
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoactivated cell-killing involving a low molecular weight, donor–acceptor diphenylacetylene†
David R.
Chisholm
a,
Rebecca
Lamb
b,
Tommy
Pallett
bc,
Valerie
Affleck
d,
Claire
Holden
ab,
Joanne
Marrison
e,
Peter
O'Toole
e,
Peter D.
Ashton
e,
Katherine
Newling
e,
Andreas
Steffen
f,
Amanda K.
Nelson
f,
Christoph
Mahler
f,
Roy
Valentine
g,
Thomas S.
Blacker
h,
Angus J.
Bain
 h,
John
Girkin
*c,
Todd B.
Marder
*f,
Andrew
Whiting
*a and
Carrie A.
Ambler
*b
h,
John
Girkin
*c,
Todd B.
Marder
*f,
Andrew
Whiting
*a and
Carrie A.
Ambler
*b
aDepartment of Chemistry, Durham University, Science Laboratories, South Road, Durham DH1 3LE, UK. E-mail: andy.whiting@durham.ac.uk
bDepartment of Biosciences, Durham University, South Road, Durham, DH1 3LE, UK
cBiophysical Sciences Institute, Department of Physics, Durham University, South Road, Durham, DH1 3LE, UK
dLightOx Limited, Wynyard Park House, Wynyard Avenue, Wynyard, Billingham, TS22 5TB, UK
eBioscience Technology Facility, Department of Biology, University of York, York, YO10 5DD, UK
fInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
gHigh Force Research Ltd., Bowburn North Industrial Estate, Bowburn, Durham, DH6 5PF, UK
hDepartment of Physics & Astronomy, University College London, Gower Street, London, WC1E 6BT, UK
First published on 21st March 2019
Abstract
Photoactivation of photosensitisers can be utilised to elicit the production of ROS, for potential therapeutic applications, including the destruction of diseased tissues and tumours. A novel class of photosensitiser, exemplified by DC324, has been designed possessing a modular, low molecular weight and ‘drug-like’ structure which is bioavailable and can be photoactivated by UV-A/405 nm or corresponding two-photon absorption of near-IR (800 nm) light, resulting in powerful cytotoxic activity, ostensibly through the production of ROS in a cellular environment. A variety of in vitro cellular assays confirmed ROS formation and in vivo cytotoxic activity was exemplified via irradiation and subsequent targeted destruction of specific areas of a zebrafish embryo.
1 Introduction
The generation and modulation of reactive oxygen species (ROS) is of huge importance in the control, maintenance, defence and death of both eukaryotic and prokaryotic cells.1,2 Indeed, ROS are produced by a number of biochemical processes in order to modulate cellular behaviour; however, they can also be generated by the action of the excited states of chemical compounds known as photosensitisers formed through absorption of light.3 Light activation of a photosensitiser typically involves photoexcitation from the ground state (S0) to a singlet excited state (S1), which can then undergo intersystem-crossing to a triplet excited state (T1).4,5 ROS formation can then occur via two pathways, type I and type II. A type I process can occur either from the S1 or the T1 state of the photosensitiser and involves either hydrogen or electron transfer between the excited photosensitiser and a substrate. The resulting radicals can react with molecular oxygen (3O2), typically producing ROS species such as superoxide (O2˙−) and hydroxyl radicals (OH˙). A type II process involves direct energy transfer from the longer-lived T1 state of the excited photosensitiser to 3O2, generating singlet oxygen (1O2).6In a cellular context, these oxygen-derived species elicit a variety of modulatory effects depending on the rate and extent of their production; at high concentrations apoptosis is observed, while at low concentrations a stimulatory response is often evident.7–9 Both pathways hold considerable therapeutic potential, but the majority of photosensitisers in the clinic are used for photodynamic therapy (PDT), in which a photosensitiser is excited near/inside a particular target tissue or condition (e.g. microbial infections, neoplasias, tumors, etc.), causing the generation of large quantities of ROS and subsequent destruction of that tissue.3,10 Photosensitisers clinically approved for PDT of various cancers include Photofrin® (Fig. 1) and 5-aminolaevulinic acid, which is metabolized to the active photosensitiser, protoporphyrin IX.11–13
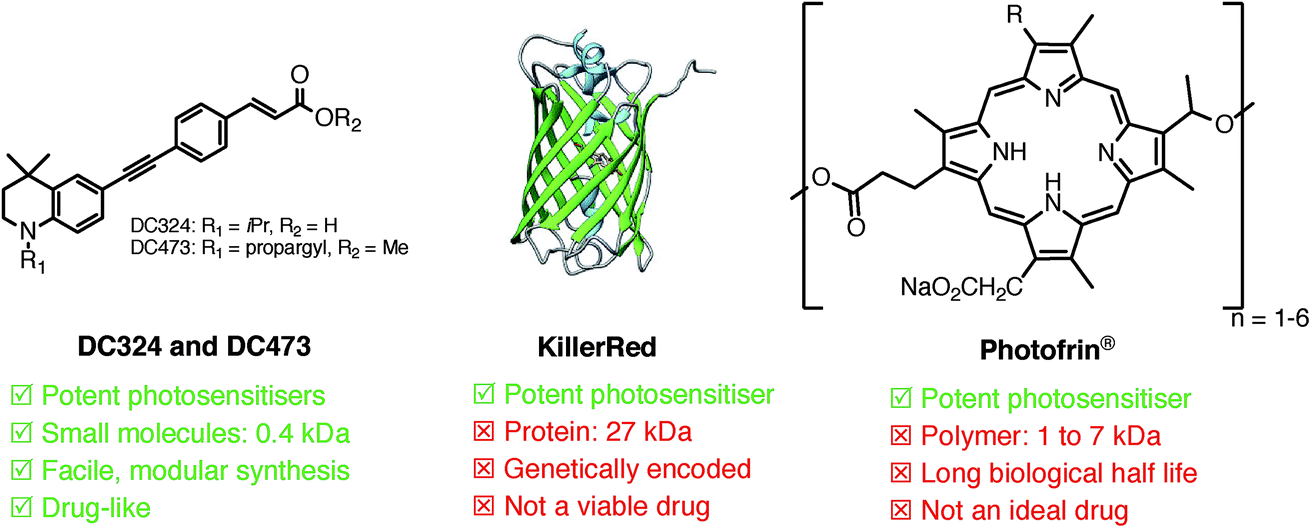 | ||
| Fig. 1 Comparison of the characteristics of DC324 and DC473 with those of existing photosensitisers, KillerRed and Photofrin®. | ||
However, these compounds exhibit inadequate pharmacological properties including poor dosage control due to the requirement for metabolic processing, and extremely long biological half-lives, which causes skin photosensitivity for weeks after treatment.3 Indeed, the vast majority of recently reported photosensitizers are high molecular weight, hydrophobic, often metal-containing porphyrin, chlorin and phthalocyanine scaffolds.14 The large size of these molecules has a significant and negative effect on cell permeability and diffusion rate into and out of the cell, and non-specific cell death is another major problem due to both the inability to target the compounds locally and issues with accumulation in off-target tissues and organs.3,15,16 Therefore, the inherent properties of these structures make them efficient photosensitisers, but far from ideal as therapeutics. A genetically encoded variant of green fluorescent protein (GFP), KillerRed (Fig. 1), is also a potent photosensitiser; however, the requirement for genetic modification probably makes KillerRed unviable for clinical PDT, though the method has great potential for biological studies.17 Due to these drawbacks, fine control of the release of ROS in a therapeutic context is not possible with current photosensitisers. Consequently, there is a major need for novel compounds and ROS generation mechanisms that are able to elicit an entirely controlled either destructive or proliferative cellular response based on the requirements of the disease, and indeed, the temporal requirements of clinical treatment. In this paper, we report a new class of low molecular weight, ‘drug-like’ compounds which exhibit the potential to elicit such controlled cytotoxic activity.
2 Results and discussion
2.1 Synthesis
Inspired by our work on the design, synthesis and applications of synthetic retinoids for controlling cellular development,18–25 we recently designed fluorescent analogues of such compounds for use in fluorescence-based biophysical studies.26,27 It is known that the addition of a strong π-donor (–NR2) and strong π-acceptor (–CO2R) to a diphenylacetylene scaffold results in efficient charge transfer from the donor moiety to the acceptor moiety upon photoexcitation.28–33 The resulting structures exhibit strong, solvatochromatic fluorescence with significant bathochromic shifts in absorption and emission spectra in polar media due to the efficient formation of intramolecular charge transfer (ICT) excited states, and we were interested in synthesising a range of such compounds that incorporated highly lipophilic and electron rich tetrahydroquinoline donor structures34,35 for use in cellular imaging studies. One such compound, DC324, was synthesised by the coupling of 6-iodo-tetrahydroquinoline (reported previously34) 5, with acceptor alkyne 4 (Fig. 2 and 3).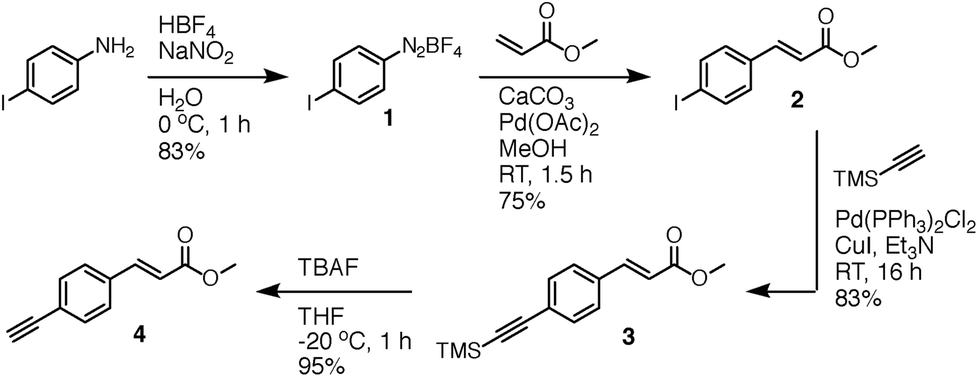 | ||
| Fig. 2 Synthesis of acceptor alkyne 4. | ||
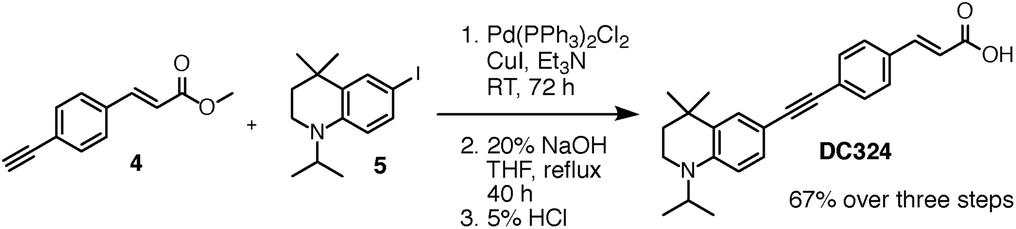 | ||
| Fig. 3 Coupling of acceptor alkyne 4 and donor tetrahydroquinoline 5 (ref. 34) to give DC324. | ||
Compound 4 was synthesised via a four-step approach, beginning with diazotisation of 4-iodoaniline to give the bench-stable diazonium tetrafluoroborate salt 1. This was suitably reactive under Heck–Matsuda conditions,36 enabling effective conversion to the desired cinnamate 2 in 75% yield with complete E-selectivity (crystal structure shown in the ESI†). Sonogashira coupling with trimethylsilylacetylene gave the protected alkyne 3,37 which was initially deprotected using K2CO3 in MeOH/DCM to give the target acceptor alkyne 4. While these conditions did remove the TMS protecting group, it was also found that the presence of traces of EtOH in the commercial grade MeOH used for the reaction caused efficient transesterification to the corresponding ethyl ester. To circumvent this, deprotection using TBAF in THF at −20 °C cleanly provided 4 with the methyl ester intact. Sonogashira coupling of acceptor 4 with donor 5 proceeded with complete conversion to the desired coupled product which, upon saponification, gave the target compound DC324 in a 67% yield over the three steps. We have also recently reported the synthesis, fluorescence properties and cellular localisation behaviour of DC473 (Fig. 1), a close analogue of DC324 with a methyl cinnamate acceptor group and N-propargyl tetrahydroquinoline donor which can be utilised for dual fluorescence/Raman imaging in cells.38
2.2 Photophysical characterisation
Photophysical and computational studies showed that DC324 exhibits two strong absorptions (Table 1, Fig. 4A) with extinction coefficients of ca. 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–41000 M−1 cm−1 in the UV and violet regions, respectively, both arising from the formation of ICT excited states. Consequently, a solvatochromatic absorption was observed, wherein polar solvents give rise to a hypsochromic shift, which indicates that the ground state S0 is more polar than the Franck–Condon S1 excited state. However, even more impressive is the highly solvatochromatic fluorescence (Table 1, Fig. 4B), with quantum yields of up to 0.92 in non-polar solvents such as toluene,28,29 and an emission lifetime of 2.0 ns. However, in more polar solvents, such as EtOH, DMSO or CH2Cl2, a second excited state process occurs, giving rise to the reproducible observation of a secondary decay starting around 500 ps after the laser pulse, which was observed on several spectrophotometers and is not an artifact of a particular instrument. DC324 shows similar photophysical properties when compared to DC473 (ref. 38) although DC324 exhibits bathochromic shifts in absorption and emission in non-polar solvents, presumably due to the increased donor strength of the N-iPr moiety in comparison to the N-propargyl group of DC473. We also measured the two-photon absorption (TPA) spectra of DC324 and DC473 in toluene (Fig. 4C), and note the TPA cross-sections of around 500 GM at 790 nm which are impressive for such a small dipolar molecule. Comparing with, for example, a simple dimethylamino donor/dimesitylboryl acceptor-substituted diphenylacetylene,39 which has similar absorption properties in toluene, the emission spectra of DC324 and DC473 are somewhat red-shifted, and the TPA cross-section is ca. 2.5 times higher for the latter two compounds. This can likely be attributed to a combination of the increased donor strength of the rigidified tetrahydroquinoline-based moiety and the extension of the conjugation length via the alkene unit in DC324 and DC473. The TPA cross-section is the typical measure of the efficiency with which a compound absorbs two photons; for one-photon absorption, the efficiency of absorption is typically reported as an extinction coefficient. In a cellular and biological imaging context, a greater TPA cross-section allows lower laser powers to be used to excite larger numbers of molecules, thus reducing the risk of thermal damage to the tissue and also enables deeper penetration in more scattering tissue samples.
000–41000 M−1 cm−1 in the UV and violet regions, respectively, both arising from the formation of ICT excited states. Consequently, a solvatochromatic absorption was observed, wherein polar solvents give rise to a hypsochromic shift, which indicates that the ground state S0 is more polar than the Franck–Condon S1 excited state. However, even more impressive is the highly solvatochromatic fluorescence (Table 1, Fig. 4B), with quantum yields of up to 0.92 in non-polar solvents such as toluene,28,29 and an emission lifetime of 2.0 ns. However, in more polar solvents, such as EtOH, DMSO or CH2Cl2, a second excited state process occurs, giving rise to the reproducible observation of a secondary decay starting around 500 ps after the laser pulse, which was observed on several spectrophotometers and is not an artifact of a particular instrument. DC324 shows similar photophysical properties when compared to DC473 (ref. 38) although DC324 exhibits bathochromic shifts in absorption and emission in non-polar solvents, presumably due to the increased donor strength of the N-iPr moiety in comparison to the N-propargyl group of DC473. We also measured the two-photon absorption (TPA) spectra of DC324 and DC473 in toluene (Fig. 4C), and note the TPA cross-sections of around 500 GM at 790 nm which are impressive for such a small dipolar molecule. Comparing with, for example, a simple dimethylamino donor/dimesitylboryl acceptor-substituted diphenylacetylene,39 which has similar absorption properties in toluene, the emission spectra of DC324 and DC473 are somewhat red-shifted, and the TPA cross-section is ca. 2.5 times higher for the latter two compounds. This can likely be attributed to a combination of the increased donor strength of the rigidified tetrahydroquinoline-based moiety and the extension of the conjugation length via the alkene unit in DC324 and DC473. The TPA cross-section is the typical measure of the efficiency with which a compound absorbs two photons; for one-photon absorption, the efficiency of absorption is typically reported as an extinction coefficient. In a cellular and biological imaging context, a greater TPA cross-section allows lower laser powers to be used to excite larger numbers of molecules, thus reducing the risk of thermal damage to the tissue and also enables deeper penetration in more scattering tissue samples.
| Solvent | λ abs(max)/nm, (ε/M−1 cm−1) | λ em(max)/nm | ϕ | τ/ns |
|---|---|---|---|---|
| MeCy | 394, 296 | 434, 460 | — | — |
| Toluene | 395(30600), 297(27700) |
498 | 0.92 | 2.0 |
| MeCN | 385(31200), 295(28000) |
513 | 0.04 | — |
| EtOH | 366(41100), 294(30800) |
533 | 0.10 | — |
| DMSO | 380(32500), 297(32200) |
512 | 0.30 | — |
| H2O | 385, 298 | 562 | — | — |
| CH2Cl2 | 403, 301 | 622 | — | — |
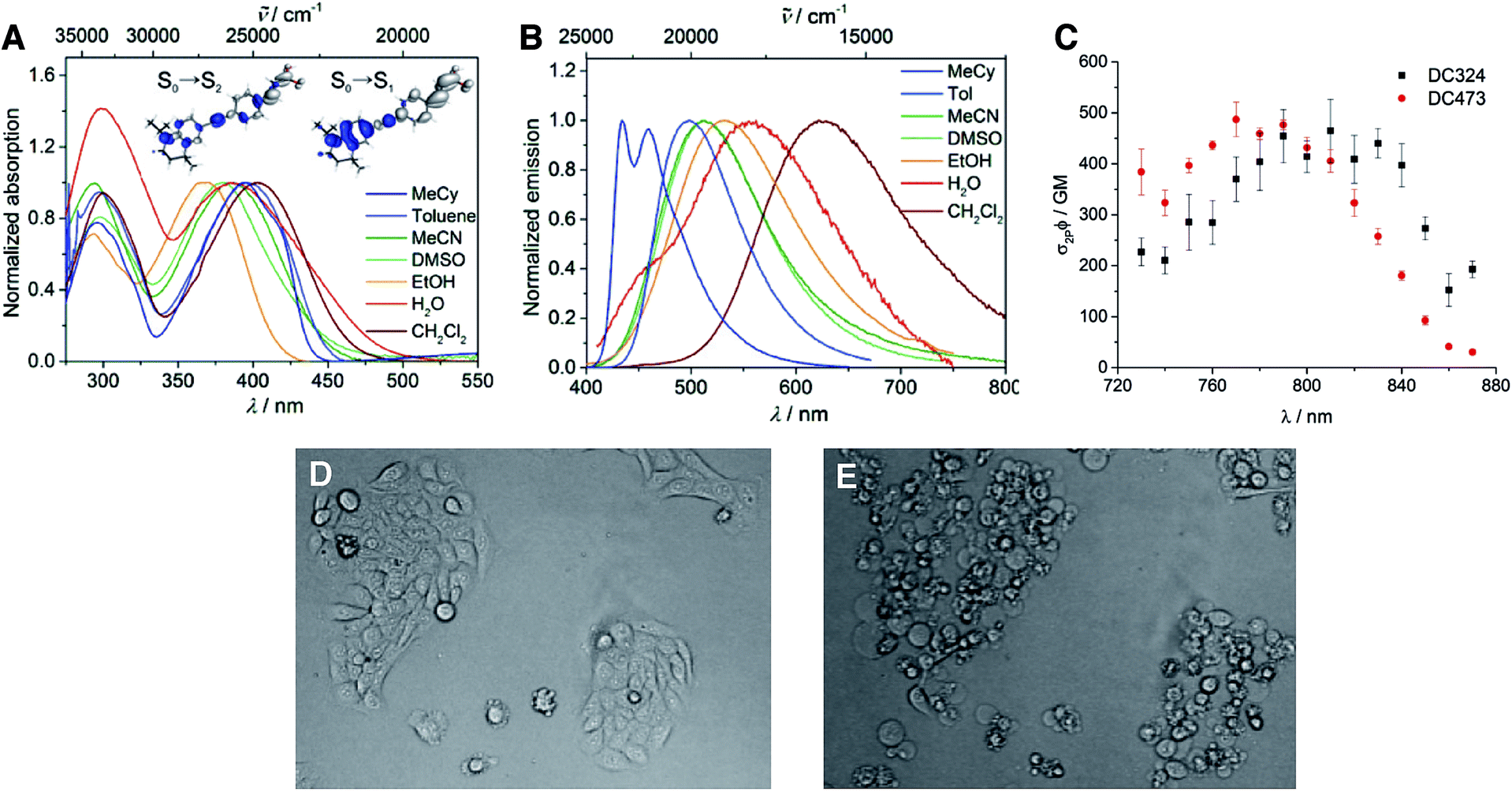 | ||
| Fig. 4 (A) Absorption spectra of DC324 in a variety of solvents and electron density difference plots for the two observed major absorption bands S0 → S1 (low energy) and S0 → S2 (high energy) obtained from TD-DFT calculations at the M06-D3-BJ/def2-TZVP level of theory. (B) Emission spectra of DC324 in a variety of solvents. (C) Two-photon absorption spectra of DC324 and DC473 in toluene. (D) HaCaT keratinocytes treated with 10 μM DC324 for 30 min, prior to irradiation. (E) HaCaT keratinocytes treated with 10 μM DC324 after multiple short irradiations with 405 nm, 12 hours after initial imaging. | ||
The most interesting finding, however, was that when DC324 and DC473 were irradiated with UV-A (350–390 nm) light in cultured epithelial cells, rapid cell death was observed that was, possibly, consistent with the production of large amounts of ROS (Fig. 4D/E).
2.3 In vitro characterisation
To investigate this observed cell death we performed a range of cellular and cuvette-based experiments to understand localisation and mode of action. According to confocal fluorescence microscopy, DC324 was found to exhibit non-specific localisation in non-polar, membrane-rich environments, including mitochondria (Pearson's correlation with MitoTracker Red, R = 0.81) and other organelles (Fig. 5A).38 This finding was supported by the fact that the emission peaks (λex = 405 nm) detected at 460–490 nm were similar to cuvette-based measurements in non-polar solvents such as toluene or methylcyclohexane. However, the observed emission wavelength varied subtly (Fig. 5B) according to the cellular environment, as a result of the solvatochromatic behaviour of the compound.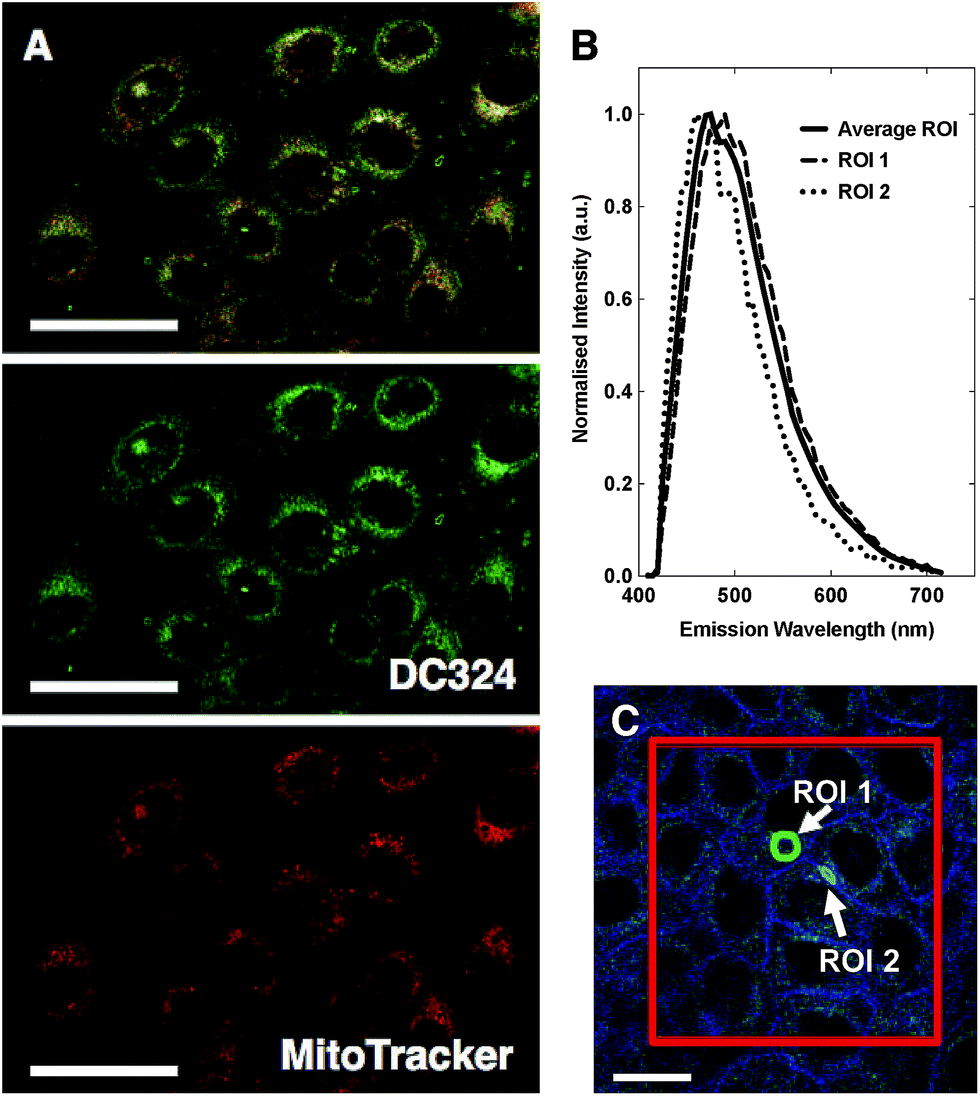 | ||
| Fig. 5 (A) HaCaT keratinocytes treated with 1 μM DC324 (green) and co-stained with MitoTracker Red (red). Scale bars equal 50 μm. (B) Peak emission wavelength of DC324 in regions of interest (ROI) of cells, with excitation at 405 nm. (C) ROI 1, green circle marks area distal to nuclear membrane (λmax = 490 nm) and ROI 2, blue circle, marks area adjacent to nucleus (λmax = 460 nm). Average emission wavelength for red square was calculated (Average ROI λmax = 475 nm). | ||
We next utilised the redox reactive dye CellRox,40 which fluoresces in response to oxidation by ROS, to determine whether ROS is produced either by, or in response to, in cellulo photoactivation of DC324. Whereas in the absence of the compound no CellRox fluorescence (and no cell death) was observed, DC324-treated cells subsequently activated with 405 nm light exhibited a strong CellRox fluorescence signal with a steady increase immediately following irradiation, particularly in intracellular organelles (Fig. 6). Similar behaviour was observed in DC473-treated cells and, therefore, this initially suggested that the compounds may act as photosensitisers to elicit ROS formation in cells.
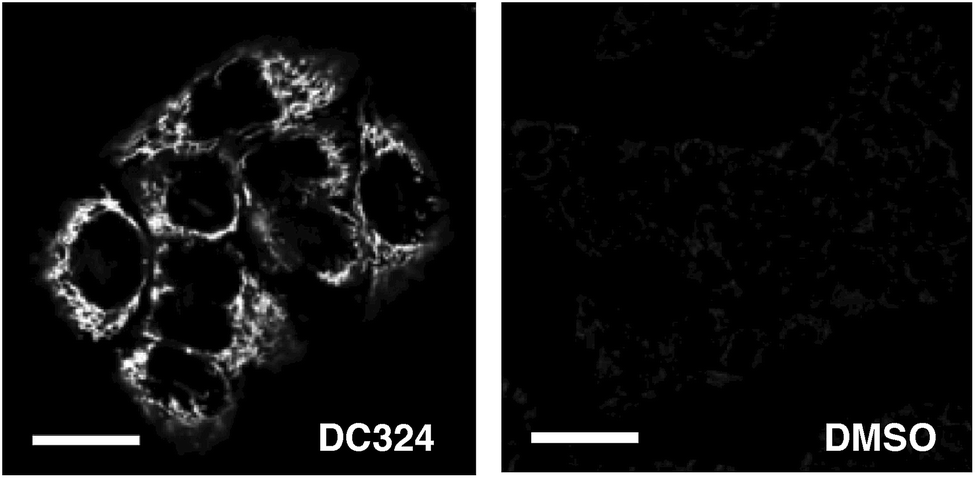 | ||
| Fig. 6 Detection of fluorescence produced by ROS reactive dye, CellRox in HaCaT keratinocytes treated with 1 μM DC324 or a DMSO control. Scale bars equal 25 μm. | ||
Because of the structural relationship with retinoids such as all-trans-retinoic acid (ATRA) and other synthetic retinoids,20 we were interested to determine whether retinoids, in general, elicit ROS production when cells are treated with light. We, therefore, chose EC23 (Fig. 7A),18 an analogue of DC324 and DC473 with retinoid biochemical activity but no absorption or photoactivity at 405 nm (since it lacks the amine donor functionality), to act as a negative control. In these experiments, treatment of cells with EC23 and subsequent irradiation did not cause activation of CellRox nor any observable cell death (Fig. 7B). It was, therefore, clear that the photophysical properties of DC324 and DC473, i.e. the ability to absorb light of 405 nm due to the addition of strong π-donor and extended π-acceptor moieties, were mandatory to initiate cell death upon photoactivation rather than an inherent biochemical or biological signalling activity of a diphenylacetylene retinoid or retinoid-like structure. Furthermore, we have previously shown that retinoid signalling activity can be completely eliminated when the compound is of a length greater than EC23/ATRA and, hence, given that DC324 and DC473 fit this non-retinoid structural criterion, we can be confident that the photoactivated cell killing activity is not related to retinoid signalling processes.21
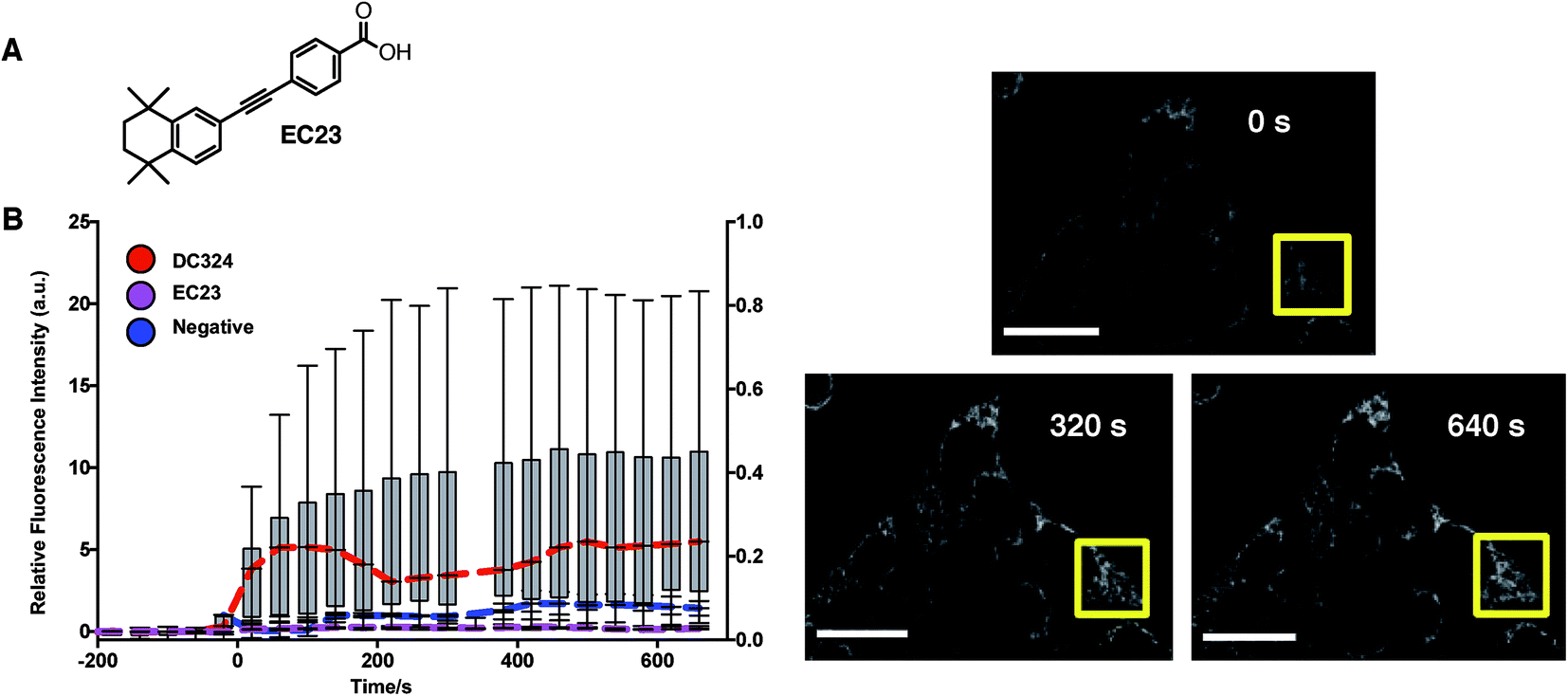 | ||
| Fig. 7 (A) Chemical structure of synthetic retinoid, EC23. (B) Quantitation of relative fluorescence of CellRox before and after irradiation using a 405 nm laser. The relative fluorescence intensity was calculated in the region of interest (ROI, yellow box) at each time point and data graphed using box-whisker plot. Graph coloured lines denote sample mean (n = 11, DC324; n = 3, EC23; n = 3, negative) from 2 experimental replicates for each treatment (red = DC324, pink = EC23, blue = negative). Time 0 equals time of irradiation. Increased fluorescence intensity in DC324-treated cells was significant as determined by one-way ANOVA (p < 0.001). Scale bars equal 50 μm. | ||
In order to identify whether DC324 and DC473 directly cause the formation of a specific ROS upon photoactivation, we performed a variety of cuvette-based photophysical experiments involving solutions of the compounds dissolved in a range of oxygen saturated solvents, at various concentrations, with irradiation at both absorption maxima (experimental details in ESI†). These included 1O2 sensitisation and phosphorescence detection thereof, hydroxyl radical detection by absorption measurements of added methylene blue, peroxyl radical detection by fluorescence measurements of added rhodamine 6G, and hydrogen peroxide detection by chemiluminescence of added luminol in the presence of a cobalt(II) catalyst (the latter three types of experiment were carried out with the addition of H2O to allow for facile formation of ROS from H2O). None of these experiments returned a positive result. Furthermore, there was no evidence, either through experimental or theoretical means, that the compounds are capable of generating triplet excited states to any significant extent.
Given the fact that we can, nevertheless, detect ROS upon photoexcitation of DC324 and DC473 in cells using CellRox, it is clear that an alternative ROS generation mechanism may be taking place. This could involve changes in the excited state properties of DC324 and DC473 and thus ROS formation by direct association with a second species of cellular origin, or that the photosensitising process involves sensitisation of another species by interaction with the compounds upon light activation. Such complex excited state modes of action would be in line with our previous observation that, in an environment where hydrogen bonding is possible, i.e. in polar solvents, the excited state lifetime decay is complicated by a secondary process starting at around 500 ps after light excitation. Alternatively, when bound to a cellular membrane (e.g. mitochondrial), a conformational change in the photoexcited state of the compound may induce significant disruption of this membrane and, therefore, the release of ROS as a biological response to this cellular stress. In either case, our experiments clearly show that the processes responsible for the observed cell death upon photoexcitation of the compounds are not trivial.
We next employed RNA sequencing to provide information on the biological and cellular responses that occur following in cellulo photoactivation of the compounds. This higher level approach enables ‘cause-and-effect’ examination of the genes and genetic pathways that are regulated in response to a stimulus. Keratinocytes were treated with 1 μM DC473 for 4 hours and were subsequently irradiated with UV-A (363–385 nm) light for 1 minute. After 1 hour, RNA was collected from cells that had been DC473-treated and irradiated, and from corresponding DMSO-treated and DC473-treated, non-irradiated controls. Gene set enrichment pathway analysis (see ESI†) of these RNA samples indicated that, among other effects, genetic responses towards the presence of oxygen species were significantly upregulated when cells treated with DC473 were irradiated in comparison to those of non-irradiated DC473-treated cells, and both irradiated and non-irradiated DMSO-treated cells. Responses towards the generation of nitrogenous compounds were also evident, perhaps indicating the generation of nitric oxide – a radical species that is heavily implicated in oxidative stress pathways resulting from PDT.41,42 Furthermore, gene regulatory pathways implicated in cell death processes were only upregulated in the irradiated, DC473-treated cells. While these experiments do not provide a precise molecular understanding of the reactive species that are generated, they do provide strong and, in many respects, more direct evidence that oxygen-containing species (and other nitrogenous species) are produced upon in cellulo photoactivation of DC324 and DC473, and that this phenomenon is likely to be the cause or at least related directly to the observed cell death activity.
In vitro cultured epithelial, mesenchymal and colorectal (see ESI†) cancer cells were next treated with a range of concentrations of DC324, and a variety of experiments were conducted in order to further quantify and characterise the observed light-activated cytotoxic activity. In all these human cell lines, irradiation with either broadband UV-A (363–385 nm) or a monochromatic 405 nm light source induced rapid cell death. Furthermore, cell death was also observed when the compound was irradiated at the corresponding two-photon absorption wavelengths (800 nm), a particularly important property, as this enables deeper tissue penetration (see ESI† for irradiation images).43 It was further found that the cell death timeline could be modulated by varying the irradiation area, photon flux and compound concentration. Fig. 8A illustrates that only the area which received UV-A irradiation was destroyed, highlighting that the cell-killing activity of the compounds is a light-activated effect, rather than one of general cytotoxicity. Furthermore, the amount of energy supplied was important; stronger irradiation per unit area expedited and increased the extent of cell death (Fig. 8B). The cytotoxic effect of the compound was also characterised by a dose–response relationship between the cell viability and the treatment concentration. Accordingly, we determined an EC50, based on the average viable fraction of the epithelial cellular population 24 hours after the initial irradiation event, of 0.20 ± 0.01 μM (Fig. 8C). The time required to induce cell death, and the change in physiology, were also significantly affected by compound concentration; i.e. irradiated cells exposed to the higher concentrations tested (10 μM) induced necrosis-like cell death in less than 60 seconds whilst cells exposed to lower concentrations (e.g. 1 μM) showed an initial cell membrane blebbing associated with cell apoptosis. Cell loss was rapid, and maximal impact on cell viability was detected approximately 400 minutes after the initial irradiation event. Importantly, cells exposed to the compound without irradiation, or irradiation treatment alone, had no impact on cell viability. These physiological and morphological changes are consistent with cell death in response to the presence of significant amounts of ROS;44 however, given the apparent inability for DC324 and DC473 to access triplet states that could lead to the generation of ROS through the paradigm set out by the porphyrins and other similar compounds,5 it is clear that an alternative cytotoxic mechanism may be operating. Whether this effect is achieved through sensitisation of a second species by the photoactivated compound or, for example, through disruption of the membrane/organelle(s) which the compound is bound to or via another biological response is unclear, and the enormous complexity of a cellular system makes interrogation of this effect inherently difficult. Nonetheless, as an orthogonal means for the generation of a photodynamic therapeutic effect, the unique activity of DC324 and DC473 is highly compelling and of great potential utility.
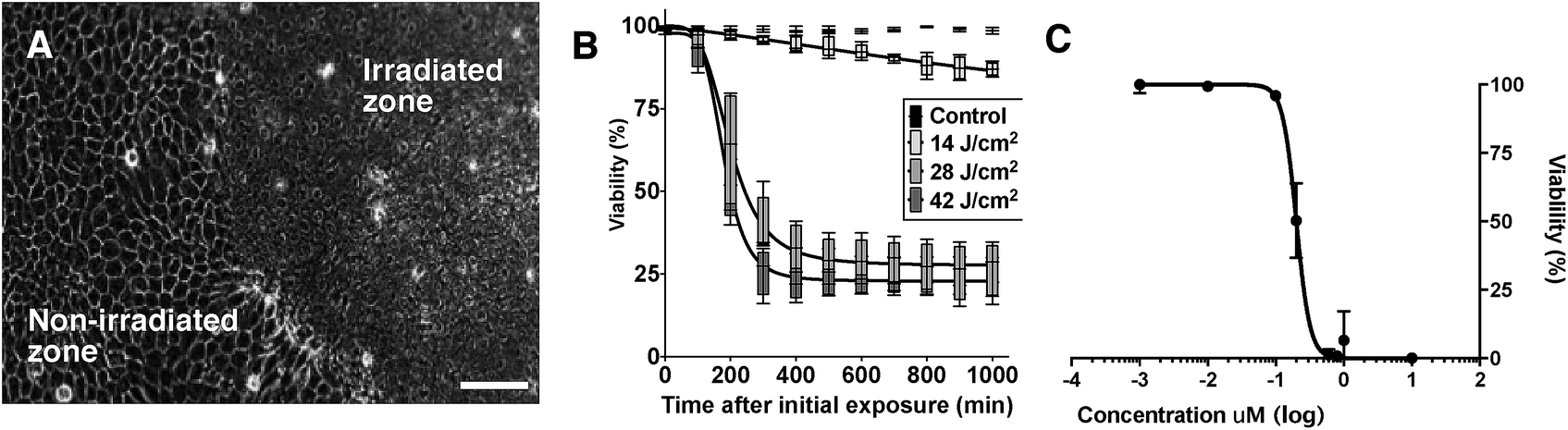 | ||
| Fig. 8 (A) Phase contrast image of confluent sheet of HaCaT epithelial cells exposed to 10 μM DC324 and the righthand area of the field irradiated with UV-A. Image captured 10 minutes post irradiation; note that cell death occurred only in the irradiated zone. Scale bars equal 100 μm. (B) Cell viability after irradiation with varying UV-A doses. Dose–response curves have been fitted to the data in a box-whisker plot graph using 4 experimental replicates (n = 4). Statistical significance was determined by one-way ANOVA (p < 0.0001) and Dunnett's multiple comparison test comparing individual dose response curves to unirradiated controls (0 J cm−2). The energy fluence of 14, 28 and 42 J cm−2 were considered statistically significant with p values of 0.0034, 0.0002 and <0.0001, respectively. (C) The average viable fraction of HaCaT epithelial cellular populations 24 hours after an initial 60 second UV-exposure, as a function of the concentration of compound administered. A non-linear regression curve is fitted to the data (experimental replicates, n = 3; R2 = 0.99). | ||
2.4 In vivo characterisation
To test the light-activated cytotoxic activity in an in vivo context, 48 hour old wild type zebrafish embryos were exposed to 1 μM DC324 for 2 hours, then transferred to E3 water without the compound and incubated for 4 hours before irradiation. Embryos irradiated (UV-A; 28 J cm−2) in the developing tail region showed evidence of cell death in the irradiated area but not in adjacent regions.45 Following irradiation, nuclei became distended and irregular in shape, a common characteristic of nuclear apoptosis (Fig. 9).46 Together, these data clearly show that DC324 and DC473 have the potential to act as a potent modality for the destruction of tissue in vitro and in vivo through a light-activated cytotoxic effect.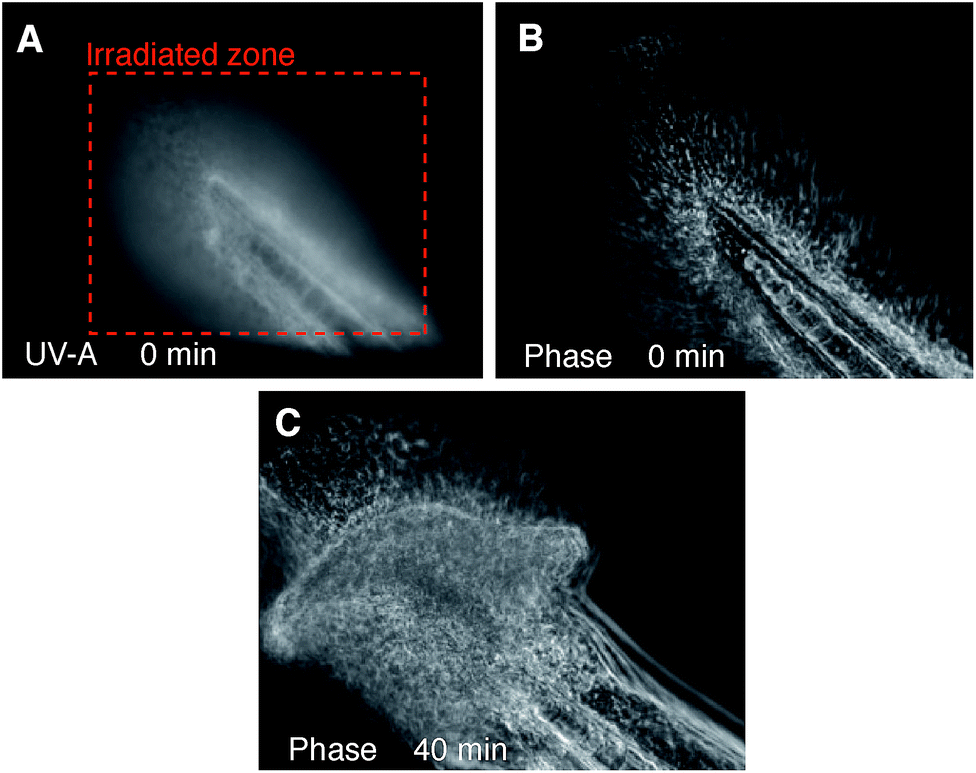 | ||
| Fig. 9 48 hour wild-type zebrafish embryo treated with 1 μM DC324 for 2 hours prior and the tail irradiated with 42 J cm−2 of UV-A (time = 0 minutes); phase contrast images captured at the time of irradiation (A/B) and 40 minutes after irradiation (C). | ||
3 Conclusions
A class of small, organic, conjugated compounds, essentially homologues of synthetic retinoids, show clear potential as a novel photosensitising agent. DC324 and DC473 possess much more ‘drug-like’ molecular weights and structures in comparison to existing photosensitisers and a thorough biological characterisation has exemplified the ability for these compounds to elicit cytotoxic activity, ostensibly through bringing about robust intracellular ROS production following UV-A or violet light absorption, or corresponding two-photon absorption via near-IR irradiation. While UV-based photodynamic therapies have typically seen only limited use in the past,47,48 in more recent years significant advances in optical and endoscope technology have enabled the production of devices including low cost portable LED-based illumination systems capable of precisely targeted irradiation at a specific wavelength. These next generation optical devices are beginning to see widespread use in the clinic and, hence, potentially pave the way for a treatment modality involving UV-activated, ‘drug-like’ photosensitisers in previously impractical or impossible treatment regimes. However, the two-photon absorption ability of the compounds would also enable use in contexts where UV-activation remains intractable, taking advantage of the greater tissue penetration of near-IR irradiation. Furthermore, the modular structures of DC324 and DC473 could be modified in further studies to incorporate targeting mechanisms, such as conjugation to an antibody to enable specific localisation to diseased tissues, or to another small molecule to influence subcellular localisation. The small molecular footprint of these novel photosensitisers would likely have a negligible effect on the efficacy of these targeting modalities, which is often not the case, for example, with antibody-conjugated porphyrin-based photosensitisers.49,50 Hence, the advantages of this new class of small, adaptable organic PDT agent, which is highly cell-permeable, potent and appears to exhibit a unique mode-of-action, could prove to be the key to realising the rich potential PDT has for tackling a wide range of disease treatment applications which, to date, has been severely limited by the inadequate properties of existing photosensitisers.4 Experimental
4.1 General synthetic information
Reagents were purchased from Sigma-Aldrich, Acros Organics, Alfa-Aesar and Fluorochem. Reagents were purified, if required, by recrystallisation or distillation/sublimation under vacuum. Solvents were used as supplied from Fisher Scientific or Sigma Aldrich, and dried before use if required with appropriate drying agents or using an Innovative Technologies Inc. Solvent Purification System. Thin-layer chromatography (TLC) was conducted using Merck Millipore silica gel 60G F254 25 glassplates and/or TLC-PET foils of aluminium oxide with fluorescent indicator 254 nm (40 × 80 mm) with visualisation by UV lamp or appropriate staining agents. Flash column chromatography was performed using SiO2 from Sigma-Aldrich (230–400 mesh, 40–63 μM, 60 Å), or activated neutral aluminium oxide (alumina) from Sigma-Aldrich, and monitored using TLC. Sublimation/distillation was performed using a Buchi Glass Oven B-585 Kugelrohr operating at a pressure between 0.2 and 2.0 torr. NMR spectra were recorded using Varian VNMRS-700, Varian VNMRS-600, Bruker Avance-400 or Varian Mercury-400 spectrometers operating at ambient probe temperature. NMR peaks are reported as singlet (s), doublet (d), triplet (t), quartet (q), broad (br), septet (sept), combinations thereof, or as a multiplet (m), with reference to the following deuterated solvent signals: CDCl3 (1H = 7.26 ppm, 13C = 77.23 ppm), (CD3)2SO (1H = 2.50 ppm, 13C = 39.50 ppm). ESMS was performed using a TQD (Waters Ltd., UK) mass spectrometer with an Acquity UPLC (Waters Ltd., UK), and accurate mass measurements were obtained using a QtoF Premier mass spectrometer with an Acquity UPLC (Waters Ltd., UK). ASAP measurements were performed using an LCT Premier XE mass spectrometer and an Acquity UPLC (Waters Ltd., UK). GCMS was performed with a QP2010-Ultra (Shimadzu) GCMS. IR spectra were recorded using a Perkin Elmer FTIR spectrometer. Melting points were obtained using a Gallenkamp melting point apparatus and are uncorrected. Elemental analysis was conducted using an Exeter Analytical CE-440 analyser.4.2 Synthesis
:DCM, 1:1, as eluent) to give compound 2 as a white solid (3.74 g, 75%): mp = 119–120 °C; 1H NMR (600 MHz, CDCl3) δ 3.81 (s, 3H), 6.44 (d, J = 16.0 Hz, 1H), 7.24 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 16.0 Hz, 1H), 7.73 (d, J = 8.4 Hz, 2H); 13C{1H} NMR (151 MHz, CDCl3) δ 52.0, 96.7, 118.8, 129.7, 134.1, 138.3, 143.8, 167.3; IR (ATR) vmax/cm−1 3080w, 3000w, 2850w, 1708s, 1636m, 1580m, 1483m, 815s, 493m; MS(ES): m/z = 289 [M + H]+; HRMS (ES) calcd for C10H10IO2 [M + H]+: 288.9726, found 288.9733; found: C, 41.86; H, 3.14. Calc. for C10H9IO2: C, 41.96; H, 3.15%.52
:EtOAc, 9:1, as eluent) to give compound 3 as a white solid (2.65 g, 83%): mp = 76–78 °C; 1H NMR (600 MHz, CDCl3) δ 0.26 (s, 9H), 3.81 (s, 3H), 6.43 (d, J = 16.0 Hz, 1H), 7.43–7.48 (m, 4H), 7.65 (d, J = 16.0 Hz, 1H); 13C{1H} NMR (151 MHz; CDCl3) δ 0.1, 52.0, 96.9, 104.7, 118.8, 125.2, 128.1, 132.6, 134.5, 144.1, 167.4; IR (ATR) vmax/cm−1 2952w, 2898w, 2156w, 1715s, 1634m, 1599w, 1442m, 1171s, 840s; MS(ES): m/z = 259 [M + H]+; HRMS (ES) calcd for C15H19SiO2 [M + H]+: 259.1154, found 259.1147.
:EtOAc, 9:1, as eluent) to give compound 4 as a white solid (1.52 g, 95%): mp = 93–95 °C; 1H NMR (600 MHz; CDCl3) δ 3.18 (s, 1H), 3.81 (s, 3H), 6.44 (d, J = 16.0 Hz, 1H), 7.46–7.51 (m, 4H), 7.66 (d, J = 16.0 Hz, 1H); 13C{1H} NMR (151 MHz; CDCl3) δ 52.0, 79.4, 83.3, 119.1, 124.2, 128.1, 132.8, 134.9, 143.9, 167.4; IR (ATR) vmax/cm−1 3260m, 2996w, 2946w, 2108w, 1700s, 1634m, 1554m, 1431m, 1206s, 831s; MS (EI): m/z = 186 [M]+; found: C, 77.40; H, 5.37. Calc. for C12H10O2: C, 77.40; H, 5.41%.53 Note: compound 4 can also be purified by Kugelrohr sublimation under vacuum (115–125 °C, 0.8 torr).
:EtOAc (8:2)). The extracts were washed with sat. NH4Cl (3×) and brine, dried (MgSO4) and evaporated to give the intermediate ester as an orange solid (0.7 g). This was dissolved in THF (20 mL), and 20% NaOH (2 mL) was added, and the resultant solution was stirred at reflux for 40 h. The mixture was cooled, acidified to pH 1 with 5% HCl, diluted with EtOAc, washed with sat. NH4Cl, H2O and brine, dried (MgSO4) and evaporated to give a crude yellow solid which was recrystallised from MeOH to give DC324 as an orange crystalline solid (0.46 g, 67% over three steps): mp = 214–215 °C (decomposition); 1H NMR (700 MHz; (CD3)2SO) δ 1.16 (d, J = 6.7 Hz, 6H), 1.22 (s, 6H, H11/12), 1.63 (t, J = 6.0 Hz, 2H), 3.18 (t, J = 6.0 Hz, 2H), 4.14 (sept, J = 6.7 Hz, 1H), 6.54 (d, J = 16.0 Hz, 1H), 6.69 (d, J = 8.7 Hz, 1H), 7.17 (dd, J = 8.7, 2.2 Hz, 1H), 7.29 (d, J = 2.2 Hz, 1H), 7.46–7.51 (m, 2H), 7.58 (d, J = 16.0 Hz, 1H), 7.66–7.72 (m, 2H), 12.41 (s, 1H); 13C{1H} NMR (176 MHz; (CD3)2SO) δ 18.6, 29.7, 31.6, 35.9, 36.1, 46.6, 86.9, 94.0, 106.8, 110.5, 119.5, 125.2, 128.4, 128.8, 130.6, 131.1, 131.2, 133.3, 143.0, 144.5, 167.5; vmax/cm−1 2968w, 2929w, 2870w, 2195m, 1683s, 1622m, 1594s, 1514s, 1421m, 1187m, 837m; MS(ES): m/z = 374 [M + H]+; HRMS (ES) calcd for C25H28NO2 [M + H]+: 374.2120, found 374.2118; found: C, 79.90; H, 7.27; N, 3.65. Calc. for C25H27NO2: C, 80.40; H, 7.29; N, 3.75%.
4.3 General photophysical information
UV-visible absorption spectra were obtained on an Agilent 1100 Series Diode Array spectrophotometer using standard 1 cm path length quartz cells. Excitation and emission spectra were recorded on an Edinburgh Instruments FLSP920 spectrophotometer, equipped with a 450 W xenon arc lamp, double monochromators for the excitation and emission pathways, a red-sensitive photomultiplier (PMT-R928) and a near-IR PMT as detectors, or on a Horiba Jobin-Yvon Fluoromax 3 spectrophotometer with single monochromators for the excitation and emission pathways. The excitation and emission spectra were corrected using the standard corrections supplied by the manufacturers for the spectral power of the excitation source and the sensitivity of the detector. The quantum yields were measured by use of integrating spheres with either an Edinburgh Instruments FLSP920 spectrophotometer or the Horiba Jobin-Yvon Fluoromax 3 spectrophotometer. The luminescence lifetimes were measured using a TCSPC module on an FLSP980 spectrometer equipped with a high speed photomultiplier tube positioned after a single emission monochromator and operating with pulsed laser diodes (376 or 274 nm, repetition rate 1–5 MHz, pulse width ca. 200 ps, instrument response function ca. 500 ps). Decays were recorded to 10000 counts in the peak channel with a record length of at least 1000 channels. The band-pass of the monochromator was adjusted to give a signal count rate of <20 kHz. Iterative reconvolution of the IRF with one decay function and nonlinear least-squares analysis were used to analyse the data. The quality of all decay fits was judged to be satisfactory, based on the calculated values of the reduced X2 and Durbin–Watson parameters and visual inspection of the weighted and autocorrelated residuals.
4.4 Computational details
Calculations (gas-phase) were performed with the ORCA 3.0.2 program suite.54 Geometry optimisations were carried out with the B3LYP55–57 functional as implemented in ORCA, and a frequency analysis ensuring that the optimized structures correspond to energy minima. The def2-TZVP58,59 basis set was used for all atoms together with the auxiliary basis set def2-TZVP/J in order to accelerate the computations within the framework of the RI approximation. van der Waals interactions have been considered by an empirical dispersion correction (Grimme-D3BJ).60,61 TD-DFT calculations for the first 100 singlet and triplet excited states were performed with the M06 functional62 and the above mentioned basis sets. Representations of electron density changes were produced with orca plot as provided by ORCA 3.0.2 and with gOpenMol 3.00.63,644.5 Cells
HaCaT human epidermal keratinocytes were purchased from a commercial supplier (Thermo Fisher) and primary human dermal fibroblasts (HDF) were isolated from human patients under ethical guidelines from National Research Ethics Service.65 Both cell lines were cultured at 37 °C/5% CO2 in DMEM (Gibco cat. no. 10566, high glucose, GlutaMAX Supplement) with 10% fetal calf serum and 1× penicillin/streptomycin. For experimental procedures, cells were seeded (1.2 × 104 to 5 × 104 per well) in 24-well or 6-well tissue culture plates (Corning) or on single or 8-well chambered coverslips for a minimum of 48 h prior to the start of an experiment. In some experiments, HaCaT (2 × 104) and HDF (4 × 104) cells were co-seeded in 8-well chambered coverslips. Cells were treated with DC324 (stock concentration 10 mM dissolved in DMSO) at a range of final concentrations (0.001–10 μM) in media with 0.1% DMSO. As controls, cells were treated with 0.1% DMSO or 1 μM EC23, a compound with similar chemical structure to DC324, but no (or negligible) absorption when excited by UV-A or violet light wavelengths.18 Cells were treated with the compound for 1–4 h before irradiation.4.6 Imaging
Live and fixed cells were imaged using a Zeiss 880 Laser Scanning Confocal Microscope (LSCM) with Airyscan detection and an environmental chamber at 37 °C and 5% CO2. Fixed cells were imaged at room temperature and in air. Samples were excited with a 405 nm, 594 nm or 633 nm laser and imaged with a Plan-Apochromat 63×/1.4 Oil DIC M27 objective lens. For irradiation-induced cell death experiments, cells were irradiated and imaged on either the Zeiss 880 LSCM or a Zeiss Axio Vert A1, wide field fluorescence microscope with environmental control chamber using image corrected 10× or 20× objective lenses designed for live cell imaging. UV-A filtered irradiation (Axio Vert A1) was carried out using an OSRAM 1× HBO 103 W/2 100 watt mercury bulb using DAPI excitation filters with a bandwidth of 335–385 nm and emission bandwidth of 420–470 nm according to manufacturer's specification; 405 nm irradiation (880 LSCM) was carried out with a 405 nm diode laser. Excitation wavelengths for both microscopes were confirmed by measuring the light spectrum with an Ocean Optics USB 2000+ Spectrometer (see ESI†). The irradiation beam was optically restricted to a limited region within the field of view for most experiments (see ESI†). A well-defined boundary was achieved using a simple mask in the illumination optical path for the widefield measurements and using ROI scanning for the laser measurements allowing for interrogation of irradiated and non-irradiated areas in the same field of view. Cells were imaged for up to 24 h on the microscope with environmental controls.4.7 MitoTracker® staining
DC324-treated cells, plated on coverslips, were incubated for 30 min with MitoTracker® Red (Thermo Fisher, cat. no. M22425), prepared according to manufacturer's directions and diluted in serum containing media (1 mL, 0.1 mM), then rinsed twice with PBS (pH 7.2) before incubation and subsequent fixation in paraformaldehyde/PBS (PFA, 4%), for 5 min. Cells were rinsed in PBS before mounting coverslips in a polyvinyl alcohol mounting media.4.8 ROS quantitation
Cells in separate wells were treated with DC324 or EC23 at 1 μM concentration (0.1% DMSO) in media and incubated in a tissue culture incubator. A negative control of 0.1% DMSO in media was also used. After 2 h, CellRox (Sigma C10422 Deep Red 640/665 nm) at a final concentration of 5 μM was added to the cells and further incubated for 30 min. Cells were then transferred to the 37 °C, 5% CO2 environmental chamber on the Zeiss 880 LCSM. To quantify ROS production detected by 633 nm excitation of CellRox dye, background fluorescence emission intensities (655–675 nm) were captured first in each experimental field for 5 min, then the field was irradiated with 405 nm and fluorescence levels (655–675 nm) were captured for an additional 10 min following irradiation. Zeiss Zen Black and FIJI imaging processing software were used to calculate the ROI relative fluorescence intensity levels. A minimum of 3 experimental replicates were performed for each treatment.4.9 RNAseq and bioinformatics
HaCaT human keratinocyte cells were seeded at a density of 2 × 105 cells per well in an uncoated, 6-well tissue culture plate. 72 h after plating, when cells were approximately 80% confluent, cells were treated with either 0.1% DMSO or 1 μM DC473 for 4 h, then some samples were irradiated with UV-A (363–385 nm with a peak at 371 nm; see ESI†) for 1 min. RNA was collected 1 h after irradiation and prepared for sequencing using a Qiagen RNeasy Mini Kit with on-column DNaseI digestion. RNA samples were quantified using the NanoDrop and fluorometric methods and the mRNA had RIN values of 8 and above. RNAseq was performed by the sequencing facility at the Institute for Genetic Medicine, Newcastle University.Illumina reads were aligned to the human reference genome (GRCh38) using HISAT2 v.2.0.5.29. The alignments were assembled and quantification performed using StringTie v.1.3.4.30. The R package Ballgown v.2.6.0 was used for all differential expression analysis. Results from using the list of genes for differential expression (pval < 0.01) was compared by gene set enrichment analysis (GSEA; http://www.software.broadinstitute.org/gsea/index.jsp) looking at the ‘GO’ (gene ontology) gene sets (see ESI†). These show the number of genes in each list which have a significant overlap with particular sets of genes.
4.10 Cell death quantitation and statistical analysis
Each well was allocated 4 set points where 3 points were subjected to 1 min of UV irradiation in a specific rectangular zone. The fourth zone was a control with no irradiation. Images were captured at 30 min intervals for 24 h. Using ImageJ,66 time zero and 24 h post irradiation numbers of viable and non-viable cells were counted from the zone areas. The viable and non-viable cell numbers for each time point were added together and the viability percentage was calculated by dividing the viable cells by the total cell count. The percentage of relative viability was then calculated by dividing the 24 h time point by the zero time point. Statistical significance was calculated by one-way ANOVA using GraphPad Prism 7 software, Macintosh, GraphPad Software, La Jolla California USA, http://www.graphpad.com.4.11 Zebrafish experiments
48 Hour, wild type zebrafish embryos (Golden) were treated with 1 μM DC324 diluted in E3 water (5.0 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) for 2 h.67 Embryos were then transferred to fresh E3 water and incubated for a further 2 h prior to irradiation. Embryos were anesthetised using buffered MS222 solution (0.1%; pH 7.0; ethyl 3-aminobenzoate methanesulfonate dissolved in E3 water and buffered with NaHCO3) and then irradiated using the Axio Vert A1 system as described above with 33 J cm−2 of UV-A with appropriate environmental controls. Phase contrast and fluorescence images were captured at the time of irradiation with only phase contrast imaged captured at subsequent time points.Conflicts of interest
A. W. and C. A. A. own shares of LightOx Limited, the company licensed to pursue commercial applications of the novel chemicals described in this manuscript.Acknowledgements
D. R. C. thanks the EPSRC, BBSRC and High Force Research Ltd. for doctoral funding. T. B. M. thanks the Julius-Maximilians-Universität Würzburg for support. A. K. N. thanks the Fulbright Foundation for a Visiting Fulbright Scholarship. We thank Dr R. M. Edkins for obtaining some of the optical spectra of DC324. We thank Dr E. Pohl for helpful discussions and proof-reading.Notes and references
- K. M. Holmström and T. Finkel, Nat. Rev. Mol. Cell Biol., 2014, 15, 411–421 CrossRef PubMed.
- X. Zhao and K. Drlica, Curr. Opin. Microbiol., 2014, 21, 1–6 CrossRef CAS PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- C. Schweitzer, Z. Mehrdad, A. Noll, E.-W. Grabner and R. Schmidt, J. Phys. Chem. A, 2003, 107, 2192–2198 CrossRef CAS.
- R. Schmidt, Photochem. Photobiol., 2007, 82, 1161–1177 CrossRef.
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181–3209 RSC.
- Y. L. Guo, S. Chakraborty, S. S. Rajan, R. Wang and F. Huang, Stem Cells Dev., 2010, 19, 1321–1331 CrossRef CAS PubMed.
- Z. Guo, S. Kozlov, M. F. Lavin, M. D. Person and T. T. Paull, Science, 2010, 330, 517–521 CrossRef CAS PubMed.
- K. Ito, A. Hirao, F. Arai, S. Matsuoka, K. Takubo, I. Hamaguchi, K. Nomiyama, K. Hosokawa, K. Sakurada, N. Nakagata, Y. Ikeda, T. W. Mak and T. Suda, Nature, 2004, 431, 997–1002 CrossRef CAS PubMed.
- N. Shirasu, S. O. Nam and M. Kuroki, Anticancer Res., 2013, 33, 2823–2831 CAS.
- R. Bonnett, Chem. Soc. Rev., 1995, 24, 19–33 RSC.
- J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS PubMed.
- B. C. Wilson, M. Olivo and G. Singh, Photochem. Photobiol., 1997, 65, 166–176 CrossRef CAS PubMed.
- J. Zhang, C. Jiang, J. P. F. Longo, R. B. Azevedo, H. Zhang and A. L. Muehlmann, Acta Pharm. Sin. B, 2018, 8, 137–146 CrossRef PubMed.
- M. Wachowska, A. Muchowicz, M. Firczuk, M. Gabrysiak, M. Winiarska, M. Wańczyk, K. Bojarczuk and J. Golab, Molecules, 2011, 16, 4140–4164 CrossRef CAS.
- A. Orenstein, G. Kostenich, L. Roitman, Y. Shechtman, Y. Kopolovic, B. Ehrenberg and Z. Malik, Br. J. Cancer, 1996, 73, 937–944 CrossRef CAS PubMed.
- M. E. Bulina, D. M. Chudakov, O. V. Britanova, Y. G. Yanushevich, D. B. Staroverov, T. V. Chepurnykh, E. M. Merzlyak, M. A. Shkrob, S. Lukyanov and K. A. Lukyanov, Nat. Biotechnol., 2006, 24, 95–99 CrossRef CAS PubMed.
- V. B. Christie, J. H. Barnard, A. S. Batsanov, C. E. Bridgens, E. B. Cartmell, J. C. Collings, D. J. Maltman, C. P. F. Redfern, T. B. Marder, S. Przyborski and A. Whiting, Org. Biomol. Chem., 2008, 6, 3497–3507 RSC.
- G. Zhou, D. M. Tams, T. B. Marder, R. Valentine, A. Whiting and S. A. Przyborski, Org. Biomol. Chem., 2013, 11, 2323–2334 RSC.
- J. H. Barnard, J. C. Collings, A. Whiting, S. A. Przyborski and T. B. Marder, Chem.–Eur. J., 2009, 15, 11430–11442 CrossRef CAS PubMed.
- G. Clemens, K. R. Flower, P. Gardner, A. P. Henderson, J. P. Knowles, T. B. Marder, A. Whiting and S. Przyborski, Mol. BioSyst., 2013, 9, 3124–3134 RSC.
- G. Clemens, K. R. Flower, A. P. Henderson, A. Whiting, S. A. Przyborski, M. Jimenez-Hernandez, F. Ball, P. Bassan, G. Cinque and P. Gardner, Mol. BioSyst., 2013, 9, 677–692 RSC.
- H. Haffez, D. R. Chisholm, R. Valentine, E. Pohl, C. P. F. Redfern and A. Whiting, Med. Chem. Commun., 2017, 8, 578–592 RSC.
- H. Haffez, T. Khatib, P. McCaffery, S. Przyborski, C. Redfern and A. Whiting, Mol. Neurobiol., 2018, 55, 1942–1950 CrossRef CAS PubMed.
- J. B. Bauer, W. P. Lippert, S. Dörrich, D. Tebbe, C. Burschka, V. B. Christie, D. M. Tams, A. P. Henderson, B. A. Murray, T. B. Marder, S. A. Przyborski and R. Tacke, ChemMedChem, 2011, 6, 1509–1517 CrossRef CAS PubMed.
- T. B. Marder and A. Whiting, UK patent application no. GB1417957.6, 2014.
- C. A. Ambler, D. R. Chisholm and A. Whiting, UK patent application no. GB1613712.7, 2016.
- J. C. Collings, A. C. Parsons, L. Porrès, A. Beeby, A. S. Batsanov, J. A. K. Howard, D. P. Lydon, P. J. Low, I. J. S. Fairlamb and T. B. Marder, Chem. Commun., 2005, 2666–2668 RSC.
- C. Dehu, F. Meyers and J. L. Bredas, J. Am. Chem. Soc., 1993, 115, 6198–6206 CrossRef CAS.
- M. Biswas, P. Nguyen, T. B. Marder and L. R. Khundkar, J. Phys. Chem. A, 1997, 101, 1689–1695 CrossRef CAS.
- P. Nguyen, Z. Yuan, L. Agocs, G. Lesley and T. B. Marder, Inorg. Chim. Acta, 1994, 220, 289–296 CrossRef CAS.
- P. Nguyen, G. Lesley, T. B. Marder, I. Ledoux and J. Zyss, Chem. Mater., 1997, 9, 406–408 CrossRef CAS.
- L. R. Khundkar, A. E. Stiegman and J. W. Perry, J. Phys. Chem., 1990, 94, 1224–1226 CrossRef CAS.
- D. R. Chisholm, G.-L. Zhou, E. Pohl, R. Valentine and A. Whiting, Beilstein J. Org. Chem., 2016, 12, 1851–1862 CrossRef CAS PubMed.
- J. H. Barnard, PhD thesis, Durham University, 2010.
- J. G. Taylor, A. V. Moro and C. R. D. Correia, Eur. J. Org. Chem., 2011, 1403–1428 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- J. Gala De Pablo, D. R. Chisholm, A. Steffen, A. K. Nelson, C. Mahler, T. B. Marder, S. A. Peyman, J. M. Girkin, C. A. Ambler, A. Whiting and S. D. Evans, Analyst, 2018, 143, 6113–6120 RSC.
- J. C. Collings, S.-Y. Poon, C. Le Droumaguet, M. Charlot, C. Katan, L.-O. Pålsson, A. Beeby, J. A. Mosely, H. M. Kaiser, D. Kaufmann, W.-Y. Wong, M. Blanchard-Desce and T. B. Marder, Chem.–Eur. J., 2009, 15, 198–208 CrossRef CAS PubMed.
- H. Choi, Z. Yang and J. C. Weisshaar, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E303–E310 CrossRef CAS PubMed.
- K. J. Reeves, M. W. R. Reed and N. J. Brown, J. Photochem. Photobiol., B, 2009, 95, 141–147 CrossRef CAS PubMed.
- R. P. Patel, J. McAndrew, H. Sellak, C. R. White, H. Jo, B. A. Freeman and V. M. Darley-Usmar, Biochim. Biophys. Acta, 1999, 1411, 385–400 CrossRef CAS.
- E. A. Wachter, W. G. Fisher, C. Dees, M. G. Petersen and M. Panjehpour, Biomedical Optical Spectroscopy and Diagnostics/Therapeutic Laser Applications, OSA Trends in Optics and Photonics Series, Optical Society of America, Orlando, Florida, USA, Orlando, Florida, 1998 Search PubMed.
- M. Redza-Dutordoir and D. A. Averill-Bates, Biochim. Biophys. Acta, 2016, 1863, 2977–2992 CrossRef CAS PubMed.
- C. Buckley, M. T. Carvalho, L. K. Young, S. A. Rider, C. McFadden, C. Berlage, R. F. Verdon, J. M. Taylor, J. M. Girkin and J. J. Mullins, Sci. Rep., 2017, 7, 5096 CrossRef CAS PubMed.
- V. L. Johnson, S. C. Ko, T. H. Holmstrom, J. E. Eriksson and S. C. Chow, J. Cell Sci., 2000, 113, 2941–2953 CAS.
- J. José Serrano-Pérez, M. Merchán and L. Serrano-Andrés, J. Phys. Chem. B, 2008, 112, 14002–14010 CrossRef PubMed.
- F. Almutawa, L. Thalib, D. Hekman, Q. Sun, I. Hamzavi and H. W. Lim, Photodermatol., Photoimmunol. Photomed., 2015, 31, 5–14 CrossRef PubMed.
- G. A. M. S. van Dongen, G. W. M. Visser and M. B. Vrouenraets, Adv. Drug Delivery Rev., 2004, 56, 31–52 CrossRef CAS PubMed.
- P. M. R. Pereira, B. Korsak, B. Sarmento, R. J. Schneider, R. Fernandes and J. P. C. Tomé, Org. Biomol. Chem., 2015, 13, 2518–2529 RSC.
- B. A. Haag, Z.-G. Zhang, J.-S. Li and P. Knochel, Angew. Chem., Int. Ed., 2010, 49, 9513–9516 CrossRef CAS PubMed.
- H. Brunner, N. Le Cousturier de Courcy and J.-P. Genêt, Tetrahedron Lett., 1999, 40, 4815–4818 CrossRef CAS.
- E. Christiansen, M. E. Due-Hansen, C. Urban, M. Grundmann, J. Schmidt, S. V. F. Hansen, B. D. Hudson, M. Zaibi, S. B. Markussen, E. Hagesaether, G. Milligan, M. A. Cawthorne, E. Kostenis, M. U. Kassack and T. Ulven, J. Med. Chem., 2013, 56, 982–992 CrossRef CAS PubMed.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- L. Laaksonen, J. Mol. Graphics, 1992, 10, 33–34 CrossRef CAS PubMed.
- D. L. Bergman, L. Laaksonen and A. Laaksonen, J. Mol. Graphics Modell., 1997, 15, 301–306 CrossRef CAS PubMed.
- K. Gledhill, A. Gardner and C. A. Jahoda, Isolation and Establishment of Hair Follicle Dermal Papilla Cell Cultures, Humana Press, Totowa, NJ, 2013, vol. 989, pp. 285–292 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- C. B. Kimmel, W. W. Ballard, S. R. Kimmel, B. Ullmann and T. F. Schilling, Dev. Dyn., 1995, 203, 253–310 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sc00199a |
| This journal is © The Royal Society of Chemistry 2019 |