
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe assemble, grow and lift-off (AGLO) strategy to construct complex gold nanostructures with pre-designed morphologies†
Xin
Luo
,
Christophe
Lachance-Brais
,
Amy
Bantle
and
Hanadi F.
Sleiman
 *
*
Department of Chemistry, McGill University, 801 Sherbrooke Street West, Montreal, Quebec H3A 0B8, Canada. E-mail: hanadi.sleiman@mcgill.ca
First published on 23rd April 2020
Abstract
The construction of metallic nanostructures with customizable morphologies and complex shapes has been an essential pursuit in nanoscience. DNA nanotechnology has enabled the fabrication of increasingly complex DNA nanostructures with unprecedented specificity, programmability and sub-nanometer precision, which makes it an ideal approach to rationally organize metallic nanostructures. Here we report an Assemble, Grow and Lift-Off (AGLO) strategy to construct robust standalone gold nanostructures with pre-designed customizable shapes in solution, using only a simple 2D DNA origami sheet as a versatile transient template. Gold nanoparticle (AuNP) seeds were firstly assembled onto the pre-designed binding sites of the DNA origami template and then additional gold was slowly deposited onto the AuNP seeds. The growing seed surfaces eventually merge with adjacent seeds to generate one continuous gold nanostructure in a pre-designed shape, which can then be lifted off the origami template. Diverse customized patterns of templated AuNP seeds were successfully transformed into corresponding gold nanostructures with the target structure transformation percentage over 80%. Moreover, the AGLO strategy can be incorporated with a magnetic bead separation platform to enable the easy recycling of the excess AuNP seeds and DNA components.
Introduction
The ability to construct metallic nanostructures with pre-designed complexity is an essential goal in nanoscience and technology. This control is particularly important in the fields of nanoplasmonics and nanoelectronics, given that the localized surface plasmonic resonance and electronic properties of metallic nanostructures profoundly depend on their morphology and spatial arrangement.1–5 Top-down lithography methods have limited resolution in fabricating 3D metallic nanoparticles, and they are unsuitable for large-scale production due to the high cost and slow stepwise processes.6–9 Over the past two decades, the field of DNA nanotechnology has enabled the bottom-up fabrication of increasingly complex one-, two- and three-dimensional structures from nano- to micro-scale with unprecedented specificity, programmability, and sub-nanometer precision.10–14 Among various techniques developed in structural DNA nanotechnology, DNA origami is one of the most promising strategies, as it not only dramatically increases the complexity of DNA nanostructures with enhanced yield, rigidity and stability,15–20 but has also been shown compatible with biotechnological mass DNA production.21 The intrinsic assets of the DNA origami make it an ideal method to rationally organize and construct hierarchical metallic nanostructures.22–26DNA structure-templated metallic nanoparticles can be generally divided into two categories: (1) individual DNA-functionalized metal nanoparticles and clusters assembled on a DNA scaffold; (2) continuous metallic structures by electroless deposition of metals templated by a DNA structure. While the first method has been well-studied and optimized over the years,27–29 strategies towards the second type of structures remain challenging. One straightforward method is DNA origami metallization,30,31 based on previous work on simple DNA double helix metallization.32–44 An impressive array of complex metallic structures have been constructed using double-stranded DNA plasmids as templates.43 However, the relationship between the DNA plasmid structure and final morphology is as yet unclear, making it difficult to construct rationally designed metal nanostructures. The use of DNA origami as a customizable template theoretically eliminated this limitation. However, due to nonspecific attachment of metal seeds on the DNA surface, this method predominantly yields a granular morphology with only approximate control of the overall metallic object shape, and little control over the surface structural details.30,31 Other methods require pre-immobilizing DNA origami structures onto flat substrates (mica, silica, etc.),45–49 which would prevent further scale-up, functionalization and application of the resulting metallic structures. Recently, a DNA origami mould method has been developed to cast metallic structures inside the DNA molds.9,50–52 To reduce mould deformation and metal leakage during the casting process, it was necessary to employ complex 3D multilayered DNA origami structures with closed cavities.9 Subsequently, each new metallic structure required the re-design of the DNA origami mould, which normally involves hundreds of new staple strands.
It is of note that all previous studies relied on the use of DNA structures as permanent templates and binding metallic particles or growing metals around them. Here we report an Assemble, Grow and Lift-Off (AGLO) strategy to construct complex pre-designed gold nanostructures in solution using only a simple 2D DNA origami sheet as a transient template. By carefully optimizing the electroless plating conditions, reagents and most importantly the surfactant, metal deposition and AuNP seed growth can occur homogeneously in solution with little aggregation. The produced gold nanostructures consist of grown and merged AuNPs. They can be lifted off the DNA origami template as robust and stable standalone particles, and stay unchanged even with several rounds of urea washing and ultrasonication treatment. This superior integrity arises from the complete merging (rather than just touching) between adjacent AuNP seeds during the gold growth process. We further investigated this process in detail and identified a number of crucial factors that dictate the merging process, as gaps or incomplete merging between AuNP seeds have been shown to tremendously affect the conductivity and plasmonic property of the gold nanostructures.48,51,53 By introducing surfactants into the gold growth process, we successfully eliminated the requirement of surface immobilization, which is necessary to prevent aggregation in previous studies.45–49 This renders the AGLO strategy a solution-based method to fabricate customizable gold nanostructures, which has a great potential for scale-up production.
Moreover, we demonstrate that the AGLO strategy can be incorporated with a magnetic bead (MB) platform to enable the easy removal and reuse of the excess AuNP seeds to reduce the cost. The gold nanostructures produced by the AGLO strategy can be released by strand displacement after the gold plating, which subsequently regenerates the DNA origami template for new AuNP seeds assemblies. This allowed us to further push the AGLO-MB system and reuse the DNA origami template for a second synthetic cycle as a proof of concept. This will help to significantly reduce the cost of DNA-templated synthesis. To the best of our knowledge, this is also the first example of recycling the DNA origami template in DNA-templated nanomaterial fabrication. More generally, this method is an important development in our ongoing efforts to use DNA nanostructures not as permanent scaffolds, but as transient templates that provide instructions for materials synthesis.54–56 In this case, much like in DNA-templated chemical synthesis, the DNA origami template assembles the gold nanoparticle structures, mediates selective gold growth to a well-defined shape, and can be reused for new cycles after nanoparticle lift-off.
Results and discussion
In the Assemble, Grow, and Lift-Off (AGLO) strategy (Fig. 1), DNA-functionalized AuNP seeds are firstly assembled onto a simple single-layered DNA origami sheet template (Fig. S1a†) to form a pre-designed AuNP pattern (a linear trimer assembly as an example). To encourage merging between adjacent AuNP seeds in the following gold growth step, the seeds must be placed as close to each other as possible; however, this can also increase the repulsion between the AuNP DNA shells, thus decreasing the yield of correctly assembled linear trimer. After a series of optimizations (Fig. S2†), AuNPs with an average diameter of around 12 nm were selected as the seeds. They are uniformly functionalized with strand BS1 (red) or BS2 (green) to hybridize to corresponding binding sites located ca. 16 nm apart on the origami template (Fig. S1b†). The origami binding sites each consist of three sequences extended from the 3′ end of staple strands (staple-BS1′ or staple-BS2′) to maximize the binding yield of the AuNP seeds. Short toehold sequences highlighted in blue in Fig. 1a are left unhybridized for the following lift-off step.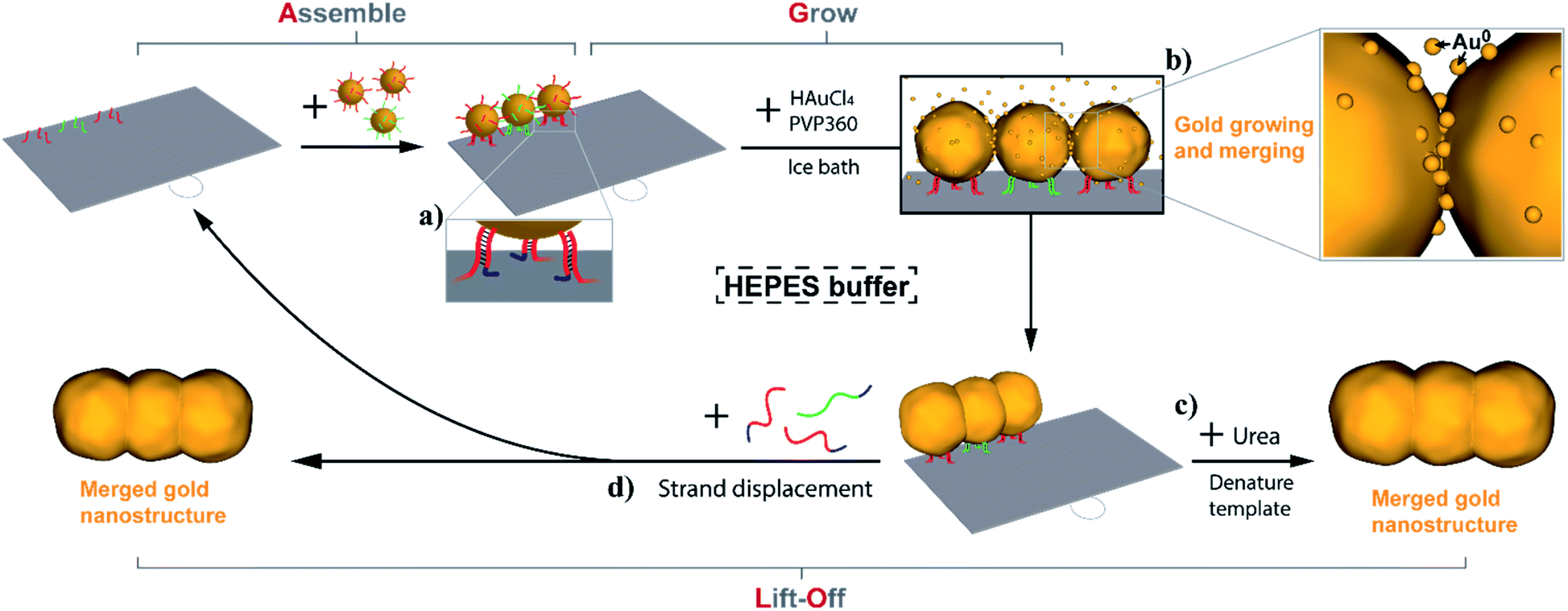 | ||
| Fig. 1 Schematic illustration of employing the Assemble, Grow and Lift-Off (AGLO) strategy to construct a pre-designed gold nanostructure. DNA-functionalized AuNP seeds are firstly assembled onto a DNA origami template to form a pre-designed AuNP pattern; supplementary gold is then slowly deposited onto the surface of the assembled AuNP seeds with the optimized conditions; eventually, the growing AuNP surface will merge with adjacent AuNPs to generate one integrated gold nanostructure (b); finally the origami template can be denatured and removed (c) or DNA strand displacement can be carried out to release the gold nanostructure and regenerate the template (a, d). | ||
Next, without further purification of the AuNP-origami assembly, 0.06% tetrachloroauric(III) acid (HAuCl4) are added as the gold source (GS) for electroless plating and the growth of the AuNP seeds. Interestingly, the HEPES (2-[4-(2-hydroxyethyl)piperazinyl]ethanesulfonic acid) buffer we use for the AuNP-origami assembly system is known to undergo oxidation through a one-electron process and thus can directly act as a mild reducer to transform the Au3+ to Au0 species,57,58 which subsequently deposit onto the AuNP seeds. The slow kinetics of the reducing process of HAuCl4 by HEPES (up to 8 min at room temperature to complete reaction) provide better control over the AuNP seed growth, resulting in uniformly-sized gold structures with roughly spherically grown AuNP seeds. Even slower kinetics (1.5 to 2 h) can be achieved by carrying out the gold growth step in an ice bath, which eliminates the random nucleation of Au0 in solution when an excess of gold source is applied (Fig. S3†). As the reduced gold slowly and continuously deposits onto the pre-assembled AuNP seeds, the growing seed surface will eventually coalesce and merge with adjacent seeds to generate one integrated gold nanostructure (Fig. 1b). Surfactant is crucial to the gold growth process, the absence of which will lead to fast aggregation of AuNPs once the gold deposition initiates (Fig. S4†). We also discovered that surfactants play an important role in the merging of adjacent AuNP seeds during growth, which will be discussed in detail below. Polyvinylpyrrolidone with an average molecular weight of 360![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 (PVP360) was found to have the highest merging yield among the surfactants investigated and was applied in all experiments unless noted otherwise.
000 g mol−1 (PVP360) was found to have the highest merging yield among the surfactants investigated and was applied in all experiments unless noted otherwise.
Given that the merged gold nanostructures after growth are integrated and stable, the DNA origami template can be removed by washing the sample with 8 M urea solution (Fig. 1c) after passivating the gold surface with a small molecule ligand (PEG7 acid disulfide, OEG). Furthermore, the AGLO product survives ultrasonication treatment which suggests the merging between AuNP seeds is complete and the structure is robust. All AGLO products reported here were washed and ultrasonicated with urea solution and water several times before TEM characterization. Alternatively, strand displacement can be carried out after the gold growth and lift-off the merged gold structure while regenerating the DNA origami template and its binding sites (Fig. 1d). This allows us to reuse the origami template based on magnetic bead separation (see below).
Fig. 2 demonstrates the characterization of the AGLO process on a AuNP seed linear trimer assembly. TEM and AFM images of the seed-origami assembly before growth show three AuNP seeds assembled to the DNA origami in a linear pattern (Fig. 2b and S5a†). In this experiment, 1.2–1.5 times of AuNP seeds were used to increase the binding yield of the assembly and the excess seeds were not removed before the gold growth, which explains the AuNP monomers in the background of the images. After electroless plating and gold growth, the AuNP seed trimer grew into larger, quasi-spherical shapes and merged into one continuous gold nanostructure, while the AuNP seeds without the DNA origami template only grew into larger AuNP monomers (Fig. 2a). AFM images of the AGLO product after gold growth (Fig. S5b†) shows that the AuNPs remained anchored on the DNA origami template throughout the gold deposition process. After urea washing and ultrasonication, both trimer and monomer samples were analyzed by agarose gel electrophoresis, which confirmed larger gold nanostructure formation in the AGLO experiment, where a clear lower mobility band can be observed (Fig. 2c). The gel bands were subsequently excised, and the products extracted and characterized by TEM. The slower-moving band (red) was mostly composed of merged gold linear trimers, and the faster-moving band contained grown AuNP monomers (Fig. S5c†). Unlike the smooth surface of a gold nanorod along the axis, the merged linear trimer structure has two “neck” junctions at the interface of merging between adjacent NPs, which have been demonstrated to have distinct plasmon coupling modes and greatly enhanced local fields.59,60 Such morphology uniformity of these merged gold nanoparticle structures and their off-template integrity in solution has not been demonstrated in previously reported strategies.
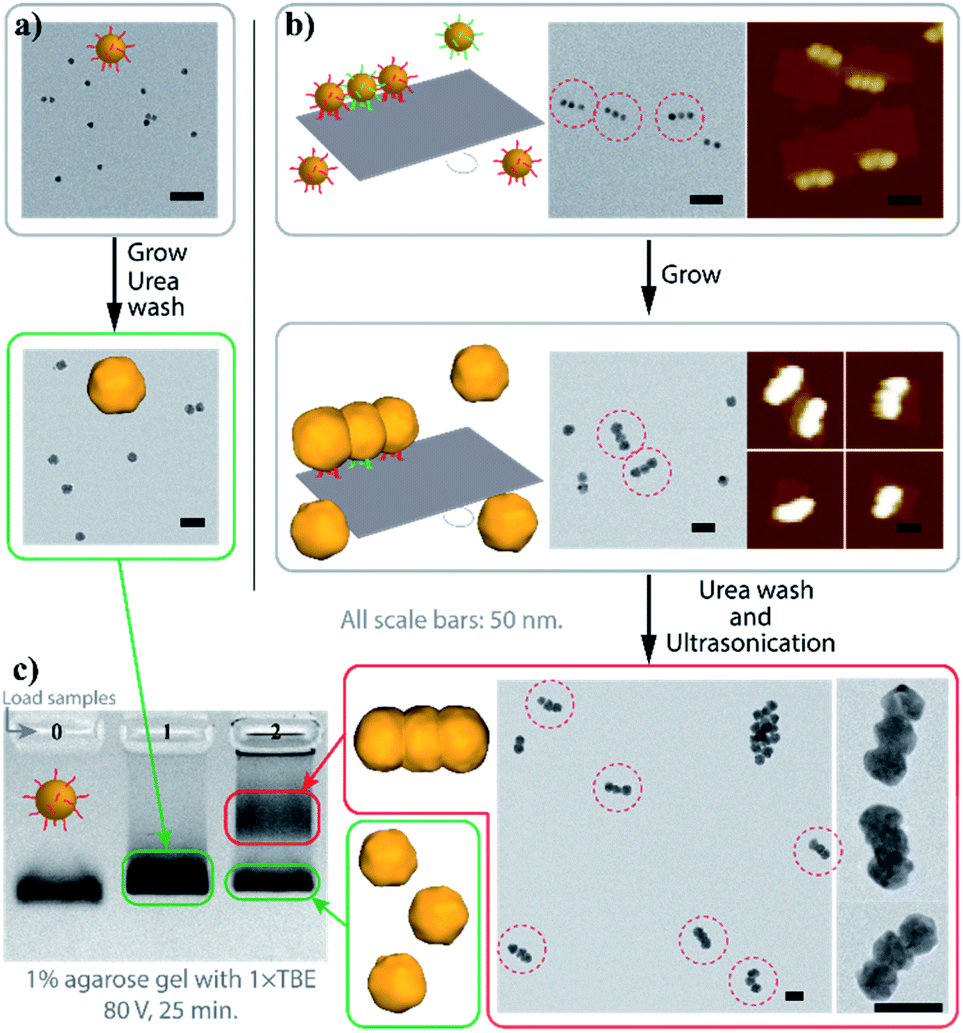 | ||
| Fig. 2 Characterization of the AGLO linear trimer process. (a) AuNP seeds growth without DNA origami template. (b) AGLO process on a linear trimer AuNP seed assembly. (c) Agarose gel electrophoresis analysis of products from (a) (lane 1) and (b) (lane 2); lane 0: AuNP seed. Gel imaged in white light channel. All scale bars are 50 nm; AFM images have the same height color scale. See Fig. S5† for more images. | ||
To gain a better understanding of this process, we examined the AuNP seed growth and merging process with increasing amounts of gold source. As shown earlier, the AGLO method normally works on unpurified AuNP seed-origami assemblies, and does not require prior removal of the excess free seeds from solution, as the product is ultimately separated by gel electrophoresis (Fig. 2c). However, to study the growth and merging of neighboring AuNP seeds and conduct statistical analyses, we used a larger excess of AuNP seeds (7 equiv.) for the assembly and an extra gel purification step was applied to remove the excess free AuNP seeds in the seed-origami assembly solution (Fig. S6†) before the gold growth. Note that this step does not separate the different AuNP seed-origami assemblies (i.e., origami templated seed monomer, dimer vs. trimer) from one another, and is only performed to remove the excess gold seed. Increasing amounts of gold source (0–6 μL, 0.06% HAuCl4) were added to 0.013 pmol purified linear trimer AuNP seed-origami assemblies (Fig. 3). TEM characterization was carried out on these AGLO samples after the growth step but before the lift-off step so that the linear trimer structure remains intact for analysis (Fig. 3a, see Fig. S7† for additional images). The AuNPs grew larger in size through electroless plating with increasing gold source, and once they reached the size threshold to be in contact with neighboring AuNPs, they started to coalesce and merge into one continuous structure. The percentage of merged interparticle gaps in the linear trimer assembly of each sample is displayed in Fig. 3b. To eliminate human analysis errors, we employed a software-based method to determine whether the gaps were merged (Fig. S8†). Statistics of the final AuNP diameter (perpendicular to the linear trimer axis if merged) distribution in each sample are also shown in Fig. 3c with their mean AuNP diameters (D) and standard deviations shown on the graph (N = 100). Based on the design, the AuNP seeds should start touching and merging with adjacent seeds when their diameter reaches 16.3 nm (Fig. S1b†). Interestingly however, Fig. 3 shows that only 6.5% of the interparticle gaps merged when the seed diameter reached 19 ± 1 nm. This could be due to the difficulty in resolving a few layers of gold coalescence from the TEM images. Alternatively, it is also possible that the AuNPs move apart to a certain extent when their surfaces are in proximity, because of their repulsive DNA shells and the bendable single layer origami template. For all samples after gold growth, the distribution of the final AuNP diameter is found to be narrow. When the final diameter of the AuNPs in the linear trimer is plotted against the amount of gold source applied, the plot fits very well into a sphere volume function (Fig. 3d and S-VI-3 in the ESI†) and the cube of the diameter is proportional to the amount of gold source (Fig. 3d inset), which indicates that the majority of the gold source added to the system deposited onto the AuNP seed and grew roughly into a quasi-spherical (or polyhedron) shape with little random background gold nucleation and formation of other gold morphologies. This is a large improvement compared to previous work in the literature.45–48 We attribute this mostly to the slow kinetics of the in situ reduction of the gold source by the HEPES buffer in solution under our AGLO experimental conditions, giving enough time for the gold source to distribute homogeneously around the AuNP assembly. The slow generation of active gold growing species are also likely to keep the system below the nucleation threshold.
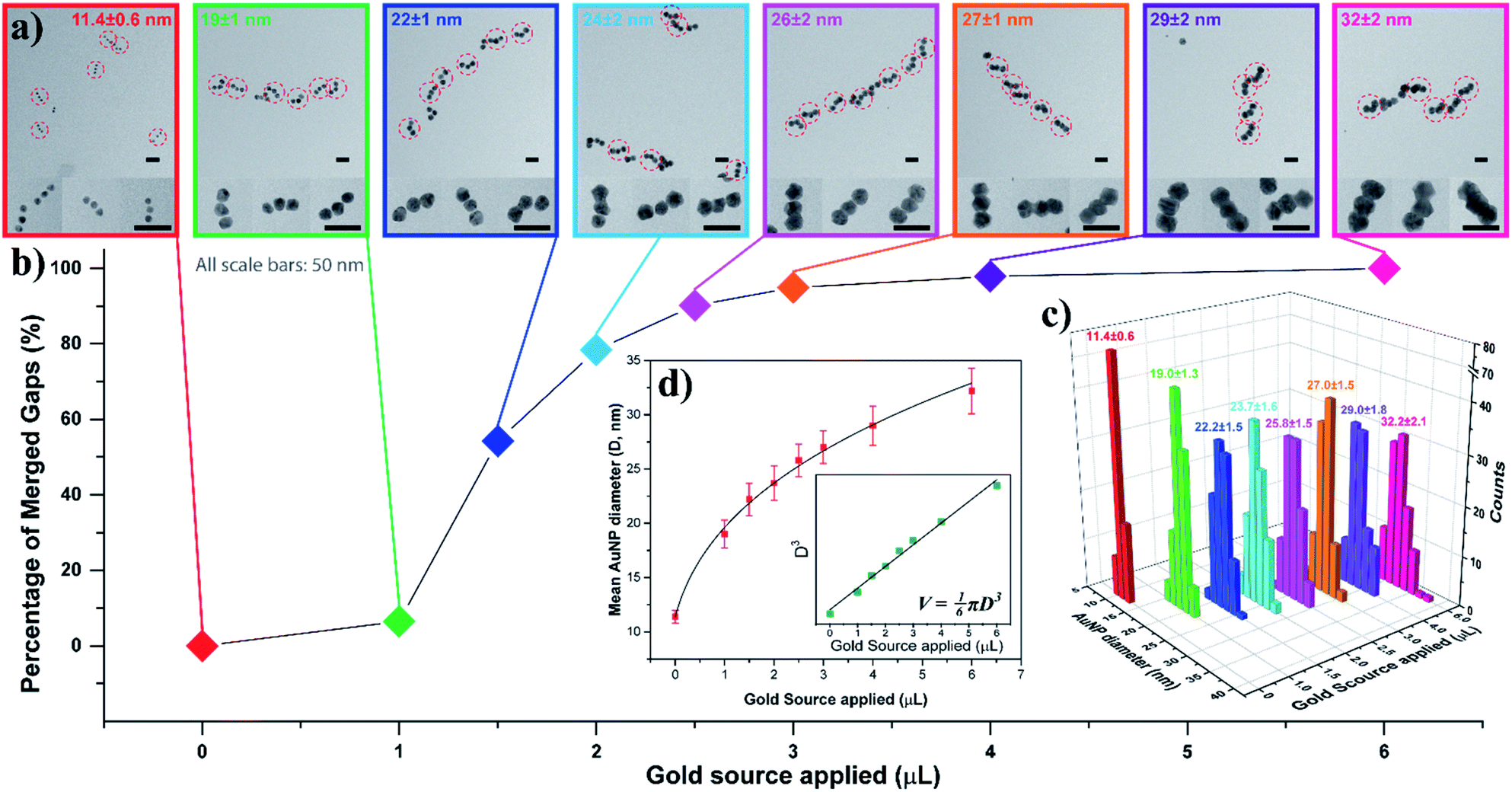 | ||
| Fig. 3 AuNP seed growth and merging in a linear trimer assembly AGLO process. 0, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0, 6.0 μL gold source (0.06% HAuCl4) was applied individually to eight purified linear trimer AuNP seed-origami assembly samples (each containing 0.013 pmol AuNP seeds) during the AGLO process. Data of the same sample are color coded for better correspondence. (a) TEM images of each sample after the grow step but before the lift-off step. All scale bars are 50 nm. See Fig. S7† for additional images. (b) The percentage of merged interparticle gaps in the linear trimer assembly of each sample. (c) Final AuNP diameter distributions, mean diameters and standard deviations of each sample (N = 100). (d) Fitting the mean diameter data (red) or the cube of the mean diameter data (green, inset) of the grown AuNP samples as a cube root function or a linear function (inset) of the amount of the gold source applied respectively. | ||
To facilitate the quantitative investigation of the experimental conditions in the AGLO strategy, we developed a software-based algorithm that counts the number of different gold nanostructures in the TEM images of the AGLO crude product based on the size and roundness of the particles (Fig. S9†). This allowed us to analyze hundreds of images in a relatively short time and provided reliable statistical results (typically N > 1000). Fig. 4a and S10a† demonstrate representative AFM images of a linear trimer AuNP seed-origami assembly (after removing the excess AuNP seeds), with an assembly yield of 52%. This seed-origami assembly was then subjected to the grow and lift-off steps as before (Fig. 4a to b), followed by TEM imaging. The AuNP seeds grew to around 32 nm in diameter with a merged target linear trimer yield of 47% (Fig. 4b and S10b†). The yields from AFM and TEM images are calculated as the total number of AuNPs in the target structure (linear trimer in this case) over the total number of AuNPs in all structures. More importantly, we define a target structure transformation percentage as (merged target structure yield from TEM)/(target structure assembly yield from AFM) × 100%. This formula measures the efficiency of the AGLO merging process, which we calculated to be as high as 91% for the linear trimer structure. Moreover, as can be observed in Fig. 4c, the distribution of the AuNP assemblies templated by the DNA origami before the gold growth (blue) were generally maintained after the complete AGLO process (red). As expected, applying less gold source (Fig. 4a to d) resulted in smaller AuNP diameter (around 24 nm) and a lower total yield (34%) of the target merged linear trimer structure after the AGLO process (Fig. 4d and S10c†). Interestingly, using the same polymer with a lower average molecular weight of 10000 g mol−1 (PVP10) as a surfactant (Fig. 4a to e) resulted in similar final AuNP diameter but significantly lower yield (12%) of the merged linear trimer structure (Fig. 4e and S10d†). The structure distribution of the crude AGLO products of the two samples (Fig. 4f) reveals that the sample with PVP10 (green) contains significantly less merged linear trimer structure and more AuNP monomers than the sample with PVP360 (red), while the percentage of the other structures are comparable. This data indicates that with PVP10 the seed-origami structures are less likely to merge during the gold growth process, and subsequently more likely to disassemble during the denaturation and ultrasonication washes. When a mixture of PVP10 and PVP360 was used as the surfactant, the merging yield was also much lower than that of using PVP360 only (Fig. S23†). These results can be accounted for by taking the hydrodynamic volume of the two PVP polymers into consideration. Dynamic light scattering (DLS) analysis revealed that the apparent hydrodynamic radius of PVP360 in HEPES buffer is 29 ± 1 nm while the radius of PVP10 is only 2.83 ± 0.05 nm (Fig. S10f†). Given that the gap size between the seeds in the seed-origami assembly is ca. 4.8 nm (Fig. S1b†), the PVP10 molecules can easily diffuse into the seed gaps and PVP360 molecules cannot. During the gold growth process, the PVP10 molecules in the gap will prevent the adjacent growing AuNP surfaces from contacting and merging (Fig. 4h). Moreover, the relatively weakly binding nature of PVP is crucial to successful particle merging. In support of this, a strongly binding and small surfactant (PEG7 acid disulfide, OEG) completely blocked the merging of adjacent AuNP seeds on the template (Fig. 4g and S10g†). These results showcase the importance of using a large, weakly bound surfactant, to surround the structure but not occupy the gaps between particles. In addition to the surfactant influence on the merging yield of the target structure in the AGLO method, we discovered that the aging of the HEPES buffer under ambient light can also play a role in the merging of gold nanostructures (S-VIII-2 in the ESI†). Our growth and merging mechanistic studies provide guidelines for future research on the templated construction of other nanomaterials, including hybrid materials (e.g. growing silver on gold seed assemblies).
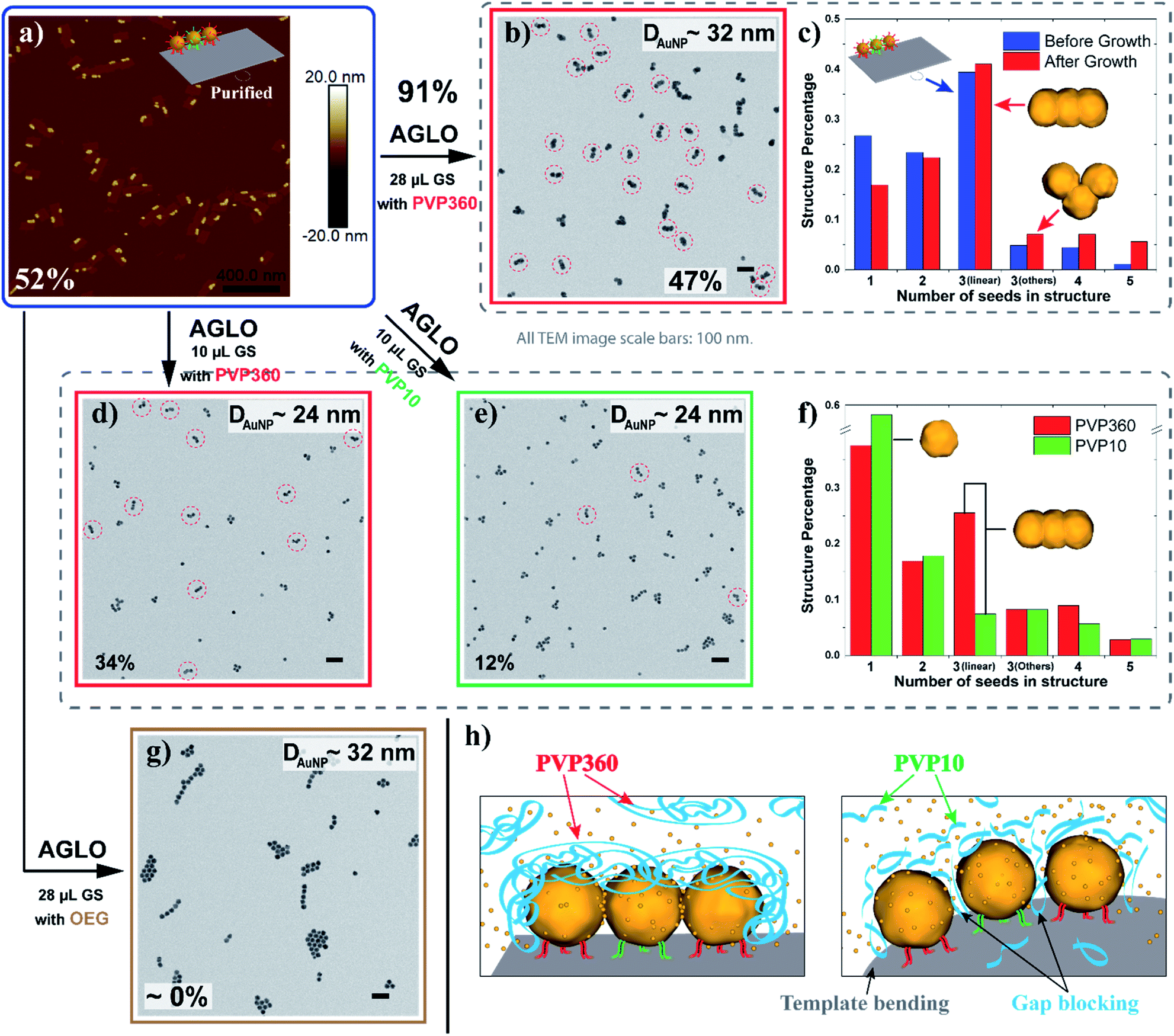 | ||
| Fig. 4 (a) A representative AFM image of the linear trimer AuNP seed-origami assembly after agarose gel purification. The assembly yield of the target linear trimer structure is 52%. (b) TEM image of the crude AGLO product with 28 μL gold source (GS) and PVP360 as the surfactant. The yield of the merged linear trimer target structure is 47%. (c) The distribution of AuNP assemblies templated by the DNA origami before the gold growth (Blue, statistics from AFM images) and the distribution of AuNP structures in the crude product after the AGLO process (red, statistics from TEM images). (d) TEM image of the crude AGLO product with 10 μL gold source and PVP360 as the surfactant. The yield of the merged linear trimer target structure is 34%. (e) TEM image of the crude AGLO product with 10 μL gold source and PVP10 as the surfactant. The yield of the merged linear trimer target structure is 12%. (f) The distribution of AuNP structures after the complete AGLO process with PVP360 (red) or PVP10 (green) as the surfactant. (g) TEM image of the crude AGLO product with 28 μL gold source and OEG as the surfactant. (h) Schematic illustration of the gold growth process with PVP360 or PVP10; smaller PVP10 molecules can diffuse into the interparticle gaps and hinder AuNP merging. Note that structure percentages in the distribution diagrams are calculated differently than the yields (S-VII in the ESI†). All TEM images scale bars: 100 nm. See Fig. S10† for additional images. | ||
With sufficient knowledge of the mechanism and the effect of the key components, the AGLO strategy can be easily generalized to construct arbitrary pre-designed complex gold nanostructures with minimum structural origami design work. As demonstrated in Fig. 5 (and Fig. S14†), a triangular trimer (a), a tetramer cluster (b), a Y-shape tetramer (c), an L-shape pentamer (d) and a complex box-shape octamer (e) were successfully synthesized with the same origami structure and AuNP seeds. The merged gold nanoparticles after AGLO give rise to a longitudinal plasmon resonance that can be observed by UV-vis spectroscopy (Fig. 5f and S21†). We moved the seed-origami assembly design more towards the center of the DNA origami template for better template robustness as the assembly became more intricate. The target structure transformation percentages of all these structures were above 80% and up to 92%, which indicated that the gold growth and merging process was highly efficient. It is worth pointing out that we did not optimize the distance of the origami binding sites in these designs individually and a decrease in assembly yield was observed as the complexity of the target gold nanostructure increased (Fig. S14†). Employing multi-layered DNA origami as the template should further increase the yield of more complex seed-origami assemblies and merged products (S-IX in the ESI†). More excitingly, it can also be foreseen that with robust 3D origami as templates,23,61 the AGLO strategy will extend the programmability and structural complexity of the gold nanostructures into the third dimension and create pre-designed and truly arbitrary, standalone hierarchical 3D gold nanostructures for various potential applications.
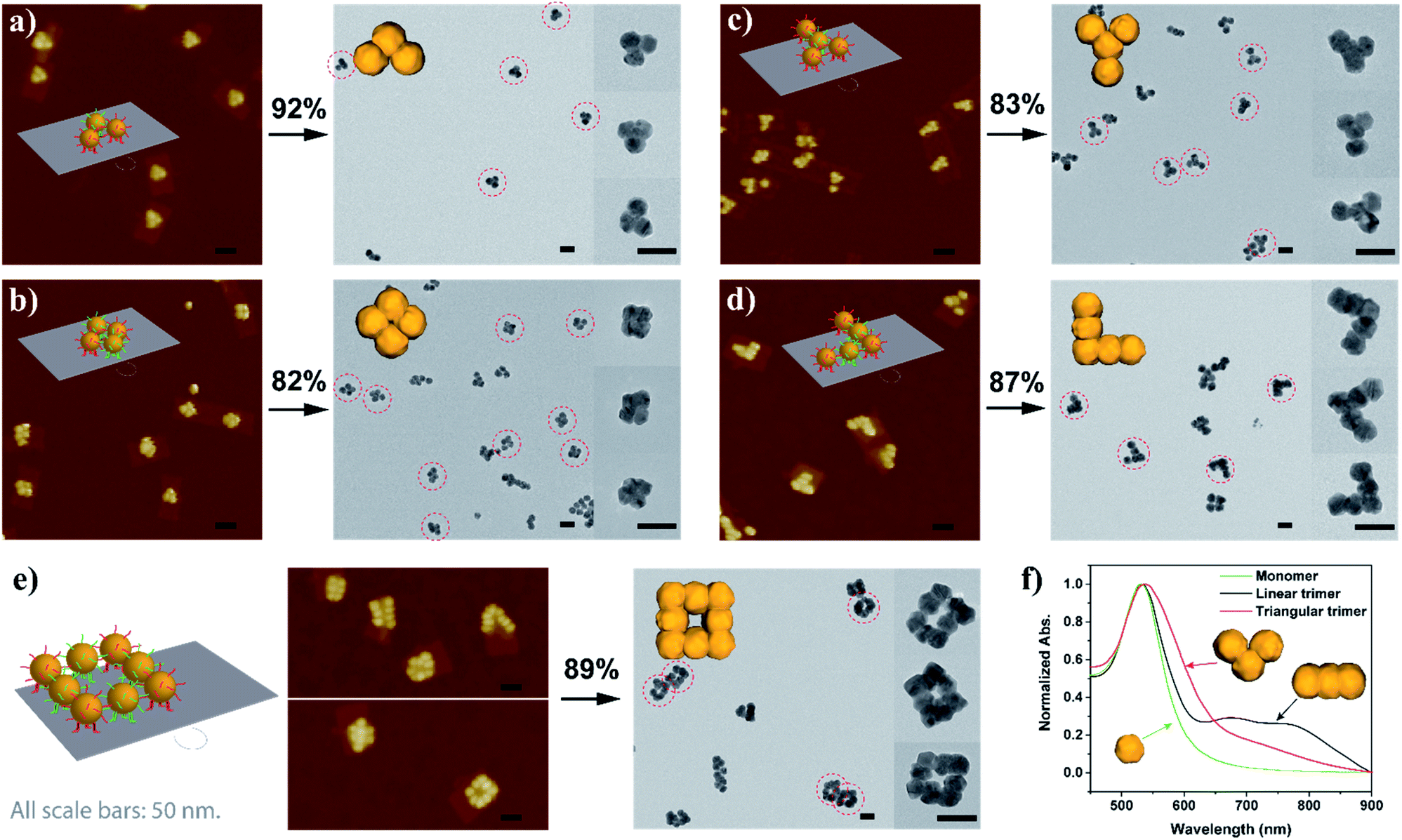 | ||
| Fig. 5 Schematic design, AuNP seed-origami assembly AFM images and AGLO crude product TEM images of the (a) triangular trimer, (b) tetramer cluster, (c) Y-shape tetramer, (d) L-shape pentamer and the (e) box-shape octamer gold nanostructures. (f) UV-vis spectra of the monomer, linear trimer and triangular trimer after AGLO. All scale bars are 50 nm. The target structure transformation percentage of each AGLO process is marked above the arrows. See Fig. S14† for additional images. | ||
Despite the recent advances in the biotechnological mass production of DNA origami, which can significantly reduce the price of DNA origami synthesis,21 the cost of DNA nanostructures, especially those with chemical modifications, is still one of the major factors limiting the practical applications of DNA nanotechnology. In terms of our AGLO strategy, the only chemical modification on DNA is the disulfide modifications on the binding strands to conjugate to the AuNP seeds. The excess amount of DNA-conjugated AuNP seeds employed, along with the binding strands conjugated to them, can undermine the cost-efficiency of the AGLO system. In order to recycle the excess AuNP seeds in a simple and straightforward manner and to avoid labor-intensive gel purification, we incorporated the AGLO strategy with a magnetic bead (MB) platform. Firstly, carboxyl group functionalized magnetic beads were decorated with an amino group functionalized linker DNA strand through a simple 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) coupling reaction. Accordingly, three edge staple strands on the DNA origami template were extended with the complementary sequence (Fig. 6a), which allowed the DNA origami to stably anchor onto the surface of the MBs after incubation (Fig. 6b). The MBs present a brown to light yellow color depending on their concentration (Fig. 6a inset) while the AuNPs seeds are intensely red when homogenously dispersed (Fig. 6b inset). The TEM image in Fig. 6b shows a MB after incubation with the crude linear trimer seed-origami assembly, where AuNP seeds assembled on the templates can be observed to attach to the MB surface while the excess AuNP seeds scattered in the background. With the help of magnetic separation, the excess AuNP seeds could be easily removed and collected for the next experiment to assemble seeds onto the DNA origami template (Fig. 6c). In the meantime, the MBs were kept and washed with the buffer several times to remove non-specifically bound AuNP seeds, resulting in a sample with recovered light-yellow color mixed with a barely detectable red color (Fig. 6d inset). The TEM image in Fig. 6d (and Fig. S16a†) shows that the origami templated linear trimer assemblies were still bound to the MB surface after washing. Next, gold source was added to the system and the templated structure grew into the merged linear trimer structure on the MBs surface (Fig. 6e and S16b†), which gave a light purple color to the solution (Fig. 6e inset). As discussed before, the merged gold structure can be lifted off the template through two methods, which subsequently creates two possible synthetic cycles with MBs (Fig. 6, Cycle I and II). The more straightforward method is to denature the DNA origami template with a urea wash and ultrasonication treatment (Fig. 6f). Subsequently, the detached AGLO product can be collected through magnetic separation. TEM characterization of such product reveals merged linear trimer gold structures with a yield of 40% (Fig. 6g and S16c†), which is close to the yield of the AGLO process without the MBs. Meanwhile, since the linking strand is covalently functionalized to the MB surface, the MBs can be recovered and reused for the next synthesis Cycle I after thorough washing with water and buffer (Fig. 6h). To further confirm the compatibility of the AGLO process and the MBs platform, the L-shape pentamer was also successfully synthesized on the MB surface (Fig. S17†).
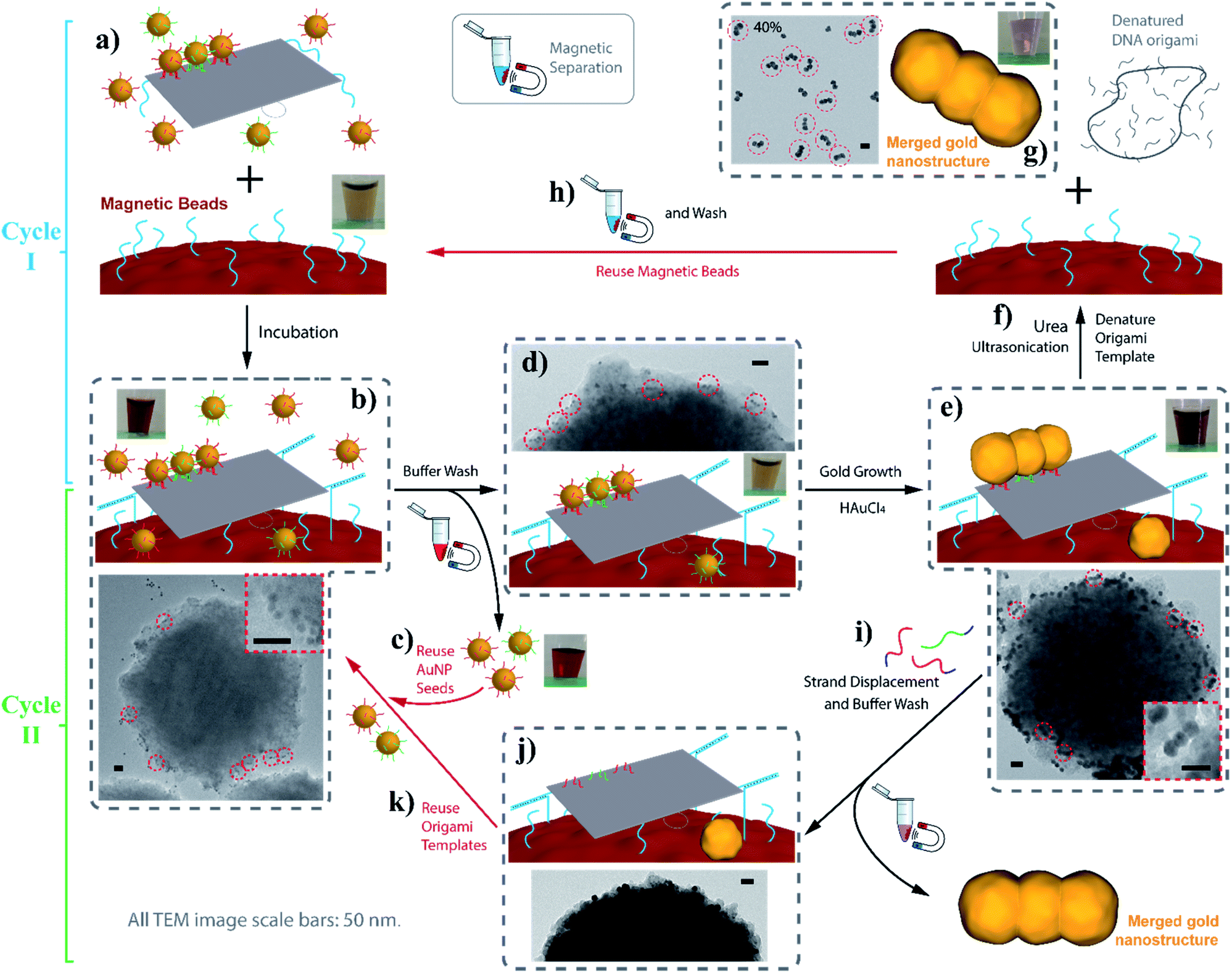 | ||
| Fig. 6 Incorporate the AGLO strategy with the magnetic bead platform. Cycle I: linking strand (LS, blue) functionalized MBs are incubated with AuNP seed-origami assembly functionalized with LS′ strands (a) to anchor the templated assembly on to the MB surface (b); after magnetic separation, excess AuNP seeds are recovered and reused (c) while origami templated AuNP assemblies remains on the MBs (d); the gold source is then added to grow and merge the seeds on the MB surface (e); the grown gold nanostructures are lift off by denaturing the DNA origami template through urea wash and ultrasonication (f); the collected AGLO crude product contains 40% merged linear trimer structure (g); the MBs can be reused for the next cycle after water and buffer wash (h). Cycle II: after the gold growth step, invading strands are added to lift off the grown gold nanostructures (i); the DNA origami templates are consequently regenerated (j) for the binding of the AuNP seeds for the next Cycle II (k). Inset pictures show the color of the experiment solution in each step of Cycle I. All TEM image scale bars are 50 nm. | ||
In addition to the easy recovery and reuse of the excess AuNP seeds and the MBs in Cycle I, the denatured origami scaffold and the staple strands can also be potentially recovered from the gold nanostructure products through centrifugation separation. In a second possible method, the grown and merged linear trimer product on the DNA origami template can be released through strand displacement (Fig. S18†). We thus designed a synthetic Cycle II with the MB platform that recycles the intact DNA origami templates anchored on the MB surface (Fig. 6, Cycle II). As the strand displacement overhangs are designed to be on the binding strands conjugated to the AuNP seeds (Fig. 1a), the binding sites of the DNA origami templates are readily available (Fig. 6j) to anchor AuNP seeds for the next synthetic Cycle II, directly after the strand displacement and buffer wash step (Fig. 6i). The excess invading strands used for the strand displacement can be easily recovered after the magnetic separation. Preliminary results showed that the current strand displacement design of the grown gold nanostructure is rather slow, as the invading strands have to diffuse into the crowded gap between the grown gold structures and the DNA origami template. Under these conditions, the MBs tend to slowly aggregate, which interferes with their recyclability and reduces the yield. After collection of the trimer product (Fig. S19†), we reused the regenerated origami template to conduct a second round of synthesis of the gold linear trimer (Fig. S20†), followed by urea wash and ultrasonication. The correctly arranged linear trimer structure synthesized from the second round can be observed in the resulting crude AGLO product (Fig. S20b,† red circle). For further possible practical applications of the Cycle II system, the MB surface can be passivated with PEG molecules to reduce non-specific binding and increase stability under the strand displacement conditions. Future strategies to increase the strand displacement releasing efficiency include incorporating azobenzene units into the binding strands for photorelease.62,63
Conclusions
In conclusion, we developed an Assemble, Grow and Lift-Off (AGLO) strategy to construct complex pre-designed gold nanostructures in solution using only a simple 2D DNA origami sheet as a versatile transient template. AuNP seeds were firstly assembled onto the predesigned binding sites of the DNA origami template and then additional gold was slowly deposited to the AuNP seeds through the reduction of HAuCl4 by HEPES buffer on an ice bath with PVP360 as the surfactant. As the reduced gold slowly and continuously deposits onto the pre-assembled AuNP seeds, the growing seed surface will eventually merge with adjacent seeds to generate one integrated gold nanostructure in a pre-designed shape. The gold nanostructures produced by the AGLO strategy consist of completely merged AuNPs and thus were robust and stable after removing the DNA origami template. The structures stay unchanged even after several rounds of urea washing and ultrasonication treatments. A number of 2D arrangements of AuNP seeds on the simple DNA origami template were successfully converted into complex and standalone gold nanostructures with the target structure transformation percentage over 80%. These structures are readily achievable from the same DNA origami template design by only changing a few staple strands. Compared to previous methods, the AGLO strategy uses DNA as a transient template in solution, which allows the recycling of materials for scalability, while offering much higher structural resolution and off-template integrity. We also believe that the AGLO strategy can be extended to 3D arrangement of AuNP seeds with 3D DNA origami structures to construct gold nanostructures with pre-designed 3D complexity. Furthermore, our mechanistic studies on nanoparticle merging (including methods to reduce aggregation, background nucleation and filling the gaps) laid down the foundation for the future DNA-templated fabrication of integrated hierarchical multicomponent nanomaterials with customizable structures, where different materials (QDs, silver NPs, iron oxide NPs, etc.) can be incorporated into one nanostructure in a pre-designed arrangement. The introduction of new materials into the AGLO strategy will fundamentally expand the application landscape of the products, and in the case of our gold particles, will result in precisely tailored nanoelectronics and nanoplasmonic properties.Moreover, we demonstrated that the AGLO strategy can be incorporated with the magnetic bead platform to enable the easy removal and reuse of the excess AuNP seeds to reduce the cost of the AGLO strategy. We further employed strand displacement to release the grown gold nanostructures from the DNA origami template and regenerated the DNA origami template for a second AGLO synthetic cycle as a proof of concept. With proper design improvement and system optimization, the AGLO-MB platform has great potential for cost-efficient synthesis of arbitrarily pre-designed gold nanostructures. To the best of our knowledge, this is also the first example of recycling the template in DNA origami-templated nanomaterials fabrication.
Experimental
AuNP seed and DNA origami assembly
To assemble AuNP seeds to the DNA origami template, 5 nM DNA origami was slowly added to concentrated AuNP-BS2 and mixed well with a pipette; after 5 min incubation at room temperature, concentrated AuNP-BS1 was added subsequently to the mixture and mixed with a pipette. The mixture was then slowly annealed over night from 37 °C to 10 °C (incubate at 37 °C for 30 min, cool down to 20 °C with a rate of 11 min per °C; then heat to 30 °C and incuabte for 30 min, followed by cooling down to 10 °C with the same rate). For experiments without agarose gel purification of the excess AuNPs before the gold growth, 1.5 equiv. of AuNP-BS2 and 1.2 equiv. of AuNP-BS1 (equivalent to the number of binding sites) were used. For experiments with a following agarose gel purification step before the gold growth step, 7 equiv. of both AuNP-BS2 and AuNP-BS1 were applied to further increase the assembly yield.Purification of seed-origami assemblies
For all experiments with subsequent statistical analysis of the AFM/TEM images, samples were purified by agarose gel electrophoresis to remove the excess gold nanoparticle seeds added. Directly after the assembly step, the annealed AuNP-origami solution was mixed with 1/5 volume of glycerol/H2O (7:1 v/v) and loaded on a 0.5–0.6% agarose gel in 1 × TAMg buffer. The gel was then run at 80 V for 2 h in a cold room (4 °C) and imaged under the white light channel. The second band from the bottom contains the target seed-origami assembly, which is extracted with a Freeze 'N Squeeze™ column. The assemblies were then re-quantified on the plate reader based on the absorbance at 450 nm.
Gold growth
In a typical AGLO experiment, 0.065 pmol (regarding to the AuNP seed) seed-origami assembly was diluted with a solution containing 150 μL 1 × HEPESMg buffer, 5 μL PVP360/H2O (5 g/100 mL) and 3.3 μL 10 × HEPESMg buffer in an Eppendorf tube. The diluted seed-origami solution was then chilled in an ice bath for 5 min, followed by adding 28 μL gold source (0.06 g/100 mL HAuCl4/H2O solution) and carefully mixing with a pipette. The total volume of the experiment solution was kept at around 200 μL and the HEPES concentration at 0.1 M (1 × HEPESMg). The solution was then left in the ice bath for 3 h without disturbance, during which time the solution color will change from light pink to dark purple/blue. After the incubation, the Eppendorf was moved from the ice bath to room temperature and 2 μL 0.1 M OEG/H2O solution was added and mixed by vortex.Lift-off standalone gold nanostructures
After a 30 min incubation following the gold growth step, 200 μL 8 M urea solution was added to the Eppendorf to denature the DNA origami template and lift-off the gold nanostructure product. The crude product was then centrifuged and washed one more time with 8 M urea and 3 times with Milli-Q water. Ultrasonication treatment was conducted after each centrifugation to re-disperse the crude AGLO product in urea solution or Milli-Q water. After washing and ultrasonication treatment, the crude product was directly imaged by TEM and analysed without further purification. To lift off the gold nanostructures by strand displacement, 500 times of both invading strands were added instead of the 8 M urea solution.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank NSERC, CFI and the Canada Research Chairs Program for funding. H. F. S. is a Cottrell Scholar of the Research Corporation.References
- V. Giannini, A. I. Fernandez-Dominguez, S. C. Heck and S. A. Maier, Chem. Rev., 2011, 111, 3888–3912 CrossRef CAS PubMed.
- N. Jiang, X. Zhuo and J. Wang, Chem. Rev., 2018, 118, 3054–3099 CrossRef CAS PubMed.
- W. P. McConnell, J. P. Novak, L. C. Brousseau, R. R. Fuierer, R. C. Tenent and D. L. Feldheim, J. Phys. Chem. B, 2000, 104, 8925–8930 CrossRef CAS.
- F. P. Zamborini, M. C. Leopold, J. F. Hicks, P. J. Kulesza, M. A. Malik and R. W. Murray, J. Am. Chem. Soc., 2002, 124, 8958–8964 CrossRef CAS PubMed.
- K. Tapio, J. Leppiniemi, B. Shen, V. P. Hytonen, W. Fritzsche and J. J. Toppari, Nano Lett., 2016, 16, 6780–6786 CrossRef CAS PubMed.
- S. Okazaki, Microelectron. Eng., 2015, 133, 23–35 CrossRef CAS.
- S. Kasani, K. Curtin and N. Wu, Nanophotonics, 2019, 8, 2065–2089 CAS.
- Y. Chen, Microelectron. Eng., 2015, 135, 57–72 CrossRef CAS.
- W. Sun, E. Boulais, Y. Hakobyan, W. L. Wang, A. Guan, M. Bathe and P. Yin, Science, 2014, 346, 1258361 CrossRef PubMed.
- A. V. Pinheiro, D. Han, W. M. Shih and H. Yan, Nat. Nanotechnol., 2011, 6, 763–772 CrossRef CAS PubMed.
- N. C. Seeman and H. F. Sleiman, Nat. Rev. Mater., 2017, 3, 17068 CrossRef.
- S. Nummelin, J. Kommeri, M. A. Kostiainen and V. Linko, Adv. Mater., 2018, 30, e1703721 CrossRef PubMed.
- P. Chidchob and H. F. Sleiman, Curr. Opin. Chem. Biol., 2018, 46, 63–70 CrossRef CAS PubMed.
- M. Madsen and K. V. Gothelf, Chem. Rev., 2019, 119, 6384–6458 CAS.
- P. W. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed.
- B. Sacca and C. M. Niemeyer, Angew. Chem., Int. Ed. Engl., 2012, 51, 58–66 CrossRef CAS PubMed.
- T. Tørring, N. V. Voigt, J. Nangreave, H. Yan and K. V. Gothelf, Chem. Soc. Rev., 2011, 40, 5636–5646 RSC.
- H. Kim, S. P. Surwade, A. Powell, C. O'Donnell and H. Liu, Chem. Mater., 2014, 26, 5265–5273 CrossRef CAS.
- F. Hong, F. Zhang, Y. Liu and H. Yan, Chem. Rev., 2017, 117, 12584–12640 CrossRef CAS PubMed.
- S. Ramakrishnan, H. Ijas, V. Linko and A. Keller, Comput. Struct. Biotechnol. J., 2018, 16, 342–349 CrossRef CAS PubMed.
- F. Praetorius, B. Kick, K. L. Behler, M. N. Honemann, D. Weuster-Botz and H. Dietz, Nature, 2017, 552, 84–87 CrossRef CAS PubMed.
- G. P. Acuna, F. M. Möller, P. Holzmeister, S. Beater, B. Lalkens and P. Tinnefeld, Science, 2012, 338, 506–510 CrossRef CAS PubMed.
- A. Kuzyk, R. Schreiber, Z. Fan, G. Pardatscher, E. M. Roller, A. Hogele, F. C. Simmel, A. O. Govorov and T. Liedl, Nature, 2012, 483, 311–314 CrossRef CAS PubMed.
- K. Vogele, J. List, G. Pardatscher, N. B. Holland, F. C. Simmel and T. Pirzer, ACS Nano, 2016, 10, 11377–11384 CrossRef CAS PubMed.
- A. Kuzyk, M. J. Urban, A. Idili, F. Ricci and N. Liu, Sci. Adv., 2017, 3, e1602803 CrossRef PubMed.
- P. Zhan, T. Wen, Z. G. Wang, Y. He, J. Shi, T. Wang, X. Liu, G. Lu and B. Ding, Angew. Chem., Int. Ed. Engl., 2018, 57, 2846–2850 CrossRef CAS PubMed.
- E. P. Gates, J. K. Jensen, J. N. Harb and A. T. Woolley, RSC Adv., 2015, 5, 8134–8141 RSC.
- F. N. Gur, F. W. Schwarz, J. Ye, S. Diez and T. L. Schmidt, ACS Nano, 2016, 10, 5374–5382 CrossRef PubMed.
- N. Li, Y. Shang, R. Xu, Q. Jiang, J. Liu, L. Wang, Z. Cheng and B. Ding, J. Am. Chem. Soc., 2019, 141, 17968–17972 CrossRef CAS PubMed.
- Y. Geng, J. Liu, E. Pound, S. Gyawali, J. N. Harb and A. T. Woolley, J. Mater. Chem., 2011, 21, 12126–12131 RSC.
- R. Schreiber, S. Kempter, S. Holler, V. Schuller, D. Schiffels, S. S. Simmel, P. C. Nickels and T. Liedl, Small, 2011, 7, 1795–1799 CrossRef CAS PubMed.
- E. Braun, Y. Eichen, U. Sivan and G. Ben-Yoseph, Nature, 1998, 391, 775–778 CrossRef CAS PubMed.
- J. Richter, R. Seidel, R. Kirsch, M. Mertig, W. Pompe, J. Plaschke and H. K. Schackert, Adv. Mater., 2000, 12, 507–510 CrossRef CAS.
- M. Mertig, L. Colombi Ciacchi, R. Seidel, W. Pompe and A. De Vita, Nano Lett., 2002, 2, 841–844 CrossRef CAS.
- K. Keren, M. Krueger, R. Gilad, G. Ben-Yoseph, U. Sivan and E. Braun, Science, 2002, 297, 72–75 CrossRef CAS PubMed.
- C. F. Monson and A. T. Woolley, Nano Lett., 2003, 3, 359–363 CrossRef CAS.
- R. Seidel, L. Colombi Ciacchi, M. Weigel, W. Pompe and M. Mertig, J. Phys. Chem. B, 2004, 108, 10801–10811 CrossRef CAS.
- Q. Gu, C. Cheng and D. T. Haynie, Nanotechnology, 2005, 16, 1358–1363 CrossRef CAS.
- C. T. Wirges, J. Timper, M. Fischler, A. S. Sologubenko, J. Mayer, U. Simon and T. Carell, Angew. Chem., Int. Ed. Engl., 2009, 48, 219–223 CrossRef CAS PubMed.
- Q. Gu and D. T. Haynie, Mater. Lett., 2008, 62, 3047–3050 CrossRef CAS.
- A. Stern, G. Eidelshtein, R. Zhuravel, G. I. Livshits, D. Rotem, A. Kotlyar and D. Porath, Adv. Mater., 2018, 30, e1800433 CrossRef PubMed.
- H. A. Becerril, R. M. Stoltenberg, D. R. Wheeler, R. C. Davis, J. N. Harb and A. T. Woolley, J. Am. Chem. Soc., 2005, 127, 2828–2829 CrossRef CAS PubMed.
- X. Ma, J. Huh, W. Park, L. P. Lee, Y. J. Kwon and S. J. Sim, Nat. Commun., 2016, 7, 12873 CrossRef CAS PubMed.
- Y. Weizmann, F. Patolsky, I. Popov and I. Willner, Nano Lett., 2004, 4, 787–792 CrossRef CAS.
- M. Pilo-Pais, S. Goldberg, E. Samano, T. H. Labean and G. Finkelstein, Nano Lett., 2011, 11, 3489–3492 CrossRef CAS PubMed.
- A. C. Pearson, J. Liu, E. Pound, B. Uprety, A. T. Woolley, R. C. Davis and J. N. Harb, J. Phys. Chem. B, 2012, 116, 10551–10560 CrossRef CAS PubMed.
- B. Uprety, E. P. Gates, Y. Geng, A. T. Woolley and J. N. Harb, Langmuir, 2014, 30, 1134–1141 CrossRef CAS PubMed.
- B. Teschome, S. Facsko, T. Schonherr, J. Kerbusch, A. Keller and A. Erbe, Langmuir, 2016, 32, 10159–10165 CrossRef CAS PubMed.
- B. Uprety, J. Jensen, B. R. Aryal, R. C. Davis, A. T. Woolley and J. N. Harb, Langmuir, 2017, 33, 10143–10152 CrossRef CAS PubMed.
- S. Helmi, C. Ziegler, D. J. Kauert and R. Seidel, Nano Lett., 2014, 14, 6693–6698 CrossRef CAS PubMed.
- T. Bayrak, S. Helmi, J. Ye, D. Kauert, J. Kelling, T. Schonherr, R. Weichelt, A. Erbe and R. Seidel, Nano Lett., 2018, 18, 2116–2123 CrossRef CAS PubMed.
- J. Ye, S. Helmi, J. Teske and R. Seidel, Nano Lett., 2019, 19, 2707–2714 CrossRef CAS PubMed.
- D. K. Lim, K. S. Jeon, J. H. Hwang, H. Kim, S. Kwon, Y. D. Suh and J. M. Nam, Nat. Nanotechnol., 2011, 6, 452–460 CrossRef CAS PubMed.
- T. G. Edwardson, K. L. Lau, D. Bousmail, C. J. Serpell and H. F. Sleiman, Nat. Chem., 2016, 8, 162–170 CrossRef CAS PubMed.
- T. Trinh, C. Liao, V. Toader, M. Barlog, H. S. Bazzi, J. Li and H. F. Sleiman, Nat. Chem., 2018, 10, 184–192 CrossRef CAS PubMed.
- T. Trinh, D. Saliba, C. Liao, D. de Rochambeau, A. L. Prinzen, J. Li and H. F. Sleiman, Angew. Chem., Int. Ed. Engl., 2019, 58, 3042–3047 CrossRef CAS PubMed.
- M. Kirsch, E. E. Lomonosova, H.-G. Korth, R. Sustmann and H. de Groot, J. Biol. Chem., 1998, 273, 12716–12724 CrossRef CAS PubMed.
- R. Chen, J. Wu, H. Li, G. Cheng, Z. Lu and C.-M. Che, Rare Met., 2010, 29, 180–186 CrossRef CAS.
- T. Atay, J.-H. Song and A. V. Nurmikko, Nano Lett., 2004, 4, 1627–1631 CrossRef CAS.
- J. H. Lee, M. H. You, G. H. Kim and J. M. Nam, Nano Lett., 2014, 14, 6217–6225 CrossRef CAS PubMed.
- S. Julin, A. Korpi, Nonappa, B. Shen, V. Liljestrom, O. Ikkala, A. Keller, V. Linko and M. A. Kostiainen, Nanoscale, 2019, 11, 4546–4551 RSC.
- B. Kou, J. Zhang, X. Huai, X. Liang and S.-J. Xiao, RSC Adv., 2015, 5, 5055–5058 RSC.
- B. Cheng, H. Kashida, N. Shimada, A. Maruyama and H. Asanuma, Chembiochem, 2017, 18, 1568–1572 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, DNA sequences and modifications, DNA origami design, seed-origami assembly and gold growth optimizations, additional experimental results, additional AFM and TEM images. See DOI: 10.1039/d0sc00553c |
| This journal is © The Royal Society of Chemistry 2020 |