
α-C–C agostic interactions and C–H bond activation in scandium cyclopropyl complexes†
Cheng
Xu
a,
Guangyu
Li
a,
Michel
Etienne
 *b,
Xuebing
Leng
a and
Yaofeng
Chen
*a
*b,
Xuebing
Leng
a and
Yaofeng
Chen
*a
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: yaofchen@mail.sioc.ac.cn
bLCC-CNRS, Université de Toulouse, CNRS, UPS, 205 route de Narbonne, 31077 Toulouse Cedex 4, France. E-mail: michel.etienne@lcc-toulouse.fr
First published on 6th November 2020
Abstract
This paper addresses the problem of the observation of the so-called C–C agostic interactions in cyclopropyl complexes of scandium. Three new cyclopropyl complexes of scandium based on the β-diketiminato ligand were synthesized including by an intramolecular C–H bond activation reaction in one case. X-ray diffraction analysis revealed distorted cyclopropyl groups in the complexes, and the distortion could be observed in solution as well for one of the complexes thanks to the natural abundance INADEQUATE NMR spectroscopy which showed a markedly reduced JC–C coupling constant. This signature of the C–C agostic interaction was further examined using DFT modelling which, together with NBO calculations, indicated that C–H and C–C agostic interactions are not exclusive but can complement each other, accounting for the distortions. The intramolecular C–H bond activation in the scandium biscyclopropyl complex was investigated by isotopic labeling experiments, which indicated a direct proton abstraction of the isopropyl group in the β-diketiminato ligand by a cyclopropyl group.
Introduction
The poor overlap of carbon-based orbitals within a strained cyclopropyl ring increases their energy and makes them spatially exposed, imparting them with unique chemical properties.1–4 One consequence in organometallic complexes [M]–(c-C3H5) is that the so-called α-C–C agostic distortions or interactions – formally three center-two electron bonds – are preferred to the much more common C–H agostic interactions, whether they be with the α- or β-C–H bonds.5,6 This has been observed especially in unsaturated early transition7–9 and alkaline10,11 metal complexes. For rare-earth metals, the bimetallic complex [(C5Me5)2Y(μ-c-C3H5)2Li(thf)] exhibits C–C agostic interactions with both lithium and yttrium centers, and the interaction with lithium is more electrostatic in nature while that with yttrium is more covalent.12 This represents a single example of a C–C agostic rare-earth metal complex since C–C agostic interactions between rare-earth metals and remote cyclopropyl rings have only been computed so far.13In this paper, we present our efforts to synthesize monometallic scandium cyclopropyl complexes. Their characterization in the solid state and in solution indicates that C–C agostic interactions are present. DFT is used to better define the nature of the interactions. An intramolecular C–H bond activation reaction degrades an ancillary ligand allowing the comparison between different types of alkyl ligands intramolecularly, confirming that cyclopropyl groups are privileged structures to observe C–C agostic interactions.
Results and discussion
Synthesis and structural aspects
Scandium dichloride [LSc(μ-Cl)Cl]2 (L = [MeC(NDIPP)CHC(Me)(NDIPP)]−, DIPP = 2,6-(iPr)2C6H3) and cyclopropyl lithium were prepared as reported.14,15 The reactions of [LSc(μ-Cl)Cl]2 with cyclopropyl lithium in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 or 1:4 molar ratio in toluene provided the scandium cyclopropyl chloride complex [LSc(μ-Cl)(c-C3H5)]2 (1) and the biscyclopropyl complex [LSc(c-C3H5)2] (2) in 38% and 80% yields, respectively (Scheme 1). Both complexes were characterized by NMR spectroscopy (1H and 13C{1H}), elemental analysis, and single-crystal X-ray crystallography. The 1H NMR spectra in C6D6 indicated a time-averaged C2v symmetry of the cyclopropyl group in 1 and 2 at room temperature. The CαH of the cyclopropyl group in 1 resonates as a triplet of triplet at δ = −0.45 ppm (3JH–H = 10.5, 8.1 Hz), and the corresponding CαH in 2 appears to be shifted slightly downfield (δ = −0.06 ppm, 3JH–H = 10.3, 8.2 Hz). The chemical shifts of ScCH3 in the scandium methyl chloride complex [LSc(μ-Cl)Me]2 and the dimethyl complex [LScMe2(THF)] are 0.29 and −0.15 ppm, respectively.14,16 All the CβH signals of the cyclopropyl groups in 1 and 2 appear as doublets, and their chemical shift values range from 0.38 to 0.61 ppm. The 13C{1H} NMR spectra confirm the C2v symmetry of the cyclopropyl group in the complexes, with only one CβH signal at δ = 9.4 ppm for 1 and δ = 7.4 ppm for 2. Due to quadrupolar relaxation, the Cα signals appear as broad singlets at δ = 33.2 and 30.8 ppm for 1 and 2, respectively; the assignment of these two signals was assisted with the HSQC spectra of the complexes.
:2 or 1:4 molar ratio in toluene provided the scandium cyclopropyl chloride complex [LSc(μ-Cl)(c-C3H5)]2 (1) and the biscyclopropyl complex [LSc(c-C3H5)2] (2) in 38% and 80% yields, respectively (Scheme 1). Both complexes were characterized by NMR spectroscopy (1H and 13C{1H}), elemental analysis, and single-crystal X-ray crystallography. The 1H NMR spectra in C6D6 indicated a time-averaged C2v symmetry of the cyclopropyl group in 1 and 2 at room temperature. The CαH of the cyclopropyl group in 1 resonates as a triplet of triplet at δ = −0.45 ppm (3JH–H = 10.5, 8.1 Hz), and the corresponding CαH in 2 appears to be shifted slightly downfield (δ = −0.06 ppm, 3JH–H = 10.3, 8.2 Hz). The chemical shifts of ScCH3 in the scandium methyl chloride complex [LSc(μ-Cl)Me]2 and the dimethyl complex [LScMe2(THF)] are 0.29 and −0.15 ppm, respectively.14,16 All the CβH signals of the cyclopropyl groups in 1 and 2 appear as doublets, and their chemical shift values range from 0.38 to 0.61 ppm. The 13C{1H} NMR spectra confirm the C2v symmetry of the cyclopropyl group in the complexes, with only one CβH signal at δ = 9.4 ppm for 1 and δ = 7.4 ppm for 2. Due to quadrupolar relaxation, the Cα signals appear as broad singlets at δ = 33.2 and 30.8 ppm for 1 and 2, respectively; the assignment of these two signals was assisted with the HSQC spectra of the complexes.
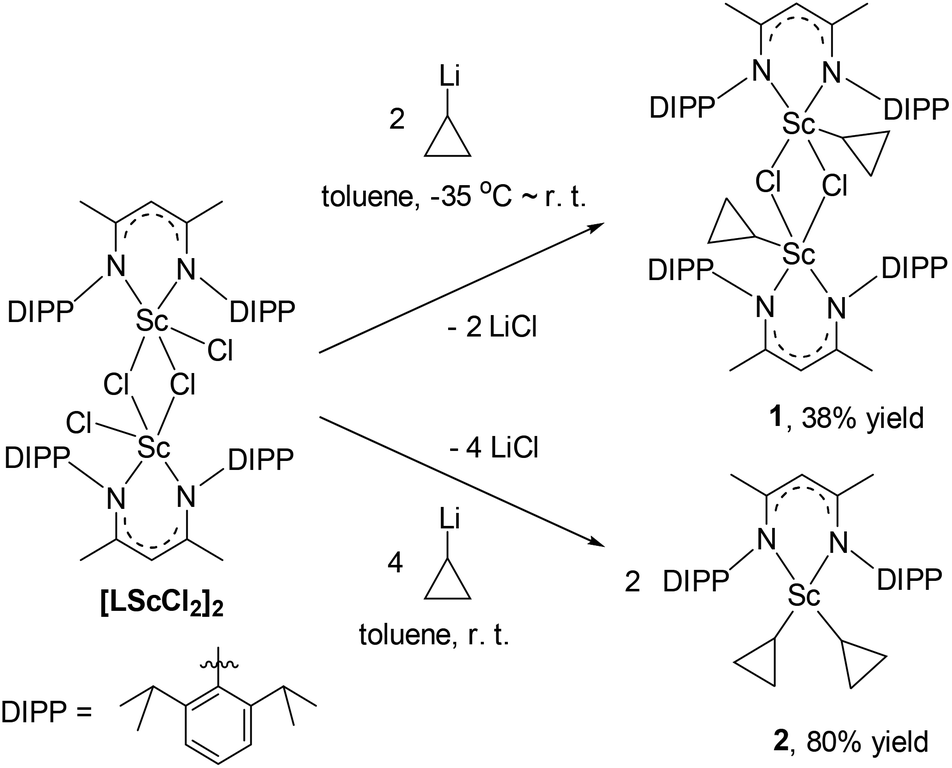 | ||
| Scheme 1 Synthesis of 1 and 2. | ||
The single-crystal X-ray diffraction analysis showed that complex 1 exists as a dimer in the solid state, in which each pentacoordinated scandium ion is coordinated by two nitrogen atoms of L, two bridging chlorides and one carbon atom of the cyclopropyl ligand (Fig. 1). The two Sc–Cl bond lengths are 2.558(1) and 2.540(1) Å. The Sc–C30 bond length is 2.195(2) Å, which is close to the Sc–C(methyl) bond lengths in [LScMe2(THF)] (2.210(9) and 2.245(9) Å).14 The distances from the scandium ion to the two Cβ atoms of the cyclopropyl ligand, 3.354 and 3.364 Å, are long, indicating no interaction between the scandium ion and the Cβ atoms. The two Sc–Cα–Cβ bond angles are close (132.3(2)° and 132.0(2)°). Complex 2 is monomeric, with the scandium ion being coordinated by two nitrogen atoms of L and two cyclopropyl carbon atoms (Fig. 1) in a pseudotetrahedral environment. The two Sc–Cα bonds, Sc–C30 and Sc–C33, are 2.180(2) and 2.195(2) Å, respectively, similar to that in 1 (2.195(2) Å). There is an inconspicuous unsymmetrical coordination of one cyclopropyl ring in 2, which can be described by the difference between the Sc⋯C31 distance (Sc⋯C31: 2.904 Å) and other Sc⋯Cβ distances (Sc⋯C32: 3.248 Å, Sc⋯C34: 3.389 Å and Sc⋯C35: 3.335 Å). Such distortion is also exhibited by the different Sc–Cα–Cβ angles, with the Sc–C30–C31 (101.7(1)°) angle being smaller than other Sc–Cα–Cβ angles [Sc–C30–C32: 122.3(1)°, Sc–C33–C34: 131.0(1)°, Sc–C33–C35: 126.6(1)°]. C30–C31 (1.526(3) Å) is barely longer than C30–32 (1.510(3) Å) which is similar to Cα–Cβ in other cyclopropyl groups [C33–C34 = 1.516(2) and C33–C35 = 1.522(2) Å].
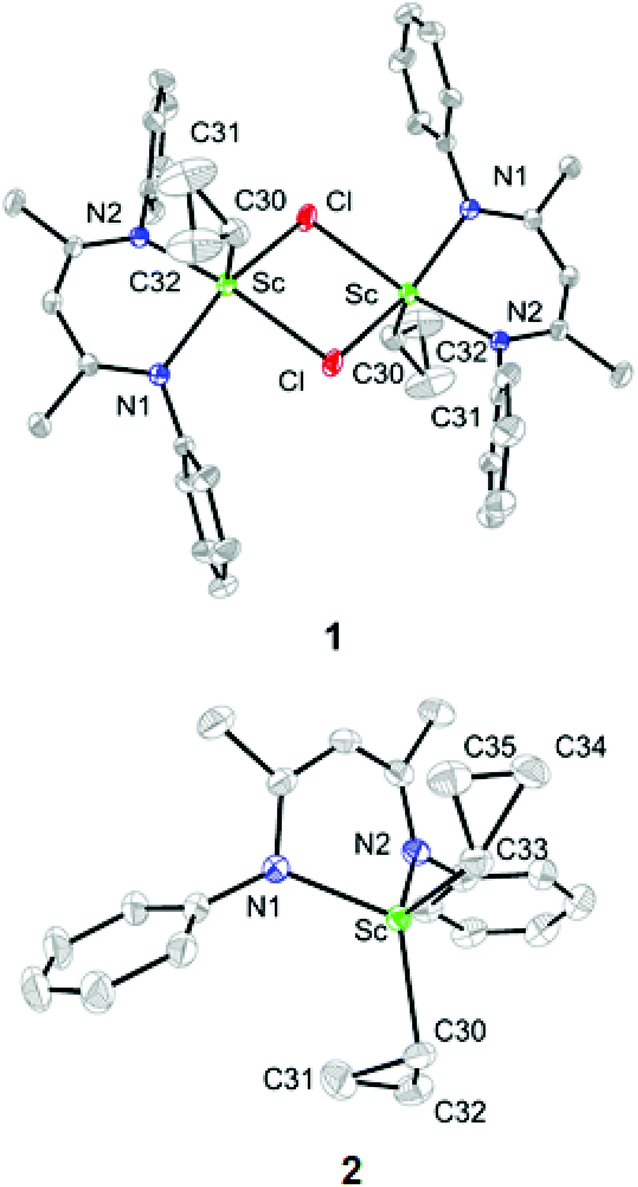 | ||
| Fig. 1 ORTEP drawings of 1 and 2 with thermal ellipsoids set at 30% probability level. Isopropyl groups of DIPP and hydrogen atoms have been omitted for clarity. | ||
As observed with other dialkyl complexes of scandium,14 complex 2 slowly eliminates cyclopropane at room temperature (δ = 0.14 ppm in the 1H NMR spectrum) to give a complicated mixture in C6D6 at room temperature. Interestingly, in the presence of THF, complex 2 nearly quantitatively converts into a new complex 3 in 24 h at 65 °C with the elimination of cyclopropane. A scaled-up reaction in hexane/THF provided 3 in 78% isolated yield as a yellow solid, in which a C–H bond of an isopropyl group in 2 has been activated to form a new dianionic ligand (L–H) as shown in Scheme 2. Complex 3 was characterized by NMR spectroscopy (1H and 13C{1H}), elemental analysis and single-crystal X-ray crystallography. The latter revealed that there are two crystallographically independent molecules in the unit cell of 3; these two molecules have very close structural parameters and one molecule was taken as the example to analyze the structural parameters (Fig. 2). In 3, the scandium ion is coordinated by two nitrogen atoms and one carbon atom of the newly formed ligand (L–H), one carbon atom of the cyclopropyl ligand and one oxygen atom of THF. The scandium atom is in a trigonal bipyramidal environment, with O and N2 forming the apical sites [N2–Sc–O = 169.94(11)°]. The distances from the scandium ion to the isopropyl carbon atom and the cyclopropyl carbon atom are 2.273(4) and 2.184(4) Å, respectively. The cyclopropyl ring and its coordination to the scandium atom are significantly distorted. The Sc–C30–C31 angle (95.9(3)°) is much smaller than the Sc–C30–C32 angle (132.7(3)°). In addition, the Sc⋯C31 distance (2.793 Å) is 0.60 Å shorter than the Sc⋯C32 distance (3.391 Å). Within 3σ, C30–C31 (1.531(6) Å) is not statistically stretched as compared to C30–C32 (1.505(5) Å).
 | ||
| Scheme 2 Thermolysis of 2 into 3. | ||
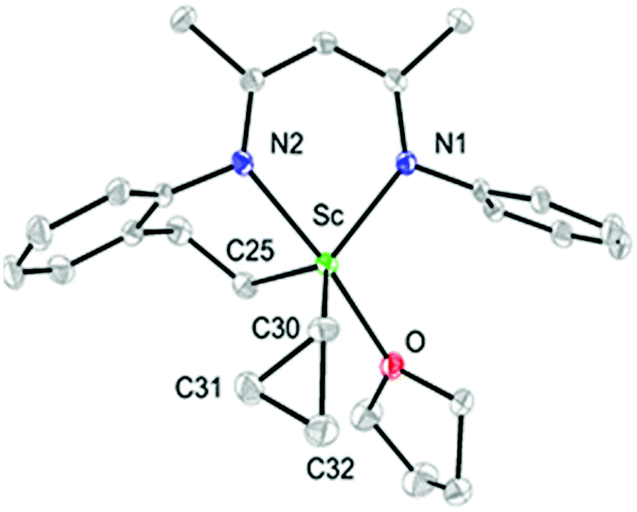 | ||
| Fig. 2 ORTEP drawing of 3 with thermal ellipsoids set at 30% probability level. Isopropyl groups of DIPP and hydrogen atoms have been omitted for clarity. | ||
The unusual distortion of the cyclopropyl ring prompted a detailed NMR investigation of the underlying agostic interaction in complex 3. First of all, a comprehensive assignment of all protons and carbons of the cyclopropyl ring in 3 was realized through 1H and 13C{1H} NMR with the aid of HSQC, COSY and NOESY spectra, as shown in Fig. 3. The assignments indicated that, in solution, complex 3 adopts a conformation fully consistent with that in the solid state, as revealed by X-ray crystallography. The 1H NMR spectrum showed five signals for the five protons of the cyclopropyl ring [C30H: −0.48 (m, 1H), C31H: 0.13, −0.16 (m, 2H), C32H: 0.55, 0.19 (m, 2H)]. The 13C{1H} NMR spectrum showed two signals for the two Cβ atoms of the cyclopropyl ring, C31 at 5.5 ppm and C32 at 8.7 ppm when Cα (C30) was observed as a broad signal due to quadrupolar relaxation at 29.0 ppm. The NOESY spectrum played a vital role in uncovering the spatial arrangement of the above protons with the guidance of the characteristic proton (C25H) of the L–H ligand. Apart from the detailed assignment of the protons in the cyclopropyl ring, the NOESY spectrum also depicted the greater intensity of the coherent signal of C25H′–C31H′ than that of C25H′–C32H′ (Fig. 4), indicating that the cyclopropyl ring in 3 presents a distorted configuration with the C30–C31 bond approaching the scandium ion in the solution phase. Subsequently, 1H–1H EXSY and variable temperature 1H NMR spectra (303–343 K) were recorded, and no proton exchange or rotation of the cyclopropyl ring in 3 was observed even at 343 K in C6D6, illustrating the relatively stable geometry conformation of the cyclopropyl ring in 3.
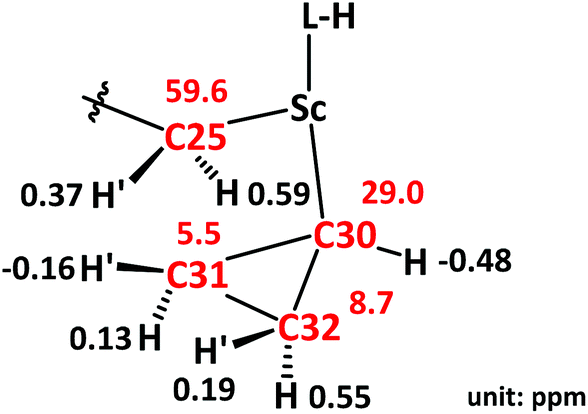 | ||
| Fig. 3 1H and 13C NMR data for key protons and carbons in 3. Data color code: black, 1H NMR data; red, 13C NMR data. | ||
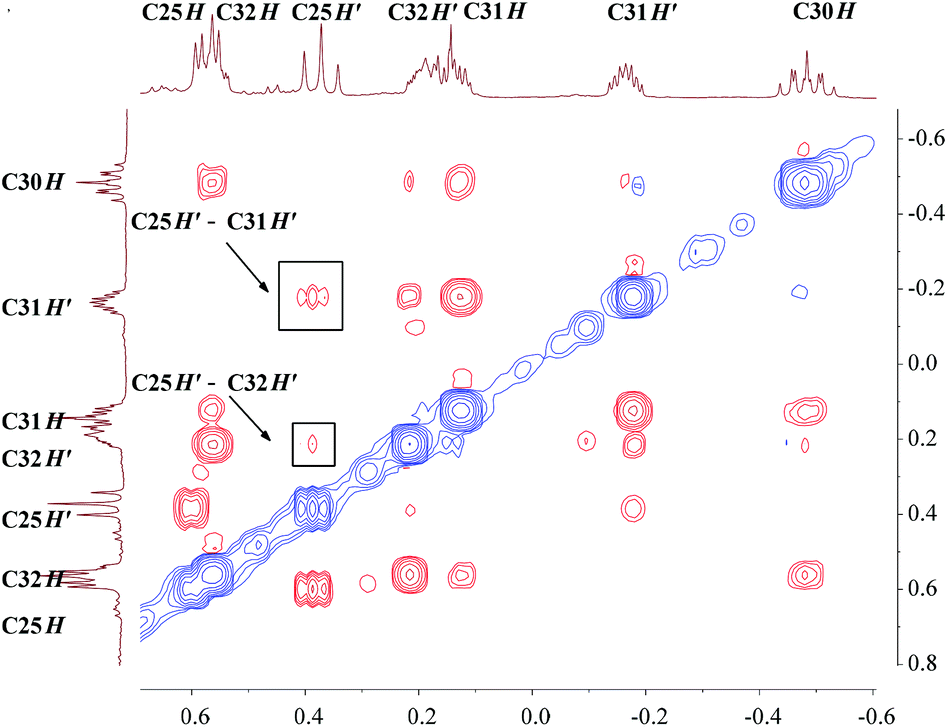 | ||
| Fig. 4 Expansion plot of the NOESY spectrum of 3 (400 MHz, C6D6). | ||
The 1JC–H values were then obtained from a gated 13C NMR spectrum. The C31 and C32 atoms resonate as triplets with 1JC–H values of 158 and 155 Hz, respectively, which are similar to the 1JC–H value of cyclopropane (161 Hz) and indicate the absence of an agostic interaction between the scandium ion and the corresponding β-C–H bonds of the cyclopropyl ring. Due to quadrupolar relaxation, 1JCα−H could not be measured. The agostic interaction between the scandium ion and the C–C bond of the cyclopropyl ring was then investigated. A remarkable decrease of the 1JC–C value is the key evidence for the presence of such agostic interaction.9,12,17 Fortunately, owing to the well-resolved resonance of Cβ signals, the good solubility of 3 (ca. 0.4 M in C6D6), and the use of a sensitivity boosted cryogenetic probe, the inherently challenging 1D INADEQUATE spectra of high quality were successfully acquired and the 1JC–C values of Cβ signals were obtained. The C32 signal displays two doublets with 1JC–C values of 14.8 and 3.2 Hz, while the C31 signal appears as a doublet with a 1JC–C value of 14.8 Hz (Fig. 5a). Therefore, the 1JC31−C32 value is 14.8 Hz and the 1JC30−C32 value is 3.2 Hz. This small value is linked to the low electronegativity of scandium. The observation of the C31 signal as a single doublet instead of two can be rationalized in two ways: (a) the 1JC31−C30 value is the same or very close to the 1JC31−C32 value, so the peaks overlap; and (b) the 1JC31−C30 value is very small and indeed less than the line width of the experiment (<2.0 Hz) and hence not detectable. Therefore, we recorded the 13C–13C INADEQUATE spectrum of 3 optimized for J = 15 Hz (Fig. 5b), which showed that the intensity of the C31 doublet is not twice but equal to that of the C32 doublet. This result clearly indicated that the 1JC31−C30 value is too small to be detected, and such a small 1JC31−C30 value revealed an agostic interaction between the scandium ion and the C30–C31 bond of the cyclopropyl ring. Unfortunately, this could not be confirmed in the absence of the C30 signal (see the Computational studies section below).
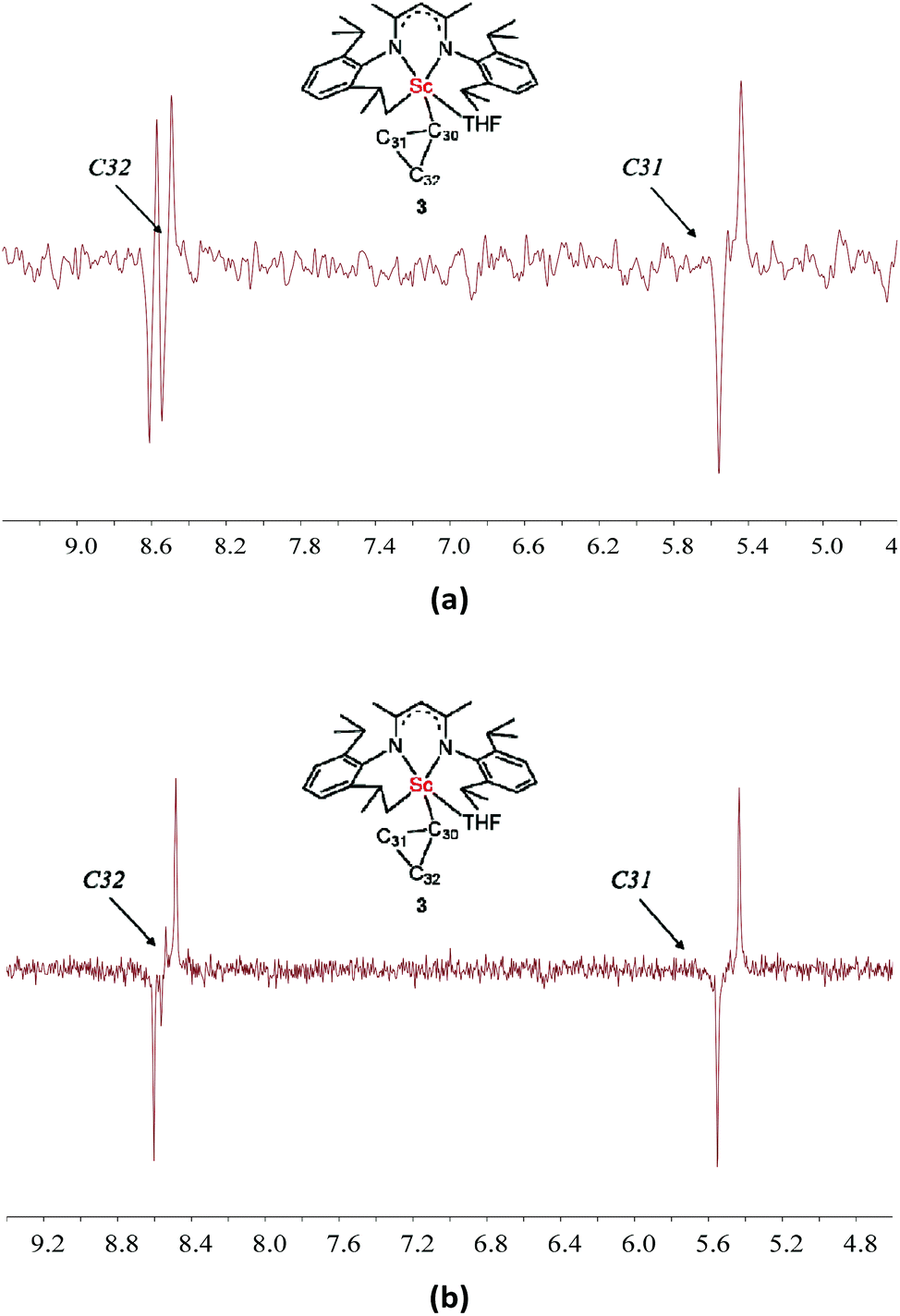 | ||
| Fig. 5 13C–13C INADEQUATE spectra of 3, optimized for J = 3 Hz (a) and J = 15 Hz (b), respectively. | ||
There are three plausible pathways by which complex 2 eliminates cyclopropane to give complex 3 (Scheme 3): (1) path A: one of the cyclopropyl ligands abstracts a proton from other cyclopropyl ligands first to form a scandium η2-cyclopropene intermediate, which is unstable and subsequently undergoes a σ-bond metathesis between the Sc–C bond and the C–H bond of the isopropyl group; (2) path B: one of the cyclopropyl ligands abstracts a proton from the isopropyl group of L; and (3) path C: an α-H abstraction to form a cyclopropylidene intermediate, followed by a deprotonation of the isopropyl group of L. To investigate the reaction pathway, we decided to synthesize complex 2-d8 which contains two 2,2,3,3-tetradeuteriocyclopropyl ligands and study its thermolysis. It can be reasonably envisioned that if complex 3 is formed via path A, the cyclopropenyl ligand in the η2-cyclopropene intermediate would abstract a proton from the isopropyl group to generate complex 3-d3, which would display the cyclopropyl CβH signal in the 1H NMR spectrum; in contrast, if complex 3 is formed via path B or C, the thermolysis of 2-d8 would give complex 3-d4, in which no signal of the CβH of the cyclopropyl would be observed in the 1H NMR spectrum.
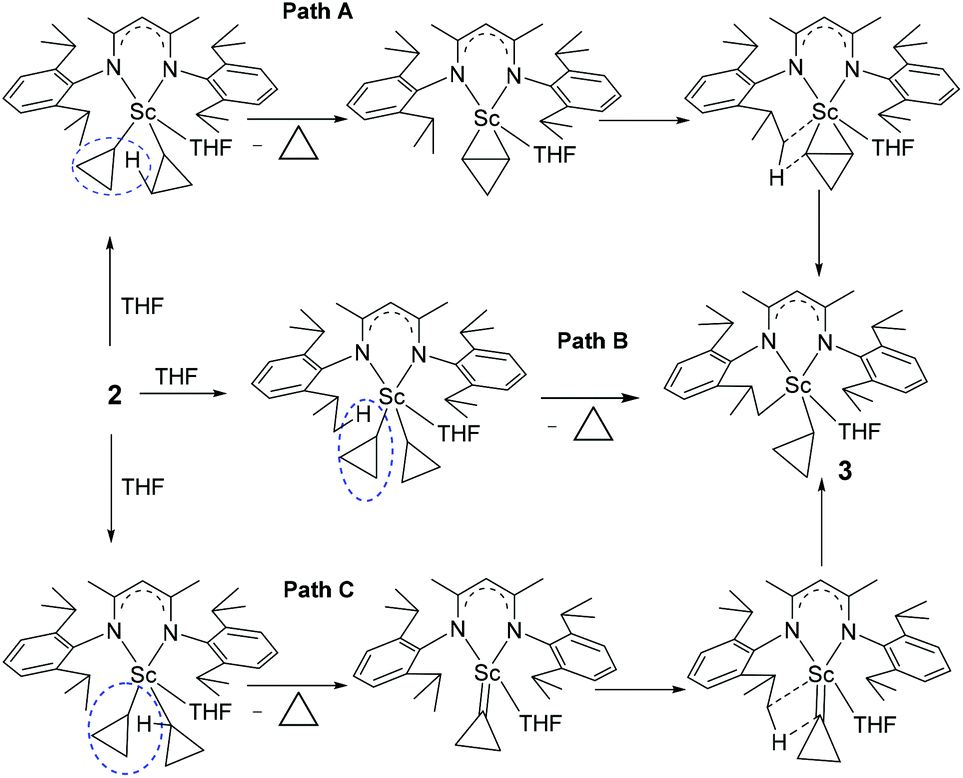 | ||
| Scheme 3 Three possible pathways for the thermolysis of 2 into 3. | ||
Tetradeuteriocyclopropyl lithium was firstly synthesized, as shown in Scheme 4a. Ethyl 2,2,3,3-tetradeuteriocyclopropanecarboxylate was prepared using THF-d8 as a starting material in three steps according to the procedures reported by de Meijere and co-workers.18 Ethyl 2,2,3,3-tetradeuteriocyclopropanecarboxylate was hydrolyzed in a H2O/MeOH solution of NaOH, and then treated with HCl aqueous solution to give 2,2,3,3-tetradeuteriocyclopropanecarboxylic acid (66% yield). With tetradeuteriocyclopropanecarboxylic acid in hand, 2,2,3,3-tetradeuteriocyclopropyl bromide was synthesized using the Cristol–Firth reaction.19 After the completion of the reaction, excess bromine was removed by treatment with sodium thiosulfate, and benzene was added. The addition of benzene helps in the distillation of 2,2,3,3-tetradeuteriocyclopropyl bromide on a small scale. The distilled tetradeuteriocyclopropyl bromide/benzene mixture was reacted with a suspension of finely divided lithium in diethylether to provide the desired 2,2,3,3-tetradeuteriocyclopropyl lithium as a white solid; the total yield of the above two steps is 13%. Following the procedure for complex 2, the salt metathesis of [LSc(μ-Cl)Cl]2 with 2,2,3,3-tetradeuteriocyclopropyl lithium in a 1:4 molar ratio in toluene provided deuterated complex 2-d8 as a yellow solid in 77% isolated yield. The 1H NMR spectrum of 2-d8 in C6D6 resembles that of 2, except the absence of resonances at 0.61 and 0.41 ppm, indicating the deuteration on Cβ atoms. In the 2H NMR spectrum of 2-d8 in C6H6, two resonances were observed at 0.52 and 0.31 ppm for the CβD of the tetradeuteriocyclopropyl ligands. The thermolysis of 2-d8 in THF-d8/C6D6 at 65 °C was investigated, which clearly indicated the formation of 3-d4 (Scheme 5a). The 1H NMR spectral monitoring showed the appearance of the CαH signal at −0.61 ppm and the absence of the CβH signal (Fig. S20 in the ESI†). In the 2H NMR spectrum of the product in C6H6, the deuterium resonances were observed in the region of −0.3–0.5 ppm (Fig. S21 in the ESI†). Therefore, complex 3 is not formed via path A, which is different from the reported cyclopropane elimination mechanism for zirconium20 and niobium21–23 cyclopropyl complexes. For example, in the biscyclopropyl complex [Cp2Zr(c-C3H5)2], one of the cyclopropyl ligands abstracts a β-proton from other cyclopropyl ligands to release cyclopropane and generate the zirconium η2-cyclopropenyl species [Cp2Zr(η2-c-C3H4)].20
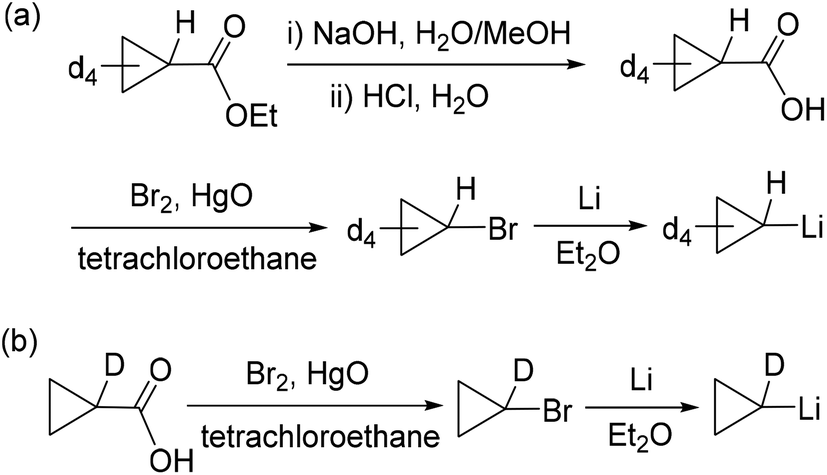 | ||
| Scheme 4 Synthesis of 2,2,3,3-tetradeuteriocyclopropyl lithium (a) and 1-monodeuteriocyclopropyl lithium (b). | ||
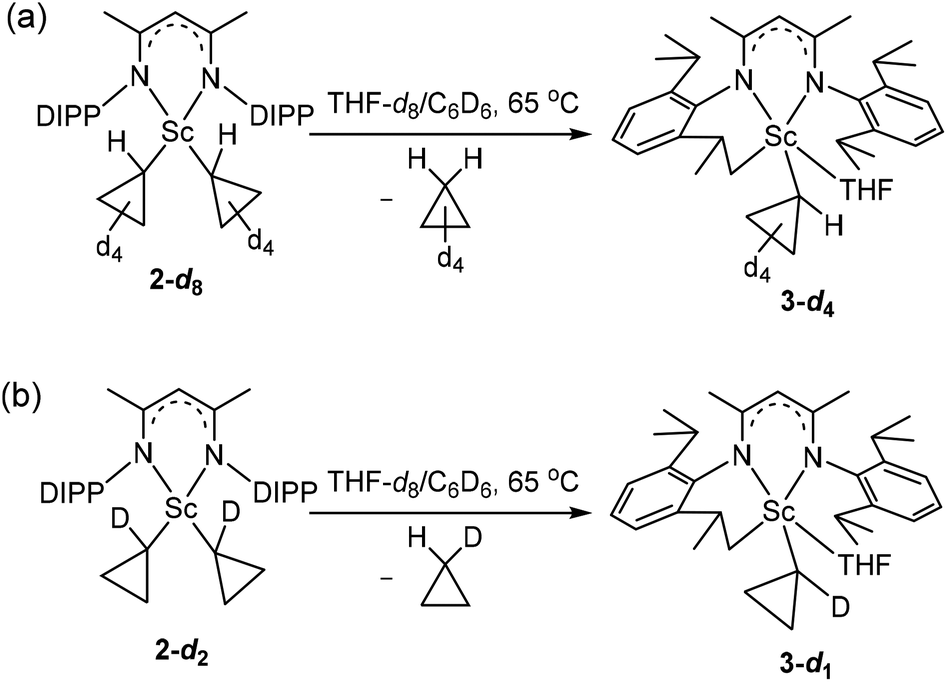 | ||
| Scheme 5 Thermolysis of 2-d8 into (a) 3-d4, and 2-d2 into (b) 3-d1. | ||
To discriminate between path B and path C, we need to synthesize complex 2-d2 which contains two 1-monodeuteriocyclopropyl ligands and study its thermolysis. If complex 3 is formed via path B, the thermolysis of 2-d2 would give complex 3-d1, in which no signal of the CαH of the cyclopropyl would be observed in the 1H NMR spectrum, and the 2H NMR spectrum of the complex would display a CαD signal; if complex 3 is formed via path C, the thermolysis of 2-d2 would give complex 3-d0, in which no CαD signal of the cyclopropyl would be observed in the 2H NMR spectrum, and the 1H NMR spectrum of the complex would show a CαH signal. Methyl cyclopropyl ketone-α,α,α,α′-d4 with a deuterium isotopic content of 90% was prepared by proton exchange of methyl cyclopropyl ketone with 40 wt% NaOD in D2O,24 and this ketone undergoes a haloform reaction25 with NaOBr to provide 1-monodeuteriocyclopropanecarboxylic acid (90 atom % D). From 1-monodeuteriocyclopropanecarboxylic acid, 1-monodeuteriocyclopropyl lithium (90 atom % D) was synthesized in a total yield of 31% using a similar approach to that of 2,2,3,3-tetradeuteriocyclopropyl lithium (Scheme 4b), and was then subjected to salt metathesis with 0.25 equiv. of [LSc(μ-Cl)Cl]2. The reaction produced complex 2-d2 as a yellow solid in 84% yield. The 1H NMR spectrum of 2-d2 in C6D6 shows ∼90% deuteration on α-C, and no 3JH–H spin–spin splitting was observed for CβH (0.61 and 0.41 ppm). In the 2H NMR spectrum of 2-d2 in C6H6, one resonance was observed at −0.10 ppm for the CαD of the 1-monodeuteriocyclopropyl ligands. The 1H and 2H NMR spectral monitoring of the thermolysis of 2-d2 in THF-d8/C6D6 or THF/C6H6 showed the transformation of 2-d2 into 3-d1 (Scheme 5b). In the 1H spectrum (Fig. S26 in the ESI†), four CβH signals were clearly observed at 0.41, 0.10, −0.04 and −0.33 ppm, while the CαH signal is very weak in line with a deuterium isotopic content of 90%. The 2H NMR spectra showed two signals, one for the CαD (δ = −0.74 ppm) in 3-d1 and the other for CHD (δ = 0.06 ppm) in eliminated monodeuteriocyclopropane (Fig. S27 in the ESI†). Therefore, complex 3 is formed via direct proton abstraction of the isopropyl group in L by a cyclopropyl group (path B).
Computational studies
We have used a computational approach to better define the C–C agostic interactions in complex 3 and possibly in complex 2. We first used the experimental data for complex 3 to benchmark our study and then considered the distorted solid state molecular structure of complex 2 whose solution structure is time averaged. Optimizations were carried out in the framework of DFT with the PBE0 functional coupled to empirical dispersion corrections. The details of basis sets can be found in the Experimental section and in the ESI.† The results for complex 3 are shown in Fig. 6, where it is seen that a remarkable agreement with the structural and spectroscopic data is reached, the Sc–Cα–Cβ angles and the dihedral angle C25–Sc–C30–C31, i.e. the conformation of the cyclopropyl ring, are correctly modelled. Considering the C–C bond lengths within the cyclopropyl ring, a small but clear elongation of the C30–C31 bond relative to the C30–C32 bond is seen which was not statistically meaningful experimentally. As to the coupling constants, not only the trend but also the absolute values are faithfully computed (IGLOOII basis set) including JC–H as references and JC–C. This approach has been validated in similar studies.9–12 For the latter, the computed values of 6 and 15 Hz are in very good agreement with the experimental values of 3 and 15 Hz found for the coupling constants involving C30–C32 and C31–C32, respectively. Therefore, the computed value of −3 Hz is consistent with our assignment of JC30−C31 < 2 Hz. The computational results together with the experimental data are consistent with the presence of an α-C–C agostic interaction in complex 3.5,6,9 The results of similar calculations for complex 2 are shown in Fig. 6 as well. The geometrical parameters are again very well reproduced. The C30 based cyclopropyl group is clearly distorted as compared to the one based on C33 which remains regular. The C30–C31 bond (1.53 Å) is indeed longer than the other Cα–Cβ bond (C30–C32 (1.50 Å)). The associated JC–C values, unavailable experimentally, are −5 and 6 Hz, respectively. The elongated C30–C31 bond associated with the reduced JC–C values is consistent with the presence of an α-C–C agostic interaction in complex 2. Examination of JβC–H values indicates that for the β-C–H bond pointing towards the scandium atom, a value of 136 Hz is computed. This value is somewhat reduced with respect to the other JβC–H values which are in the narrow, normal range of 147–151 Hz. In complex 2, an α-C–C agostic interaction is accompanied by a β-C–H agostic interaction. In order to probe the nature of these distortions, we carried out a Natural Bond Orbital (NBO)26 study of the electronic structure of complex 2. The NBO analysis shows two significant interactions at the second order perturbation theory involving (i) the σ-orbital of the β-C–H bond on C31 with the empty antibonding Sc–C33 with a strong d character of 17 kJ mol−1, and (ii) the σ-C30–C31 bond with the d-orbitals of the scandium atom of 33 kJ mol−1.‡ These interactions are also nicely pictured in NLMOs which reveal the participation of scandium d-orbitals in the C–H (ca. 1.0%) and C–C (ca. 1.3%) based NLMOs, respectively (Fig. 7). Although the absolute values might be questioned, interactions of similar magnitude have been observed in zirconium20 and yttrium12 cyclopropyl complexes. The involvement of both C–C and C–H bonds in the interaction with the rare-earth metal has been observed previously in the bimetallic complex [(C5Me5)2Y(μ-c-C3H5)2Li(thf)] albeit with different weights.12 This reinforces the view that there is a continuum between pure β-C–H and α-C–C agostic structures with both types of interactions complementing each other depending on the metals,27 the type of ligands, electron count, coordination number and geometry. The significant involvement of the C–C orbital is a distinctive property of the cyclopropyl group.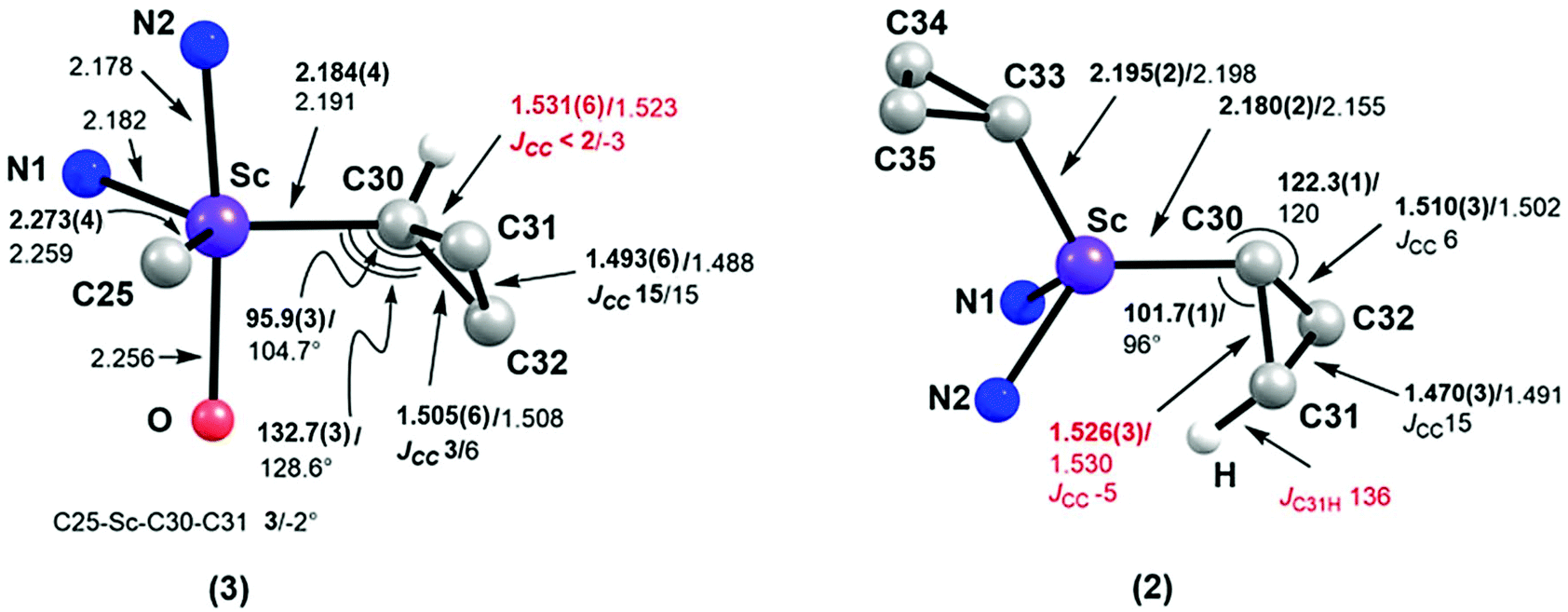 | ||
| Fig. 6 Computed structures and coupling constants J of complexes 3 and 2 and their comparison with experimental values when available. Only the core coordination sphere and the cyclopropyl groups are drawn for clarity. Experimental values (distance in Å, coupling constants in Hz) are boldface. Key data for agostic interactions are highlighted in red. Element color code: violet, Sc; blue, N; red, O; grey, C; and off white, H. | ||
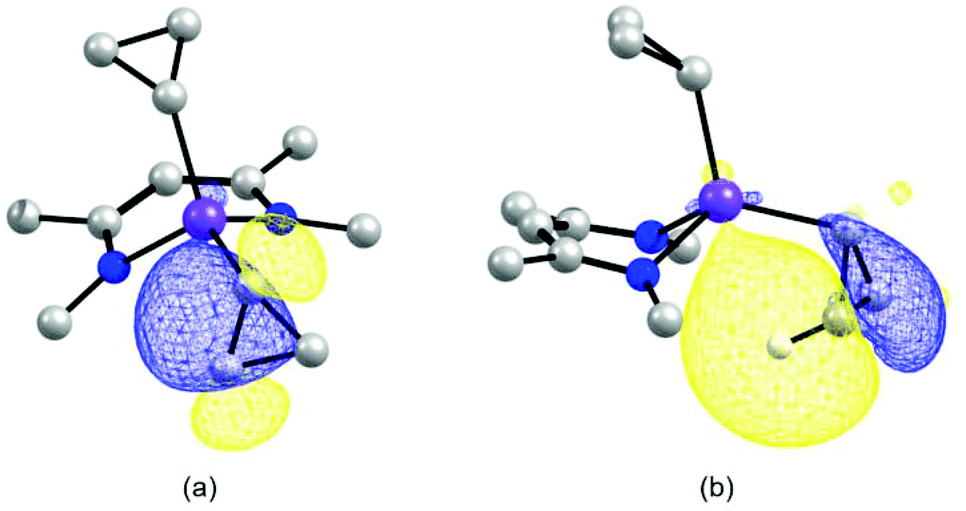 | ||
| Fig. 7 NLMOs describing the (a) α-C–C interaction and the (b) α-C–H interaction in complex 2. | ||
Conclusions
Discrete electron deficient scandium cyclopropyl complexes have been synthesized and fully characterized for the first time. They show distortions of their coordination sphere in the solid state. In one case, solution NMR studies allow the direct measurement of the reduced JC–C coupling constant. Together with DFT modelling, α-C–C agostic interactions possibly accompanied by β-C–H agostic interactions have been established. They reinforce the view that a continuum of stabilizing secondary interactions exists in the strongly electron deficient species. For the thermolysis of the scandium biscyclopropyl complex, the cyclopropyl ligand prefers to abstract a proton from the isopropyl group of the β-diketiminato ligand rather than from other cyclopropyl ligands in the complex.Experimental
General methods
All operations were carried out under an atmosphere of argon using Schlenk techniques or in nitrogen or argon filled gloveboxes. Toluene, THF, hexane, THF-d8 and C6D6 were dried over a Na/K alloy, transferred under vacuum, and stored in the gloveboxes. Scandium dichloride [LSc(μ-Cl)Cl]2 (L = [MeC(NDIPP)CHC(Me)(NDIPP)]−, DIPP = 2,6-(iPr)2C6H3),14 cyclopropyl lithium,15 ethyl 2,2,3,3-tetradeuteriocyclopropanecarboxylate18 and 1-monodeuteriocyclopropanecarboxylic acid25 were prepared as reported. 1H, 2H and 13C NMR spectra were recorded on a Bruker AV III 400 spectrometer, a Varian MR400 spectrometer or an Agilent MR400 spectrometer. Variable-temperature NMR, 1H–1H EXSY and NOESY spectra were acquired on a Bruker NEO 600 spectrometer. INADEQUATE experiments were performed on a Bruker NEO 500 spectrometer equipped with a BBO cryoprobe. All chemical shifts were reported in δ units with references to the residual solvent resonances of the deuterated solvents for proton and carbon chemical shifts, and 2H NMR was referred to CDCl3 in chloroform, C6D6 in benzene and THF-d8 in THF, respectively. Elemental analysis was performed by the Analytical Laboratory of Shanghai Institute of Organic Chemistry.Synthesis
1: A solution of [LSc(μ-Cl)Cl]2 (100 mg, 0.094 mmol) in 8 mL of toluene was added by a slurry of 28.4 mg of cyclopropyl lithium·lithium bromide (cyclopropyl lithium: 28.5% by weight, 0.17 mmol) in 3 mL of toluene at −35 °C under stirring, and then the reaction mixture was gradually warmed to room temperature. After stirring at room temperature for 4 h, the precipitate was removed by centrifugation. The volatiles of the solution were removed under vacuum to give a yellow solid. The solid was washed with hexane (2 mL × 3) and toluene (2 mL), and dried under vacuum to give 1 as a pale yellow solid (35 mg, 38% yield). Due to the poor solubility of 1 in C6D6, its 1H and 13C{1H} NMR spectra were recorded in THF-d8. 1H NMR (400 MHz, THF-d8, 25 °C): δ (ppm) 7.20 (m, 6H, ArH), 5.39 (s, 1H, MeC(N)CH), 3.39 (sept, 4H, ArCHMe2), 1.82 (s, 6H, MeC(N)), 1.30 (d, 3JH–H = 6.8 Hz, 12H, ArCHMe2), 1.19 (d, 3JH–H = 6.6 Hz, 12H, ArCHMe2), 0.48 (d, 3JH–H = 8.1 Hz, 2H, CβH2), 0.38 (d, 3JH–H = 10.5 Hz, 2H, CβH′2), −0.45 (tt, 3JH–H = 10.5, 8.1 Hz, 1H, CαH). 13C{1H} NMR (100 MHz, THF-d8, 25 °C): δ (ppm) 169.1 (MeC(N)CH), 147.5, 144.4 (i-ArC and o-ArC), 127.2, 125.3 (p-ArC and m-ArC), 99.1(MeC(N)CH), 33.2 (br, CαH), 29.5 (ArCHMe2), 26.0, 25.7 (MeC(N) and ArCHMe2), 9.4 (CβH2). Anal. calcd (%) for C32H46ClN2Sc: C, 71.29; H, 8.60; N, 5.20. Found: C, 70.53; H, 8.12; N, 5.03.2: A solution of [LSc(μ-Cl)Cl]2 (500 mg, 0.47 mmol) in 10 mL toluene was added by a slurry of 284 mg cyclopropyl lithium·lithium bromide (cyclopropyl lithium: 28.5% by weight, 1.69 mmol) in 5 mL of toluene at room temperature under stirring. After stirring at room temperature for 1.5 h, the precipitate was removed by centrifugation. The volatiles of the solution were removed under vacuum to give a yellow solid. This solid was extracted with 2 mL of hexane, and the extract was stored at −35 °C to give 2 as yellow needle crystals (365 mg, 80% yield). 1H NMR (400 MHz, C6D6, 25 °C): δ (ppm) 7.15 (m, 6H, ArH), 4.91 (s, 1H, MeC(N)CH), 3.43 (sept, 4H, ArCHMe2), 1.56 (s, 6H, MeC(N)), 1.44 (d, 3JH–H = 6.8 Hz, 12H, ArCHMe2), 1.19 (d, 3JH–H = 6.8 Hz, 12H, ArCHMe2), 0.61 (d, 3JH–H = 10.7 Hz, 2 H, CβH2), 0.41 (d, 3JH–H = 8.2 Hz, 2H, CβH′2), −0.06 (tt, 3JH–H = 10.3, 8.2 Hz, 1H, CαH). 13C{1H} NMR (100 MHz, C6D6, 25 °C): δ (ppm) 168.0 (MeC(N)CH), 143.6, 143.0 (i-ArC and o-ArC), 127.3, 124.9 (p-ArC and m-ArC), 94.9 (MeC(N)CH), 30.8 (br, CαH), 29.5 (ArCHMe2), 26.1, 25.1, 24.6 (ArCHMe2 and MeC(N)), 7.4 (CβH2). 13C NMR (100 MHz, C6D6, 25 °C): δ (ppm) 30.8 (br, CαH, 1JC–H was not observed due to quadrupolar relaxation), 7.4 (1JC–H = 159 Hz, CβH2). Anal. calcd (%) for C35H51N2Sc: C, 77.17; H, 9.44; N, 5.14. Found: C, 76.75; H, 9.01; N, 5.09.
3: Complex 2 (500 mg, 0.92 mmol) in a mixed solvent of hexane (10 mL) and THF (200 mg) was heated at 65 °C for 24 h. After being cooled to room temperature, the volatiles of the reaction solution were removed under vacuum. The residue was washed with hexane (1 mL × 3), and dried under vacuum to give 3 as a yellow solid (360 mg, 78% yield).
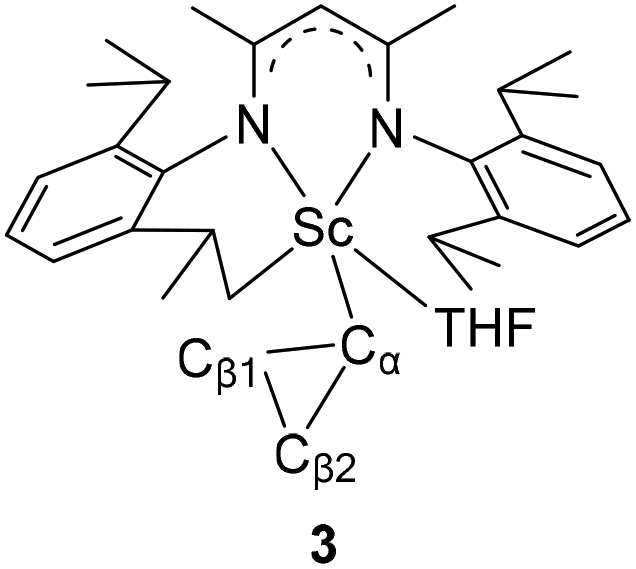
X-ray crystallography
Single crystals of 1 were grown from a toluene solution; single crystals of 2 and 3 were grown from hexane solutions. The suitable single crystals were mounted under a nitrogen atmosphere on a glass fiber, and data collection was performed at 170(2) K on a Bruker D8 Venture diffractometer with graphite-monochromated Ga Kα radiation (λ = 1.34139 Å). The SMART program package was used to determine the unit cell parameters. The absorption correction was applied using the SADABS program.28 The structures were solved by direct methods and refined on F2 by full-matrix least-squares techniques with anisotropic thermal parameters for non-hydrogen atoms. Hydrogen atoms were placed at calculated positions and were included in the structure calculation. Calculations were carried out using the SHELXL-2015 and Olex2 programs.29 Crystallographic data and refinement parameters are listed in Table S1 of the ESI.†Computational details
Calculations were performed at the DFT level using the software Gaussian09, revision D.01.30 Geometry optimizations were carried out without symmetry constraints in the gas phase using the PBE0 functional31 and including dispersion corrections (GD3-BJ).32 The scandium atom was described using the Stuttgart/Dresden ECP (SDD) pseudo-potential and its associated basis set33 to which was added an f polarization function.34 All other atoms were described with the def2-SVP basis set except those directly bonded to the scandium atom (N, O) and the carbon atoms of the cyclopropyl groups for which the def2-TZVP basis set was used.35 The nature of the stationary points was ascertained by vibrational analysis within the harmonic approximation (1 atm and 298 K). Minima were identified by a full set of real frequencies. Computation of the NMR coupling constants was realized with the IGLOII basis set for carbon and hydrogen atoms.36 NBO calculations were carried out using NBO6 as implemented in Gaussian09.37 Drawings were produced using the software Chemcraft.38Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21732007, 21890721, 21821002), the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000), and the Program of Shanghai Academic Research Leader.Notes and references
- A. de Meijere, Bonding Properties of Cyclopropane and Their Chemical Consequences, Angew. Chem., Int. Ed. Engl., 1979, 18, 809–826 CrossRef.
- K. B. Wiberg, Bent bonds in organic compounds, Acc. Chem. Res., 1996, 29, 229–234 CrossRef CAS.
- P. Rademacher, Photoelectron spectra of cyclopropane and cyclopropene compounds, Chem. Rev., 2003, 103, 933–976 CrossRef CAS.
- A. de Meijere, Introduction: Cyclopropanes and Related Rings, Chem. Rev., 2003, 103, 931–932 CrossRef CAS.
- M. Etienne and A. S. Weller, Intramolecular C–C agostic complexes: C–C sigma interactions by another name, Chem. Soc. Rev., 2014, 43, 242–259 RSC.
- B. G. Harvey and R. D. Ernst, Transition-Metal Complexes with (C–C)→M Agostic Interactions, Eur. J. Inorg. Chem., 2017, 2017, 1205–1226 CrossRef CAS.
- J. Jaffart, M. Etienne, M. Reinhold, J. E. McGrady and F. Maseras, An unprecedented α-C–C agostic interaction in a cyclopropyl tris(pyrazolyl)boratoniobium complex, Chem. Commun., 2003, 876–877 RSC.
- J. Jaffart, M. L. Cole, M. Etienne, M. Reinhold, J. E. McGrady and F. Maseras, C–H and C–C agostic interactions in cycloalkyl tris(pyrazolyl)boratoniobium complexes, Dalton Trans., 2003, 4057–4064 RSC.
- C. Boulho, T. Keys, Y. Coppel, L. Vendier, M. Etienne, A. Locati, F. Bessac, F. Maseras, D. A. Pantazis and J. E. McGrady, C–C Coupling Constants, JCC, Are Reliable Probes for α-C–C Agostic Structures, Organometallics, 2009, 28, 940–943 CrossRef CAS.
- Q. Dufrois, J.-C. Daran, L. Vendier, C. Dinoi and M. Etienne, Triangles and Squares for a Unique Molecular Crystal Structure: Unsupported Two-Coordinate Lithium Cations and CC Agostic Interactions in Cyclopropyllithium Derivatives, Angew. Chem., Int. Ed., 2018, 57, 1786–1791 CrossRef CAS.
- Q. Dufrois, L. Vendier and M. Etienne, α-CC agostic structures and aggregation diversity in cyclopropyllithium derivatives, Chem. Commun., 2016, 52, 6781–6784 RSC.
- Y. Escudie, C. Dinoi, O. Allen, L. Vendier and M. Etienne, An Unsymmetrical bis C–C Agostic Heterobimetallic Lithium Yttrium Complex, Angew. Chem., Int. Ed., 2012, 51, 2461–2464 CrossRef CAS.
- S. Tobisch, Organolanthanide-Mediated Ring-Opening Ziegler Polymerization (ROZP) of Methylenecycloalkanes: A Theoretical Mechanistic Investigation of Alternative Mechanisms for Chain Initiation of the Samarocene-Promoted ROZP of 2-Phenyl-1-methylenecyclopropane, Chem. – Eur. J., 2005, 11, 3113–3126 CrossRef CAS.
- P. G. Hayes, W. E. Piers, L. W. M. Lee, L. K. Knight, M. Parvez, M. R. J. Elsegood and W. Clegg, Dialkylscandium Complexes Supported by β-Diketiminato Ligands: Synthesis, Characterization, and Thermal Stability of a New Family of Organoscandium Complexes, Organometallics, 2001, 20, 2533–2544 CrossRef CAS.
- D. B. Denney and F. J. Gross, Preparation and some chemistry of tricyclopropylphosphine, J. Org. Chem., 1967, 32, 2445–2447 CrossRef CAS.
- L. K. Knight, W. E. Piers, P. Fleurat-Lessard, M. Parvez and R. McDonald, β-Diketiminato Scandium Chemistry: Synthesis, Characterization, and Thermal Behavior of Primary Amido Alkyl Derivatives, Organometallics, 2004, 23, 2087–2094 CrossRef CAS.
- T. N. Valadez, J. R. Norton and M. C. Neary, Reaction of Cp*(Cl)M(Diene) (M = Ti, Hf) with Isonitriles, J. Am. Chem. Soc., 2015, 137, 10152–10155 CrossRef CAS.
- M. von Seebach, S. I. Kozhushkov, H. Schill, D. Frank, R. Boese, J. Benet-Buchholz, D. S. Yufit and A. de Meijere, Stereoselective Preparation of Six Diastereomeric Quatercyclopropanes from Bicyclopropylidene and Some Derivatives, Chem. – Eur. J., 2007, 13, 167–177 CrossRef CAS.
- J. S. Meek and D. T. Osuda, BROMOCYCLOPROPANE, Org. Synth., 1963, 43, 9 CrossRef CAS.
- Y. Hu, N. Romero, C. Dinoi, L. Vendier, S. Mallet-Ladeira, J. E. McGrady, A. Locati, F. Maseras and M. Etienne, β-H Abstraction/1,3-CH Bond Addition as a Mechanism for the Activation of CH Bonds at Early Transition Metal Centers, Organometallics, 2014, 33, 7270–7278 CrossRef CAS.
- P. Oulié, C. Dinoi, C. Li, A. Sournia-Saquet, K. Jacob, L. Vendier and M. Etienne, CH Bond Activation of Unsaturated Hydrocarbons by a Niobium Methyl Cyclopropyl Precursor. Cyclopropyl Ring Opening and Alkyne Coupling Reaction, Organometallics, 2017, 36, 53–63 CrossRef.
- C. Boulho, P. Oulié, L. Vendier, M. Etienne, V. Pimienta, A. Locati, F. Bessac, F. Maseras, D. A. Pantazis and J. E. McGrady, C–H Bond Activation of Benzene by Unsaturated η2-Cyclopropene and η2-Benzyne Complexes of Niobium, J. Am. Chem. Soc., 2010, 132, 14239–14250 CrossRef CAS.
- C. Li, C. Dinoi, Y. Coppel and M. Etienne, CH Bond Activation of Methane by a Transient η2-Cyclopropene/Metallabicyclobutane Complex of Niobium, J. Am. Chem. Soc., 2015, 137, 12450–12453 CrossRef CAS.
- R. A. Wolf, M. J. Migliore, P. H. Fuery, P. R. Gagnier, I. C. Sabeta and R. J. Trocino, Properties of small-ring free radicals. 2. Thermal decomposition of alicyclic percarboxylates, J. Am. Chem. Soc., 1978, 100, 7967–7976 CrossRef CAS.
- A. I. Vogel, A Textbook of Practical Organic Chemistry, 5th edn, 1989, p. 859 Search PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- M. Montag, I. Efremenko, Y. Diskin-Posner, Y. Ben-David, J. M. L. Martin and D. Milstein, Exclusive C–C Oxidative Addition in a Rhodium Thiophosphoryl Pincer Complex and Computational Evidence for an η3-C–C–H Agostic Intermediate, Organometallics, 2012, 31, 505–512 CrossRef CAS.
- G. M. Sheldrick, SADABS: An Empirical Absorption Correction Program for Area Detector Data, University of Göttingen: Göttingen, Germany, 1996 Search PubMed.
- (a) G. M. Scheldrick, SHELXS-97 and SHELXL-97, University of Göttingen, Göttingen, Germany, 1997 and 2008 Search PubMed; (b) G. M. Sheldrick, SHELXS-2014, University of Göttingen, Göttingen, Germany, 2014 Search PubMed; (c) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: A Complete Structure Solution, Refinement and Analysis Program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS; (d) SMART, Version 5.628, Bruker AXS Inc., Madison, WI, 2002 Search PubMed; (e) SAINT+, Version 6.22a, Bruker AXS Inc., Madison, WI, 2002 Search PubMed; (f) SAINT+, Version v7.68A, Bruker AXS Inc., Madison, WI, 2009 Search PubMed; (g) SHELXTL NT/2000, Version 6.1, Bruker AXS Inc., Madison, WI, 2002 Search PubMed; (h) G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, Energy-adjusted ab initio pseudopotentials for the first row transition elements, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc-Cu, Y-Ag and La-Au, Chem. Phys. Lett., 1993, 208, 111–114 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. Schindler and W. Kutzelnigg, Theory of magnetic susceptibilities and NMR chemical shifts in terms of localized quantities. II. Application to some simple molecules, J. Chem. Phys., 1982, 76, 1919–1933 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
- Chemcraft - graphical software for visualization of quantum chemistry computations, https://www.chemcraftprog.com Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data and refinement parameters for 1–3, NMR spectra. Cartesian coordinates, energy, full drawing and a summary of the main NBO interactions for the optimized structures of 2 and 3. CCDC 2033496 (1), 2033489 (2) and 2033488 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qi01163k |
| ‡ As a reference, we used the NBO delocalizations of the σ-C–C orbitals in the σ*-C–C orbitals within a cyclopropyl ring which describes the bonding in any cyclopropane derivative. These interactions are in the range of 17–21 kJ mol−1 for the present study (see the ESI†). |
| This journal is © the Partner Organisations 2020 |