
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMixing Aβ(1–40) and Aβ(1–42) peptides generates unique amyloid fibrils†
Linda
Cerofolini
 a,
Enrico
Ravera
ab,
Sara
Bologna
ab,
Thomas
Wiglenda
c,
Annett
Böddrich
c,
Bettina
Purfürst
d,
Iryna
Benilova‡
e,
Magdalena
Korsak§
f,
Gianluca
Gallo¶
ab,
Domenico
Rizzo
ab,
Leonardo
Gonnelli
ab,
Marco
Fragai
ab,
Bart
De Strooper
*eg,
Erich E.
Wanker
*c and
Claudio
Luchinat
*abf
a,
Enrico
Ravera
ab,
Sara
Bologna
ab,
Thomas
Wiglenda
c,
Annett
Böddrich
c,
Bettina
Purfürst
d,
Iryna
Benilova‡
e,
Magdalena
Korsak§
f,
Gianluca
Gallo¶
ab,
Domenico
Rizzo
ab,
Leonardo
Gonnelli
ab,
Marco
Fragai
ab,
Bart
De Strooper
*eg,
Erich E.
Wanker
*c and
Claudio
Luchinat
*abf
aMagnetic Resonance Center (CERM), University of Florence and Interuniversity Consortium for Magnetic Resonance of Metalloproteins (CIRMMP), Via L. Sacconi 6, 50019, Sesto Fiorentino (FI), Italy. E-mail: claudioluchinat@cerm.unifi.it
bDepartment of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 3, 50019, Sesto Fiorentino (FI), Italy
cNeuroproteomics, Max Delbrück Center for Molecular Medicine, Robert-Roessle-Strasse 10, 13125 Berlin, Germany. E-mail: ewanker@mdc-berlin.de
dCore Facility Electron Microscopy, Max-Delbrück Center for Molecular Medicine, Robert-Roessle-Strasse 10, 13125 Berlin, Germany
eVIB Center for Brain and Disease Research, Herestraat 49, 3000 Leuven, Belgium
fGiotto Biotech S.R.L., Via Madonna del Piano 6, 50019 Sesto Fiorentino (FI), Italy
gKULeuven, Department of Neurology, Herestraat 49, 3000 Leuven, Belgium. E-mail: bart.destrooper@kuleuven.vib.be
First published on 6th July 2020
Abstract
Recent structural studies show distinct morphologies for the fibrils of Aβ(1–42) and Aβ(1–40), which are believed not to co-fibrillize. We describe here a novel, structurally-uniform 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixed fibrillar species, which differs from both pure fibrils. It forms preferentially even when Aβ(1–42):Aβ(1–40) peptides are mixed in a non-stoichiometric ratio.
:1 mixed fibrillar species, which differs from both pure fibrils. It forms preferentially even when Aβ(1–42):Aβ(1–40) peptides are mixed in a non-stoichiometric ratio.
Among the major unknowns in Alzheimer's disease research are the mechanisms by which different Aβ(1–42) and/or Aβ(1–40) aggregate species cause toxicity in mammalian cells. Most biophysical studies on Aβ peptides reported in the literature only deal with the behavior of a single alloform of the peptide, and do not consider the many Aβ peptides that coexist in vivo.1–6 However, it has been widely demonstrated that increasing amounts of Aβ(1–42) relative to Aβ(1–40) speed up the aggregation kinetics and also alter the pattern of spontaneously formed oligomeric species,7–11 which are considered the main toxic species.12–14 The rate of formation of these species is markedly different between the two main isoforms.15,16
Kuperstein et al. have previously reported that all mixtures of Aβ(1–42) and Aβ(1–40) peptides with ratios higher than 3:7 are equally prone to aggregation, and show a similar lag-phase.10 Based on this observation, it was concluded that toxicity results from an increase of the Aβ(1–42)/Aβ(1–40) ratio,10 suggesting that the properties of mixture do not match the sum of the properties of the two individual components, therefore implying the formation of mixed species. The formation of mixed intermediate species has been proposed,17 and can be considered the result of the diverse conversion and aggregation pathways of these peptides.15,18,19 However, it is widely believed that Aβ(1–42) and Aβ(1–40) do not co-fibrillize.17 Whether the two alloforms interplay or act separately instead is an important question, as this has implications for the propagation of fibrillar seeds in the brain.20,21
We have prepared fibrils in the same experimental conditions as those previously used to obtain well-shaped fibrils of pure Aβ(1–40),22 using a 1:1 ratio of the two isoforms (Fig. S1 and S2, ESI†). A new single species is spontaneously formed. The mixtures before fibrillization show a marked toxicity to cultured neurons (see for the characterization Fig. S3, ESI†). When a 3:7 Aβ(1–42):Aβ(1–40) ratio (previously found to be the most toxic mixture10) is used, the same single species is observed, but with the excess Aβ(1–40) simultaneously forming the same pure fibrillar species previously characterized by Bertini et al.22 (Fig. S4, ESI†). No cross-peaks among the two species are observable. The ratio between the two species has been estimated from the intensity of the signals in the 2D 13C–13C correlation spectra and found to be approximately 4:3, in line with the expectation (see ESI†)||.
We have acquired solid-state NMR spectra on two samples of the species obtained at the 1:1 ratio with either one of the peptides uniformly 13C–15N labeled. The spectra of the labeled Aβ(1–42) and the Aβ(1–40) components in the two 1:1 mixed samples are superimposable (Fig. 1). The spectra of the Aβ(1–42) component show some extra peaks (particularly for S8 and G9), suggesting that the Aβ(1–42) may be more rigid than the Aβ(1–40) in the N-terminal loop, as well as a few minor peaks attributable to other species, possibly linked to a slight imbalance in the concentration of the two isoforms. When assigned23,24 (Fig. S6 and S7, ESI†), the spectra yield the same intra- and intermolecular contacts, showing that the conformation of the two peptides is identical. Signals correlating the side chains of Leu17 with Leu34/Val36, Phe19 with Gly33/Leu34, Ala21 with Ile32, and His13 with Val40 were detected and assigned unambiguously on the 13C–13C correlation25 spectra at different mixing times on both samples (see Table S1, ESI†). These contacts are only consistent with a U-shaped conformation of the monomer typical of Aβ(1–40) and not with the characteristic S-shaped conformation of Aβ(1–42) (Scheme S1, ESI†).
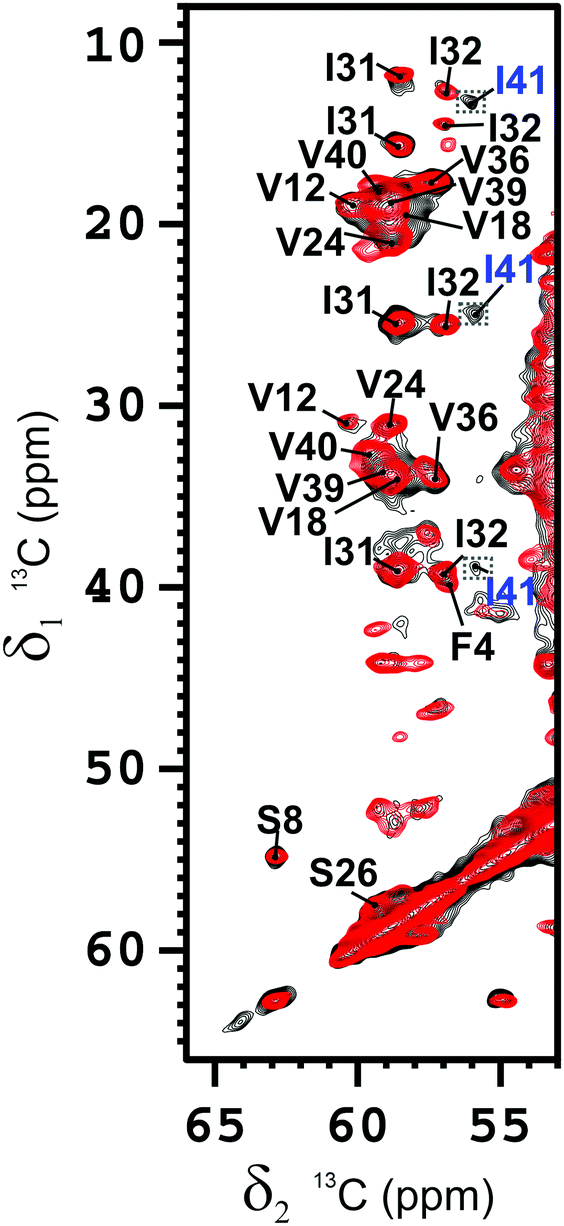 | ||
| Fig. 1 Section of the overlaid 2D 13C–13C-correlation spectra of the Aβ(1–42) component (black) and of the Aβ(1–40) component (red) in the 1:1 Aβ(1–42):Aβ(1–40) mixed fibrils. Mixing time = 100 ms. Magnetic field: 700 MHz (16.4 T), dimension of rotor: 3.2 mm (∼14 mg of fibrils), 12 kHz spinning, 100 kHz 1H decoupling, T = 283 K. The resonances are assigned as indicated. The crosspeaks corresponding to I41 are magnified by a factor 2. | ||
When the unambiguous contacts are reported on the topology of the monomer, it is clear that in the β-arch the reciprocal packing of the two β-strands (β1 and β2) (Fig. S8A, ESI†), is different from that of pure Aβ(1–40) obtained in the same conditions22 (Fig. S8B, ESI,† and Scheme 1) and, instead, resembles that reported for fibrils of pure Aβ(1–40) or Aβ(1–42) obtained under different conditions by Tycko and Smith and coworkers2,26,27 (Scheme S1, see ESI,† for the details of structure calculations), and has also the same register of the highly toxic oligomers stabilized by an intramolecular disulfide bond between residues 21 and 30, mutated to cysteine.28
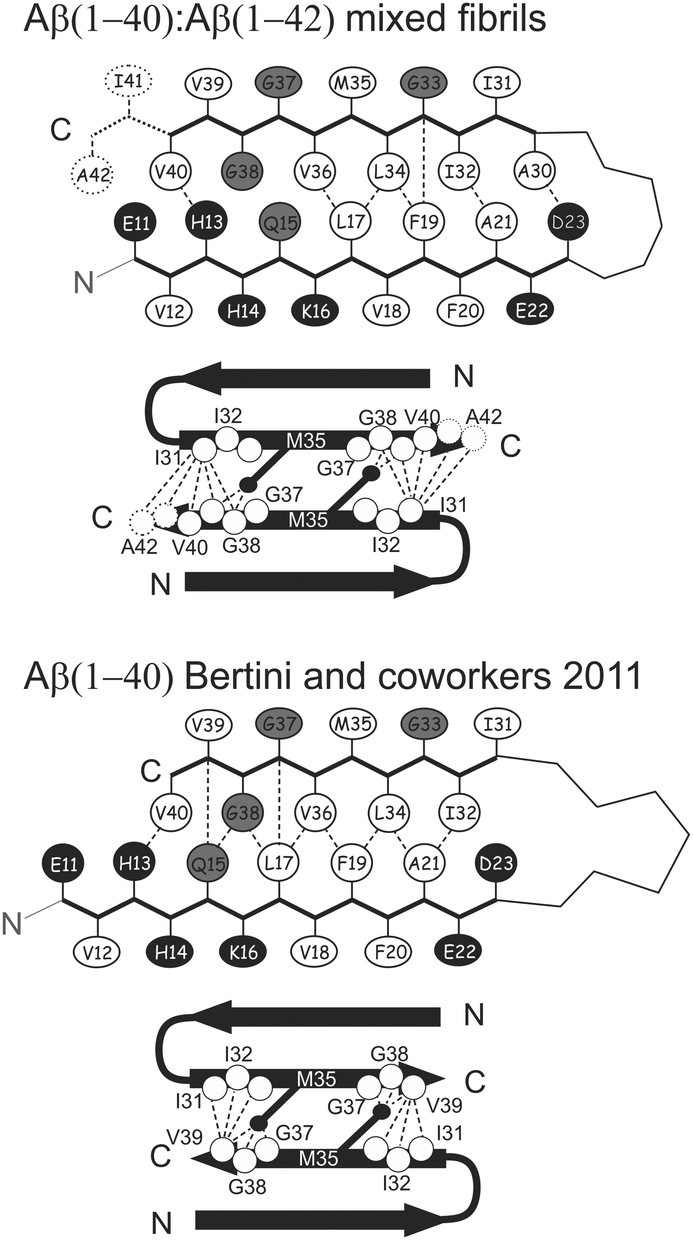 | ||
| Scheme 1 Topologies of monomer and the interprotofilament interface identified in the present work and in previously studied pure Aβ(1–40).22 The dashed/dotted lines represent unambiguous experimental restraints used to derive the corresponding topology. In the schematic description of the monomer, the hydrophobic, acidic/basic, and other types of residues are shown in white, black, and gray, respectively. The filled black circles represent the Cε of the Met35 residue. Other residues included in SS-NMR-observed structural restraints for linking the protofilaments are shown as hollow circles. | ||
As previously observed,22 Lys28 is exposed to the solvent and not involved in the formation of salt-bridges.29–32 The analysis of the cross-peaks in the 13C–13C correlation spectra supports the presence of a parallel arrangement of the protein molecules along the β-spine. No cross-peaks correlating the N-terminus and C-terminus of β1 or β2 strands have been observed in the spectrum of either sample. This indicates that the β-strand-turn-β-strand motif is organized in parallel cross-β sheets as reported in the literature for mature fibrils of Aβ(1–40).2,22,26,27,33,34 This model is further supported by the presence of a single pattern of signals for each residue in the SS-NMR spectra. For symmetry considerations, this is consistent only with the presence of a parallel in-registry β-spine.35 Each of the β-spines constituting the sides of the cross-β sheet arrangement is called “protofilament” for simplicity.
More specifically, the β1–β2 arrangement of the 1–40 filaments of both Aβ(1–40) and Aβ(1–42) are identical in the mixed fibrils.
Homogeneous protofilaments of either Aβ(1–40) or Aβ(1–42) can be excluded by the presence in the spectra of cross-peaks between N-terminus and C-terminus of the β2 strand, which would not be present if all the labeled peptide molecules were in the same protofibril (Fig. S9 and S10, ESI†). We are thus left with the possibility of an interlaced arrangement. To further prove this, fibrils from 1:1 mixtures of 15N-enriched Aβ(1–42) and 13C-enriched Aβ(1–40) were prepared, in such a way as to have NMR signals only if 15N and 13C nuclei are in close proximity. In particular, a two-dimensional nitrogen-carbon correlation experiment, 2D 15N–13C hNhhC36 shows good signal intensity in several parts of the spectrum and particularly in the NH-carbonyl region, thus demonstrating direct, short range contacts between Aβ(1–40) and Aβ(1–42) filaments (Fig. 2), further confirmed by a 1D TEDOR experiment (Fig. S11, ESI†).37 These data demonstrate beyond any doubt that Aβ(1–40) and Aβ(1–42) can co-fibrillize in a 1:1 ratio to form an interlaced fibril (Fig. 3 and Fig. S12, ESI†).
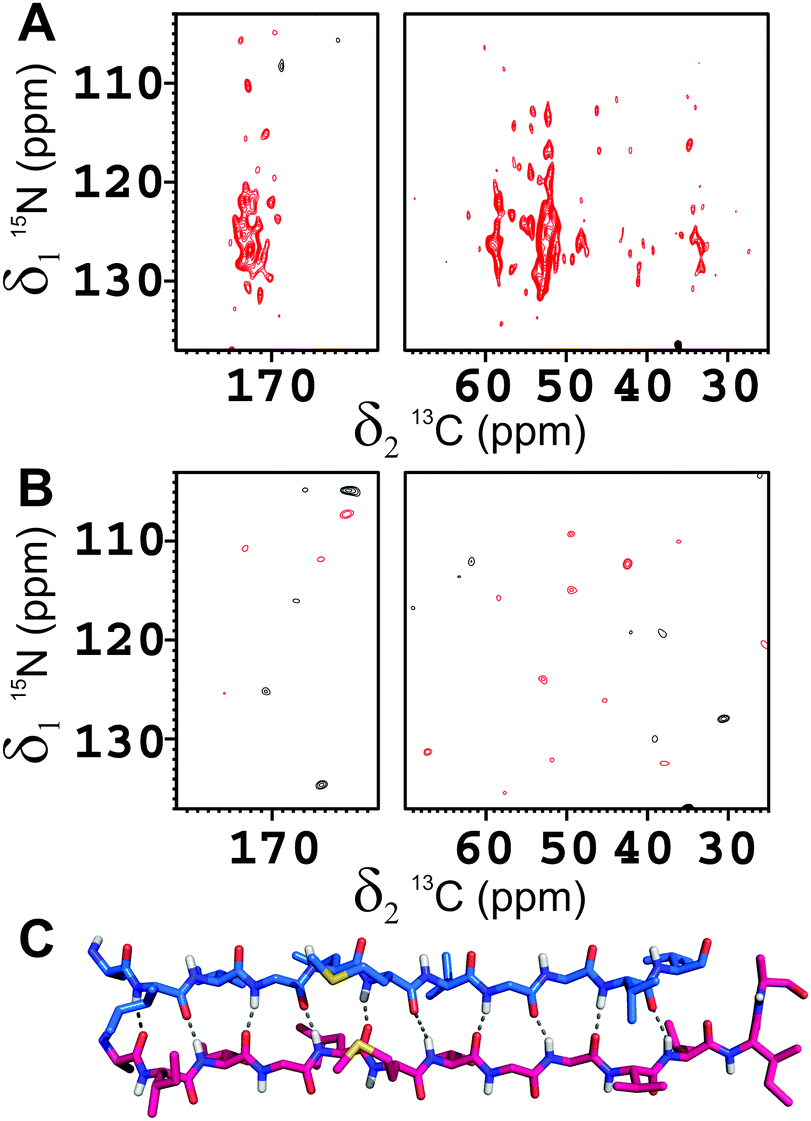 | ||
| Fig. 2 2D 15N–13C hNhhC spectra of the Aβ(1–42):Aβ(1–40) mixed fibrils in the 1:1 molar ratio, where (A) Aβ(1–42) is 15N-enriched and Aβ(1–40) is 13C-enriched, (B) Aβ(1–42) is 15N-enriched and Aβ(1–40) is in natural abundance. Magnetic field: 800 MHz (19 T, 201.2 MHz 13C Larmor frequency), dimension of rotor: 3.2 mm, 16 kHz spinning, 80 kHz 1H decoupling; number of scans: 2048. The strong cross peaks in the carbonyl and Cα regions in (A) and the total absence of signals in (B) clearly demonstrates that the transfer in (A) is occurring between the two alloforms. (C) The H-bonds pattern of Aβ(1–42) interlaced with Aβ(1–40) in the β-spine is displayed. | ||
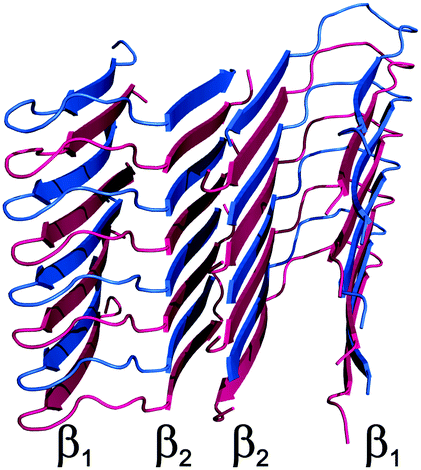 | ||
| Fig. 3 Structural model of Aβ(1–40)/Aβ(1–42) interlaced mixed fibrils. The Aβ(1–42) polypeptide is colored in magenta while the Aβ(1–40) polypeptide in blue. | ||
The heterogeneity observed in the SS-NMR spectra of pure Aβ(1–42) under the present conditions may reflect the endpoint of a fast aggregation reaction, which is instead prevented by the formation of a 1:1 product when Aβ(1–40) and Aβ(1–42) are present simultaneously in solution and which also favors a conformation with the turn at positions G25 and S26 over the one with the turn at positions E22 and D23, which are putatively involved in the toxicity of early aggregates.38,39 In the present interlaced fibrils, the observed U-shape register ideally accommodates the requirements of both filaments, and is likely to provide an extra stabilization by preventing the steric clashes potentially caused by Ile41 and Ala42 because these two residues are alternatively present and absent in the interlaced fibrils. The buried surface area is maximum for the mixture in this arrangement, see Table S5 (ESI†).
The present observation that a single fibrillary species is obtained from mixtures of Aβ(1–42) and Aβ(1–40) indicates that the interplay between the two alloforms may contribute to extend the number of possible polymorphs formed by these peptides, increasing the complexity of the structural landscape of the amyloid aggregates, which may correspond to phenotypic differences.40 We expect that the availability of a structural model for this mixed-species will be useful for a better understanding of the variable nature of cross-seeding,29,41,42 as well as in the development of potential drugs.43,44
This research activity has been supported by EC funding initiative ERA-NET NEURON ABETA ID (“Novel Methods and Approaches towards the Understanding of Brain Diseases”), funded by the German Federal Ministry for Education and Research (BMBF), grant no. 01W1301, by the Fonds voor Wetenschappelijk Onderzoek (FWO) and the Italian Ministero della Salute. BDS is supported by the FWO, the KU Leuven and VIB, a Methusalem grant of the KU Leuven/Flemish Government and by the Bax-Vanluffelen Chair for Alzheimer's Disease and “Opening the Future” of the Leuven Universiteit Fonds. Support has been provided also by: Fondazione CRFirenze, MIUR (“Progetto Dipartimenti di Eccellenza 2018–2022” and PRIN 2017A2KEPL), the Helmholtz Validation Fund grant no. HVF-0013 “Enabling Technologies for Drug Discovery against Protein Misfolding Diseases” by the Helmholtz Association, Germany and the Berlin Institute of Health Collaborative Research Grant no. 1.1.2.a.3 “Elucidating the proteostasis network to control Alzheimer's disease”, funded by the BMBF. The Italy CERM/CIRMMP centre of INSTRUCT-ERIC is also acknowledged. This work was also supported by Instruct-ULTRA (Grant 731005), an EU H2020 project to further develop the services of Instruct-ERIC.
Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Tycko, Protein Sci., 2014, 23, 1528–1539 CrossRef CAS PubMed.
- A. K. Paravastu, R. D. Leapman, W.-M. Yau and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18349–18354 CrossRef CAS PubMed.
- Y. Qi-Takahara, M. Morishima-Kawashima, Y. Tanimura, G. Dolios, N. Hirotani, Y. Horikoshi, F. Kametani, M. Maeda, T. C. Saido, R. Wang and Y. Ihara, J. Neurosci., 2005, 25, 436–445 CrossRef CAS PubMed.
- D. M. Bolduc, D. R. Montagna, M. C. Seghers, M. S. Wolfe and D. J. Selkoe, eLife, 2016, 5, e17578 CrossRef PubMed.
- J.-P. Colletier, A. Laganowsky, M. Landau, M. Zhao, A. B. Soriaga, L. Goldschmidt, D. Flot, D. Cascio, M. R. Sawaya and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16938–16943 CrossRef CAS PubMed.
- D. M. Bolduc and M. S. Wolfe, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 14643–14644 CrossRef CAS PubMed.
- Y. Yoshiike, D.-H. Chui, T. Akagi, N. Tanaka and A. Takashima, J. Biol. Chem., 2003, 278, 23648–23655 CrossRef CAS PubMed.
- A. Jan, O. Gokce, R. Luthi-Carter and H. A. Lashuel, J. Biol. Chem., 2008, 283, 28176–28189 CrossRef CAS PubMed.
- A. Jan, D. M. Hartley and H. A. Lashuel, Nat. Protocols, 2010, 5, 1186–1209 CAS.
- I. Kuperstein, K. Broersen, I. Benilova, J. Rozenski, W. Jonckheere, M. Debulpaep, A. Vandersteen, I. Segers-Nolten, K. Van Der Werf, V. Subramaniam, D. Braeken, G. Callewaert, C. Bartic, R. D’Hooge, I. C. Martins, F. Rousseau, J. Schymkowitz and B. De Strooper, EMBO J., 2010, 29, 3408–3420 CrossRef CAS PubMed.
- K. Pauwels, T. L. Williams, K. L. Morris, W. Jonckheere, A. Vandersteen, G. Kelly, J. Schymkowitz, F. Rousseau, A. Pastore, L. C. Serpell and K. Broersen, J. Biol. Chem., 2012, 287, 5650–5660 CrossRef CAS PubMed.
- B. R. Sahoo, S. J. Cox and A. Ramamoorthy, Chem. Commun., 2020, 56, 4627–4639 RSC.
- D. M. Walsh, I. Klyubin, J. V. Fadeeva, W. K. Cullen, R. Anwyl, M. S. Wolfe, M. J. Rowan and D. J. Selkoe, Nature, 2002, 416, 535–539 CrossRef CAS PubMed.
- K. Ono, Neurochem. Int., 2018, 119, 57–70 CrossRef CAS PubMed.
- G. Bellomo, S. Bologna, L. Gonnelli, E. Ravera, M. Fragai, M. Lelli and C. Luchinat, Chem. Commun., 2018, 54, 7601–7604 RSC.
- T. C. T. Michaels, A. Šarić, S. Curk, K. Bernfur, P. Arosio, G. Meisl, A. J. Dear, S. I. A. Cohen, C. M. Dobson, M. Vendruscolo, S. Linse and T. P. J. Knowles, Nat. Chem., 2020, 12, 445–451 CrossRef CAS PubMed.
- R. Cukalevski, X. Yang, G. Meisl, U. Weininger, K. Bernfur, B. Frohm, T. P. J. Knowles and S. Linse, Chem. Sci., 2015, 6, 4215–4233 RSC.
- Y. Lin, B. R. Sahoo, D. Ozawa, M. Kinoshita, J. Kang, M. H. Lim, M. Okumura, Y. H. Huh, E. Moon, J. H. Jang, H.-J. Lee, K.-Y. Ryu, S. Ham, H.-S. Won, K.-S. Ryu, T. Sugiki, J. K. Bang, H.-S. Hoe, T. Fujiwara, A. Ramamoorthy and Y.-H. Lee, ACS Nano, 2019, 13, 8766–8783 CrossRef CAS PubMed.
- I. Bertini, G. Gallo, M. Korsak, C. Luchinat, J. Mao and E. Ravera, ChemBioChem, 2013, 14, 1891–1897 CrossRef CAS PubMed.
- R. Tycko, Nature, 2016, 537, 492–493 CrossRef CAS PubMed.
- M. Jucker and L. C. Walker, Nat. Neurosci., 2018, 21, 1341–1349 CrossRef CAS PubMed.
- I. Bertini, L. Gonnelli, C. Luchinat, J. Mao and A. Nesi, J. Am. Chem. Soc., 2011, 133, 16013–16022 CrossRef CAS PubMed.
- A. Schuetz, C. Wasmer, B. Habenstein, R. Verel, J. Greenwald, R. Riek, A. Böckmann and B. H. Meier, ChemBioChem, 2010, 11, 1543–1551 CrossRef CAS PubMed.
- S. Sun, Y. Han, S. Paramasivam, S. Yan, A. E. Siglin, J. C. Williams, I.-J. L. Byeon, J. Ahn, A. M. Gronenborn and T. Polenova, in Protein NMR Techniques, ed. A. Shekhtman and D. S. Burz, Humana Press, Totowa, NJ, 2012, pp. 303–331 Search PubMed.
- B. Hu, O. Lafon, J. Trébosc, Q. Chen and J.-P. Amoureux, J. Magn. Reson., 2011, 212, 320–329 CrossRef CAS PubMed.
- A. T. Petkova, W.-M. Yau and R. Tycko, Biochemistry, 2006, 45, 498–512 CrossRef CAS PubMed.
- M. Ahmed, J. Davis, D. Aucoin, T. Sato, S. Ahuja, S. Aimoto, J. I. Elliott, W. E. Van Nostrand and S. O. Smith, Nat. Struct. Mol. Biol., 2010, 17, 561–567 CrossRef CAS PubMed.
- A. Sandberg, L. M. Luheshi, S. Söllvander, T. P. de Barros, B. Macao, T. P. J. Knowles, H. Biverstål, C. Lendel, F. Ekholm-Petterson, A. Dubnovitsky, L. Lannfelt, C. M. Dobson and T. Härd, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15595–15600 CrossRef CAS PubMed.
- Y. Xiao, B. Ma, D. McElheny, S. Parthasarathy, F. Long, M. Hoshi, R. Nussinov and Y. Ishii, Nat. Struct. Mol. Biol., 2015, 22, 499–505 CrossRef CAS PubMed.
- M. T. Colvin, R. Silvers, Q. Z. Ni, T. V. Can, I. Sergeyev, M. Rosay, K. J. Donovan, B. Michael, J. Wall, S. Linse and R. G. Griffin, J. Am. Chem. Soc., 2016, 138, 9663–9674 CrossRef CAS PubMed.
- J.-X. Lu, W. Qiang, W.-M. Yau, C. D. Schwieters, S. C. Meredith and R. Tycko, Cell, 2013, 154, 1257–1268 CrossRef CAS PubMed.
- A. T. Petkova, R. D. Leapman, Z. Guo, W.-M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262–265 CrossRef CAS PubMed.
- A. T. Petkova, Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16742–16747 CrossRef CAS PubMed.
- W. Qiang, W.-M. Yau, Y. Luo, M. P. Mattson and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4443–4448 CrossRef CAS PubMed.
- J. T. Nielsen, M. Bjerring, M. D. Jeppesen, R. O. Pedersen, J. M. Pedersen, K. L. Hein, T. Vosegaard, T. Skrydstrup, D. E. Otzen and N. C. Nielsen, Angew. Chem., Int. Ed., 2009, 48, 2118–2121 CrossRef CAS PubMed.
- A. Lange, S. Becker, K. Seidel, K. Giller, O. Pongs and M. Baldus, Angew. Chem., Int. Ed., 2005, 44, 2089–2092 CrossRef CAS PubMed.
- C. P. Jaroniec, C. Filip and R. G. Griffin, J. Am. Chem. Soc., 2002, 124, 10728–10742 CrossRef CAS PubMed.
- Y. Matsushima, R. C. Yanagita and K. Irie, Chem. Commun., 2020, 56, 4118–4121 RSC.
- A. R. Foley, H.-W. Lee and J. A. Raskatov, J. Org. Chem., 2020, 85, 1385–1391 CrossRef CAS PubMed.
- J. Stöhr, C. Condello, J. C. Watts, L. Bloch, A. Oehler, M. Nick, S. J. DeArmond, K. Giles, W. F. DeGrado and S. B. Prusiner, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10329–10334 CrossRef PubMed.
- M. Törnquist, R. Cukalevski, U. Weininger, G. Meisl, T. P. J. Knowles, T. Leiding, A. Malmendal, M. Akke and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(21), 11265–11273 CrossRef PubMed.
- J. Tran, D. Chang, F. Hsu, H. Wang and Z. Guo, FEBS Lett., 2017, 591, 177–185 CrossRef CAS PubMed.
- M. Zhang, J. Zheng, R. Nussinov and B. Ma, Antibodies, 2018, 7(3), 25 CrossRef CAS PubMed.
- J. Zhao, R. Nussinov and B. Ma, J. Biol. Chem., 2017, 292, 18325–18343 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cc02463e |
| ‡ Current address: MRC Prion Unit at UCL, Institute of Prion diseases, Courtauld building, 33 Cleveland Street, London W1W7FF, UK. |
| § Current address: Roche Polska Sp. zo. o. Domaniewska 28, 02-672 Warszawa, Poland. |
| ¶ Current address: Fresenius Kabi, Via Camagri 41, Verona, Italy. |
| || Structures formed by a molar excess of Aβ(1–42) were not analyzed, but Aβ(1–42) by itself yields a strongly heterogeneous mixture under the present conditions (Fig. S5, ESI†). |
| This journal is © The Royal Society of Chemistry 2020 |