
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStability of radical-functionalized gold surfaces by self-assembly and on-surface chemistry†
Tobias
Junghoefer‡
 a,
Ewa Malgorzata
Nowik-Boltyk‡
a,
J. Alejandro
de Sousa
bd,
Erika
Giangrisostomi
c,
Ruslan
Ovsyannikov
c,
Thomas
Chassé
a,
Jaume
Veciana
b,
Marta
Mas-Torrent
b,
Concepció
Rovira
b,
Núria
Crivillers
b and
Maria Benedetta
Casu
*a
a,
Ewa Malgorzata
Nowik-Boltyk‡
a,
J. Alejandro
de Sousa
bd,
Erika
Giangrisostomi
c,
Ruslan
Ovsyannikov
c,
Thomas
Chassé
a,
Jaume
Veciana
b,
Marta
Mas-Torrent
b,
Concepció
Rovira
b,
Núria
Crivillers
b and
Maria Benedetta
Casu
*a
aInstitute of Physical and Theoretical Chemistry, University of Tübingen, 72076 Tübingen, Germany. E-mail: benedetta.casu@uni-tuebingen.de
bInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Networking Research Center on Bioengineering Biomaterials and Nanomedicine (CIBER-BBN), Campus de la UAB, 08193 Bellaterra, Spain
cHelmholtz-Zentrum Berlin für Materialien und Energie (HZB), 12489 Berlin, Germany
dLaboratorio de Electroquímica, Departamento de Química, Facultad de Ciencias, Universidad de los Andes, 5101 Mérida, Venezuela
First published on 13th August 2020
Abstract
We have investigated the radical functionalization of gold surfaces with a derivative of the perchlorotriphenylmethyl (PTM) radical using two methods: by chemisorption from the radical solution and by on-surface chemical derivation from a precursor. We have investigated the obtained self-assembled monolayers by photon-energy dependent X-ray photoelectron spectroscopy. Our results show that the molecules were successfully anchored on the surfaces. We have used a robust method that can be applied to a variety of materials to assess the stability of the functionalized interface. The monolayers are characterized by air and X-ray beam stability unprecedented for films of organic radicals. Over very long X-ray beam exposure we observed a dynamic nature of the radical–Au complex. The results clearly indicate that (mono)layers of PTM radical derivatives have the necessary stability to withstand device applications.
Introduction
Molecular systems are materials that intersect with many different promising fields such as organic/molecular spintronics, electronics, and organic magnetism.1–8 In this framework, organic radicals are exceptionally promising in various fields, and the research on radical thin films and interfaces has recently flourished, due to their potential use in applications from quantum computing to organic electronics and spintronics.8–13We have recently demonstrated that a Blatter radical derivative is a potential quantum bit and we attached it to copper contacts to investigate the influence of a substrate on the radical magnetic moment.9 Our work indicated the need for identifying strategies in order to attach the radical to the surface preserving its magnetic moment at the interface by using different methods ranging from evaporation to preparation in a wet environment. However, the radical functionalization of a substrate is eased by choosing a specific chemical group that has a high chemical affinity for the selected substrate. Usually thiols and disulfides are chosen to covalently modify gold surfaces, including gold nanoparticles, with organic radicals by adsorption from solution. More recently, alkyne terminated derivatives have started to play a role. Nitroxides (TEMPO),14–17 nitronyl nitroxides18–20 and tripheylmethyl21–23 radicals have been successfully employed to prepare such paramagnetic hybrid materials. In this work, we capitalize our knowledge of radical thin films and interfaces by studying the functionalization of gold surfaces with derivatives of the perchlorotriphenylmethyl (PTM) radical. PTM is a very persistent and stable radical that shows a long coherence time at room temperature, being a strong potential candidate for quantum technologies.24 Previously, self-assembled monolayers (SAMs) of PTM on gold substrates have been investigated to study their transport properties.21–23,25 The radical character of the layers was proved by several techniques (UV-vis, cyclic voltammetry, EPR, NEXAFS and UPS); however, a careful and in-depth characterization of the stability of these radical SAMs has not been carried out so far. Such a stability is a necessary precondition to use radical-based SAMs for any practical application. Here, we used a ferrocene functionalized PTM derivative with an alkyne termination (Fig. 1) that covalently attaches to a gold substrate spontaneously.26–31 The ferrocene functionalization makes the molecules interesting for current rectification, as seen in SAMs incorporating ferrocene acting as a redox-active moiety.32–34 We investigated also the formation of radical self-assembled monolayers (SAMs) obtained by using on-surface chemistry.
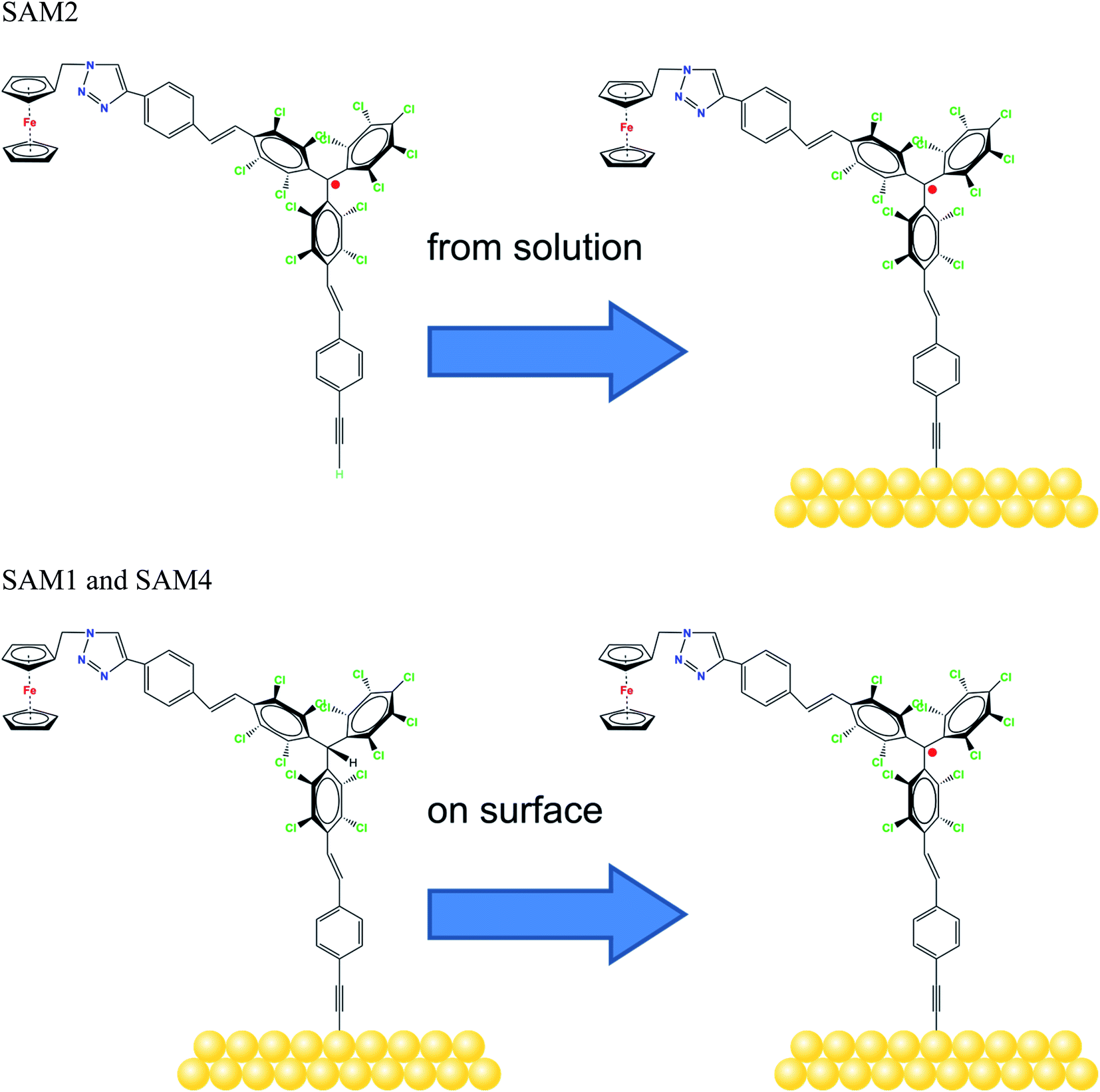 | ||
| Fig. 1 Molecular structure of the radicals, as indicated, and the schematic sketch of the different SAM preparations. | ||
Our investigations were performed using X-ray photoelectron spectroscopy (XPS). While XPS is a well-established technique to investigate the electronic structure of materials, this is not the only aspect that can be examined.35 Because of its high sensitivity, it is also possible to quantitatively calculate the stoichiometry of the investigated systems. Further aspects can be explored: it is very sensitive to the chemical environment of the elements, allowing the occurring chemical bonds and the charge transfer from/to surfaces to be revealed. It is possible to gain information on film stability (e.g., under X-ray beam or air exposure) and on post-growth phenomena. It is extremely well suited to investigate radical thin films (including their radical character) when evaporated by using controlled conditions.36 We proved in our previous work that XPS in combination with a careful and robust best fit procedure allows investigation of the radical character, with the results being in perfect agreement with electron paramagnetic resonance (EPR) measurements.9,36–41 EPR is the technique typically used for radical characterization. However, its use for films is limited (1) by the fact that it is an ex situ technique. Radical thin films might not be stable enough outside the ultra-high vacuum environment where they are deposited or obtained by on-surface reaction.42–44 (2) By the choice of the substrate that might contribute to the EPR signal.41 (3) By the substrate dimensions that are often over-dimensioned for standard spectrometers. (4) By the fact that standard EPR spectrometers do not have the necessary sensitivity to measure (sub)monolayers. Conversely, XPS has a high sensitivity further beyond many other conventional chemical techniques, as it can detect less than 1013 atoms,45 allowing investigations in the monolayer and submonolayer regime without requiring advanced “state-of-the-art” spectrometers, as it is the case for EPR, but a standard, commercially available, monochromatized laboratory XPS station is sufficient.
In this work, we investigate the chemistry of the SAM/gold interface, demonstrating that the SAMs were successfully attached to the substrate, using also on-surface chemistry. We also show that it is possible to identify the spectroscopic lines associated with the radical character versus its diamagnetic counterpart. The work focuses on the SAM stability, under X-ray and air exposure, using a method that can be applied to any material to explore any kind of stability issue, such as gas exposure, humidity, aging, and temperature that are of paramount importance for technological applications.
Experimental section
SAM1 and SAM2 were prepared following the protocol thoroughly described in ref. 34. SAM4 was grown following a two-step reaction: (1) SAM1 was immersed in a 2 mM solution of Bu4NOH/THF (freshly distilled) under an argon atmosphere. The solution was left with a gentle stirring for 8 h at room temperature in the dark. Then, the substrates were removed from the flask and thoroughly rinsed with THF (distilled). (2) Immediately afterwards, the substrates were immersed in a 4 mM p-chloranil/THF (distilled) solution under an argon atmosphere. The solution was left for 12 h at room temperature in the dark. Finally, the substrates were removed from the flask, thoroughly rinsed with THF (distilled) and dried with a nitrogen stream. Coverage and radical formation were checked with cyclic voltammetry.An XPS Ultra High Vacuum (UHV) system (2 × 10−10 mbar base pressure) equipped with a monochromatic Al Kα source (SPECS Focus 500) and a SPECS Phoibos 150 hemispherical electron analyzer was used. Survey spectra were measured at 50 eV pass energy and individual core level spectra at 20 eV pass energy. Both were subsequently calibrated to the Au 4f signal at 84 eV. To minimize potential radiation damage, freshly prepared films were measured, and radiation exposure was minimized unless differently stated in the text (i.e., stability measurements). For measurements probing air stability, X-ray beam exposure was further limited after air exposure to attribute the observed changes exclusively to the degradation by air exposure.
Photon-energy dependent XPS measurements were performed at the third-generation synchrotron radiation source BESSY II (Berlin, Germany) at the Low-Dose PES end station installed at the PM4 beamline (E/ΔE = 6000 at 400 eV). They were carried out in multibunch hybrid mode with a SCIENTA ArTOF electron energy analyzer (ring current in top up mode = 300 mA).
Results and discussion
We examined two different layer preparations using the PTM radical derivative (SAM2 and SAM4) and we compared them with those obtained by depositing the diamagnetic counterpart, SAM1 (Fig. 1). The PTM radical and the diamagnetic derivative shown in Fig. 1 were synthesized as previously reported:34 SAM2 is obtained by depositing the radical on a gold substrate from its solution. SAM4, in contrast, is obtained by first depositing the analogous diamagnetic molecules on gold and following a two-step synthesis (i.e., anion generation and oxidation), and thus, the PTM radical is formed on the surface.46Fig. 2 shows the SAM2 XPS spectra of the important core levels (for the survey and the stoichiometric analysis, see Fig. S1, Tables S1 and S2 in the ESI†). The spectra are characterized by the predominance of gold signals in agreement with the deposition of a monolayer. Apart from a carbon concentration that slightly exceeds the theoretical values, which is usual in samples prepared ex situ with wet-environment techniques, the films are remarkably clean, and no significant amounts of contaminants are visible. In XPS, the integrated area of the main lines corresponding to photoelectrons emitted from a given element, together with their satellites, is proportional to the concentration of that same element in the investigated system.35,47,48 In highly resolved XPS spectra, the rich fine structure allows fitting the lines including contributions from different atomic sites of the same element which, due to a different chemical environment, are expected to show differences in their binding energies.35,47,48
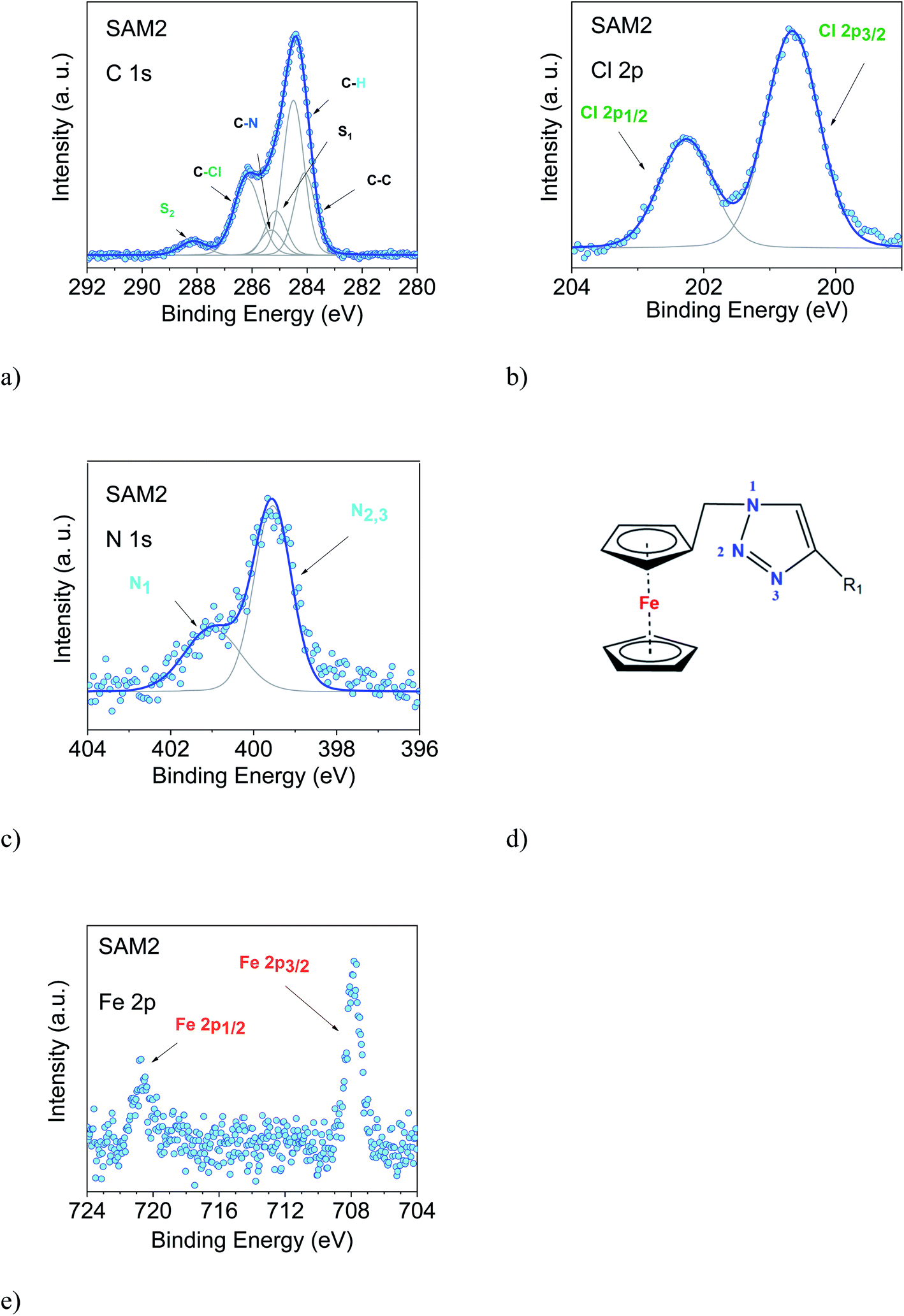 | ||
| Fig. 2 SAM2. (a) C 1s, (b) Cl 2p, (c) N 1s (together with their best fit) and (e) Fe 2p XPS spectra (photon energy: 1486.6 eV). In (d) the chemical environment of the triazole derivative is shown in detail. | ||
The film stoichiometry agrees with the expected values, confirming that the radical derivative was indeed attached to the gold substrate. The C 1s spectroscopic line is characterized by a main peak at around 284.5 eV and a feature at around 286 eV. The C 1s intensity is due to photoelectrons emitted from the carbon atoms. The contributions mirror several different chemical environments. In fact, carbon atoms are not only bound to other carbon atoms, but to hydrogen, nitrogen, and chlorine atoms. Each different environment leads to a slightly different binding energy that can be identified by using a best fit procedure (Fig. 2a).34,37,49 The fitting procedure in XPS is driven by specific and detailed chemical and physical arguments, and not by a mere mathematical approach. The curves are described using a Voigt profile, i.e., a convolution of a Gaussian and a Lorentzian profile. This is because different contributions influence the line shape of the XPS main features: intrinsic lifetime broadening, vibronic and inhomogeneous broadening lead to a Lorentzian profile, while experimental contributions have a Gaussian profile. The lifetime of the core hole is determined basically using the Heisenberg uncertainty principle and consequently the intrinsic peak width is determined, too. For example, the Lorentzian width for the C 1s orbital is around 80 meV, and for the N 1s orbital it is around 100 meV in organic materials.50 The experimental setup gives a contribution assumed to have a Gaussian lineshape due to the resolution of the analyser, the non-perfect monochromaticity of the X-rays, and inhomogeneities of different nature. The fit that we use is based on the procedure adopted for closed-shell molecules.14 The final fit is the result of several self-consistent iteractions of sequential fits performed considering all physical and chemical information and adding more constraints at each iteraction, with the goal of keeping the parameter dependency very low (dependency values of the last fits in this work were very close to zero). The constraints in our fit are based on the element concentration, and the binding energy constraints must adhere to electronegativity and known values in the literature, so we use the published or measured core-hole lifetimes for each element. The fit procedure must systematically hold for all samples of a specific system, prepared and measured under the same conditions. Our procedure revealed to be extremely robust giving results in very good agreement with EPR and ab initio calculations, both for open-shell and closed-shell systems, as well.9,36–41,51–55 To reach this result, we work on sets of samples that are large enough to be statistically significant. In this way, we can also identify the samples that do not correspond to the expected stoichiometry.37,39,56,57
In the spectra, we observe the presence of shake-up satellite intensities (Fig. 2). As a result of the core-hole formation, the symmetry is reduced, and a larger number of non-equivalent carbon atoms should be considered.58,59 The ionization at different carbon sites may give different contributions to the shake-up spectra. The S1 satellite can be related to the first HOMO–LUMO shake-up.60 Its energy position with respect to the main line is lower than the optical gap, a typical effect in the HOMO–LUMO shake-up satellites of polyaromatic molecules caused by the enhanced screening of the core-hole due to its delocalization.60–63 A large number of satellite features is expected upon a photoemission event. However, their assignment is very complicated, especially for large molecules because they are not completely described by theoretical models. Such a detailed description is outside the goal of this work; therefore, we have identified most of the higher binding energy satellite intensities under a single component, S2. This component is correlated with the C–Cl feature from a stoichiometric point of view. This assignment is further corroborated by the fact that the C–Cl feature and S2 change simultaneously depending on the photon energy, as it can be easily seen in Fig. 2 and 3.
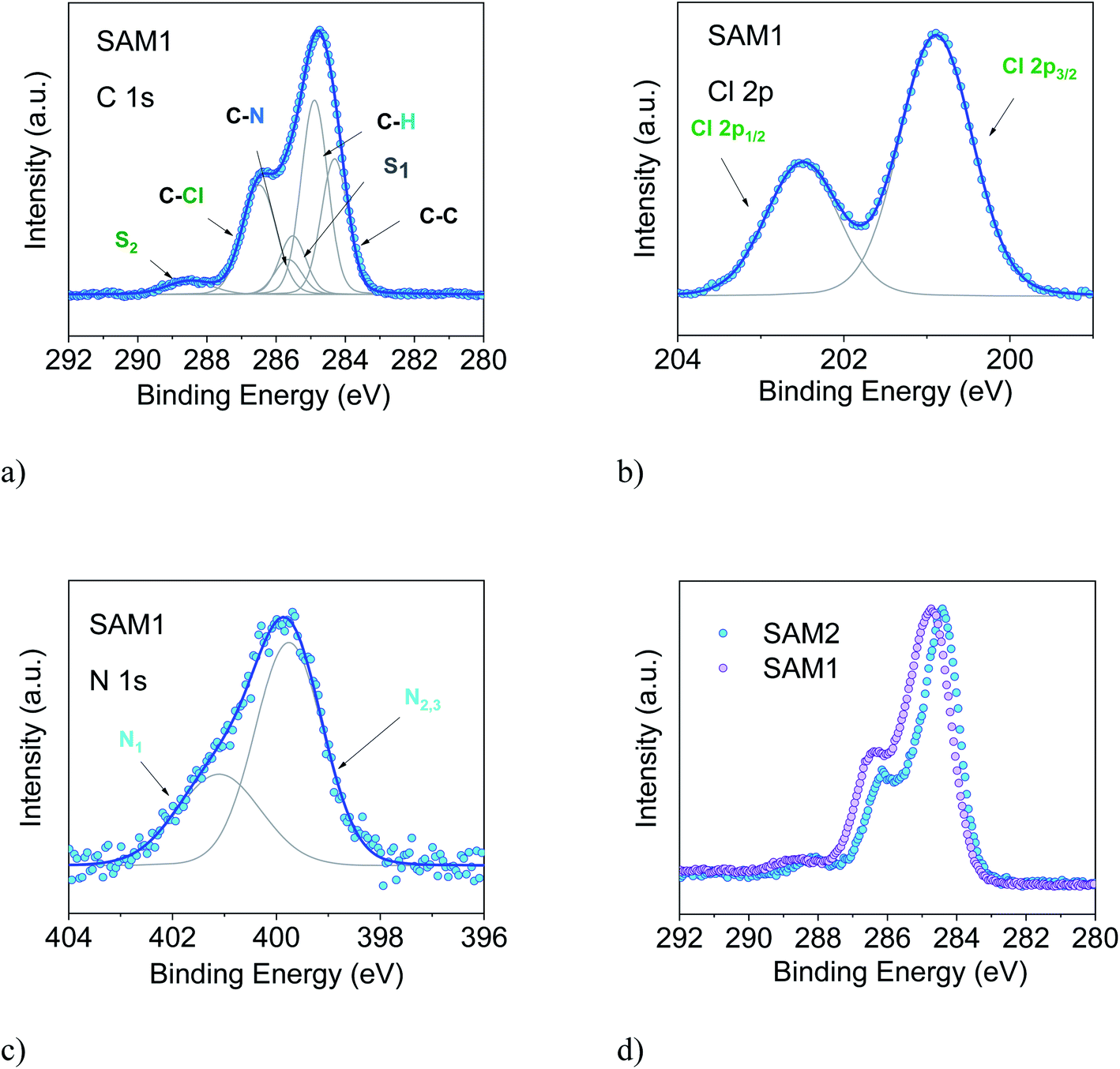 | ||
| Fig. 3 SAM1. (a) C 1s, (b) Cl 2p, and (c) N 1s, together with their best XPS spectra (photon energy: 1486.6 eV). (d) Comparison of the C 1s XPS line of SAM1 (diamagnetic) and SAM2 (radical). Intensities are normalized to the peak maximum to allow comparison. | ||
The intensities of the various contributions agree with the expected stoichiometry, confirming once more that the SAM2 carbon line corresponds to the radical derivative (Table S2 in the ESI†). The Cl 2p, N 1s and Fe 2p core level spectra are also shown. Their features confirm the presence of an intact molecule: Cl 2p core level lines show the typical doublet feature (spin–orbit splitting = 1.6 eV, as in the literature64), and the N 1s spectrum (Fig. 2c) is characterized by contributions due to photoelectrons emitted from three different chemical environments, confirming the intactness of the triazole derivative (Fig. 2c and d). The signal of the Fe 2p shows the expected doublet (spin–orbit splitting = 12.8 eV, concomitant with the values in the literature64,65), and the noteworthy absence of further intensities indicates that the signal is due to electrons emitted from iron atoms in the +2 oxidation state, as it is the case for ferrocene.65–67 Note that, for the monolayers of clean ferrocene, the Fe 2p spectrum does not show any satellite intensity.65–69 In fact, this intensity depends on the ligands and it varies with their electronegativity.70 Additionally, in the case of monolayers on metal substrates, the image-charge formed at the interface71 may further screen the satellite intensities. The XPS intensities and line shapes indicate that the radical was attached to the surface preserving the expected stoichiometry. Thus, we can confidently infer that the synthesis and the preparation of SAM2 were successful.
To support this conclusion and explore the use of XPS to identify the PTM radical, we investigated SAM1, i.e., the SAM obtained from the diamagnetic counterpart of the PTM radical derivative (Fig. 1).
The essential core level spectra are shown in Fig. 3 (for the survey and the stoichiometric analysis, see Fig. S2, Tables S3 and S4 in the ESI†). In our discussion, we focus on the C 1s core level spectroscopic line. This is the line that is directly correlated with the radical character (see Fig. 1) because the unpaired electron mainly resides in the central radical carbon atom of the perchlorinated triphenylmethyl unit. The stoichiometry for SAM2 and SAM1 is different. In SAM1 the central methyl carbon atom of the PTM is bound to hydrogen. Therefore, we expect a different C 1s line broadening with respect to the radical spectra. Indeed, we observe a larger line for SAM1 (Full Width at Half Maximum (FWHM) = 1.8 eV versus 1.4 eV for SAM2, under the same experimental conditions). This difference is mirrored by a larger Gaussian width required in the fit procedure (see Table S4 in the ESI†). We also observe a different binding energy. The SAM1 C 1s main line is at higher binding energy than the SAM2 main line. This indicates that the core-hole created upon photoemission is more efficiently screened in SAM2 than in the diamagnetic molecule. This can be explained considering the donor–acceptor character of SAM2 (ref. 34) where the simultaneous presence of the radical and the azidomethyl ferrocene unit stands for faster charge delocalization of the core-hole. These differences in the C 1s main line, binding energy and broadening between SAM1 and SAM2 allow using XPS to identify the radical character of the SAMs.
In an XPS experiment it is possible to probe different sampling depths:72 when changing the photon energy, the materials emit electrons with different kinetic energy which is equivalent to emitting photoelectrons with different inelastic mean free path (λ). Thus, we performed a photon-energy-dependent experiment on SAM1, SAM2 and SAM4 using 460 and 640 eV photon energy, respectively. This corresponds to varying λ between 0.17 and 0.28 nm (ref. 73 and 74) (Fig. 4 and S3†). The experiment at 460 eV is very surface-sensitive (note that both experiments at 460 and 640 eV are very surface-sensitive with respect to the measurements so far discussed, which were performed at 1486.6 eV). We observe that, by varying the photon energy, the relative intensities of the main line and the line due to photoelectrons emitted from carbon atoms bound to the electronegative nitrogen and chlorine atoms change: the feature at higher binding energy has higher intensity at 640 eV. What is also important is that these changes are accompanied by changes in the S2 satellite, indicating, as mentioned, that these two components are strongly correlated, corroborating our fit assignments. This change in the intensity depends on the photon energy and, thus, on the inelastic mean free path, and it is due to the surface core level shift effect,75–78i.e., the difference of the core level photoemission between a surface atom/molecule and a bulk atom/molecule.76,78,79 This effect is visible in organic thin films when the molecules are not planar and carry electronegative atoms.49,80,81 In fact, electronegative atoms shift the electronic cloud, causing a different screening of the core-hole created upon photoemission. However, this screening is different when it occurs at different depths where structural differences are significant, for example, in the case of upright versus flat lying molecules.80 In the present case, the C–Cl components are stronger at 640 eV when the experiment is less surface-sensitive. We can infer structural information from this dependence: the XPS results indicate that the PTM radical is closer to the substrate with respect to the azidomethyl-ferrocene unit (as sketched in Fig. 1); therefore, its contribution is stronger when λ is longer. For the photon energy of 1486.6 eV, λ is comparable with the dimensions of the molecule (λ = 0.81 nm (ref. 73)), in which case the stoichiometry information plays the major role against the structural information, as seen in closed-shell systems like phthalocyanines.82
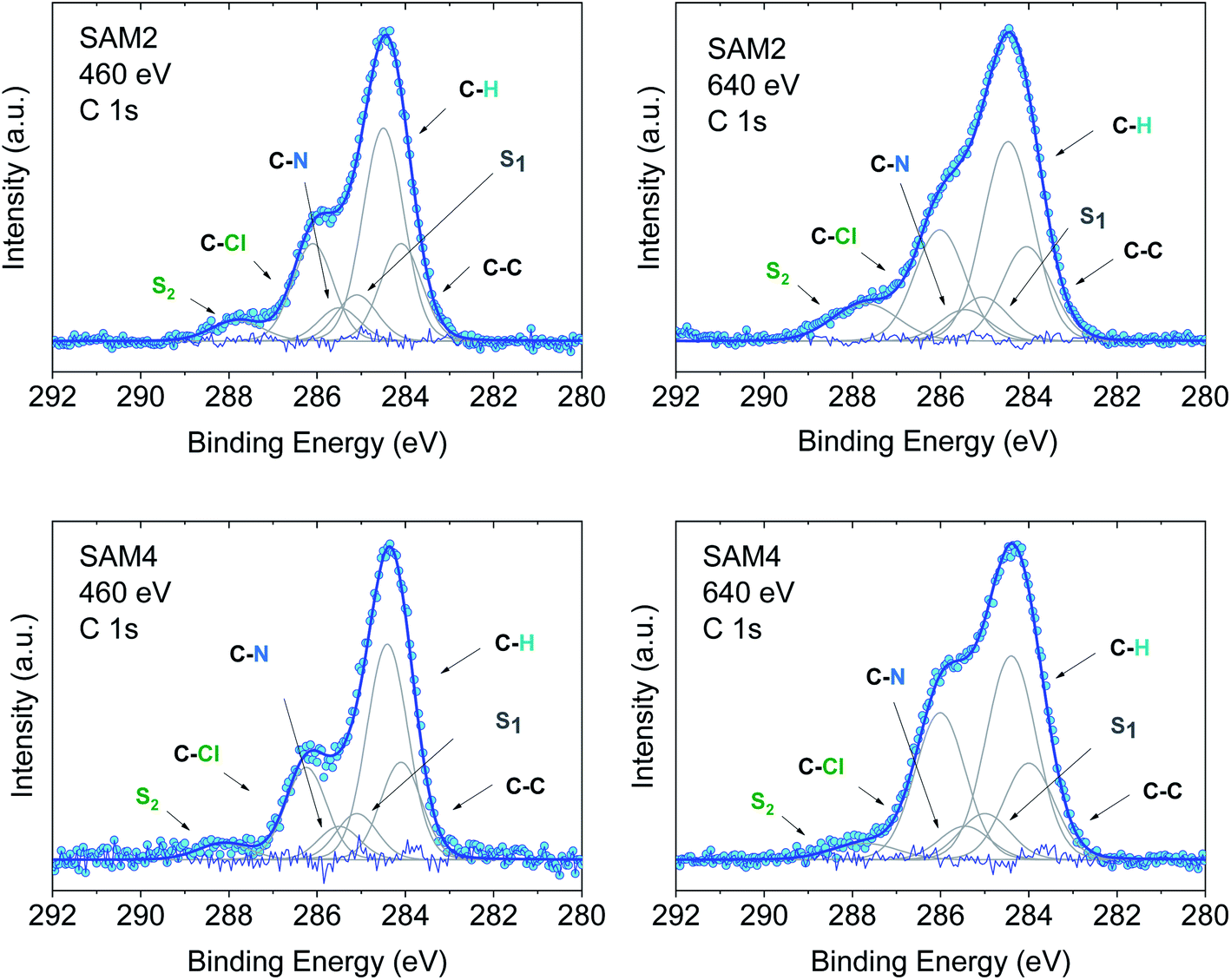 | ||
| Fig. 4 C 1s core level spectra at 460 and 640 eV, as indicated, together with their best fit and residual. Upper panel: SAM2. Lower panel: SAM4. Intensities are normalized to the peak maximum to allow comparison. For the curve fits see the ESI.† | ||
Using the above results as a reference, we investigated SAM4 (Fig. 4, lower panel). This monolayer has the same theoretical stoichiometry as SAM2, but it has been obtained via on-surface radical formation from the diamagnetic molecule. We focused once more on the C 1s core level spectra. First, from the point of view of the stoichiometry, as previously performed for SAM2 and SAM1, we observe that the C 1s line shape has the same features as in the SAM2 core spectra. In this case also, we observe the same photon energy dependence at 460 and 640 eV, hinting at a similar structural adjustment of the molecule units with respect to the substrate. What is most important is that the FWHM of the C 1s line is narrower than in the case of the diamagnetic molecule, i.e., SAM4 has a narrower main line than SAM1 (see Fig. S3†). Following our above discussion, this effect indicates a radical character of the film. Since the radical generation occurred on the surface, this result hints at and supports the successful on-surface preparation of the radical. A fit procedure backs these observations: the same best fit procedure leads to the same intensities and binding energies for the C 1s contributions of the spectra of SAM2 and SAM4 (Fig. 4 and Tables S5–S8 in the ESI†). Cyclic voltammetry experiments support the radical character of the layers, too (see Fig. S4 in the ESI†). The redox peaks corresponding to the PTM radical ↔ PTM anion and ferrocene ↔ ferrocenium redox process are clearly observed.
A change in photon energy as performed in the present XPS experiments also implies a change in the C 1s cross-section increasing the complexity of the screening effects. Looking at the fit results, we note that the S1 intensity decreases with increasing the photon energy while the intensities of the S2 satellite show the opposite behaviour (Tables S5–S8 in the ESI†). This gives a hint about the fact that the S1 intensity is related to the dipole excitation of a core electron to the lowest unoccupied molecular orbital (LUMO) accompanied by the monopole ionization of the valence electron: this shake up contribution is near the ionization threshold region and decreases with the increase in energy,83,84 as observed in our fits.
An important aspect that we intend to address here is the stability of the monolayer in the real environment. While the PTM radical is known to be chemically stable both in solution and in powder if visible light is avoided, there is no report on the chemical and structural stability of its films where single radical molecules are exposed to air. To tackle this issue, we kept SAM2 monolayers under air in darkness and measured them again 128 days later, always minimizing X-ray exposure during measurements. The results are shown in Fig. 5. The C 1s core level spectrum comparison between the fresh monolayer and the “aged” monolayer shows a small difference in the relative intensity of the main feature with respect to the feature at higher binding energy, while the Cl 2p spectra do not show major differences. Post-growth phenomena, such as desorption and ripening, are expected and well-known in the case of organic molecules, and expected also in radical films, especially for those systems having low vapor pressure at room temperature and physisorbed on surfaces.36,37,40 To investigate the origin of the difference in the C 1s core level spectra we performed a best fit analysis, following two hypotheses. In one case, we performed the fit considering that PTM might switch to the perchlorophenylfluorenyl radical (PPF) (Fig. S5 and Table S9 in the ESI†). This is a known derivative of the PTM radical generated both by heating over 300 °C (ref. 85) and by photoirradiation.86 In the second case, we considered that the stoichiometry of the monolayer stays unchanged but the carbon intensity increases due to the adsorption of carbon impurities from the environment (Fig. S6 and Table S10 in the ESI†). Both fits are plausible. A closer inspection of the survey spectra helps to interpret the results (Fig. 5, lower panel). Initially, the gold signal is stronger, i.e., its intensity decreases with time. Simultaneously the carbon signal increases, while the chlorine signal does not change. From the stoichiometric analysis of the spectra, we found that in the fresh monolayer the carbon to gold ratio (C/Au) and the chlorine to gold ratio (Cl/Au) are 0.37 and 0.04, respectively. After 128 days, they are 0.40 and 0.04, respectively. This clearly indicates that the chlorine content does not diminish and that the phenomenon playing the major role is carbon adsorption. This means that not only the PTM radical is chemically stable, but also its monolayers are stable under prolonged air exposure. This is a result of great significance because it fully supports the use in devices of the PTM radical and its derivatives grafted on surfaces.
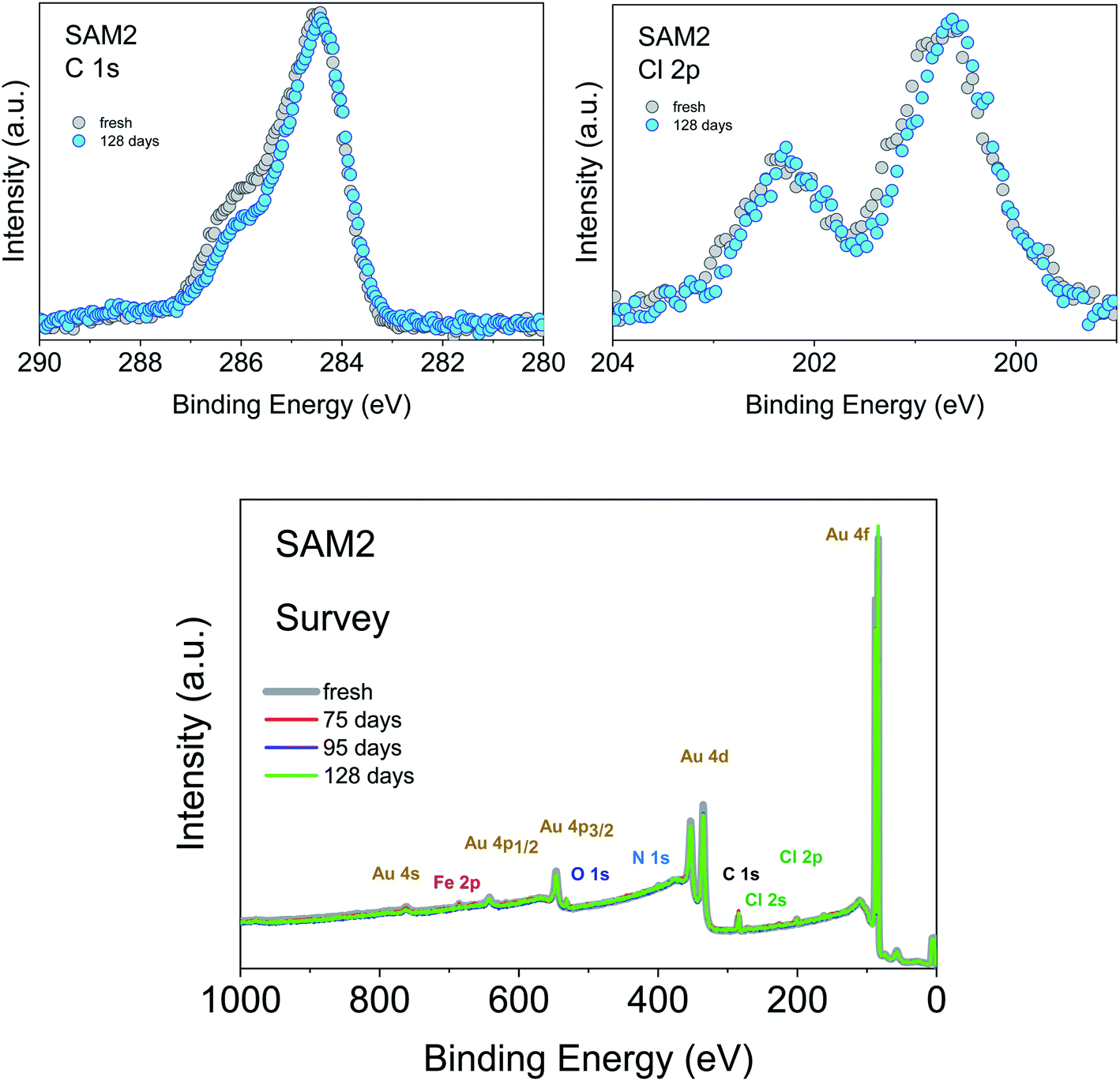 | ||
| Fig. 5 Upper panel: (left) C 1s and (right) Cl 2p core level spectra of a freshly prepared monolayer and after 128 days of exposure to air and kept in darkness, as indicated (photon energy: 1486.6 eV). Intensities are normalized to the peak maximum to allow comparison. Lower panel: SAM2 survey XPS spectra under air exposure, as indicated (photon energy: 1486.6 eV). | ||
We also studied the stability of SAM2 against X-rays. As previously, we focus our discussion on the PTM radical analysing the C 1s and the Cl 2p core level spectra (Fig. 6). We could observe first small changes in the spectroscopic lines after 18 hours of X-ray exposure, a 0.1 eV shift of the binding energy towards higher values and a difference in the satellite intensities. The fit analysis performed on the C 1s line confirms that these are not significant stoichiometric changes (Fig. S7, Tables S11 and S12 in the ESI†). We crosschecked this finding also using synchrotron radiation and monitoring the film in real-time over around 8 hours (Fig. 6, lower panel, photon energy: 640 eV, flux: 1 × 109 to 1 × 1010 photons per s). No changes were detected.
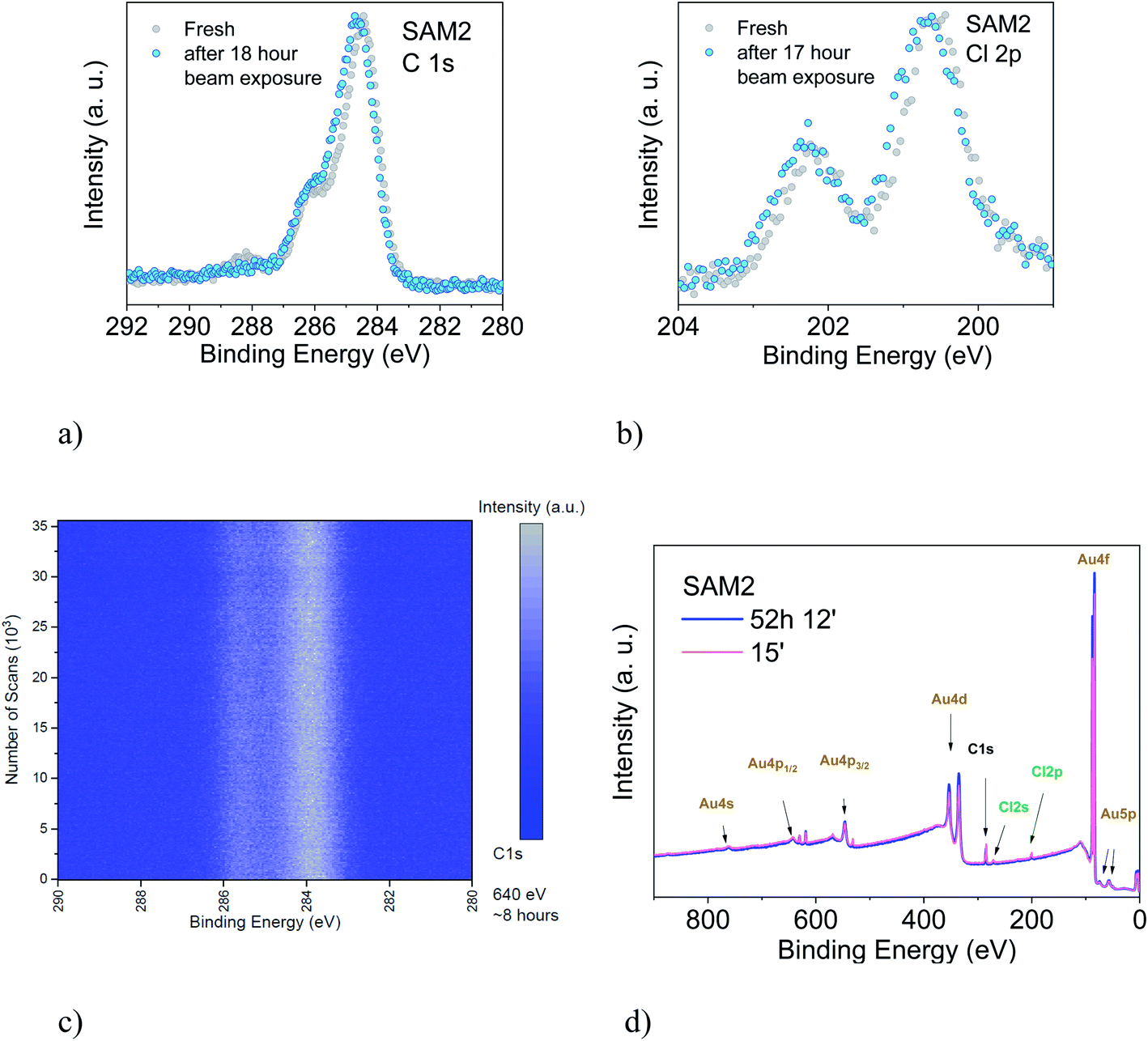 | ||
| Fig. 6 Upper panel: (a) C 1s and (b) Cl 2p core level spectra of a freshly prepared monolayer and after 18 hours of X-ray exposure, as indicated (photon energy: 1486.6 eV). Intensities are normalized to the peak maximum to allow comparison. Lower panel: (c) time-dependent C 1s core level signal. Color scale: blue represents the background signal; white the initial peak intensity (photon energy: 640 eV). (d) Survey XPS spectra of a freshly prepared monolayer and after 52 hours of X-ray exposure, as indicated (photon energy: 1486.6 eV). | ||
To understand what happens under very long X-ray exposure, we exposed the films to X-rays for 52 hours and we looked at the effects (Fig. 6d). After such a long exposure, the gold signal is more intense, while the C 1s and Cl 2p lines show no decrease in the intensity. This indicates that the gold substrate is more exposed with time. Usually this result hints at changes in the film morphology due to post-growth phenomena, such as desorption, dewetting or Ostwald ripening, which lead to the coalescence of small islands into big islands leaving a larger area of the substrate surface free. The result indicates, also in this case, some degree of dynamics, suggesting a change in the layer morphology. These experimental observations seem puzzling in the case of a strong adsorbate–substrate chemical bond. To help in understanding this phenomenon, we can look at one of the most investigated SAM systems: thiolates on gold. Investigations of thiolate–Au surfaces have demonstrated a clear dynamic nature of these surfaces, where the mobility of the adsorbate–Au complex plays an important role, both on flat surfaces as well as on nanoparticles, upon mild annealing and even at room temperature.87 The mobility is explained in terms of the presence of defects on gold surfaces.87–90 At defect sites, the interaction between a single gold atom and a covalently attached molecule is stronger than the interaction with the environment (gold atoms and surrounding molecules, respectively) causing the motion of the complete adsorbate + Au assembly on the surface, giving rise to ripening, and even to desorption. This is a very general mechanism of surface diffusion occurring when an adsorbate is strongly bound to coinage metals such as gold.87,89 The behaviour of the PTM-based SAMs on gold and the resulting XPS spectra observed during prolonged X-ray beam exposure would hint at the fact that such a mechanism also occurs in the present case, favoured or induced by the prolonged X-ray exposure.
Conclusions
Once more, XPS has proved to be a very powerful tool to investigate radical films and radical/metal interfaces, uncovering phenomena not yet known. Furthermore, our XPS method to assess the stability of radical/inorganic interfaces can be applied to any system. In this work, we have investigated the stability of chemically functionalized gold surfaces with a PTM radical, either by preparing the self-assembled monolayers directly from the radical solution or, alternately, by chemical means obtaining the radical on the surface from its diamagnetic precursor. While the chemical stability of the PTM radical is well-known (PTM is considered an inert radical) here we show that the radical monolayers have unprecedented stability under ambient conditions and aggressive X-ray exposure. Extremely prolonged X-ray exposure indicates a dynamic nature of the radical–Au complex, analogously to the case of thiolate–Au surfaces. To our knowledge, this phenomenon has not yet been reported for this class of adsorbate–Au systems. Therefore, further investigations, including annealing experiments and theoretical modelling, are necessary to deepen the understanding of the dynamical aspects of this surface. We cannot exclude that similar phenomena might occur at room temperature also upon prolonged air exposure, with a reaction time of weeks, as seen for thiolate–Au nanoparticles.87 Although further investigations on the long-term aging pattern of the PTM radical-based layers also depending on different parameters, such as temperature and visible light, are necessary, our results point out that carbon absorption from the ambient environment plays the major role when the monolayer is exposed to air for a long time.The PTM radical and its derivatives form monolayers that have unprecedented stability properties, confirming that these systems are suitable candidates for market-oriented applications.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Helmholtz-Zentrum Berlin (HZB) for providing beamtime at BESSY II (Berlin, Germany), and Hilmar Adler, Elke Nadler, and Sergio Naselli for technical support. J. A. de S. is enrolled in the Materials Science PhD program of UAB. J. A. de S. thanks the Spanish Ministry for an FPI fellowship. This work was funded by the Spanish Ministry project FANCYCTQ2016-80030-R and GENESIS PID2019-111682RB-100, the Generalitat de Catalunya (2017SGR918) and the Spanish Ministry of Economy and Competitiveness, through the “Severo Ochoa” Programme for Centers of Excellence in R&D (SEV-2015-0496), the CSIC with the i-Link+ 2018 (Ref. LINKA20128) and CIBERBBN. Financial support from HZB and German Research Foundation (DFG) under the contract CA852/11-1 is gratefully acknowledged.References
- F. Troiani and M. Affronte, Chem. Soc. Rev., 2011, 40, 3119–3129 RSC.
- K. Bader, D. Dengler, S. Lenz, B. Endeward, S.-D. Jiang, P. Neugebauer and J. van Slageren, Nat. Commun., 2014, 5, 5304 CrossRef CAS PubMed.
- K. S. Pedersen, A.-M. Ariciu, S. McAdams, H. Weihe, J. Bendix, F. Tuna and S. Piligkos, J. Am. Chem. Soc., 2016, 138, 5801–5804 CrossRef CAS PubMed.
- A. Fernandez, E. Moreno Pineda, C. A. Muryn, S. Sproules, F. Moro, G. A. Timco, E. J. L. McInnes and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2015, 54, 10858–10861 CrossRef CAS PubMed.
- A. Fernandez, J. Ferrando-Soria, E. M. Pineda, F. Tuna, I. J. Vitorica-Yrezabal, C. Knappke, J. Ujma, C. A. Muryn, G. A. Timco, P. E. Barran, A. Ardavan and R. E. P. Winpenny, Nat. Commun., 2016, 7, 10240 CrossRef CAS PubMed.
- M. Shiddiq, D. Komijani, Y. Duan, A. Gaita-Ariño, E. Coronado and S. Hill, Nature, 2016, 531, 348 CrossRef CAS PubMed.
- J. M. Zadrozny, J. Niklas, O. G. Poluektov and D. E. Freedman, ACS Cent. Sci., 2015, 1, 488–492 CrossRef CAS PubMed.
- M. Mas-Torrent, N. Crivillers, C. Rovira and J. Veciana, Chem. Rev., 2011, 112, 2506–2527 CrossRef PubMed.
- F. Ciccullo, A. Calzolari, K. Bader, P. Neugebauer, N. M. Gallagher, A. Rajca, J. van Slageren and M. B. Casu, ACS Appl. Mater. Interfaces, 2019, 11, 1571–1578 CrossRef CAS PubMed.
- H. Guo, Q. Peng, X.-K. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Credgington, V. Coropceanu, R. H. Friend, J.-L. Brédas and F. Li, Nat. Mater., 2019, 18, 977–984 CrossRef CAS PubMed.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef CAS PubMed.
- M. Mas-Torrent, N. Crivillers, V. Mugnaini, I. Ratera, C. Rovira and J. Veciana, J. Mater. Chem., 2009, 19, 1691–1695 RSC.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- O. Swiech, R. Bilewicz and E. Megiel, RSC Adv., 2013, 3, 5979–5986 RSC.
- O. Swiech, N. Hrynkiewicz-Sudnik, B. Palys, A. Kaim and R. Bilewicz, J. Phys. Chem. C, 2011, 115, 7347–7354 CrossRef CAS.
- A. Kaim, J. Szydłowska, P. Piotrowski and E. Megiel, Polyhedron, 2012, 46, 119–123 CrossRef CAS.
- E. Megiel, Adv. Colloid Interface Sci., 2017, 250, 158–184 CrossRef CAS PubMed.
- M. Mannini, L. Sorace, L. Gorini, F. M. Piras, A. Caneschi, A. Magnani, S. Menichetti and D. Gatteschi, Langmuir, 2007, 23, 2389–2397 CrossRef CAS PubMed.
- L. Poggini, G. Cucinotta, L. Sorace, A. Caneschi, D. Gatteschi, R. Sessoli and M. Mannini, Rendiconti Lincei. Scienze Fisiche e Naturali, 2018, 29, 623–630 CrossRef.
- V. Lloveras, E. Badetti, J. Veciana and J. Vidal-Gancedo, Nanoscale, 2016, 8, 5049–5058 RSC.
- N. Crivillers, C. Munuera, M. Mas-Torrent, C. Simão, S. T. Bromley, C. Ocal, C. Rovira and J. Veciana, Adv. Mater., 2009, 21, 1177–1181 CrossRef CAS.
- L. Yuan, C. Franco, N. Crivillers, M. Mas-Torrent, L. Cao, C. S. S. Sangeeth, C. Rovira, J. Veciana and C. A. Nijhuis, Nat. Commun., 2016, 7, 12066 CrossRef CAS PubMed.
- C. Simão, M. Mas-Torrent, J. Veciana and C. Rovira, Nano Lett., 2011, 11, 4382–4385 CrossRef PubMed.
- Y.-Z. Dai, B.-W. Dong, Y. Kao, Z.-Y. Wang, H.-I. Un, Z. Liu, Z.-J. Lin, L. Li, F.-B. Xie, Y. Lu, M.-X. Xu, T. Lei, Y.-J. Sun, J.-Y. Wang, S. Gao, S.-D. Jiang and J. Pei, ChemPhysChem, 2018, 19, 2972–2977 CrossRef CAS PubMed.
- M. R. Ajayakumar, I. Alcón, S. T. Bromley, J. Veciana, C. Rovira and M. Mas-Torrent, RSC Adv., 2017, 7, 20076–20083 RSC.
- T. Heinrich, C. H. H. Traulsen, E. Darlatt, S. Richter, J. Poppenberg, N. L. Traulsen, I. Linder, A. Lippitz, P. M. Dietrich, B. Dib, W. E. S. Unger and C. A. Schalley, RSC Adv., 2014, 4, 17694–17702 RSC.
- Y.-Q. Zhang, N. Kepčija, M. Kleinschrodt, K. Diller, S. Fischer, A. C. Papageorgiou, F. Allegretti, J. Björk, S. Klyatskaya, F. Klappenberger, M. Ruben and J. V. Barth, Nat. Commun., 2012, 3, 1286 CrossRef PubMed.
- T. Zaba, A. Noworolska, C. M. Bowers, B. Breiten, G. M. Whitesides and P. Cyganik, J. Am. Chem. Soc., 2014, 136, 11918–11921 CrossRef CAS PubMed.
- N. J. Tao, Nat. Nanotechnol., 2006, 1, 173 CrossRef CAS PubMed.
- D. Fracasso, S. Kumar, P. Rudolf and R. C. Chiechi, RSC Adv., 2014, 4, 56026–56030 RSC.
- F. Bejarano, I. J. Olavarria-Contreras, A. Droghetti, I. Rungger, A. Rudnev, D. Gutiérrez, M. Mas-Torrent, J. Veciana, H. S. J. van der Zant, C. Rovira, E. Burzurí and N. Crivillers, J. Am. Chem. Soc., 2018, 140, 1691–1696 CrossRef CAS PubMed.
- M. Souto, V. Díez-Cabanes, L. Yuan, A. R. Kyvik, I. Ratera, C. A. Nijhuis, J. Cornil and J. Veciana, Phys. Chem. Chem. Phys., 2018, 20, 25638–25647 RSC.
- L. Yuan, N. Nerngchamnong, L. Cao, H. Hamoudi, E. del Barco, M. Roemer, R. K. Sriramula, D. Thompson and C. A. Nijhuis, Nat. Commun., 2015, 6, 6324 CrossRef CAS PubMed.
- J. A. de Sousa, F. Bejarano, D. Gutiérrez, Y. R. Leroux, E. M. Nowik-Boltyk, T. Junghoefer, E. Giangrisostomi, R. Ovsyannikov, M. B. Casu, J. Veciana, M. Mas-Torrent, B. Fabre, C. Rovira and N. Crivillers, Chem. Sci., 2020, 11, 516–524 RSC.
- K. M. Siegbahn, Nobel lecture: electron spectroscopy for atoms, molecules and condensed matter, 2020, http://www.nobelprize.org/nobel_prizes/physics/laureates/1981/siegbahn-lecture.html, accessed 09 June 2020 Search PubMed.
- M. B. Casu, Acc. Chem. Res., 2018, 51, 753–760 CrossRef CAS PubMed.
- S.-A. Savu, I. Biswas, L. Sorace, M. Mannini, D. Rovai, A. Caneschi, T. Chassé and M. B. Casu, Chem.–Eur. J., 2013, 19, 3445–3450 CrossRef CAS PubMed.
- R. Kakavandi, S.-A. Savu, L. Sorace, D. Rovai, M. Mannini and M. B. Casu, J. Phys. Chem. C, 2014, 118, 8044–8049 CrossRef CAS.
- R. Kakavandi, P. Ravat, S. A. Savu, Y. B. Borozdina, M. Baumgarten and M. B. Casu, ACS Appl. Mater. Interfaces, 2015, 7, 1685–1692 CrossRef CAS PubMed.
- F. Ciccullo, N. M. Gallagher, O. Geladari, T. Chasse, A. Rajca and M. B. Casu, ACS Appl. Mater. Interfaces, 2016, 8, 1805–1812 CrossRef CAS PubMed.
- F. Ciccullo, M. Glaser, M. S. Sättele, S. Lenz, P. Neugebauer, Y. Rechkemmer, J. van Slageren and M. B. Casu, J. Mater. Chem. C, 2018, 6, 8028–8034 RSC.
- N. Gallagher, H. Zhang, T. Junghoefer, E. Giangrisostomi, R. Ovsyannikov, M. Pink, S. Rajca, M. B. Casu and A. Rajca, J. Am. Chem. Soc., 2019, 141, 4764–4774 CrossRef CAS PubMed.
- S. Tebi, M. Paszkiewicz, H. Aldahhak, F. Allegretti, S. Gonglach, M. Haas, M. Waser, P. S. Deimel, P. C. Aguilar, Y.-Q. Zhang, A. C. Papageorgiou, D. A. Duncan, J. V. Barth, W. G. Schmidt, R. Koch, U. Gerstmann, E. Rauls, F. Klappenberger, W. Schöfberger and S. Müllegger, ACS Nano, 2017, 11, 3383–3391 CrossRef CAS PubMed.
- M. R. Ajayakumar, C. Moreno, I. Alcón, F. Illas, C. Rovira, J. Veciana, S. T. Bromley, A. Mugarza and M. Mas-Torrent, J. Phys. Chem. Lett., 2020, 11, 3897–3904 CrossRef CAS PubMed.
- Surface analysis – the principal techniques, ed. J. C. Vickerman and I. S. Gilmore, Wiley, 2nd edn, 2009 Search PubMed.
- N. Crivillers, M. Mas-Torrent, S. Perruchas, N. Roques, J. Vidal-Gancedo, J. Veciana, C. Rovira, L. Basabe-Desmonts, B. J. Ravoo, M. Crego-Calama and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2007, 46, 2215–2219 CrossRef CAS PubMed.
- O. Travnikova, K. J. Børve, M. Patanen, J. Söderström, C. Miron, L. J. Sæthre, N. Mårtensson and S. Svensson, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 191–197 CrossRef CAS.
- K. Siegbahn, C. Nordling, A. Fahlman, R. Nordberg, K. Hamrin, J. Hedman, G. Johansson, T. Bergmark, S.-E. Karlsson, I. Lindgren and B. Lindberg, ESCA, atomic, molecular and solid state structure studied by means of electron spectroscopy, Almqvist and Wiksells, Uppsala, 1967 Search PubMed.
- S.-A. Savu, M. B. Casu, S. Schundelmeier, S. Abb, C. Tonshoff, H. F. Bettinger and T. Chassé, RSC Adv., 2012, 2, 5112–5118 RSC.
- A. Schöll, Y. Zou, M. Jung, T. Schmidt, R. Fink and E. Umbach, J. Chem. Phys., 2004, 121, 10260–10267 CrossRef PubMed.
- F. Ciccullo, A. Calzolari, I. Píš, S. A. Savu, M. Krieg, H. F. Bettinger, E. Magnano, T. Chassé and M. B. Casu, J. Phys. Chem. C, 2016, 120, 17645–17651 CrossRef CAS.
- M. Glaser, F. Ciccullo, E. Giangrisostomi, R. Ovsyannikov, A. Calzolari and M. B. Casu, J. Mater. Chem. C, 2018, 6, 2769–2777 RSC.
- R. Kakavandi, A. Calzolari, Y. B. Borozdina, P. Ravat, T. Chassé, M. Baumgarten and M. B. Casu, Nano Res., 2016, 9, 3515–3527 CrossRef CAS.
- R. Kakavandi, S.-A. Savu, A. Caneschi and M. B. Casu, J. Phys. Chem. C, 2013, 117, 26675–26679 CrossRef CAS.
- S. A. Savu, G. Biddau, L. Pardini, R. Bula, H. F. Bettinger, C. Draxl, T. Chasse and M. B. Casu, J. Phys. Chem. C, 2015, 119, 12538–12544 CrossRef CAS.
- A. Caneschi and M. B. Casu, Chem. Commun., 2014, 50, 13510–13513 RSC.
- R. Kakavandi, S.-A. Savu, A. Caneschi, T. Chasse and M. B. Casu, Chem. Commun., 2013, 49, 10103–10105 RSC.
- C. Enkvist, S. Lunell, B. Sjögren, P. A. Brühwiler and S. Svensson, J. Chem. Phys., 1995, 103, 6333–6342 CrossRef CAS.
- B. Sjögren, S. Svensson, A. N. d. Brito, N. Correia, M. P. Keane, C. Enkvist and S. Lunell, J. Chem. Phys., 1992, 96, 6389–6398 CrossRef.
- M. L. M. Rocco, M. Haeming, D. R. Batchelor, R. Fink, A. Schöll and E. Umbach, J. Chem. Phys., 2008, 129, 074702 CrossRef CAS PubMed.
- B.-E. Schuster, M. B. Casu, I. Biswas, A. Hinderhofer, A. Gerlach, F. Schreiber and T. Chassé, Phys. Chem. Chem. Phys., 2009, 11, 9000–9004 RSC.
- M. B. Casu, S.-A. Savu, P. Hoffmann, B.-E. Schuster, T. O. Menteş, M. A. Niño, A. Locatelli and T. Chassé, CrystEngComm, 2011, 13, 4139–4144 RSC.
- M. B. Casu, B.-E. Schuster, I. Biswas, C. Raisch, H. Marchetto, T. Schmidt and T. Chassé, Adv. Mater., 2010, 22, 3740–3744 CrossRef CAS PubMed.
- Handbook of X-ray photoelectron spectroscopy, ed. J. Chastain, 1992 Search PubMed.
- A. W. Taylor and P. Licence, ChemPhysChem, 2012, 13, 1917–1926 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- C. M. Woodbridge, D. L. Pugmire, R. C. Johnson, N. M. Boag and M. A. Langell, J. Phys. Chem. B, 2000, 104, 3085–3093 CrossRef CAS.
- Y. Yokota, Y. Mino, Y. Kanai, T. Utsunomiya, A. Imanishi, M. A. Wolak, R. Schlaf and K.-i. Fukui, J. Phys. Chem. C, 2014, 118, 10936–10943 CrossRef CAS.
- Y. Yokota, Y. Mino, Y. Kanai, T. Utsunomiya, A. Imanishi and K.-i. Fukui, J. Phys. Chem. C, 2015, 119, 18467–18480 CrossRef CAS.
- A. P. Grosvenor, B. A. Kobe, M. C. Biesinger and N. S. McIntyre, Surf. Interface Anal., 2004, 36, 1564–1574 CrossRef CAS.
- H. Ishii, K. Sugiyama, E. Ito and K. Seki, Adv. Mater., 1999, 11, 605–625 CrossRef CAS.
- S. Hüfner, Photoelectron spectroscopy, Springer-Verlag, Berlin, Heidelberg, 3rd edn, 2003 Search PubMed.
- T. Graber, F. Forster, A. Schöll and F. Reinert, Surf. Sci., 2011, 605, 878–882 CrossRef CAS.
- S. Tanuma, C. J. Powell and D. R. Penn, Surf. Interface Anal., 1991, 17, 911–926 CrossRef CAS.
- S. F. Alvarado, M. Campagna and W. Gudat, J. Electron Spectrosc. Relat. Phenom., 1980, 18, 43–49 CrossRef CAS.
- D. E. Eastman, T. C. Chiang, P. Heimann and F. J. Himpsel, Phys. Rev. Lett., 1980, 45, 656–659 CrossRef CAS.
- R. E. Watson, J. W. Davenport, M. L. Perlman and T. K. Sham, Phys. Rev. B: Condens. Matter Mater. Phys., 1981, 24, 1791–1797 CrossRef CAS.
- W. F. Egelhoff Jr, Surf. Sci. Rep., 1987, 6, 253–415 CrossRef.
- P. S. Bagus, C. J. Nelin, X. Zhao, S. V. Levchenko, E. Davis, X. Weng, F. Späth, C. Papp, H. Kuhlenbeck and H.-J. Freund, Phys. Rev. B, 2019, 100, 115419 CrossRef CAS.
- M. B. Casu, Y. Zou, S. Kera, D. Batchelor, T. Schmidt and E. Umbach, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 193311 CrossRef.
- M. B. Casu, Phys. Status Solidi RRL, 2008, 2, 40–42 CrossRef CAS.
- L. Zhang, H. Peisert, I. Biswas, M. Knupfer, D. Batchelor and T. Chassé, Surf. Sci., 2005, 596, 98–107 CrossRef CAS.
- E. E. Rennie, B. Kempgens, H. M. Köppe, U. Hergenhahn, J. Feldhaus, B. S. Itchkawitz, A. L. D. Kilcoyne, A. Kivimäki, K. Maier, M. N. Piancastelli, M. Polcik, A. Rüdel and A. M. Bradshaw, J. Chem. Phys., 2000, 113, 7362–7375 CrossRef CAS.
- K. Ueda, M. Hoshino, T. Tanaka, M. Kitajima, H. Tanaka, A. De Fanis, Y. Tamenori, M. Ehara, F. Oyagi, K. Kuramoto and H. Nakatsuji, Phys. Rev. Lett., 2005, 94, 243004 CrossRef.
- M. Ballester, J. Castaner, J. Riera, J. Pujadas, O. Armet, C. Onrubia and J. A. Rio, J. Org. Chem., 1984, 49, 770–778 CrossRef CAS.
- M. A. Fox, E. Gaillard and C. C. Chen, J. Am. Chem. Soc., 1987, 109, 7088–7094 CrossRef CAS.
- T. Bürgi, Nanoscale, 2015, 7, 15553–15567 RSC.
- S. J. Stranick, A. N. Parikh, Y. T. Tao, D. L. Allara and P. S. Weiss, J. Phys. Chem., 1994, 98, 7636–7646 CrossRef CAS.
- S. J. Stranick, A. N. Parikh, D. L. Allara and P. S. Weiss, J. Phys. Chem., 1994, 98, 11136–11142 CrossRef CAS.
- R. K. Smith, S. M. Reed, P. A. Lewis, J. D. Monnell, R. S. Clegg, K. F. Kelly, L. A. Bumm, J. E. Hutchison and P. S. Weiss, J. Phys. Chem. B, 2001, 105, 1119–1122 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Survey spectra of SAM2A and fit results for the photoemission lines in the SAM2A C 1s spectra. SAM1 survey, stoichiometric analysis, and fit results for the photoemission lines in the SAM1 C 1s spectra. C 1s core level spectra at 460 and 640 eV. Fit results for SAM2 and SAM4 at 460 eV. Fit results for SAM2 and SAM4 at 640 eV. Electrochemical measurements, stability under air exposure, and stability under X-ray beam exposure. See DOI: 10.1039/d0sc03399e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |