
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFirst-principles microkinetic study of methane and hydrogen sulfide catalytic conversion to methanethiol/dimethyl sulfide on Mo6S8 clusters: activity/selectivity of different promoters†
Adam A.
Arvidsson
 a,
William
Taifan
b,
Anders
Hellman
*a and
Jonas
Baltrusaitis
*b
a,
William
Taifan
b,
Anders
Hellman
*a and
Jonas
Baltrusaitis
*b
aDepartment of Physics, Chalmers University of Technology, 412 96 Gothenburg, Sweden. E-mail: anders.hellman@chalmers.se
bDepartment of Chemical and Biomolecular Engineering, Lehigh University, B336 Iacocca Hall, 111 Research Drive, Bethlehem, PA 18015, USA. E-mail: job314@lehigh.edu
First published on 17th July 2019
Abstract
A large fraction of the global natural gas reserves is in the form of sour gas, i.e. contains hydrogen sulfide (H2S) and carbon dioxide (CO2), and needs to be sweetened before utilization. The traditional amine-based separation process is energy-intensive, thereby lowering the value of the sour gas. Thus, there is a need to find alternative processes to remove, e.g., hydrogen sulfide. Mo6S8 clusters are promising candidates for transforming methane (CH4) and hydrogen sulfide into methanethiol (CH3SH) and dimethyl sulfide (CH3SCH3), which are high-value sulfur-containing products that can be further used in the chemical industry. Here first-principles microkinetics is used to investigate the activity and selectivity of bare and promoted (K, Ni, Cl) Mo6S8. The results show that methanethiol is produced via two different pathways (direct and stepwise), while dimethyl sulfide is formed via a competing pathway in the stepwise formation of methanethiol. Moreover, there is an increase in activity and a decrease in selectivity when adding an electropositive promoter (K), whereas the reverse behaviour is observed when adding an electronegative promoter (Cl). When adding Ni there is also a decrease in activity and an increase in selectivity; however, Ni is acting as an electron donor. The results provide insights and guidance as to what catalyst formulation is preferred for the removal of hydrogen sulfide in sour gas.
1 Introduction
Rational design of catalysts to obtain new or improved catalyst formulations with a highly selective conversion of hydrocarbon gas, such as methane (CH4), is of utmost importance in solving seminal petroleum industry processing problems. These problems include everything from energy carrier generation (e.g., steam reforming and water gas shift on the supported Ni and CuO/ZnO/Al2O3 catalysts, respectively),1 obtaining feedstock for a variety of commodity chemicals (e.g., olefin metathesis of ethylene and 2-butene to produce propylene on supported metal oxide catalysts)2,3 and environmental remediation of refinery tail gases (e.g., H2S oxidation on Al2O3 or TiO2 in the Claus process).4 Yet, many fundamental aspects governing hydrocarbon catalyst conversion and selectivity are still not resolved because the underlying complex selectivity descriptors are still lacking.5 Of particular interest is understanding the structure–selectivity relationships in metal oxide and metal sulfide catalysts for sour natural gas catalytic conversion.6 Sour gas – mainly CO2 and H2S – molecules are invariably present in natural gas and present significant challenges for conventional oxide-based catalysts including catalyst degradation and loss of activity. The Society of Petroleum Engineers (SPE) estimates that about 40% of the world's total accessible natural gas reserves are acidic, containing large concentrations of CO2 and H2S, with over 10% H2S totaling up to 350 Tcf.7 The concentrations of acidic gases can range up to 90% by volume and this sub-quality natural gas accounts for ∼30% of U.S. natural gas resources8 with the consequence that most of the gas is not utilized.9 A conventional approach to sweeten natural gas includes a priori removal of these gases using alkanolamines, with the two most common being monoethanolamine (MEA) and diethanolamine (DEA), via solvent recycling and adsorption/desorption processes. Typically, the energy consumed during the amine regeneration step is 3.65 MJ kg−1 CO2,10–12 which significantly increases the cost of natural gas.A conceptually new approach recently emerged to catalytically transform both hydrocarbon and sour gas molecules into valuable products simultaneously via reactive separation.6 In particular, hydrogen sulfide methane reforming13 has been proposed at temperatures above 1000 °C with considerable amounts of carbonyl sulfide (COS) being formed. While H2S poisons conventional catalysts for steam reforming,14 the overall process is thermodynamically limited and a series of supported catalysts have been utilized for this reaction, including metal oxides (Pt/Al2O3,15 Mo,Cr/ZrO2–SBA15 (ref. 16), Mo,Cr/ZrO2–La2O3 (ref. 16) and Fe2O3/γ-Al2O3 (ref. 17)) and metal sulfides (MoS2,Cr2S3 and Ce2S3).18–20 The Langmuir–Hinshelwood–Hougen–Watson (LHHW) reaction mechanism on MoS2 catalysts was proposed with the rate-determining step (RDS) being the reaction among the adsorbed species ( , H2S* and S*) on the catalyst surface with the participation of three catalytic sites.19 These catalysts have been used instead of thermal decomposition taking place at high (>70% for both CH4 and H2S) conversion above 1250 °C.21 In turn, CS2 and COS have been shown to selectively react to form CH3SH on Ni-, K-, and Co-promoted MoS2/SiO2, providing indirect routes of acidic gas processing to CH3SH.22–25 On the other hand, CH3SH, which is isostructural with CH3OH,26 can be further converted into higher hydrocarbons. Chang and Silvestri27 utilized H-ZSM-5 at 480 °C to convert CH3SH into a mixture of hydrocarbons and H2S with 7.0% selectivity towards C2 and C3 olefins with a significant amount of the carbon feed converted to dimethyl sulfide (DMS). Butter et al.28 claimed overall 96.1% conversion of CH3SH with significant amounts of CH4 generated at 258 °C on Al-exchanged H-ZSM-5. Similarly, Mashkina et al.29 identified the presence of CH4 when CH3SH was reacted on various acid catalysts between 350 and 400 °C, while at lower temperatures DMS was the only product observed. In general, large amounts of CH4, DMS and coke were observed in addition to the BTX (benzene, toluene, xylene) products formed.30–32 Insights from density functional theory (DFT) calculations have suggested less favorable free energy reactive pathways for CH3SH as compared with CH3OH for the same acidity catalysts due to the differences in charge transfer between the organic species and the zeolite framework.33–35 DMS, on the other hand, was shown to yield 15.4% C2H4 in the product stream using WO3/Al2O3, with CH4 and C3H6 comprising the rest at 380 °C and 32% conversion.36 Hence, obtaining partially activated CH4 analogues, such as CH3SH and CH3SCH3, to be converted to higher olefins as opposed to a complete reforming from sour gas streams is of direct interest.
, H2S* and S*) on the catalyst surface with the participation of three catalytic sites.19 These catalysts have been used instead of thermal decomposition taking place at high (>70% for both CH4 and H2S) conversion above 1250 °C.21 In turn, CS2 and COS have been shown to selectively react to form CH3SH on Ni-, K-, and Co-promoted MoS2/SiO2, providing indirect routes of acidic gas processing to CH3SH.22–25 On the other hand, CH3SH, which is isostructural with CH3OH,26 can be further converted into higher hydrocarbons. Chang and Silvestri27 utilized H-ZSM-5 at 480 °C to convert CH3SH into a mixture of hydrocarbons and H2S with 7.0% selectivity towards C2 and C3 olefins with a significant amount of the carbon feed converted to dimethyl sulfide (DMS). Butter et al.28 claimed overall 96.1% conversion of CH3SH with significant amounts of CH4 generated at 258 °C on Al-exchanged H-ZSM-5. Similarly, Mashkina et al.29 identified the presence of CH4 when CH3SH was reacted on various acid catalysts between 350 and 400 °C, while at lower temperatures DMS was the only product observed. In general, large amounts of CH4, DMS and coke were observed in addition to the BTX (benzene, toluene, xylene) products formed.30–32 Insights from density functional theory (DFT) calculations have suggested less favorable free energy reactive pathways for CH3SH as compared with CH3OH for the same acidity catalysts due to the differences in charge transfer between the organic species and the zeolite framework.33–35 DMS, on the other hand, was shown to yield 15.4% C2H4 in the product stream using WO3/Al2O3, with CH4 and C3H6 comprising the rest at 380 °C and 32% conversion.36 Hence, obtaining partially activated CH4 analogues, such as CH3SH and CH3SCH3, to be converted to higher olefins as opposed to a complete reforming from sour gas streams is of direct interest.
In this work, we utilize DFT to investigate the activity and selectivity of bare and promoted Mo6S8 clusters toward methanethiol (CH3SH) and DMS (CH3SCH3) using first-principles microkinetics. Earlier studies have shown that the stable and highly symmetric Mo6S8 cluster has many promising properties, e.g. large CO binding energy37 and large dipole moment38 and a number of other optical and reactive properties.39–43 We expand our previous work44 by adding diffusion steps in a phenomenological manner in the reaction mechanism in order to consider multiple reaction pathways and thereby selectivity. We utilize bare and K-, Cl- or Ni-promoted Mo6S8 clusters as a versatile and tunable H2S-tolerant catalyst material.37,39,40,44,45
2 Computational details
2.1 Electronic structure calculations
Periodic DFT calculations were performed as implemented in the VASP code.46–49 The projector augmented wave (PAW) method of Blöchl,50 as adapted by Kresse and Joubert,51 was used to describe the effective interaction between the valance electrons and the core. The exchange-correlation energy was described by the spin-polarized version of the PBE.52 The Brillouin zone was sampled using the Γ-point only. The Kohn–Sham equations have been solved self-consistently in a plane-wave basis set with a cutoff of 400 eV. The convergence criterion for the electronic self-consistency cycle, measured by the change in the total energy between successive iterations, was set to 10−6 eV. The electronic structures of the different clusters were analyzed and are shown in Fig. S10† using the crystal orbital Hamiltonian populations projected on the nearest neighbour orbitals using the LOBSTER software.53–562.2 Structural optimization calculations
Transition states have been identified using the DIMER method,57 as improved by Heyden et al.58 The atomic positions were considered relaxed if all forces acting on the atoms were less than 0.005 eV Å−1. Transition states were proven to be first-order saddle points of the potential energy surface using vibrational analysis. The intrinsic reaction coordinates (IRCs)59,60 for the forward and backward reaction steps were identified using the damped velocity Verlet algorithm.61 The structures corresponding to potential energy minima along the IRC were further relaxed using a conjugate-gradient algorithm in order to satisfy the same optimization criterion as for transition states. Vibrational analysis was performed to ensure that the relaxed structures correspond to true potential energy minima. This procedure guarantees that reactant and product states are linked through one single transition state. Optimized structures are provided in the ESI† (Fig. S1–S3).2.3 Microkinetic modelling
The reaction kinetics were investigated using energies and vibrational frequencies as calculated from DFT. The rate constants for the reaction between reaction intermediates were expressed using the conventional transition state theory (TST),62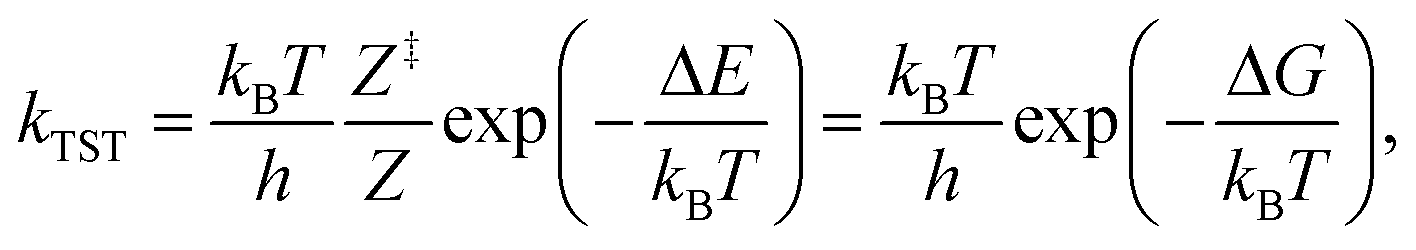 | (1) |
Adsorption of CH4, H2S, CH3SH, H2, and DMS was considered to be direct with rate constants expressed as
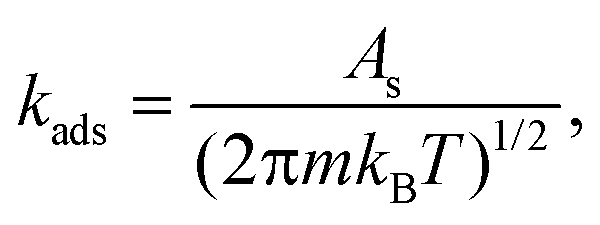 | (2) |
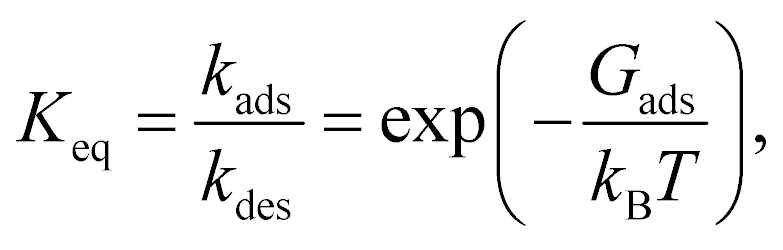 | (3) |
| Eads = EA* − (E* + EA(g)) | (4) |
The CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2S pressure ratio was kept at 1:12 in the kinetic calculations in order to simulate conditions necessary to avoid coke formation.16
:H2S pressure ratio was kept at 1:12 in the kinetic calculations in order to simulate conditions necessary to avoid coke formation.16
In addition to the transition state (TS) steps and the barrierless adsorption/desorption (ads/des) steps, we also considered the diffusion steps between different clusters (diffusion) in a parametric way. From practical consideration this was done by introducing a low barrier (0.05 eV) and treating diffusion using TST.
Here we will assume a Langmuir–Hinshelwood reaction mechanism62 over a double site, using a double site notation [A*,B*], symbolizing adsorption on a –S–Mo– site with A adsorbed on S and B on Mo; all reaction steps are described in Table 1. We ignore the possibility of an Eley–Rideal mechanism62 since Eley–Rideal is usually less probable62,65 as compared to Langmuir–Hinshelwood. In addition, we do not include a Mars–Van Krevelen mechanism62 since under industrial reaction conditions sulfur is in excess and the cost of creating a sulfur vacancy (Mo6S7) is calculated to be 2.4 eV, while the energy cost of adding one sulfur to Mo6S8 is 0.2 eV (here the reference state for sulfur is H2S–H2), which indicates that a reaction mechanism including Mo6S7 clusters is unlikely. The inclusion of Eley–Rideal and Mars–Van Krevelen mechanisms is a future challenge that is beyond the scope of this study.
| Reaction | Type |
|---|---|
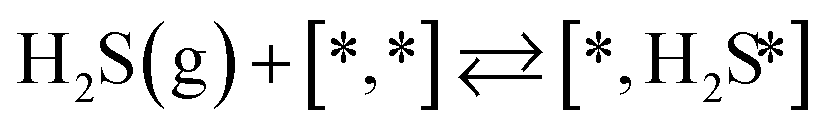
|
Ads/des |
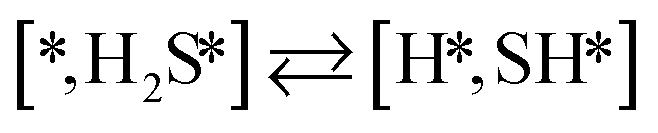
|
TS |
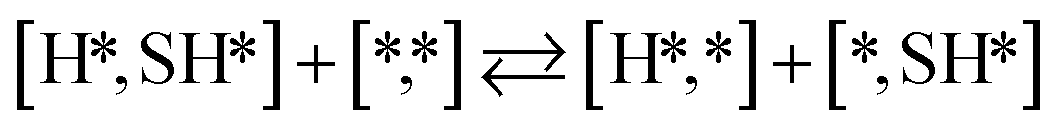
|
Diffusion |
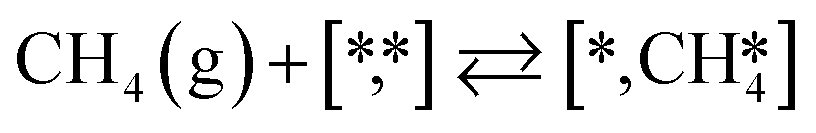
|
Ads/des |
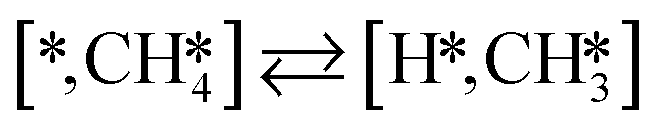
|
TS |
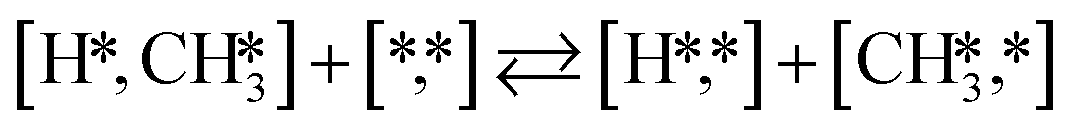
|
Diffusion |
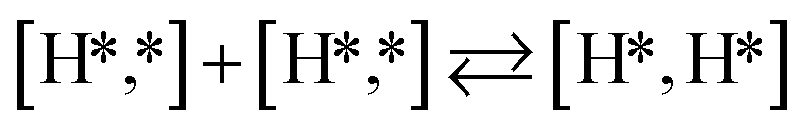
|
Diffusion |
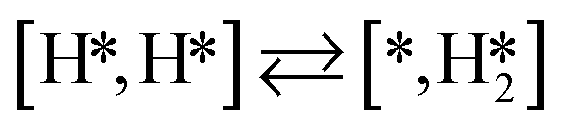
|
TS |
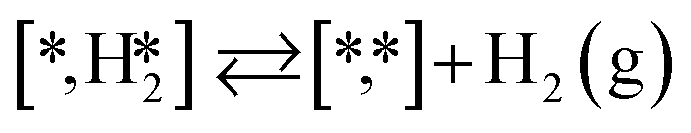
|
Ads/des |
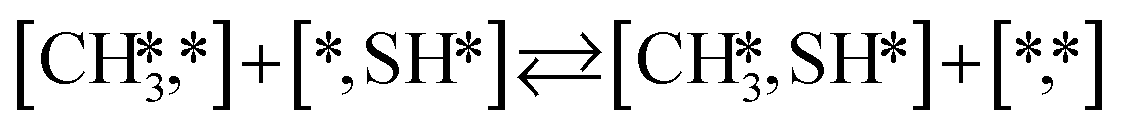
|
Diffusion |
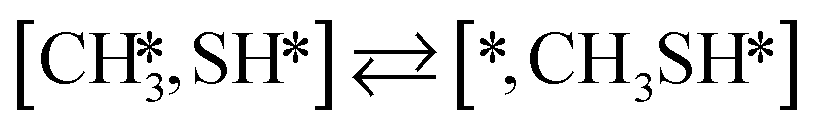
|
TS |
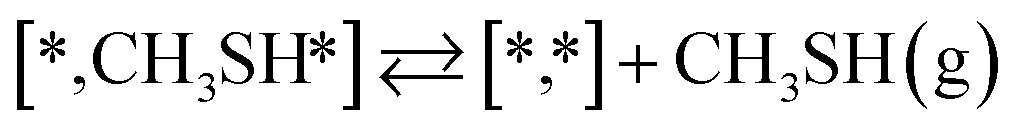
|
Ads/des |
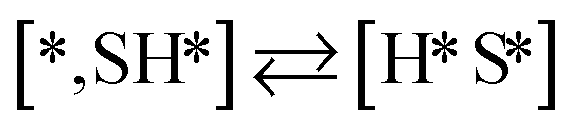
|
TS |
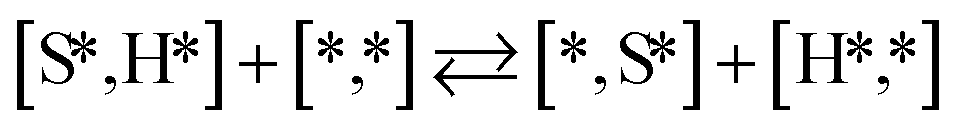
|
Diffusion |
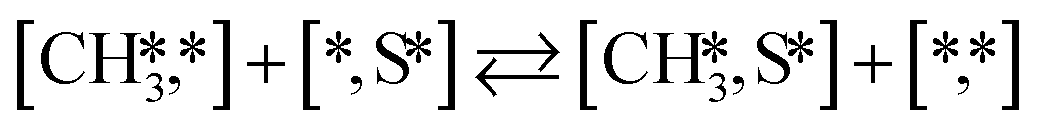
|
Diffusion |
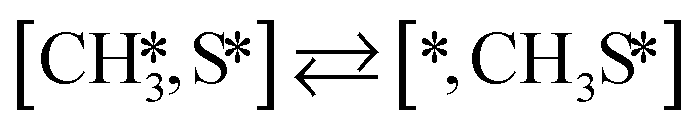
|
TS |
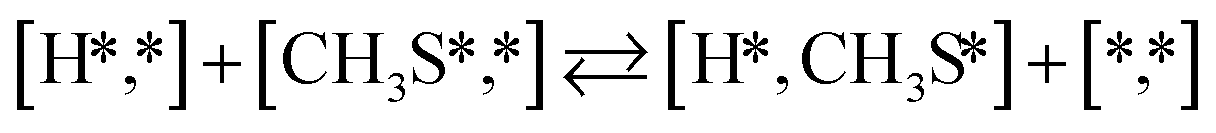
|
Diffusion |
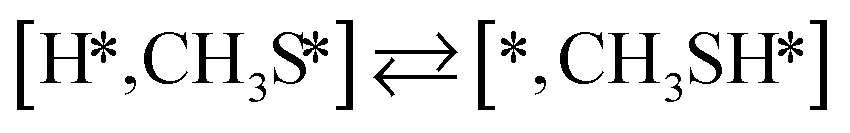
|
TS |
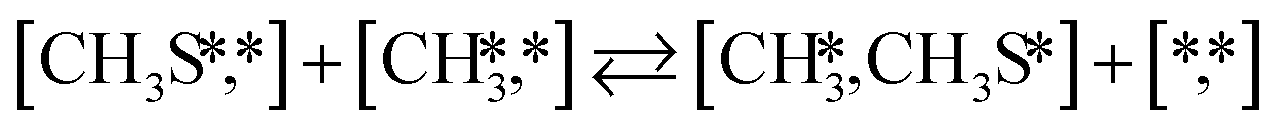
|
Diffusion |
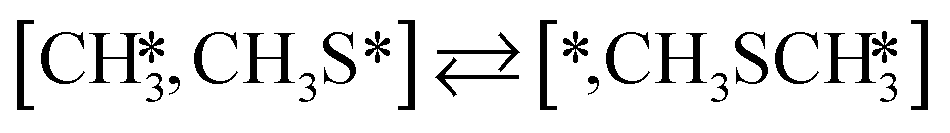
|
TS |

|
Ads/des |
Selectivity between DMS and methanethiol is defined as the ratio between the TOF of DMS and the sum of the direct and stepwise TOF of methanethiol.
The ensuing non-linear differential equations were integrated numerically using the SciPy Python package, which relies on the ODEPACK Fortran library.66,67
3 Results and discussion
The calculated formation energies of the reactions CH4 + H2S → CH3SH + H2 and 2CH4 + H2S → CH3SCH3 + 2H2 are −0.84 eV and −1.57 eV, respectively. This is in fair agreement with the experimental formation energies,68 which are −0.75 eV and −1.37 eV, respectively.The bare Mo6S8 cluster forms a face-capped octahedral cluster where the Mo atoms sits in octahedral positions and the S atoms occupy the face of the octahedron. The Mo–Mo and Mo–S distances are 2.62 Å and 2.44 Å, respectively. There is a charge transfer from the molybdenum atoms to the more electronegative sulfur atoms. A Bader analysis69–72 shows a 0.7–0.8 excess of electrons per atom on the sulfur atoms, while the molybdenum atoms lose close to one whole electron per atom.
The promoters (K, Ni, and Cl) affect the structural and electronic properties of the Mo6S8 cluster in different ways. K adsorbs in a bridge position between two S atoms, with a bond distance of 3.00 Å, but there is no noticeable distortion in the Mo6S8 cluster owing to the adsorption. However, K acts as an electron donor, and via a Bader charge analysis we observe an additional charge of 0.2 and 0.1 electrons per atom to the sulfur and molybdenum atoms close to the adsorption site. Ni also adsorbs in the bridge position (bond distance of 2.15 Å) between two sulfur atoms. However, there are noticeable structural changes in the cluster; in particular, the sulfur atoms adjust so that the Ni promoter gains access to the two underlying molybdenum atoms. The Ni–Mo distances (2.43 Å and 2.60 Å) are not symmetric. However, according to the Bader charge analysis, Ni transfers 0.2 electrons almost symmetrically between the two coordinating molybdenum atoms. In marked difference with the other promoters, Cl coordinates to one molybdenum atom (bond distance 2.33 Å). There is a charge transfer to Cl (0.4 electrons) which comes from the molybdenum atom (0.2 electrons) coordinated to Cl and symmetrically from all sulfur atoms in the cluster.
The proposed reaction mechanism of CH4 and H2S conversion to methanethiol and DMS is shown in Fig. 1 for the bare cluster (see Fig. S1–S3† for K-, Ni-, and Cl-promoted clusters). The reaction mechanism can be divided into three parts: (i) activation of reactants (CH4 and H2S), (ii) formation of methanethiol where two different pathways are considered (one direct route, SH* + 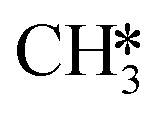 , and one associative route that involves atomic S*), and finally (iii) coupling of CH3S* and
, and one associative route that involves atomic S*), and finally (iii) coupling of CH3S* and 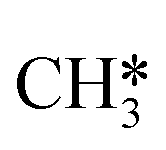 to DMS. The stepwise methanethiol and DMS pathway has one common reaction intermediate, namely CH3S* adsorbed on the Mo site. Depending on which reaction intermediate, either hydrogen or methyl, occupies the accompanying sulfur site, methanethiol or DMS will be formed. There is also a number of diffusion steps included in the reaction mechanism, which are important as this allows relevant reaction intermediates to pair up according to the dual site notation shown in Fig. 1.
to DMS. The stepwise methanethiol and DMS pathway has one common reaction intermediate, namely CH3S* adsorbed on the Mo site. Depending on which reaction intermediate, either hydrogen or methyl, occupies the accompanying sulfur site, methanethiol or DMS will be formed. There is also a number of diffusion steps included in the reaction mechanism, which are important as this allows relevant reaction intermediates to pair up according to the dual site notation shown in Fig. 1.
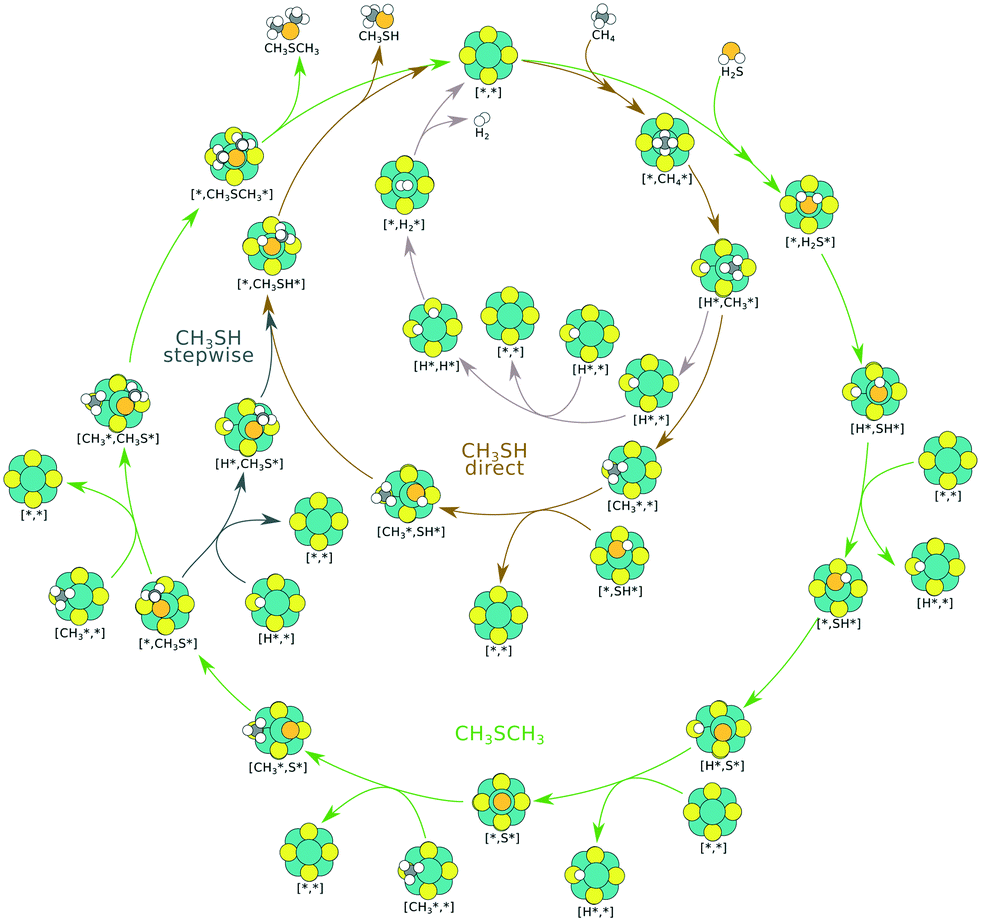 | ||
| Fig. 1 Proposed reaction mechanism for the conversion of CH4 and H2 to methanethiol and DMS. The production of methanethiol occurs along two different pathways (direct and stepwise), while the production of DMS is a competing path in the stepwise reaction. Color code: hydrogen, white; carbon, black; sulfur, yellow (Mo6S8) or orange (adsorbed); and molybdenum, green. | ||
The reaction energy landscape of CH4 and H2S conversion to methanethiol and dimethyl sulfide is shown in Fig. 2, where all energies are referenced to the empty cluster with CH4 and H2S in the gas phase. The free energy landscape at 1400 K is shown in Fig. S4† and the rate constants and free energy barriers are listed in Tables S1–S4.† The activation of CH4 on the bare and promoted Mo6S8 cluster occurs through a weak physisorption state with an adsorption energy of −0.21 eV, −0.09 eV, −0.13 eV and −0.21 eV on Mo6S8, K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8, respectively. From that state the C–H bond that coordinates to the cluster becomes activated and finally reaches a transition state with an energy of 1.32 eV (Mo6S8), 1.16 eV (K–Mo6S8), 1.53 eV (Ni–Mo6S8) and 1.22 eV (Cl–Mo6S8). The C–H bond distance at the transition state ranges between 1.73 Å and 1.91 Å, where the bond distance correlates with the transition state energy. The dissociative state [H*,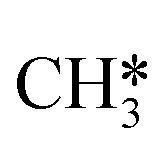 ] is endothermic with respect to the physisorbed state, with comparable energies (1.34 eV and 1.28 eV) for the bare and Ni–Mo6S8, while K–Mo6S8 and Cl–Mo6S8 are slightly less destabilized (1.13 eV and 1.15 eV).
] is endothermic with respect to the physisorbed state, with comparable energies (1.34 eV and 1.28 eV) for the bare and Ni–Mo6S8, while K–Mo6S8 and Cl–Mo6S8 are slightly less destabilized (1.13 eV and 1.15 eV).
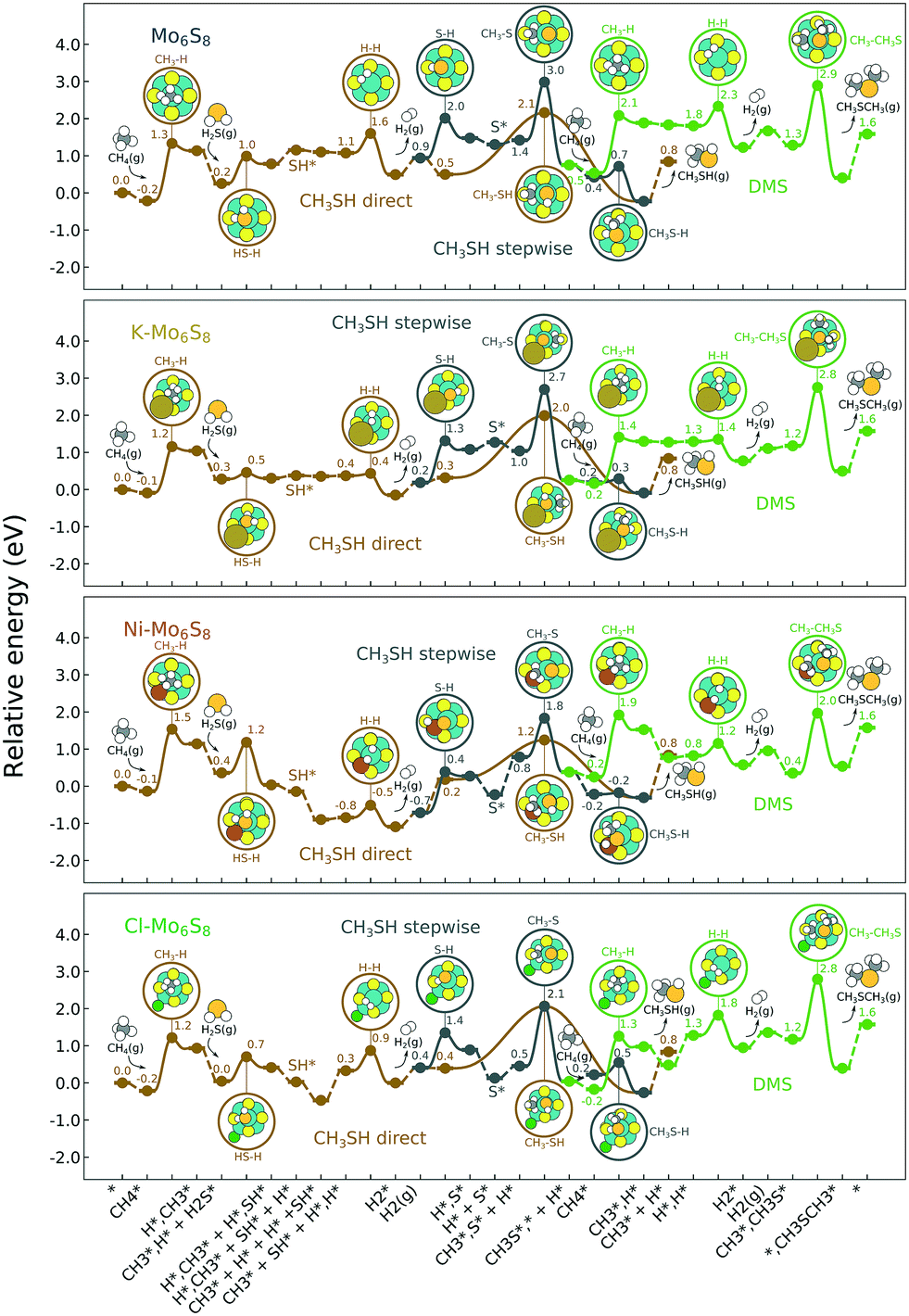 | ||
| Fig. 2 Reaction energy landscape for the conversion of CH4 and H2S to methanethiol and DMS. The production of methanethiol occurs along two different pathways (direct and stepwise), while the production of DMS is another path of the stepwise reaction. Energy (eV) with gas-phase H2S and CH4 as reference is shown for the most important steps. The insets show the transition states for all the clusters and also the adsorption site of the different promoters and intermediates. All energies are referenced to the empty cluster with CH4 and H2S in the gas phase. Color code: hydrogen, white; carbon, black; sulfur, yellow (Mo6S8) or orange (adsorbed); molybdenum, green; potassium, brown; nickel, red; and chlorine, dark green. | ||
On the other hand, the activation of H2S occurs through a rather strong chemisorption state with an adsorption energy of −0.88 eV, −0.76 eV, −0.78 eV and −0.89 eV on Mo6S8, K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8, respectively. The transition state involves the activation of one of the H–S bonds. K–Mo6S8 has the lowest barrier (0.18 eV), followed by Cl–Mo6S8 (0.66 eV), Mo6S8 (0.73 eV) and finally Ni–Mo6S8 (0.82 eV). The dissociation is endothermic on Mo6S8 and Cl–Mo6S8 with an energy of 0.53 eV and 0.37 eV, respectively. This is very different as compared to Ni–Mo6S8 where the dissociation is exothermic with an energy of −0.32 eV. On K–Mo6S8 the reaction is endothermic but with an energy of only 0.02 eV.
The next part involves the diffusion of hydrogen away from the sulfur part of the dual site. In the reaction energy landscape the separation of SH* and H* to two separate dual sites is shown first. Here it is clear that for the bare Mo6S8 this process is strongly endothermic (0.47 eV), while for the Cl- and Ni-promoted clusters this process is exothermic with −0.39 eV and −0.18 eV, respectively. On K–Mo6S8 the separation is slightly endothermic (0.07 eV). Similar results are found for the separation of 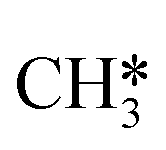 and H* to two separate dual sites, where the reaction energy is 0.03 eV, −0.02 eV, −0.76 eV and −0.50 eV for Mo6S8, K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8, respectively. However, the diffusion of H* to the same dual site ([H*,H*]) displays a reverse trend where on the bare Mo6S8 this process is exothermic (−0.11 eV), while the same process is endothermic on K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8 with an energy of 0.02 eV, 0.05 eV and 0.80 eV, respectively. The two H* can associate and form
and H* to two separate dual sites, where the reaction energy is 0.03 eV, −0.02 eV, −0.76 eV and −0.50 eV for Mo6S8, K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8, respectively. However, the diffusion of H* to the same dual site ([H*,H*]) displays a reverse trend where on the bare Mo6S8 this process is exothermic (−0.11 eV), while the same process is endothermic on K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8 with an energy of 0.02 eV, 0.05 eV and 0.80 eV, respectively. The two H* can associate and form 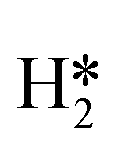 on the molybdenum part of the dual site with a reaction barrier of 0.52 eV (Mo6S8), 0.06 eV (K–Mo6S8), 0.33 eV (Ni–Mo6S8) and 0.54 eV (Cl–Mo6S8). The formation of
on the molybdenum part of the dual site with a reaction barrier of 0.52 eV (Mo6S8), 0.06 eV (K–Mo6S8), 0.33 eV (Ni–Mo6S8) and 0.54 eV (Cl–Mo6S8). The formation of 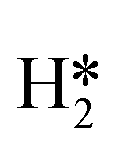 is endothermic irrespective of whether the cluster is promoted or not.
is endothermic irrespective of whether the cluster is promoted or not.
The direct pathway to methanethiol involves diffusion of 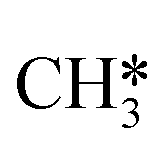 and SH* to the same dual site. In the case of Mo6S8 this step is exothermic with −0.53 eV, and −0.01 eV on Cl–Mo6S8, while for the K- and Ni-promoted clusters the diffusion is endothermic, 0.13 eV and 0.90 eV on K–Mo6S8 and Ni–Mo6S8, respectively. The next step in the formation of methanethiol occurs through a transition state that involves the formation of an S–C bond. The bond distance is calculated to be 2.53 Å, 2.60 Å, 2.52 Å, 2.61 Å with an energy barrier of 1.65 eV, 1.67 eV, 1.06 eV and 1.68 eV on Mo6S8, K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8, respectively.
and SH* to the same dual site. In the case of Mo6S8 this step is exothermic with −0.53 eV, and −0.01 eV on Cl–Mo6S8, while for the K- and Ni-promoted clusters the diffusion is endothermic, 0.13 eV and 0.90 eV on K–Mo6S8 and Ni–Mo6S8, respectively. The next step in the formation of methanethiol occurs through a transition state that involves the formation of an S–C bond. The bond distance is calculated to be 2.53 Å, 2.60 Å, 2.52 Å, 2.61 Å with an energy barrier of 1.65 eV, 1.67 eV, 1.06 eV and 1.68 eV on Mo6S8, K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8, respectively.
Besides the direct pathway there is also the possibility to further decompose SH* into elementary S*. The barrier of the transition state is 1.08 eV, 1.13 eV, 1.10 eV and 0.94 eV on Mo6S8, K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8, respectively. The S–H bond distance at the transition state ranges between 1.74 Å and 2.76 Å and the different bond distances correlate with the transition state energy. The dissociation of SH* is endothermic on all clusters. The diffusion of H* from the same dual site is endothermic, with the exception of K–Mo6S8 where this process is endothermic with 0.19 eV. The reverse behaviour in energy is observed for the diffusion of 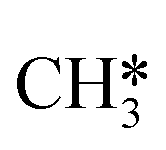 to the sulfur part of the dual site occupied by S*. The reaction between S* and methyl occurs through a rather high energy barrier of 1.54 eV, 1.66 eV, 1.05 eV and 1.61 eV on Mo6S8, K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8, respectively. The S–C bond distance at the transition state ranges between 2.53 Å and 2.60 Å. The formation of CH3S* is exothermic on all clusters. Depending on which species diffuse to the empty sulfur site, the methylthiol can react to form methanethiol (stepwise pathway) or dimethyl sulfide. In the case of diffusion of H* the reaction pathway is the same as the direct pathway but with an additional diffusion step, while in the case of diffusion of methyl the reaction can proceed to form dimethyl sulfide. In the reaction energy landscape (Fig. 2), to form dimethyl sulfide two additional steps (methane dissociation and hydrogen desorption) are necessary to keep the reaction stoichiometry. However, these are the same as described earlier. The diffusion of
to the sulfur part of the dual site occupied by S*. The reaction between S* and methyl occurs through a rather high energy barrier of 1.54 eV, 1.66 eV, 1.05 eV and 1.61 eV on Mo6S8, K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8, respectively. The S–C bond distance at the transition state ranges between 2.53 Å and 2.60 Å. The formation of CH3S* is exothermic on all clusters. Depending on which species diffuse to the empty sulfur site, the methylthiol can react to form methanethiol (stepwise pathway) or dimethyl sulfide. In the case of diffusion of H* the reaction pathway is the same as the direct pathway but with an additional diffusion step, while in the case of diffusion of methyl the reaction can proceed to form dimethyl sulfide. In the reaction energy landscape (Fig. 2), to form dimethyl sulfide two additional steps (methane dissociation and hydrogen desorption) are necessary to keep the reaction stoichiometry. However, these are the same as described earlier. The diffusion of 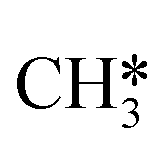 to the same dual site as that of CH3S* is exothermic, with the exception of K–Mo6S8 where the reaction is slightly endothermic (0.07 eV). The barrier at the transition state is high (1.60 eV, 1.57 eV, 1.61 eV and 1.62 eV on Mo6S8, K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8, respectively) and there is only a small change due to the addition of promoters. The formation is exothermic with the exception of Ni–Mo6S8. This is also reflected in the lower adsorption energy of dimethyl sulfide on Ni–Mo6S8 (−1.04 eV) as compared to the other clusters, namely −1.18 eV (Mo6S8), −1.08 eV (K–Mo6S8) and −1.18 eV (Cl–Mo6S8).
to the same dual site as that of CH3S* is exothermic, with the exception of K–Mo6S8 where the reaction is slightly endothermic (0.07 eV). The barrier at the transition state is high (1.60 eV, 1.57 eV, 1.61 eV and 1.62 eV on Mo6S8, K–Mo6S8, Ni–Mo6S8 and Cl–Mo6S8, respectively) and there is only a small change due to the addition of promoters. The formation is exothermic with the exception of Ni–Mo6S8. This is also reflected in the lower adsorption energy of dimethyl sulfide on Ni–Mo6S8 (−1.04 eV) as compared to the other clusters, namely −1.18 eV (Mo6S8), −1.08 eV (K–Mo6S8) and −1.18 eV (Cl–Mo6S8).
The turnover frequencies (TOFs) (Fig. 3) under industrially relevant conditions for forming methanethiol and DMS are calculated in a microkinetic model based on the reaction mechanism shown in Fig. 1 and the energy landscape shown in Fig. 2. It is clear that the formation of methanethiol occurs mainly through the stepwise mechanism. The direct pathway becomes more important at lower temperature but consequently with a rather low TOF. It is also clear that the TOF for DMS is always low as compared to the TOF for methanethiol, the exception being Ni–Mo6S8 at low temperatures (∼900 K). K–Mo6S8 displays a higher activity for methanethiol formation as compared to bare Mo6S8 at temperatures higher than 1200 K (see Fig. S5†); however, the production of DMS is lower over the whole temperature range. The reason behind the crossover in activity can be seen in the coverage of the active sites (Fig. S6†) where the number of free sites becomes larger on K–Mo6S8 as compared to that on Mo6S8 at 1300 K. This in turn is due to the weaker adsorption energy of S* on K–Mo6S8 as compared to Mo6S8. Ni–Mo6S8 and Cl–Mo6S8 are less active with respect to both methanethiol and DMS and also here the reason can be found in the coverage of the active site, as both clusters bind S* too strongly (Fig. S6†), which thereby poisons the active site and prevents the reaction from proceeding. It is possible that the high coverage of S* will lead to higher S-containing clusters, i.e., Mo6Sx, x > 8; however, this remains as a challenge for future investigation.
 | ||
| Fig. 3 The turnover frequency as a function of temperature for the direct (dashed line and square point) and stepwise (solid line and circles) formation of methanethiol and DMS (dashed line and circles) for the bare Mo6S8 (blue), K–Mo6S8 (yellow), Ni–Mo6S8 (red) and Cl–Mo6S8 (green). | ||
In contrast to a recent study44 of the formation of methanethiol on bare and promoted Mo6S8, diffusion is included in the reaction mechanism but it is treated parametrically with an energy barrier of 0.05 eV. The sensitivity of the actual diffusion barrier is evaluated by changing the barrier to 0.005 eV and 0.5 eV. The effect on the TOF and the selectivity (see Fig. S7 and S8†) is only minor, which indicates that the exact height of the diffusion barrier is not important, although the addition of the diffusion step is crucial for the reaction mechanisms. A degree of rate control analysis (shown in Fig. S9†) confirms that the diffusion steps are crucial for the reactions to happen.
Selectivity between methanethiol and DMS is an important property when deciding the catalyst formulation. The selectivity between DMS and methanethiol, calculated as the ratio between the TOF for DMS and the TOF for methanethiol, is shown in Fig. 4. It is clear from the results that what benefit the K-promoted cluster has in activity is the reverse in selectivity. In contrast, the Ni-promoted and Cl-promoted clusters display higher selectivity as compared to the bare Mo6S8. There is a noticeable difference between the two promoted clusters as Ni–Mo6S8 is several orders of magnitude higher in selectivity at low temperature, while Cl–Mo6S8 surpasses the bare Mo6S8 in selectivity first at a temperature of 1400 K (in fact, Cl–Mo6S8 even surpasses Ni–Mo6S8 at a temperature of 1500 K). The improved selectivity is closely connected with the poisoning effect of S* (Fig. S6†).
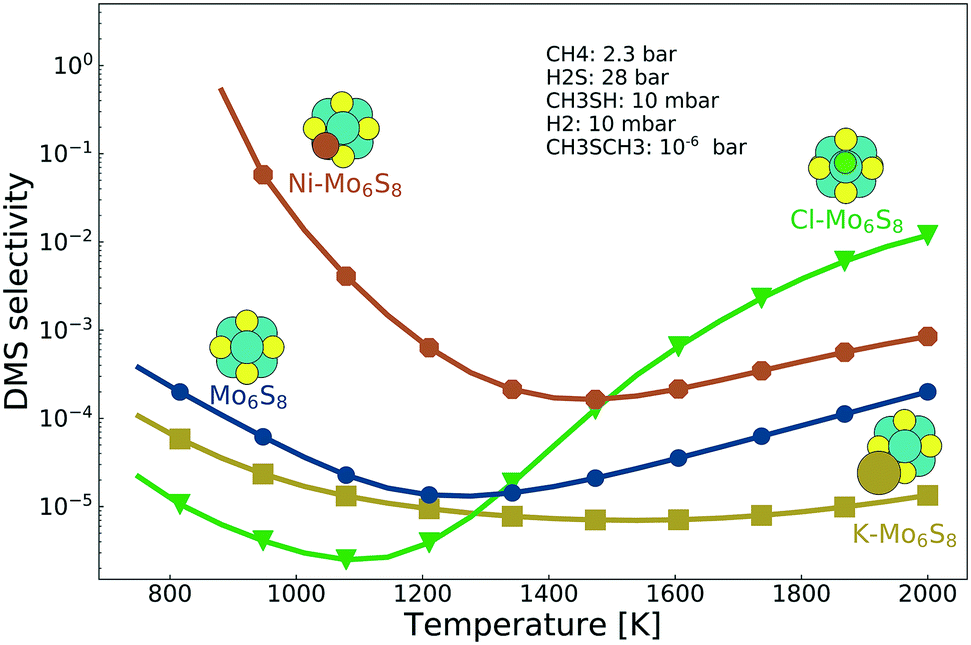 | ||
| Fig. 4 The selectivity between methanethiol and DMS as a function of temperature for the bare Mo6S8 (blue), K–Mo6S8 (yellow), Ni–Mo6S8 (red) and Cl–Mo6S8 (green). | ||
4 Conclusions
The activity and selectivity of CH4 and H2S to methanethiol and DMS are investigated by first-principles calculations using a microkinetic model. The proposed reaction mechanism includes a dual site (sulfur and molybdenum) and phenomenological diffusion between the dual sites. The reaction landscape involves several transition states and in particular there are two different pathways to form methanethiol (direct and stepwise). The stepwise pathway is part of the pathway to form DMS, which has adsorbed sulfur as a reaction intermediate.The promoters fall into two categories; either the activity of the main product, methanethiol, is increased (K–Mo6S8) or there is a loss in activity but with a gain in selectivity (Ni–Mo6S8 and Cl–Mo6S8). In the case of K and Cl, the effect is due to the electron-donor and electron-acceptor properties of the promoters. In the case of Ni the underlying reason is not as clear. Ni acts as an electron donor but still stabilizes the reaction intermediates to the extent that the dual site becomes poisoned. The results presented here highlight the balance between activity and selectivity when using promoters on Mo6S8 clusters and can thus guide future catalyst formulation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work by A. A. and A. H. was supported as part of the Röntgen-Ångström collaboration “Time-resolved in situ methods for design of catalytic sites within sustainable chemistry” and the Swedish Research Council. The Knut and Alice Wallenberg Foundation via the project Atomistic Design of Catalysts is also gratefully acknowledged. The microkinetic simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at C3SE. The work by W. T. and J. B. was supported as part of the UNCAGE-ME, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Basic Energy Sciences under Award No. DE-SC0012577. This research used resources of the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility operated under Contract No. DE-AC02-05CH11231.References
- V. Subramani, P. Sharma, L. Zhang and K. Liu, in Hydrogen and Syngas Production and Purification Technologies, John Wiley & Sons, Inc., 2009, pp. 14–126 Search PubMed.
- S. Lwin and I. E. Wachs, ACS Catal., 2014, 4, 2505–2520 CrossRef CAS.
- A. M. Rouhi, Chem. Eng. News, 2002, 80, 29 CrossRef.
- M. J. Pearson, Ind. Eng. Chem. Prod. Res. Dev., 1977, 16, 154–158 CrossRef CAS.
- W. Taifan and J. Baltrusaitis, Appl. Catal., B, 2016, 198, 525–547 CrossRef CAS.
- W. Taifan and J. Baltrusaitis, Catal. Sci. Technol., 2017, 7, 2919–2929 RSC.
- F. Lallemand, A. Rocher and N. Aimard, SPE International Oil & Gas Conference and Exhibition, Beijing, China, 2006, p. SPE 103802 Search PubMed.
- R. H. Hugman, E. H. Vidas and P. S. Springer, Chemical Composition of Discovered and Undiscovered Natural Gas in the Lower-48 United States. Project Summary. Final Report, 1 November 1988-31 March 1990, Technical Report PB-91-144600/XAB, Energy and Environmental Analysis, Inc., Arlington, VA (USA), 1990 Search PubMed.
- C. Huang and A. T-Raissi, J. Power Sources, 2007, 163, 645–652 CrossRef CAS.
- H. P. Mangalapally and H. Hasse, Energy Procedia, 2011, 4, 1–8 CrossRef CAS.
- H. P. Mangalapally, R. Notz, S. Hoch, N. Asprion, G. Sieder, H. Garcia and H. Hasse, Energy Procedia, 2009, 1, 963–970 CrossRef CAS.
- M. Lucquiaud and J. Gibbins, Chem. Eng. Res. Des., 2011, 89, 1553–1571 CrossRef CAS.
- C. Huang and A. T-Raissi, J. Power Sources, 2007, 163, 645–652 CrossRef CAS.
- P. Sadooghi and R. Rauch, Int. J. Hydrogen Energy, 2015, 40, 10418–10426 CrossRef CAS.
- S. K. Megalofonos and N. G. Papayannakos, Appl. Catal., A, 1996, 138, 39–55 CrossRef CAS.
- A. L. Martínez-Salazar, J. A. Melo-Banda, A. I. Reyes de la Torre, Y. Salazar-Cerda, M. A. Coronel-García, B. Portales Martínez, V. H. Martínez-Sifuentes, J. M. Domínguez-Esquivel and R. Silva Rodrigo, Int. J. Hydrogen Energy, 2015, 40, 17272–17283 CrossRef.
- F. Galindo-Hernández, J. M. Domínguez and B. Portales, J. Power Sources, 2015, 287, 13–24 CrossRef.
- F. Q. Miao and E. J. Erekson, Method for Direct Production of Carbon Disulfide and Hydrogen from Hydrocarbons and Hydrogen Sulfide Feedstock, Technical Report US Pat., A9018957, Federal Energy Technology Center, Pittsburgh, PA (US), 1998 Search PubMed.
- S. K. Megalofonos and N. G. Papayannakos, Appl. Catal., A, 1997, 165, 249–258 CrossRef CAS.
- S. K. Megalofonos and N. G. Papayannakos, Int. J. Hydrogen Energy, 1991, 16, 319–327 CrossRef CAS.
- Y. Li, X. Yu, Q. Guo, Z. Dai, G. Yu and F. Wang, Energy Procedia, 2017, 142, 1065–1070 CrossRef CAS.
- O. Y. Gutiérrez, L. Zhong, Y. Zhu and J. A. Lercher, ChemCatChem, 2013, 5, 3249–3259 CrossRef.
- O. Y. Gutiérrez, C. Kaufmann and J. A. Lercher, ChemCatChem, 2011, 3, 1480–1490 CrossRef.
- O. Y. Gutiérrez, C. Kaufmann and J. A. Lercher, ACS Catal., 2011, 1, 1595–1603 CrossRef.
- O. Y. Gutiérrez, C. Kaufmann, A. Hrabar, Y. Zhu and J. A. Lercher, J. Catal., 2011, 280, 264–273 CrossRef.
- J. Baltrusaitis, C. de Graaf, R. Broer and E. V. Patterson, ChemPhysChem, 2013, 14, 3960–3970 CrossRef CAS PubMed.
- C. D. Chang and A. J. Silvestri, J. Catal., 1977, 47, 249–259 CrossRef CAS.
- S. A. Butter, A. T. Jurewicz and W. W. Kaeding, Conversion of alcohols, mercaptans, sulfides, halides and/or amines, US Pat. 3894107, 1975 Search PubMed.
- A. V. Mashkina, V. R. Grunvald, V. I. Nasteka, B. P. Borodin, V. N. Yakovleva and L. N. Khairulina, React. Kinet. Catal. Lett., 1990, 41, 357–362 CrossRef CAS.
- V. Hulea, E. Huguet, C. Cammarano, A. Lacarriere, R. Durand, C. Leroi, R. Cadours and B. Coq, Appl. Catal., B, 2014, 144, 547–553 CrossRef CAS.
- E. Huguet, B. Coq, R. Durand, C. Leroi, R. Cadours and V. Hulea, Appl. Catal., B, 2013, 134–135, 344–348 CrossRef CAS.
- C. Cammarano, E. Huguet, R. Cadours, C. Leroi, B. Coq and V. Hulea, Appl. Catal., B, 2014, 156–157, 128–133 CrossRef CAS.
- Q.-M. Li, M. Zhang, C.-M. Wang, Y.-A. Zhu, X.-G. Zhou and Z.-K. Xie, Mol. Catal., 2018, 446, 106–114 CrossRef CAS.
- J. Baltrusaitis, T. Bučko, W. Michaels, M. Makkee and G. Mul, Appl. Catal., B, 2016, 187, 195–203 CrossRef CAS.
- M. Reina, A. Martinez, C. Cammarano, C. Leroi, V. Hulea and T. Mineva, ACS Omega, 2017, 2, 4647–4656 CrossRef CAS PubMed.
- G. A. Olah, Acc. Chem. Res., 1987, 20, 422–428 CrossRef CAS.
- X. Zheng, L. Guo, W. Li, Z. Cao, N. Liu, Q. Zhang, M. Xing, Y. Shi and J. Guo, Mol. Catal., 2017, 439, 155–162 CrossRef CAS.
- J. Zhou, J. Zhou, N. Camillone and M. G. White, Phys. Chem. Chem. Phys., 2012, 14, 8105–8110 RSC.
- P. Liu, Y. Choi, Y. Yang and M. G. White, J. Phys. Chem. A, 2010, 114, 3888–3895 CrossRef CAS PubMed.
- C. Liu and P. Liu, ACS Catal., 2015, 5, 1004–1012 CrossRef CAS.
- M. J. Patterson, J. M. Lightstone and M. G. White, J. Phys. Chem. A, 2008, 112, 12011–12021 CrossRef CAS PubMed.
- J. M. Lightstone, M. J. Patterson and M. G. White, Chem. Phys. Lett., 2005, 413, 429–433 CrossRef CAS.
- P. Murugan, V. Kumar, Y. Kawazoe and N. Ota, Phys. Rev. A: At., Mol., Opt. Phys., 2005, 71, 063203 CrossRef.
- W. Taifan, A. A. Arvidsson, E. Nelson, A. Hellman and J. Baltrusaitis, Catal. Sci. Technol., 2017, 7, 3546–3554 RSC.
- T. J. Paskach, G. L. Schrader and R. E. McCarley, J. Catal., 2002, 211, 285–295 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- R. Dronskowski and P. E. Bloechl, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2013, 34, 2557–2567 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- A. Heyden, A. T. Bell and F. J. Keil, J. Chem. Phys., 2005, 123, 224101 CrossRef PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- H. P. Hratchian and H. B. Schlegel, J. Phys. Chem. A, 2002, 106, 165–169 CrossRef CAS.
- I. Chorkendorff and J. W. Niemantsverdriet, Concepts of Modern Catalysis and Kinetics, John Wiley & Sons, 2006 Search PubMed.
- S. R. Bahn and K. W. Jacobsen, Comput. Sci. Eng., 2002, 4, 56–66 CrossRef CAS.
- A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dułak, J. Friis, M. N. Groves, B. Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, K. Kaasbjerg, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schütt, M. Strange, K. S. Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng and K. W. Jacobsen, J. Phys.: Condens. Matter, 2017, 29, 273002 CrossRef PubMed.
- A. C. Luntz, in Chemical Bonding at Surfaces and Interfaces, 2008, pp. 143–255 Search PubMed.
- A. Hindmarsh, IMACS Transactions on Scientific Computation, 1983, vol. 1, pp. 55–64 Search PubMed.
- E. Hairer, G. Wanner and S. P. Nørsett, Solving Ordinary Differential Equations I, Springer Berlin Heidelberg, Berlin, Heidelberg, 1993, vol. 8 Search PubMed.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD, 20899, DOI:10.18434/T4D303, (retrieved February 12, 2019).
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- E. Sanville, S. D. Kenny, R. Smith and G. Henkelman, J. Comput. Chem., 2007, 28, 899–908 CrossRef CAS PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- M. Yu and D. R. Trinkle, J. Chem. Phys., 2011, 134, 064111 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A zip file with all structures and figures of the reaction mechanism for the K-, Ni-, and Cl-promoted clusters, a free energy landscape, a comparison of the TOF for all clusters, tables with kinetic information, the steady-state coverages, the effect of the diffusion barrier on the TOF and selectivity, a degree of rate control analysis for all clusters, and the projected crystal orbital Hamiltonian populations. See DOI: 10.1039/c9cy00375d |
| This journal is © The Royal Society of Chemistry 2019 |