oxyhydroxides, J. Solid State Chem., 1995, 114(1), 6–14 CrossRef CAS.
- Y.-F. Li and A. Selloni, Mechanism and activity of water oxidation on selected surfaces of pure and Fe-doped NiOx, ACS Catal., 2014, 4(4), 1148–1153 CrossRef CAS.
- B. S. Yeo and A. T. Bell, In situ Raman study of nickel oxide and gold-supported nickel oxide catalysts for the electrochemical evolution of oxygen, J. Phys. Chem. C, 2012, 116(15), 8394–8400 CrossRef CAS.
- J. D. Michael, E. L. Demeter, S. M. Illes, Q. Fan, J. R. Boes and J. R. Kitchin, Alkaline electrolyte and Fe impurity effects on the performance and active-phase structure of NiOOH thin films for OER catalysis applications, J. Phys. Chem. C, 2015, 119(21), 11475–11481 CrossRef CAS.
- D. K. Bediako, Y. Surendranath and D. G. Nocera, Mechanistic Studies of the Oxygen Evolution Reaction Mediated by a Nickel–Borate Thin Film Electrocatalyst, J. Am. Chem. Soc., 2013, 135(9), 3662–3674 CrossRef CAS PubMed.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces, ChemCatChem, 2011, 3(7), 1159–1165 CrossRef CAS.
- C. Roy, B. Sebok, S. B. Scott, E. M. Fiordaliso, J. E. Sørensen, A. Bodin, D. B. Trimarco, C. D. Damsgaard, P. C. K. Vesborg, O. Hansen, I. E. L. Stephens, J. Kibsgaard and I. Chorkendorff, Impact of nanoparticle size and lattice oxygen on water oxidation on NiFeOxHy, Nat. Catal., 2018, 1(11), 820–829 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0se00977f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |