
An engineered exosome for delivering sgRNA:Cas9 ribonucleoprotein complex and genome editing in recipient cells†
Yangyang
Ye‡
,
Xiang
Zhang‡
,
Fei
Xie
,
Bin
Xu
,
Ping
Xie
,
Ting
Yang
,
Qian
Shi
,
Chen-Yu
Zhang
,
Yujing
Zhang
,
Jiangning
Chen
*,
Xiaohong
Jiang
* and
Jing
Li
 *
*
State Key Laboratory of Pharmaceutical Biotechnology, Collaborative Innovation Centre of Chemistry for Life Sciences, Jiangsu Engineering Research Centre for MicroRNA Biology and Biotechnology, NJU Advanced Institute for Life Sciences (NAILS), School of Life Sciences, Nanjing University, 163 Xianlin Avenue, Nanjing, Jiangsu 210023, China. E-mail: jingli220@nju.edu.cn; xiaohongjiang@nju.edu.cn; jnchen@nju.edu.cn
First published on 17th April 2020
Abstract
CRISPR-Cas9 is a versatile genome-editing technology that is a promising gene therapy tactic. However, the delivery of CRISPR-Cas9 is still a major obstacle to its broader clinical application. Here, we confirm that the components of CRISPR-Cas9—sgRNA and Cas9 protein—can be packaged into exosomes, where sgRNA and Cas9 protein exist as a sgRNA:Cas9 ribonucleoprotein complex. Although exosomal CRISPR-Cas9 components can be delivered into recipient cells, they are not adequate to abrogate the target gene in recipient cells. To solve this, we engineered a functionalized exosome (M-CRISPR-Cas9 exosome) that could encapsulate CRISPR-Cas9 components more efficiently. To improve the loading efficiency of Cas9 proteins into exosomes, we artificially engineered exosomes by fusing GFP and GFP nanobody with exosomal membrane protein CD63 and Cas9 protein, respectively. Therefore, Cas9 proteins could be captured selectively and efficiently loaded into exosomes due to the affinity of GFP-GFP nanobody rather than random loading. sgRNA and Cas9 protein exist as a complex in functionalized exosomes and can be delivered into recipient cells. To show the function of modified exosomes-delivered CRISPR-Cas9 components in recipient cells visually, we generated a reporter cell line (A549stop-DsRed) that produced a red fluorescent signal when the stop element was deleted by the sgRNA-guided endonuclease. Using A549stop-DsRed reporter cells, we showed that modified exosomes loaded with CRISPR-Cas9 components abrogated the target gene more efficiently in recipient cells. Our study reports an alternative tactic for CRISPR-Cas9 delivery.
Introduction
Gene therapy is a promising tactic to treat cancers or genetic disorders. More than 3000 genes are related to disease-causing mutations, which cannot be fixed by pharmaceutical treatments.1 Nonetheless, the intended gene therapy effects are not always accomplished due to the complexity of gene therapy. CRISPR (clustered regularly-interspaced short palindromic) and the CRISPR-associated system (Cas) are part of the adaptive prokaryotic immune system.2–4 Recently, the type II CRISPR system has been harnessed as a versatile genome editing technology.5–7 Endonuclease Cas9 protein is guided by single guide RNA (sgRNA) and interrogates the genome.8,9 sgRNA:Cas9 complex binds to complementary genomic sequence if there is a protospacer adjacent motif next to the target sequence.10,11 Then, endonuclease Cas9 generates the DNA double-strand breaks, resulting in an indel mutation while the genome is being fixed by nonhomologous end-joining12 or homology-directed repair.13,14 By using customized sgRNA, Cas9 can be reprogrammed to perform site-specific genome manipulation on intended target genes.15 CRISPR-Cas9 has been used to generate an animal model15 as well as to treat genetic disorders16 and cancer17 and in invading pathogen therapy.9,18 However, the broader clinical use of the CRISPR-Cas9 system has been prevented by the lack of a feasible delivery vehicle.To facilitate the clinical use of CRISPR-Cas9 technology to advance gene therapy, many delivery vehicles have been studied, of which adeno-associated virus (AAV) has attracted the most attention. This virus efficiently integrates into the genome of host cells and persistently expresses CRISPR-Cas9 elements in vitro or in vivo.15,19 Although AAV vehicles have been used to generate an animal model15 and treat metabolic disease20 in the laboratory, there are still concerns about their clinical use, such as cell toxicity caused by the capsid,21 dysregulation of the expression of CRISPR-Cas9 elements,22 and the unexpected activation of oncogenes triggered by the insertion of viral vectors.22 Thus, alternative means to deliver CRISPR-Cas9 components are still required.
Exosomes, which consist of extracellular vesicles, are a class of double-membrane vesicles with diameters of 50–150 nm that are released by all types of cells under both physiological and pathological conditions.23,24 Exosomes transfer small RNAs and convey bioinformation between tissues in vivo, serving as an alternative mediator of cell-cell communications.25,26 Exosomes could be used to transfer artificially designed siRNA to treat various diseases, including pancreatic carcinoma27 and morphine relapse.28 Given that exosomes are naturally released from cells, they provoke little adaptive immune response and toxicity compared with other vesicles. Thus, these properties of exosomes may be suitable for the clinical application of the CRISPR-Cas9 system.
Here, we confirm that CRISPR-Cas9 elements—sgRNA and Cas9 protein—can be loaded into exosomes and transferred into recipient cells in the sgRNA:Cas9 ribonucleoprotein (RNP) complex. To increase the efficiency of loading, we engineered a modified exosome by fusing the exosomal membrane protein CD63 with GFP, which can bind to the GFP antibody (GFP nanobody) fused with Cas9 protein.29 Because of the high affinity of GFP protein with its nanobody, Cas9 proteins were captured and efficiently loaded into exosomes rather than a random package. We demonstrate proof-of-principle of a method to deliver sgRNA:Cas9 RNP and provide an alternative tool to achieve the clinical application of the CRISPR-Cas9 system.
Results
sgRNA and Cas9 protein can be packaged into exosomes
To investigate whether sgRNA and Cas9 protein can be sorted into exosomes in vitro, a plasmid that encoded sgRNA and SpCas9 from Streptococcus pyogenes was generated (Fig. 1A). sgRNA was partitioned into CRISPR RNA (crRNA) and trans-activating (tracrRNA). A guide sequence that was integrated in crRNA was designed using CRISPR Design Tool (http://zlab.bio/guide-design-resources) and synthesized artificially. The guide sequence was then cloned into the lentiCRISPRv2 plasmid vector containing the tracrRNA sequence (plasmid 1). This plasmid was then transfected transiently into HEK 293T cells (CRISPR-Cas9 cells). As expected, sgRNAs could be detected using quantitative real-time PCR (qRT-PCR) in CRISPR-Cas9 cells but not in cells untreated or transfected with scrambled plasmids (Fig. 1B). Agarose gel electrophoresis of the DNA also showed that sgRNAs specifically were detected in CRISPR-Cas9 cells (Fig. 1C). Furthermore, mRNA levels and protein levels of Cas9 were detected in CRISPR-Cas9 cells but not in cells untreated or treated with scrambled plasmids (Fig. 1D and E). These results demonstrate that the CRISPR-Cas9 plasmids were expressed in the cells. Next, we investigated whether sgRNAs and Cas9 proteins can be loaded into exosomes. Exosomes derived from untreated cells and CRISPR-Cas9 cells were collected. The transmission electron microscope (TEM) analysis showed that the morphology of exosomes derived from CRISPR-Cas9 cells was clearly discernible, showing a double-membrane-bound structure (Fig. 1F). Nanoparticle tracking analysis (NTA) results showed that exosomes derived from normal cells and CRISPR-Cas9 cells had similar diameters, which peaked at approximately 143 ± 2.2 nm and 149 ± 1.2 nm respectively (Fig. 1G). The results of qRT-PCR and DNA agarose gel electrophoresis showed that an abundant amount of sgRNAs were packaged into exosomes derived from CRISPR-Cas9 cells (Fig. 1H and I). Intriguingly, Cas9 mRNA levels in CRISPR-Cas9 exosomes could not be detected (Fig. 1J). However, a considerable amount of Cas9 proteins were detected in CRISPR-Cas9 exosomes (Fig. 1K). These results demonstrate sgRNA can be packaged into exosomes, and Cas9 protein rather than mRNA can be packaged into exosomes preferentially.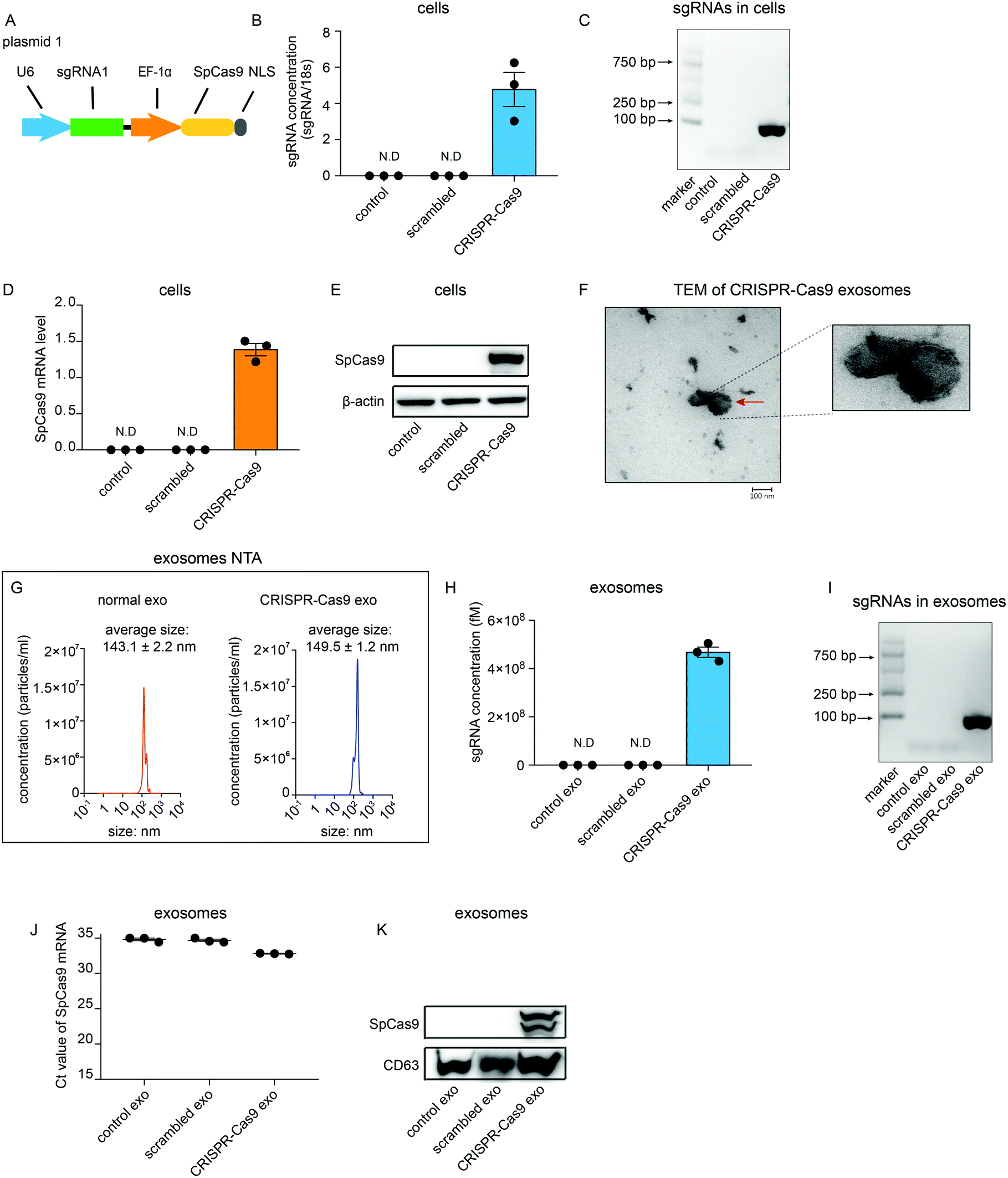 | ||
| Fig. 1 A. Schematic of LentiCRISPR-sgRNA1-Cas9 vector (plasmid 1) design. B. qRT-PCR analysis of sgRNA concentrations in cells untreated or transfected with scrambled or CRISPR-Cas9 plasmids. C. Levels of sgRNA expression in cells untreated or transfected with scrambled or CRISPR-Cas9 plasmids. D. qRT-PCR analysis of Cas9 mRNA levels in cells untreated or transfected with scrambled or CRISPR-Cas9 plasmids. E. Western blot analysis of Cas9 protein levels in cells untreated or transfected with scrambled or CRISPR-Cas9 plasmids. F. TEM analysis of exosomes derived from cells transfected with CRISPR-Cas9 plasmids. G. Nanoparticle tracking analysis of exosomes. H. qRT-PCR analysis of sgRNA concentrations in exosomes derived from cells untreated or transfected with scrambled or CRISPR-Cas9 plasmids. I. Levels of sgRNA concentrations in exosomes derived from cells untreated or transfected with scrambled or CRISPR-Cas9 plasmids. J. qRT-PCR analysis of Cas9 mRNA levels in exosomes derived from cells untreated or transfected with scrambled or CRISPR-Cas9 plasmids. K. Western blot analysis of Cas9 protein levels in exosomes derived from cells untreated or transfected with scrambled or CRISPR-Cas9 plasmids. n = 3. Error bars, mean ± s.d. | ||
CRISPR-Cas9 components exist in exosomes as the sgRNA:Cas9 ribonucleoprotein complex
To investigate whether exosomal CRISPR-Cas9 components function in recipient cells, we examined whether the sgRNAs were physically associated with Cas9 proteins in exosomes. Cells were transfected with plasmids (plasmid 2) expressing sgRNAs and FLAG-tagged Cas9 proteins (CRISPR-Cas9-FLAG cells) (Fig. 2A). RNA-immunoprecipitation assays were performed with antibodies raised against FLAG or IgG followed by western blotting analysis with FLAG antibody. In the whole-cell lysates of untreated cells, FLAG-tagged Cas9 proteins were not detected, whereas a considerable level of FLAG-tagged Cas9 proteins was detected in CRISPR-Cas9-FLAG cells (Fig. 2B, input). Furthermore, FLAG-tagged Cas9 could be pulled down using the FLAG antibody (Fig. 2B, IP) and qRT-PCR analysis showed sgRNAs were detected in the eluted immunoprecipitants using the FLAG antibody, demonstrating Cas9 proteins and sgRNAs were associated in cells (Fig. 2C). These results confirmed that CRISPR-Cas9 components were expressed in exosomes donor cells and formed a sgRNA:Cas9 RNP complex. Next, the interaction of sgRNA and Cas9 protein was assayed in the exosomes. The FLAG-tagged Cas9 proteins were detected in lysates of the exosomes derived from CRISPR-Cas9-FLAG cells (Fig. 2D, input). In the immunoprecipitants of CRISPR-Cas9-FLAG exosomes, FLAG-tagged Cas9 was clearly detected with the FLAG antibody but not with the IgG antibody or in untreated cells (Fig. 2D, IP). qRT-PCR was preformed to determine the sgRNA levels in immunoprecipitants. sgRNA levels were clearly detected in the immunoprecipitants of CRISPR-Cas9-FLAG exosomes co-immunoprecipitated with the FLAG antibody (Fig. 2E). These results demonstrate that sgRNA and Cas9 existed in exosomes as an RNP complex.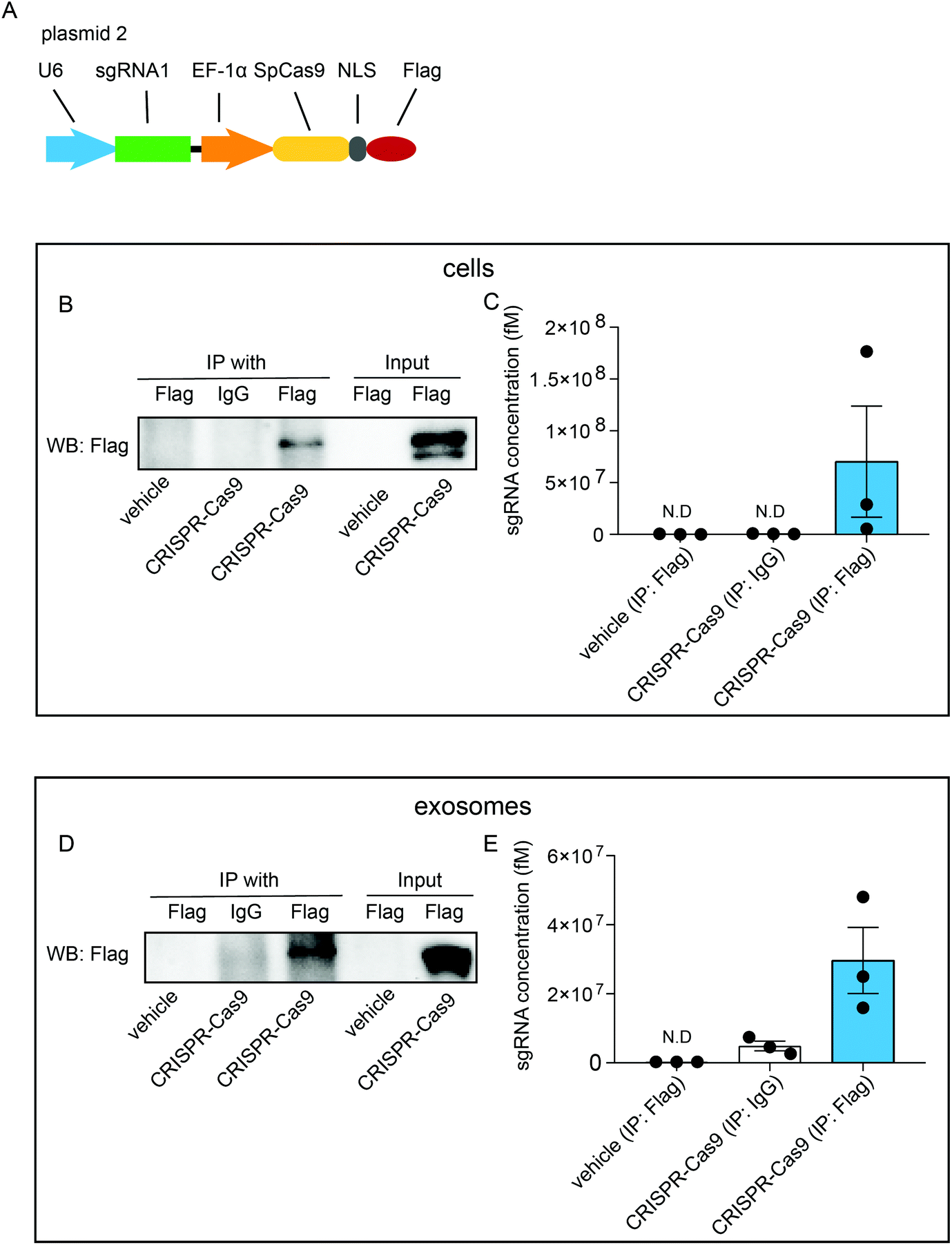 | ||
| Fig. 2 A. Schematic of LentiCRISPR-sgRNA1-Cas9-FLAG vector (plasmid 2) design. B. Immunoprecipitation assay of cells untreated or treated with CRISPR-Cas9-FLAG plasmids. Lysates of cells were blotted using FLAG antibody (input) or immunoprecipitated with FLAG and blotted using FLAG antibody (IP with). IP with anti-IgG served as the control. C. sgRNA analysis of immunoprecipitants following the IP assay in B. D. Immunoprecipitation assay of exosomes derived from cells untreated or treated with CRISPR-Cas9-FLAG plasmids. Lysates of exosomes were blotted using FLAG antibody (input) or immunoprecipitated with FLAG and blotted using FLAG antibody (IP with). IP with anti-IgG served as the control. E. sgRNA analysis of immunoprecipitants following the IP assay in D. n = 3. Error bars, mean ± s.d. | ||
Exosomal sgRNAs and Cas9 proteins are taken up by recipient cells
Previous studies have demonstrated that exosomes can deliver RNA bio-cargos into recipient cells. Therefore, we investigated whether CRISPR-Cas9 components can be delivered into recipient cells via the transmission of exosomes. The lung adenocarcinoma cell line A549 was treated with normal exosomes and exosomes loaded with CRISPR-Cas9 components. The levels of sgRNA were significantly increased in cells treated with CRISPR-Cas9 exosomes (Fig. 3A), and Cas9 proteins were clearly detected in recipient cells (Fig. 3B). Thus, the exosomes delivered CRISPR-Cas9 components into recipient cells.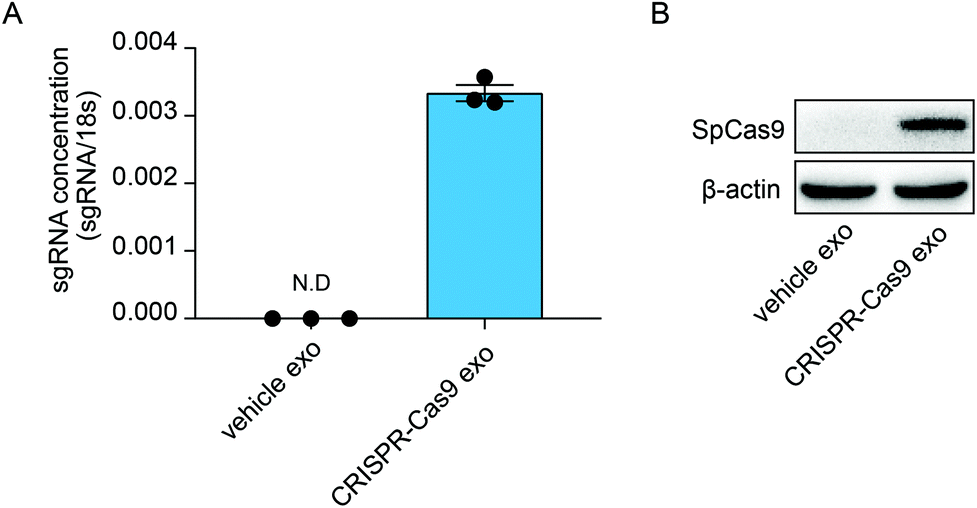 | ||
| Fig. 3 A. qRT-PCR analysis of sgRNA concentrations in cells untreated or treated with exosomes loaded with CRISPR-Cas9 components. B. Western blot analysis of Cas9 protein levels in cells untreated or treated with exosomes loaded with CRISPR-Cas9 components. n = 3. Error bars, mean ± s.d. | ||
Enrichment of sgRNAs and Cas9 proteins in exosomes using GFP-binding nanobody
Although sgRNAs and Cas9 proteins are loaded in exosomes as a ribonucleoprotein complex, these exosomes cannot efficiently alter the expression of the target gene. Thus, we examined whether the enrichment of CRISPR-Cas9 components in exosomes resulted in more efficient deletion of the target gene in the recipient cells. We generated two plasmids that expressed CD63-GFP fusion protein (plasmid 3) and GFP nanobody-fused Cas9 (plasmid 4). These plasmids were co-transfected into donor cells (Fig. 4A). GFP was fused with CD63 protein, which is a member of the tetraspanin family and expressed on the surface of the inner side of exosome membrane, whereas Cas9 protein was fused with GFP-binding nanobody, which binds readily with GFP protein and can be efficiently loaded into exosomes (Fig. 4B). TEM analysis showed that the modified CRISPR-Cas9 exosomes (M-CRISPR-Cas9 exo) had the standard exosome morphology with a diameter of around 100 nm (Fig. 4C). Next, we confirmed that the GFP was expressed in exosomes derived from co-transfected cells (Fig. 4D). In modified CRISPR-Cas9 exosomes, the levels of sgRNA and Cas9 proteins were significantly increased compared with those in CRISPR-Cas9 exosomes (Fig. 4E–G). This indicated that some of the CRISPR-Cas9 components may be packaged into exosomes as an intact sgRNA:Cas9 RNP complex from donor cells, and increased sgRNA levels may stem from the increased Cas9 proteins loaded into modified exosomes. Intriguingly, in addition to the protein levels, the levels of Cas9 mRNA in M-CRISPR-Cas9 exosomes increased (Fig. 4H). The loading of CRISPR-Cas9 components was significantly improved using the GFP-binding nanobody system. Next, we investigated whether the sgRNAs and Cas9-GFP nanobody proteins interacted in the exosomes. An RNA-immunoprecipitation assay showed that FLAG-tagged Cas9-GFP nanobody proteins could be pulled down with the antibody raised against FLAG (Fig. 4I). qRT-PCR analysis of the immunoprecipitants showed that sgRNAs were strongly associated with the Cas9-GFP nanobody protein (Fig. 4J). The GFP-GFP nanobody system improved the loading efficiency of the CRISPR-Cas9 components into the exosomes, and this method did not alter the function of the CRISPR-Cas9 system.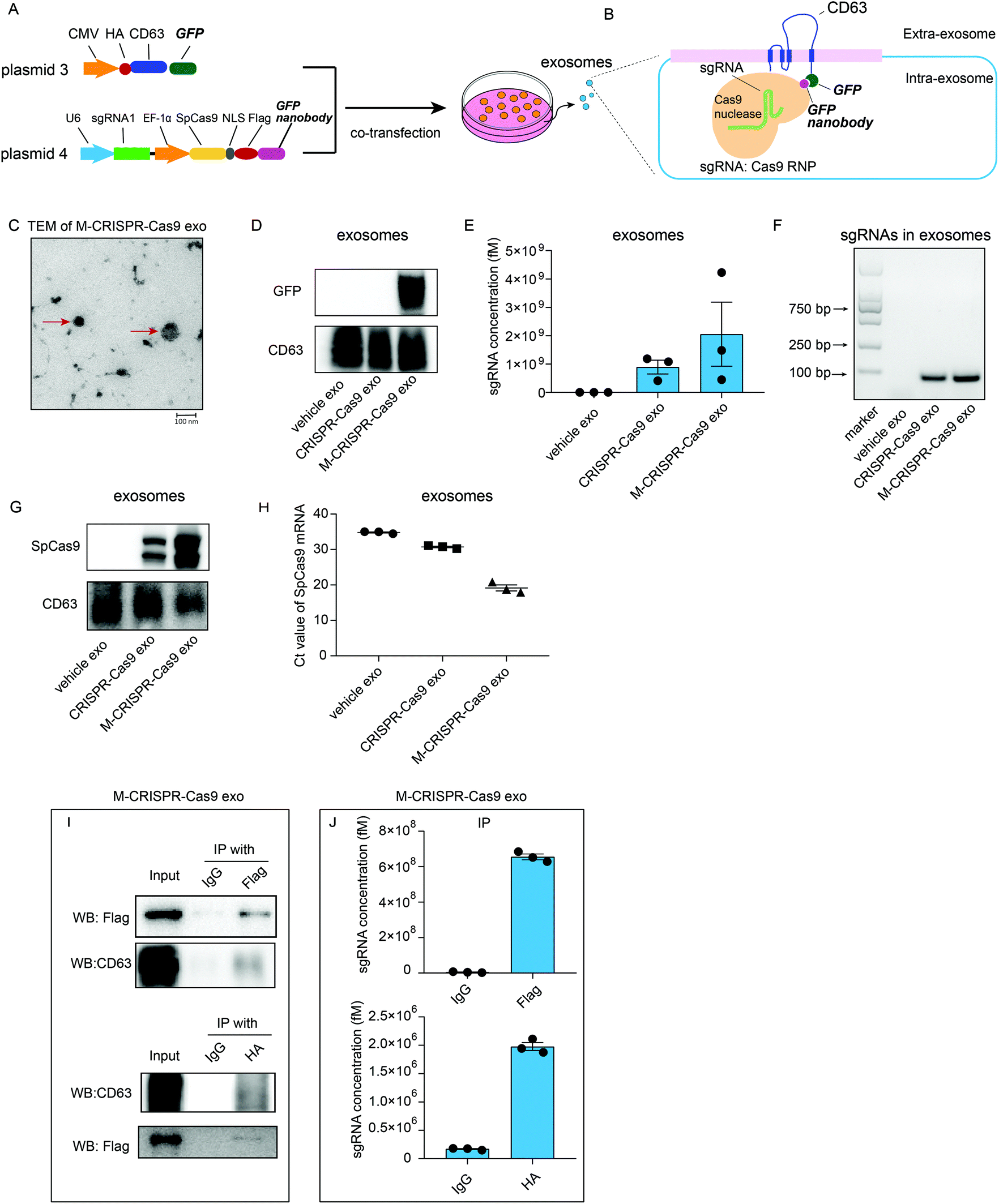 | ||
| Fig. 4 A. Schematic of pEGFP-HA-CD63 (plasmid 3) and LentiCRISPRV2-sgRNA1-Cas9 + FLAG-GFP nanobody (plasmid 4) vector designs. B. Schematic of the mechanism underlying GFP-GFP nanobody-mediated CRISPR-Cas9 component loading. C. TEM analysis of modified CRISPR-Cas9 exosomes. D. Protein levels of GFP and CD63 in exosomes derived from cells untreated or treated with normal CRISPR-Cas9 exosomes or modified CRISPR-Cas9 exosomes. E. qRT-PCR analysis of sgRNA concentrations in vehicle exosomes, normal exosomes loaded with CRISPR-Cas9 components, or modified exosomes loaded with CRISPR-Cas9 components. F. Levels of sgRNA expression in vehicle exosomes, normal exosomes loaded with CRISPR-Cas9 components, or modified exosomes loaded with CRISPR-Cas9 components. G. Western blot analysis of Cas9 protein levels in vehicle exosomes, normal exosomes loaded with CRISPR-Cas9 components, or modified exosomes loaded with CRISPR-Cas9 components. H. qRT-PCR analysis of Cas9 mRNA levels in vehicle exosomes, normal exosomes loaded with CRISPR-Cas9 components, or modified exosomes loaded with CRISPR-Cas9 components. I. Immunoprecipitation assay of modified exosomes loaded with CRISPR-Cas9 components. Lysates of exosomes were blotted using FLAG antibody or CD63 antibody (input) or immunoprecipitated with FLAG or HA and blotted using FLAG antibody or HA antibody (IP with). IP with anti-IgG served as the control. J. An sgRNA analysis of immunoprecipitants following IP assay in H. n = 3. Error bars, mean ± s.d. | ||
Exosomal CRISPR-Cas9 components abrogate the target gene in recipient cells
To investigate whether the CRISPR-Cas9 components loaded in the modified CRISPR-Cas9 exosomes were functional in recipient cells, we generated a reporter cell line in which a stop-DsRed sequence was stably infected into the genome of A549 cells (A549stop-DsRed cells) (Fig. 5A). DsRed was not expressed due to a frameshift mutation caused by the stop sequence (49 nucleotides) (Fig. 5C). sgRNA1 and sgRNA2 targeting stop sequences were designed and cloned into lentiCRISPRv2 vectors as well as the Cas9-GFP nanobody sequence (plasmids 4 and 4*) (Fig. 5A and B). First, A549stop-DsRed cells were transiently co-transfected with plasmids 3, 4, and plasmid 4* (Fig. 5A and C) to assess the function of the recipient reporter cells and plasmids. Confocal images showed GFP was expressed in cells (Fig. S5A,† row 3, lane 2). Furthermore, A549stop-DsRed cells exhibited red fluorescent signals (Fig. S5A,† row 3, lane 3), demonstrating that the reporter cells and plasmids were set up successfully. To yield the modified CRISPR-Cas9 exosomes, plasmids 3, 4, and 4* were co-transfected into cells and the exosomes were harvested from the conditioned medium. After incubating A549stop-DsRed cells with CRISPR-Cas9 exosomes, a high level of sgRNAs was detected in recipient A549stop-DsRed cells (Fig. 5D). Modified CRISPR-Cas9 exosomes transferred a larger amount of sgRNAs into recipient cells than normal CRISPR-Cas9 exosomes (Fig. 5D). Because GFP was fused with exosomal membrane protein CD63, the expression of GFP determined in recipient cells confirmed the internalization of the exosomes (Fig. 5E, row 2). Modified CRISPR-Cas9 exosomes transferred a considerable amount of Cas9 proteins into recipient cells compared with normal CRISPR-Cas9 exosomes (Fig. 5E, row 1). Next, we investigated whether exosomal CRISPR-Cas9 components functioned in recipient cells by assessing the fluorescent signals with a confocal microscope. GFP fused on the membrane of exosomes was detected in recipient cells, visually confirming that the exosomes were internalized in recipient reporter cells (Fig. 5F, row 3, lane 2). Notably, recipient A549stop-DsRed cells treated with normal exosomes that transferred fewer CRISPR-Cas9 components into recipient cells exhibited weak red fluorescent signals (Fig. 5F, row 2, lane 3), whereas the cells treated with modified CRISPR-Cas9 exosomes exhibited detectable scattered red fluorescent signals (Fig. 5F, row 3, lane 3). Sanger sequencing confirmed that 37 nucleotides in the stop sequence were removed by sgRNA1/sgRNA2-guided Cas9 endonuclease (Fig. 5C and G). These data demonstrate that exosomal CRISPR-Cas9 elements functioned in recipient cells as an endogenous CRISPR-Cas9 system.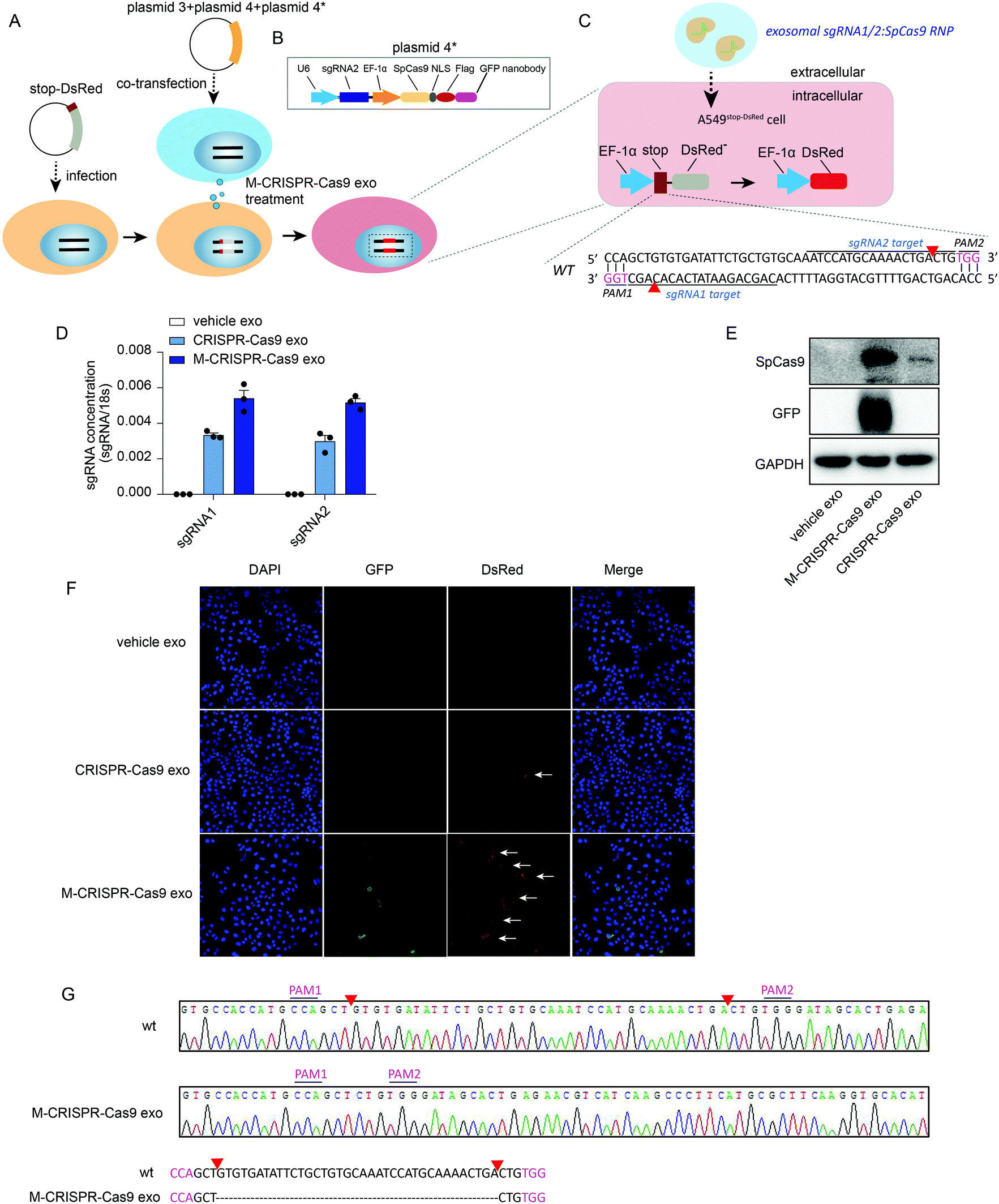 | ||
| Fig. 5 A. Schematic of the generation of A549stop-DsRed cells and exosome treatment. B. Schematic of LentiCRISPRV2-sgRNA2-Cas9 + FLAG-GFP nanobody (plasmid 4*) vector designs. C. Schematic of the mechanism underlying sgRNA-guided endonuclease genome editing in A549stop-DsRed cells. D. qRT-PCR analysis of sgRNA concentrations in cells treated with vehicle exosomes, normal exosomes loaded with CRISPR-Cas9 components, or modified exosomes loaded with CRISPR-Cas9 components. E. Western blot analysis of Cas9 protein levels in recipient cells treated with vehicle exosomes, normal exosomes loaded with CRISPR-Cas9 components, or modified exosomes loaded with CRISPR-Cas9 components. F. Confocal images showing red fluorescent expression in A549stop-DsRed cells treated with vehicle exosomes, normal exosomes loaded with CRISPR-Cas9 components, or modified exosomes loaded with CRISPR-Cas9 components. G. Sanger sequencing of A549stop-DsRed cells untreated or treated with modified exosomes loaded with CRISPR-Cas9 components. | ||
Discussion
In this study, we developed an engineered exosome for the more efficient delivery of CRISPR-Cas9 components. Exosomes offer great advantages as couriers for drug delivery.30 Exosomes are naturally occurring carriers that can deliver bio-information intercellularly; thus, they are more compatible with the immune system and exhibit little immunogenicity and toxicity.31 In addition, exosomes can be conveniently equipped with the target peptide, which may facilitate the tissue/cell-specific delivery or the crossing of biological barriers, such as the blood brain barrier.28 Furthermore, because exosomes are released from natural cells, they bear natural exosomal proteins, such as CD47, which protects them from being phagocytosed by monocytes and macrophages and makes them stable in blood.27Owing to their advantages, exosomes have been already combined with CRISPR-Cas9 delivery. Plasmids were loaded into cancer-derived exosomes and delivered into cancer cells32 and provided more effective delivery than epithelial cell-derived exosomes.32 However, plasmid DNA tends to integrate randomly into the host genome, which may result in constitutive expression of Cas9.33 This can be either beneficial or problematic for gene therapy. The manipulation of the CRISPR-Cas9 system in target cells is vital for the host's health after treatment. Thus, delivering CRISPR-Cas9 components instead of vectors may provide a better means of CRISPR-Cas9 system regulation in vivo. Other approaches for CRISPR-Cas9 delivery have also been studied. Most focus on the engineering of delivery carriers, including the generation of lipid-like nanoparticles18,34 or exosome-liposome hybrid nanoparticles,35 which still raise concerns about immunogenicity.
Small RNAs can be efficiently loaded into exosomes.23 However, there is little evidence to show that sufficient protein can be loaded into exosomes and function in recipient cells. In addition, loading mRNA of interest into exosomes always has challenges. Therefore, these problems need to be overcome in delivering Cas9. In our study, we confirmed that Cas9 protein can be loaded into exosomes instead of its mRNA. To improve the efficiency of Cas9 protein loading into exosomes, we used the GFP-GFP nanobody system. We confirmed that engineering the GFP-GFP nanobody system did not alter the function of the CRISPR-Cas9 system in recipient cells. Furthermore, the improved loading of the CRISPR-Cas9 components facilitated efficient genome editing presented in our A549Stop-DsRed reporter cells. By increasing the loading of Cas9 proteins into modified exosomes, the levels of sgRNA were also increased in exosomes, suggesting that part of CRISPR-Cas9 components may be packaged into exosomes as an intact functional unit, the sgRNA:Cas9 ribonucleoprotein complex, from the donor cells. However, according to our results and other studies, sgRNA and Cas9 also can be delivered separately and form the sgRNA:Cas9 ribonucleoprotein complex in exosomes or recipient cells.34
We should also ask why Cas9 mRNA cannot be packaged into exosomes. A special tactic is always needed to facilitate the packaging of mRNA. Cas9 mRNA engineered with AU-rich elements can be enriched into exosomes that bear RNA binding proteins.36 In addition, a ZIP-code-like 25 nucleotide sequence in the 3′-untranslated region of mRNA facilitates the enrichment of this mRNA in exosomes.37 The bio-cargoes of exosomes consist of miRNA, other non-coding RNA, mRNA, and cytoplasmic and membrane proteins. Importantly, owing to their highly regulated biogenesis, exosomes selectively encapsulate some defined components. RNA cargo sorting to generate exosomes shows a precise and unique biochemical composition.38 A study performed on MDA-MB-231 tumour cells revealed the enrichment of mRNA molecules in extracellular vesicles compared with that in donor cells, suggesting that, instead of a random package, there is a sorting mechanism that facilitates the selection and packaging of mRNA into exosomes.39 Several mechanisms have provided some insights into how cells regulate the biogenesis of exosomes and select cargo, although the precise mechanism remains unknown. Thus, the fact that SpCas9 mRNA cannot be sorted into exosomes may be because SpCas9 are from S. pyogenes, the mRNA of which cannot be recognized or selected into exosomes by mammal donor cells. Interestingly, Cas9 mRNA was enriched in our modified exosomes, although the mechanism was not clear. One putative explanation is that artificial engineering may change the sorting preference of exosomes. Further work is needed to explain the Cas9 mRNA sorting into GFP nanobody-modified exosomes.
Our study provides proof-of-principle of an alternative method to facilitate the delivery of the CRISPR-Cas9 system, which may help advance the clinical use of CRISPR-Cas9 technology.
Materials and methods
Cells, reagents and antibodies
The HEK 293T cell line was purchased from the Institute of Biochemistry and Cell Biology at the Shanghai Institute for Biological Science at the Chinese Academy of Science (Shanghai, China). HEK 293T cells were maintained in Dulbecco's modified Eagle's medium (Gibco, CA, USA) supplemented with 10% FBS (Gibco), 1% penicillin–streptomycin (Gibco). The adenocarcinomic human alveolar basal epithelial (A549) cell line was purchased from the Institute of Biochemistry and Cell Biology (Shanghai, China). The A549 cells were maintained in DMEM (Gibco) supplemented with 10% FBS, 100 units mL−1 penicillin, and 100 μg mL−1 streptomycin (Gibco). STR profiling and detection of mycoplasma contamination were performed to authenticate all cell lines. Antibodies raised against SpCas9, GAPDH, CD63, β-actin, and EGFP were purchased from Cell Signalling Technology (MA, USA); the FLAG antibody was purchased from Thermo Fisher Scientific (MA, USA); and the TSG101 antibody was purchased from Santa Cruz Biotechnology (CA, USA).Guide RNA design
Guide RNAs sequences were designed using the CRISPR Design Tool (http://zlab.bio/guide-design-resources). Guide RNAs sequences are listed in Table S1.†DNA constructs
The cDNA of sgRNA, SpCas9, and FLAG-tagged SpCas9 were cloned into lentiCRISPRv2 vectors (Addgene, MA, USA). The GFP nanobody was synthesized by Genescript (Nanjing, China) and cloned into the lentiCRISPRv2 vector (Addgene) using a BamHI restriction site. Human U6 promoter was used for driving the sgRNA transcription, and the EF-1α promoter was used for driving the expression of SpCas9, FLAG-tagged SpCas9, and SpCas9-GFP nanobody. HA-CD63 cDNA was amplified by PCR using primers for flanking, and the amplicons were cloned into the pEGFP-N1 vector (Addgene) between XhoI and BamHI restriction sites, with the resultant correct plasmid designated as pEGFP-N1-HA-CD63. The DsRed sequence was amplified using PCR from Rosa tdTomato mice (The Jackson Laboratory, ME, USA), and the stop element sequence was synthesized by Genescript. Stop element and DsRed (stop-DsRed) were finally cloned into the pSin-Puro vector (Addgene) using MLUI and SpeI restriction sites. The sequences of all the plasmids are listed in Table S1.†Lentivirus construction and stable cell lines
The plasmid expressing stop-DsRed was transfected into HEK 293T cells together with plasmids psPAX2 and pMD2G with Lipofectamine 2000 (Thermo Fisher Scientific). The three plasmids were mixed at a molar ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:1, dissolved in Opti-MEM, and incubated at room temperature for 15 min. Then, HEK 293T cells at 70–80% confluence were transfected. After 6 h, the medium was changed to medium supplemented with 2% FBS (Gibco). Lentivirus particles were collected from the medium supernatant after 48 h and filtered using a 0.22 μm filter. Virus particles were stored at −80 °C. An A549stop-DsRed reporter cell line was generated using a standard lentiviral transduction protocol. For viral infection, A549 cells were seeded in 6-well plates and incubated with stop-DsRed virus supplemented with 4 μg mL−1 polybrene (HANBIO, Shanghai, China). Afterwards, cells were cultured as a polyclonal population and kept under selection using 1 μg mL−1 puromycin (Sangon Biotech, Shanghai, China).
:5:1, dissolved in Opti-MEM, and incubated at room temperature for 15 min. Then, HEK 293T cells at 70–80% confluence were transfected. After 6 h, the medium was changed to medium supplemented with 2% FBS (Gibco). Lentivirus particles were collected from the medium supernatant after 48 h and filtered using a 0.22 μm filter. Virus particles were stored at −80 °C. An A549stop-DsRed reporter cell line was generated using a standard lentiviral transduction protocol. For viral infection, A549 cells were seeded in 6-well plates and incubated with stop-DsRed virus supplemented with 4 μg mL−1 polybrene (HANBIO, Shanghai, China). Afterwards, cells were cultured as a polyclonal population and kept under selection using 1 μg mL−1 puromycin (Sangon Biotech, Shanghai, China).
DNA construction transfection
Cells were seeded in 60 cm2 dishes overnight and transfected the next day using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's instructions. Cells and exosomes were harvested 24 h after transfection for the following assays.Ultracentrifugation exosomes isolation
Exosomes were isolated and purified from the cell supernatant as described previously. Briefly, cells were cultured in the medium containing exosome-depleted FBS. The culture medium was collected from an equivalent number of cells 24 h after cell culture or transfection. Exosomes were isolated by the following sequential centrifugation steps: 5 min at 300g to remove the cells; 30 min at 3000g to remove cell debris; 30 min at 10000g to remove larger microvesicles; and ultracentrifugation at 120000g for 70 min using a rotor (70 Ti, Beckman, Bremen, Germany). The pellets were resuspended in a convenient volume of phosphate-buffered saline (PBS). Exosomes were quantified by protein level using the BCA method and kept at −80 °C for long-term storage.
Exosome isolation using the total exosomes isolation kit
The cell culture medium was harvested and centrifuged at 2000g for 30 min to remove the cells and debris. The cell-free medium was transferred to a new tube and 0.5 volumes of the Total Exosomes Isolation reagent (Thermo Fisher Scientific) was added. The solution was mixed by vortexing or pipetting until it was homogenous. After incubating overnight at 4 °C, the homogenous solution was centrifuged at 10000g for 1 h at 4 °C. The pellets were resuspended in a convenient volume of PBS and either used or stored at −80 °C.
NTA
The distribution of nanoparticle sizes was analysed by NTA (NanoSight, Malvern Panalytical, UK). Samples were manually injected into the sample chamber at ambient temperature. Each sample was measured in triplicate at camera setting 10 with acquisition time of 20 s and a detection threshold setting of 7. At least 200 completed tracks were analysed per video. NTA analytical software version 3.2 was used for capturing and analysing data.Fluorescent imaging analysis
A549stop-DsRed cells treated with control exosomes or modified CRISPR-Cas9 exosomes were analysed. After treatment, cells were rinsed twice with PBS and seeded onto glass coverslips in 12-well plates. The cells were fixed with 4% paraformaldehyde at room temperature for 15 min and stained with Hoechst 33342 (Thermo Fisher Scientific) at room temperature for 15 min. Slides were imaged using a confocal imaging system (TCS SP8-MaiTai M, Leica, Wetzler, Germany) and images processed using TCS SP8 software (Leica). Results are from three independent experiments.RNA immunoprecipitation assay
Cells and exosomes were lysed using lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 0.5% Nonidet P-40, 2 mM EDTA, 0.5 mM DTT, 1 mM NaF, 1 mM PMSF, 1% protease inhibitor cocktail, and 10 U mL−1 RNase inhibitor) for 30 min on ice, followed by centrifugation at 16000g for 10 min at 4 °C. Supernatants were incubated with anti-FLAG antibody, anti-HA antibody, or IgG at 4 °C overnight. Then, supernatants were incubated with 40 μL Protein A/G PLUS-Agarose beads (SantaCruz Biotechnology) for 2 h at 4 °C. Beads were collected by centrifugation at 2500 rpm for 5 min at 4 °C. Immunoprecipitants were eluted from the beads using lysis buffer, and treated with RIPA for western blot analysis or TRIzol reagent (TaKaRa, Dalian, China) for RNA analysis.
Sanger sequencing
PCR was performed in a 20 μL reaction mixture consisting of 1 μL template DNA, 1 μL forward primer (10 μM), 1 μL reverse primer (10 μM), 10 μL SuperStar High-Fidelity Polymerase (TSINGKE, Nanjing, China), and 7 μL ddH2O. Amplification was carried out with the following programme: 98 °C for 60 s; 35 cycles of 98 °C for 10 s; 58 °C for 15 s; 72 °C for 15 s; hold at 4 °C. Agarose gel (2%) was used for separating DNA fragments. The PCR products of expected length were cloned into pClone Blunt plasmids (TSINGKE) and sequenced using primer M13F.RNA extraction and qRT-PCR
Total RNA was harvested from cells or exosomes using TRIzol Reagent (TaKaRa) according to the manufacturer's instructions. qRT-PCR was performed using optimized primers. Briefly, cDNA was synthesized from 5 μL of total RNA using AMV reverse transcriptase (TaKaRa). qRT-PCR was then performed on a sequence detection system (7300, Applied Biosystems, CA, USA). All reactions were run in triplicate. Ct values were determined using fixed threshold settings. mRNA gene expression was normalized to 18s RNA. To calculate the absolute expression levels of sgRNA, a series of synthetic sgRNAs with known concentration was also reverse-transcribed and amplified. The absolute amount of sgRNA was then calculated referring to the standard curve. The expressions of the sgRNA in the cells were normalized to the expression of 18s RNAWestern blot assay
Total protein from the cells or exosomes was extracted using RIPA lysis buffer (Beyotime, Shanghai China). Protein concentrations were determined with a bicinchoninic acid protein assay kit (Thermo Fisher Scientific). Lysates were separated on SDS-PAGE (10%) gel and transferred to polyvinylidene fluoride membranes. After blocking, membranes were incubated with primary antibodies overnight at 4 °C, followed by incubation with peroxidase-conjugated secondary antibody at room temperature for 1 h. Protein expression was determined using an enhanced chemiluminescence western blot kit (Supersignal West Pico, Thermo Fisher Scientific). Normalisations were performed after blotting the same samples with an antibody against β-actin or GADPH.Statistical analysis
Data are expressed as mean ± SD. Technical and biological triplicates of each experiment were performed. Student's t-test was used for comparing two groups, and one-way ANOVA was used to compare the differences among three or more groups (GraphPad Prism 8.0). P < 0.05 was considered statistically significant.Author contributions
Yangyang Ye conducted all the experiments with assistance from the other authors. Xiang Zhang designed the plasmids and assisted in the establishing the exosome engineering method. Fei Xie, Bin Xu, Ping Xie, Ting Yang, and Qian Shi performed the cell culturing and plasmid generation. Yujing Zhang conducted the TEM analysis. Jiangning Chen and Chen-Yu Zhang oversaw the experiments. Jing Li conceived the study. Jing Li and Xiaohong Jiang wrote the manuscript with contributions from Yangyang Ye and Fei Xie.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is supported by grants from National Natural Science Foundation of China (31741066, 31771666, 31972912) and the Fundamental Research Funds for the Central Universities (020814380094, 020814380087, 020814380137).References
- D. B. Cox, R. J. Platt and F. Zhang, Therapeutic genome editing: prospects and challenges, Nat. Med., 2015, 21, 121–131, DOI:10.1038/nm.3793.
- A. Bolotin, B. Quinquis, A. Sorokin and S. D. Ehrlich, Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin, Microbiology, 2005, 151, 2551–2561, DOI:10.1099/mic.0.28048-0.
- F. J. Mojica, C. Diez-Villasenor, J. Garcia-Martinez and E. Soria, Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements, J. Mol. Evol., 2005, 60, 174–182, DOI:10.1007/s00239-004-0046-3.
- C. Pourcel, G. Salvignol and G. Vergnaud, CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies, Microbiology, 2005, 151, 653–663, DOI:10.1099/mic.0.27437-0.
- M. Jinek, et al., A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity, Science, 2012, 337, 816–821, DOI:10.1126/science.1225829.
- S. W. Cho, S. Kim, J. M. Kim and J. S. Kim, Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease, Nat. Biotechnol., 2013, 31, 230–232, DOI:10.1038/nbt.2507.
- L. Cong, et al., Multiplex genome engineering using CRISPR/Cas systems, Science, 2013, 339, 819–823, DOI:10.1126/science.1231143.
- K. S. Makarova, N. V. Grishin, S. A. Shabalina, Y. I. Wolf and E. V. Koonin, A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action, Biol. Direct, 2006, 1, 7, DOI:10.1186/1745-6150-1-7.
- P. D. Hsu, E. S. Lander and F. Zhang, Development and applications of CRISPR-Cas9 for genome engineering, Cell, 2014, 157, 1262–1278, DOI:10.1016/j.cell.2014.05.010.
- S. H. Sternberg, S. Redding, M. Jinek, E. C. Greene and J. A. Doudna, DNA interrogation by the CRISPR RNA-guided endonuclease Cas9, Nature, 2014, 507, 62–67, DOI:10.1038/nature13011.
- H. Nishimasu, et al., Crystal structure of Cas9 in complex with guide RNA and target DNA, Cell, 2014, 156, 935–949, DOI:10.1016/j.cell.2014.02.001.
- M. Bibikova, M. Golic, K. G. Golic and D. Carroll, Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases, Genetics, 2002, 161, 1169–1175 CAS.
- M. Bibikova, et al., Stimulation of homologous recombination through targeted cleavage by chimeric nucleases, Mol. Cell Biol., 2001, 21, 289–297, DOI:10.1128/MCB.21.1.289-297.2001.
- M. Bibikova, K. Beumer, J. K. Trautman and D. Carroll, Enhancing gene targeting with designed zinc finger nucleases, Science, 2003, 300, 764, DOI:10.1126/science.1079512.
- R. J. Platt, et al., CRISPR-Cas9 knockin mice for genome editing and cancer modeling, Cell, 2014, 159, 440–455, DOI:10.1016/j.cell.2014.09.014.
- S. Demirci, A. Leonard, J. J. Haro-Mora, N. Uchida and J. F. Tisdale, CRISPR/Cas9 for Sickle Cell Disease: Applications, Future Possibilities, and Challenges, Adv. Exp. Med. Biol., 2019, 1144, 37–52, DOI:10.1007/5584_2018_331.
- S. Yao, Z. He and C. Chen, CRISPR/Cas9-Mediated Genome Editing of Epigenetic Factors for Cancer Therapy, Hum. Gene Ther., 2015, 26, 463–471, DOI:10.1089/hum.2015.067.
- C. Jiang, et al., A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo, Cell Res., 2017, 27, 440–443, DOI:10.1038/cr.2017.16.
- Z. Wu, H. Yang and P. Colosi, Effect of genome size on AAV vector packaging, Mol. Ther., 2010, 18, 80–86, DOI:10.1038/mt.2009.255.
- F. A. Ran, et al., In vivo genome editing using Staphylococcus aureus Cas9, Nature, 2015, 520, 186–191, DOI:10.1038/nature14299.
- R. J. Samulski and N. Muzyczka, AAV-Mediated Gene Therapy for Research and Therapeutic Purposes, Annu. Rev. Virol., 2014, 1, 427–451, DOI:10.1146/annurev-virology-031413-085355.
- D. R. Deyle and D. W. Russell, Adeno-associated virus vector integration, Curr. Opin. Mol. Ther., 2009, 11, 442–447 CAS.
- X. Chen, H. Liang, J. Zhang, K. Zen and C. Y. Zhang, Secreted microRNAs: a new form of intercellular communication, Trends Cell Biol., 2012, 22, 125–132, DOI:10.1016/j.tcb.2011.12.001.
- G. Turturici, R. Tinnirello, G. Sconzo and F. Geraci, Extracellular membrane vesicles as a mechanism of cell-to-cell communication: advantages and disadvantages. American journal of physiology, Cell Physiol., 2014, 306, C621–C633, DOI:10.1152/ajpcell.00228.2013.
- Y. Zhang, et al., Secreted monocytic miR-150 enhances targeted endothelial cell migration, Mol. Cell, 2010, 39, 133–144, DOI:10.1016/j.molcel.2010.06.010.
- G. van Niel, G. D'Angelo and G. Raposo, Shedding light on the cell biology of extracellular vesicles., Nat. Rev. Mol. Cell Biol., 2018, 19, 213–228, DOI:10.1038/nrm.2017.125.
- S. Kamerkar, et al., Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer, Nature, 2017, 546, 498–503, DOI:10.1038/nature22341.
- Y. Liu, et al., Targeted exosome-mediated delivery of opioid receptor Mu siRNA for the treatment of morphine relapse, Sci. Rep., 2015, 5, 17543, DOI:10.1038/srep17543.
- M. H. Kubala, O. Kovtun, K. Alexandrov and B. M. Collins, Structural and thermodynamic analysis of the GFP:GFP-nanobody complex, Protein Sci., 2010, 19, 2389–2401, DOI:10.1002/pro.519.
- P. Vader, E. A. Mol, G. Pasterkamp and R. M. Schiffelers, Extracellular vesicles for drug delivery, Adv. Drug Delivery Rev., 2016, 106, 148–156, DOI:10.1016/j.addr.2016.02.006.
- F. Villa, R. Quarto and R. Tasso, Extracellular Vesicles as Natural, Safe and Efficient Drug Delivery Systems, Pharmaceutics, 2019, 11, 557, DOI:10.3390/pharmaceutics11110557.
- S. M. Kim, et al., Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting, J. Controlled Release, 2017, 266, 8–16, DOI:10.1016/j.jconrel.2017.09.013.
- H. Yang, et al., One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering, Cell, 2013, 154, 1370–1379, DOI:10.1016/j.cell.2013.08.022.
- H. Yin, et al., Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo, Nat. Biotechnol., 2016, 34, 328–333, DOI:10.1038/nbt.3471.
- Y. Lin, et al., Exosome-Liposome Hybrid Nanoparticles Deliver CRISPR/Cas9 System in MSCs, Adv. Sci., 2018, 5, 1700611, DOI:10.1002/advs.201700611.
- Z. Li, et al., In Vitro and in Vivo RNA Inhibition by CD9-HuR Functionalized Exosomes Encapsulated with miRNA or CRISPR/dCas9, Nano Lett., 2019, 19, 19–28, DOI:10.1021/acs.nanolett.8b02689.
- M. F. Bolukbasi, et al., miR-1289 and “Zipcode”-like Sequence Enrich mRNAs in Microvesicles, Mol. Ther. – Nucleic Acids, 2012, 1, e10, DOI:10.1038/mtna.2011.2.
- C. Thery, L. Zitvogel and S. Amigorena, Exosomes: composition, biogenesis and function, Nat. Rev. Immunol., 2002, 2, 569–579, DOI:10.1038/nri855.
- A. Zomer, et al., In Vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior, Cell, 2015, 161, 1046–1057, DOI:10.1016/j.cell.2015.04.042.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm00427h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |